
Recent Advances in Bioactive Flavonoid Hybrids Linked by 1,2,3-Triazole Ring Obtained by Click Chemistry





Abstract
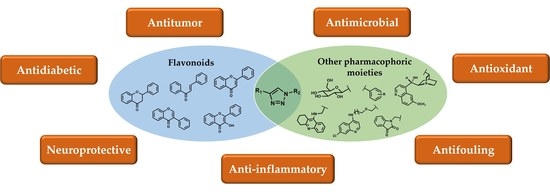
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
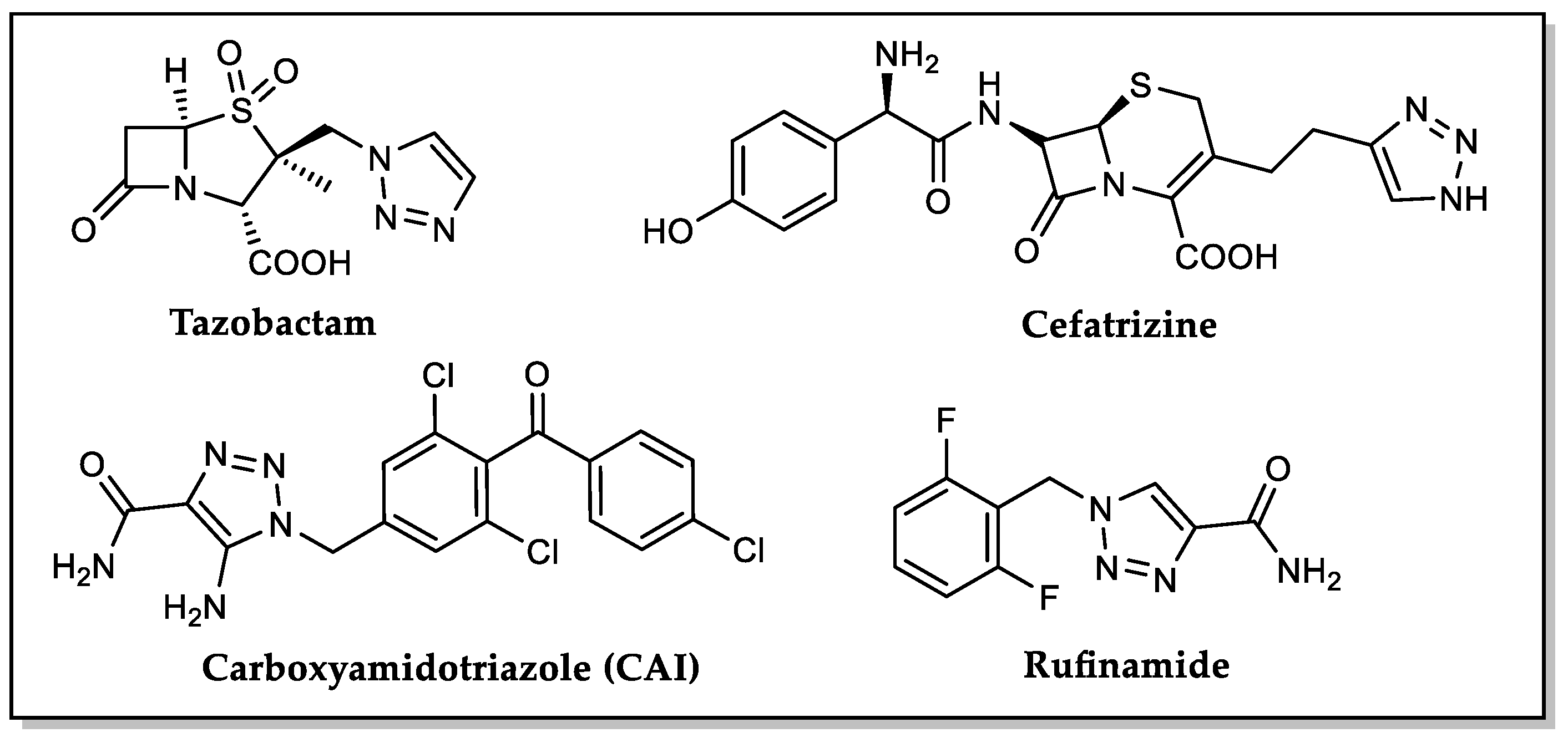
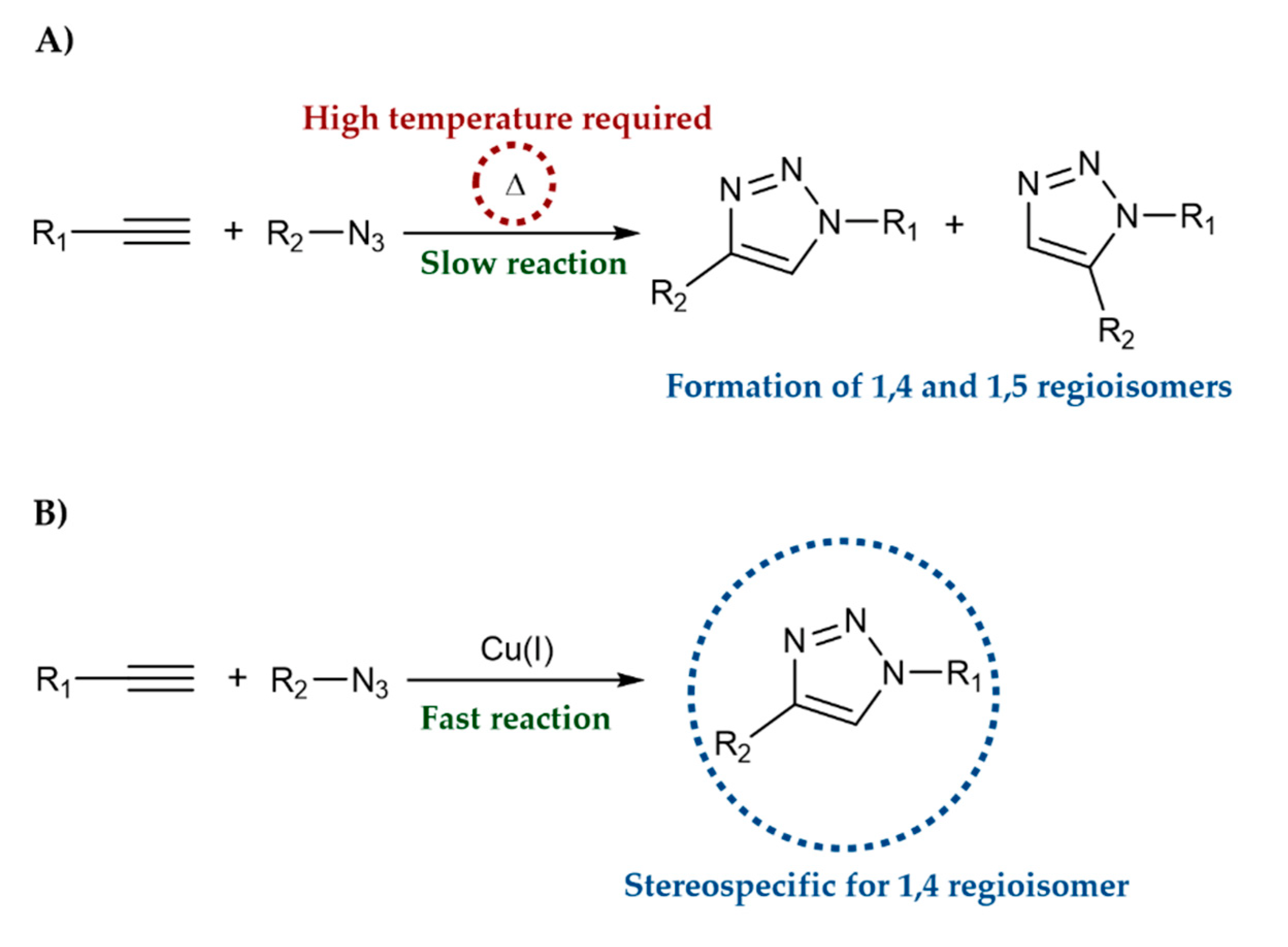
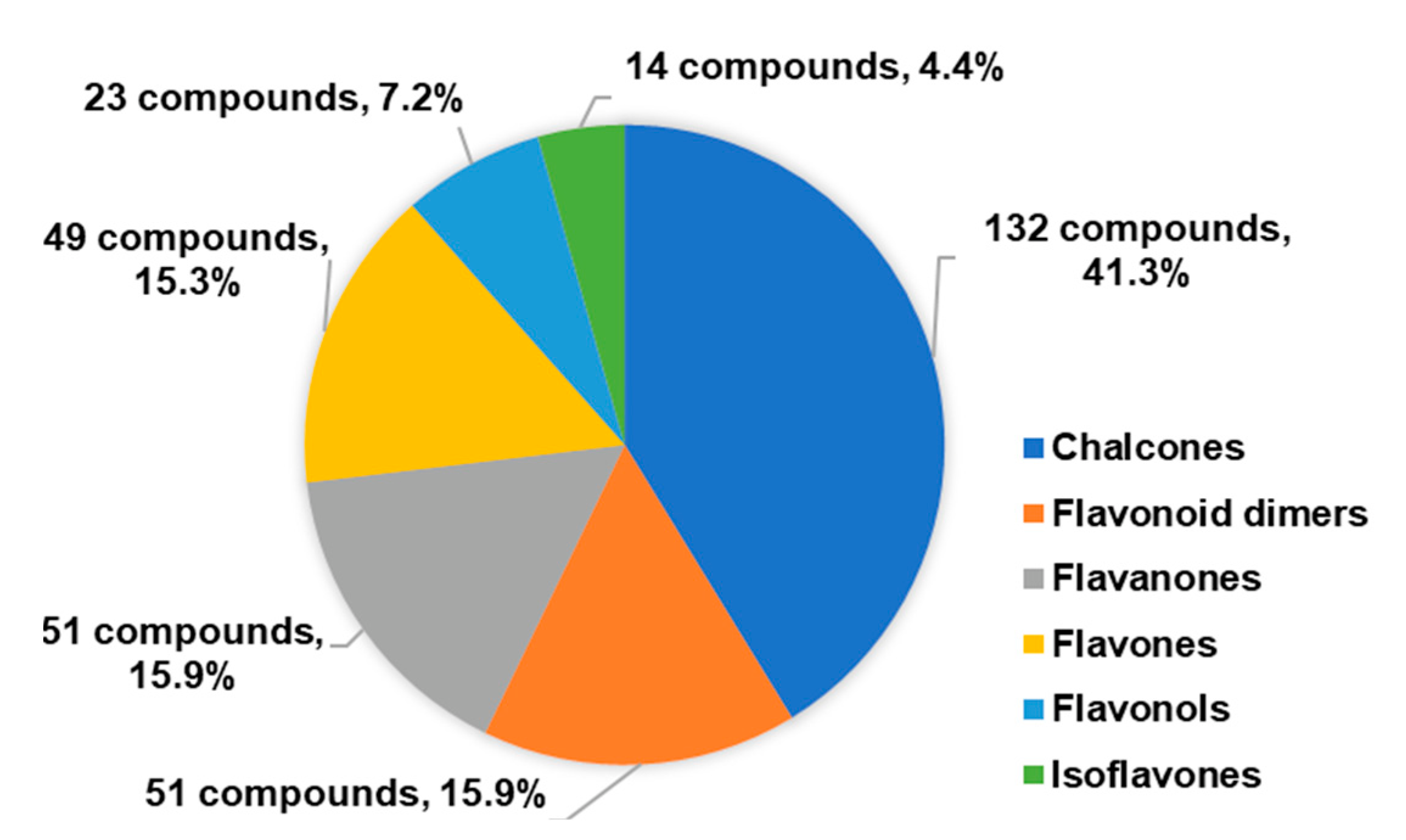
1. Introduction
2. 1,2,3-Triazole-Linked Flavonoid Hybrids with Antitumor Activity
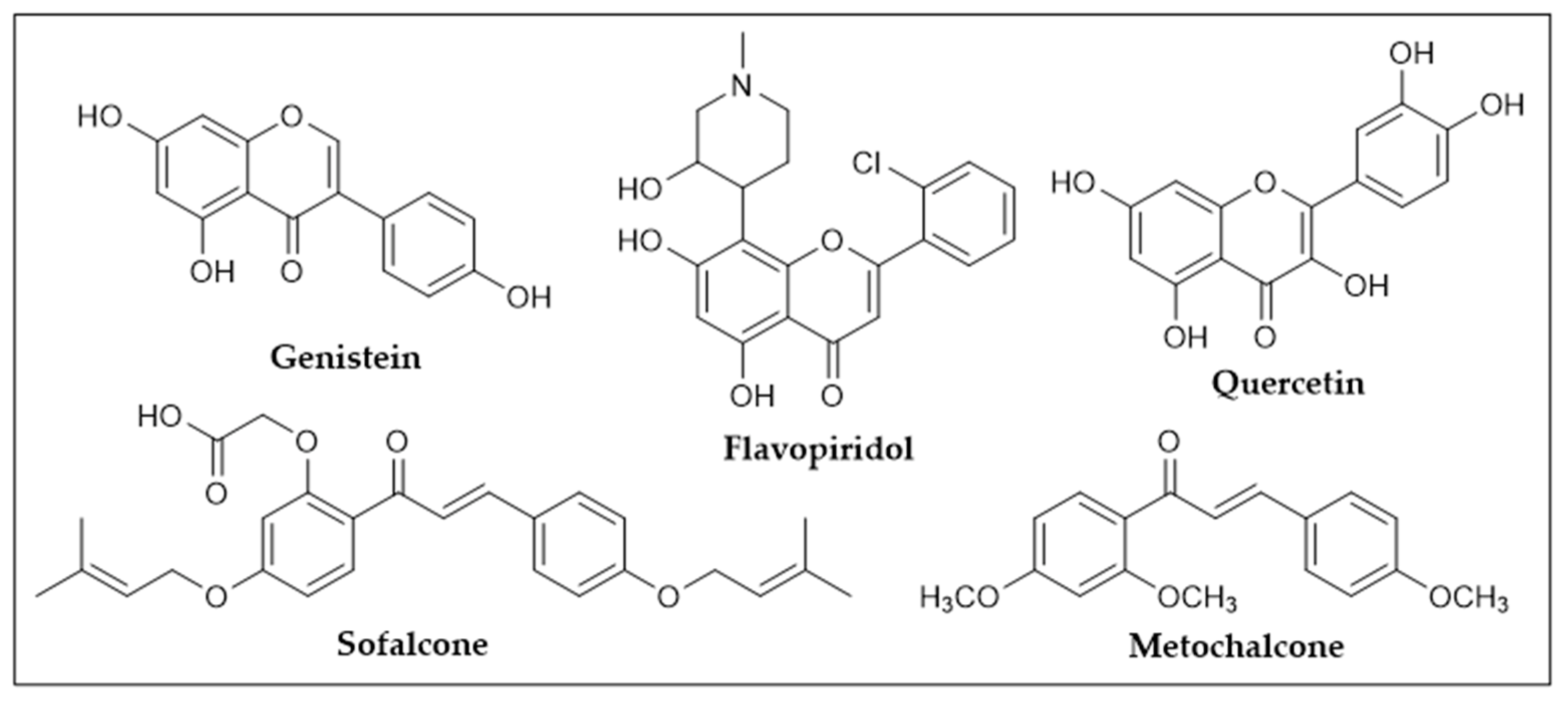
2.1. 1,2,3-Triazole-Linked Chalcone Hybrids
2.2. 1,2,3-Triazole-Linked Flavonoid Dimers
2.3. 1,2,3-Triazole-Linked Flavanone Hybrids
2.4. 1,2,3-Triazole-Linked Flavone Hybrids
2.5. 1,2,3-Triazole-Linked Flavonol Hybrids
2.6. 1,2,3-Triazole-Linked Isoflavone Hybrids
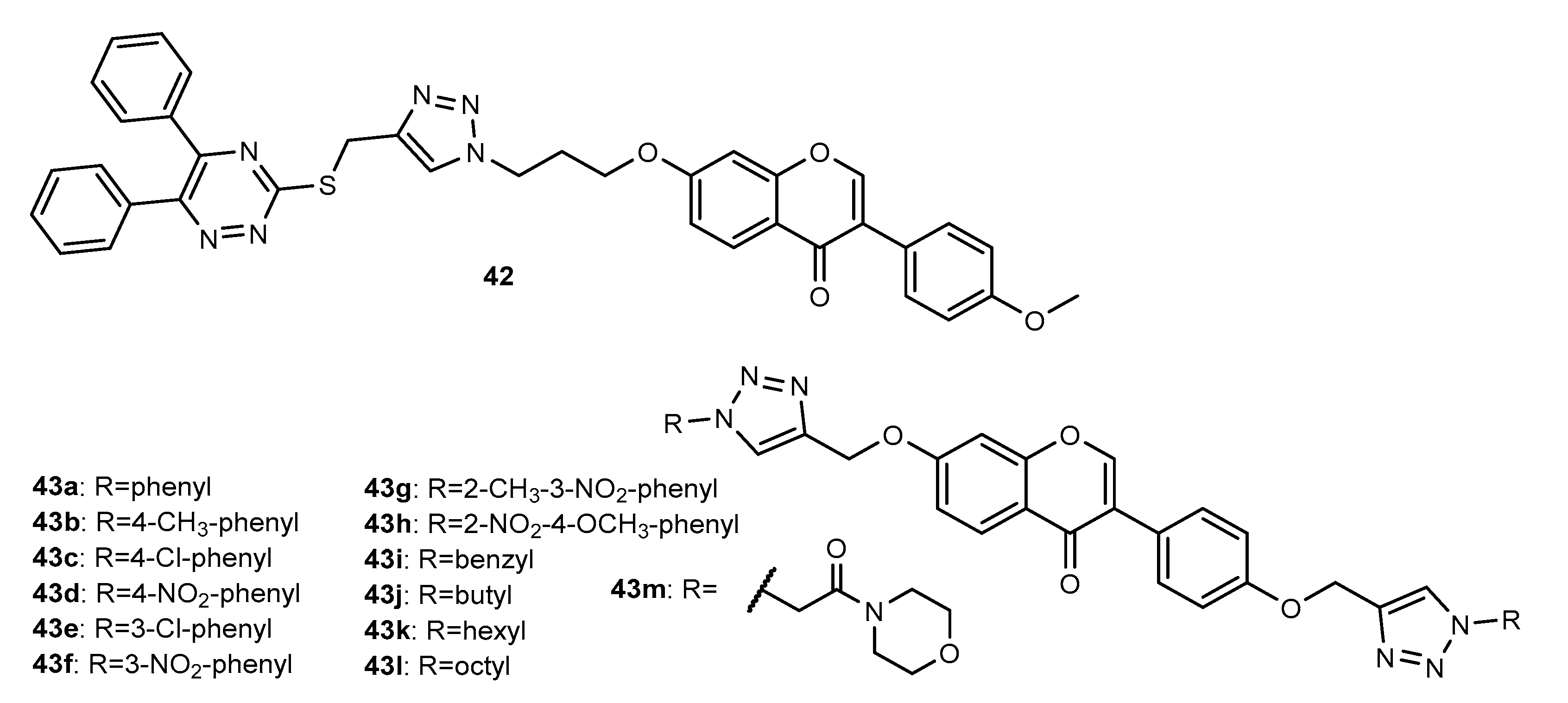
3. 1,2,3-Triazole-Linked Flavonoid Hybrids with Antimicrobial Activity
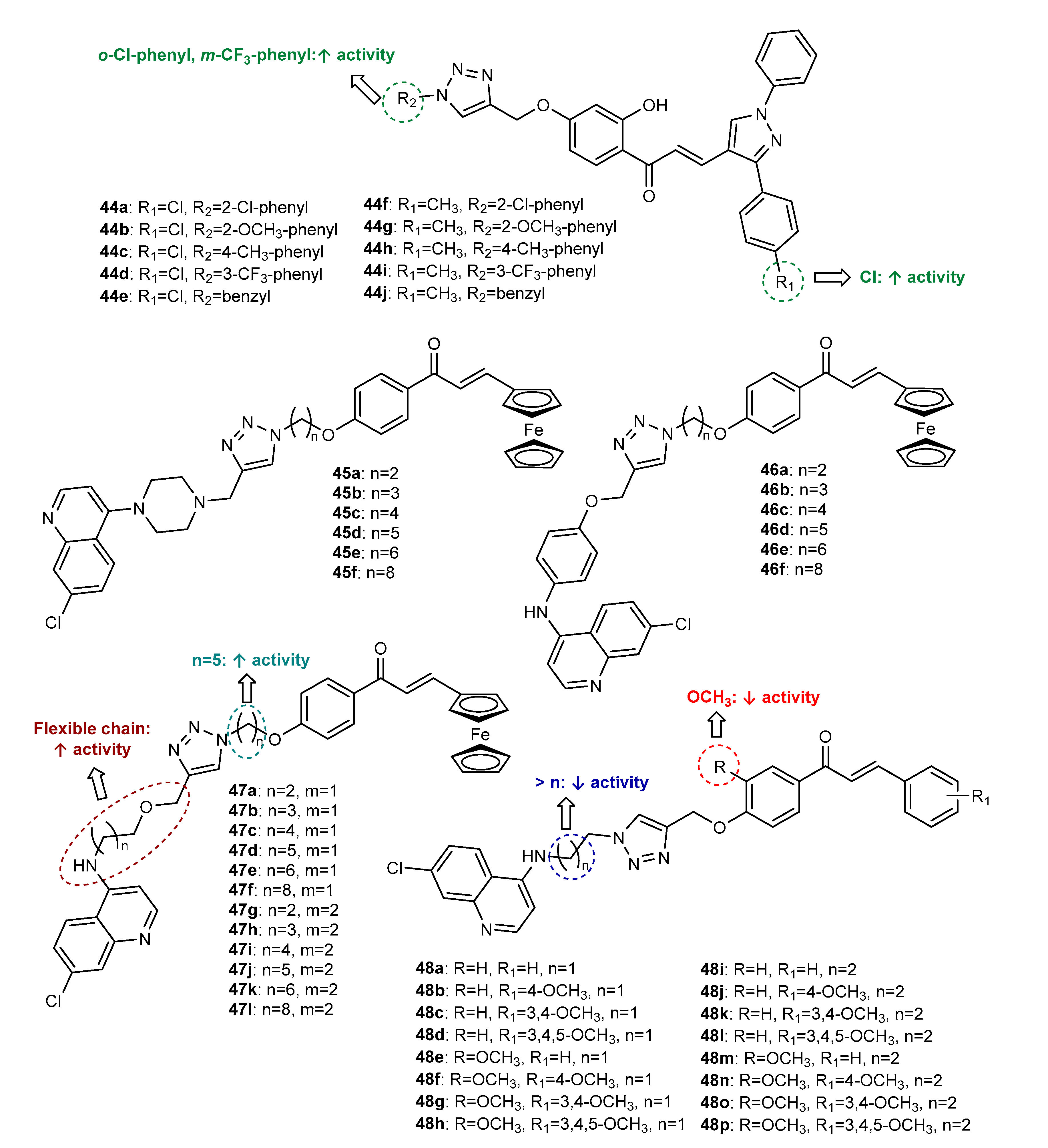
3.1. 1,2,3-Triazole-Linked Chalcone Hhybrids
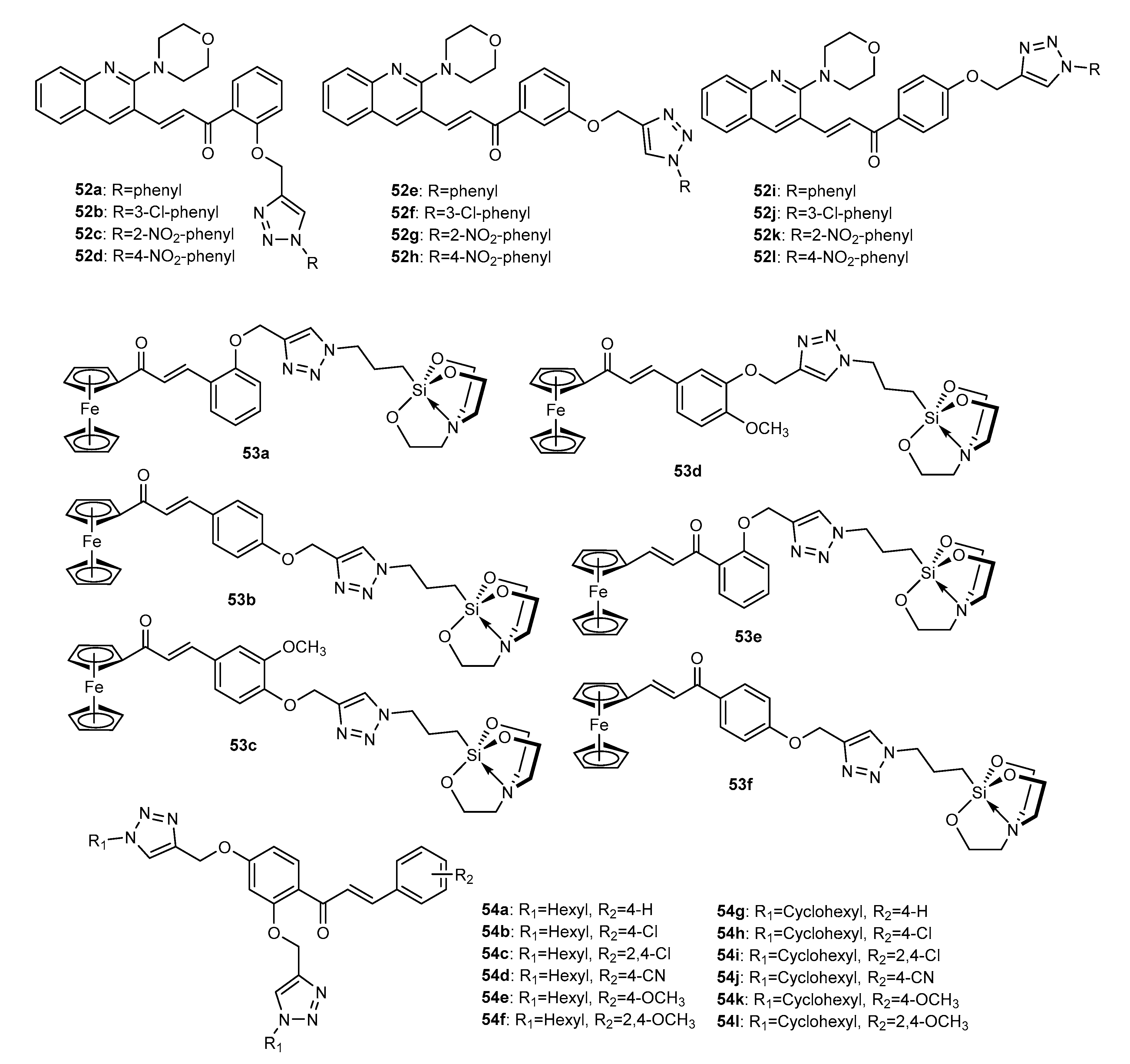
3.2. 1,2,3-Triazole-Linked Flavanone Hybrids
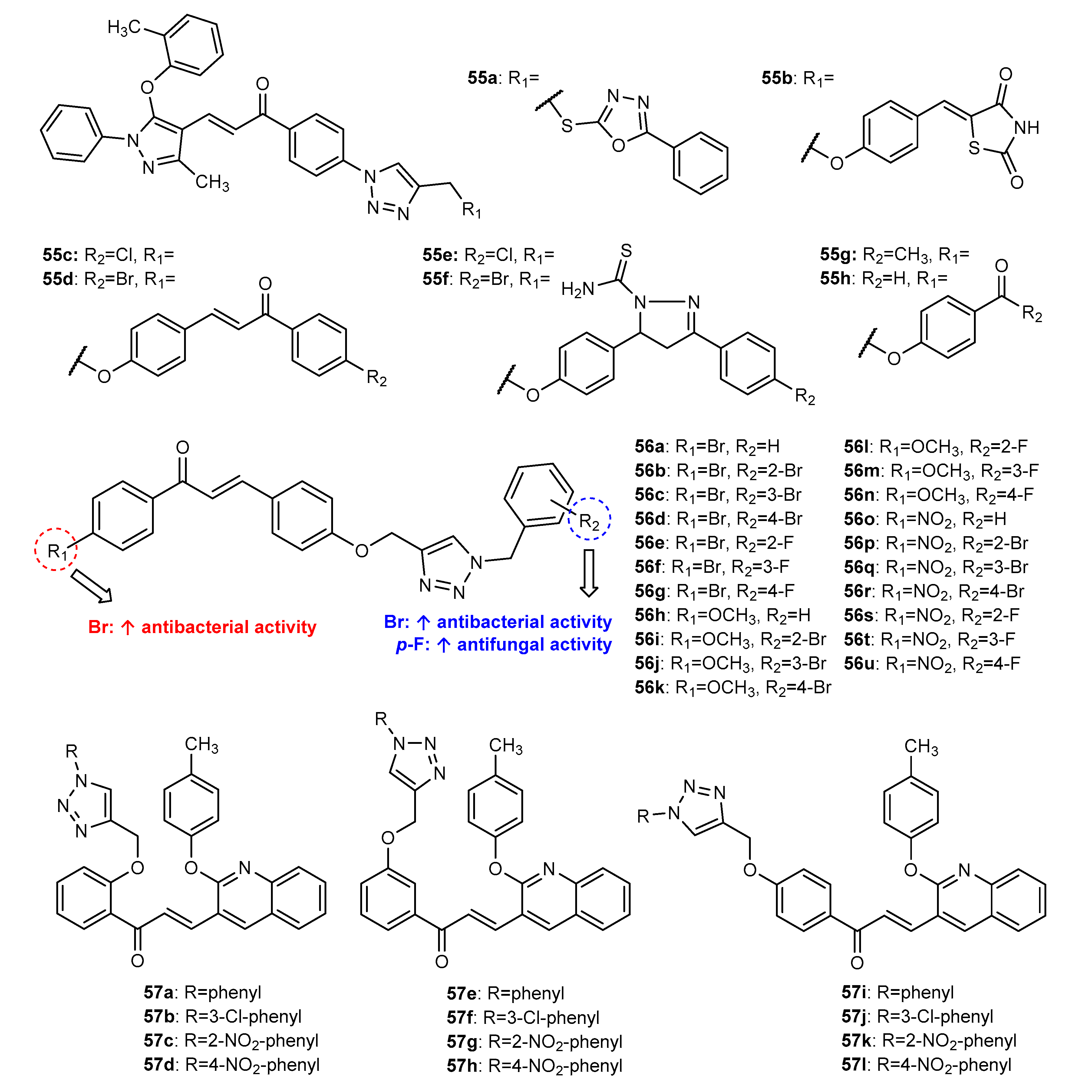
3.3. 1,2,3-Triazole-Linked Flavone Hybrids
3.4. 1,2,3-Triazole-Linked Isoflavone Hybrids
4. Other Activities
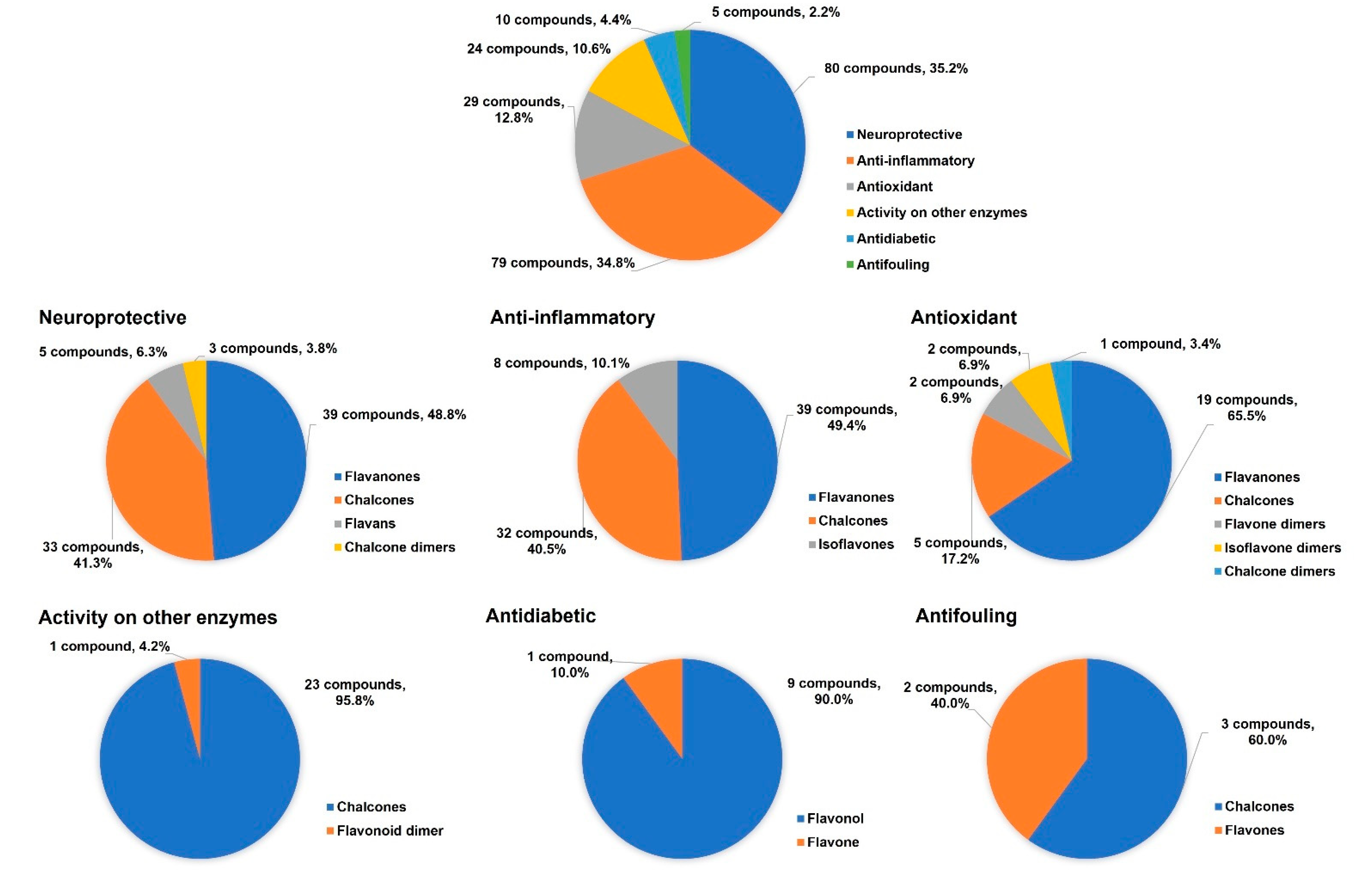
4.1. Neuroprotective Activity
4.2. Anti-Inflammatory Activity
4.3. Antioxidant Activity
4.4. Activity on Other Enzymes
