
Functionalization of Biphenylcarbazole (CBP) with Siloxane-Hybrid Chains for Solvent-Free Liquid Materials

 , ,
, ,  ,
,  and
and
Abstract
:1. Introduction
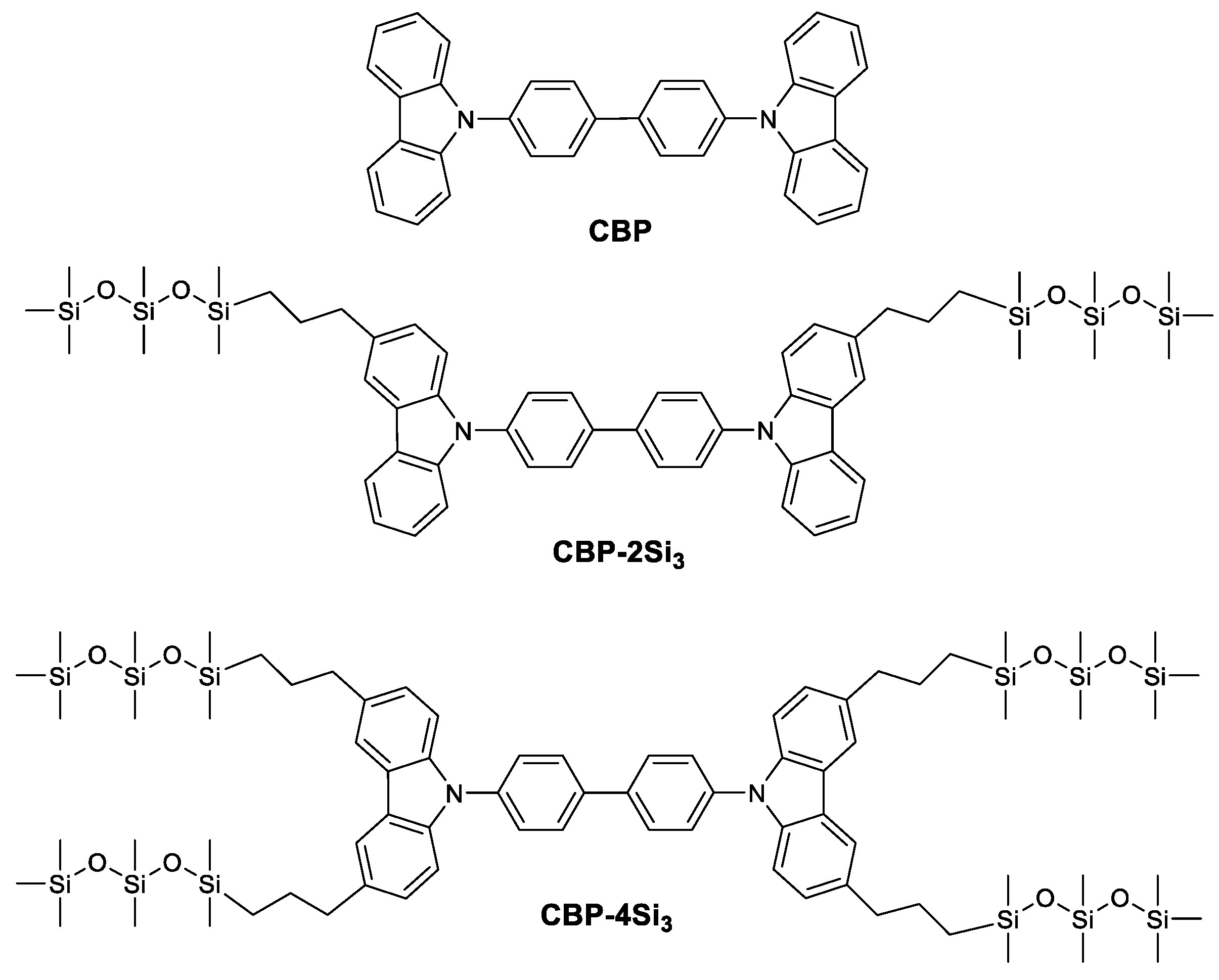
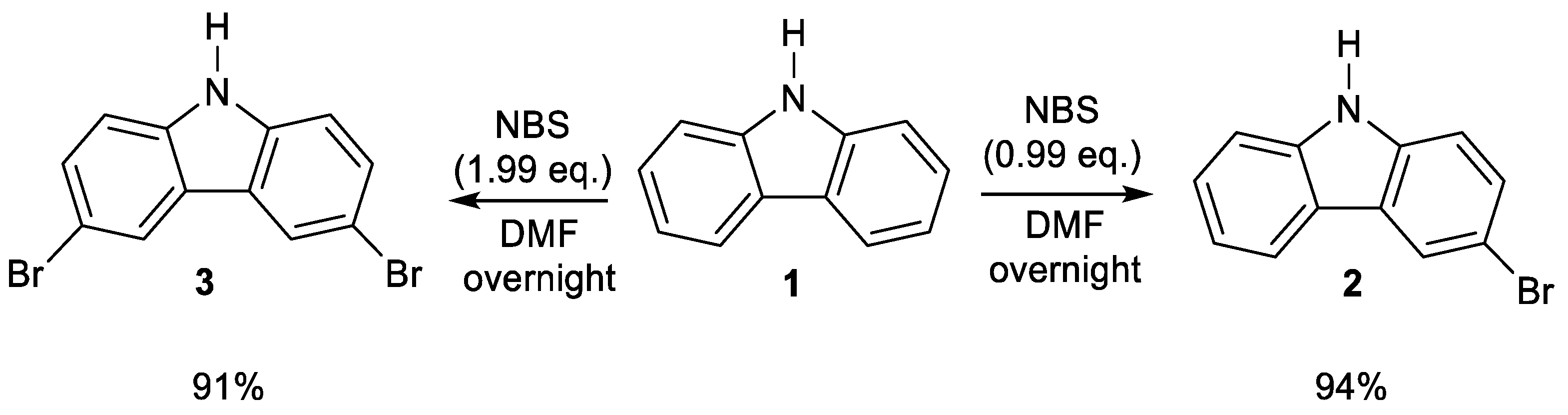
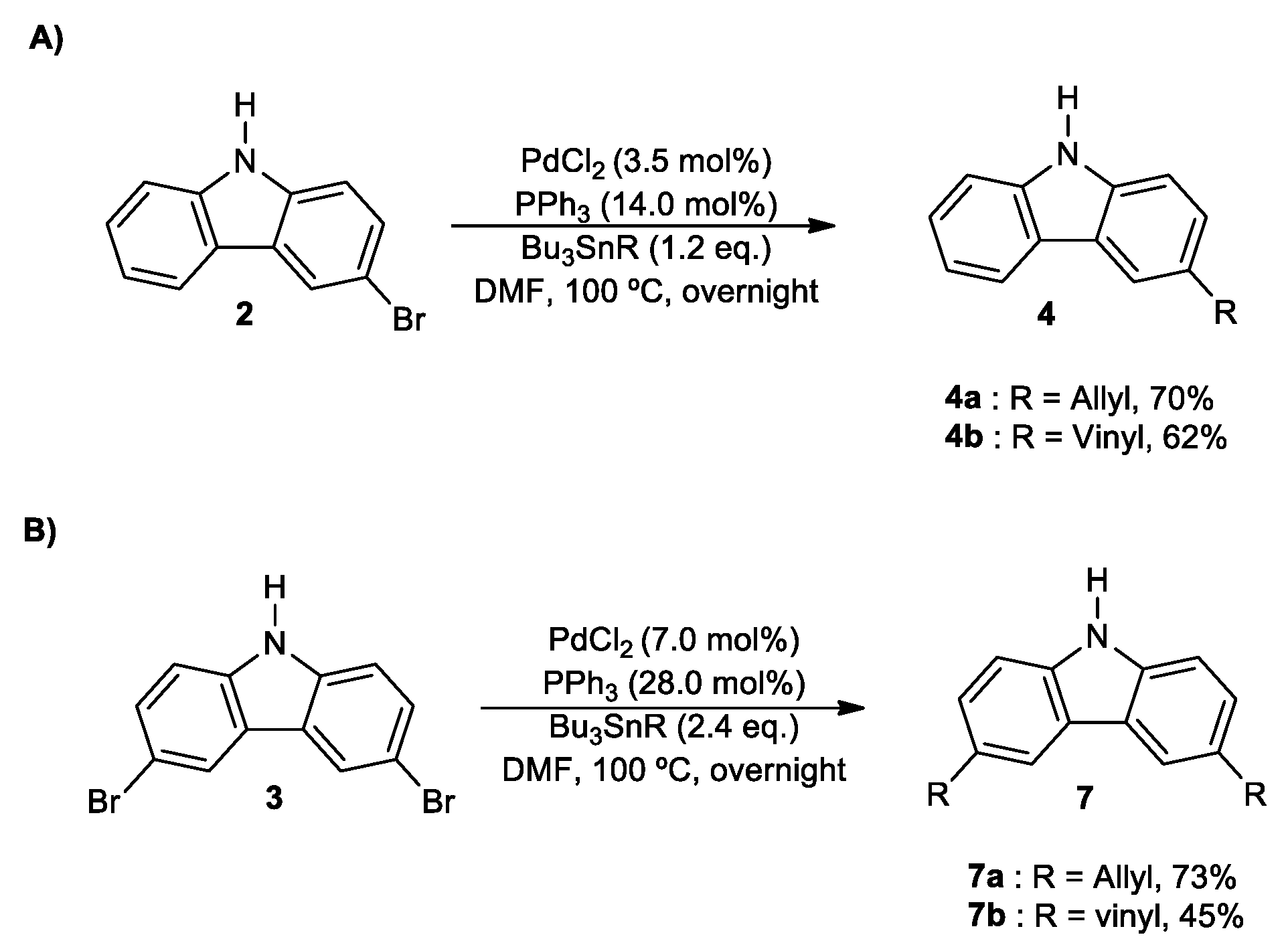
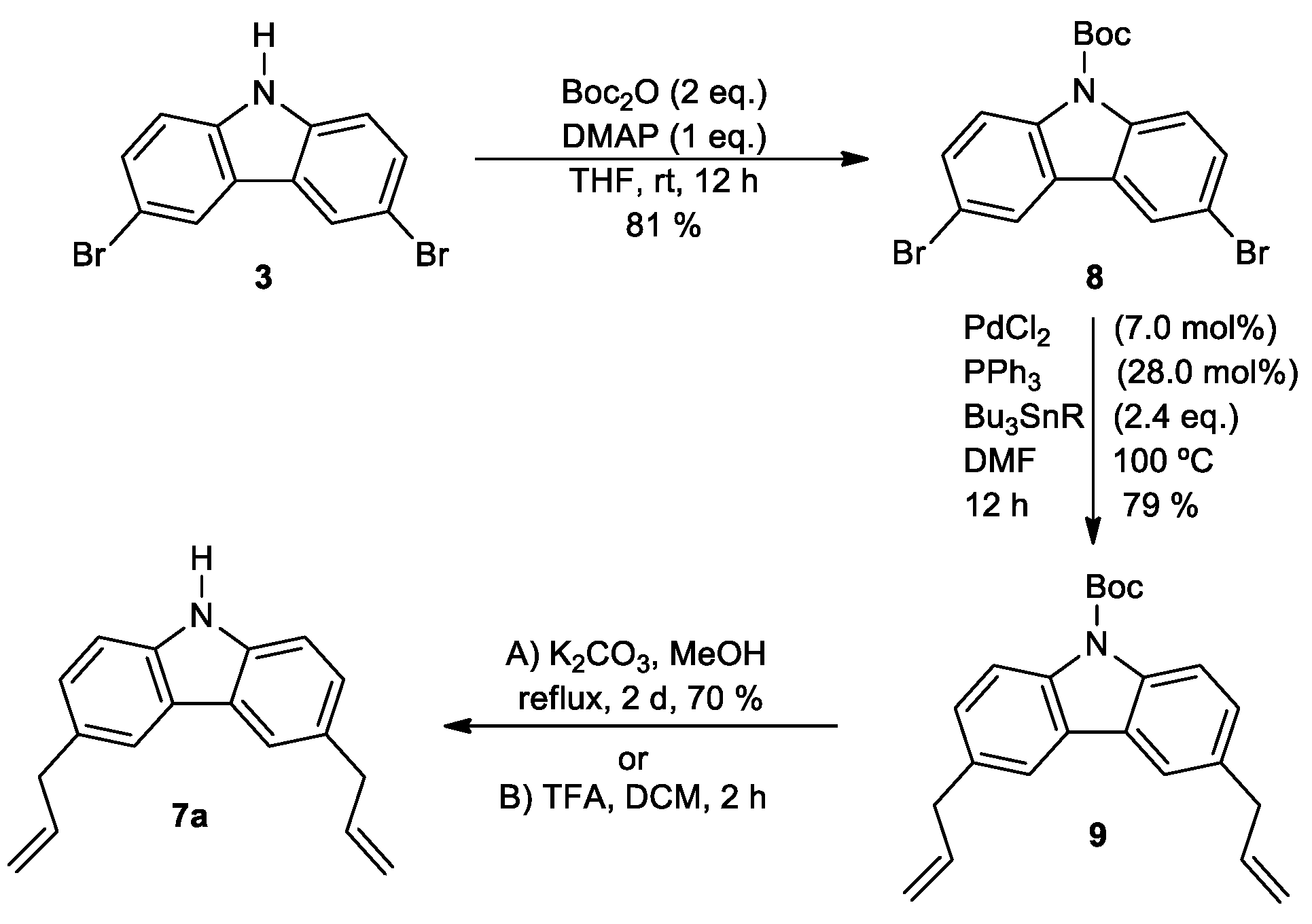
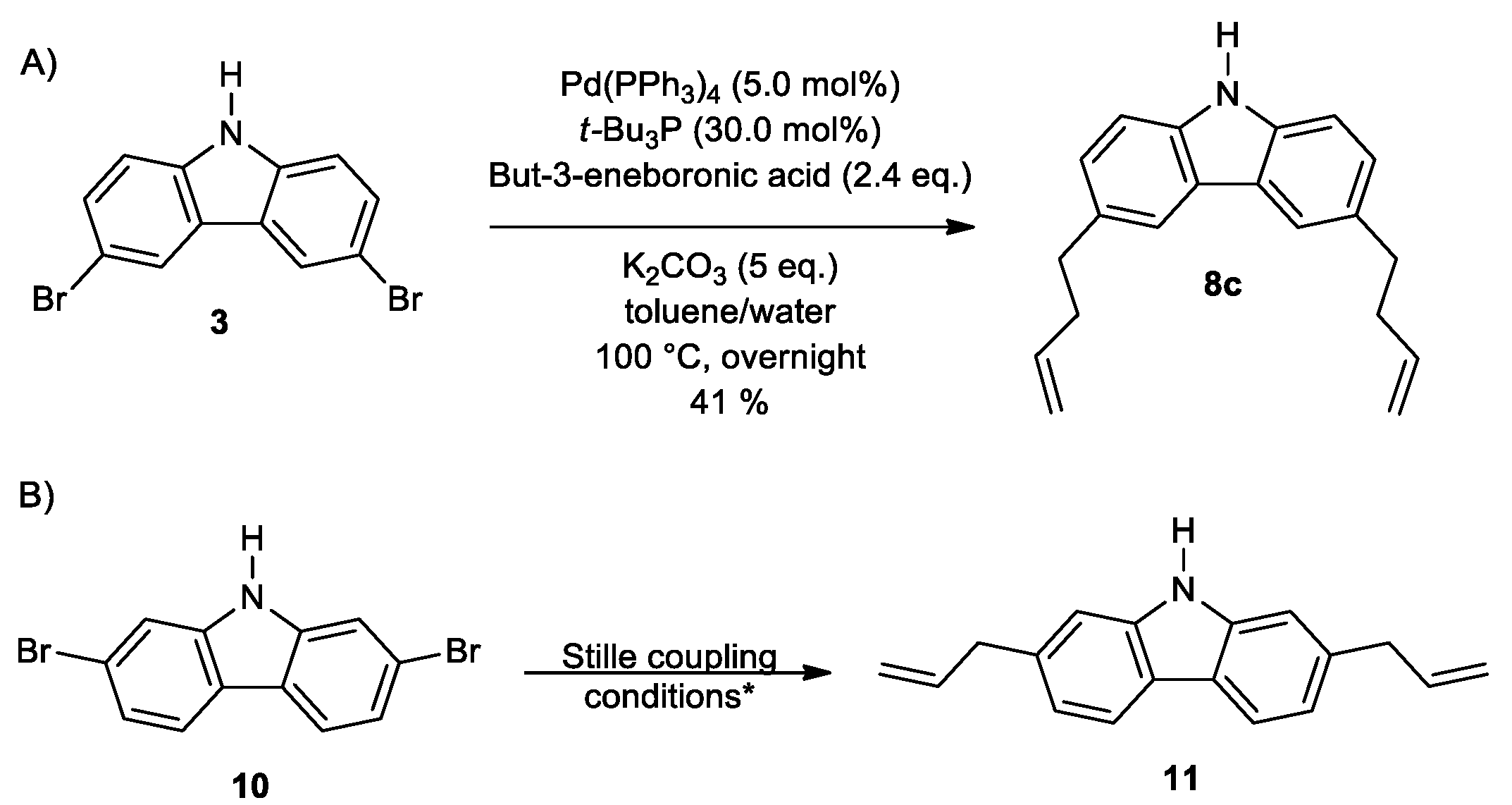
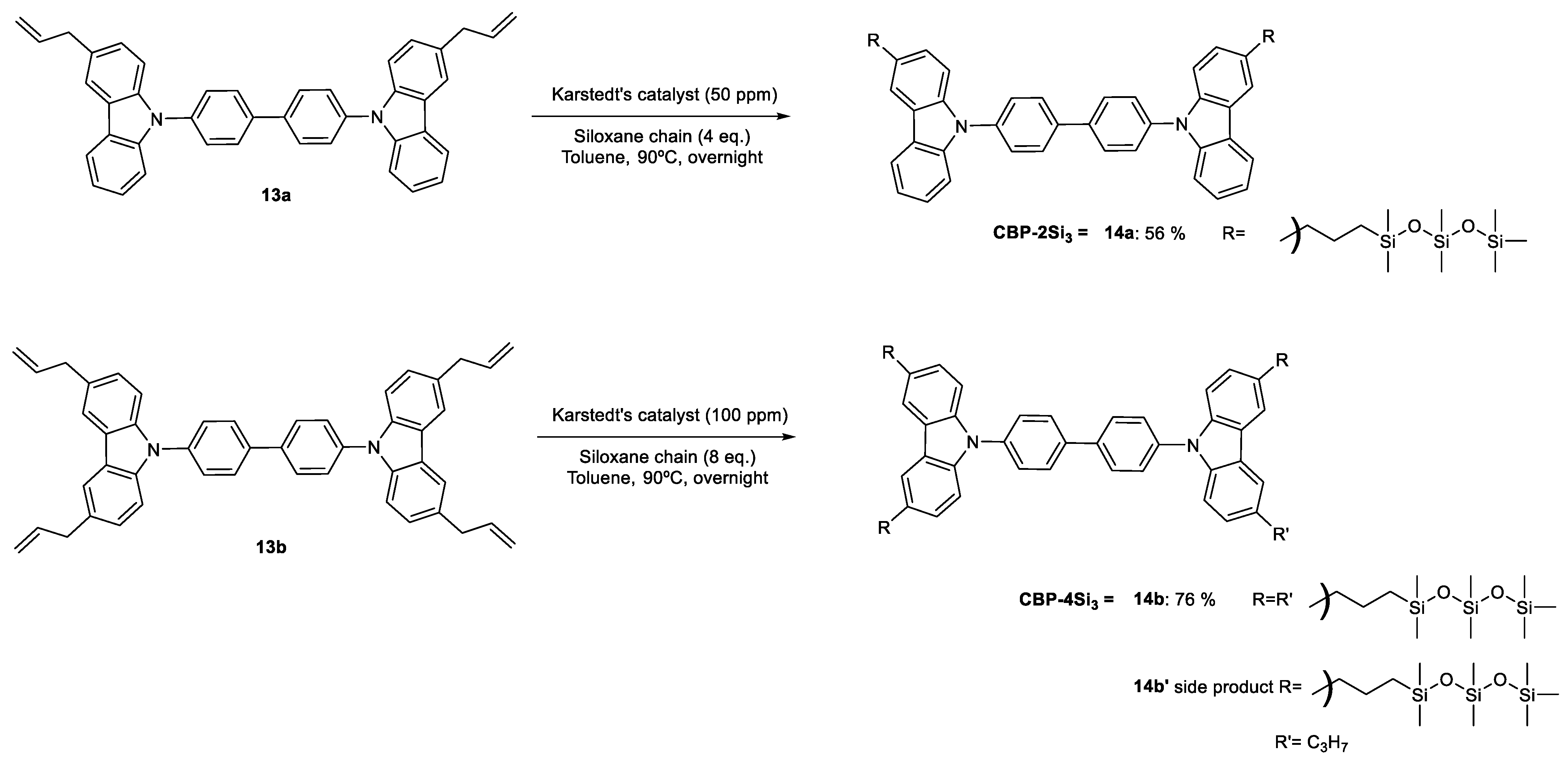
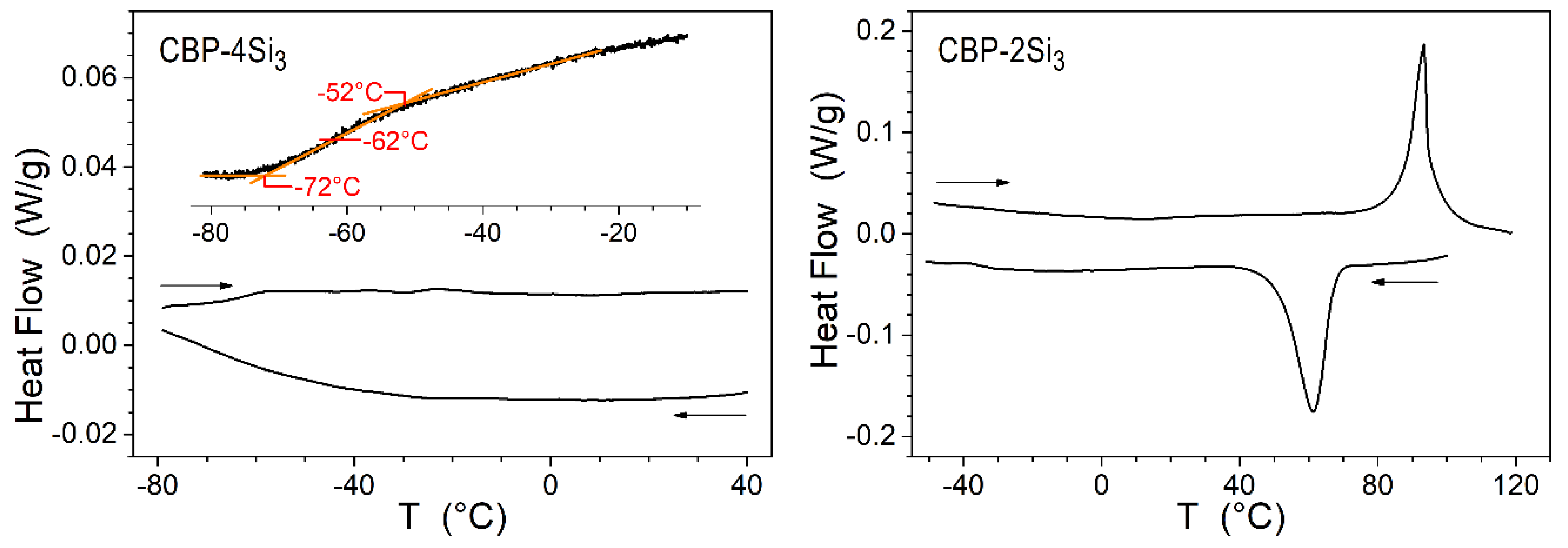
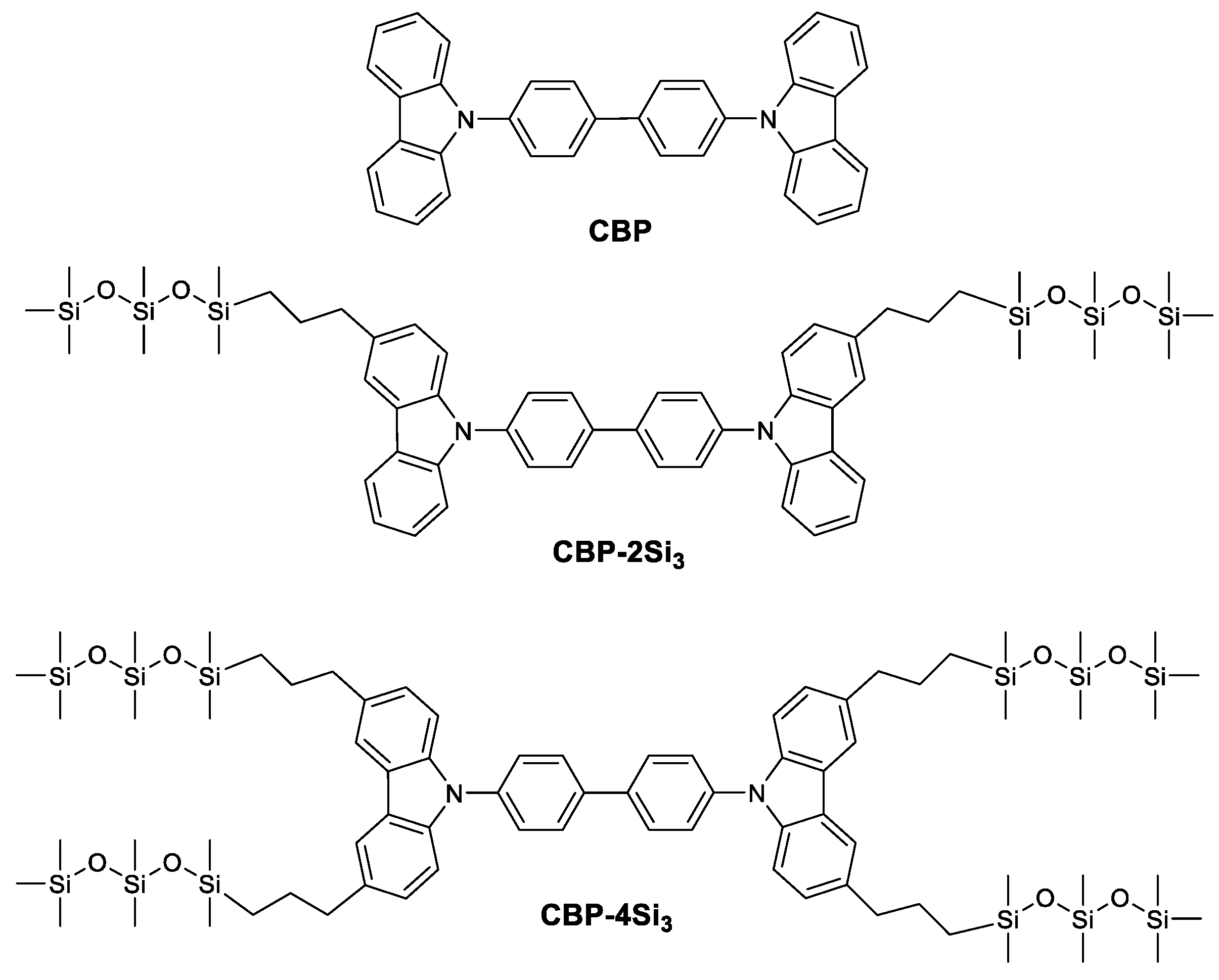
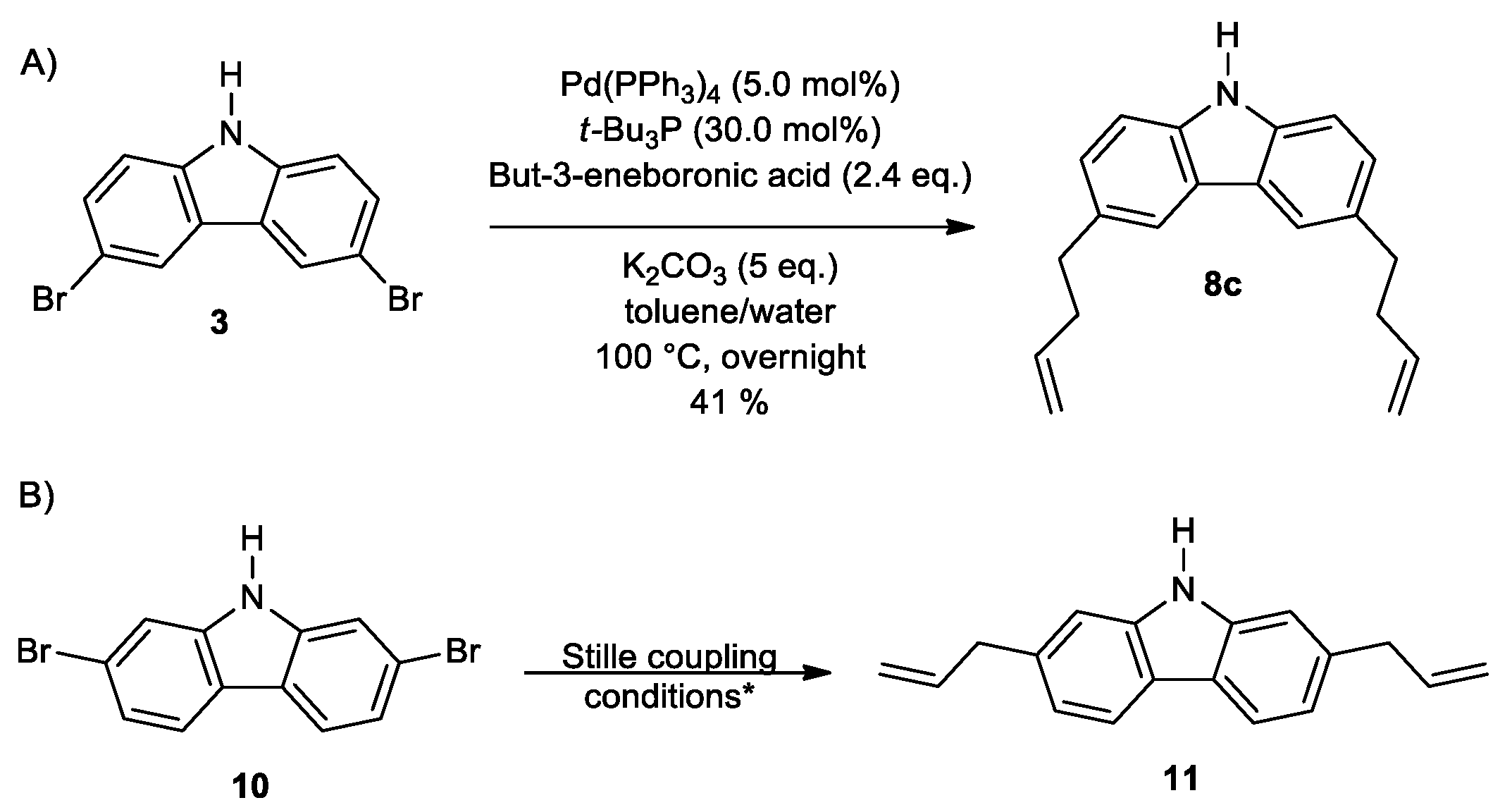
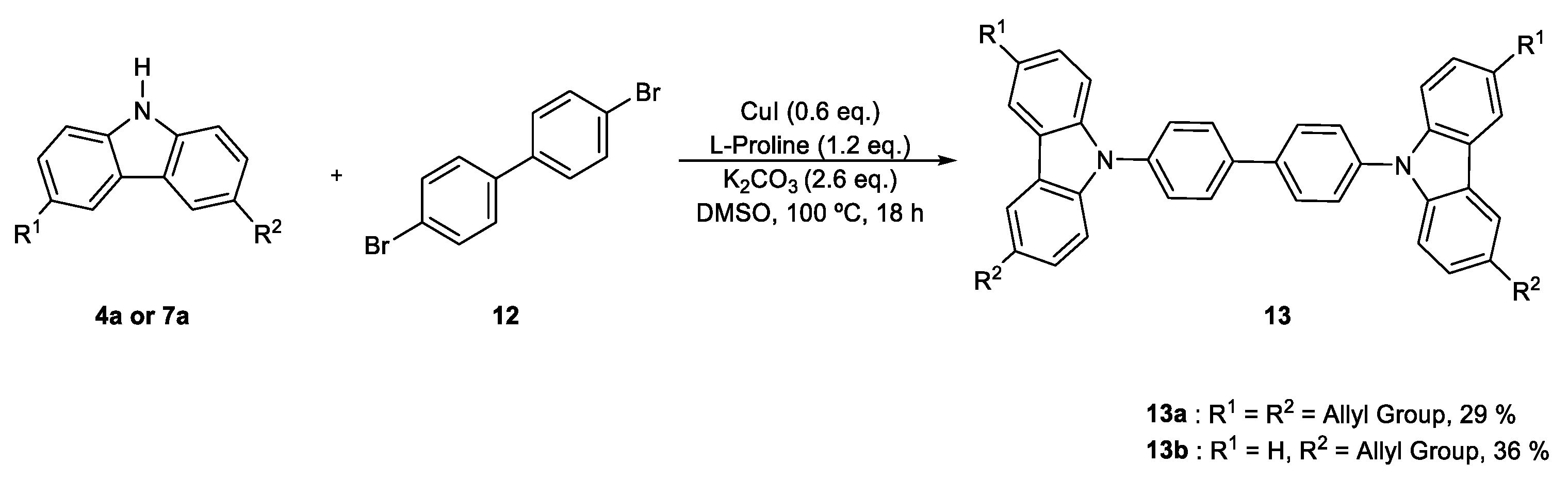
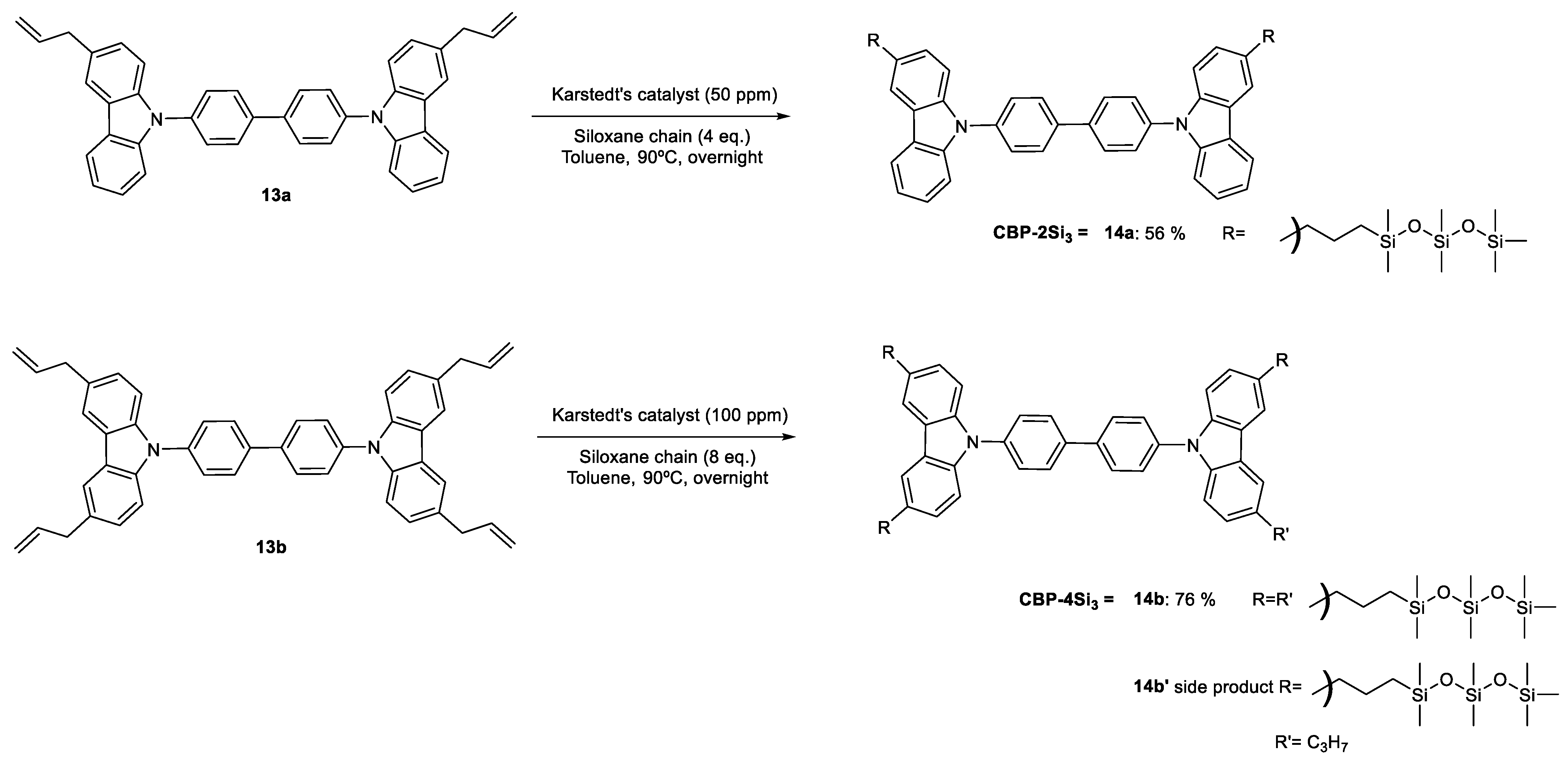
2. Results and Discussion
3. Conclusions
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Jones, R.G.; Ando, W.; Chojnowski., J. (Eds.) Silicon-Containing Polymers: The Science and Technology of Their Synthesis and Applications; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2000. [Google Scholar]
- Yilgör, E.; Yilgör, I. Silicone Containing Copolymers: Synthesis, Properties and Applications. Prog. Polym. Sci. 2014, 39, 1165–1195. [Google Scholar] [CrossRef] [Green Version]
- Eduok, U.; Faye, O.; Szpunar, J. Recent Developments and Applications of Protective Silicone Coatings: A Review of PDMS Functional Materials. Prog. Org. Coat. 2017, 111, 124–163. [Google Scholar] [CrossRef]
- Babu, S.S.; Nakanishi, T. Nonvolatile Functional Molecular Liquids. Chem. Commun. 2013, 49, 9373–9382. [Google Scholar] [CrossRef] [PubMed]
- Kamino, B.A.; Bender, T.P.; Klenkler, R.A. Hole Mobility of a Liquid Organic Semiconductor. J. Phys. Chem. Lett. 2012, 3, 1002–1006. [Google Scholar] [CrossRef]
- Kamino, B.A.; Bender, T.P. The Use of Siloxanes, Silsesquioxanes, and Silicones in Organic Semiconducting Materials. Chem. Soc. Rev. 2013, 42, 5119–5130. [Google Scholar] [CrossRef]
- Sepehrifard, A.; Kamino, B.A.; Bender, T.P.; Morin, S. Siliconized Triarylamines as Redox Mediator in Dye-Sensitzed Solar Cell. ACS Appl. Mater. Interfaces 2012, 4, 6211–6215. [Google Scholar] [CrossRef]
