
Development of Phenothiazine Hybrids with Potential Medicinal Interest: A Review





{kind=link}
{kind=link}
{kind=link}
{kind=link}
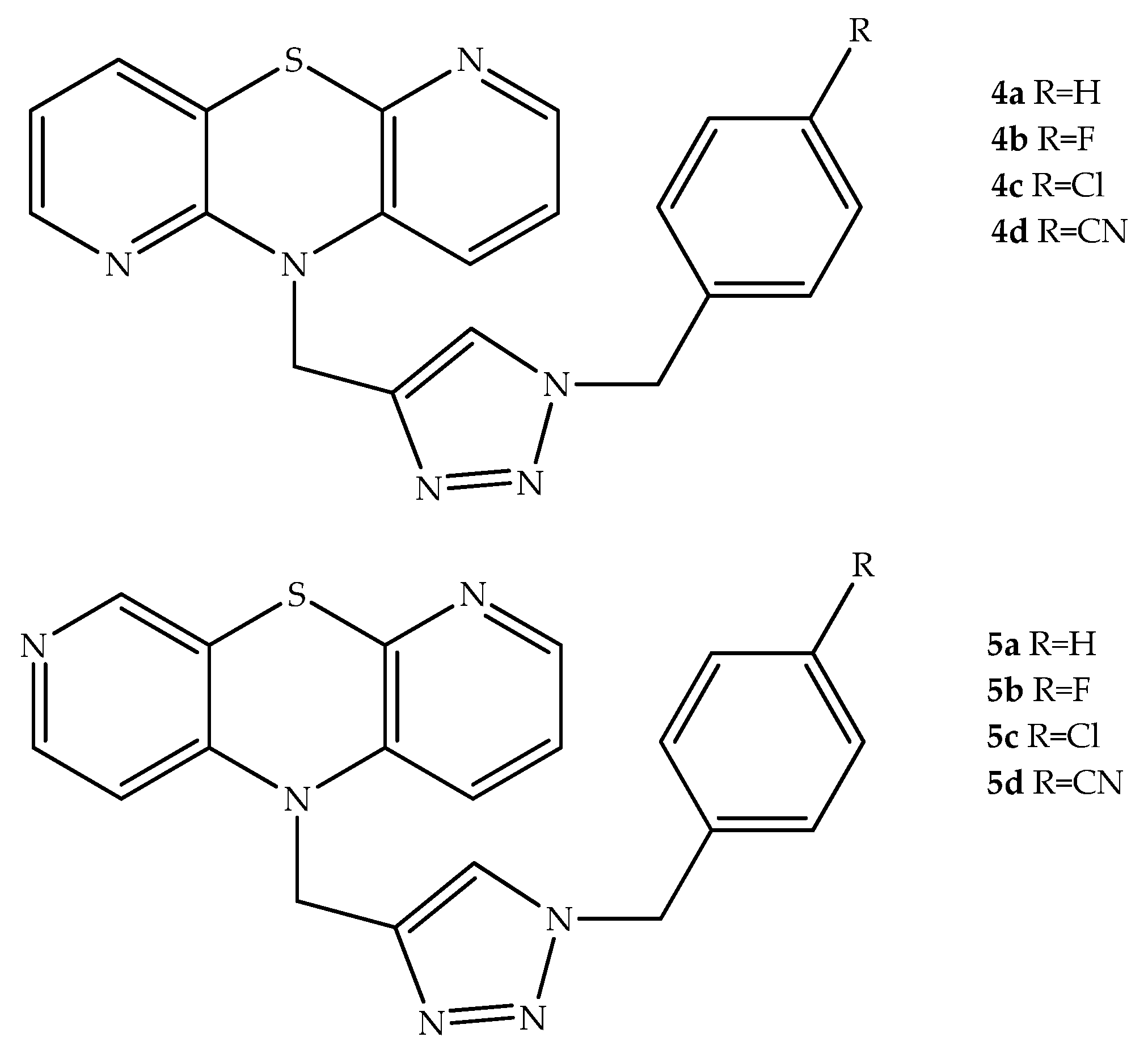{kind=link}
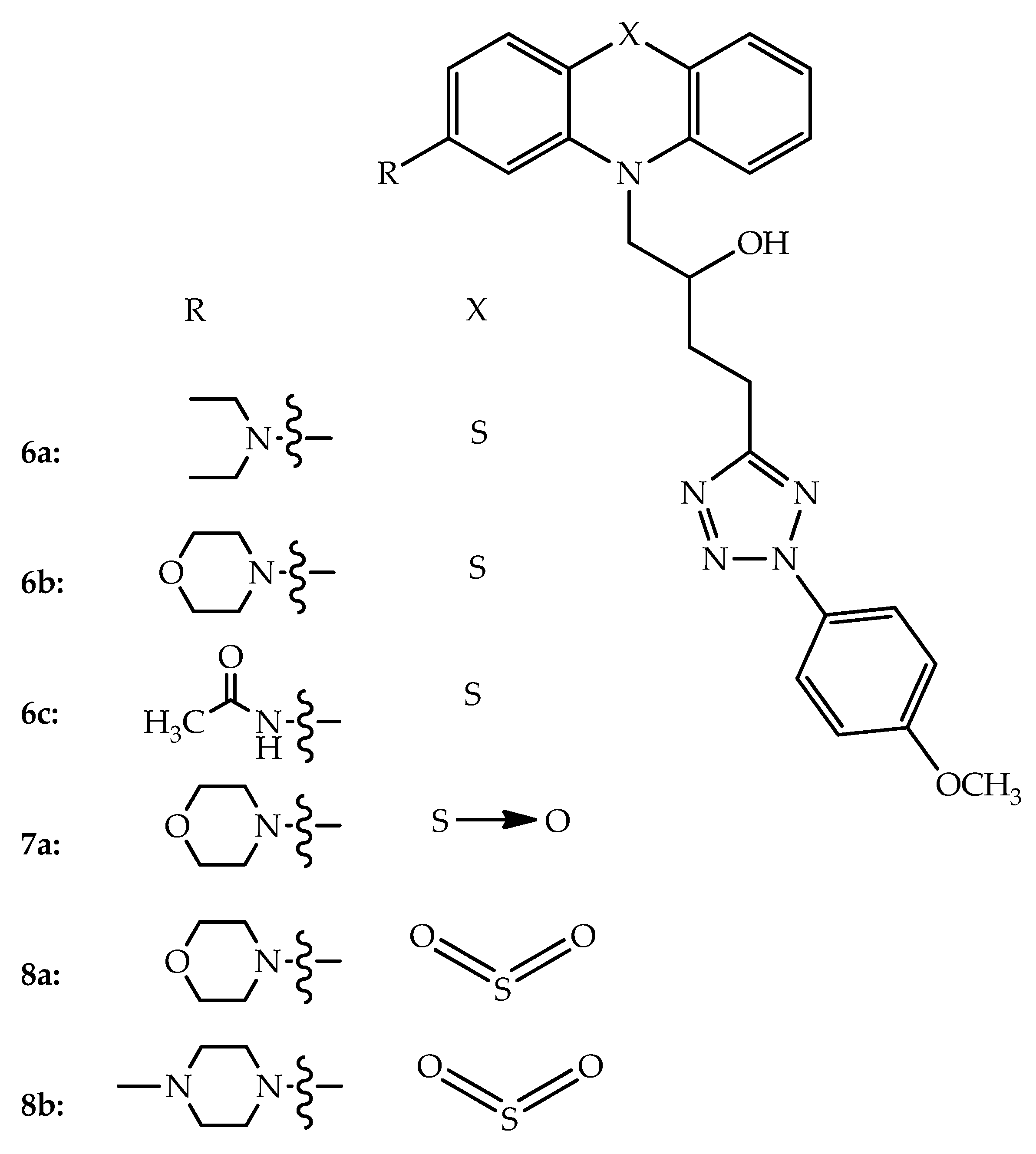{kind=link}
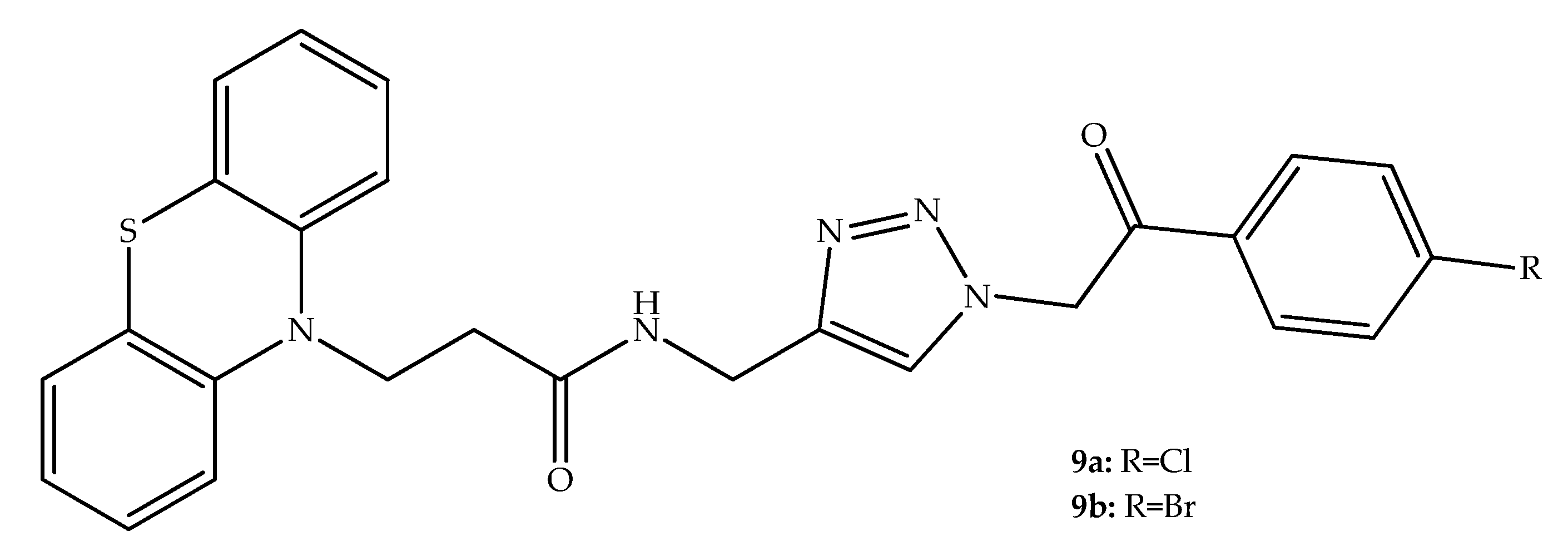{kind=link}
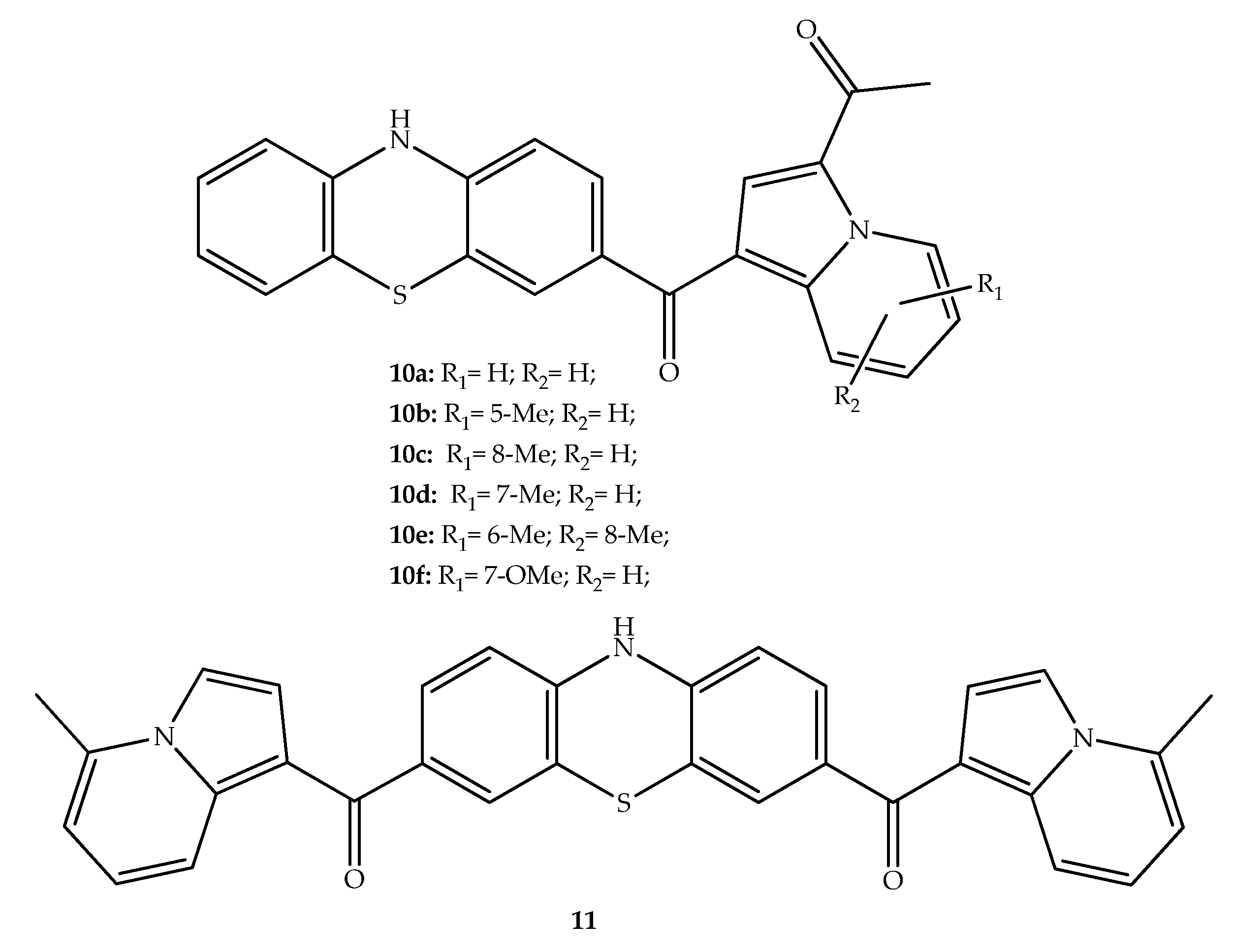{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
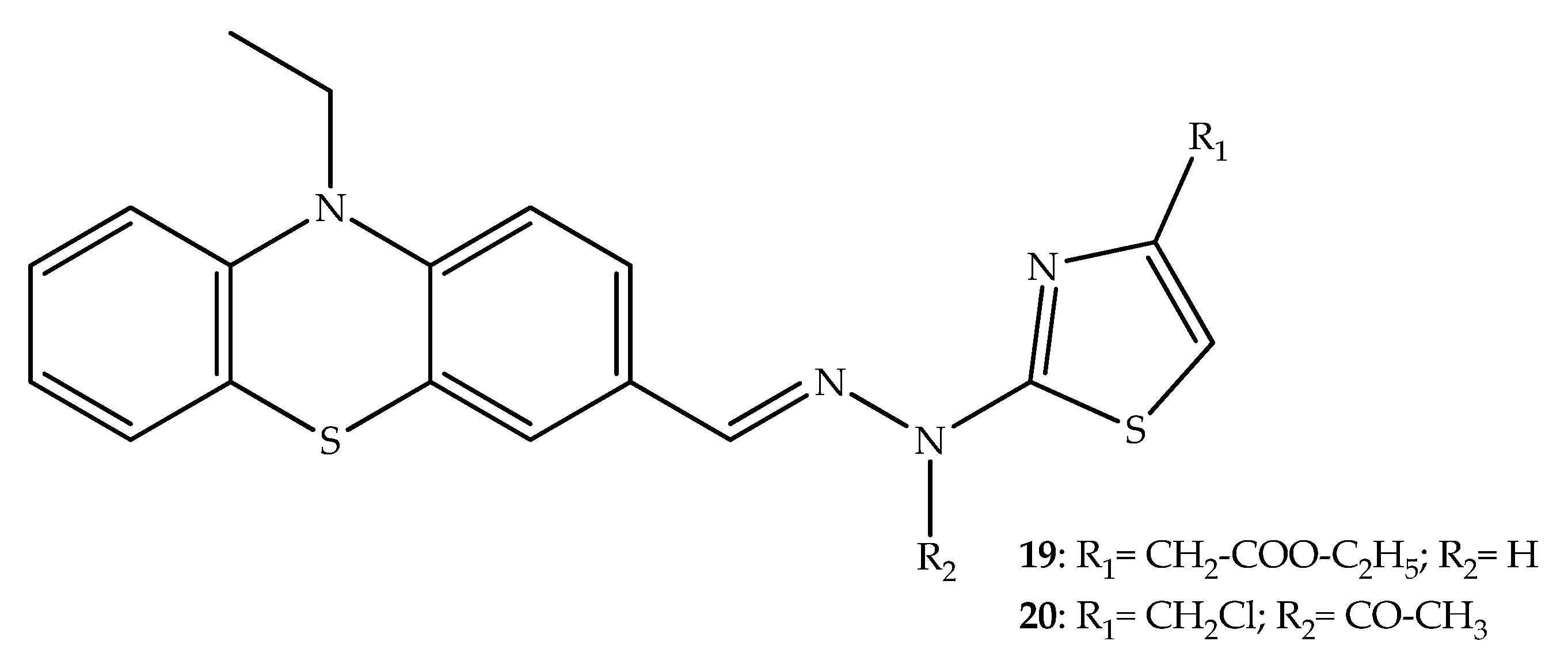{kind=link}
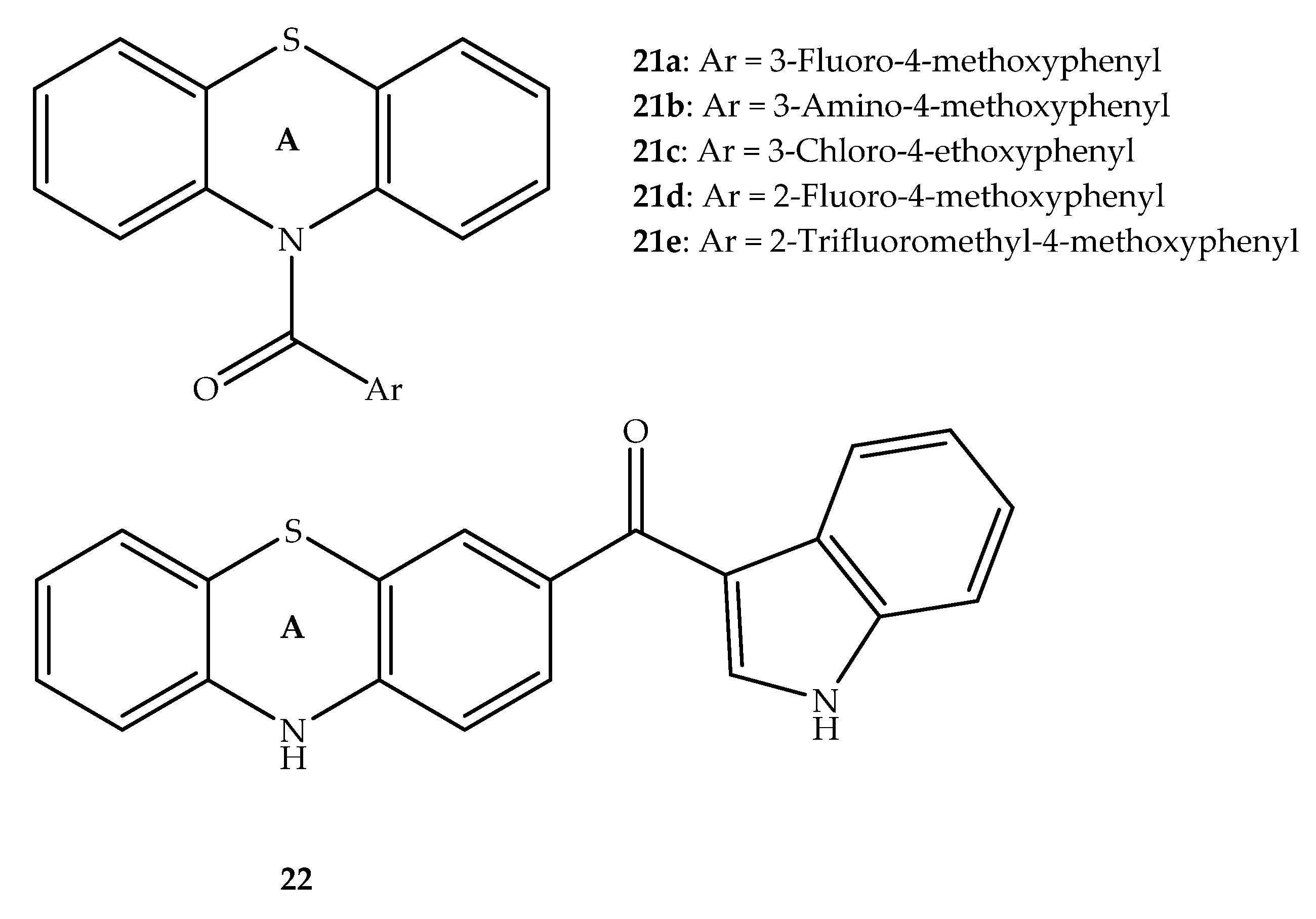{kind=link}
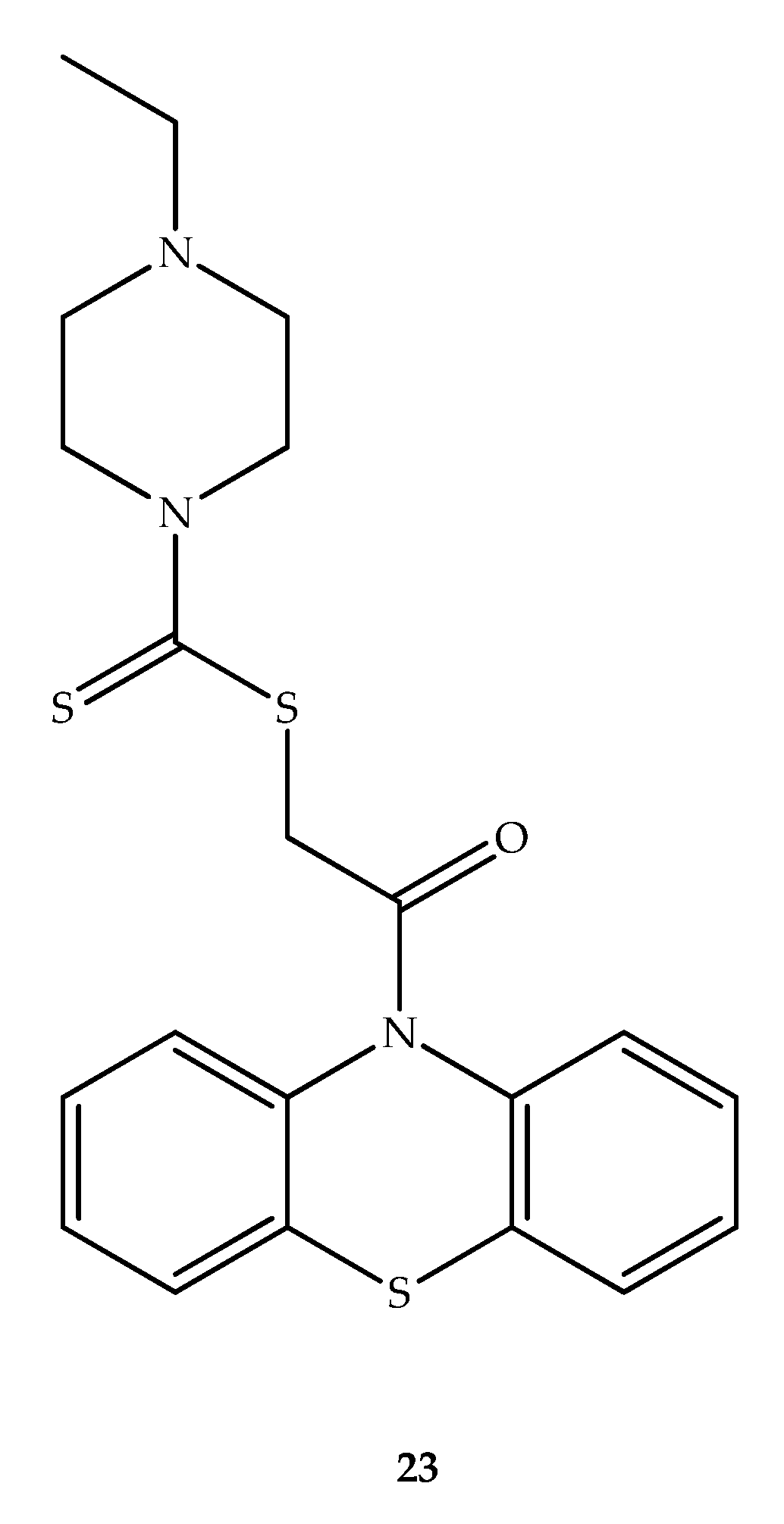{kind=link}
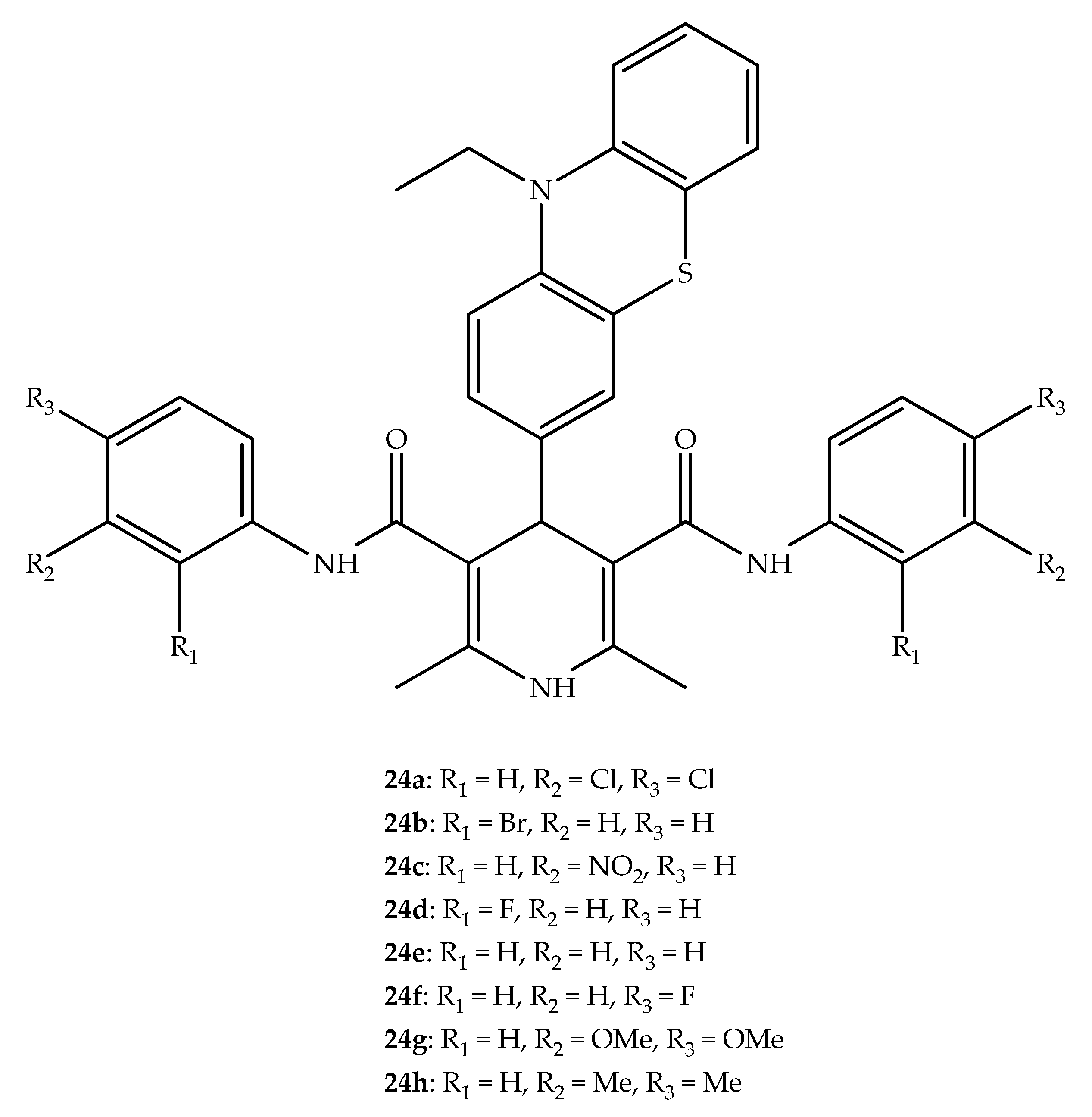{kind=link}
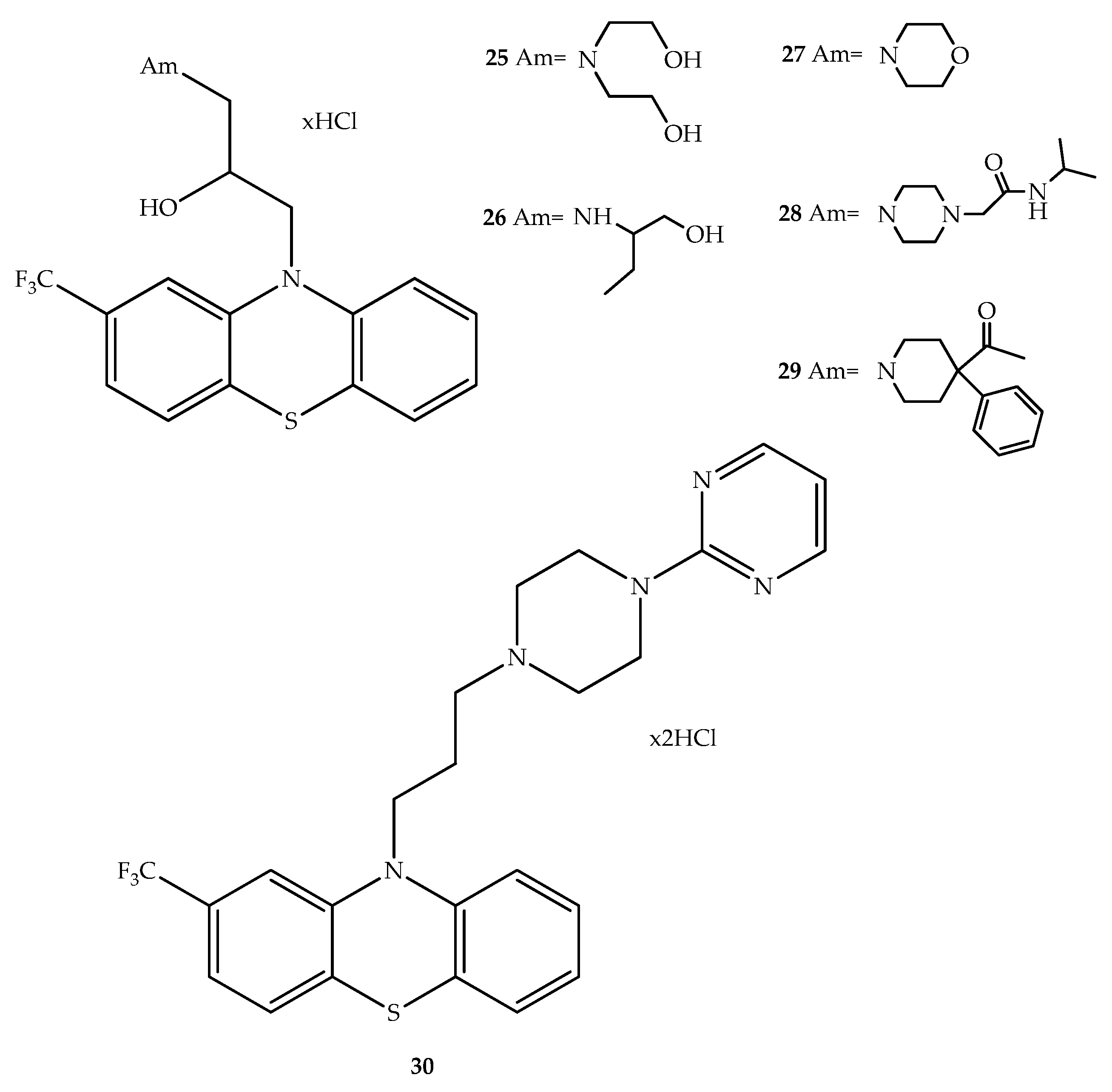{kind=link}
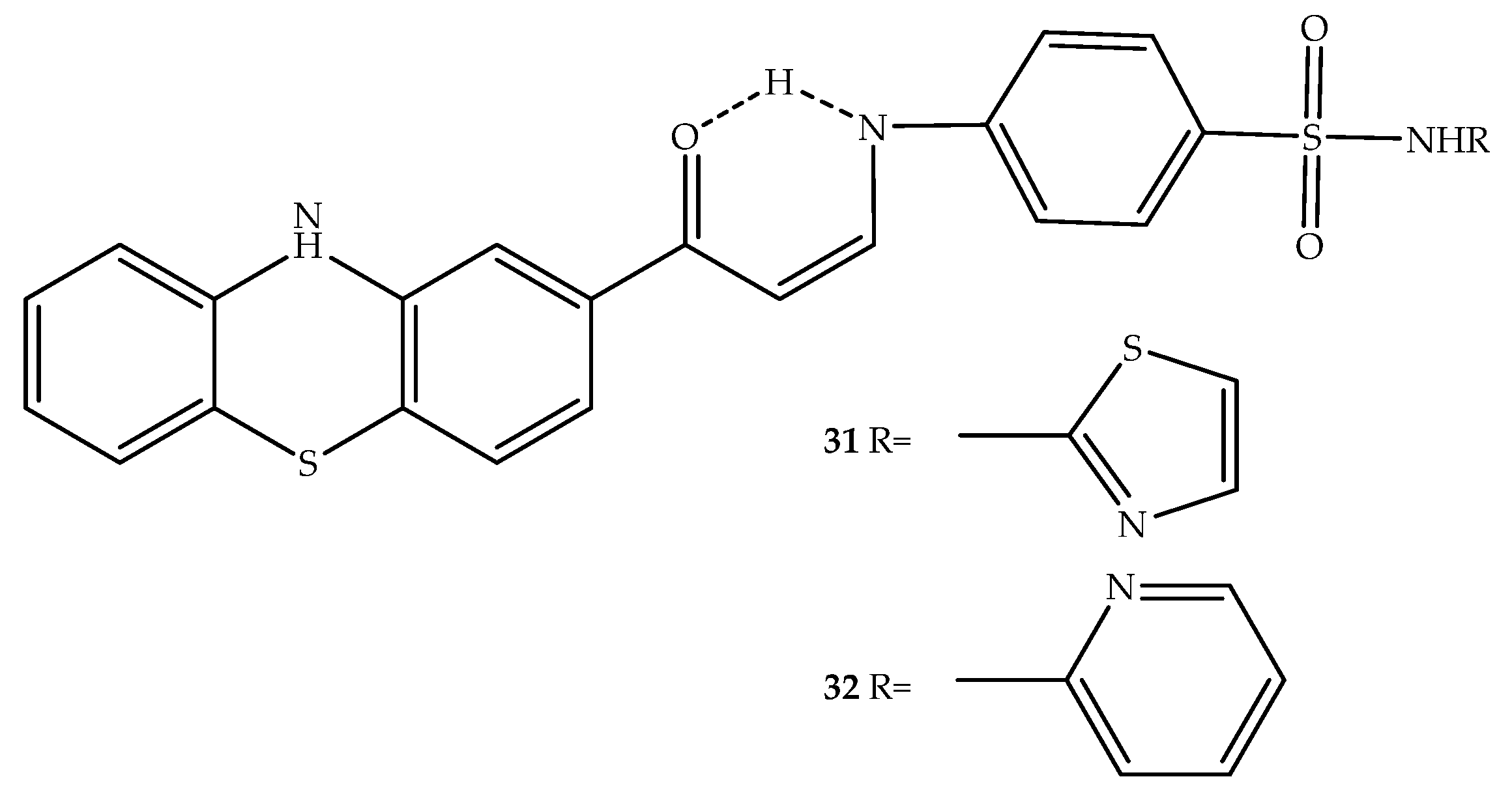{kind=link}
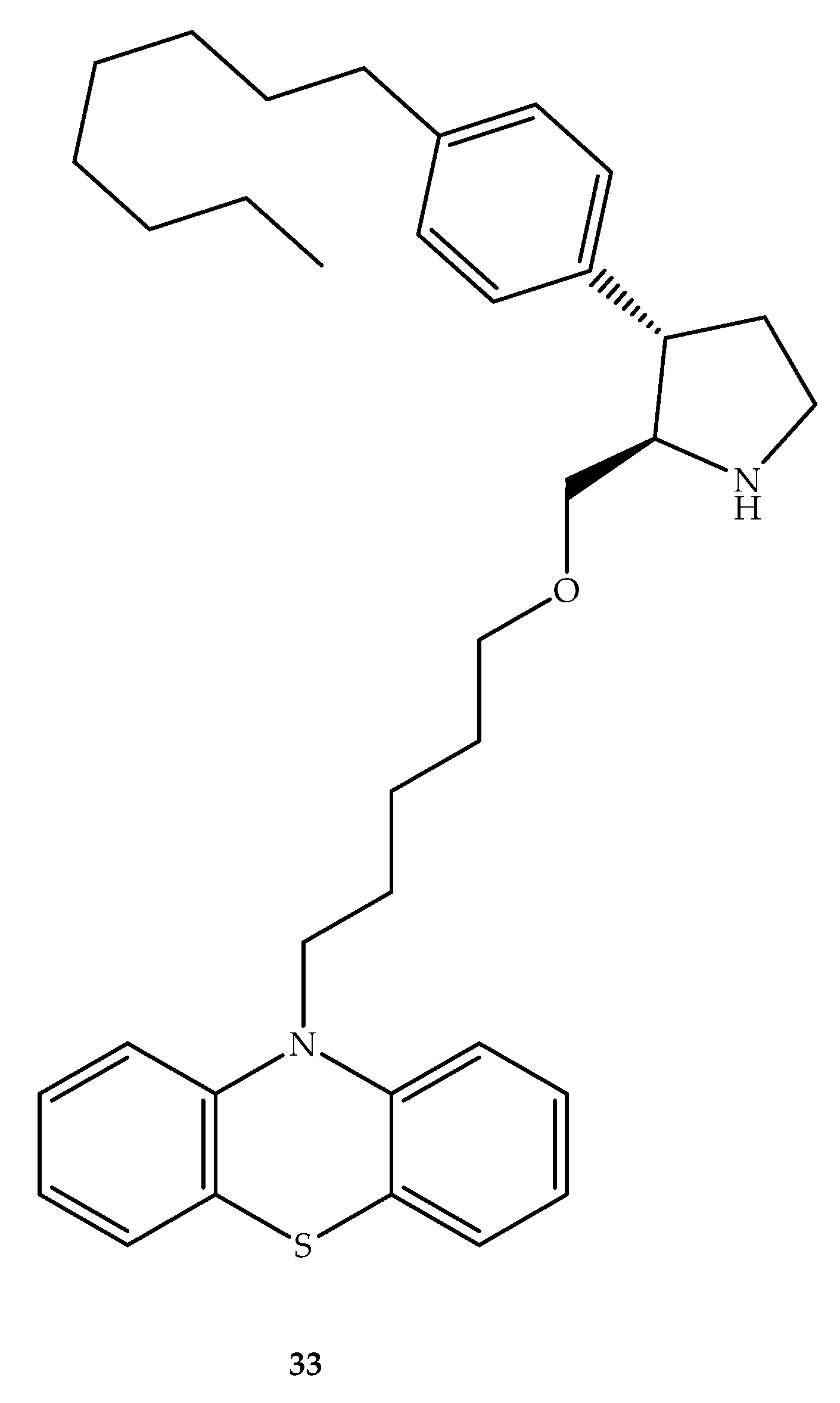{kind=link}
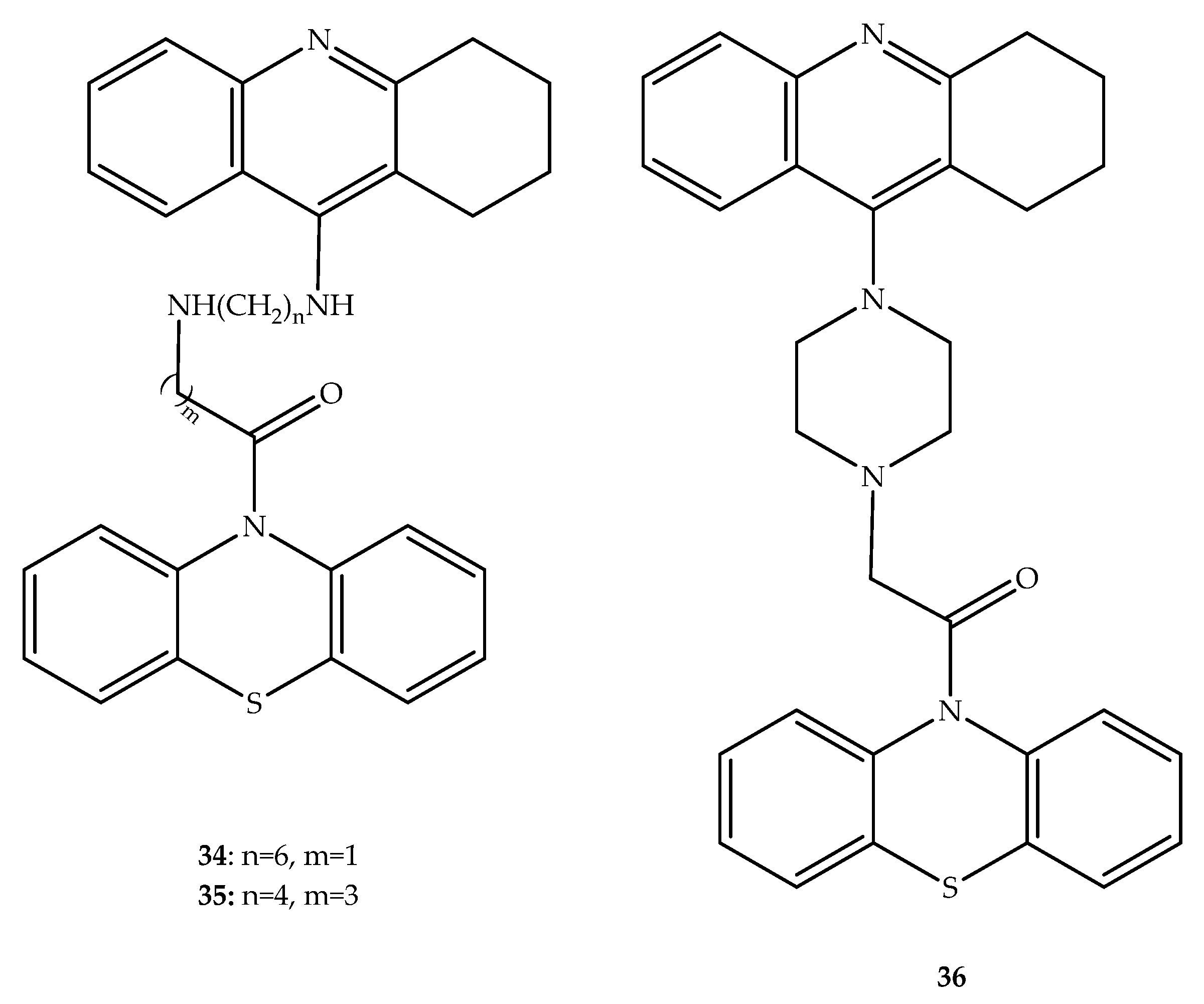{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
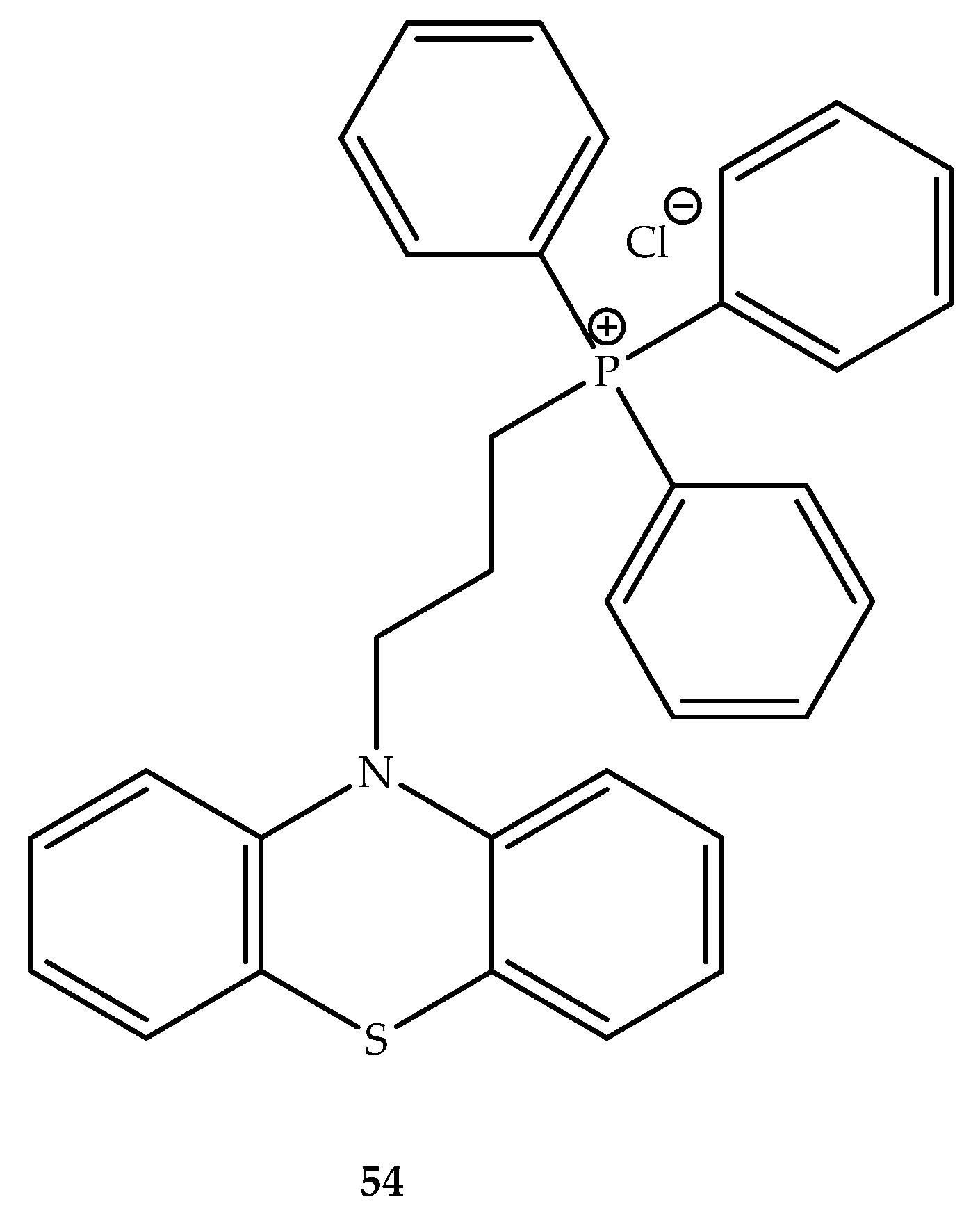{kind=link}
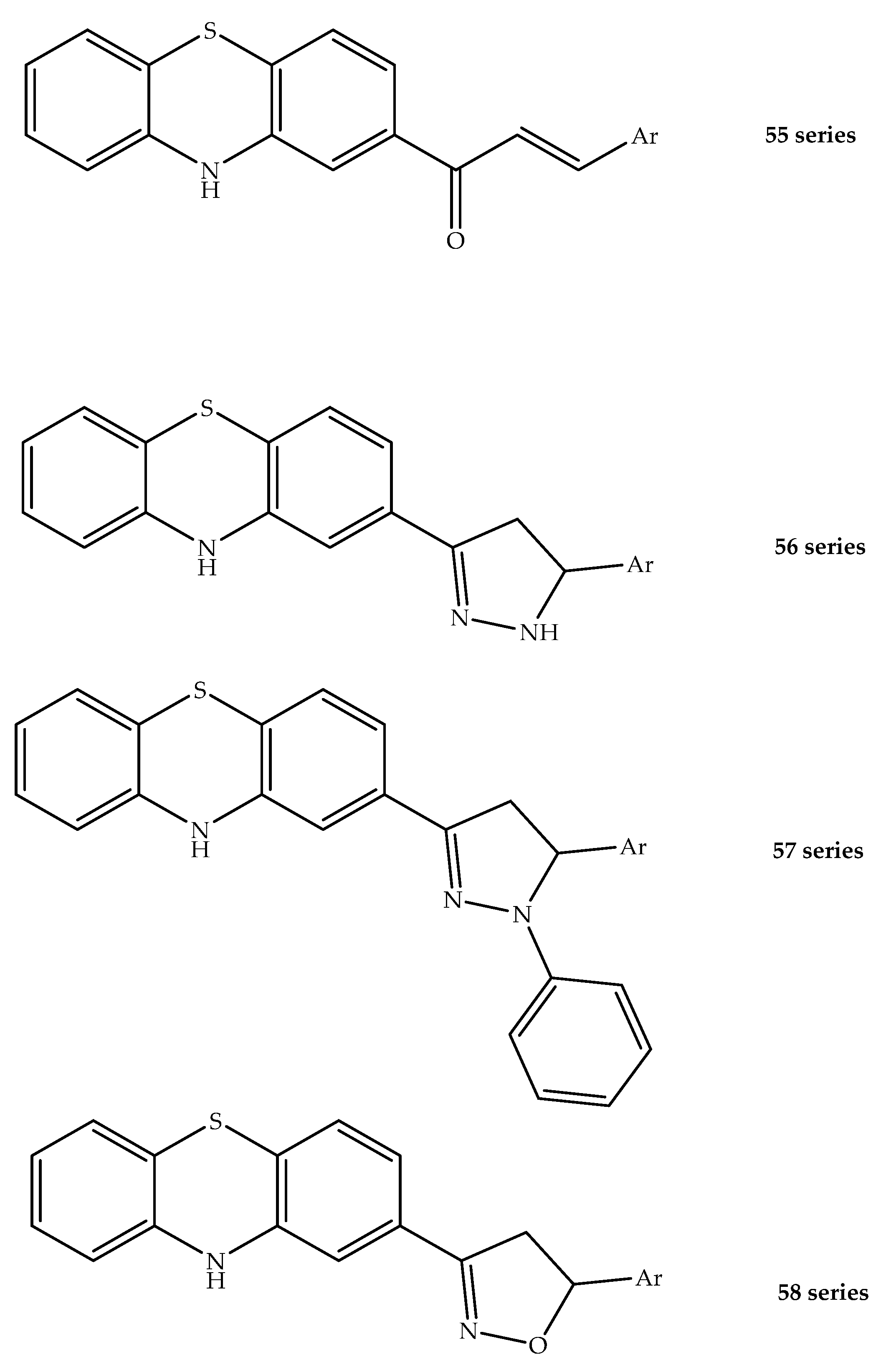{kind=link}
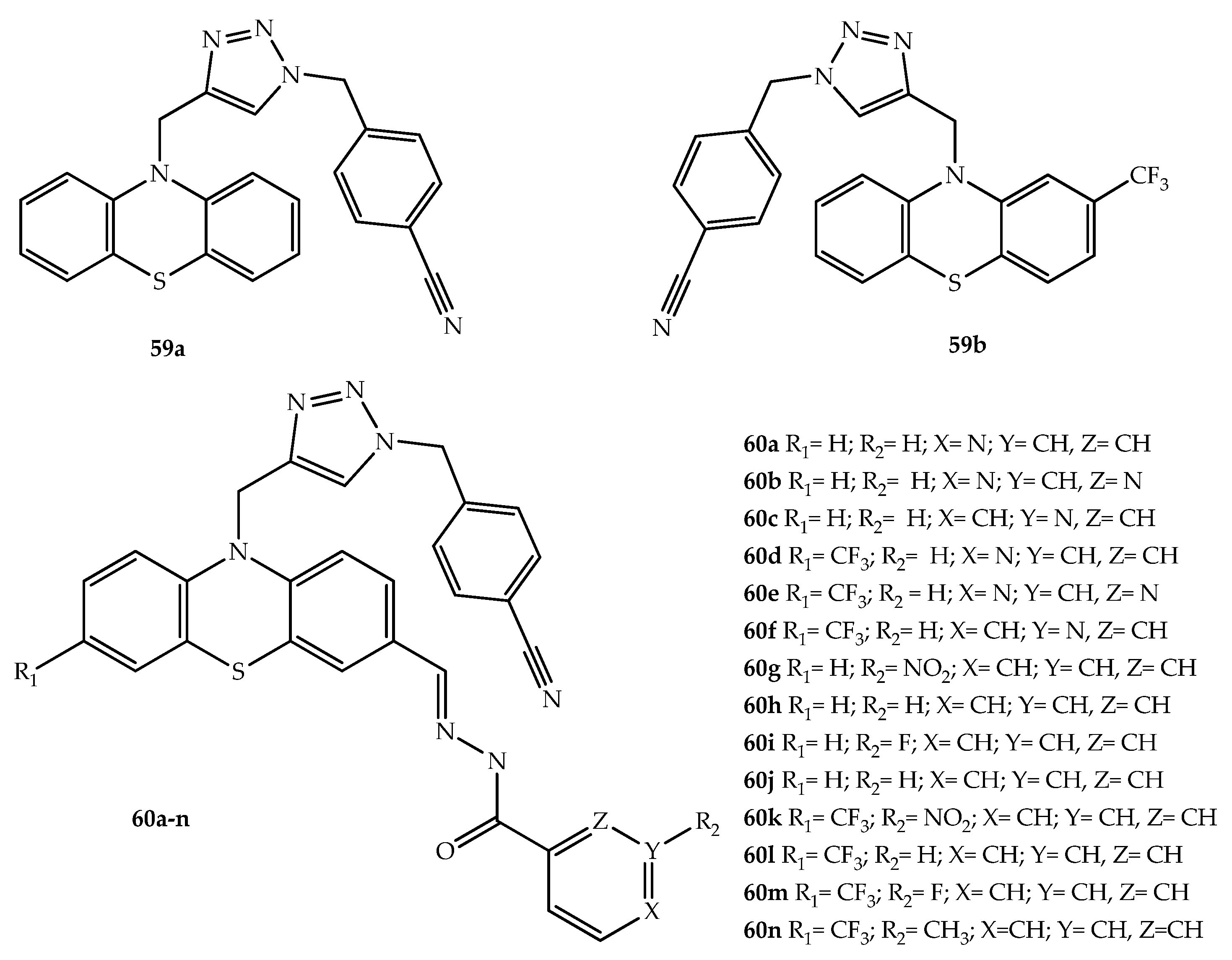{kind=link}
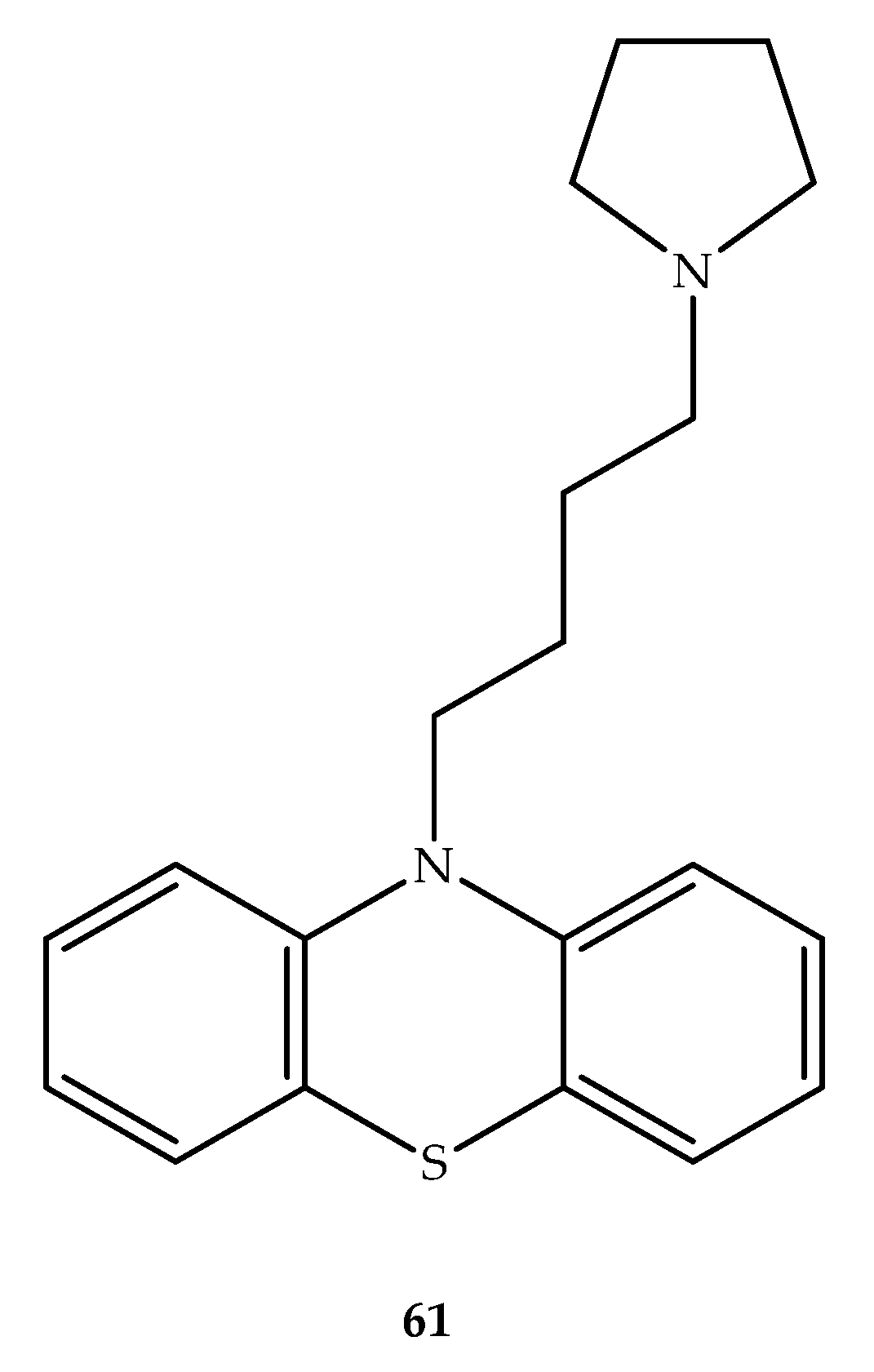{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Anticancer Hybrids
3. Anti-Alzheimer Hybrids
4. Antihistaminic Hybrids
5. Antimicrobial Hybrids
5.1. Antibacterial Hybrids
5.2. Antitubercular Hybrids
5.3. Antimalarial Hybrids
5.4. Antifungal Hybrids
6. Anti-Obesity Hybrids
7. Analgesic and Anti-Inflammatory Properties
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ramos, R.; Olden, K. Gene-Environment Interactions in the Development of Complex Disease Phenotypes. Int. J. Environ. Res. Public Health 2008, 5, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Makhoba, X.H.; Viegas, C., Jr.; Mosa, R.A.; Viegas, F.P.D.; Pooe, O.J. Potential Impact of the Multi-Target Drug Approach in the Treatment of Some Complex Diseases. Drug Des. Devel. Ther. 2020, 14, 3235–3249. [Google Scholar] [CrossRef]
- He, B.; Lu, C.; Zheng, G.; He, X.; Wang, M.; Chen, G.; Zhang, G.; Lu, A. Combination Therapeutics in Complex Diseases. J. Cell. Mol. Med. 2016, 20, 2231–2240. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.; Singh, P.K.; Verma, H.; Singh, H.; Silakari, O. Success Stories of Natural Product-based Hybrid Molecules for Multi-factorial Diseases. Eur. J. Med. Chem. 2018, 151, 62–97. [Google Scholar] [CrossRef]
- Fink, D.M.; Palermo, M.G.; Bores, G.M.; Huger, F.P.; Kurys, B.E.; Merriman, M.C.; Olsen, G.E.; Petko, W.; O’Malley, G.J. Imino 1,2,3,4-tetrahydrocyclopent[b]indole carbamates as Dual Inhibitors of Acetylcholinesterase and Monoamine Oxidase. Bioorg. Med. Chem. Lett. 1996, 6, 625–630. [Google Scholar] [CrossRef]
- Bérubé, G. An Overview of Molecular Hybrids in Drug Discovery. Expert Opin. Drug Discov. 2016, 11, 281–305. [Google Scholar] [CrossRef] [PubMed]
- Shaveta; Mishra, S.; Singh, P. Hybrid Molecules: The Privileged Scaffolds for Various Pharmaceuticals. Eur. J. Med. Chem. 2016, 124, 500–536. [Google Scholar] [CrossRef]
- Ohlow, M.J.; Moosmann, B. Phenothiazine: The Seven Lives of Pharmacology’s First Lead Structure. Drug Discov. Today 2011, 16, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Berneth, H. Azine Dyes. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008. [Google Scholar]