4.5. Antidiabetic Activity
4.6. Antifouling Activity
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Silva, F.; Cardoso, M.; Ferreira, P.; Ferreira, V. Biological Properties of 1H-1,2,3- and 2H-1,2,3-Triazoles. In Chemistry of 1,2,3-triazoles; Dehaen, W., Bakulev, V.A., Eds.; Springer International Publishing: Berlin/Heidelberg, Germany, 2014; pp. 117–165. [Google Scholar]
- Bonandi, E.; Christodoulou, M.S.; Fumagalli, G.; Perdicchia, D.; Rastelli, G.; Passarella, D. The 1,2,3-triazole ring as a bioisostere in medicinal chemistry. Drug Discov. Today 2017, 22, 1572–1581. [Google Scholar] [CrossRef] [PubMed]
- Dheer, D.; Singh, V.; Shankar, R. Medicinal attributes of 1,2,3-triazoles: Current developments. Bioorg. Chem. 2017, 71, 30–54. [Google Scholar] [CrossRef] [PubMed]
- Bi, F.; Ji, S.; Venter, H.; Liu, J.; Semple, S.J.; Ma, S. Substitution of terminal amide with 1H-1,2,3-triazole: Identification of unexpected class of potent antibacterial agents. Bioorg. Med. Chem. Lett. 2018, 28, 884–891. [Google Scholar] [CrossRef] [PubMed]
- Lakkakula, R.; Roy, A.; Mukkanti, K.; Sridhar, G. Synthesis and Anticancer Activity of 1,2,3-Triazole Fused N-Arylpyrazole Derivatives. Russ. J. Gen. Chem. 2019, 89, 831–835. [Google Scholar] [CrossRef]
