
New Chemotypes for the Inhibition of (p)ppGpp Synthesis in the Quest for New Antimicrobial Compounds

 , , , and
, , , and
Abstract
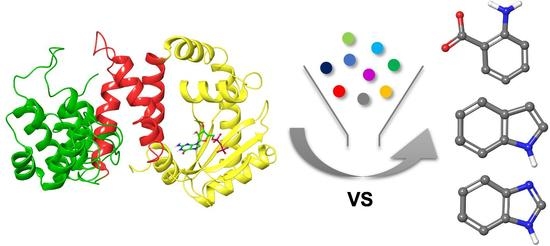
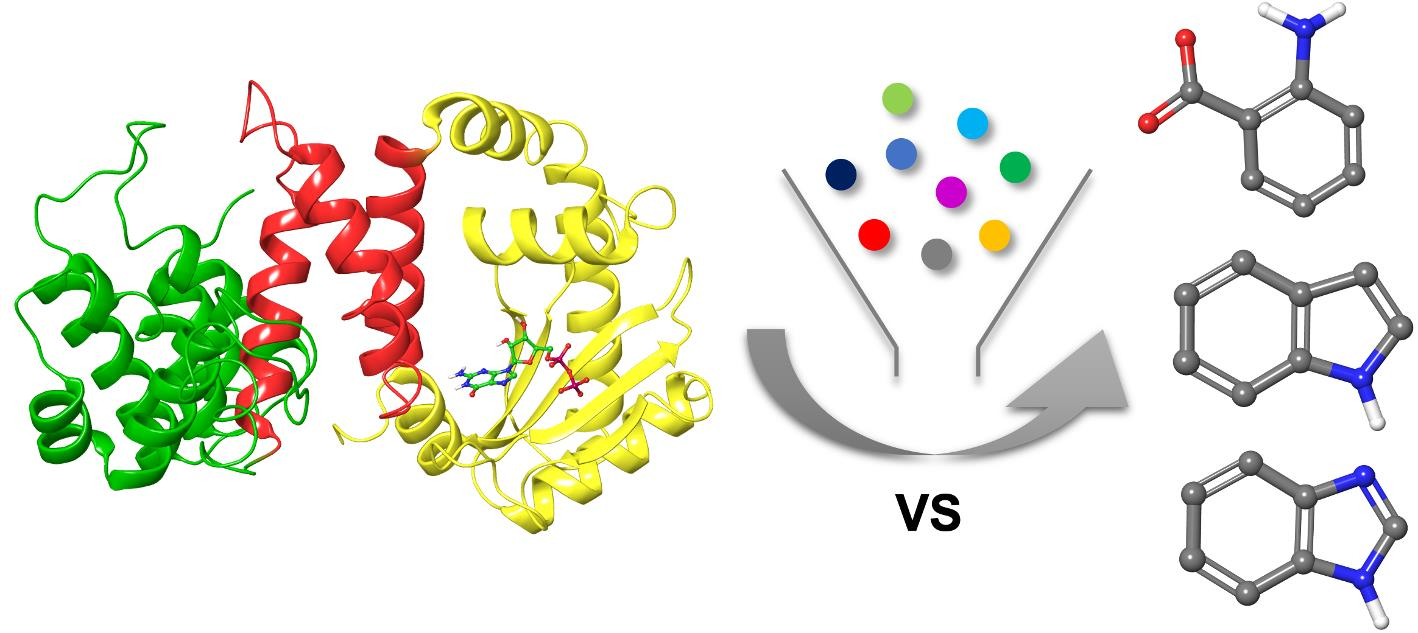
1. Introduction
2. Results and Discussion
2.1. Fragment Libraries Virtual Screening in RelSeq Synthetase Site
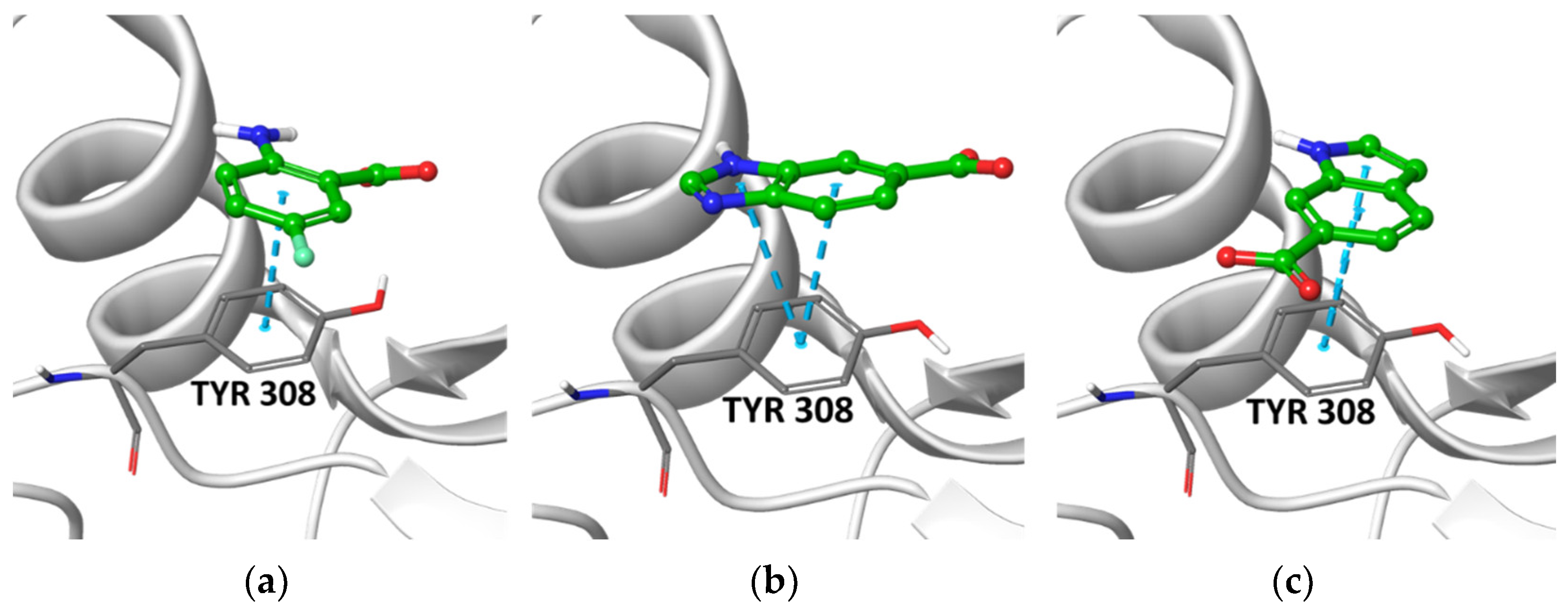
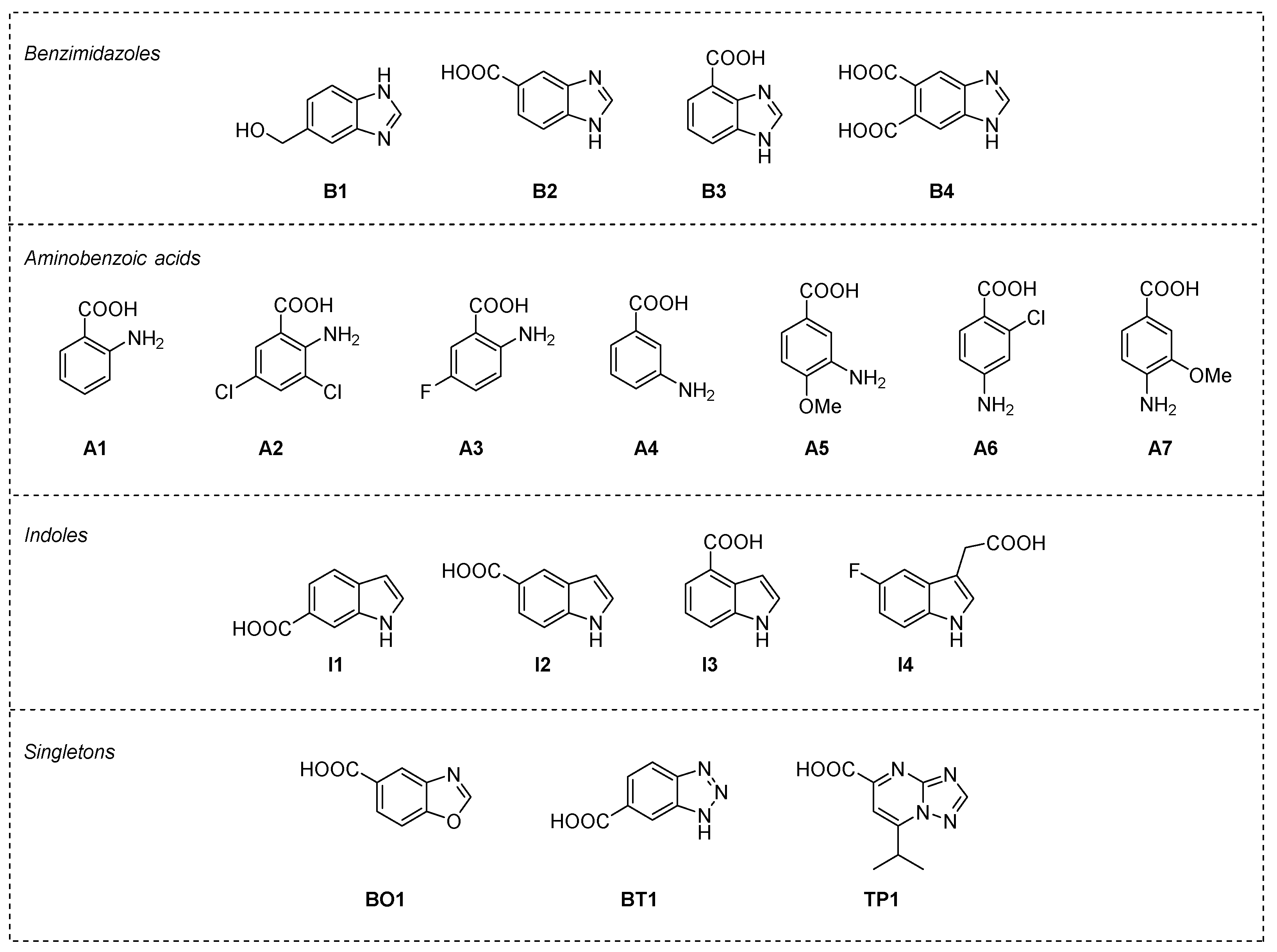
2.2. Post-Docking Analysis and Chemotype Selection
2.3. Thermal Shift Assay on Selected Fragments vs. RelSeq Protein Constructs
2.4. STD-NMR Protein–Fragment Interaction Experiments
3. Materials and Methods
3.1. Computational Methods
- Maybridge Ro3 Diversity Set (2500 fragments) (www.maybridge.com, accessed on 9 June 2015);
- Asinex-Fragments-21872 (21,872 fragments) (www.asinex.com, accessed on 18 January 2019);
- ‘Fragment Libraries with Experimental Solubility Data’, two datasets from Life Chemicals (11,667 and 2921 fragments, respectively) (www.lifechemicals.com, accessed on 1 February 2019);
- OTAVA Solubility Fragment Library (1021 fragments) (www.otavachemicals.com, accessed on 4 February 2019);
- FragmentLibrary_sdf_13808 (13,808 fragments) from CHEMBRIDGE (www.chembridge.com, accessed on 18 April 2019)
- Preplated Fragment-Based Library (4532 fragments) from SPECS (www.specs.net, accessed on 11 February 2019).
3.2. Experimental Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fleming, A. On the Antibacterial Action of Cultures of a Penicillium, with Special Reference to their Use in the Isolation of B. influenzæ. Br. J. Exp. Pathol. 1929, 10, 226–236. [Google Scholar] [CrossRef]
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations in the Review on Antimicrobial Resistance. 2016, pp. 1–81. Available online: https://apo.org.au/node/63983 (accessed on 19 April 2022).