- Hu, W.; Zhang, S. Method for the Synthesis of Phenothiazines via a Domino Iron-Catalyzed C–S/C–N Cross-Coupling Reaction. J. Org. Chem. 2015, 80, 6128–6132. [Google Scholar] [CrossRef]
- Otręba, M.; Kośmider, L.; Rzepecka-Stojko, A. Antiviral Activity of Chlorpromazine, Fluphenazine, Perphenazine, Prochlorperazine, and Thioridazine Towards RNA-Viruses. A Review. Eur. J. Pharmacol. 2020, 887, 173553. [Google Scholar] [CrossRef]
- Sudeshna, G.; Parimal, K. Multiple Non-psychiatric Effects of Phenothiazines: A Review. Eur. J. Pharmacol. 2010, 648, 6–14. [Google Scholar] [CrossRef]
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M. The Global Cancer Observatory-All cancers. Available online: https://gco.iarc.fr/today/home (accessed on 28 October 2021).
- Whiteman, D.C.; Wilson, L.F. The Fractions of Cancer Attributable to Modifiable Factors: A Global Review. Cancer Epidemiol. 2016, 44, 203–221. [Google Scholar] [CrossRef]
- Martínez-Reyes, I.; Chandel, N.S. Cancer Metabolism: Looking Forward. Nat. Rev. Cancer 2021, 21, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, H.; Chen, X. Drug Resistance and Combating Drug Resistance in Cancer. Cancer Drug Resist. 2019. [Google Scholar] [CrossRef] [PubMed]
- Zhelev, Z.; Ohba, H.; Bakalova, R.; Hadjimitova, V.; Ishikawa, M.; Shinohara, Y.; Baba, Y. Phenothiazines Suppress Proliferation and Induce Apoptosis in Cultured Leukemic Cells Without Any Influence on the Viability of Normal Lymphocytes. Cancer Chemother. Pharmacol. 2004, 53, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Pulkoski-Gross, A.; Li, J.; Zheng, C.; Li, Y.; Ouyang, N.; Rigas, B.; Zucker, S.; Cao, J. Repurposing the Antipsychotic Trifluoperazine as an Antimetastasis Agent. Mol. Pharmacol. 2015, 87, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Jhou, A.J.; Chang, H.C.; Hung, C.C.; Lin, H.C.; Lee, Y.C.; Liu, W.; Han, K.F.; Lai, Y.W.; Lin, M.Y.; Lee, C.H. Chlorpromazine, an Antipsychotic Agent, Induces G2/M Phase Arrest and Apoptosis Via Regulation of the PI3K/AKT/mTOR-mediated Autophagy Pathways in Human Oral Cancer. Biochem. Pharmacol. 2021, 184, 114403. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-H.; Bai, L.-Y.; Tsai, M.-H.; Chu, P.-C.; Chiu, C.-F.; Chen, M.Y.; Chiu, S.-J.; Chiang, J.-H.; Weng, J.-R. Pharmacological Exploitation of the Phenothiazine Antipsychotics to Develop Novel Antitumor Agents–A Drug Repurposing Strategy. Sci. Rep. 2016, 6, 27540. [Google Scholar] [CrossRef] [PubMed]
- Sachdeva, T.; Low, M.L.; Mai, C.; Cheong, S.L.; Liew, Y.K.; Milton, M.D. Design, Synthesis and Characterisation of Novel Phenothiazine-Based Triazolopyridine Derivatives: Evaluation of Anti-Breast Cancer Activity on Human Breast Carcinoma. ChemistrySelect 2019, 4, 12701–12707. [Google Scholar] [CrossRef]
- Kellogg, E.H.; Hejab, N.M.A.; Howes, S.; Northcote, P.; Miller, J.H.; Díaz, J.F.; Downing, K.H.; Nogales, E. Insights into the Distinct Mechanisms of Action of Taxane and Non-Taxane Microtubule Stabilizers from Cryo-EM Structures. J. Mol. Biol. 2017, 429, 633–646. [Google Scholar] [CrossRef]
