
Glutathione in Brain Disorders and Aging



Abstract
1. Introduction
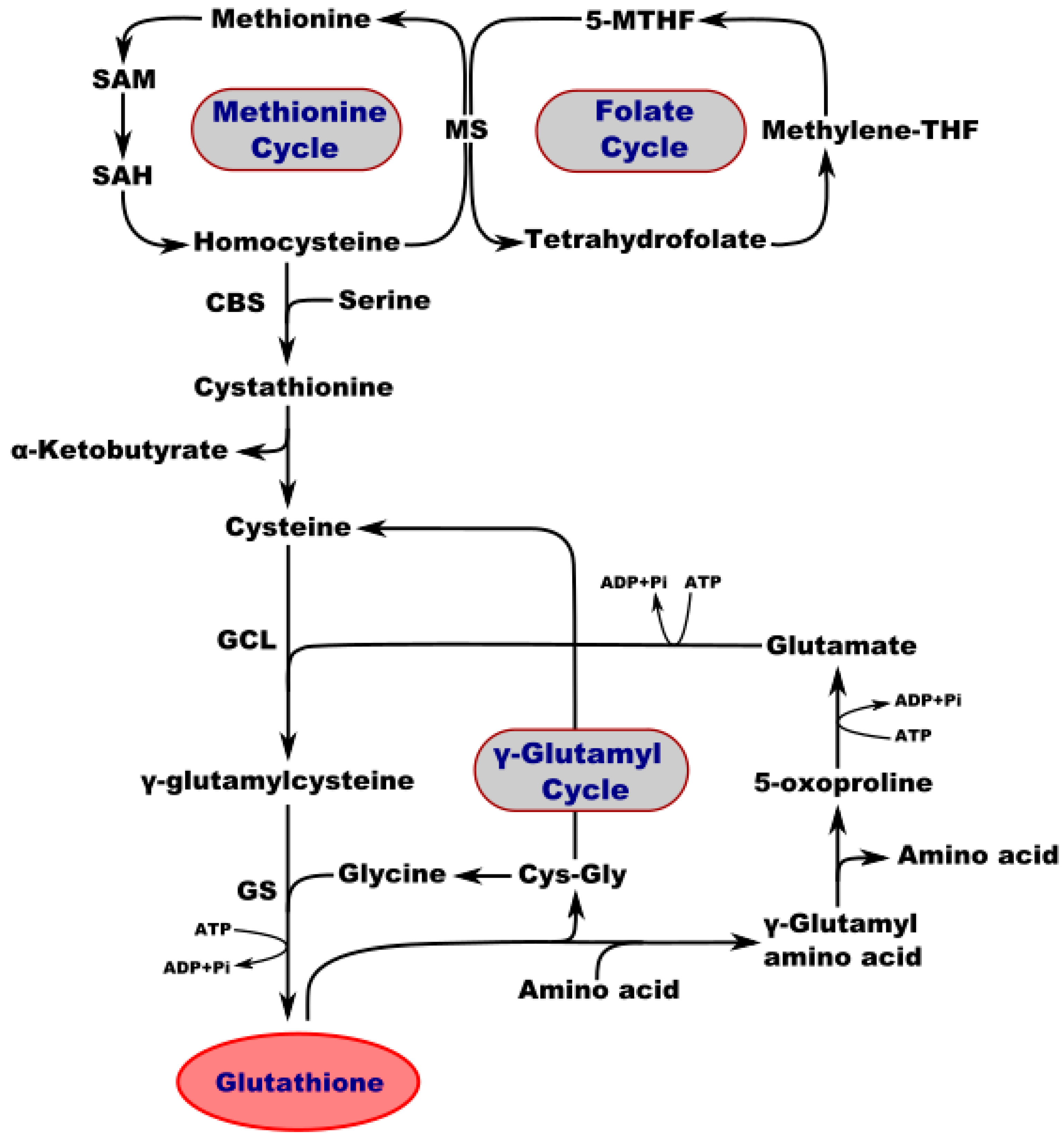
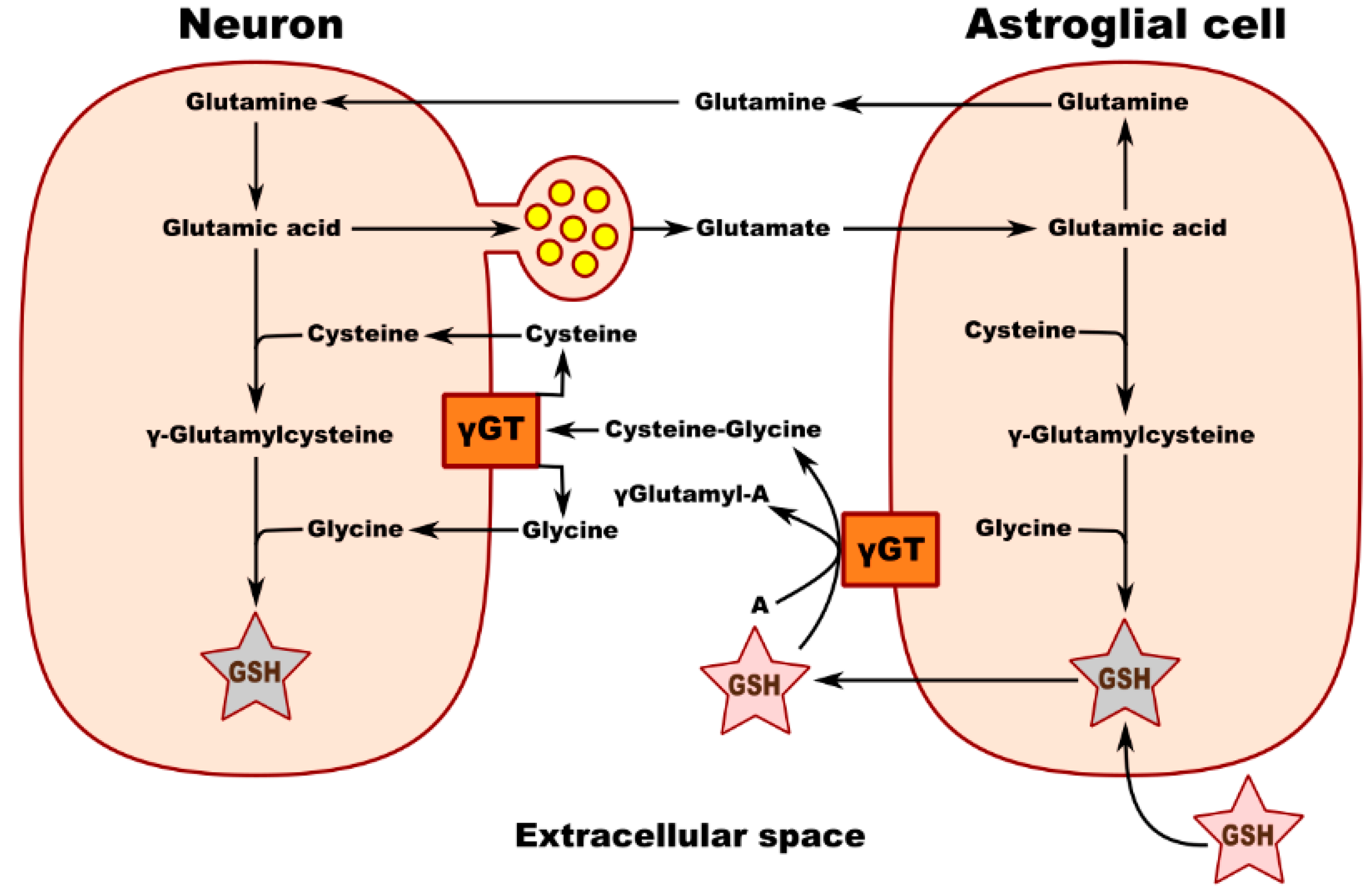
2. A Pivotal Role for GSH in the Regulation of Homeostasis and Metabolism in the Nervous System
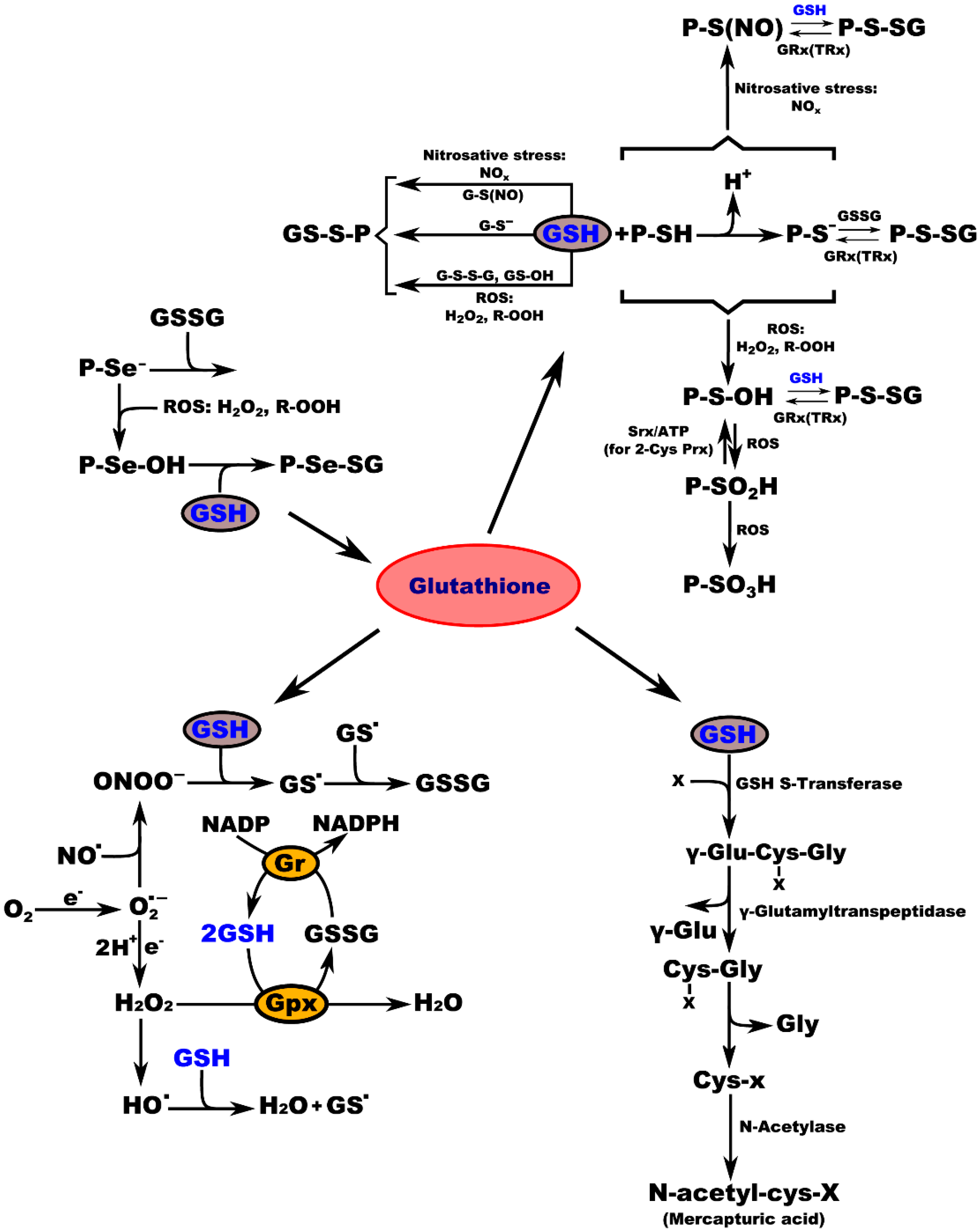
3. Glutathione Regulates Aging and Neurodegeneration
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
Abbreviations
| GSSG | oxidized glutathione |
| GSH | reduced glutathione |
| ROS | reactive oxygen species |
| H2O2 | hydrogen peroxide |
| P-SH | protein sulfhydryl group |
| P-S-SG | protein-S-glutathione |
| GRx | glutaredoxin |
| TRx | thioredoxin |
| P-S-OH | protein-sulfenic acid |
| Srx | sulfiredoxin |
| ATP | adenosine triphosphate |
| NO | nitric oxide radical |
| ONOO− | peroxynitrite |
| GS. | glutathione radical |
| Gr | glutathione reductase |
| GPx | glutathione peroxidase |
| NADP | nicotinamide adenine dinucleotide |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| PFF | pre-formed fibrils |
References
- Noctor, G.; Queval, G.; Mhamdi, A.; Chaouch, S.; Foyer, C.H. Glutathione. Arab. Book 2011, 9, e0142. [Google Scholar] [CrossRef] [PubMed]
- Lian, G.; Gnanaprakasam, J.R.; Wang, T.; Wu, R.; Chen, X.; Liu, L.; Shen, Y.; Yang, M.; Yang, J.; Chen, Y.; et al. Glutathione de novo synthesis but not recycling process coordinates with glutamine catabolism to control redox homeostasis and directs murine T cell differentiation. eLife 2018, 7, e36158. [Google Scholar] [CrossRef] [PubMed]
- Circu, M.L.; Aw, T.Y. Glutathione and apoptosis. Free Radic. Res. 2008, 42, 689–706. [Google Scholar] [CrossRef]
- Lushchak, V.I. Glutathione Homeostasis and Functions: Potential Targets for Medical Interventions. J. Amino Acids 2012, 2012, 736837. [Google Scholar] [CrossRef]
- Gu, F.; Chauhan, V.; Chauhan, A. Glutathione redox imbalance in brain disorders. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 89–95. [Google Scholar] [CrossRef]
- Morris, G.; Anderson, G.; Dean, O.; Berk, M.; Galecki, P.; Martin-Subero, M.; Maes, M. The glutathione system: A new drug target in neuroimmune disorders. Mol. Neurobiol. 2014, 50, 1059–1084. [Google Scholar] [CrossRef] [PubMed]
- Dröge, W.; Breitkreutz, R. Glutathione and immune function. Proc. Nutr. Soc. 2000, 59, 595–600. [Google Scholar] [CrossRef]
- Ghezzi, P. Role of glutathione in immunity and inflammation in the lung. Int. J. Gen. Med. 2011, 4, 105–113. [Google Scholar] [CrossRef]
- Marí, M.; Morales, A.; Colell, A.; García-Ruiz, C.; Fernández-Checa, J.C. Mitochondrial glutathione, a key survival antioxidant. Antioxid. Redox Signal 2009, 11, 2685–2700. [Google Scholar] [CrossRef]
- Sastre, J.; Pallardó, F.V.; Viña, J. Glutathione, oxidative stress and aging. AGE 2006, 19, 129–139. [Google Scholar] [CrossRef]
- Margalit, A.; Hauser, S.D.; Zweifel, B.S.; Anderson, M.A.; Isakson, P.C. Regulation of prostaglandin biosynthesis in vivo by glutathione. Am. J. Physiol. 1998, 274, R294–R302. [Google Scholar] [CrossRef]
- Ballatori, N. Glutathione mercaptides as transport forms of metals. Adv. Pharmacol. 1994, 27, 271–298. [Google Scholar] [CrossRef] [PubMed]
- Ballatori, N.; Krance, S.M.; Marchan, R.; Hammond, C.L. Plasma membrane glutathione transporters and their roles in cell physiology and pathophysiology. Mol. Asp. Med. 2009, 30, 13–28. [Google Scholar] [CrossRef]
