
One-Pot and Catalyst-Free Transformation of N-Protected 1-Amino-1-Ethoxyalkylphosphonates into Bisphosphonic Analogs of Protein and Non-Protein α-Amino Acids


Abstract
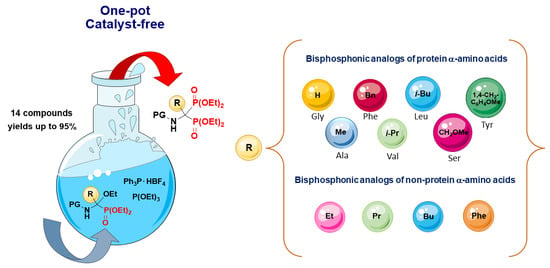
:
1. Introduction
2. Results and Discussion
2.1. Optimization of Conditions for the Synthesis of α-Ethoxy Derivatives of Phosphorus Analogs of α-Amino Acids
2.2. Development of an Optimized One-Pot Procedure for the Synthesis of Bisphosphonate Analogs of α-Amino Acids
2.3. Scope of the Reaction
3. Materials and Methods
3.1. General Information
3.2. Substrate Synthesis
- Ethyl N-(benzyloxycarbonyl)acetimidate (2a). Pale yellow oil; 90% yield (1.592 g). 1H-NMR (400 MHz, CDCl3): δ 7.40–7.32 (m, 5H), 5.19 (s, 2H), 4.16 (q, J = 7.2 Hz, 2H), 2.05 (s, 3H) 1.28 (t, J = 7.2 Hz, 3H). 13C-NMR (100 MHz, CDCl3): δ 167.9, 161.5, 135.9, 128.5, 128.4, 128.3, 68.1, 63.2, 18.4, 13.8. HRMS (ESI) m/z: calcd for C12H15NO3Na [M + Na]+ 244.0950, found 244.0951.
- Ethyl N-(pivaloyl)acetimidate (2b) [41]. Pale yellow oil; 80% yield (1.095 g). 1H-NMR (400 MHz, CDCl3): δ 4.12 (q, J = 7.0 Hz, 2H), 1.98 (s, 3H), 1.29 (t, J = 7.2 Hz, 3H), 1.18 (s, 9H). 13C-NMR (100 MHz, CDCl3): δ 191.7, 161.3, 62.5, 41.4, 27.1, 18.0, 14.0. HMRS (ESI) m/z: calcd for C9H18NO2 [M + H]+ 172.1338, found 172.1343.
- EthylN-(benzyloxycarbonyl)formimidate (2c). Pale yellow oil; 34% yield (565 mg). 1H-NMR (400 MHz, CDCl3): δ 8.45 (s, 1H), 7.54–7.28 (m, 5H), 5.12 (s, 2H), 4.34 (q, J = 7.0 Hz, 2H), 1.34 (t, J = 7.0 Hz, 3H). 13C-NMR (100 MHz, CDCl3): δ 167.1, 161.9, 135.6, 128.53, 128.51, 128.3, 68.4, 64.5, 13.8. HRMS (ESI) m/z: calcd for C11H14NO3 [M + H]+ 208.0974, found 208.0977.
- EthylN-(benzyloxycarbonyl)-2-phenylacetimidate (2d). Pale yellow oil; 88% yield (2.095 g). 1H-NMR (400 MHz, CDCl3): δ 7.35–7.19 (m, 10H), 5.13 (s, 2H), 4.16 (q, J = 7.2 Hz, 2H), 3.68 (s, 2H), 1.25 (t, J = 7.2 Hz, 3H). 13C-NMR (100 MHz, CDCl3): δ 167.9, 161.1, 135.7, 134.2, 129.2, 128.6, 128.5, 128.3, 127.0, 68.2, 63.5, 38.8, 13.8. HRMS (ESI) m/z: calcd for C18H20NO3 [M + H]+ 298.1443, found 298.1443.
- Ethyl N-(acetyl)-2-phenylacetimidate (2e) [42]. Pale yellow oil; 85% yield (1.389 g). 1H-NMR (400 MHz, CDCl3): δ 7.26–7.53 (m, 5H), 4.10 (q, J = 7.0 Hz, 2H), 3.66 (s, 2H), 1.96 (s, 3H), 1.17 (t, J = 7.2 Hz, 3H). 13C-NMR (100 MHz, CDCl3): δ183.4, 161.1, 134.5, 129.3, 128.6, 127.1, 62.9, 38.3, 26.3, 13.8. HRMS (ESI) m/z: calcd for C12H16NO2 [M + H]+ 206.1181, found 206.1184.
- Ethyl N-(benzyloxycarbonyl)propanimidate (2f). Pale yellow oil; 80% yield (1.507 g). 1H-NMR (400 MHz, CDCl3): δ 7.42–7.30 (m, 5H), 5.19 (s, 2H), 4.15 (q, J = 7.2 Hz, 2H), 2.36 (q, J = 7.6 Hz, 2H), 1.28 (t, J = 7.2 Hz, 3H), 1.12 (t, J = 7.6 Hz, 3H). 13C-NMR (100 MHz, CDCl3): δ 170.7, 161.4, 135.9, 128.5, 128.4, 128.3, 68.1, 63.0, 26.1, 13.8, 10.5. HRMS (ESI) m/z: calcd for C13H17NO3Na [M + Na]+ 258.1106, found 258.1108.
- Ethyl N-(benzyloxycarbonyl)butanimidate (2g) [43]. Pale yellow oil; 95% yield (1.899 g). 1H-NMR: (400 MHz, CDCl3): δ 7.42–7.28 (m, 5H), 5.19 (s, 2H), 4.15 (q, J = 7.2 Hz, 2H), 2.30 (t, J = 7.6 Hz, 2H), 1.57 (sext, J = 7.6 Hz, 2H), 1.28 (t, J = 7.2 Hz, 3H), 0.86 (t, J = 7.4 Hz, 3H). 13C-NMR: (100 MHz, CDCl3): δ 169.8, 161.3, 135.9, 128.5, 128.3, 68.0, 62.9, 34.4, 19.5, 13.8, 13.6. HMRS (ESI) m/z: calcd for C14H19NO3Na [M + Na]+ 272.1263, found 272.1264.
- Ethyl N-(benzyloxycarbonyl)-2-methylpropanimidate (2h). Pale yellow oil; 79% yield (1.580 g). 1H-NMR (400 MHz, CDCl3): δ 7.41–7.30 (m, 5H), 5.18 (s, 2H), 4.12 (q, J = 7.2 Hz, 2H), 2.72 (sept, J = 7.0 Hz, 1H), 1.27 (t, J = 7.2 Hz, 3H), 1.12 (d, J = 6.8 Hz, 6H).13C-NMR (100 MHz, CDCl3): δ 172.2, 161.2, 136.0, 128.5, 128.4, 128.2, 68.1, 62.9, 32.8, 19.5, 13.7. HMRS (ESI) m/z: calcd for C14H20NO3 [M + H]+ 250.1443 found 250.1445.
- Ethyl N-(benzyloxycarbonyl)pentanimidate (2i) [43]. Yellow oil; 91% yield (1.912 g). 1H-NMR (400 MHz, CDCl3): δ 7.42–7.29 (m, 5H), 5.18 (s, 2H), 4.14 (q, J = 7.2 Hz, 2H), 2.32 (t, J = 7.8 Hz, 2H), 1.53 (qu, J = 7.6 Hz, 2H), 1.28 (t, J = 7.2 Hz, 3H), 1.25 (sext, J = 7.2 Hz, 2H) 0.85 (t, J = 7.2 Hz, 3H) 13C-NMR (100 MHz, CDCl3): δ 170.0, 161.4, 135.9, 128.5, 128.3, 68.1, 63.0, 32.4, 28.2, 22.3, 13.9, 13.6. HMRS (ESI) m/z: calcd for C15H21NO3Na [M + Na]+ 286.1419, found 286.1417.
- Ethyl N-(benzyloxycarbonyl)-3-methylbutanimidate (2j). Yellow oil; 99% yield (2.083 g). 1H-NMR (400 MHz, CDCl3): δ 7.42–7.26 (m, 5H), 5.18 (s, 2H), 4.15 (q, J = 7.0 Hz, 2H), 2.02 (d, J = 7.2 Hz, 2H), 1.97 (sept, J = 6.8 Hz, 1H). 1.28 (t, J = 7.2 Hz, 3H), 0.85 (d, J = 6.8 Hz, 6H). 13C-NMR (100 MHz, CDCl3): δ 169.1, 161.3, 135.9, 128.6, 128.5, 128.4, 68.0, 62.9, 41.1, 26.3, 22.2, 13.9. HMRS (ESI) m/z: calcd for C15H22NO3 [M + H]+ 264.1600, found 264.1599.
- Ethyl N-(acetyl)-3-methylbutanimidate (2k). Pale yellow oil; 99% yield (1.358 g). 1H-NMR (400 MHz, CDCl3): δ 4.09 (q, J = 7.1 Hz, 2H), 2.20 (d, J = 7.2 Hz, 2H), 2.16 (s, 3H) 2.05 (m, 1H), 1.28 (t, J = 7.0 Hz, 3H), 0.95 (d, J = 6.8 Hz, 6H). 13C-NMR (100 MHz, CDCl3): δ 183.4, 162.2, 62.4, 41.0, 26.7, 26.1, 22.3, 13.9. HRMS (ESI) m/z: calcd for C9H18NO2 [M + H]+ 172.1338, found 172.1343.