- Kumar, B.; Kumar, R.; Skvortsova, I.; Kumar, V. Mechanisms of Tubulin Binding Ligands to Target Cancer Cells: Updates on their Therapeutic Potential and Clinical Trials. Curr. Cancer Drug Targets 2017, 17, 357–375. [Google Scholar] [CrossRef]
- Liu, N.; Jin, Z.; Zhang, J.; Jin, J. Antitumor Evaluation of Novel Phenothiazine Derivatives that Inhibit Migration and Tubulin Polymerization Against Gastric Cancer MGC-803 cells. Investig. New Drugs 2019, 37, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.-H.; Liu, N.; Lu, J.-L.; Zhao, J.; Zhang, X.-J. Design, Synthesis and Antiproliferative Activity of Novel Phenothiazine-1,2,3-Triazole Analogues. J. Chem. Res. 2017, 41, 696–698. [Google Scholar] [CrossRef]
- Zhang, J.-X.; Guo, J.-M.; Zhang, T.-T.; Lin, H.-J.; Qi, N.-S.; Li, Z.-G.; Zhou, J.-C.; Zhang, Z.-Z. Antiproliferative Phenothiazine Hybrids as Novel Apoptosis Inducers Against MCF-7 Breast Cancer. Molecules 2018, 23, 1288. [Google Scholar] [CrossRef] [PubMed]
- Morak-Młodawska, B.; Pluta, K.; Latocha, M.; Jeleń, M.; Kuśmierz, D. Design, Synthesis, and Structural Characterization of Novel Diazaphenothiazines with 1,2,3-Triazole Substituents as Promising Antiproliferative Agents. Molecules 2019, 24, 4388. [Google Scholar] [CrossRef] [PubMed]
- Takács, D.; Egyed, O.; Drahos, L.; Szabó, P.; Jemnitz, K.; Szabó, M.; Veres, Z.; Visy, J.; Molnár, J.; Riedl, Z.; et al. Synthesis and Pharmacological Investigation of New N-hydroxyalkyl-2-aminophenothiazines Exhibiting Marked MDR Inhibitory Effect. Bioorg. Med. Chem. 2013, 21, 3760–3779. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Takács, D.; Csonka, Á.; Horváth, Á.; Windt, T.; Gajdács, M.; Riedl, Z.; Hajós, G.; Amaral, L.; Molnár, J.; Spengler, G. Reversal of ABCB1-related Multidrug Resistance of Colonic Adenocarcinoma Cells by Phenothiazines. Anticancer Res. 2015, 35, 3245–3251. [Google Scholar]
- Rhett, J.M.; Khan, I.; O’Bryan, J.P. Biology, pathology, and therapeutic targeting of RAS. Adv Cancer Res. 2020, 148, 69–146. [Google Scholar] [CrossRef]
- Mullard, A. The FDA Approves a First Farnesyltransferase Inhibitor. Nat. Rev. Drug Discov. 2021, 20, 8. [Google Scholar] [CrossRef]
- Belei, D.; Dumea, C.; Samson, A.; Farce, A.; Dubois, J.; Bîcu, E.; Ghinet, A. New farnesyltransferase inhibitors in the phenothiazine series. Bioorg. Med. Chem. Lett. 2012, 22, 4517–4522. [Google Scholar] [CrossRef]
- Moise, I.-M.; Bîcu, E.; Farce, A.; Dubois, J.; Ghinet, A. Indolizine-phenothiazine Hybrids as the First Dual Inhibitors of Tubulin Polymerization and Farnesyltransferase with Synergistic Antitumor Activity. Bioorg. Chem. 2020, 103, 104184. [Google Scholar] [CrossRef]
- Baciu-Atudosie, L.; Ghinet, A.; Farce, A.; Dubois, J.; Belei, D.; Bîcu, E. Synthesis and Biological Evaluation of New Phenothiazine Derivatives Bearing a Pyrazole Unit as Protein Farnesyltransferase Inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 6896–6902. [Google Scholar] [CrossRef] [PubMed]
- Klimpel, A.; Stillger, K.; Wiederstein, J.L.; Krüger, M.; Neundorf, I. Cell-Permeable CaaX-Peptides Affect K-Ras Downstream Signaling and Promote Cell Death in Cancer Cells. FEBS J. 2021, 288, 2911–2929. [Google Scholar] [CrossRef] [PubMed]