- Forman, H.J. Glutathione—From antioxidant to post-translational modifier. Arch. Biochem. Biophys. 2016, 595, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A. Reduced glutathione: A radioprotector or a modulator of DNA-repair activity? Nutrients 2013, 5, 525–542. [Google Scholar] [CrossRef]
- Evans, J.W.; Taylor, Y.C.; Brown, J.M. The role of glutathione and DNA strand break repair in determining the shoulder of the radiation survival curve. Br. J. Cancer Suppl. 1984, 6, 49–53. [Google Scholar] [PubMed]
- Suthanthiran, M.; Anderson, M.E.; Sharma, V.K.; Meister, A. Glutathione regulates activation-dependent DNA synthesis in highly purified normal human T lymphocytes stimulated via the CD2 and CD3 antigens. Proc. Natl. Acad. Sci. USA 1990, 87, 3343–3347. [Google Scholar] [CrossRef]
- Griffith, O.W.; Bridges, R.J.; Meister, A. Transport of gamma-glutamyl amino acids: Role of glutathione and gamma-glutamyl transpeptidase. Proc. Natl. Acad. Sci. USA 1979, 76, 6319–6322. [Google Scholar] [CrossRef] [PubMed]
- Jaspers, C.; Penninckx, M. On the role of glutathione in the transport of amino acid in the yeast Saccharomyces cerevisiae: Contradictory results. FEBS Lett. 1981, 132, 41–44. [Google Scholar] [CrossRef]
- Damgaard, D.; Bjørn, M.E.; Steffensen, M.A.; Pruijn, G.J.; Nielsen, C.H. Reduced glutathione as a physiological co-activator in the activation of peptidylarginine deiminase. Arthritis Res. Ther. 2016, 18, 102. [Google Scholar] [CrossRef]
- Takusagawa, F. Microsomal prostaglandin E synthase type 2 (mPGES2) is a glutathione-dependent heme protein, and dithiothreitol dissociates the bound heme to produce active prostaglandin E2 synthase in vitro. J. Biol. Chem. 2013, 288, 10166–10175. [Google Scholar] [CrossRef]
- Sipes, I.G.; Wiersma, D.A.; Armstrong, D.J. The role of glutathione in the toxicity of xenobiotic compounds: Metabolic activation of 1,2-dibromoethane by glutathione. Adv. Exp. Med. Biol. 1986, 197, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of glutathione in cancer progression and chemoresistance. Oxid. Med. Cell. Longev. 2013, 2013, 972913. [Google Scholar] [CrossRef] [PubMed]
- Hanigan, M.H.; Ricketts, W.A. Extracellular glutathione is a source of cysteine for cells that express gamma-glutamyl transpeptidase. Biochemistry 1993, 32, 6302–6306. [Google Scholar] [CrossRef] [PubMed]
- Lushchak, V.I. Environmentally induced oxidative stress in aquatic animals. Aquat. Toxicol. 2011, 101, 13–30. [Google Scholar] [CrossRef]
- Valko, M.; Morris, H.; Cronin, M.T. Metals, toxicity and oxidative stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef] [PubMed]
- Baxter, P.S.; Bell, K.F.; Hasel, P.; Kaindl, A.M.; Fricker, M.; Thomson, D.; Cregan, S.P.; Gillingwater, T.H.; Hardingham, G.E. Synaptic NMDA receptor activity is coupled to the transcriptional control of the glutathione system. Nat. Commun. 2015, 6, 6761. [Google Scholar] [CrossRef]
- Freitas, H.R.; Ferraz, G.; Ferreira, G.C.; Ribeiro-Resende, V.T.; Chiarini, L.B.; do Nascimento, J.L.; Matos Oliveira, K.R.; Pereira Tde, L.; Ferreira, L.G.; Kubrusly, R.C.; et al. Glutathione-Induced Calcium Shifts in Chick Retinal Glial Cells. PLoS ONE 2016, 11, e0153677. [Google Scholar] [CrossRef]
- Nazıroğlu, M.; Özgül, C.; Çiğ, B.; Doğan, S.; Uğuz, A.C. Glutathione modulates Ca2+ influx and oxidative toxicity through TRPM2 channel in rat dorsal root ganglion neurons. J. Membr. Biol. 2011, 242, 109–118. [Google Scholar] [CrossRef]