- Ethyl N-(benzyloxycarbonyl)-2-methoxyacetimidate (2l). Pale yellow oil; 74% yield (1.481 g). 1H-NMR (400 MHz, CDCl3): δ 7.42–7.30 (m, 5H), 5,17 (s, 2H), 4.20 (q, J = 7.2 Hz, 2H), 4.10 (s, 2H), 3.24 (s, 3H), 1.30 (t, J = 7.2 Hz, 3H). 13C-NMR (100 MHz, CDCl3): δ 164.4, 160.6, 135.9, 128.7, 128.4, 128.2, 69.7, 68.0, 63.5, 59.6, 13.8. HMRS (ESI) m/z: calcd for C13H17NO4Na [M + Na]+ 274.1055, found 274.1057.
- EthylN-(benzyloxycarbonyl)benzimidate (2m). Pale yellow oil; 58% yield (1.316 g). 1H-NMR (400 MHz, CDCl3): δ 7.55–7.50 (m, 5H), 5.12 (s, 2H), 4.34 (q, J = 7.0 Hz, 2H), 1.34 (t, J = 7.0 Hz, 3H). 13C-NMR (100 MHz, CDCl3): δ 167.1, 161.9, 135.6, 128.53, 128.51, 128.3, 68.4, 64.5, 13.8. HRMS (ESI) m/z: calcd for C17H18NO3 [M + H]+ 284.1287, found 284.1290.
- Ethyl N-(benzyloxycarbonyl)-2-(4-methoxyphenyl)acetimidate (2n). Yellow oil; 96% yield (2.519 g). 1H-NMR (400 MHz, CDCl3): δ 7.36–7.32 (m, 5H), 7.11–7.09 (m, 2H), 6.80–6.76 (m, 2H), 5.13 (s, 2H), 4.15 (q, J = 7.2 Hz, 2H), 3.77 (s, 3H), 3.61 (s, 2H), 1.25 (t, J = 7.2 Hz, 3H). 13C-NMR (100 MHz, CDCl3): δ 168.06, 161.19, 158.62, 135.76, 130.26, 128.57, 128.51, 128.31, 113.90, 68.17, 63.42, 55.20, 37.96, 13.79. HMRS (ESI) m/z: calcd for C19H22NO4 [M + H]+ 328.1549, found 328.1546.
- Diethyl 1-(N-benzyloxycarbonylamino)-1-ethoxyethylphosphonate (3a). White solid; 94% yield (338 mg); mp 69.4 to 71.0 °C. 1H-NMR (400 MHz, CDCl3): δ 7.39–7.30 (m, 5H), 5.71 (br d, J = 7.9 Hz, 1H), 5.09 (ABq, J = 12.2 Hz, 2H), 4.24–4.12 (m, 4H)a, 3.67–3.60 (m, 2H), 1.90 (d, J = 15.0 Hz, 3H), 1.34 (t, J = 7.2 Hz, 3H) and 1.32 (t, J = 7.2 Hz, 3H)b, 1.17 (t, J = 7.0, 3H). 13C-NMR (100 MHz, CDCl3): δ 154.6 (d, J = 16.4 Hz), 136.2, 128.5, 128.2, 128.1, 84.4 (d, J = 196.8 Hz), 66.7, 63.8 (d, J = 6.9 Hz), 63.4 (d, J = 6.9 Hz), 58.5 (d, J = 8.0 Hz), 18.9, 16.4 (d, J = 5.3 Hz), 15.4. 31P-NMR (162 MHz, CDCl3): δ 18.5. IR (ATR): 3203, 1717, 1541, 1224, 1047, 960, 750 cm−1. HMRS (ESI) m/z: calcd for C16H27NO6P [M + H]+ 360.1576, found 360.1578. aOverlapping signals of P(O)(OCH2CH3)2 groups. bOverlapping signals of P(O)(OCH2CH3)2 groups.
- Diethyl 1-(N-pivaloylamino)-1-ethoxyethylphosphonate (3b). Colorless oil; 74% yield (230 mg). 1H-NMR (400 MHz, CDCl3): δ 6.41 (br d, J = 7.5 Hz, 1H), 4.26–4.14 (m, 4H)a, 3.67–3.59 (m, 2H), 1.95 (d, J = 15.4 Hz, 3H), 1.35 (t, J = 7.0 Hz, 3H) and 1.34 (t, J = 7.0 Hz, 3H)b, 1.22 (s, 9H), 1.19 (t, J = 7.0 Hz, 3H). 13C-NMR (100 MHz, CDCl3): δ 178.8 (d, J = 9.9 Hz), 84.9 (d, J = 194.5 Hz), 63.9 (d, J = 6.9 Hz), 63.1 (d, J = 6.9 Hz), 58.6 (d, J = 9.5 Hz), 39.9, 27.5, 18.7, 16.5 (d, J = 5.3 Hz), 16.4 (d, J = 5.4 Hz), 15.5. 31P-NMR (162 MHz, CDCl3): δ 19.1. IR (ATR): 3283, 1676, 1519, 1244, 1021, 958 cm−1. HMRS (ESI) m/z: calcd for C13H29NO5P [M + H]+ 310.1783, found 310.1790. aOverlapping signals of P(O)(OCH2CH3)2 groups. bOverlapping signals of P(O)(OCH2CH3)2 groups.
- Diethyl 1-(N-benzyloxycarbonylamino)-1-ethoxymethylphosphonate (3c). Colorless oil; 93% yield (321 mg). 1H-NMR (400 MHz, CDCl3): δ 7.39–7.32 (m, 5H), 5.65 (dd, J = 10.8, 4.3 Hz, 1H), 5.24 (dd, J1 = 10.8 Hz, J2 = 9.3 Hz, 1H), 5.15 (ABq, J = 12.2 Hz, 2H), 4.24–4.11 (m, 4H)a, 3.81–3.74 (m, 1H), 3.65–3.57 (m, 1H), 1.33 (t, J = 7.2 Hz, 3H) and 1.29 (t, J = 7.0 Hz, 3H)b, 1.22 (t, J = 7.2 Hz, 3H). 13C-NMR (100 MHz, CDCl3): δ 156.0 (d, J = 12.2 Hz), 135.9, 128.6, 128.3, 128.1, 77.4 (d, J = 201.1 Hz), 67.4, 65.4 (d, J = 12.9 Hz), 63.7 (d, J = 6.5 Hz), 63.2 (d, J = 6.9 Hz), 16.38 (d, J = 5.3 Hz) and 16.36 (d, J = 5.3 Hz)a, 14.9. 31P-NMR (162 MHz, CDCl3): δ 15.9. IR (ATR): 3221, 1720, 1526, 1229, 1026, 977, 752 cm−1. HMRS (ESI) m/z: calcd for C15H25NO6P [M + H]+ 346.1419, found 346.1426. aOverlapping signals of P(O)(OCH2CH3)2 groups. bOverlapping signals of P(O)(OCH2CH3)2 groups.
- Diethyl 1-(N-benzyloxycarbonylamino)-1-ethoxy-2-phenylethylphosphonate (3d). White solid; 88% yield (383 mg); mp 78.2 to 79.5 °C. 1H-NMR (400 MHz, CDCl3): δ 7.38–7.17 (m, 10H), 5.81 (br d, J = 10.6 Hz, 1H), 5.14 (ABq, J = 12 Hz, 2H), 4.09–3.68 (m, 7H)a, 3.41 (dd, J1 = 14.4 Hz, J2 = 11.1 Hz, 2H), 1.23 (t, J = 7.2 Hz, 3H) and 1.21 (t, J = 7.2 Hz, 3H)b, 1.05 (t, J = 7.0 Hz, 3H). 13C-NMR (100 MHz, CDCl3): δ 154.6 (d, J = 16.4 Hz), 136.1, 135.6 (d, J = 3.6 Hz), 131.2, 128.5, 128.24, 128.19, 127.6, 126.5, 87.1 (d, J = 186.5 Hz), 66.8, 63.2 (d, J = 7.2 Hz), 62.9 (d, J = 7.2 Hz), 59.4 (d, J = 4.6 Hz), 39.1 (d, J = 2.9 Hz), 16.3 (d, J = 5.8 Hz), 16.0 (d, J = 6.1 Hz), 15.2. 31P-NMR (162 MHz, CDCl3): δ 17.5. IR (ATR): 3204, 1726, 1548, 1245, 1019, 959, 754, 701 cm−1. HMRS (ESI) m/z: calcd for C22H30NO6NaP [M + Na]+ 458.1708, found 458.1704. aOverlapping signals of CαCH2C6H5 and P(O)(OCH2CH3)2 groups. bOverlapping signals of P(O)(OCH2CH3)2 groups.
- Diethyl 1-(N-acetylamino)-1-ethoxy-2-phenylethylphosphonate (3e). White solid; 53% yield (183 mg); mp 92.8 to 94.3 °C. 1H-NMR (400 MHz, CDCl3): δ 7.33–7.20 (m, 5H), 6.12 (br d, J = 11.4 Hz, 1H), 4.12–3.92 (m, 5H)a, 3.87–3.71 (m, 2H), 3.39 (dd, J1 = 14.5 Hz, J2 = 9.1 Hz, 1H), 2.04 (s, 3H), 1.29 (t, J = 7.0 Hz, 3H), 1.23 (t, J = 7.0 Hz, 3H), 1.10 (t, J = 7.0 Hz, 3H). 13C-NMR (100 MHz, CDCl3): δ 170.3 (d, J = 9.2 Hz), 135.6 (d, J = 4.0 Hz), 131.1, 127.7, 126.6, 87.6 (d, J = 185.9 Hz), 63.4 (d, J = 7.2 Hz), 62.8 (d, J = 7.2 Hz), 59.9 (d, J = 5.3 Hz), 38.9, 24.5, 16.4 (d, J = 6.1 Hz), 16.1 (d, J = 6.1 Hz), 15.2. 31P-NMR (162 MHz, CDCl3): δ 17.9. IR (ATR): 3185, 1670, 1548, 1218, 1029, 962, 749, 696 cm−1. HMRS (ESI) m/z: calcd for C16H27NO5P [M + H]+ 344.1627, found 344.1627. aOverlapping signals of CαCH2C6H5 and P(O)(OCH2CH3)2 groups.