- Dumitriu, G.-M.; Bîcu, E.; Belei, D.; Rigo, B.; Dubois, J.; Farce, A.; Ghinet, A. Phenothiazine-based CaaX Competitive Inhibitors of Human Farnesyltransferase Bearing a Cysteine, Methionine, Serine or Valine Moiety as a New Family of Antitumoral Compounds. Bioorg. Med. Chem. Lett. 2015, 25, 4447–4452. [Google Scholar] [CrossRef] [PubMed]
- Al Zahrani, N.A.; El-Shishtawy, R.M.; Elaasser, M.M.; Asiri, A.M. Synthesis of Novel Chalcone-Based Phenothiazine Derivatives as Antioxidant and Anticancer Agents. Molecules 2020, 25, 4566. [Google Scholar] [CrossRef]
- Brem, B.; Gal, E.; Găină, L.; Silaghi-Dumitrescu, L.; Fischer-Fodor, E.; Tomuleasa, C.I.; Grozav, A.; Zaharia, V.; Filip, L.; Cristea, C. Novel Thiazolo[5,4-b]phenothiazine Derivatives: Synthesis, Structural Characterization, and In Vitro Evaluation of Antiproliferative Activity against Human Leukaemia. Int. J. Mol. Sci. 2017, 18, 1365. [Google Scholar] [CrossRef]
- Ignat, A.; Lovasz, T.; Vasilescu, M.; Fischer-Fodor, E.; Tatomir, C.B.; Cristea, C.; Silaghi-Dumitrescu, L.; Zaharia, V. Heterocycles 27. Microwave Assisted Synthesis and Antitumour Activity of Novel Phenothiazinyl-Thiazolyl-Hydrazine Derivatives. Arch. Pharm. 2012, 345, 574–583. [Google Scholar] [CrossRef]
- Chen, I.S.; Liang, W.Z.; Wang, J.L.; Kuo, C.C.; Hao, L.J.; Chou, C.T.; Jan, C.R. Exploration of Thioridazine-Induced Ca2+ Signaling and Non-Ca2+-Triggered Cell Death in HepG2 Human Hepatocellular Carcinoma Cells. Chin. J. Physiol. 2020, 187–194. [Google Scholar] [CrossRef]
- Karatoprak, G.Ş.; Küpeli Akkol, E.; Genç, Y.; Bardakci, H.; Yücel, Ç.; Sobarzo-Sánchez, E. Combretastatins: An Overview of Structure, Probable Mechanisms of Action and Potential Applications. Molecules 2020, 25, 2560. [Google Scholar] [CrossRef]
- Abuhaie, C.-M.; Bîcu, E.; Rigo, B.; Gautret, P.; Belei, D.; Farce, A.; Dubois, J.; Ghinet, A. Synthesis and anticancer activity of analogues of phenstatin, with a phenothiazine A-ring, as a new class of microtubule-targeting agents. Bioorg. Med. Chem. Lett. 2013, 23, 147–152. [Google Scholar] [CrossRef]
- Verones, V.; Flouquet, N.; Lecoeur, M.; Lemoine, A.; Farce, A.; Baldeyrou, B.; Mahieu, C.; Wattez, N.; Lansiaux, A.; Goossens, J.F.; et al. Synthesis, antiproliferative activity and tubulin targeting effect of acridinone and dioxophenothiazine derivatives. Eur. J. Med. Chem. 2013, 59, 39–47. [Google Scholar] [CrossRef]
- Prinz, H.; Ridder, A.K.; Vogel, K.; Böhm, K.J.; Ivanov, I.; Ghasemi, J.B.; Aghaee, E.; Müller, K. N-heterocyclic (4-phenylpiperazin-1-yl)methanones derived from phenoxazine and phenothiazine as highly potent inhibitors of tubulin polymerization. J. Med. Chem. 2017, 60, 749–766. [Google Scholar] [CrossRef]
- Prinz, H.; Chamasmani, B.; Vogel, K.; Böhm, K.J.; Aicher, B.; Gerlach, M.; Günther, E.G.; Amon, P.; Ivanov, I.; Müller, K. N-Benzoylated Phenoxazines and Phenothiazines: Synthesis, Antiproliferative Activity, and Inhibition of Tubulin Polymerization. J. Med. Chem. 2011, 54, 4247–4263. [Google Scholar] [CrossRef] [PubMed]