- Mager, L.; Méry, S. Low-Tg Photorefractive Materials Based on Bifunctional Molecules. Mol. Cryst. Liq. Cryst. 1998, 322, 21–28. [Google Scholar] [CrossRef]
- Ribierre, J.-C.; Zhao, L.; Inoue, M.; Schwartz, P.-O.; Kim, J.-H.; Yoshida, K.; Sandanayaka, A.S.D.; Nakanotani, H.; Mager, L.; Mery, S.; et al. Low Threshold Amplified Spontaneous Emission and Ambipolar Charge Transport in Non-Volatile Liquid Fluorene Derivatives. Chem. Commun. 2016, 52, 3103–3106. [Google Scholar] [CrossRef]
- Sun, D.; Ren, Z.; Bryce, M.R.; Yan, S. Arylsilanes and Siloxanes as Optoelectronic Materials for Organic Light-Emitting Diodes (OLEDs). J. Mater. Chem. C 2015, 3, 9496–9508. [Google Scholar] [CrossRef] [Green Version]
- Mei, J.; Kim, D.H.; Ayzner, A.L.; Toney, M.; Bao, Z. Siloxane—Terminated Solubilizing Side Chains: Bringing Conjugated Polymer Backbones Closer and Boosting Hole Mobilities in Thin-Film Transistors. J. Am. Chem. Soc. 2011, 133, 20130–20133. [Google Scholar] [CrossRef]
- Fan, B.; Ying, L.; Zhu, P.; Pan, F.; Liu, F.; Chen, J.; Huang, F.; Cao, Y. All-Polymer Cells Based on a Conjugated Polymer Containing Siloxane-Functionalized Side Chains with Efficiency over 10%. Adv. Mater. 2017, 29, 1703906. [Google Scholar] [CrossRef]
- Ding, Y.; Yuan, N.; Wang, X.; Zhang, G.; Qiu, L. Intrinsically Stretchable n-Type Polymer Semiconductor Through Side Chains Engineering. Macromolecules 2021, 54, 8849–8859. [Google Scholar] [CrossRef]
- Kamatham, N.; Ibraikulov, O.A.; Durand, P.; Wang, J.; Boyron, O.; Heinrich, B.; Heiser, T.; Lévêque, P.; Leclerc, N.; Méry, S. On the Impact of Linear Siloxanated Side Chains on the Molecular Self-Assembling and Charge Transport Properties of Conjugated Polymers. Adv. Funct. Mater. 2021, 31, 2007734. [Google Scholar] [CrossRef]
- Ghosh, A.; Nakanishi, T. Frontiers of Solvent-free- Functional Molecular Liquids. Chem. Commun. 2017, 53, 10344–10357. [Google Scholar] [CrossRef]
- Lu, F.; Nakanishi, T. Solvent-free Luminous Molecular Liquids. Adv. Opt. Mater. 2019, 7, 1900176. [Google Scholar] [CrossRef]
