
Borylated 2,3,4,5-Tetrachlorophthalimide and Their 2,3,4,5-Tetrachlorobenzamide Analogues: Synthesis, Their Glycosidase Inhibition and Anticancer Properties in View to Boron Neutron Capture Therapy

 , and
, and 
Abstract
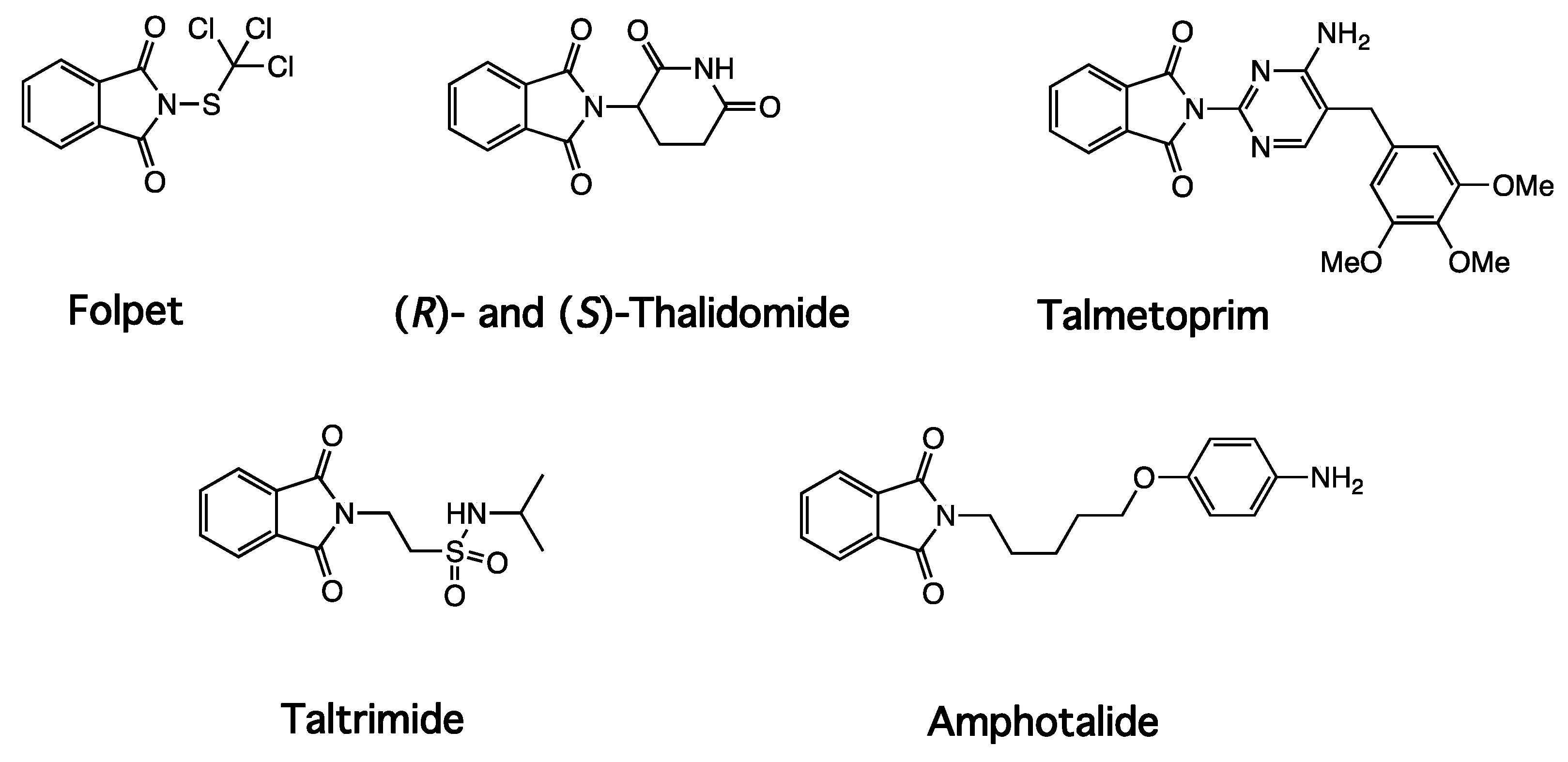
1. Introduction
2. Results and Discussion
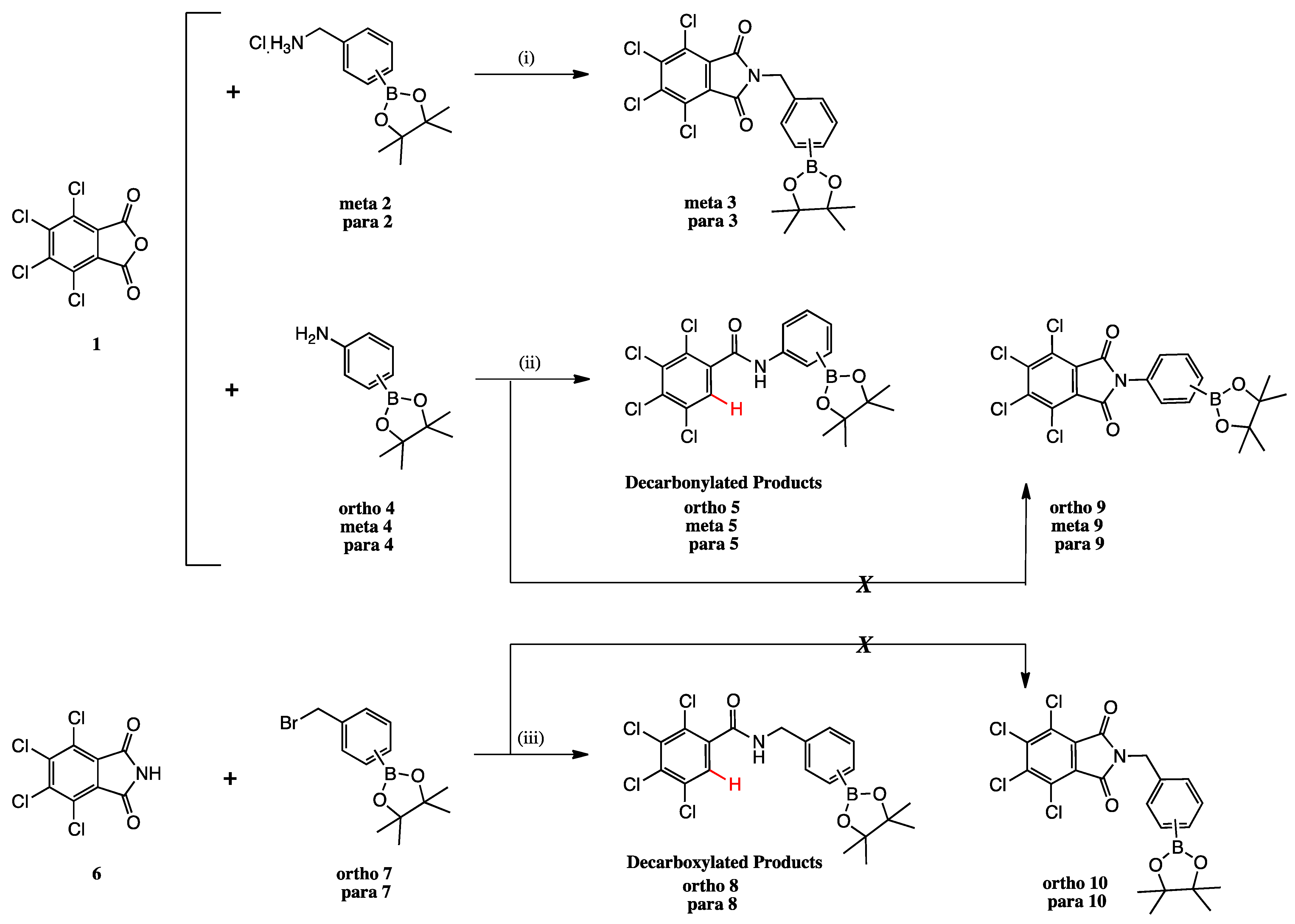
2.1. Summary of Synthetic Work
2.2. Decarbonylation Reaction
2.3. Decarboxylation Reaction to Benzamides
3. X-ray Crystallography Commentary
4. Glycosidase Assay
4.1. Glycosidases
4.2. Biological Activities for Phthalimides and Benzamides in the Literature
4.2.1. Phthalamides
4.2.2. 2,3,4,5-Tetrachlorophthalimides
4.2.3. Benzamides
4.3. Biological Activities for Our Drug Library Screened in Methanol
4.3.1. Controls
4.3.2. 2,3,4,5-Tetrachlorophthalimides
4.3.3. 2,3,4,5-Tetrachlorobenzamides
- (a)
- In the inhibition of bovine liver β-galactosidase, which is inhibited weakly by ortho 8 (IC50 278 μM), but para 8 inhibits the same enzyme with a good IC50 74.7 μM. This is the most potent drug in the small libraries reported in this communication.
- (b)
- In the inhibition of almond β-glucosidase by ortho 8, which is on the edge of moderate/weak, with an IC50 of 254 μM.
4.4. Biological Activities for Our Drug Library Screened in Water
5. Cancer Assay and Structure Activity Relationships
5.1. Analysis of the Percent Cell Growth Inhibition Data
5.2. Analysis of the GI50 Data
6. Experimental
6.1. Glycosidase Inhibition Experimental from Laboratory 1
6.2. Glycosidase Inhibition Experimental from Laboratory 2
6.3. Cancer Screening Experimental
6.4. Chemistry Experimental
6.4.1. General Experimental
6.4.2. Experimental
From Synthetic Strategy 1





From Synthetic Strategy 2


7. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hashimoto, Y. Thalidomide as a Multi-Template for Development of Biologically Active Compounds. Arch. Pharm. Chem. Life Sci. 2008, 341, 536–547. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Zhai, S.; Wang, H.; Tao, C.; Zhao, H.; Zhai, H. Efficient Synthesis of Phthalimides via Cobalt-Catalyzed C(sp2)−H Carbonylation of Benzoyl Hydrazides with Carbon Monoxide. Adv. Synth. Cat. 2018, 360, 3271–3276. [Google Scholar] [CrossRef]
- Bian, X.; Wang, Q.; Ke, C.; Zhao, G.; Li, Y. A new series of N2-substituted-5-(p-toluenesulfonylamino)phthalimide analogues as α-glucosidase inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 2022–2026. [Google Scholar] [CrossRef] [PubMed]
- Pascale, R.; Carocci, A.; Catalano, A.; Lentini, G.; Spagnoletta, A.; Cavalluzzi, M.M.; Santis, F.D.; Palma, A.D.; Scalera, V.; Franchini, C. New N-(phenoxydecyl)phthalimide derivatives displaying potent inhibition activity towards α-glucosidase. Bioorg. Med. Chem. 2010, 18, 5903–5914. [Google Scholar] [CrossRef]
- Sou, S.; Takahashi, H.; Yamasaki, R.; Kagechika, H.; Endo, Y.; Hashimoto, Y. α-Glucosidase Inhibitors with a 4,5,6,7-Tetrachlorophthalimide Skeleton Pendanted with a Cycloalkyl or Dicarba-closo-dodecaborane Group. Chem. Pharm. Bull. 2001, 49, 791–793. [Google Scholar] [CrossRef]
- Sou, S.; Mayumi, S.; Takahashi, H.; Yamasaki, R.; Kadoya, S.; Sodeoka, M.; Hashimoto, Y. Novel α-Glucosidase Inhibitors with a Tetrachlorophthalimide Skeleton. Bioorg. Med. Chem. Lett. 2000, 10, 1081–1084. [Google Scholar] [CrossRef]
- Baker, S.J.; Ding, C.Z.; Akama, T.; Zhang, Y.K.; Hernandez, V.; Xia, Y. Therapeutic potential of boron-containing compounds. Future Med. Chem. 2009, 1, 1275–1288. [Google Scholar] [CrossRef]
- Leśnikowski, Z.J. Recent developments with boron as a platform for novel drug design. Expert Opin. Drug Discov. 2016, 11, 569–578. [Google Scholar] [CrossRef]