- Mashayekh, K.; Shiri, P. An Overview of Recent Advances in the Applications of Click Chemistry in the Synthesis of Bioconjugates with Anticancer Activities. ChemistrySelect 2019, 4, 13459–13478. [Google Scholar] [CrossRef]
- Xu, Z.; Zhao, S.-J.; Liu, Y. 1,2,3-Triazole-containing hybrids as potential anticancer agents: Current developments, action mechanisms and structure-activity relationships. Eur. J. Med. Chem. 2019, 183, 111700. [Google Scholar] [CrossRef]
- Angajala, K.K.; Vianala, S.; Macha, R.; Raghavender, M.; Thupurani, M.K.; Pathi, P.J. Synthesis, anti-inflammatory, bactericidal activities and docking studies of novel 1,2,3-triazoles derived from ibuprofen using click chemistry. SpringerPlus 2016, 5, 423. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.-Y.; Chen, Z.-A.; Shen, Q.-K.; Quan, Z.-S. Application of triazoles in the structural modification of natural products. J. Enzyme Inhib. Med. Chem. 2021, 36, 1115–1144. [Google Scholar] [CrossRef]
- Jiang, X.; Hao, X.; Jing, L.; Wu, G.; Kang, D.; Liu, X.; Zhan, P. Recent applications of click chemistry in drug discovery. Expert Opin. Drug Discov. 2019, 14, 779–789. [Google Scholar] [CrossRef]
- Totobenazara, J.; Burke, A.J. New click-chemistry methods for 1,2,3-triazoles synthesis: Recent advances and applications. Tetrahedron Lett. 2015, 56, 2853–2859. [Google Scholar] [CrossRef]