- Diethyl 1-(N-benzyloxycarbonylamino)-1-ethoxypropylphosphonate (3f). White solid; 82% yield (306 mg); mp 59.4 to 60.8 °C. 1H-NMR (400 MHz, CDCl3): δ 7.38–7.30 (m, 5H), 5.77 (d, J = 8.0 Hz, 1H), 5.09 (ABq, J = 10.0 Hz, 2H), 4.24–4.11 (m, 4H)a, 3.68–3.56 (m, 2H), 2.61 (ddq, J1 = 23.4 Hz, J2 = 15.0 Hz, J3 = 7.5 Hz, 1H), 2.25 (tq, J1 = 14.9 Hz, J2 = 7.5 Hz, 1H), 1.33 (t, J = 7.0 Hz, 3H) and 1.32 (t, J = 7.0 Hz, 3H)b, 1.17 (t, J = 7.0 Hz, 3H), 1.02 (t, J = 7.6 Hz, 3H), 13C-NMR (100 MHz, CDCl3): δ 154.4 (d, J = 16.2), 136.2, 128.5, 128.3, 128.1, 87.7 (d, J = 189.5 Hz), 66.7, 63.6 (d, J = 7.2 Hz), 63.1 (d, J = 7.1 Hz), 58.4 (d, J = 7.2 Hz), 25.2, 16.4 (d, J = 5.7 Hz), 15.3, 8.5 (d, J = 2.1 Hz). 31P-NMR (162 MHz, CDCl3): δ 19.0. IR (ATR): 3253, 1231, 1024, 773 cm−1. HMRS (ESI) m/z: calcd for C17H28NO6NaP [M + Na]+ 396.1552, found 396.1545. aOverlapping signals of P(O)(OCH2CH3)2 groups. bOverlapping signals of P(O)(OCH2CH3)2 groups.
- Diethyl 1-(N-benzyloxycarbonylamino)-1-ethoxybutylphosphonate (3g). Colorless oil; 68% yield (263 mg). 1H-NMR (400 MHz, CDCl3): δ 7.39–7.31 (m, 5H), 5.78 (br d, J = 8.0 Hz, 1H), 5.08 (ABq, J = 12.2 Hz, 2H), 4.23–4.11 (m, 4H)a, 3.68–3.55 (m, 2H), 2.61–2.47 (m, 1H), 2.23–2.11 (m, 1H), 1.54–1.44 (m, 2H), 1.33 (t, J = 7.0 Hz, 3H) and 1.32 (t, J1 = 7.0 Hz, 3H)b, 1.16 (t, J = 7.0 Hz, 3H), 0.93 (t, J = 7.4 Hz, 3H). 13C-NMR (100 MHz, CDCl3): δ 154.4 (d, J = 16.1 Hz), 136.2, 128.5, 128.1, 128.0, 87.3 (d, J = 189.7 Hz), 66.6, 63.6 (d, J = 7.1 Hz), 63.1 (d, J = 7.0 Hz), 58.4 (d, J = 7.4 Hz), 34.4, 17.2 (d, J = 2.0 Hz), 16.4 (d, J = 5.5 Hz), 15.3, 14.4. 31P-NMR (162 MHz, CDCl3): δ 19.0. IR (ATR): 2976, 1737, 1499, 1240, 1019, 969, 742 cm−1. HMRS (ESI) m/z: calcd for C18H30NO6NaP [M + Na]+ 410.1708, found 410.1706. aOverlapping signals of P(O)(OCH2CH3)2 groups. bOverlapping signals of P(O)(OCH2CH3)2 groups.
- Diethyl 1-(N-benzyloxycarbonylamino)-1-ethoxy-2-methylpropylphosphonate (3h). White solid; 53% yield (205 mg); mp 54.2 to 55.5 °C. 1H-NMR (400 MHz, CDCl3): δ 7.37–7.31 (m, 5H), 5.91 (br d, J = 10.8 Hz, 1H), 5.09 (ABq, J = 12.2 Hz, 2H), 4.21–4.12 (m, 4H)a, 3.68–3.56 (m, 2H), 3.19 (dsept, J1 = 32.6 Hz, J2 = 7.0 Hz 1H), 1.33 (t, J = 7.0 Hz, 3H), 1.15 (t, J = 7.0 Hz, 3H), 1.12 (d, J = 6.8 Hz, 3H), 1.06 (d, J = 6.8 Hz, 3H). 13C-NMR (100 MHz, CDCl3): δ 154.6 (d, J = 18.2 Hz), 136.3, 128.5, 128.2, 128.1, 90.2 (d, J = 185.8 Hz), 66.7, 63.4 (d, J = 7.2 Hz), 62.9 (d, J = 7.6 Hz), 58.6 (d, J = 6.5 Hz), 31.4, 17.7 (d, J = 3.1 Hz), 17.5, 16.4 (d, J = 5.3 Hz), 15.3. 31P-NMR (162 MHz, CDCl3): δ 19.7. IR (ATR): 3218, 1723, 1544, 1239, 1023, 976, 745 cm−1. HMRS (ESI) m/z: calcd for C18H31NO6P [M + H]+ 388.1889, found 388.1890. aOverlapping signals of P(O)(OCH2CH3)2 groups.
- Diethyl 1-(N-benzyloxycarbonylamino)-1-ethoxypentylphosphonate (3i). White solid; 54% yield (217 mg); mp 53.1 to 54.7 °C. 1H-NMR (400 MHz, CDCl3): δ 7.36–7.28 (m, 5H), 5.78 (br d, J = 8.4 Hz, 1H), 5.08 (ABq, J = 12.2 Hz, 2H), 4.23–4.11 (m, 4H)a, 3.68–3.56 (m, 2H), 2.61–2.47 (m, 1H), 2.26–2.14 (m, 1H), 1.49–1.41 (m, 2H), 1.35–1.26 (m, 2H), 1.33 (t, J = 7.0 Hz, 3H), 1.32 (t, J = 7.0 Hz, 3H), 1.16 (t, J = 7.0 Hz, 3H), 0.91 (t, J = 7.2 Hz, 3H). 13C-NMR (100 MHz, CDCl3): δ 154.3 (d, J = 16.1 Hz), 136.2, 128.4, 128.1, 128.0, 87.3 (d, J = 189.5 Hz), 66.6, 63.5 (d, J = 7.2 Hz) and 63.1 (d, J = 7.2 Hz), 58.3 (d, J = 7.2 Hz), 31.9, 25.8 (d, J = 1.9 Hz), 22.9, 16.3 (d, J = 5.6 Hz), 15.2, 13.9. 31P-NMR (162 MHz, CDCl3): δ 19.0. IR (ATR): 2973, 1722, 1545, 1240, 1022, 985, 754 cm−1. HMRS (ESI) m/z: calcd for C19H32NO6NaP [M + Na]+ 424.1865, found 424.1863. aOverlapping signals of CαCH2CH2CH2CH3 and P(O)(OCH2CH3)2 groups.
- Diethyl 1-(N-benzyloxycarbonylamino)-1-ethoxy-3-methylbutylphosphonate (3j). Colorless oil; 32% yield (135.7 mg). 1H-NMR (400 MHz, CDCl3): δ 7.36–7.31 (m, 5H), 5.88 (br d, J = 8.8 Hz, 1H), 5.08 (ABq, J = 12.2 Hz, 2H), 4.25–4.11 (m, 4H)a, 3.61 (qd, J = 7.0, 1.0 Hz, 2H), 2.62 (ddd, J1 = 26.3 Hz, J2 = 15.0 Hz, J3 = 7.9 Hz, 1H), 2.09–2.03 (m, 1H), 1.96 (ddd, J1 = 15.0 Hz, J2 = 9.1 Hz, J3 = 4.3 Hz, 1H), 1.34 (t, J = 7.2 Hz, 3H) and 1.32 (t, J = 7.2 Hz, 3H)b, 1.16 (t, J = 7.0 MHz, 3H), 1.00 (d, J = 6.8 Hz, 3H), 0.94 (d, J = 6.8 Hz, 3H). 13C-NMR (100 MHz, CDCl3): δ 154.4 (d, J = 16.7 Hz), 136.2, 128.5, 128.14, 128.06, 87.9 (d, J = 188.2 Hz), 66.6, 63.8 (d, J = 7.2 Hz), 62.9 (d, J = 7.2 Hz), 58.5 (d, J = 7.2 Hz), 40.3, 24.5 (d, J = 4.6 Hz), 23.2, 16.40 (d, J = 5.7 Hz) and 16.38 (d, J = 5.7 Hz)b, 15.1. 31P-NMR (162 MHz, CDCl3): δ 19.2. IR (ATR): 3248, 1739, 1499, 1242, 1021, 967, 749 cm−1. HMRS (ESI) m/z: calcd for C19H32NO6NaP [M + Na]+ 424.1865, found 424.1862. aOverlapping signals of P(O)(OCH2CH3)2 groups. aOverlapping signals of P(O)(OCH2CH3)2 groups. bOverlapping signals of P(O)(OCH2CH3)2 groups.