- Jenkinson, S.F.; Thompson, A.L.; Simone, M.I. Methyl 2-(5,5-dimethyl-1,3,2-dioxaborinan-2-yl)-4-nitrobenzoate. Acta Cryst. 2012, E68, o2699–o2700. [Google Scholar] [CrossRef]
- Simone, M.I.; Houston, T.A. A brief overview of some latest advances in the applications of boronic acids. J. Glycom. Lipidom. 2014, 4, e124–e129. [Google Scholar] [CrossRef]
- Pappin, B.B.; Levonis, S.M.; Healy, P.C.; Kiefel, M.J.; Simone, M.I.; Houston, T.A. Crystallization Induced Amide Bond Formation Creates a Boron-Centred Spirocyclic System. Heterocycl. Commun. 2017, 23, 167–169. [Google Scholar] [CrossRef]
- Pappin, B.B.; Garget, T.A.; Healy, P.C.; Simone, M.I.; Kiefel, M.J.; Houston, T.A. Facile amidinations of 2-aminophenylboronic acid promoted by boronate ester formation. Org. Biomol. Chem. 2019, 17, 803–806. [Google Scholar] [CrossRef]
- Legge, W.J.; Shimadate, Y.; Sakoff, J.; Houston, T.A.; Kato, A.; Bernhardt, P.V.; Simone, M.I. Borylated methyl cinnamates: Green synthesis, characterization, crystallographic analysis and biological activities in glycosidase inhibition and in cancer cells lines—Of (E)-methyl 3-(2/3/4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)acrylates. Beil. Arch. 2021, 2021, 4. [Google Scholar]
- Sarazin, H.; Prudent, S.; Defoin, A.; Tarnus, C. Evaluation of 6-Deoxy–amino–sugars as Potent Glycosidase Inhibitors. Importance of the CH2OH(6) Group for Enzyme-Substrate Interaction. ChemistrySelect 2017, 2, 1484–1490. [Google Scholar] [CrossRef]
- Bonduelle, C.; Huang, J.; Mena-Barraga, T.; Mellet, C.O.; Decroocq, C.; Etame, E.; Heise, A.; Compain, P.; Lecommandoux, S. Iminosugar-based glycopolypeptides: Glycosidase inhibition with bioinspired glycoprotein analogue micellar self-assemblies. Chem. Commun. 2014, 50, 3350–3352. [Google Scholar] [CrossRef] [PubMed]
- Glawar, A.F.G.; Martínez, R.F.; Ayers, B.J.; Hollas, M.A.; Ngo, N.; Nakagawa, S.; Kato, A.; Butters, T.D.; Fleet, G.W.J.; Jenkinson, S.F. Structural essentials for β-N-acetylhexosaminidase inhibition by amides of prolines, pipecolic and azetidine carboxylic acids. Org. Biomol. Chem. 2016, 14, 10371–10385. [Google Scholar] [CrossRef] [PubMed]
- Cruz, F.P.D.; Newberry, S.; Jenkinson, S.F.; Wormald, M.R.; Butters, T.D.; Alonzi, D.S.; Nakagawa, S.; Becq, F.; Norez, C.; Nash, R.J.; et al. 4-C-Methyl-DAB and 4-C-Me-LAB—Enantiomeric alkyl branched pyrrolidine iminosugars—Are specific and potent α-glucosidase inhibitors; acetone as the sole protecting group. Tetrahedron Lett. 2011, 52, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Simone, M.I.; Soengas, R.G.; Jenkinson, S.F.; Evinson, E.L.; Nash, R.J.; Fleet, G.W.J. Synthesis of three branched iminosugars [(3R,4R,5S)-3-(hydroxymethyl)piperidine-3,4,5-triol, (3R,4R,5R)-3-(hydroxymethyl)piperidine-3,4,5-triol and (3S,4R,5R)-3-(hydroxymethyl)piperidine-3,4,5-triol] and a branched trihydroxynipecotic acid [(3R,4R,5R)-3,4,5-trihydroxypiperidine-3-carboxylic acid] from sugar lactones with a carbon substituent at C-2. Tetrahedron Asymm. 2012, 23, 401–408. [Google Scholar]
- Soengas, R.G.; Simone, M.I.; Hunter, S.; Nash, R.J.; Fleet, G.W.J. Hydroxymethyl-branched piperidines from hydroxymethyl-branched lactones: Synthesis and biological evaluation of 1,5-dideoxy-2-C-hydroxymethyl-1,5-imino-D-mannitol, 1,5-dideoxy-2-hydroxymethyl-1,5-imino-L-gulitol and 1,5-dideoxy-2-hydroxymethyl-1,5-imino-D-talitol. Eur. J. Org. Chem. 2012, 12, 2394–2402. [Google Scholar]
- Simone, M.I.; Mares, L.; Eveleens, C.; McCluskey, A.; Pappin, B.; Kiefel, M.; Houston, T.A. Back to (non)-Basic: An Update on Neutral and Charge-Balanced Glycosidase Inhibitors. Mini-Rev. Med. Chem. 2017, 18, 812–827. [Google Scholar] [CrossRef]