- Shim, C.-H.; Hirata, S.; Oshima, J.; Edura, T.; Hattori, R.; Adachi, C. Uniform and refreshable liquid electroluminescent device with a back side reservoir. Appl. Phys. Lett. 2012, 101, 113302. [Google Scholar] [CrossRef]
- Kobayashi, N.; Kasahara, T.; Edura, T.; Oshima, J.; Ishimatsu, R.; Tsuwaki, M.; Imato, T.; Shoji, S.; Mizuno, J. Microfluidic White Organic Light-Emitting Diode Based on Integrated Patterns of Greenish-Blue and Yellow Solvent-Free Liquid Emitters. Sci. Rep. 2015, 5, 14822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajima, Y.; Shimada, S. Hydrosilylation Reaction of Olefins: Recent Advances and Perspectives. RSC Adv. 2015, 5, 20603–20616. [Google Scholar] [CrossRef]
- Marciniec, B. Comprehensive Handbook of Hydrosilylation; Pergamon Press: Oxford, UK, 1992. [Google Scholar]
- de Meijere, A.; Bräse, S.; Oestreich, M. (Eds.) Metal Catalyzed Cross-Coupling Reactions and More; Wiley VCH: Weinheim, Germany, 2014; Volume 3, pp. 1–1551. [Google Scholar]
- Hassam, M.; Taher, A.; Arnott, G.E.; Green, I.R.; van Otterlo, W.A.L. Isomerization of Allylbenzenes. Chem. Rev. 2015, 115, 5462–5569. [Google Scholar] [CrossRef]
- Wex, B.; Kaafarani, B.R. Perspective on Carbazole-based Organic Compounds as Emitters and Hosts in TADF Applications. J. Mater. Chem. C 2017, 5, 8622–8653. [Google Scholar] [CrossRef] [Green Version]
- Sathiyan, G.; Sivakumar, E.K.T.; Ganesamoorthy, R.; Thangamuthu, R.; Sakthivel, P. Review of Carbazole Based Conjugated Molecules for highly Efficient Organic Solar Cell Application. Tet. Lett. 2017, 57, 243–252. [Google Scholar] [CrossRef]
- Berton, N.; Nakar, R.; Schmaltz, B. DMPA-Containing Carbazole-based Hole Transporting Materials for Perovskite Solar Cells: Recent Advances and Perspectives. Synt. Met. 2019, 252, 91–106. [Google Scholar] [CrossRef]
- Shaya, J.; Srour, H.; Karamé, I. An outline of carbon dioxide chemistry, uses and technology. In Carbon Dioxide Chemistry, Capture and Oil Recovery; Karamé, I., Shaya, J., Srour, H., Eds.; InTech Open: London, UK, 2018; Online publication; Chapter 1; pp. 4–13. [Google Scholar]