- Monin, A.; Baumann, P.S.; Griffa, A.; Xin, L.; Mekle, R.; Fournier, M.; Butticaz, C.; Klaey, M.; Cabungcal, J.H.; Steullet, P.; et al. Glutathione deficit impairs myelin maturation: Relevance for white matter integrity in schizophrenia patients. Mol. Psychiatry 2015, 20, 827–838. [Google Scholar] [CrossRef]
- Janáky, R.; Ogita, K.; Pasqualotto, B.A.; Bains, J.S.; Oja, S.S.; Yoneda, Y.; Shaw, C.A. Glutathione and signal transduction in the mammalian CNS. J. Neurochem. 1999, 73, 889–902. [Google Scholar] [CrossRef]
- Koga, M.; Serritella, A.V.; Messmer, M.M.; Hayashi-Takagi, A.; Hester, L.D.; Snyder, S.H.; Sawa, A.; Sedlak, T.W. Glutathione is a physiologic reservoir of neuronal glutamate. Biochem. Biophys. Res. Commun. 2011, 409, 596–602. [Google Scholar] [CrossRef]
- Sagara, J.; Makino, N. Glutathione induces neuronal differentiation in rat bone marrow stromal cells. Neurochem. Res. 2008, 33, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Kurdi, M.; Sivakumaran, V.; Duhé, R.J.; Aon, M.A.; Paolocci, N.; Booz, G.W. Depletion of cellular glutathione modulates LIF-induced JAK1-STAT3 signaling in cardiac myocytes. Int. J. Biochem. Cell Biol. 2012, 44, 2106–2115. [Google Scholar] [CrossRef]
- Lee, H.M.; Kim, J.S.; Kang, S.O. Glutathione upregulates cAMP signalling via G protein alpha 2 during the development of Dictyostelium discoideum. FEBS Lett. 2016, 590, 4361–4371. [Google Scholar] [CrossRef][Green Version]
- Mak, T.W.; Grusdat, M.; Duncan, G.S.; Dostert, C.; Nonnenmacher, Y.; Cox, M.; Binsfeld, C.; Hao, Z.; Brüstle, A.; Itsumi, M.; et al. Glutathione Primes T Cell Metabolism for Inflammation. Immunity 2017, 46, 675–689. [Google Scholar] [CrossRef] [PubMed]
- Limón-Pacheco, J.H.; Hernández, N.A.; Fanjul-Moles, M.L.; Gonsebatt, M.E. Glutathione depletion activates mitogen-activated protein kinase (MAPK) pathways that display organ-specific responses and brain protection in mice. Free Radic. Biol. Med. 2007, 43, 1335–1347. [Google Scholar] [CrossRef]
- Chin, T.Y.; Chueh, S.H.; Tao, P.L. S-Nitrosoglutathione and glutathione act as NMDA receptor agonists in cultured hippocampal neurons. Acta Pharmacol. Sin. 2006, 27, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Soh, H.; Jung, W.; Uhm, D.Y.; Chung, S. Modulation of large conductance calcium-activated potassium channels from rat hippocampal neurons by glutathione. Neurosci. Lett. 2001, 298, 115–118. [Google Scholar] [CrossRef]
- Luberda, Z. The role of glutathione in mammalian gametes. Reprod. Biol. 2005, 5, 5–17. [Google Scholar]
- Winkler, A.; Njålsson, R.; Carlsson, K.; Elgadi, A.; Rozell, B.; Abraham, L.; Ercal, N.; Shi, Z.Z.; Lieberman, M.W.; Larsson, A.; et al. Glutathione is essential for early embryogenesis—Analysis of a glutathione synthetase knockout mouse. Biochem. Biophys. Res. Commun. 2011, 412, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Chi, L.; Ke, Y.; Luo, C.; Gozal, D.; Liu, R. Depletion of reduced glutathione enhances motor neuron degeneration in vitro and in vivo. Neuroscience 2007, 144, 991–1003. [Google Scholar] [CrossRef]
- Hansen, J.M.; Harris, C. Glutathione during embryonic development. Biochim. Biophys. Acta 2015, 1850, 1527–1542. [Google Scholar] [CrossRef] [PubMed]
- Zitka, O.; Skalickova, S.; Gumulec, J.; Masarik, M.; Adam, V.; Hubalek, J.; Trnkova, L.; Kruseova, J.; Eckschlager, T.; Kizek, R. Redox status expressed as GSH:GSSG ratio as a marker for oxidative stress in paediatric tumour patients. Oncol. Lett. 2012, 4, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Guo, N.; McIntosh, C.; Shaw, C. Glutathione: New candidate neuropeptide in the central nervous system. Neuroscience 1992, 51, 835–842. [Google Scholar] [CrossRef]