- Prichard, K.; Campkin, D.; O’Brien, N.; Kato, A.; Fleet, G.W.J.; Simone, M.I. Biological Activities of 3,4,5-Trihydroxypiperidines and their O- and N-Alkylated Derivatives. Chem. Biol. Drug Des. 2018, 2018, 1171–1197. [Google Scholar] [CrossRef]
- Brás, N.F.; Cerqueira, N.M.F.S.A.; Ramos, M.J.; Fernandes, P.A. Glycosidase inhibitors: A patent review (2008–2013). Expert Opin. Ther. Pat. 2014, 24, 857–874. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Nakai, K.; Matsumura, A. Boron Neutron Capture Therapy for Glioblastoma. Cancer Lett. 2008, 262, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Zavjalov, E.; Zaboronok, A.; Kanygin, V.; Kasatova, A.; Kichigin, A.; Mukhamadiyarov, R.; Razumov, I.; Sycheva, T.; Mathis, B.J.; Maezono, S.E.B.; et al. Accelerator-based Boron Neutron Capture Therapy for Malignant Glioma: A Pilot Neutron Irradiation Study using Boron Phenylalanine, Sodium Borocaptate and Liposomal Borocaptate with a Heterotopic U87 Glioblastoma Model in SCID Mice. Int. J. Radiat. Biol. 2020, 96, 868–878. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, Y.; Pooh, K.; Kobayashi, T.; Kageji, T.; Uyama, S.; Matsumura, A.; Kumada, H. Clinical Review of the Japanese Experience with Boron Neutron Capture Therapy and a Proposed Strategy using Epithermal Neutron Beams. J. Neuro-Oncol. 2003, 62, 87–99. [Google Scholar] [CrossRef]
- Nordlander, B.W.; Cass, W.E. The Esterification of tetrachlorophthalic Anhydride. J. Am. Chem. Soc. 1947, 69, 2679. [Google Scholar] [CrossRef]
- Harvey, P.G.; Smith, F.; Stacey, M.; Tatlow, J.C. Studies on the Nuclear Chlorination of Aromatic Compounds. J. Appl. Chem. 1954, 4, 325. [Google Scholar] [CrossRef]
- Tust, P. Ueber Tetrachlorobenzoësäure und einige Derivate derselben. Ber. Dtsch. Chem. Ges. Berl. 1887, 20, 2439–2442. [Google Scholar] [CrossRef]
- Beilstein, F.; Kuhlberg, A. Untersuchungen über Isomerie in der Benzoëreihe. Justus Liebigs Ann. Chem. 1869, 152, 224–246. [Google Scholar] [CrossRef][Green Version]
- Pagani, G.; Baruffini, A.; Mazza, M.; Vicarini, L.; Caccialanza, G. Sull’attività fitotossica selettiva di N,N-di,sec.butilamidi di acidi benzoici variamente alogenati. Il Farm.-Ed. Sc. 1973, 28, 570–589. [Google Scholar]
- Medina, I.-M.R.Y.; Rohdenburg, M.; Mostaghimi, F.; Grabowsky, S.; Swiderek, P.; Beckmann, J.; Hoffmann, J.; Dorcet, V.; Hissler, M.; Staubitz, A. Tuning the Optoelectronic Properties of Stannoles by the Judicious Choice of the Organic Substituents. Inorg. Chem. 2018, 57, 12562–12575. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Winands, T.; Doltsinis, N.L.; Li, Y.; Wang, Z. A Decatwistacene with an Overall 170° Torsion. Angew. Chem. Int. Ed. 2017, 56, 15373–15377. [Google Scholar] [CrossRef] [PubMed]
- Hanefeld, M.; Schaper, F. The role of alpha-glucosidase inhibitors (Acarbose). In Pharmacotherapy of Diabetes: New Developments; Mogesen, C.E., Ed.; Springer: Boston, MA, USA, 2007; pp. 143–152. [Google Scholar]
- Laar, F.A.V.D.; Lucassen, P.L.; Akkermans, R.P.; Lisdonk, E.H.V.D.; Rutten, G.E.; Weel, C.V. α-Glucosidase Inhibitors for Patients with Type 2 Diabetes. Diabetes Care 2005, 28, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhang, X.; Soloveva, V.; Warren, T.; Guo, F.; Wu, S.; Lu, H.; Guo, J.; Su, Q.; Shen, H.; et al. Enhancing the antiviral potency of ER α-glucosidase inhibitor IHVR-19029 against hemorrhagic fever viruses in vitro and in vivo. Antivir. Res. 2018, 150, 112–122. [Google Scholar] [CrossRef]