- Singh, M.S.; Chowdhury, S.; Koley, S. Advances of azide-alkyne cycloaddition-click chemistry over the recent decade. Tetrahedron 2016, 72, 5257–5283. [Google Scholar] [CrossRef]
- Haldón, E.; Nicasio, M.C.; Pérez, P.J. Copper-catalysed azide–alkyne cycloadditions (CuAAC): An update. Org. Biomol. Chem. 2015, 13, 9528–9550. [Google Scholar] [CrossRef]
- Liang, L.; Astruc, D. The copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC) “click” reaction and its applications. An overview. Coord. Chem. Rev. 2011, 255, 2933–2945. [Google Scholar] [CrossRef]
- Keivanloo, A.; Fakharian, M.; Sepehri, S. 1,2,3-Triazoles based 3-substituted 2-thioquinoxalines: Synthesis, anti-bacterial activities, and molecular docking studies. J. Mol. Struct. 2020, 1202, 127262. [Google Scholar] [CrossRef]
- Qiu, J.; Yuan, C.-M.; Wen, M.; Li, Y.-N.; Chen, J.; Jian, J.-Y.; Huang, L.-J.; Gu, W.; Li, Y.-M.; Hao, X.-J. Design, synthesis, and cytotoxic activities of novel hybrids of parthenolide and thiazolidinedione via click chemistry. J. Asian Nat. Prod. Res. 2020, 22, 425–433. [Google Scholar] [CrossRef]
- Du, Z.; Yu, D.; Du, X.; Scott, P.; Ren, J.; Qu, X. Self-triggered click reaction in an Alzheimer’s disease model: In situ bifunctional drug synthesis catalyzed by neurotoxic copper accumulated in amyloid-β plaques. Chem. Sci. 2019, 10, 10343–10350. [Google Scholar] [CrossRef] [Green Version]