- Ogita, K.; Yoneda, Y. Temperature-dependent and -independent apparent binding activities of [3H]glutathione in brain synaptic membranes. Brain Res. 1988, 463, 37–46. [Google Scholar] [CrossRef]
- Dringen, R.; Gutterer, J.M.; Hirrlinger, J. Glutathione metabolism in brain metabolic interaction between astrocytes and neurons in the defense against reactive oxygen species. Eur. J. Biochem. 2000, 267, 4912–4916. [Google Scholar] [CrossRef]
- Dringen, R.; Hirrlinger, J. Glutathione pathways in the brain. Biol. Chem. 2003, 384, 505–516. [Google Scholar] [CrossRef]
- Anderson, M.E.; Meister, A. Dynamic state of glutathione in blood plasma. J. Biol. Chem. 1980, 255, 9530–9533. [Google Scholar] [CrossRef]
- Meister, A.; Anderson, M.E. Glutathione. Annu. Rev. Biochem. 1983, 52, 711–760. [Google Scholar] [CrossRef]
- Hargreaves, K.M.; Pardridge, W.M. Neutral amino acid transport at the human blood-brain barrier. J. Biol. Chem. 1988, 263, 19392–19397. [Google Scholar] [CrossRef]
- Wade, L.A.; Brady, H.M. Cysteine and cystine transport at the blood-brain barrier. J. Neurochem. 1981, 37, 730–734. [Google Scholar] [CrossRef]
- Belrose, J.C.; Xie, Y.F.; Gierszewski, L.J.; MacDonald, J.F.; Jackson, M.F. Loss of glutathione homeostasis associated with neuronal senescence facilitates TRPM2 channel activation in cultured hippocampal pyramidal neurons. Mol. Brain 2012, 5, 11. [Google Scholar] [CrossRef] [PubMed]
- Beiswanger, C.M.; Diegmann, M.H.; Novak, R.F.; Philbert, M.A.; Graessle, T.L.; Reuhl, K.R.; Lowndes, H.E. Developmental changes in the cellular distribution of glutathione and glutathione S-transferases in the murine nervous system. Neurotoxicology 1995, 16, 425–440. [Google Scholar] [PubMed]
- Huang, J.; Philbert, M.A. Distribution of glutathione and glutathione-related enzyme systems in mitochondria and cytosol of cultured cerebellar astrocytes and granule cells. Brain Res. 1995, 680, 16–22. [Google Scholar] [CrossRef]
- Griffith, O.W.; Meister, A. Origin and turnover of mitochondrial glutathione. Proc. Natl. Acad. Sci. USA 1985, 82, 4668–4672. [Google Scholar] [CrossRef]
- Ribas, V.; García-Ruiz, C.; Fernández-Checa, J.C. Glutathione and mitochondria. Front. Pharmacol. 2014, 5, 151. [Google Scholar] [CrossRef]
- Oja, S.S.; Janáky, R.; Varga, V.; Saransaari, P. Modulation of glutamate receptor functions by glutathione. Neurochem. Int. 2000, 37, 299–306. [Google Scholar] [CrossRef]
- Shen, X.M.; Dryhurst, G. Oxidation chemistry of (−)-norepinephrine in the presence of L-cysteine. J. Med. Chem. 1996, 39, 2018–2029. [Google Scholar] [CrossRef]
- Aizenman, E.; Lipton, S.A.; Loring, R.H. Selective modulation of NMDA responses by reduction and oxidation. Neuron 1989, 2, 1257–1263. [Google Scholar] [CrossRef]
- Choi, Y.B.; Lipton, S.A. Redox modulation of the NMDA receptor. Cell. Mol. Life Sci. 2000, 57, 1535–1541. [Google Scholar] [CrossRef]
- Gozlan, H.; Ben-Ari, Y. NMDA receptor redox sites: Are they targets for selective neuronal protection? Trends Pharmacol. Sci. 1995, 16, 368–374. [Google Scholar] [CrossRef]