- Chang, J.; Block, T.M.; Guo, J.-T. Antiviral therapies targeting host ER α-glucosidases: Current status and future directions. Antivir. Res. 2013, 99, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Karpas, A.; Fleet, G.W.J.; Dwek, R.A.; Petursson, S.; Namgoong, S.K.; Ramsden, N.G.; Jacob, G.S.; Rademacher, T.W. Aminosugar derivatives as potential anti-human immunodeficiency virus agents. Proc. Natl. Acad. Sci. USA 1988, 85, 9229–9233. [Google Scholar] [CrossRef]
- Sunkara, P.S.; Bowlin, T.L.; Liu, P.S.; Sjoerdsma, A. Antiretroviral activity of castanospermine and deoxynojirimycin, specific inhibitors of glycoprotein processing. Biochem. Biophys. Res. Commun. 1987, 148, 206–210. [Google Scholar] [CrossRef]
- Bernacki, R.; Niedbala, M.; Korytnyk, W. Glycosidases in cancer and invasion. Cancer Metastasis 1985, 4, 81–102. [Google Scholar] [CrossRef]
- Martín-Banderas, L.; Holgado, M.A.; Durán-Lobato, M.; Infante, J.J.; Álvarez-Fuentes, J.; Fernández-Arévalo, M. Role of Nanotechnology for Enzyme Replacement Therapy in Lysosomal Diseases. A Focus on Gaucher’s Disease. Curr. Med. Chem. 2016, 23, 929–952. [Google Scholar] [CrossRef]
- Raben, N.; Fukuda, T.; Gilbert, A.L.; Jong, D.D.; Thurberg, B.L.; Mattaliano, R.J.; Meikle, P.; Hopwood, J.J.; Nagashima, K.; Nagaraju, K.; et al. Replacing Acid α-Glucosidase in Pompe Disease: Recombinant and Transgenic Enzymes are Equipotent, but Neither Completely Clears Glycogen from Type II Muscle Fibers. Mol. Ther. 2005, 11, 48–56. [Google Scholar] [CrossRef]
- Cox, T.M. Gaucher’s Disease; Academic Press: Cambridge, MA, USA, 2001; pp. 753–757. [Google Scholar]
- Mena-Barragán, T.; García-Moreno, M.I.; Sevšek, A.; Okazaki, T.; Nanba, E.; Higaki, K.; Martin, N.I.; Pieters, R.J.; Fernández, J.M.G.; Mellet, C.O. Probing the Inhibitor versus Chaperone Properties of sp2-Iminosugars towards Human β-Glucocerebrosidase: A Picomolar Chaperone for Gaucher Disease. Molecules 2018, 23, 927. [Google Scholar] [CrossRef] [PubMed]
- Hinek, A.; Zhang, S.; Smith, A.C.; Callahan, J.W. Impaired Elastic-Fiber Assembly by Fibroblasts from Patients with Either Morquio B Disease or Infantile GM1-Gangliosidosis Is Linked to Deficiency in the 67-kD Spliced Variant of β-Galactosidase. Am. J. Hum. Genet. 2000, 67, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.G. Chapter 33—Disorders of Glycoprotein Degradation: Sialidosis, Fucosidosis, α-Mannosidosis, β-Mannosidosis, and Aspartylglycosaminuria. In Rosenberg’s Molecular and Genetic Basis of Neurological and Psychiatric Disease, 5th ed.; Academic Press: Cambridge, MA, USA, 2015; pp. 369–383. [Google Scholar]
- Stütz, A.E.; Wrodnigg, T.M. Chapter Four—Carbohydrate-Processing Enzymes of the Lysosome: Diseases Caused by Misfolded Mutants and Sugar Mimetics as Correcting Pharmacological Chaperones. In Advances in Carbohydrate Chemistry and Biochemistry; Academic Press: Cambridge, MA, USA, 2016; Volume 73, pp. 225–302. [Google Scholar]
- Malm, D.; Nilssen, Ø. Alpha-mannosidosis. Orphanet J. Rare Dis. 2008, 3, 21. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Clavijo, E.; Carmona, A.T.; Moreno-Vargas, A.J.; Molina, L.; Robina, I. Syntheses and Biological Activities of Iminosugars as α-L-Fucosidase Inhibitors. Curr. Org. Synth. 2011, 8, 102–133. [Google Scholar] [CrossRef]
- Mohamed, F.E.; Al-Gazali, L.; Al-Jasmi, F.; Ali, B.R. Pharmaceutical Chaperones and Proteostasis Regulators in the Therapy of Lysosomal Storage Disorders: Current Perspective and Future Promises. Front. Pharmacol. 2017, 8, 448. [Google Scholar] [CrossRef]
- National Centre for Advancing Translational Sciences, Genetic and Rare Diseases Information Centre. Available online: https://rarediseases.info.nih.gov/diseases/10372/trehalase-deficiency (accessed on 21 May 2022).