- Theuretzbacher, U.; Gottwalt, S.; Beyer, P.; Butler, M.; Czaplewski, L.; Lienhardt, C.; Moja, L.; Paul, M.; Paulin, S.; Rex, J.H.; et al. Analysis of the clinical antibacterial and antituberculosis pipeline. Lancet Infect. Dis. 2019, 19, e40–e50. [Google Scholar] [CrossRef]
- Schrader, S.M.; Vaubourgeix, J.; Nathan, C. Biology of antimicrobial resistance and approaches to combat it. Sci. Transl. Med. 2020, 12, eaaz6992. [Google Scholar] [CrossRef] [PubMed]
- Cashel, M.; Gallant, J. Two Compounds implicated in the Function of the RC Gene of Escherichia coli. Nature 1969, 221, 838–841. [Google Scholar] [CrossRef]
- Pulschen, A.A.; Fernandes, A.Z.N.; Cunha, A.F.; Sastre, D.E.; Matsuguma, B.E.; Gueiros-Filho, F.J. Many birds with one stone: Targeting the (p)ppGpp signaling pathway of bacteria to improve antimicrobial therapy. Biophys. Rev. 2021, 13, 1039–1051. [Google Scholar] [CrossRef]
- Kamarthapu, V.; Epshtein, V.; Benjamin, B.; Proshkin, S.; Mironov, A.; Cashel, M.; Nudler, E. ppGpp couples transcription to DNA repair in E. coli. Science 2016, 352, 993–996. [Google Scholar] [CrossRef]
- Molodtsov, V.; Sineva, E.; Zhang, L.; Huang, X.; Cashel, M.; Ades, S.E.; Murakami, K.S. Allosteric Effector ppGpp Potentiates the Inhibition of Transcript Initiation by DksA. Mol. Cell 2018, 69, 828–839.e5. [Google Scholar] [CrossRef]
- Diez, S.; Ryu, J.; Caban, K.; Gonzalez, R.L., Jr.; Dworkin, J. The alarmones (p)ppGpp directly regulate translation initiation during entry into quiescence. Proc. Natl. Acad. Sci. USA 2020, 117, 15565–15572. [Google Scholar] [CrossRef]
- Steinchen, W.; Zegarra, V.; Bange, G. (p)ppGpp: Magic Modulators of Bacterial Physiology and Metabolism. Front. Microbiol. 2020, 11, 2072. [Google Scholar] [CrossRef]
- Irving, S.E.; Choudhury, N.R.; Corrigan, R.M. The stringent response and physiological roles of (pp)pGpp in bacteria. Nat. Rev. Microbiol. 2021, 19, 256–271. [Google Scholar] [CrossRef]
- Buke, F.; Grilli, J.; Cosentino Lagomarsino, M.; Bokinsky, G.; Tans, S.J. ppGpp is a bacterial cell size regulator. Curr. Biol. 2022, 32, 870–877.e5. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Islam, M.; Jung, H.W.; Lim, D.; Kim, K.; Lee, S.G.; Park, C.; Lee, J.C.; Shin, M. ppGpp signaling plays a critical role in virulence of Acinetobacter baumannii. Virulence 2021, 12, 2122–2132. [Google Scholar] [CrossRef] [PubMed]
- Kalia, D.; Merey, G.; Nakayama, S.; Zheng, Y.; Zhou, J.; Luo, Y.; Guo, M.; Roembke, B.T.; Sintim, H.O. Nucleotide, c-di-GMP, c-di-AMP, cGMP, cAMP, (p)ppGpp signaling in bacteria and implications in pathogenesis. Chem. Soc. Rev. 2013, 42, 305–341. [Google Scholar] [CrossRef] [PubMed]
- Hauryliuk, V.; Atkinson, G.C.; Murakami, K.S.; Tenson, T.; Gerdes, K. Recent functional insights into the role of (p)ppGpp in bacterial physiology. Nat. Rev. Microbiol. 2015, 13, 298–309. [Google Scholar] [CrossRef]
- Bigger, J.W. Treatment of staphylococcal infections with penicillin by intermittent sterilisation. Lancet 1944, 244, 497–500. [Google Scholar] [CrossRef]
- Cohen, N.R.; Lobritz, M.A.; Collins, J.J. Microbial persistence and the road to drug resistance. Cell Host Microbe 2013, 13, 632–642. [Google Scholar] [CrossRef]
- Huemer, M.; Mairpady Shambat, S.; Brugger, S.D.; Zinkernagel, A.S. Antibiotic resistance and persistence—Implications for human health and treatment perspectives. EMBO Rep. 2020, 21, e51034. [Google Scholar] [CrossRef]