- Diethyl 1-(N-acetylamino)-1-ethoxy-3-methylbutylphosphonate (3k). White solid; 64% yield (198 mg); mp 58.6 to 59.7 °C. 1H-NMR (400 MHz, CDCl3): δ 6.30 (br d, J = 7.6 Hz, 1H), 4.28–4.13 (m, 4H)a, 3.70–3.58 (m, 2H), 2.81–2.70 (m, 1H), 1.98–1.92 (m, 2H), 2.02 (s, 3H), 1.35 (t, J = 7.0 Hz, 3H) and 1.34 (t, J = 7.0 Hz, 3H)b, 1.18 (t, J = 7.2 Hz, 3H), 1.00 (d, J = 6.7 Hz, 3H), 0.95 (d, J = 6.7 Hz, 3H). 13C-NMR (100 MHz, CDCl3): δ 170.1 (d, J = 12.9 Hz), 88.8 (d, J = 187.3 Hz), 64.1 (d, J = 7.2 Hz), 62.7 (d, J = 7.2 Hz), 59.0 (d, J = 8.0 Hz), 39.7, 24.9, 24.7 (d, J = 3.0 Hz), 24.5, 23.1, 16.43 (d, J = 6.1 Hz) and 16.40 (d, J = 5.3 Hz)b, 15.1. 31P-NMR (162 MHz, CDCl3): δ 19.6. IR (ATR): 3197, 1670, 1541, 1224, 1070, 956, 759 cm−1. HMRS (ESI) m/z: calcd for C13H29NO5P [M + H]+ 310.1783, found 310.1776. aOverlapping signals of P(O)(OCH2CH3)2 groups. aOverlapping signals of P(O)(OCH2CH3)2 groups. bOverlapping signals of P(O)(OCH2CH3)2 groups.
- Diethyl 1-(N-benzyloxycarbonylamino)-1-ethoxy-2-methoxyethylphosphonate (3l). White solid; 91% yield (354 mg); mp 60.1 to 62.1 °C. 1H-NMR (400 MHz, CDCl3): δ 7.37–7.30 (m, 5H), 5.97 (br d, J = 11.0 Hz, 1H), 5.10 (ABq, J = 12.0 Hz, 2H), 4.26–4.15 (m, 5H)a, 3.92 (dd, J1 = 10.6 Hz, J2 = 9.3 Hz, 2H), 3.75–3.61 (m, 2H), 3.41 (s, 3H), 1.34 (td, J1 = 7.2 Hz, J2 = 0.4 Hz, 3H) and 1.33 (td, J1 = 7.2 Hz, J2 = 0.4 Hz, 3H)b, 1.18 (t, J = 7.2 Hz, 3H). 13C-NMR (100 MHz, CDCl3): δ 154.4 (d, J = 14.3 Hz), 136.1, 128.5, 128.2, 128.1, 86.0 (d, J = 188.8 Hz), 72.5, 67.0, 63.6 (d, J = 7.2 Hz), 63.5 (d, J = 6.9 Hz), 59.4, 59.3 (d, J = 6.0 Hz), 16.4 (d, J = 5.7 Hz), 15.4. 31P-NMR (162 MHz, CDCl3): δ 17.9. IR (ATR): 3227, 2985, 1733, 1528, 1245, 1027, 987, 758 cm−1. HMRS (ESI) m/z: calcd for C17H28NO7NaP [M + Na]+ 412.1501, found 412.1494. aOverlapping signals of CαCH2OMe and P(O)(OCH2CH3)2 groups.
- Diethyl 1-(N-benzyloxycarbonylamino)-1-ethoxy-1-phenylmethylphosphonate (3m). White solid; 82% yield (345 mg); mp 96.6 to 97.6 °C. 1H-NMR (400 MHz, CDCl3): δ 7.55–7.52 (m, 2H) and 7.35–7.28 (m, 8H)a, 6.23 (d, J = 10. Hz, 1H), 5.04 (ABq, J = 12.4 Hz, 2H), 4.14–3.67 (m, 6H)b, 1.26 (t, J = 7.0 Hz, 3H) and 1.25 (td, J1 = 7.0 Hz, J2 = 0.8 Hz, 3H)c, 1.17 (t, J = 7.2 Hz, 3H). 13C-NMR (100 MHz, CDCl3): δ 154.4 (d, J = 20.9 Hz), 136.0, 128.4, 128.14, 128.11, 127.75, 127.72, 127.41, 127.37, 87.3 (d, J = 185.8 Hz), 67.0, 64.5 (d, J = 7.2 Hz), 63.8 (d, J = 7.7 Hz), 59.8 (d, J = 6.1 Hz), 16.3 (d, J = 5.7 Hz), 16.2 (d, J = 5.6 Hz), 15.3. 31P-NMR (162 MHz, CDCl3): δ 15.4. IR (ATR): 3195, 1729, 1541, 1234,1027, 957, 737 cm−1. HMRS (ESI) m/z: calcd for C21H28NO6NaP [M + Na]+ 445.1552, found 444.1546. aOverlapping signals of PhCH2O and CαPh groups. aOverlapping signals of PhCH2O and P(O)(OCH2CH3)2 groups. cOverlapping signals of P(O)(OCH2CH3)2 groups.
- Diethyl 1-(N-benzyloxycarbonylamino)-1-ethoxy-2-(4-methoxyphenyl)ethylphosphonate (3n). White solid; 70% yield (326 mg); mp 80.1 to 81.6 °C. 1H-NMR (400 MHz, CD3CN): δ 7.40–7.33 (m, 5H), 7.20–7.16 (m, 2H), 6.79–6.75 (m, 2H), 5.91 (br d, J = 10.0 Hz, 1H), 5.11 (s, 2H), 4.05–3.87 (m, 4H)a, 3.80–3.72 (m, 1H)b, 3.74 (s, 3H)b, 3.67–3.57 (m, 2H), 1.17 (td, J1 = 7.0, J2 = 0.5 Hz, 3H), 1.13 (t, J = 7.0 Hz, 3H), 1.10 (td, J1 = 7.0 Hz, J2 = 0.5 Hz, 3H). 13C-NMR (100 MHz, CDCl3): δ 159.5, 155.4 (d, J = 14.1 Hz), 138.0, 133.1, 129.5, 129.1, 129.0, 128.5 (d, J = 3.8 Hz), 114.0, 88.3 (d, J = 187.3 Hz), 67.2, 63.78 (d, J = 7.2 Hz) and 62.73 (d, J = 7.2 Hz)a, 60.1 (d, J = 4.2 Hz), 55.8, 39.1 (d, J = 5.0 Hz), 15.7 (d, J = 5.7 Hz) and 15.6 (d, J = 5.7 Hz), 14.6. 31P-NMR (162 MHz, CD3CN): δ 17.29. IR (ATR): 3197, 2973, 1720, 1514, 1255, 1019, 972, 738, 697 cm−1. HMRS (ESI) m/z: calcd for C23H32NO7NaP [M + Na]+ 488.1814, found 488.1812. aOverlapping signals of P(O)(OCH2CH3)2 groups. bOverlapping signals of C6H4OCH3 and CαCH2C6H4CH3 groups.
- Synthesis of diethyl 1-(N-benzyloxycarbonylamino)-1-triphenylphosphonium-methylphosphonate tetrafluoroborate 4c.
- Diethyl 1-(N-benzyloxycarbonylamino)-1-triphenylphosphoniummethylphosphonate (4c) Colorless crystals; 95% yield (615 mg), mp 163.7 to 164.9 °C. 1H-NMR (400 MHz, CDCl3): δ 7.85–7.58 (m, 16H)a 7.33–7.25 (m, 5H), 5.96 (ddd, J = 22.7, 16.6, 9.9 Hz, 1H), 4.96 (ABq, J = 12.6 Hz, 2H), 4.17–4.07 (m, 2H), 3.95–3.84 (m, 2H), 1.23 (t, J = 7.1 Hz, 3H), 1.15 (t, J = 7.1 Hz, 3H). 13C-NMR (100 MHz, CDCl3): δ 156.3, 135.7, 135.1 (d, J = 3.1 Hz), 134.8 (d, J = 10.3 Hz), 130.1 (d, J = 13.0 Hz), 128.4, 128.1, 128.0, 116.9 (d, J = 84.7 Hz), 67.9, 65.1 (d, J = 7.6 Hz), 64.9 (d, J = 6.9 Hz), 48.1 (dd, J = 152.8, 48.5 Hz), 16.1 (d, J = 6.1 Hz), 16.0 (d, J = 5.0 Hz). 31P-NMR (162 MHz, CDCl3): 27.5 (d, J = 37.5 Hz), 11.2 (d, J = 37.5 Hz). IR (ATR) 3213, 1712, 1522, 1273, 1008, 747, 688. HRMS (ESI) m/z: calcd for C31H34NO5P2 [M + H]+ 562.1912, found 562.1912. aOverlapping signals of +PPh3 and NH groups.