- Gibson, R.P.; Gloster, T.M.; Roberts, S.; Warren, R.A.J.; Gracia, I.S.D.; García, Á.; Chiara, J.L.; Davies, G.J. Molecular Basis for Trehalase Inhibition Revealed by the Structure of Trehalase in Complex with Potent Inhibitors. Angew. Chem. Int. Ed. 2007, 46, 4115–4119. [Google Scholar] [CrossRef]
- Matalon, R.; Matalon, K.M. Mucopolysacharidoses. In Encyclopedia of the Neurological Sciences; Academic Press: Cambridge, MA, USA, 2003; pp. 237–241. [Google Scholar]
- Batool, F.; Khan, M.A.; Shaikh, N.N.; Iqbal, S.; Akbar, S.; Fazal-ur-rehman, S.; Choudhary, M.I.; Basha, F.Z. New Benzamide Analogues of Metronidazole-tethered Triazoles as Non-sugar Based Inhibitors of β-Glucuronidase. ChemistrySelect 2019, 4, 8634–8637. [Google Scholar] [CrossRef]
- Guillotin, L.; Kim, H.; Traore, Y.; Moreau, P.; Lafite, P.; Coquoin, V.; Nuccio, S.; Vaumas, R.D.; Daniellou, R. Biochemical Characterization of the α-L-Rhamnosidase DtRha from Dictyoglomus thermophilum: Application to the Selective Derhamnosylation of Natural Flavonoids. ACS Omega 2019, 4, 1916–1922. [Google Scholar] [CrossRef]
- Yadav, V.; Yadav, P.K.; Yadav, S.; Yadav, K.D.S. α-L-Rhamnosidase: A review. Proc. Biochem. 2010, 45, 1226–1235. [Google Scholar] [CrossRef]
- Souza, P.M.D.; Sales, P.M.D.; Simeoni, L.A.; Silva, E.C.; Silveira, D.; Magalhães, P.D.O. Inhibitory Activity of α-Amylase and α-Glucosidase by Plant Extracts from the Brazilian Cerrado. Planta Med. 2012, 78, 393–399. [Google Scholar] [CrossRef]
- Aoyama, T.; Kojima, F.; Imada, C.; Muraoka, Y.; Naganawa, H.; Okami, Y.; Takeuchi, T.; Aoyagi, T. Pyrostatins A and B, New inhibitors of N-Acetyl-β-D-Glucosaminidase, produced by Streptomyces sp. SA-3501. J. Enz. Inhib. 1995, 8, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Shitara, E.; Takeuchi, T. Enantioselective synthesis of a new family of α-L-fucosidase inhibitors. Tetrahedron Lett. 1999, 40, 2351–2354. [Google Scholar] [CrossRef]
- Takahashi, H.; Sou, S.; Yamasaki, R.; Sodeoka, M.; Hashimoto, Y. α-Glucosidase inhibitors with a phthalimide skeleton: Structure-activity relationship study. Chem. Pharm. Bull. 2000, 48, 1494–1499. [Google Scholar] [CrossRef]
- Pluempanupat, W.; Adisakwattana, S.; Yibchok-Anun, S.; Chavasiri, W. Synthesis of N-phenylphthalimide Derivatives as alpha-Glucosidase Inhibitors. Arch. Pharm. Res. 2007, 30, 1501–1506. [Google Scholar] [CrossRef] [PubMed]
- Motoshima, K.; Noguchi-Yachide, T.; Sugita, K.; Hashimoto, Y.; Ishikawa, M. Separation of a-glucosidase-inhibitory and liver X receptor-antagonistic activities of phenethylphenyl phthalimide analogs and generation of LXRa-selective antagonists. Bioorg. Med. Chem. 2009, 17, 5001–5014. [Google Scholar] [CrossRef]
- Dodo, K.; Aoyama, A.; Noguchi-Yachide, T.; Makishima, M.; Miyachi, H.; Hashimoto, Y. Co-existence of α-glucosidase-inhibitory and liver X receptor-regulatory activities and their separation by structural development. Bioorg. Med. Chem. 2008, 16, 4272–4285. [Google Scholar] [CrossRef]
- Noguchi-Yachide, T.; Aoyama, A.; Makishima, M.; Miyachi, H.; Hashimoto, Y. Liver X receptor antagonists with a phthalimide skeleton derived from thalidomide-related glucosidase inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 3957–3961. [Google Scholar] [CrossRef]
- Noguchi-Yachide, T.; Miyachi, H.; Aoyama, H.; Aoyama, A.; Makishima, M.; Hashimoto, Y. Structural Development of Liver X Receptor (LXR) Antagonists Derived from Thalidomide-Related Glucosidase Inhibitors. Chem. Pharm. Bull. 2007, 55, 1750–1754. [Google Scholar] [CrossRef][Green Version]
- Mbarki, S.; Hallaoui, M.E.; Dguigui, K. 3D-QSAR for α-glucosidase inhibitory activity of n-(phenoxyalkyl) phthalimide derivatives. Int. J. Rec. Res. Appl. Stud. 2012, 11, 395–401. [Google Scholar]
- Wang, G.; Peng, Y.; Xie, Z.; Wang, J.; Chen, M. Synthesis, α-glucosidase inhibition and molecular docking studies of novel thiazolidine-2,4-dione or rhodanine derivatives. MedChemComm 2017, 8, 1477–1484. [Google Scholar] [CrossRef]