- Wilmaerts, D.; Windels, E.M.; Verstraeten, N.; Michiels, J. General Mechanisms Leading to Persister Formation and Awakening. Trends Genet 2019, 35, 401–411. [Google Scholar] [CrossRef]
- Atkinson, G.C.; Tenson, T.; Hauryliuk, V. The RelA/SpoT homolog (RSH) superfamily: Distribution and functional evolution of ppGpp synthetases and hydrolases across the tree of life. PLoS ONE 2011, 6, e23479. [Google Scholar] [CrossRef]
- Kaspy, I.; Glaser, G. Escherichia coli RelA Regulation via Its C-Terminal Domain. Front. Microbiol. 2020, 11, 572419. [Google Scholar] [CrossRef]
- Winther, K.S.; Roghanian, M.; Gerdes, K. Activation of the Stringent Response by Loading of RelA-tRNA Complexes at the Ribosomal A-Site. Mol. Cell. 2018, 70, 95–105.e4. [Google Scholar] [CrossRef] [PubMed]
- Takada, H.; Roghanian, M.; Caballero-Montes, J.; Van Nerom, K.; Jimmy, S.; Kudrin, P.; Trebini, F.; Murayama, R.; Akanuma, G.; Garcia-Pino, A.; et al. Ribosome association primes the stringent factor Rel for tRNA-dependent locking in the A-site and activation of (p)ppGpp synthesis. Nucleic Acids Res. 2021, 49, 444–457. [Google Scholar] [CrossRef] [PubMed]
- Pausch, P.; Abdelshahid, M.; Steinchen, W.; Schafer, H.; Gratani, F.L.; Freibert, S.A.; Wolz, C.; Turgay, K.; Wilson, D.N.; Bange, G. Structural Basis for Regulation of the Opposing (p)ppGpp Synthetase and Hydrolase within the Stringent Response Orchestrator Rel. Cell Rep. 2020, 32, 108157. [Google Scholar] [CrossRef] [PubMed]
- Hogg, T.; Mechold, U.; Malke, H.; Cashel, M.; Hilgenfeld, R. Conformational Antagonism between Opposing Active Sites in a Bifunctional RelA/SpoT Homolog Modulates (p)ppGpp Metabolism during the Stringent Response. Cell 2004, 117, 57–68. [Google Scholar] [CrossRef]
- Avarbock, A.; Avarbock, D.; Teh, J.-S.; Buckstein, M.; Wang, Z.-m.; Rubin, H. Functional Regulation of the Opposing (p)ppGpp Synthetase/Hydrolase Activities of RelMtb from Mycobacterium tuberculosis †. Biochemistry 2005, 44, 9913–9923. [Google Scholar] [CrossRef] [PubMed]
- Tamman, H.; Van Nerom, K.; Takada, H.; Vandenberk, N.; Scholl, D.; Polikanov, Y.; Hofkens, J.; Talavera, A.; Hauryliuk, V.; Hendrix, J.; et al. A nucleotide-switch mechanism mediates opposing catalytic activities of Rel enzymes. Nat. Chem. Biol. 2020, 16, 834–840. [Google Scholar] [CrossRef]
- Sinha, A.K.; Winther, K.S. The RelA hydrolase domain acts as a molecular switch for (p)ppGpp synthesis. Commun. Biol. 2021, 4, 434. [Google Scholar] [CrossRef]
- Wexselblatt, E.; Oppenheimer-Shaanan, Y.; Kaspy, I.; London, N.; Schueler-Furman, O.; Yavin, E.; Glaser, G.; Katzhendler, J.; Ben-Yehuda, S. Relacin, a novel antibacterial agent targeting the Stringent Response. PLoS Pathog. 2012, 8, e1002925. [Google Scholar] [CrossRef]
- Wexselblatt, E.; Katzhendler, J.; Saleem-Batcha, R.; Hansen, G.; Hilgenfeld, R.; Glaser, G.; Vidavski, R.R. ppGpp analogues inhibit synthetase activity of Rel proteins from Gram-negative and Gram-positive bacteria. Bioorg. Med. Chem. 2010, 18, 4485–4497. [Google Scholar] [CrossRef]
- Wexselblatt, E.; Kaspy, I.; Glaser, G.; Katzhendler, J.; Yavin, E. Design, synthesis and structure-activity relationship of novel Relacin analogs as inhibitors of Rel proteins. Eur. J. Med. Chem. 2013, 70, 497–504. [Google Scholar] [CrossRef]