3.3. General Procedure for the One-Pot Synthesis of Tetraethyl 1-(N-acylamino)alkylene-1,1-bisphosphonates 5
- Tetraethyl 1-(N-benzyloxycarbonylamino)ethylene-1,1-bisphosphonate (5a). Colorless crystals; 95% yield (430 mg), mp 47.1 to 48.7 °C. 1H-NMR (400 MHz, CDCl3): δ 7.36–7.31 (m, 5H), 5.40 (br t, J = 3.4 Hz, 1H), 5.07 (s, 2H), 4.25–4.12 (m, 8H)a, 1.98 (t, J = 17.0 Hz, 3H), 1.33 (t, J = 7.2 Hz, 6H) and 1.31 (t, J = 7.2 Hz, 6H)b. 13C-NMR (100 MHz, CDCl3): 154.3, 136.3, 128.4, 128.11, 128.09, 66.7, 63.83 (d, J = 3.4 Hz) and 63.80 (d, J = 3.4 Hz) and 63.75 (d, J = 3.4 Hz) and 63.72 (d, J = 3.4 Hz)a, 55.8 (t, J = 146.9 Hz), 16.5–16.3 (m)b, 16.2 (br t, J = 4.1 Hz). 31P-NMR (162 MHz, CDCl3): 19.6. IR (ATR) 3218, 1714, 1537, 1229, 1016, 958, 750. HRMS (ESI) m/z: calcd for C18H32NO8P2 [M + H]+ 452.1603, found 452.1610. aOverlapping signals of P(O)(OCH2CH3)2 groups. bOverlapping signals of P(O)(OCH2CH3)2 groups.
- Tetraethyl 1-(N-pivaloylamino)ethylene-1,1-bisphosphonate (5b). Colorless crystals; 62% yield (247 mg), mp 50.8 to 52.3 °C. 1H-NMR (400 MHz, CDCl3): δ 6.19 (br t, J = 4.6 Hz, 1H), 4.28–4.19 (m, 8H)a, 2.01 (t, J = 17.0 Hz, 3H), 1.35 (t, J = 7.0 Hz, 12H), 1.20 (s, 9H). 13C-NMR (100 MHz, CDCl3): δ 177.7 (t, J = 5.1 Hz), 63.76 (d, J = 3.4 Hz) and 63.73 (d, J = 3.4 Hz) and 63.67 (d, J = 3.4 Hz) and 63.64 (d, J = 3.4 Hz)a, 56.7 (t, J = 144.9 Hz), 39.8, 27.4, 16.7 (t, J = 4.5 Hz), 16.5–16.4 (m)b. 31P-NMR (162 MHz, CDCl3): 20.0. IR (ATR) 3276, 1677, 1515, 1233, 1016, 945. HRMS (ESI) m/z: calcd for C15H34NO7P2 [M + H]+ 402.1811, found 402.1813. aOverlapping signals of P(O)(OCH2CH3)2 groups. bOverlapping signals of P(O)(OCaH2CH3)2 groups.
- Tetraethyl 1-(N-benzyloxycarbonylamino)methylene-1,1-bisphosphonate (5c). Colorless crystals; 82% yield (357 mg), mp 59.8 to 60.7 °C. 1H-NMR (400 MHz, CDCl3): δ 7.36–7.31 (m, 5H), 5.32 (br d, J = 10.4 Hz, 1H), 5.15 (s, 2H), 4.59 (td J1 = 21.9, J2 = 10.4 Hz), 4.25–4.12 (m, 8H)a, 1.32 (t, J = 7.0 Hz, 6H) and 1.29 (t, J = 7.0 Hz, 6H)b. 13C-NMR (100 MHz, CDCl3): δ 155.5 (t, J = 4.9 Hz), 135.9, 128.5, 128.3, 128.1, 67.6, 63.5, 46.0 (t, J = 146.8 Hz), 16.3–16.2 (m)b. 31P-NMR (162 MHz, CDCl3): 16.3. IR (ATR) 3354, 1717, 1528, 1266, 1019, 977, 736. HRMS (ESI) m/z: calcd for C17H29NO8NaP2 [M + Na]+ 460.1266, found 460.1261. aOverlapping signals of P(O)(OCH2CH3)2 groups. bOverlapping signals of P(O)(OCH2CH3)2 groups.
- Tetraethyl 1-(N-benzyloxycarbonylamino)-2-phenylethylene-1,1-bisphosphonate (5d). Colorless crystals; 86% yield (455 mg), mp 60.2 to 61.5 °C. 1H-NMR (400 MHz, CDCl3) δ 7.44–7.34 (m, 5H), 7.26–7.16 (m, 5H), 5.44 (t, J = 12.7 Hz, 1H), 5.18 (s, 2H), 4.30–4.15 (m, 4H)a, 4.13–4.03 (m, 2H), 3.98–3.88 (m, 2H), 3.58 (dd, J = 15.3, 11.7 Hz, 2H), 1.30 (t, J = 7.2 Hz, 6H), 1.19 (t, J = 7.2 Hz, 6H). 13C-NMR (100 MHz, CDCl3) δ 154.9 (t, J = 8.8 Hz), 136.4, 135.3 (t, J = 8.6 Hz), 131.2, 128.49, 128.45, 128.2, 127.7, 126.7, 67.1, 63.9 (d, J = 7.5 Hz), 63.0 (d, J = 7.4 Hz), 61.2 (t, J = 143.1 Hz), 35.5, 16.3 (d, J = 6.3 Hz), 16.2 (d, J = 6.2 Hz). 31P-NMR (162 MHz, CDCl3): 18.8. IR (ATR) 3224, 1711, 1534, 1266, 1022, 963, 752. HRMS (ESI) m/z: calcd for C24H35NO8NaP2 [M + Na]+ 550.1736, found 550.1732. aOverlapping signals of P(O)(OCH2CH3)2 groups.
- Tetraethyl 1-(N-acetylamino)-2-phenylethylene-1,1-bisphosphonate (5e). Colorless crystals; 59% yield (255 mg); mp 91.0 to 92.3 °C. 1H-NMR (400 MHz, CDCl3): δ 7.28–7.19 (m, 5H), 6.04 (br t, J = 13.3 Hz, 1H), 4.33–4.25 (m, 4H)a, 4.16–4.06 (m, 2H), 4.04–3.94 (m, 2H), 3.57 (dd, J = 15.3, 12.0 Hz, 2H), 2.05 (s, 3H), 1.35 (t, J = 7.1, Hz, 6H), 1.23 (t, J = 7.1 Hz, 6H). 13C-NMR (100 MHz, CDCl3): δ 169.9 (t, J = 7.3 Hz), 135.5 (t, J = 8.2 Hz), 131.1, 127.7, 126.9, 64.1 (d, J = 7.3 Hz), 62.9 (d, J = 7.6 Hz), 61.4 (t, J = 143.1 Hz), 35.2, 23.9, 16.4 (d, J = 6.2 Hz), 16.1 (d, J = 6.5 Hz). 31P-NMR (162 MHz, CDCl3): 19.2. IR (ATR) 3305, 2989, 1684, 1537, 1245, 1065, 1008, 962. HRMS (ESI) m/z: calcd for C18H31NO7NaP2 [M + Na]+ 458.1473, found 458.1467. aOverlapping signals of P(O)(OCH2CH3)2 groups.
- Tetraethyl 1-(N-benzyloxycarbonylamino)propylene-1,1-bisphosphonate (5f). Colorless crystals; 90% yield (417 mg), mp 61.6 to 62.8 °C. 1H-NMR (400 MHz, CDCl3) δ 7.37–7.31 (m, 5H), 5.47 (br t, J = 8.3 Hz, 1H), 5.08 (s, 2H), 4.27–4.15 (m, 8H)a, 2.42 (tq, J1 = 16.0 Hz, J2 = 7.7 Hz, 2H), 1.34 (t, J = 7.0 Hz, 6H) and 1.32 (t, J = 7.0 Hz, 6H)b, 1.11 (t, J = 7.4 Hz, 3H). 13C-NMR (100 MHz, CDCl3) δ 154.3 (t, J = 8.0 Hz), 136.4, 128.4, 128.1, 66.8, 63.67 (d, J = 3.5 Hz) and 63.64 (d, J = 3.5 Hz)b, 63.42 (d, J = 3.5 Hz) and 63.38 (d, J = 3.5 Hz)a, 60.4 (t, J = 144.4 Hz), 23.8 (t, J = 3.0 Hz), 16.5–16.3 (m)b, 9.1(t, J = 6.5 Hz). 31P-NMR (162 MHz, CDCl3): 20.0. IR (ATR) 3422, 1737, 1503, 1248, 1022, 968, 770. HRMS (ESI) m/z: calcd for C19H33NO8NaP2 [M + Na]+ 488.1579, found 488.1577. aOverlapping signals of P(O)(OCH2CH3)2 groups. bOverlapping signals of P(O)(OCH2CH3)2 groups.
- Tetraethyl 1-(N-benzyloxycarbonylamino)butylene-1,1-bisphosphonate (5g). Colorless crystals; 95% yield (457 mg), mp 66.7 to 68.5 °C. 1H-NMR (400 MHz, CDCl3) δ 7.37–7.30 (m, 5H), 5.46 (br t, J = 8.2 Hz, 1H), 5.08 (s, 2H), 4.26–4.14 (m, 8H)a, 2.36–2.24 (m, 2H), 1.61–1.55 (m, 2H), 1.33 (t, J = 7.0 Hz, 6H) and 1.32 (t, J = 7.0 Hz, 6H)b, 0.93 (t, J = 7.3 Hz, 3H). 13C-NMR (100 MHz, CDCl3): δ 154.3 (t, J = 7.6 Hz), 136.4, 128.4, 128.1, 66.8, 63.66 (d, J = 3.5 Hz) and 63.63 (d, J = 3.5 Hz)a, 63.43 (d, J = 3.5 Hz) and 63.40 (d, J = 3.5 Hz)a, 60.1 (t, J = 143.5 Hz), 32.7 (t, J = 3.0 Hz), 17.7 (t, J = 6.2 Hz), 16.45 (d, J = 2.9 Hz) and 16.43 (d, J = 2.6 Hz) and 16.40 (d, J = 2.7 Hz) and 16.37 (d, J = 2.9 Hz)b, 14.5. 31P-NMR (162 MHz, CDCl3): 20.1. IR (ATR) 1735, 1499, 1243, 1017, 958, 740. HRMS (ESI) m/z: calcd for C20H35NO8NaP2 [M + Na]+ 502.1736, found 502.1731. aOverlapping signals of P(O)(OCH2CH3)2 groups. bOverlapping signals of P(O)(OCH2CH3)2 groups.