- Shehayeb, S.; Zaher, S.; Ghannam, L.; Srour, H.; Kanj, A.; Shaya, J.; Karamé, I. Sustainable valorization of the abundant biodiesel byproduct—The glycerol. In Handbook of Greener Synthesis of Nanomaterials and Compounds; Kharisov, B., Kharissova, O., Eds.; Elsevier, S&T: Amsterdam, The Netherlands, 2021; Volume 1, Chapter 26; pp. 807–860. [Google Scholar]
- Zhang, X.; Lu, J.; Zhang, J. Porosity Enhancement of Carbazolic Porous Organic Frameworks Using Dendritic Building Blocks for Gas Storage and Separation. Chem. Mater. 2014, 26, 4023–4029. [Google Scholar] [CrossRef]
- Young, M.Y.; Zysman-Colman, E. Purely Organic Thermally Activated Delayed Fluorescence Materials for Organic Light-Emitting Diodes. Adv. Mater. 2017, 29, 1605444. [Google Scholar]
- Fukagawa, H.; Shimizu, T.; Kamada, T.; Yui, S.; Hasegawa, M.; Morii, K.; Yamamoto, T. Highly Efficient and Stable Organic Light-Emitting Diodes with a Greatly Reduced Amount of Phosphorescent Emitter. Sci. Rep. 2015, 5, 9855. [Google Scholar] [CrossRef] [PubMed]
- Uoyama, H.; Goushi, K.; Shizu, K.; Nomura, H.; Adachi, C. Highly Efficient Organic Light-Emitting Diodes from Delayed Fluorescence. Nature 2012, 492, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Schrögel, P.; Tomkevičienė, A.; Strohriegl, P.; Hoffmann, S.T.; Köhler, A.; Lennartz, C. A Series of CBP-Derivatives as Host Materials for Blue Phosphorescent Organic Light-Emitting Diodes. J. Mater. Chem. 2011, 21, 2266–2273. [Google Scholar] [CrossRef]
- Shaya, J.; Deschamps, M.-A.; Michel, B.Y.; Burger, A. Air-Stable Pd Catalytic Systems for Sequential One-Pot Synthesis of Challenging Unsymmetrical Aminoaromatics. J. Org. Chem. 2016, 81, 7566–7573. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-C.; Shaya, J.; Polychronopoulou, K.; Golovko, V.B.; Tesana, S.; Wang, S.-Y.; Lu, C.-S. Photocatalytic Degradation of Ethiofencarb by a Visible Light-Driven SnIn4S8 Photocatalyst. Nanomaterials 2021, 11, 1325. [Google Scholar] [CrossRef]