- Ghinet, A.; Moise, I.-M.; Rigo, B.; Homerin, G.; Farce, A.; Dubois, J.; Bîcu, E. Studies on Phenothiazines: New Microtubule-interacting Compounds with Phenothiazine A-ring as Potent Antineoplastic Agents. Bioorg. Med. Chem. 2016, 24, 2307–2317. [Google Scholar] [CrossRef]
- Fu, D.-J.; Zhao, R.-H.; Li, J.-H.; Yang, J.-J.; Mao, R.-W.; Wu, B.-W.; Li, P.; Zi, X.-L.; Zhang, Q.-Q.; Cai, H.-J.; et al. Molecular Diversity of Phenothiazines: Design and Synthesis of Phenothiazine–Dithiocarbamate Hybrids as Potential Cell Cycle Blockers. Mol. Divers. 2017, 21, 933–942. [Google Scholar] [CrossRef]
- Chu, C.-W.; Ko, H.-J.; Chou, C.-H.; Cheng, T.-S.; Cheng, H.-W.; Liang, Y.-H.; Lai, Y.-L.; Lin, C.-Y.; Wang, C.; Loh, J.-K.; et al. Thioridazine Enhances P62-Mediated Autophagy and Apoptosis Through Wnt/β-Catenin Signaling Pathway in Glioma Cells. Int. J. Mol. Sci. 2019, 20, 473. [Google Scholar] [CrossRef] [PubMed]
- Sivaramakarthikeyan, R.; Iniyaval, S.; Padmavathy, K.; Liew, H.-S.; Looi, C.-K.; Mai, C.-W.; Ramalingan, C. Phenothiazine and Amide-ornamented Novel Nitrogen Heterocyclic Hybrids: Synthesis, Biological and Molecular Docking Studies. New J. Chem. 2019, 43, 17046–17057. [Google Scholar] [CrossRef]
- Zyta, J.; Jaszczyszyn, A.; Swiatek, P.; Gasiorowski, K.; Malinka, W. Synthesis, Pro-apoptotic Activity and 2D-QSAR Studies of New Analogues of Fluphenazine. Acta Pol. Pharm. 2014, 71, 301–309. [Google Scholar]
- Ghorab, M.M.; Alsaid, M.S.; Samir, N.; Abdel-Latif, G.A.; Soliman, A.M.; Ragab, F.A.; Abou El Ella, D.A. Aromatase Inhibitors and Apoptotic Inducers: Design, Synthesis, Anticancer Activity and Molecular Modeling Studies of Novel Phenothiazine Derivatives Carrying Sulfonamide Moiety as Hybrid Molecules. Eur. J. Med. Chem. 2017, 134, 304–315. [Google Scholar] [CrossRef]
- Garsi, J.-B.; Vece, V.; Sernissi, L.; Auger-Morin, C.; Hanessian, S.; McCracken, A.N.; Selwan, E.; Ramirez, C.; Dahal, A.; Romdhane, N.B.; et al. Design, Synthesis and Anticancer Activity of Constrained Sphingolipid-phenoxazine/phenothiazine Hybrid Constructs Targeting Protein Phosphatase 2A. Bioorg. Med. Chem. Lett. 2019, 29, 2681–2685. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, A.; Pan, L.; Groen, R.W.J.; Baleydier, F.; Kentsis, A.; Marineau, J.; Grebliunaite, R.; Kozakewich, E.; Reed, C.; Pflumio, F.; et al. Phenothiazines Induce PP2A-Mediated Apoptosis in T Cell Acute Lymphoblastic Leukemia. J. Clin. Investig. 2014, 124, 644–655. [Google Scholar] [CrossRef]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef]
- Montine, T.J.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; Mirra, S.S.; et al. National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: A practical approach. Acta Neuropathol. 2012, 123, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Tong, G.; Ballard, C. Treatment Combinations for Alzheimer’s Disease: Current and Future Pharmacotherapy Options. J. Alzheimer’s Dis. 2019, 67, 779–794. [Google Scholar] [CrossRef] [PubMed]
- Hui, A.; Chen, Y.; Zhu, S.; Gan, C.; Pan, J.; Zhou, A. Design and Synthesis of Tacrine-phenothiazine Hybrids as Multitarget Drugs for Alzheimer’s Disease. Med. Chem. Res. 2014, 23, 3546–3557. [Google Scholar] [CrossRef]