- Tetraethyl 1-(N-benzyloxycarbonylamino)-2-methylpropylene-1,1-bisphosphonate (5h). Colorless oil; 72% yield (344 mg). 1H-NMR (400 MHz, CDCl3): δ 7.37–7.28 (m, 5H), 5.71 (t, J = 10.1 Hz, 1H), 5.09 (s, 2H), 4.28–4.13 (m, 8H)a, 3.06 (tsept, J1 = 23.6, J2 = 7.0 Hz 1H), 1.33 (t, J = 7.0 Hz, 6H) and 1.32 (t, J = 7.0 Hz, 6H)b, 1.22 (d, J = 6.9 Hz, 6H). 13C-NMR (100 MHz, CDCl3): δ 154.3 (t, J = 8.2 Hz), 136.4, 128.4, 128.0, 66.8, 64.6 (t, J = 139.2 Hz), 63.40 (d, J = 3.6 Hz) and 63.36 (d, J = 3.6 Hz)a, 63.11 (d, J = 3.5 Hz) and 63.08 (d, J = 3.6 Hz)a, 30.7, 18.8 (t, J = 4.3 Hz), 16.40 (d, J = 3.0 Hz) and 16.37 (d, J = 2.9 Hz) and 16.34 (d, J = 2.9 Hz) and 16.31 (d, J = 3.0 Hz)b. 31P-NMR (162 MHz, CDCl3): 20.7. IR (ATR) 3433, 1743, 1500, 1244, 1019, 966, 741. HRMS (ESI) m/z: calcd for C20H36NO8P2 [M + H]+ 480.1916, found 480.1917. aOverlapping signals of P(O)(OCH2CH3)2 groups. bOverlapping signals of P(O)(OCH2CH3)2 groups.
- Tetraethyl 1-(N-benzyloxycarbonylamino)pentylene-1,1-bisphosphonate (5i). Colorless crystals; 90% yield (444 mg), mp 56.0 to 57.2 °C. 1H-NMR (400 MHz, CDCl3) δ 7.37–7.29 (m, 5H), 5.47 (br t, J = 8.5 Hz, 1H), 5.08 (s, 2H), 4.26–4.14 (m, 8H)a, 2.38–2.26 (m, 2H), 1.57–1.49 (m, 2H), 1.37–1.25 (m, 2H) and 1.33 (t, J = 7.0 Hz, 6H) and 1.32 (t, J = 7.0 Hz, 6H)b, 0.90 (t, J = 7.3 Hz, 3H). 13C-NMR (100 MHz, CDCl3) δ 154.3 (t, J = 7.3 Hz), 136.4, 128.4, 128.0, 66.7, 63.59 (d, J = 3.5 Hz) and 63.55 (d, J = 3.5 Hz)b, 63.38 (d, J = 3.4 Hz) and 63.34 (d, J = 3.5 Hz)b, 60.0 (t, J = 144.4 Hz), 30.4 (t, J = 3.0 Hz), 26.2 (t, J = 6.0 Hz), 23.0, 16.39 (d, J = 2.7 Hz) and 16.36 (d, J = 2.6 Hz) and 16.33 (d, J = 2.6 Hz) and 16.31 (d, J = 2.7 Hz)a, 13.9. 31P-NMR (162 MHz, CDCl3): 20.1. IR (ATR) 3224, 1736, 1498, 1233, 1019, 953, 772. HRMS (ESI) m/z: calcd for C21H37NO8NaP2 [M + Na]+ 516.1892, found 516.1889. aOverlapping signals of P(O)(OCH2CH3)2 groups. bOverlapping signals of CαCH2CH2CH2CH3 and P(O)(OCH2CH3)2 groups. cOverlapping signals of P(O)(OCH2CH3)2 groups.
- Tetraethyl 1-(N-benzyloxycarbonylamino)-3-methylbutylene-1,1-bisphosphonate (5j). Colorless crystals; 74% yield (367 mg), mp 54.9 to 55.5 °C. 1H-NMR (400 MHz, CDCl3) δ 7.35–7.30 (m, 5H), 5.55 (br t, J = 12.0 Hz, 1H), 5.09 (s, 2H), 4.28–4.16 (m, 8H)a, 2.19–2.11 (m, 3H), 1.33 (t, J = 7.1 Hz, 12H), 0.95 (d, J = 6.2 Hz, 6H). 13C-NMR (100 MHz, CDCl3): 154.4 (t, J = 8.0 Hz), 136.4, 128.4, 128.14, 128.06, 66.9, 63.56 (d, J = 3.5 Hz) and 63.52 (d, J = 3.5 Hz)a, 63.34 (d, J = 3.5 Hz) and 63.30 (d, J = 3.5 Hz)a, 60.7 (t, J = 142.9 Hz), 38.6 (t, J = 2.2 Hz), 25.2 (t, J = 7.8 Hz), 24.2, 16.44–16.26 (m)b. 31P-NMR (162 MHz, CDCl3): 20.4. IR (ATR) 3231, 1716, 1528, 1250, 1026, 967, 749. HRMS (ESI) m/z: calcd for C21H37NO8NaP2 [M + Na]+ 516.1892, found 516.1891. aOverlapping signals of P(O)(OCH2CH3)2 groups. bOverlapping signals of P(O)(OCH2CH3)2 groups.
- Tetraethyl 1-(N-acetylamino)-3-methylbutylene-1,1-bisphosphonate (5k). Colorless crystals; 69% yield (278 mg), mp 108.8 to 110.3 °C. 1H-NMR (400 MHz, CDCl3): δ 6.25 (br t, J = 13.0 Hz, 1H), 4.28–4.17 (m, 8H)a, 2.18–2.07 (m, 3H), 2.02 (s, 3H), 1.349 (t, J = 7.0 Hz, 6H) and 1.345 (t, J = 7.0 Hz, 6H)b, 0.96 (d, J = 6.3 Hz, 6H). 13C-NMR (100 MHz, CDCl3): δ 169.2 (t, J = 6.9 Hz), 63.6 (d, J = 7.3 Hz), 63.1 (d, J = 7.2 Hz), 60.9 (t, J = 143.2 Hz), 38.4 (t, J = 2.6 Hz), 25.4 (t, J = 8.0 Hz), 24.2, 23.9, 16.4 (d, J = 6.0 Hz) and 16.3 (d, J = 6.3 Hz)b. 31P-NMR (162 MHz, CDCl3): 20.9. IR (ATR) 3441, 1682, 1541, 1234, 1023, 971. HRMS (ESI) m/z: calcd for C15H34NO7P2 [M + H]+ 402.1811, found 402.1811. aOverlapping signals of P(O)(OCH2CH3)2 groups. bOverlapping signals of P(O)(OCH2CH3)2 groups.
- Tetraethyl 1-(N-benzyloxycarbonylamino)-2-methoxyethylene-1,1-bisphosphonate (5l). Colorless oil; 52% yield (248 mg), 1H-NMR (400 MHz, CDCl3) δ 7.36–7.28 (m, 5H), 5.56 (br t, J = 8.0 Hz, 1H), 5.09 (s, 2H), 4.26–4.12 (m, 10H)a, 3.39 (s, 3H), 1.32 (t, J = 7.0 Hz, 12H). 13C-NMR (100 MHz, CDCl3): δ 154.4 (t, J = 6.2 Hz), 136.3, 128.4, 128.1, 128.0, 70.1, 66.9, 63.59 (d, J = 3.5 Hz) and 63.55 (d, J = 3.6 Hz) and 63.52 (d, J = 3.6 Hz)b, 60.5 (t, J = 142.7 Hz), 59.1, 16.4–16.3 (m)c. 31P-NMR (162 MHz, CDCl3): 18.0. IR (ATR) 1741, 1498, 1254, 1023, 973, 733. HRMS (ESI) m/z: calcd for C19H33NO9NaP2 [M + Na]+ 504.1528, found 504.1526. aOverlapping signals of and CαCH2OCH3 and P(O)(OCH2CH3)2 groups. aOverlapping signals of P(O)(OCH2CH3)2 groups. bOverlapping signals of P(O)(OCH2CH3)2 groups.
- Tetraethyl 1-(N-benzyloxycarbonylamino)phenylmethylene-1,1-bisphosphonate (5m). Colorless crystals; 40% yield (204 mg); mp 66.1 to 68.0 °C. 1H-NMR (400 MHz, CDCl3): δ 7.70–7.66 (m, 2H) and 7.38–7.25 (m, 8H)a, 5.93 (br s, 1H), 5.12 (s, 2H), 4.16–3.94 (m, 8H)b, 1.21 (t, J = 7.0 Hz, 12H). 13C-NMR (100 MHz, CDCl3) δ 154.4 (br s), 136.3, 132.0, 128.4, 128.1 (t, J = 5.0 Hz), 127.6 (t, J = 2.6 Hz), 127.5 (t, J = 2.3 Hz), 67.1, 64.25 (d, J = 3.7 Hz) and 64.21 (d, J = 3.6 Hz) and 64.08 (d, J = 3.6 Hz) and 64.05 (d, J = 3.8 Hz)b, 64.15 (t, J = 140.4 Hz), 16.26 (d, J = 3.1 Hz) and 16.23 (d, J = 3.0 Hz)c. 31P-NMR (162 MHz, CDCl3): 17.1. IR (ATR) 3218, 1723, 1530, 1014, 949, 750, 697. HRMS (ESI) m/z: calcd for C23H33NO8NaP2 [M + Na]+ 536.1579, found 536.1570. aOverlapping signals of PhCH2O and CαPh groups. aOverlapping signals of P(O)(OCH2CH3)2 groups. cOverlapping signals of P(O)(OCH2CH3)2 groups.