- Sadat-Ebrahimi, S.E.; Rahmani, A.; Mohammadi-Khanaposhtani, M.; Jafari, N.; Mojtabavi, S.; Faramarzi, M.A.; Emadi, M.; Yahya-Meymandi, A.; Larijani, B.; Biglar, M.; et al. New phthalimide-benzamide-1,2,3-triazole hybrids; design, synthesis, α-glucosidase inhibition assay, and docking study. Med. Chem. Res. 2020, 29, 868–876. [Google Scholar] [CrossRef]
- Bian, X.; Fan, X.; Ke, C.; Luan, Y.; Zhao, G.; Zeng, A. Synthesis and alpha-glucosidase inhibitory activity evaluation of N-substituted aminomethyl-b-D-glucopyranosides. Bioorg. Med. Chem. 2013, 21, 5442–5450. [Google Scholar] [CrossRef] [PubMed]
- Hoos, R.; Naughton, A.B.; Thiel, W.; Vasella, A.; Weber, W.; Rupitz, K.; Withers, S.G. D-Gluconhydroximo-1,5-Lactam And Related N-Arylcarbamates Theoretical Calculations, Structure, Synthesis, and Inhibitory Effect on Beta-Glucosidases. Helv. Chim. Acta 1993, 76, 2666–2686. [Google Scholar] [CrossRef]
- Awolade, P.; Cele, N.; Kerru, N.; Gummidi, L.; Oluwakemi, E.; Singh, P. Therapeutic significance of b-glucuronidase activity and its inhibitors: A review. Eur. J. Med. Chem. 2020, 187, 111921. [Google Scholar] [CrossRef] [PubMed]
- Ferhati, X.; Matassini, C.; Fabbrini, M.G.; Goti, A.; Morrone, A.; Cardona, F.; Moreno-Vargas, A.J.; Paoli, P. Dual targeting of PTP1B and glucosidases with new bifunctional iminosugar inhibitors to address type 2 diabetes. Bioorg. Chem. 2019, 87, 534–549. [Google Scholar] [CrossRef] [PubMed]
- Glenister, A.; Simone, M.I.; Hambley, T.W. A Warburg effect targeting vector designed to increase the uptake of compounds by cancer cells demonstrates glucose and hypoxia dependent uptake. PLoS ONE 2019, 14, e0217712. [Google Scholar] [CrossRef]
- Glenister, A.; Chen, C.K.J.; Renfrew, A.K.; Simone, M.I.; Hambley, T.W. Warburg Effect Targeting Cobalt(III) Cytotoxin Chaperone Complexes. J. Med. Chem. 2021, 64, 2678–2690. [Google Scholar] [CrossRef]
- Venhuizen, J.R. Idaho National Engineering Laboratory, INEL BNCT Research Program Annual Report; 1995. Available online: https://www.osti.gov/biblio/421334 (accessed on 20 February 2022).
- Koo, M.-S.; Ozawa, T.; Santos, R.A.; Lamborn, K.R.; Bollen, A.W.; Deen, D.F.; Kahl, S.B. Synthesis and Comparative Toxicology of a Series of Polyhedral Borane Anion-Substituted Tetraphenyl Porphyrins. J. Med. Chem. 2007, 50, 820–827. [Google Scholar] [CrossRef]
- Cano, W.G.; Solares, G.R.; Dipetrillo, T.A.; Meylaerts, S.A.G.; Lin, S.C.; Zamenhof, R.G.; Saris, S.C.; Duker, J.S.; Goad, E.; Madoc-Jones, H.; et al. Toxicity Associated with Boronophenylalanine and Cranial Neutron Irradiation. Radiat. Oncol. Investig. 1995, 3, 108–118. [Google Scholar] [CrossRef]
- Pereira, G.L.; Siqueira, J.A.; Batista-Silva, W.; Cardoso, F.B.; Nunes-Nesi, A.; Araújo, W.L. Boron: More Than an Essential Element for Land Plants? Front. Plant Sci. 2021, 11, 610307. [Google Scholar] [CrossRef]
- Warington, K. The effect of boric acid and borax on the broad bean and certain other plants. Ann. Bot. 1923, 37, 629–672. [Google Scholar] [CrossRef]
- Uluisika, I.; Karakaya, H.C.; Koc, A. The Importance of Boron in Biological Systems. J. Trace Elem. Med. Biol. 2018, 45, 156–162. [Google Scholar] [CrossRef] [PubMed]
- U.S. Department of Health and Human Services; Public Health Service Agency for Toxic Substances and Disease Registry. Division of Toxicology and Environmental Medicine/Applied Toxicology Branch 1600 Clifton Road NE, Mailstop F-32, Atlanta, Georgia 30333, Toxicological Profile for Benzene. 2007. Available online: https://www.atsdr.cdc.gov/ (accessed on 20 February 2022).
- Centers for Disease Control and Prevention; The National Institute for Occupational Safety and Health (NIOSH). Benzene (Immediately Dangerous to Life or Health Concentrations (IDLH)). Available online: https://www.cdc.gov/niosh/idlh/71432.html (accessed on 20 February 2022).
- Thermo Fisher Scientific, Safety Data Sheet, Phenylboronic Acid. Available online: https://www.fishersci.com/store/msds?partNumber=AC130360100&productDescription=PHENYLBORIC+ACID%2C+98%2B%25+10GR&vendorId=VN00032119&countryCode=US&language=en (accessed on 20 February 2022).