- Frenzel, D.; Glück, J.M.; Brener, O.; Oesterhelt, F.; Nagel-Steger, L.; Willbold, D. Immobilization of Homogeneous Monomeric, Oligomeric and Fibrillar Aβ Species for Reliable SPR Measurements. PLoS ONE 2014, 9, e89490. [Google Scholar] [CrossRef]
- González-Muñoz, G.C.; Arce, M.P.; López, B.; Pérez, C.; Romero, A.; del Barrio, L.; Martín-de-Saavedra, M.D.; Egea, J.; León, R.; Villarroya, M.; et al. N-Acylaminophenothiazines: Neuroprotective Agents Displaying Multifunctional Activities for a Potential Treatment of Alzheimer’s Disease. Eur. J. Med. Chem. 2011, 46, 2224–2235. [Google Scholar] [CrossRef]
- González-Muñoz, G.C.; Arce, M.P.; López, B.; Pérez, C.; Villarroya, M.; López, M.G.; García, A.G.; Conde, S.; Rodríguez-Franco, M.I. Old Phenothiazine and Dibenzothiadiazepine Derivatives for Tomorrow’s Neuroprotective Therapies Against Neurodegenerative Diseases. Eur. J. Med. Chem. 2010, 45, 6152–6158. [Google Scholar] [CrossRef] [PubMed]
- Darvesh, S.; Macdonald, I.R.; Martin, E. Selectivity of Phenothiazine Cholinesterase Inhibitors for Neurotransmitter Systems. Bioorg. Med. Chem. Lett. 2013, 23, 3822–3825. [Google Scholar] [CrossRef]
- Ohsawa, Y.; Hirasawa, N. The Role of Histamine H1 and H4 Receptors in Atopic Dermatitis: From Basic Research to Clinical Study. Allergol. Int. 2014, 63, 533–542. [Google Scholar] [CrossRef]
- Kubota, K.; Kurebayashi, H.; Miyachi, H.; Tobe, M.; Onishi, M.; Isobe, Y. Synthesis and Structure–activity Relationships of Phenothiazine Carboxylic Acids Having Pyrimidine-dione as Novel Histamine H1 Antagonists. Bioorg. Med. Chem. Lett. 2009, 19, 2766–2771. [Google Scholar] [CrossRef]
- Burnham, C.-A.D.; Leeds, J.; Nordmann, P.; O’Grady, J.; Patel, J. Diagnosing Antimicrobial Resistance. Nat. Rev. Microbiol. 2017, 15, 697–703. [Google Scholar] [CrossRef]
- Grimsey, E.M.; Piddock, L.J. V Do Phenothiazines Possess Antimicrobial and Efflux Inhibitory Properties? FEMS Microbiol. Rev. 2019, 43, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Grimsey, E.M.; Fais, C.; Marshall, R.L.; Ricci, V.; Ciusa, M.L.; Stone, J.W.; Ivens, A.; Malloci, G.; Ruggerone, P.; Vargiu, A.V.; et al. Chlorpromazine and Amitriptyline Are Substrates and Inhibitors of the AcrB Multidrug Efflux Pump. MBio 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Tegos, G.P.; Masago, K.; Aziz, F.; Higginbotham, A.; Stermitz, F.R.; Hamblin, M.R. Inhibitors of bacterial multidrug efflux pumps potentiate antimicrobial photoinactivation. Antimicrob. Agents Chemother. 2008, 52, 3202–3209. [Google Scholar] [CrossRef] [PubMed]
- Tegos, G.P.; Hamblin, M.R. Phenothiazinium Antimicrobial Photosensitizers Are Substrates of Bacterial Multidrug Resistance Pumps. Antimicrob. Agents Chemother. 2006, 50, 196–203. [Google Scholar] [CrossRef]
- Rineh, A.; Dolla, N.K.; Ball, A.R.; Magana, M.; Bremner, J.B.; Hamblin, M.R.; Tegos, G.P.; Kelso, M.J. Attaching the NorA Efflux Pump Inhibitor INF55 to Methylene Blue Enhances Antimicrobial Photodynamic Inactivation of Methicillin-Resistant Staphylococcus aureus in Vitro and in Vivo. ACS Infect. Dis. 2017, 3, 756–766. [Google Scholar] [CrossRef] [PubMed]