- Sullivan, J.M.; Traynelis, S.F.; Chen, H.S.; Escobar, W.; Heinemann, S.F.; Lipton, S.A. Identification of two cysteine residues that are required for redox modulation of the NMDA subtype of glutamate receptor. Neuron 1994, 13, 929–936. [Google Scholar] [CrossRef]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free radicals: Properties, sources, targets, and their implication in various diseases. Indian J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef]
- Wilkins, H.M.; Kirchhof, D.; Manning, E.; Joseph, J.W.; Linseman, D.A. Mitochondrial glutathione transport is a key determinant of neuronal susceptibility to oxidative and nitrosative stress. J. Biol. Chem. 2013, 288, 5091–5101. [Google Scholar] [CrossRef]
- Iskusnykh, I.Y.; Popova, T.N.; Agarkov, A.A.; Pinheiro de Carvalho, M.; Rjevskiy, S.G. Expression of Glutathione Peroxidase and Glutathione Reductase and Level of Free Radical Processes under Toxic Hepatitis in Rats. J. Toxicol. 2013, 2013, 870628. [Google Scholar] [CrossRef] [PubMed]
- Njålsson, R. Glutathione synthetase deficiency. Cell. Mol. Life Sci. 2005, 62, 1938–1945. [Google Scholar] [CrossRef]
- Njålsson, R.; Ristoff, E.; Carlsson, K.; Winkler, A.; Larsson, A.; Norgren, S. Genotype, enzyme activity, glutathione level, and clinical phenotype in patients with glutathione synthetase deficiency. Hum. Genet. 2005, 116, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Ristoff, E.; Larsson, A. Inborn errors in the metabolism of glutathione. Orphanet J. Rare Dis. 2007, 2, 16. [Google Scholar] [CrossRef]
- Ristoff, E.; Mayatepek, E.; Larsson, A. Long-term clinical outcome in patients with glutathione synthetase deficiency. J. Pediatr. 2001, 139, 79–84. [Google Scholar] [CrossRef]
- Bannon, M.J.; Goedert, M.; Williams, B. The possible relation of glutathione, melanin and 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine (MPTP) to Parkinson’s disease. Biochem. Pharmacol. 1984, 33, 2697–2698. [Google Scholar] [CrossRef]
- Perry, T.L.; Yong, V.W. Idiopathic Parkinson’s disease, progressive supranuclear palsy and glutathione metabolism in the substantia nigra of patients. Neurosci. Lett. 1986, 67, 269–274. [Google Scholar] [CrossRef]
- Riederer, P.; Sofic, E.; Rausch, W.D.; Schmidt, B.; Reynolds, G.P.; Jellinger, K.; Youdim, M.B. Transition metals, ferritin, glutathione, and ascorbic acid in parkinsonian brains. J. Neurochem. 1989, 52, 515–520. [Google Scholar] [CrossRef]
- Smeyne, M.; Smeyne, R.J. Glutathione metabolism and Parkinson’s disease. Free Radic. Biol. Med. 2013, 62, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.G.; Trabesinger, A.H.; Boesiger, P.; Wieser, H.G. Brain glutathione levels in patients with epilepsy measured by in vivo (1)H-MRS. Neurology 2001, 57, 1422–1427. [Google Scholar] [CrossRef]
- Abbott, L.C.; Nejad, H.H.; Bottje, W.G.; Hassan, A.S. Glutathione levels in specific brain regions of genetically epileptic (tg/tg) mice. Brain Res. Bull. 1990, 25, 629–631. [Google Scholar] [CrossRef]
- Hu, H.L.; Bennett, N.; Holton, J.L.; Nolan, C.C.; Lister, T.; Cavanagh, J.B.; Ray, D.E. Glutathione depletion increases brain susceptibility to m-dinitrobenzene neurotoxicity. Neurotoxicology 1999, 20, 83–90. [Google Scholar]
- Ravindranath, V.; Shivakumar, B.R.; Anandatheerthavarada, H.K. Low glutathione levels in brain regions of aged rats. Neurosci. Lett. 1989, 101, 187–190. [Google Scholar] [CrossRef]
- Haddad, J.J.; Harb, H.L. L-gamma-Glutamyl-L-cysteinyl-glycine (glutathione; GSH) and GSH-related enzymes in the regulation of pro- and anti-inflammatory cytokines: A signaling transcriptional scenario for redox(y) immunologic sensor(s)? Mol. Immunol. 2005, 42, 987–1014. [Google Scholar] [CrossRef]