- Shankar, E.P.S.; Bahulayan, D. Chemistry, chemical biology and photophysics of certain new chromene–triazole–coumarin triads as fluorescent inhibitors of CDK2 and CDK4 induced cancers. New J. Chem. 2019, 43, 13863–13872. [Google Scholar] [CrossRef]
- Yue, P.; Zhang, Y.; Guo, Z.F.; Cao, A.C.; Lu, Z.L.; Zhai, Y.G. Synthesis of bifunctional molecules containing [12]aneN3 and coumarin moieties as effective DNA condensation agents and new non-viral gene vectors. Org. Biomol. Chem. 2015, 13, 4494–4505. [Google Scholar] [CrossRef]
- Araújo, M.; Bidarra, S.J.; Alves, P.M.; Valcarcel, J.; Vázquez, J.A.; Barrias, C.C. Coumarin-grafted blue-emitting fluorescent alginate as a potentially valuable tool for biomedical applications. J. Mater. Chem. B 2020, 8, 813–825. [Google Scholar] [CrossRef]
- Huang, Y.; Liu, Y.; Muhammad, S.; Jia, Q.; Ding, Y.; Chen, Y. A facile end-functionalization of polystyrene by ATRP and click chemistry: Chain end effect on the glass transition temperature. React. Funct. Polym. 2020, 151, 104566. [Google Scholar] [CrossRef]
- Zeng, M.; Cao, X.; Xu, H.; Gan, W.; Smith, B.D.; Gao, H.; Yuan, J. Synthesis and direct assembly of linear–dendritic copolymers via CuAAC click polymerization-induced self-assembly (CPISA). Polym. Chem. 2020, 11, 936–943. [Google Scholar] [CrossRef]