- Andresen, L.; Varik, V.; Tozawa, Y.; Jimmy, S.; Lindberg, S.; Tenson, T.; Hauryliuk, V. Auxotrophy-based High Throughput Screening assay for the identification of Bacillus subtilis stringent response inhibitors. Sci. Rep. 2016, 6, 35824. [Google Scholar] [CrossRef] [PubMed]
- Beljantseva, J.; Kudrin, P.; Jimmy, S.; Ehn, M.; Pohl, R.; Varik, V.; Tozawa, Y.; Shingler, V.; Tenson, T.; Rejman, D.; et al. Molecular mutagenesis of ppGpp: Turning a RelA activator into an inhibitor. Sci. Rep. 2017, 7, 41839. [Google Scholar] [CrossRef] [PubMed]
- Dutta, N.K.; Klinkenberg, L.G.; Vazquez, M.J.; Segura-Carro, D.; Colmenarejo, G.; Ramon, F.; Rodriguez-Miquel, B.; Mata-Cantero, L.; Porras-De Francisco, E.; Chuang, Y.M.; et al. Inhibiting the stringent response blocks Mycobacterium tuberculosis entry into quiescence and reduces persistence. Sci. Adv. 2019, 5, eaav2104. [Google Scholar] [CrossRef] [PubMed]
- Conti, G.; Minneci, M.; Sattin, S. Optimised Synthesis of the Bacterial Magic Spot (p)ppGpp Chemosensor PyDPA. ChemBioChem 2019, 20, 1717–1721. [Google Scholar] [CrossRef]
- Civera, M.; Sattin, S. Homology Model of a Catalytically Competent Bifunctional Rel Protein. Front. Mol. Biosci. 2021, 8, 628596. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Erlanson, D.A.; Fesik, S.W.; Hubbard, R.E.; Jahnke, W.; Jhoti, H. Twenty years on: The impact of fragments on drug discovery. Nat. Rev. Drug Discov. 2016, 15, 605–619. [Google Scholar] [CrossRef]
- Lamoree, B.; Hubbard, R.E. Using Fragment-Based Approaches to Discover New Antibiotics. SLAS Discov. 2018, 23, 495–510. [Google Scholar] [CrossRef]
- Schrödinger. Maestro, Schrödinger Release 2018-1; Schrödinger, LLC: New York, NY, USA, 2018. [Google Scholar]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pK(a) prediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Duan, J.; Dixon, S.L.; Lowrie, J.F.; Sherman, W. Analysis and comparison of 2D fingerprints: Insights into database screening performance using eight fingerprint methods. J. Mol. Graph. Model. 2010, 29, 157–170. [Google Scholar] [CrossRef]
- Sastry, M.; Lowrie, J.F.; Dixon, S.L.; Sherman, W. Large-Scale Systematic Analysis of 2D Fingerprint Methods and Parameters to Improve Virtual Screening Enrichments. J. Chem. Inf. Model. 2010, 50, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2020, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossváry, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the ACM/IEEE Conference on Supercomputing (SC06), Tampa, FL, USA, 11–17 November 2006. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J.M.; Lu, C.; Dahlgren, M.K.; Mondal, S.; Chen, W.; Wang, L.; et al. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. [Google Scholar] [CrossRef] [PubMed]
- Huynh, K.; Partch, C.L. Analysis of protein stability and ligand interactions by thermal shift assay. Curr. Protoc. Protein. Sci. 2015, 79, 28.9.1–28.9.14. [Google Scholar] [CrossRef]
- Mechold, U.; Murphy, H.; Brown, L.; Cashel, M. Intramolecular Regulation of the Opposing (p)ppGpp Catalytic Activities of RelSeq, the Rel/Spo Enzyme from Streptococcus equisimilis. J. Bacteriol. 2002, 184, 2878–2888. [Google Scholar] [CrossRef]
- Meyer, B.; Peters, T. NMR Spectroscopy Techniques for Screening and Identifying Ligand Binding to Protein Receptors. Angew. Chem. Int. Ed. 2003, 42, 864–890. [Google Scholar] [CrossRef]