- Beutler, E.; Gelbart, T.; Kondo, T.; Matsunaga, A.T. The Molecular Basis of a Case of γ-Glutamylcysteine Synthetase Deficiency. Blood 1999, 94, 2890–2894. [Google Scholar] [CrossRef]
- Walsh, A.C.; Feulner, J.A.; Reilly, A. Evidence for functionally significant polymorphism of human glutamate cysteine ligase catalytic subunit: Association with glutathione levels and drug resistance in the National Cancer Institute tumor cell line panel. Toxicol. Sci. 2001, 61, 218–223. [Google Scholar] [CrossRef]
- Gysin, R.; Kraftsik, R.; Sandell, J.; Bovet, P.; Chappuis, C.; Conus, P.; Deppen, P.; Preisig, M.; Ruiz, V.; Steullet, P.; et al. Impaired glutathione synthesis in schizophrenia: Convergent genetic and functional evidence. Proc. Natl. Acad. Sci. USA 2007, 104, 16621–16626. [Google Scholar] [CrossRef]
- Lu, S.C. Regulation of glutathione synthesis. Mol. Asp. Med. 2009, 30, 42–59. [Google Scholar] [CrossRef] [PubMed]
- Simon, E.; Vogel, M.; Fingerhut, R.; Ristoff, E.; Mayatepek, E.; Spiekerkötter, U. Diagnosis of glutathione synthetase deficiency in newborn screening. J. Inherit. Metab. Dis. 2009, 32 (Suppl. 1), S269–S272. [Google Scholar] [CrossRef]
- Atwal, P.S.; Medina, C.R.; Burrage, L.C.; Sutton, V.R. Nineteen-year follow-up of a patient with severe glutathione synthetase deficiency. J. Hum. Genet. 2016, 61, 669–672. [Google Scholar] [CrossRef]
- Esteban, L.; Decker, S.; Riddle, D.; Caputo, A.; Zhang, B.; Cole, T.; Caswell, C.; Xie, S.; Lee, V.; Luk, K. Differential α-synuclein expression contributes to selective vulnerability of hippocampal neuron subpopulations to fibril-induced toxicity. Acta Neuropathol. 2018, 135, 855–875. [Google Scholar] [CrossRef]
- Kuo, Y.; Ng, I.W.; Rajesh, R. Glutathione- and apolipoprotein E-grafted liposomes to regulate mitogen-activated protein kinases and rescueneurons in Alzheimer’s disease. Mater. Sci. Eng. C Mater. Biol. Appl. 2021, 127, 112233. [Google Scholar] [CrossRef]
- Mudher, A.; Colin, M.; Dujardin, S.; Medina, M.; Dewachter, I.; Alavi Naini, S.; Mandelkow, E.; Mandelkow, E.; Buée, L.; Goedert, M.; et al. What is the evidence that tau pathology spreads through prion-like propagation? Acta Neuropathol. Commun. 2017, 5, 99. [Google Scholar] [CrossRef]
- Van Den Berge, N.; Ferreira, N.; Gram, H.; Werenberg Mikkelsen, T.; Olsen Alstrup, A.; Casadei, N.; Tsung-Pin, P.; Riess, O.; Randel Nyengaard, J.; Tamgüney, G.; et al. Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol. 2019, 138, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, N.; Pereira Gonçalves, N.; Jan, A.; Møller Jensen, N.; Laan, A.; Mohseni, S.; Bjerggaard Vægter, C.; Henning Jensen, P. Trans-synaptic spreading of alpha-synuclein pathology through sensory afferents leads to sensory nerve degeneration and neuropathic pain. Acta Neuropathol. Commun. 2021, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, N.; Gram, H.; Sorrentino, Z.; Gregersen, E.; Schmidt, S.; Reimer, L.; Betzer, C.; Perez-Gozalbo, C.; Beltoja, M.; Nagaraj, M.; et al. Multiple system atrophy-associated oligodendroglial protein p25α stimulates formation of novel α-synuclein strain with enhanced neurodegenerative potential. Acta Neuropathol. 2021, 142, 87–115. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.; Woerman, A.; Mordes, D.; Watts, J.; Rampersaud, R.; Berry, D.; Patel, S.; Oehler, A.; Lowe, J.; Kravitz, S.; et al. Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc. Natl. Acad. Sci. USA 2015, 112, E5308–E5317. [Google Scholar] [CrossRef] [PubMed]