- Concellón, A.; Termine, R.; Golemme, A.; Romero, P.; Marcos, M.; Serrano, J.L. High hole mobility and light-harvesting in discotic nematic dendrimers prepared via ‘click’ chemistry. J. Mater. Chem. C 2019, 7, 2911–2918. [Google Scholar] [CrossRef]
- Rajavelu, K.; Rajakumar, P. Synthesis, photophysical, and electrochemical properties of triazolyl dendrimers with thiazolylchalcone surface unit. Synth. Commun. 2018, 48, 38–49. [Google Scholar] [CrossRef]
- Puthiyedath, T.; Bahulayan, D. A click derived triazole-coumarin derivative as fluorescence on-off PET based sensor for Ca2+ and Fe3+ ions. Sens. Actuators B Chem. 2018, 272, 110–117. [Google Scholar] [CrossRef]
- Brandão, P.; Loureiro, J.B.; Carvalho, S.; Hamadou, M.H.; Cravo, S.; Moreira, J.; Pereira, D.; Palmeira, A.; Pinto, M.; Saraiva, L.; et al. Targeting the MDM2-p53 protein-protein interaction with prenylchalcones: Synthesis of a small library and evaluation of potential antitumor activity. Eur. J. Med. Chem. 2018, 156, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Jucá, M.M.; Cysne Filho, F.M.S.; de Almeida, J.C.; Mesquita, D.d.S.; Barriga, J.R.d.M.; Dias, K.C.F.; Barbosa, T.M.; Vasconcelos, L.C.; Leal, L.K.A.M.; Ribeiro, J.E.; et al. Flavonoids: Biological activities and therapeutic potential. Nat. Prod. Res. 2020, 34, 692–705. [Google Scholar] [CrossRef] [PubMed]