- Chen, C.-C.; Chen, T.-T.; Shaya, J.; Wu, C.-L.; Lu, C.-S. Bi12SiO20/g-C3N4 Heterojunctions: Synthesis, Characterization, Photocatalytic Activity for Organic Pollutant Degradation, and Mechanism. J. Taiwan Inst. Chem. E. 2021, 123, 228–244. [Google Scholar] [CrossRef]
- Chen, C.; Fan, H.; Shaya, J.; Chang, Y.; Golovko, V.B.; Toulemonde, O.; Huang, C.; Song, Y.; Lu, C. Accelerated ZnMoO4 Photocatalytic Degradation of Pirimicarb under UV Light Mediated by Peroxymonosulfate. Appl. Organomet. Chem. 2019, 33, e5113. [Google Scholar]
- Tsai, H.; Shaya, J.; Tesana, S.; Golovko, V.B.; Wang, S.-Y.; Liao, Y.-Y.; Lu, C.-S.; Chen, C.-C. Visible-Light Driven Photocatalytic Degradation of Pirimicarb by Pt-Doped AgInS2 Nanoparticles. Catalysts 2020, 10, 857. [Google Scholar] [CrossRef]
- Barnoin, G.; Shaya, J.; Richert, L.; Le, H.-N.; Vincent, S.; Guérineau, V.; Mély, Y.; Michel, B.Y.; Burger, A. Intermolecular Dark Resonance Energy Transfer (DRET): Upgrading Fluorogenic DNA Sensing. Nucleic Acids Res. 2021, 49, e72. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Ojha, H.; Bharadwaj, A.; Pathak, D.P.; Sharma, R.K. Preparation and Catalytic Applications of Nanomaterials: A Review. RSC Adv. 2015, 5, 53381–53403. [Google Scholar] [CrossRef]
- Smith, C.J.; Tsang, M.W.S.; Holmes, A.B.; Danheiser, R.L.; Tester, J.W. Palladium Catalysed Aryl Amination Reactions in Supercritical Carbon Dioxide. Org. Biomol. Chem. 2005, 3, 3767–3781. [Google Scholar] [CrossRef] [PubMed]
- Zeng, G.; Ouyang, X.; Yang, D.; Zeng, H. A Promising Strategy for Two-Photon Absorption Materials by Novel Dicarbazole-Conjugated C60/C70 Fullerene Derivatives. Opt. Mater. 2010, 32, 637–642. [Google Scholar] [CrossRef]
- Fors, B.P.; Buchwald, S.L. A Multiligand Based Pd Catalyst for C–N Cross-Coupling Reactions. J. Am. Chem. Soc. 2010, 132, 15914–15917. [Google Scholar] [CrossRef] [Green Version]
- Anémian, R.; Morel, Y.; Baldeck, P.L.; Paci, B.; Kretsch, K.; Nunzi, J.-M.; Andraud, C. Optical Limiting in the Visible Range: Molecular Engineering Around N 4,N 4′-Bis(4-methoxyphenyl)-N 4,N 4-Diphenyl-4,4-Diaminobiphenyl. J. Mater. Chem. 2003, 13, 2157–2163. [Google Scholar] [CrossRef]