- Tetraethyl 1-(N-benzyloxycarbonylamino)-2-(4-methoxyphenyl)ethylene-1,1-bisphosphonate (5n). Colorless crystals; 74% yield (412 mg), mp 71.7 to 73.5 °C. 1H-NMR (400 MHz, CDCl3) δ 7.44–7.34 (m, 5H), 7.14–7.09 (m, 2H), 6.71–6.69 (m, 2H), 5.43 (br t, J = 12.8 Hz), 5.17 (s, 2H), 4.29–4.17 (m, 4H), 4.13–4.08 (m, 2H), 4.05–3.91 (m, 2H), 3.75 (s, 3H), 3.52 (dd, J1 = 15.3, J2 = 11.8 Hz, 2H) 1.31 (t, J = 7.1 Hz, 6H), 1.21 (t, J = 7.1 Hz, 6H). 13C-NMR (100 MHz, CDCl3) δ 158.5, 154.9 (t, J = 8.8 Hz), 136.3, 132.1, 128.46, 128.45, 128.2, 127.2 (t, J = 8.6 Hz), 113.1, 67.0, 63.9 (d, J = 7.4 Hz), 63.0 (d, J = 7.6 Hz), 61.1 (t, J = 143.0 Hz), 55.2, 34.7, 16.3 (d, J = 6.3 Hz), 16.2 (d, J = 6.2 Hz). 31P-NMR (162 MHz, CDCl3): 19.0. IR (ATR) 3251, 1715, 1513, 1247, 1028, 953, 774. HRMS (ESI) m/z: calcd for C25H37NO9NaP2 [M + Na]+ 580.1841, found 580.1839.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Romanenko, V.D.; Kukhar, V.P. 1-Amino-1,1-Bisphosphonates. Fundamental Syntheses and New Developments. Arkivoc 2012, 4, 127–166. [Google Scholar] [CrossRef] [Green Version]
- Kafarski, P.; Lejczak, B. Aminophosphonic Acids of Potential Medical Importance. Curr. Med. Chem.-Anti-Cancer Agents 2001, 1, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Mucha, A.; Kafarski, P.; Berlicki, Ł. Remarkable Potential of the α-Aminophosphonate/Phosphinate Structural Motif in Medicinal Chemistry. J. Med. Chem. 2011, 54, 5955–5980. [Google Scholar] [CrossRef]
- Orsini, F.; Sello, G.; Sisti, M. Aminophosphonic Acids and Derivatives. Synthesis and Biological Applications. Curr. Med. Chem. 2010, 17, 264–289. [Google Scholar] [CrossRef]
- Kukhar, V.P.; Hudson, H.R. Aminophosphonic and Aminophosphinic Acids: Chemistry and Biological Activity; Kukhar, V.P., Hudson, H.R., Eds.; John Wiley & Sons Ltd.: New York, NY, USA, 2000; ISBN 978-0-471-89149-9. [Google Scholar]
- Russell, R.G.G. Bisphosphonates: The First 40 years. Bone 2011, 49, 2–19. [Google Scholar] [CrossRef]
- Widler, L.; Jaeggi, K.A.; Glatt, M.; Müller, K.; Bachmann, R.; Bisping, M.; Born, A.-R.; Cortesi, R.; Guiglia, G.; Jeker, H.; et al. Highly Potent Geminal Bisphosphonates. From Pamidronate Disodium (Aredia) to Zoledronic Acid (Zometa). J. Med. Chem. 2002, 45, 3721–3738. [Google Scholar] [CrossRef]
- Zhang, S.; Gangal, G.; Uludağ, H. ‘Magic Bullets’ for Bone Diseases: Progress in Rational Design of Bone-Seeking Medicinal Agents. Chem. Soc. Rev. 2007, 36, 507–531. [Google Scholar] [CrossRef]
- Hiraga, T.; Tanaka, S.; Yamamoto, M.; Nakajima, T.; Ozawa, H. Inhibitory Effects of Bisphosphonate (YM175) on Bone Resorption Induced by a Metastatic Bone Tumor. Bone 1996, 18, 1–7. [Google Scholar] [CrossRef]
- Wang, L.; Kamath, A.; Das, H.; Li, L.; Bukowski, J.F. Antibacterial Effect of Human Vγ2Vδ2 T Cells In Vivo. J. Clin. Investig. 2001, 108, 1349–1357. [Google Scholar] [CrossRef]
- Chmielewska, E.; Kafarski, P. Physiologic Activity of Bisphosphonates—Recent Advances. Open Pharm. Sci. J. 2016, 3, 56–78. [Google Scholar] [CrossRef] [Green Version]
- Szajnman, S.H.; Ravaschino, E.L.; Docampo, R.; Rodriguez, J.B. Synthesis and Biological Evaluation of 1-Amino-1,1-Bisphosphonates Derived from Fatty Acids against Trypanosoma Cruzi Targeting Farnesyl Pyrophosphate Synthase. Bioorg. Med. Chem. Lett. 2005, 15, 4685–4690. [Google Scholar] [CrossRef] [PubMed]
- Yajima, S.; Hara, K.; Sanders, J.M.; Yin, F.; Ohsawa, K.; Wiesner, J.; Jomaa, H.; Oldfield, E. Crystallographic Structures of Two Bisphosphonate:1-Deoxyxylulose-5-Phosphate Reductoisomerase Complexes. J. Am. Chem. Soc. 2004, 126, 10824–10825. [Google Scholar] [CrossRef] [PubMed]
- Occhipinti, A.; Berlicki, Ł.; Giberti, S.; Dziȩdzioła, G.; Kafarski, P.; Forlani, G. Effectiveness and Mode of Action of Phosphonate Inhibitors of Plant Glutamine Synthetase: Phosphonate Inhibitors of Plant Glutamine Synthetase. Pest. Manag. Sci. 2010, 66, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Ferlazzo, V.; Sferrazza, C.; Caccamo, N.; Di Fede, G.; Di Lorenzo, G.; D’Asaro, M.; Meraviglia, S.; Dieli, F.; Rini, G.; Salerno, A. In Vitro Effects of Aminobisphosphonates on Vγ9Vδ2 T Cell Activation and Differentiation. Int. J. Immunopathol. Pharmacol. 2006, 19, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Farrell, K.B.; Karpeisky, A.; Thamm, D.H.; Zinnen, S. Bisphosphonate Conjugation for Bone Specific Drug Targeting. Bone Rep. 2018, 9, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Uludag, H. Bisphosphonates as a Foundation of Drug Delivery to Bone. Curr. Pharm. Des. 2002, 8, 1929–1944. [Google Scholar] [CrossRef] [PubMed]
- Kuźnik, A.; Październiok-Holewa, A.; Jewula, P.; Kuźnik, N. Bisphosphonates—Much More than Only Drugs for Bone Diseases. Eur. J. Pharm. 2020, 866, 172773. [Google Scholar] [CrossRef]
- Kubíček, V.; Rudovský, J.; Kotek, J.; Hermann, P.; Vander Elst, L.; Muller, R.N.; Kolar, Z.I.; Wolterbeek, H.T.; Peters, J.A.; Lukeš, I. A Bisphosphonate Monoamide Analogue of DOTA: A Potential Agent for Bone Targeting. J. Am. Chem. Soc. 2005, 127, 16477–16485. [Google Scholar] [CrossRef]
- Kaboudin, B.; Esfandiari, H.; Moradi, A.; Kazemi, F.; Aoyama, H. ZnCl2-Mediated Double Addition of Dialkylphosphite to Nitriles for the Synthesis of 1-Aminobisphosphonates. J. Org. Chem. 2019, 84, 14943–14948. [Google Scholar] [CrossRef]
- Kaboudin, B.; Alipour, S. A Microwave-Assisted Solvent- and Catalyst-Free Synthesis of Aminomethylene Bisphosphonates. Tetrahedron Lett. 2009, 50, 4243–4245. [Google Scholar] [CrossRef]
- Balakrishna, A.; Narayana Reddy, M.V.; Rao, P.V.; Kumar, M.A.; Kumar, B.S.; Nayak, S.K.; Reddy, C.S. Synthesis and Bio-Activity Evaluation of Tetraphenyl(Phenylamino) Methylene Bisphosphonates as Antioxidant Agents and as Potent Inhibitors of Osteoclasts In Vitro. Eur. J. Med. Chem. 2011, 46, 1798–1802. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-S.; Park, J.; De Schutter, J.W.; Huang, X.F.; Berghuis, A.M.; Sebag, M.; Tsantrizos, Y.S. Design and Synthesis of Active Site Inhibitors of the Human Farnesyl Pyrophosphate Synthase: Apoptosis and Inhibition of ERK Phosphorylation in Multiple Myeloma Cells. J. Med. Chem. 2012, 55, 3201–3215. [Google Scholar] [CrossRef] [PubMed]
- Yokomatsu, T.; Yoshida, Y.; Nakabayashi, N.; Shibuya, S. Simple and Efficient Method for Preparation of Conformationally Constrained Aminomethylene Gem-Diphosphonate Derivatives via Beckmann Rearrangement. J. Org. Chem. 1994, 59, 7562–7564. [Google Scholar] [CrossRef]
- Wang, A.-E.; Chang, Z.; Sun, W.-T.; Huang, P.-Q. General and Chemoselective Bisphosphonylation of Secondary and Tertiary Amides. Org. Lett. 2015, 17, 732–735. [Google Scholar] [CrossRef] [PubMed]