- Maher, P. The Potential of Flavonoids for the Treatment of Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 3056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, B.T.; Correia da Silva, M.; Pinto, M.; Cidade, H.; Kijjoa, A. Marine natural flavonoids: Chemistry and biological activities. Nat. Prod. Res. 2019, 33, 3260–3272. [Google Scholar] [CrossRef]
- Mendanha, D.; Vieira de Castro, J.; Moreira, J.; Costa, B.M.; Cidade, H.; Pinto, M.; Ferreira, H.; Neves, N.M. A New Chalcone Derivative with Promising Antiproliferative and Anti-Invasion Activities in Glioblastoma Cells. Molecules 2021, 26, 3383. [Google Scholar] [CrossRef]
- Moreira, J.; Almeida, J.; Saraiva, L.; Cidade, H.; Pinto, M. Chalcones as Promising Antitumor Agents by Targeting the p53 Pathway: An Overview and New Insights in Drug-Likeness. Molecules 2021, 26, 3737. [Google Scholar] [CrossRef]
- Moreira, J.; Ribeiro, D.; Silva, P.M.A.; Nazareth, N.; Monteiro, M.; Palmeira, A.; Saraiva, L.; Pinto, M.; Bousbaa, H.; Cidade, H. New Alkoxy Flavone Derivatives Targeting Caspases: Synthesis and Antitumor Activity Evaluation. Molecules 2019, 24, 129. [Google Scholar] [CrossRef] [Green Version]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, e47. [Google Scholar] [CrossRef] [Green Version]