- Rineh, A.; Bremner, J.B.; Hamblin, M.R.; Ball, A.R.; Tegos, G.P.; Kelso, M.J. Attaching NorA Efflux Pump Inhibitors to Methylene Blue Enhances Antimicrobial Photodynamic Inactivation of Escherichia coli and Acinetobacter baumannii In Vitro and In Vivo. Bioorg. Med. Chem. Lett. 2018, 28, 2736–2740. [Google Scholar] [CrossRef]
- Guguloth, V.; Thirukovela, N.S.; Paidakula, S.; Vadde, R.; Guguloth, V.; Thirukovela, N.S.; Paidakula, S.; Vadde, R. One-Pot Regioselective Synthesis of Some Novel Isoxazole-Phenothiazine Hybrids and Their Antibacterial Activity. Russ. J. Gen. Chem. 2020, 90, 470–475. [Google Scholar] [CrossRef]
- Benson, T.E.; Harris, M.S.; Choi, G.H.; Cialdella, J.I.; Herberg, J.T.; Martin, J.P.J.; Baldwin, E.T. A Structural Variation for MurB: X-ray Crystal Structure of Staphylococcus aureus UDP-N-acetylenolpyruvylglucosamine Reductase (MurB). Biochemistry 2001, 40, 2340–2350. [Google Scholar] [CrossRef]
- Ramprasad, J.; Nayak, N.; Dalimba, U. Design of New Phenothiazine-thiadiazole Hybrids Via Molecular Hybridization Approach for the Development of Potent Antitubercular Agents. Eur. J. Med. Chem. 2015, 106, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Khawbung, J.L.; Nath, D.; Chakraborty, S. Drug Resistant Tuberculosis: A Review. Comp. Immunol. Microbiol. Infect. Dis. 2021, 74, 101574. [Google Scholar] [CrossRef]
- Schurig-Briccio, L.A.; Yano, T.; Rubin, H.; Gennis, R.B. Characterization of the Type 2 NADH: Menaquinone Oxidoreductases from Staphylococcus aureus and the Bactericidal Action of Phenothiazines. Biochim. Biophys. Acta-Bioenerg. 2014, 1837, 954–963. [Google Scholar] [CrossRef]
- Addla, D.; Jallapally, A.; Gurram, D.; Yogeeswari, P.; Sriram, D.; Kantevari, S. Rational Design, Synthesis and Antitubercular Evaluation of Novel 2-(trifluoromethyl)phenothiazine-[1,2,3]triazole Hybrids. Bioorg. Med. Chem. Lett. 2014, 24, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Singh, K.; Naran, K.; Hamzabegovic, F.; Hoft, D.F.; Warner, D.F.; Ruminski, P.; Abate, G.; Chibale, K. Design, Synthesis, and Evaluation of Novel Hybrid Efflux Pump Inhibitors for Use against Mycobacterium tuberculosis. ACS Infect. Dis. 2016, 2, 714–725. [Google Scholar] [CrossRef]
- Dunn, E.A.; Roxburgh, M.; Larsen, L.; Smith, R.A.J.; McLellan, A.D.; Heikal, A.; Murphy, M.P.; Cook, G.M. Incorporation of Triphenylphosphonium Functionality Improves the Inhibitory Properties of Phenothiazine Derivatives in Mycobacterium tuberculosis. Bioorg. Med. Chem. 2014, 22, 5320–5328. [Google Scholar] [CrossRef]
- Pemmadi, R.V.; Kurre, P.N.; Guruvelli, P.V.S.; Jamullamudi, R.N.; Muthyala, M.K.K.; Pemmadi, R.V.; Kurre, P.N.; Guruvelli, P.V.S.; Jamullamudi, R.N.; Muthyala, M.K.K. Microwave Assisted Synthesis and SAR Studies of Novel Hybrid Phenothiazine Analogs as Potential Antitubercular Agents. INDIAN J. Chem. Sect. B-Org. Chem. Incl. Med. Chem. 2018, 57, 556–566. [Google Scholar]
- Reddyrajula, R.; Dalimba, U.; Madan Kumar, S. Molecular Hybridization Approach for Phenothiazine Incorporated 1,2,3-triazole Hybrids as Promising Antimicrobial Agents: Design, Synthesis, Molecular Docking and In Silico ADME Studies. Eur. J. Med. Chem. 2019, 168, 263–282. [Google Scholar] [CrossRef]