- Kessler, M.; Acuto, O.; Storelli, C.; Murer, H.; Müller, M.; Semenza, G. A modified procedure for the rapid preparation of efficiently transporting vesicles from small intestinal brush border membranes. Their use in investigating some properties of D-glucose and choline transport systems. Biochim. Biophys. Acta 1978, 506, 136–154. [Google Scholar] [CrossRef]
- Gottlieb, H.E.; Kolyar, V.; Nudelman, A. NMR chemical shifts of common laboratory solvents as trace impurities. J. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Cryst. 2020, 53, 226–235. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Enzyme | α-Glucosidase | β-Glucosidase | α-Galactosidase | β-Galactosidase | α-Mannosidase | β-Mannosidase | α-L-Rhamnosidase | α-L-Fucosidase | β-Glucuronidase | Trehalase | Amyloglucosidase | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Rice | Yeast | Rat Intestinal Maltase | Almond | Bovine Liver | Coffee Beans | Bovine Liver | Jack Bean | Snail | Penicillium decumbens | Bovine Kidney | E. coli | Bovine Liver | Porcine Kidney | A. niger | ||
 | BSH | aNI b(0%) | aNI b(6.9%) | aNI b(0%) | aNI b (0%) | aNI b (15%) | aNI b (12.3%) | aNI b (0%) | aNI b (7.9%) | aNI b (19.2%) | aNI b (0.2%) | aNI b (0%) | aNI b (6%) | aNI b (19.6%) | aNI b (4.2%) | aNI b (0%) |
| 10B-BSH | aNI b(0%) | aNI b (5.6%) | aNI b(0%) | aNI b (0%) | aNI b (11.9%) | aNI b (3.9%) | aNI b (22.3%) | aNI b (7.7%) | aNI b (15%) | aNI b (0%) | aNI b (6.5%) | aNI b (3.2%) | aNI b (12.5%) | aNI b (2.3%) | aNI b (0%) | |
 | BPA | cNI d(0%) | cNI d (0%) | cNI d(0%) | cNI d (0%) | cNI d (0%) | cNI d (4.3%) | cNI d (10.9%) | cNI d (1.1%) | cNI d (2.1%) | cNI d (0%) | cNI d (0%) | cNI d (0.5%) | cNI d (7.4%) | cNI d (0%) | cNI d (0%) |
| 10B-BPA | cNI d(0%) | cNI d (0%) | cNI d (0%) | cNI d (0%) | cNI d (14.2%) | cNI d (1.2%) | cNI d (0%) | cNI d (0%) | cNI d (0.3%) | cNI d (0%) | cNI d (0%) | cNI d (2.4%) | cNI d (0%) | cNI d (0%) | cNI d (0%) | |
 para 3 | aNI b(5.6%) | aNI b (0%) | aNI b (12.2%) | aNI b (0.7%) | aNI b (4.0%) | aNI b (0.2%) | aNI b (8,2%) | aNI b (0%) | aNI b (0%) | aNI b (0%) | aNI b (0%) | aNI b (2.2%) | aNI b (1.0%) | aNI b (0%) | aNI b (1.0%) | |
 meta 3 | cNI d(5.1%) | cNI d (0%) | cNI d (22.1%) | cNI d (5.0%) | cNI d (18.8%) | cNI d (3.5%) | cNI d (12.4%) | cNI d (2.7%) | cNI d (1.1%) | cNI d (0%) | cNI d (4.7%) | cNI d (6.1%) | cNI d (5.7%) | aNI b (0%) | cNI d (3.2%) | |
 para 5 | aNI b(0%) | aNI b(0%) | 188 | aNI b (17.1%) | 543 | aNI b (35.8%) | 333 | aNI b (19.3%) | aNI b (30.4%) | aNI b (14.4%) | aNI b (5.1%) | aNI b (45.5%) | aNI b (36.5%) | aNI b (0%) | aNI b (0%) | |
 meta 5 | aNI b(45.5%) | aNI b(12.8%) | 274 | aNI b (30.2%) | 175 | aNI b (30.2%) | 213 | aNI b (7.6%) | aNI b (39.1%) | aNI b (2.2%) | aNI b (3.3%) | 932 | aNI b (6.0%) | aNI b (0%) | aNI b (2.8%) | |
 ortho 5 | cNI d(0%) | cNI d(0%) | cNI d(3%) | cNI d (11.6%) | cNI d (21.2%) | cNI d (0%) | cNI d (0%) | cNI d (3.4%) | cNI d (0%) | cNI d (0%) | cNI d (0%) | cNI d (3.8%) | cNI d (0%) | cNI d (1.2%) | cNI d (0%) | |
 para 8 | aNI b(2.1%) | aNI b(0%) | 207 | aNI b (29.6%) | 702 | aNI b (18.0%) | 74.7 | aNI b (4.5%) | aNI b (34.7%) | aNI b (3.3%) | aNI b (7.2%) | aNI b (34.7%) | aNI b (18.8%) | aNI b (4.1%) | aNI b (0%) | |
 ortho 8 | aNI b (0%) | aNI b (0%) | 305 | 254 | 922 | aNI b (114.4%) | 278 | aNI b (12.5%) | aNI b (39%) | aNI b (6.2%) | aNI b (19.9%) | aNI b (41.7%) | aNI b (16.3%) | aNI b (16.8%) | aNI b (0%) | |
| For BSH and BPA. aNI: No inhibition (less than 50% inhibition at 1000 μM). | cNI: No inhibition (less than 50% inhibition at 100 μM). | For our drugs: aNI: No inhibition (less than 50% inhibition at 100 μM). | cNI: No inhibition (less than 50% inhibition at 10 μM). | |||||||||||||
| b ( ): inhibition % at 1000 μM. | d ( ): inhibition % at 100 μM. | b ( ): inhibition % at 100 μM. | d ( ): inhibition % at 10 μM. | |||||||||||||
| Compound | Enzyme | Appearance of the 1 mg/mL Aqueous Solution Tested | α-Glucosidase | β-Glucosidase | α-Mannosidase | N-Acetyl-β-Glucosaminidase | β-Glucuronidase | |
|---|---|---|---|---|---|---|---|---|
| Yeast | Bacillus | Almond | JACK BEAN | Bovine Kidney | Bovine Liver | |||
 | BSH | in solution | 59 | 48.1 | 47.3 | −18.7 | 37.1 | 31.6 |
| 10B-BSH | in solution | 65.9 | 53 | 49.9 | −16.8 | 40.9 | 44.1 | |
 | BPA | some in solution with undissolved sediment | 2.9 | 19.9 | 3.9 | 0.6 | 6.7 | −0.7 |