- Midrier, C.; Lantsoght, M.; Volle, J.-N.; Pirat, J.-L.; Virieux, D.; Stevens, C.V. Hydrophosphonylation of Alkenes or Nitriles by Double Radical Transfer Mediated by Titanocene/Propylene Oxide. Tetrahedron Lett. 2011, 52, 6693–6696. [Google Scholar] [CrossRef]
- Islas, R.E.; García, J.J. Nickel-Catalyzed Hydrophosphonylation and Hydrogenation of Aromatic Nitriles Assisted by Lewis Acid. ChemCatChem 2019, 11, 1337–1345. [Google Scholar] [CrossRef]
- Abdou, W.M.; Shaddy, A.A. The Development of Bisphosphonates for Therapeutic Uses, and Bisphosphonate Structure-Activity Consideration. Arkivoc 2008, 9, 143–182. [Google Scholar] [CrossRef] [Green Version]
- Schrader, T.; Steglich, W.; Kober, R. Synthese von 1-Aminophosphonsäure-Derivaten Über Acyliminophosphonsäure-Ester. Synthesis 1986, 5, 372–375. [Google Scholar] [CrossRef]
- Kuźnik, A.; Mazurkiewicz, R.; Grymel, M.; Zielińska, K.; Adamek, J.; Chmielewska, E.; Bochno, M.; Kubica, S. A New Method for the Synthesis of α-Aminoalkylidenebisphosphonates and Their Asymmetric Phosphonyl-Phosphinyl and Phosphonyl-Phosphinoyl Analogues. Beilstein J. Org. Chem. 2015, 11, 1418–1424. [Google Scholar] [CrossRef] [Green Version]
- Mazurkiewicz, R.; Adamek, J.; Październiok-Holewa, A.; Zielińska, K.; Simka, W.; Gajos, A.; Szymura, K. α-Amidoalkylating Agents from N-Acyl-α-Amino Acids: 1-(N-Acylamino)Alkyltriphenylphosphonium Salts. J. Org. Chem. 2012, 77, 1952–1960. [Google Scholar] [CrossRef]
- Mazurkiewicz, R.; Październiok-Holewa, A.; Kononienko, A. A Novel Synthesis of 1-Aminoalkanephosphonic Acid Derivatives from 1-(N-Acylamino)-Alkyltriphenylphosphonium Salts. Phosphorus Sulfur Silicon Relat. Elem. 2010, 185, 1986–1992. [Google Scholar] [CrossRef]
- Kuźnik, A.; Mazurkiewicz, R.; Zięba, M.; Erfurt, K. 1-(N-Acylamino)-1-Triphenylphosphoniumalkylphosphonates: General Synthesis and Prospects for Further Synthetic Applications. Tetrahedron Lett. 2018, 59, 3307–3310. [Google Scholar] [CrossRef]
- Neilson, D.G. The Chemistry of Amidines and Imidates; Patai, S., Ed.; John Wiley & Sons Ltd.: New York, NY, USA, 1975; pp. 385–489. [Google Scholar] [CrossRef]
- Roger, R.; Neilson, D.G. The Chemistry of Imidates. Chem. Rev. 1961, 61, 179–211. [Google Scholar] [CrossRef]
- Rassukana, Y.; Kolotylo, M.; Sinitsa, O.; Pirozhenko, V.; Onys’ko, P. α-Iminotrifluoroethylphosphonates: The First Representatives of N-H Imidoyl Phosphonates. Synthesis 2007, 17, 2627–2630. [Google Scholar] [CrossRef]
- Adamek, J.; Październiok-Holewa, A.; Zielińska, K.; Mazurkiewicz, R. Comparative Studies on the Amidoalkylating Properties of N-(1-Methoxyalkyl)Amides and 1-(N-Acylamino)Alkyltriphenylphosphonium Salts in the Michaelis–Arbuzov-Like Reaction: A New One-Pot Transformation of N-(1-Methoxyalkyl)Amides into Phosphonic or Phosphinic Analogs of N-Acyl-α-Amino Acids. Phosphorus Sulfur Silicon Relat. Elem. 2013, 188, 967–980. [Google Scholar] [CrossRef]
- Adamek, J.; Węgrzyk, A.; Kończewicz, J.; Walczak, K.; Erfurt, K. 1-(N-Acylamino)Alkyltriarylphosphonium Salts with Weakened Cα-P+ Bond Strength—Synthetic Applications. Molecules 2018, 23, 2453. [Google Scholar] [CrossRef] [Green Version]
- Ohme, R.; Schmitz, E. A Simple Synthesis of Alkyl Formimidates. Angew. Chem. Int. Ed. Engl. 1967, 6, 566. [Google Scholar] [CrossRef]
- Yadav, V.K.; Babu, K.G. A Remarkably Efficient Markovnikov Hydrochlorination of Olefins and Transformation of Nitriles into Imidates by Use of AcCl and an Alcohol. Eur. J. Org. Chem. 2005, 2005, 452–456. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Morita, Y.; Minami, K. 1,3-Oxazines and related compounds. XII. Facile synthesis of 2,4-disubstituted 6H-1,3-oxazin-6-ones. Chem. Pharm. Bull. 1986, 34, 1980–1986. [Google Scholar] [CrossRef] [Green Version]
- Harizi, A. Synthese Originale de 5-Aryl (ou 5-benzyl)-2-[(1-Diethoxyphosphonyl)methyl]-1,3,4-oxadiazoles par Action du Phosphonomethylhydrazide sur les Imidates N-Acyles. Phosphorus Sulfur Silicon Relat. Elem. 2006, 181, 2377–2385. [Google Scholar] [CrossRef]
- Emura, T.; Kimura, N.; Nagafuji, T. Preparation of Benzene Derivatives Having NOS Inhibitory Activity. Patent WO 9746515, 11 December 1997. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Comp. 3 | PG | R2 | Nu (eq.) | Time [days] | Temperature [°C] | Yield 2 |
|---|---|---|---|---|---|---|---|
| 1 | 3a | Cbz | Me | 1.2 | 2 | rt | 94 |
| 2 | 3b | Piv | Me | 1.2 | 3 | rt | 74 |
| 3 | 3c | Cbz | H | 1.2 | 2 | −10 | 93 |
| 4 | 3d | Cbz | CH2Ph | 2 | 4 | −20 | 88 |
| 5 | 3e | Ac | CH2Ph | 1.2 | 4 | rt | 53 |
| 6 | 3f | Cbz | Et | 1.2 | 3 | −5 | 82 |
| 7 | 3g | Cbz | Pr | 2 | 4 | −10 | 68 |
| 8 | 3h | Cbz | i-Pr | 2 | 7 | −40 | 53 |
| 9 | 3i | Cbz | Bu | 2 | 4 | −10 | 54 |
| 10 | 3j | Cbz | i-Bu | 3 | 7 | −40 | 32 |
| 11 | 3k | Ac | i-Bu | 2 | 7 | −40 | 65 |
| 12 | 3l | Cbz | CH2OMe | 1.2 | 4 | −10 | 91 |
| 13 | 3m | Cbz | Ph | 1.2 | 3 | rt | 82 |
| 14 | 3n | Cbz | 1,4-CH2-C6H4OMe | 3 | 3 | −25 | 70 |
| Entry | Molar Ratios | MePh3P+ I− | Temperature [°C] | Time [h] | Yield [%] 1 | |
|---|---|---|---|---|---|---|
| 3d | Ph3P·HBF4 | |||||
| 1 | 1 | 1.06 | + | 0–5 for 45 min. then rt | 24 | 52 |
| 2 | 1 | 1.08 | − | 0–5 for 45 min. then rt | 24 | 73 |
| 3 | 1 | 1.05 | − | rt | 24 | 77 |
| 4 | 1 | 1.27 | − | rt | 24 | 75 |
| 5 | 1 | 1.06 | − | rt | 6 | 86 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuźnik, A.; Kozicka, D.; Hawranek, W.; Socha, K.; Erfurt, K. One-Pot and Catalyst-Free Transformation of N-Protected 1-Amino-1-Ethoxyalkylphosphonates into Bisphosphonic Analogs of Protein and Non-Protein α-Amino Acids. Molecules 2022, 27, 3571. https://doi.org/10.3390/molecules27113571
Kuźnik A, Kozicka D, Hawranek W, Socha K, Erfurt K. One-Pot and Catalyst-Free Transformation of N-Protected 1-Amino-1-Ethoxyalkylphosphonates into Bisphosphonic Analogs of Protein and Non-Protein α-Amino Acids. Molecules. 2022; 27(11):3571. https://doi.org/10.3390/molecules27113571
Chicago/Turabian StyleKuźnik, Anna, Dominika Kozicka, Wioleta Hawranek, Karolina Socha, and Karol Erfurt. 2022. "One-Pot and Catalyst-Free Transformation of N-Protected 1-Amino-1-Ethoxyalkylphosphonates into Bisphosphonic Analogs of Protein and Non-Protein α-Amino Acids" Molecules 27, no. 11: 3571. https://doi.org/10.3390/molecules27113571
APA StyleKuźnik, A., Kozicka, D., Hawranek, W., Socha, K., & Erfurt, K. (2022). One-Pot and Catalyst-Free Transformation of N-Protected 1-Amino-1-Ethoxyalkylphosphonates into Bisphosphonic Analogs of Protein and Non-Protein α-Amino Acids. Molecules, 27(11), 3571. https://doi.org/10.3390/molecules27113571