- Vaquer-Alicea, J.; Diamond, M.; Joachimiak, L. Tau strains shape disease. Rev. Acta Neuropathol. 2021, 142, 57–71. [Google Scholar] [CrossRef]
- Bjorklund, G.; Peano, M.; Maes, M.; Dadar, M.; Severin, B. The glutathione system in Parkinson’s disease and its progression. Neurosci. Biobehav. Rev. 2021, 120, 470–478. [Google Scholar] [CrossRef]
- Mandal, P.; Shukla, D.; Tripathi, M.; Ersland, L. Cognitive improvement with glutathione supplement in Alzheimer’s disease: A way forward. J. Alzheimers Dis. 2019, 68, 531–535. [Google Scholar] [CrossRef] [PubMed]
- Kim, K. Glutathione in the nervous system as a potential therapeutic target to control the development and progression of amyotrophic lateral sclerosis. Antioxidants 2021, 10, 1011. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Function | Role of GSH | Reference |
|---|---|---|
| Enhancement of immune system function | Protects against inflammatory pathologies Impaired immunological function caused by cysteine and glutathione deficiency is restored when supplemented with cysteine | [7,8] |
| Prevention of oxidative cell damage | Serves as an antioxidant | [9,10] |
| Prostaglandin synthesis | Inhibits prostaglandin synthesis at elevated levels | [11] |
| Transport of metals across membranes | Chelates reactive metals and facilitates their transport across cell membranes | [12] |
| Transfers metals between ligands | Forms coordinate-covalent adducts with several transition metals and transports them between ligands | [13] |
| Protein synthesis | Is involved in post-translational modification of proteins | [14] |
| DNA synthesis and repair | Efficient scavenging of OH• and secondary radicals; participates in rejoining of X-ray induced DNA strand breaks; activates T-cell proliferation; provides a source of cysteine moieties | [15,16,17] |
| Amino acid transport | Participates in amino acid permeation system; assists with transmembrane transport of amino acids by acting as a donor of g-glutamyl groups to amino acids, catalyzed by membrane-bound g-glutamyl transpeptidase | [18,19] |
| Enzyme activation | As a reducing agent, is important for activation of peptidyl-arginine deiminase; binds to microsomal prostaglandin E synthase type 2 (mPGES2), a heme group is attached to GSH via an iron-sulfur bond, and the resulting enzyme catalyzes degradation of PGH2 | [20,21] |
| Metabolism of toxins and carcinogens | Detoxification of electrophilic xenobiotics and endogenous compounds after spontaneous or enzymatic (GSH-S-transferase) GSH conjugation; conjugation of 1,2-dibromoethane | [22,23] |
| Redox reactions | Neutralizes charges on ROS, RNS, and other reactive species | [4] |
| Source of cysteine | Serves as an extracellular source of cysteine | [24] |
| Metal Homeostasis | Reduction of Cr6+ | [25,26] |
| NMDAR responses, neuronal activity | Enhances NMDAR responses and neuronal activation by calcium or electrical signals, while its depletion or oxidation results in NMDAR hypofunction | [27] |
| Calcium signaling | Participates in calcium signaling (mM range), especially in Müller glia; can regulate GABA release with protective effects on the retinal neuron-glial circuit; modulates influx of Ca2+ and oxidative toxicity through the TRPM2 channel in rat dorsal root ganglion neurons | [28,29] |