| 10B-BPA | some in solution with undissolved sediment | 3.4 | 19.5 | 3 | −0.7 | 6.3 | −1 | |
 para 3 | some in solution with undissolved sediment | 29.3 | 17.9 | 15.4 | 16.5 | 8.3 | 2.1 | |
 meta 3 | opaque suspension | 32.2 | 31.7 | 19.3 | 30.3 | 17.1 | 5.8 | |
 para 5 | some in solution with undissolved sediment | 38.4 | 20 | 11.3 | −8.2 | 20.6 | −2.2 | |
 meta 5 | some in solution with undissolved sediment | 82.8 | 54.1 | 18.7 | 2.2 | 30.6 | −5.7 | |
 ortho 5 | some in solution with undissolved sediment | 17.3 | 14.4 | 21 | 0 | 28.8 | −3.5 | |
 para 8 | some in solution with undissolved sediment | 99.5 | 62.2 | 19.1 | −3.3 | 15 | −1.1 | |
 ortho 8 | some in solution with undissolved sediment | 68.9 | 23.7 | 83.4 | −6.1 | 28.3 | −6.3 | |
| Compound | HT29 | U87 | MCF-7 | A2780 | H460 | A431 | Du145 | BE2-C | SJ-G2 | MIA-Pa-Ca2 | MCF10A | Mode of Action | Selective |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Colon Carcinoma | Glioblastoma | Breast Carcinoma | Ovarian Carcinoma | Lung Carcinoma | Skin Carcinoma | Prostate Carcinoma | Neuroblastoma | Glioblastoma | Pancreatic Carcinoma | Breast (Normal) | |||
| BSH | 3 ± 2 | <0 | 15 ± 3 | 2 ± 5 | 8 ± 2 | <0 | 0 ± 8 | 10 ± 6 | 3 ± 8 | 2 ± 6 | 8 ± 3 | NA | |
| 10B-BSH | 5 ± 1 | 0 ± 2 | 5 ± 3 | 5 ± 4 | 4 ± 2 | <0 | 7 ± 7 | 8 ± 7 | 1 ± 9 | 2 ± 4 | 13 ± 4 | NA | |
| BPA | 14 ± 0 | <0 | <0 | 4 ± 1 | 7 ± 8 | 4 ± 6 | 19 ± 10 | 13 ± 10 | 5 ± 8 | 3 ± 3 | 4 ± 1 | NA | |
| 10B-BPA | 15 ± 4 | <0 | 1 ± 3 | 8 ± 4 | 8 ± 5 | 4 ± 4 | 15 ± 9 | 10 ± 6 | 5 ± 10 | 11 ± 3 | <0 | NA | |
 para 3 | 99 ± 2 | >100 | >100 | >100 | 100 ± 2 | 85 ± 8 | >100 | >100 | >100 | 98 ± 3 | >100 | A | |
| 5.3 ± 1.3 | 4.2 ± 1.0 | 3.0 ± 1.3 | 11 ± 1.1 | 13 ± 0.33 | 18 ± 2.3 | 5.5 ± 0.23 | 8.1 ± 1.7 | 6.6 ± 2.4 | 9.2 ± 1.9 | 12 ± 0.58 | |||
 meta 3 | 53 ± 3 | 70 ± 8 | 67 ± 7 | 70 ± 2 | 42 ± 6 | 48 ± 3 | 43 ± 13 | 45 ± 2 | 35 ± 4 | 29 ± 2 | 69 ± 7 | A | |
| 26 ± 2.6 | 19 ± 1.5 | 23 ± 3.7 | 19 ± 0.33 | 33 ± 2.3 | 28 ± 0.85 | 30 ± 0.41 | 29 ± 0.58 | 35 ± 1.7 | 38 ± 2.9 | 18 ± 1.0 | |||
 para 5 | 69 ± 7 | 21 ± 16 | 42 ± 5 | 50 ± 1 | 54 ± 2 | 64 ± 4 | 1 ± 13 | 54 ± 1 | 28 ± 6 | 52 ± 1 | 2 ± 11 | A | Y |
 meta 5 | 10 ± 7 | <0 | 16 ± 4 | 49 ± 4 | 18 ± 6 | 9 ± 9 | <0 | 3 ± 4 | <0 | 8 ± 5 | <0 | A | Y |
 ortho 5 | 98 ± 2 | 58 ± 5 | >100 | 97 ± 2 | >100 | 100 ± 2 | >100 | >100 | 92 ± 5 | 100 ± 3 | 58 ± 9 | B | Y |
| 14 ± 1.7 | 25 ± 2.6 | 12 ± 1.2 | 16 ± 1.2 | 13 ± 1.0 | 11 ± 1.8 | 16 ± 1.9 | 16 ± 0.58 | 16 ± 0.91 | 14 ± 0.25 | 27 ± 2.8 | |||
 para 8 | 36 ± 9 | 14 ± 9 | 18 ± 5 | 58 ± 2 | 21 ± 3 | <0 | <0 | 15 ± 5 | 8 ± 10 | 23 ± 6 | 2 ± 6 | A | y |
 ortho 8 | 5 ± 10 | <0 | <0 | 19 ± 10 | 0 ± 8 | <0 | <0 | 2 ± 5 | <0 | 6 ± 10 | <0 | A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campkin, D.M.; Shimadate, Y.; Bartholomew, B.; Bernhardt, P.V.; Nash, R.J.; Sakoff, J.A.; Kato, A.; Simone, M.I. Borylated 2,3,4,5-Tetrachlorophthalimide and Their 2,3,4,5-Tetrachlorobenzamide Analogues: Synthesis, Their Glycosidase Inhibition and Anticancer Properties in View to Boron Neutron Capture Therapy. Molecules 2022, 27, 3447. https://doi.org/10.3390/molecules27113447
Campkin DM, Shimadate Y, Bartholomew B, Bernhardt PV, Nash RJ, Sakoff JA, Kato A, Simone MI. Borylated 2,3,4,5-Tetrachlorophthalimide and Their 2,3,4,5-Tetrachlorobenzamide Analogues: Synthesis, Their Glycosidase Inhibition and Anticancer Properties in View to Boron Neutron Capture Therapy. Molecules. 2022; 27(11):3447. https://doi.org/10.3390/molecules27113447
Chicago/Turabian StyleCampkin, David M., Yuna Shimadate, Barbara Bartholomew, Paul V. Bernhardt, Robert J. Nash, Jennette A. Sakoff, Atsushi Kato, and Michela I. Simone. 2022. "Borylated 2,3,4,5-Tetrachlorophthalimide and Their 2,3,4,5-Tetrachlorobenzamide Analogues: Synthesis, Their Glycosidase Inhibition and Anticancer Properties in View to Boron Neutron Capture Therapy" Molecules 27, no. 11: 3447. https://doi.org/10.3390/molecules27113447
APA StyleCampkin, D. M., Shimadate, Y., Bartholomew, B., Bernhardt, P. V., Nash, R. J., Sakoff, J. A., Kato, A., & Simone, M. I. (2022). Borylated 2,3,4,5-Tetrachlorophthalimide and Their 2,3,4,5-Tetrachlorobenzamide Analogues: Synthesis, Their Glycosidase Inhibition and Anticancer Properties in View to Boron Neutron Capture Therapy. Molecules, 27(11), 3447. https://doi.org/10.3390/molecules27113447