- Guzzetti, I.; Civera, M.; Vasile, F.; Araldi, E.M.; Belvisi, L.; Gennari, C.; Potenza, D.; Fanelli, R.; Piarulli, U. Determination of the binding epitope of RGD-peptidomimetics to αvβ3 and αIIbβ3 integrin-rich intact cells by NMR and computational studies. Org. Biomol. Chem. 2013, 11, 3886–3893. [Google Scholar] [CrossRef]
- Sattin, S.; Panza, M.; Vasile, F.; Berni, F.; Goti, G.; Tao, J.H.; Moroni, E.; Agard, D.; Colombo, G.; Bernardi, A. Synthesis of Functionalized 2-(4-Hydroxyphenyl)-3-methylbenzofuran Allosteric Modulators of Hsp90 Activity. Eur. J. Org. Chem. 2016, 2016, 3349–3364. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins 2004, 55, 351–367. [Google Scholar] [CrossRef]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins 2011, 79, 2794–2812. [Google Scholar] [CrossRef] [PubMed]
- Ponder, J.W.; Richards, F.M. An efficient newton-like method for molecular mechanics energy minimization of large molecules. J. Comput. Chem. 1987, 8, 1016–1024. [Google Scholar] [CrossRef]
- Grest, G.S.; Kremer, K. Molecular dynamics simulation for polymers in the presence of a heat bath. Phys. Rev. A 1986, 33, 3628–3631. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Vangunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular-Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Compound | Kd RelSeq 1–385 | Kd RelSeq SYNTH | Kd RelSeq HYD |
|---|---|---|---|---|
| 1 | ATP | 0.49 ± 0.09 (0.39 ± 0.04) 1 | ||
| 2 | GDP | 0.26 ± 0.06 (0.15 ± 0.01) 1 | ||
| 3 | B1 | No binding | No binding | No binding |
| 4 | B2 | Biphasic | No binding | 1.9 ± 0.7 |
| 5 | B3 | 3.4 ± 0.3 | 4.3 ± 0.4 | 3.3 ± 0.9 |
| 6 | B4 | No binding | No binding | No binding |
| 7 | A1 | 1.2 ± 0.3 | 10.8 ± 2.2 | No binding |
| 8 | A2 | No binding | No binding | No binding |
| 9 | A3 | 1.5 ± 0.1 | 5.5 ± 0.9 | No binding |
| 10 | A4 | 6.6 ± 0.1 | 9.8 ± 2.8 | No binding |
| 11 | A5 | 1.1 ± 0.2 | 2.2 ± 0.4 | No binding |
| 12 | A6 | 4.3 ± 1.1 | 6.5 ± 1.2 | No binding |
| 13 | A7 | 4.0 ± 0.9 | 4.3 ± 0.5 | No binding |
| 14 | I1 | 6.5 ± 1.1 | 9.6 ± 1.5 | No binding |
| 15 | I2 | 2.5 ± 0.6 | 5.5 ± 0.9 | No binding |
| 16 | I3 | 4.0 ± 0.5 | 9.9 ± 4.5 | No binding |
| 17 | I4 | 3.2 ± 0.7 | >15 | No binding |
| 18 | BO1 | 2.2 ± 0.3 | 2.7 ± 0.6 | No binding |
| 19 | BT1 | No binding | No binding | No binding |
| 20 | TP1 | 3.4 ± 0.8 | 8.3 ± 1.6 | No binding |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coppa, C.; Sorrentino, L.; Civera, M.; Minneci, M.; Vasile, F.; Sattin, S. New Chemotypes for the Inhibition of (p)ppGpp Synthesis in the Quest for New Antimicrobial Compounds. Molecules 2022, 27, 3097. https://doi.org/10.3390/molecules27103097
Coppa C, Sorrentino L, Civera M, Minneci M, Vasile F, Sattin S. New Chemotypes for the Inhibition of (p)ppGpp Synthesis in the Quest for New Antimicrobial Compounds. Molecules. 2022; 27(10):3097. https://doi.org/10.3390/molecules27103097
Chicago/Turabian StyleCoppa, Crescenzo, Luca Sorrentino, Monica Civera, Marco Minneci, Francesca Vasile, and Sara Sattin. 2022. "New Chemotypes for the Inhibition of (p)ppGpp Synthesis in the Quest for New Antimicrobial Compounds" Molecules 27, no. 10: 3097. https://doi.org/10.3390/molecules27103097
APA StyleCoppa, C., Sorrentino, L., Civera, M., Minneci, M., Vasile, F., & Sattin, S. (2022). New Chemotypes for the Inhibition of (p)ppGpp Synthesis in the Quest for New Antimicrobial Compounds. Molecules, 27(10), 3097. https://doi.org/10.3390/molecules27103097