- Hu, X.; Ma, X.; Jian, Z. Coordination–Insertion Polymerization of Polar Allylbenzene Monomers. Polym. Chem. 2019, 10, 1912–1919. [Google Scholar] [CrossRef]
- Cui, L.; Chen, M.; Chen, C.; Liu, D.; Jian, Z. Systematic Studies on (Co)Polymerization of Polar Styrene Monomers with Palladium Catalysts. Macromolecules 2019, 52, 7197–7206. [Google Scholar] [CrossRef]
- Kathe, P.M.; Fleischer, I. Palladium-Catalyzed Tandem Isomerization/Hydrothiolation of Allylarenes. Org. Lett. 2019, 21, 2213–2217. [Google Scholar] [CrossRef] [PubMed]
- De, S.; Sivendran, N.; Maity, B.; Pirkl, N.; Koley, D.; Gooßen, L.J. Dinuclear PdI Catalysts in Equilibrium Isomerizations: Mechanistic Understanding, in Silico Casting, and Catalyst Development. ACS Catal. 2020, 10, 4517–4533. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Niu, Y.-N.; Xia, X.-F. Pd-Catalyzed Tandem Isomerization/Cyclization for the Synthesis of Aromatic Oxazaheterocycles and Pyrido[3,4-b]Indoles. J. Org. Chem. 2021, 86, 10032–10042. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Kim, E.H.; Wei, A.; Negishi, E. Pd- and Ni-Catalyzed Cross-Coupling Reactions in the Synthesis of Organic Electronic Materials. Sci. Technol. Adv. Mater. 2014, 15, 044201. [Google Scholar] [CrossRef] [PubMed]
- El-Maiss, J.; Dine, T.M.E.; Lu, C.-S.; Karamé, I.; Kanj, A.; Polychronopoulou, K.; Shaya, J. Recent Advances in Metal-Catalyzed Alkyl–Boron (C(Sp3)–C(Sp2)) Suzuki-Miyaura Cross-Couplings. Catalysts 2020, 10, 296. [Google Scholar] [CrossRef] [Green Version]
- Stein, J.; Lewis, L.N.; Gao, Y.; Scott, R.A. In-Situ Determination of the Active Catalyst in Hydrosilylation Reactions Using Highly Reactive Pt(0) Catalyst Precursors. J. Am. Chem. Soc. 1999, 121, 3693–3703. [Google Scholar] [CrossRef]
- Meister, T.K.; Riener, K.; Gigler, P.; Stohhrer, J.; Herrmann, W.A.; Kühn, F.E. Platinum Catalysis Revisited—Unraveling Principles of Catalytic Olefin Hydrosilylation. ACS Catal. 2016, 6, 1274–1284. [Google Scholar] [CrossRef]