| Myelin maturation | Its deficit impairs maturation of myelin | [30] |
| Neuro-modulator, neurotransmitter | Has binding sites for a putative receptor, likely the glutamate receptor; can serve as a neuromodulator or neurotransmitter | [31] |
| Source of neuronal glutamate | Serves as a physiologic reservoir of neuronal glutamate | [32] |
| Neuronal differentiation | Induces neuronal differentiation in rat bone marrow stromal cells | [33] |
| Apoptosis | Intracellular levels decrease after activation of the mitochondrial death receptor, drug exposure, or oxidative stress | [3] |
| JAK1-STAT3 signaling | Reducing its intracellular pool enhances LIF-induced JAK1-STAT3 signaling | [34] |
| cAMP signaling | Upregulates cAMP signaling via G protein alpha 2 | [35] |
| T-cell metabolism | Primes T cell metabolism for inflammation | [36] |
| Activates MAPK pathways | Reducing its intracellular pool activates mitogen-activated protein kinase (MAPK) pathways | [37] |
| NMDA-R agonist | Act as an agonist of the NMDA receptor | [27,38] |
| Ca2+-activated K+ channels | Reduced form increases channel activities, while oxidized form inhibits channel activities | [39] |
| Mammalian development | Essential for embryonic development, as knockout of homozygous glutathione synthetase is lethal in mice; implicated in maintenance of meiotic spindle morphology in oocytes | [40,41] |
| Prevention of motor neuron degeneration | Decreased levels promote degeneration of motor neurons in vitro and in vivo | [42] |
| Disease/Disorder | Blood | Brain |
|---|---|---|
| Alzheimer’s disease (AD) | Decreased GSH | Decreased GSH |
| Decreased GSH/GSSG | Decreased GST | |
| Increased GSSG | - | |
| Decreased Gpx activity | - | |
| Amyotrophic lateral sclerosis (ALS) | Decreased GSH (erythrocytes) | Decreased GSH (motor cortex) |
| - | Decreased GST (motor cortex) | |
| Autism | Decreased GSH | Decreased GSH in (celebellum and temporal cortex) |
| Increased GSSG | Decreased Gpx activity | |
| Decreased Gpx activity (erythrocytes) | Decreased GST activity | |
| Decreased GST activity (erythrocytes) | Decreased GCL activity | |
| Decreased GSH/GSSG | Decreased GSH/GSSG (cerebellum and temporal cortex) | |
| Bipolar disorder | Decreased GSH | Decreased GSH (hippocampus and anterior cortex) |
| Increased GSSG | - | |
| Decreased GSH/GSSG | - | |
| Huntington’s disease (HD) | Decreased GSH | - |
| Multiple sclerosis | Decreased GSH | Decreased GSH |
| Parkinson’s disease (PD) | Decreased GSH | Decreased GSH |
| - | Increased Gpx protein | |
| - | Increased GST protein | |
| Schizophrenia | Decreased GSH | Decreased GSH |
| Decreased GSH/GSSG | - | |
| Increased GSSG | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iskusnykh, I.Y.; Zakharova, A.A.; Pathak, D. Glutathione in Brain Disorders and Aging. Molecules 2022, 27, 324. https://doi.org/10.3390/molecules27010324
Iskusnykh IY, Zakharova AA, Pathak D. Glutathione in Brain Disorders and Aging. Molecules. 2022; 27(1):324. https://doi.org/10.3390/molecules27010324
Chicago/Turabian StyleIskusnykh, Igor Y., Anastasia A. Zakharova, and Dhruba Pathak. 2022. "Glutathione in Brain Disorders and Aging" Molecules 27, no. 1: 324. https://doi.org/10.3390/molecules27010324
APA StyleIskusnykh, I. Y., Zakharova, A. A., & Pathak, D. (2022). Glutathione in Brain Disorders and Aging. Molecules, 27(1), 324. https://doi.org/10.3390/molecules27010324