- Ashley, E.A.; Pyae Phyo, A.; Woodrow, C.J. Malaria. Lancet 2018, 391, 1608–1621. [Google Scholar] [CrossRef]
- Guan, J.; Kyle, D.E.; Gerena, L.; Zhang, Q.; Milhous, W.K.; Lin, A.J. Design, Synthesis, and Evaluation of New Chemosensitizers in Multi-Drug-Resistant Plasmodium falciparum. J. Med. Chem. 2002, 45, 2741–2748. [Google Scholar] [CrossRef] [PubMed]
- Brilhante, R.S.N.; Gotay, W.J.P.; Pereira, V.S.; de Oliveira, J.S.; Pereira-Neto, W.A.; Castelo-Branco, D.d.S.C.M.; de Aguiar Cordeiro, R.; Sidrim, J.J.C.; Rocha, M.F.G. Antifungal Activity of Promethazine and Chlorpromazine Against Planktonic Cells and Biofilms of Cryptococcus neoformans/Cryptococcus gattii Complex Species. Med. Mycol. 2020, 58, 906–912. [Google Scholar] [CrossRef]
- Montoya, M.C.; DiDone, L.; Heier, R.F.; Meyers, M.J.; Krysan, D.J. Antifungal Phenothiazines: Optimization, Characterization of Mechanism, and Modulation of Neuroreceptor Activity. ACS Infect. Dis. 2018, 4, 499–507. [Google Scholar] [CrossRef]
- De Azua, I.R.; Mancini, G.; Srivastava, R.K.; Rey, A.A.; Cardinal, P.; Tedesco, L.; Zingaretti, C.M.; Sassmann, A.; Quarta, C.; Schwitter, C.; et al. Adipocyte cannabinoid receptor CB1 regulates energy homeostasis and alternatively activated macrophages. J. Clin. Investig. 2017, 127, 4148–4162. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Bátkai, S.; Kunos, G. The Endocannabinoid System as an Emerging Target of Pharmacotherapy. Pharmacol. Rev. 2006, 58, 389–462. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.K.; Machhi, J.; Murumkar, P.; Yadav, M.R. New Role of Phenothiazine Derivatives as Peripherally Acting CB1 Receptor Antagonizing Anti-obesity Agents. Sci. Rep. 2018, 8, 1650. [Google Scholar] [CrossRef] [PubMed]
- Wiley, J. Cannabinoid Pharmacological Properties Common to Other Centrally Acting Drugs. Eur. J. Pharmacol. 2003, 471, 185–193. [Google Scholar] [CrossRef]
- Silva, G.A.; Costa, L.M.M.; Brito, F.C.F.; Miranda, A.L.P.; Barreiro, E.J.; Fraga, C.A.M. New Class of Potent Antinociceptive and Antiplatelet 10H-phenothiazine-1-acylhydrazone Derivatives. Bioorg. Med. Chem. 2004, 12, 3149–3158. [Google Scholar] [CrossRef]



































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Posso, M.C.; Domingues, F.C.; Ferreira, S.; Silvestre, S. Development of Phenothiazine Hybrids with Potential Medicinal Interest: A Review. Molecules 2022, 27, 276. https://doi.org/10.3390/molecules27010276
Posso MC, Domingues FC, Ferreira S, Silvestre S. Development of Phenothiazine Hybrids with Potential Medicinal Interest: A Review. Molecules. 2022; 27(1):276. https://doi.org/10.3390/molecules27010276
Chicago/Turabian StylePosso, Marina C., Fernanda C. Domingues, Susana Ferreira, and Samuel Silvestre. 2022. "Development of Phenothiazine Hybrids with Potential Medicinal Interest: A Review" Molecules 27, no. 1: 276. https://doi.org/10.3390/molecules27010276
APA StylePosso, M. C., Domingues, F. C., Ferreira, S., & Silvestre, S. (2022). Development of Phenothiazine Hybrids with Potential Medicinal Interest: A Review. Molecules, 27(1), 276. https://doi.org/10.3390/molecules27010276