- Siraj, N.; Hasan, F.; Das, S.; Kiruri, L.W.; Gall, K.E.S.; Baker, G.A.; Warner, I.M. Carbazole-Derived Group of Uniform Materials Based on Organic Salts: Solid State Fluorescent Analogues of Ionic Liquids for Potential Applications in Organic-Based Blue Light-Emitting Diodes. J. Phys. Chem. C. 2014, 118, 2312–2320. [Google Scholar] [CrossRef]
- Bag, P.P.; Wang, D.; Chen, Z.; Cao, R. Outstanding drug loading capacity by water stable microporous MOF: A potential drug carrier. Chem. Commun. 2016, 52, 3669–3672. [Google Scholar] [CrossRef]
- Casado, A.L.; Espinet, P.; Gallego, A.M. Mechanism of the Stille Reaction. 2. Couplings of Aryl Triflates with Vinyltributyltin. Observation of Intermediates. A More Comprehensive Scheme. J. Am. Chem. Soc. 2000, 122, 11771–11782. [Google Scholar] [CrossRef]
- Cordovilla, C.; Bartolomé, C.; Martinez-Ilarduya, J.M.; Espinet, P. The Stille Reaction, 38 Years Later. ACS Catal. 2015, 5, 3040–3053. [Google Scholar] [CrossRef] [Green Version]
- Amatore, C.; Jutand, A. Anionic Pd(0) and Pd(II) Intermediates in Palladium-Catalyzed Heck and Cross-Coupling Reactions. Acc. Chem. Res. 2000, 33, 314–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bremner, J.B.; Coates, J.A.; Keller, P.A.; Pyne, S.G.; Witchard, H.M. Synthesis of Carbazole-Linked Cyclic and Acyclic Peptoids with Antibacterial Activity. Tetrahedron 2003, 59, 8741–8755. [Google Scholar] [CrossRef]
- Kang, M.S.; Sung, S.D.; Choi, I.T.; Kim, H.; Hong, M.; Kim, J.; Lee, W.I.; Kim, H.K. Novel Carbazole-Based Hole-Transporting Materials with Star-Shaped Chemical Structures for Perovskite-Sensitized Solar Cells. ACS Appl. Mater. Interfaces 2015, 7, 22213–22217. [Google Scholar] [CrossRef]
- Fujisawa, S.; Kadoma, Y.; Yokoe, I. Radical-Scavenging Activity of Butylated Hydroxytoluene (BHT) and Its Metabolites. Chem. Phys. Lipids 2004, 130, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Diep, V.; Dannenberg, J.J.; Franck, R.W. An O-Iminothioquinone: Its Cycloaddition To Produce an Indologlycoside and Its Self-Dimerization To Form a Dithio-Diazocycloctane, the Structure Assignment of Which Is Based on the DFT Prediction of Its IR Spectrum. J. Org. Chem. 2003, 68, 7907–7910. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Wang, M.; Chen, X.; Hong, B. Synthesis of OLED Materials of Several Triarylamines by Palladium Catalysts and their Emitting Property. J. Chem. Res. 2005, 9, 558–560. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Catalytic System | Solvent | 1 | 4a | 5 | 6 |
|---|---|---|---|---|---|---|
| 1 | 5.0 mol% Pd(PPh3)4 | DMF | - | - | - | Mixture of N,3-bisallyl isomers |
| 2 | 2.5 mol% PdCl2 10.0 mol% PPh3 | DMF | 4% | 74% | <1% | - |
| 3 | 3.5 mol% PdCl2 14.0 mol% PPh3 | DMF | 1% | 98% | <1% | - |
| 4 | 6.0 mol% PdCl2 20.0 mol% PPh3 | DMF | 1% | 91% | 8% | - |
| 5 | 3.5 mol% PdCl2 14.0 mol% PPh3 | Toluene | 2% | 77% | 8% | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shaya, J.; Correia, G.; Heinrich, B.; Ribierre, J.-C.; Polychronopoulou, K.; Mager, L.; Méry, S. Functionalization of Biphenylcarbazole (CBP) with Siloxane-Hybrid Chains for Solvent-Free Liquid Materials. Molecules 2022, 27, 89. https://doi.org/10.3390/molecules27010089
Shaya J, Correia G, Heinrich B, Ribierre J-C, Polychronopoulou K, Mager L, Méry S. Functionalization of Biphenylcarbazole (CBP) with Siloxane-Hybrid Chains for Solvent-Free Liquid Materials. Molecules. 2022; 27(1):89. https://doi.org/10.3390/molecules27010089
Chicago/Turabian StyleShaya, Janah, Gabriel Correia, Benoît Heinrich, Jean-Charles Ribierre, Kyriaki Polychronopoulou, Loïc Mager, and Stéphane Méry. 2022. "Functionalization of Biphenylcarbazole (CBP) with Siloxane-Hybrid Chains for Solvent-Free Liquid Materials" Molecules 27, no. 1: 89. https://doi.org/10.3390/molecules27010089
APA StyleShaya, J., Correia, G., Heinrich, B., Ribierre, J.-C., Polychronopoulou, K., Mager, L., & Méry, S. (2022). Functionalization of Biphenylcarbazole (CBP) with Siloxane-Hybrid Chains for Solvent-Free Liquid Materials. Molecules, 27(1), 89. https://doi.org/10.3390/molecules27010089