- Pereira, D.; Lima, R.T.; Palmeira, A.; Seca, H.; Soares, J.; Gomes, S.; Raimundo, L.; Maciel, C.; Pinto, M.; Sousa, E.; et al. Design and synthesis of new inhibitors of p53–MDM2 interaction with a chalcone scaffold. Arab. J. Chem. 2019, 12, 4150–4161. [Google Scholar] [CrossRef]
- Pinto, P.; Machado, C.M.; Moreira, J.; Almeida, J.D.P.; Silva, P.M.A.; Henriques, A.C.; Soares, J.X.; Salvador, J.A.R.; Afonso, C.; Pinto, M.; et al. Chalcone derivatives targeting mitosis: Synthesis, evaluation of antitumor activity and lipophilicity. Eur. J. Med. Chem. 2019, 184, 111752. [Google Scholar] [CrossRef]
- Ravishankar, D.; Rajora, A.K.; Greco, F.; Osborn, H.M.I. Flavonoids as prospective compounds for anti-cancer therapy. Int. J. Biochem. Cell Biol. 2013, 45, 2821–2831. [Google Scholar] [CrossRef]
- Zhou, B.; Xing, C. Diverse Molecular Targets for Chalcones with Varied Bioactivities. Med. Chem. 2015, 5, 388–404. [Google Scholar] [CrossRef]
- Almeida, J.R.; Moreira, J.; Pereira, D.; Pereira, S.; Antunes, J.; Palmeira, A.; Vasconcelos, V.; Pinto, M.; Correia-da-Silva, M.; Cidade, H. Potential of synthetic chalcone derivatives to prevent marine biofouling. Sci. Total Environ. 2018, 643, 98–106. [Google Scholar] [CrossRef]
- de Lima Cherubim, D.J.; Buzanello Martins, C.V.; Oliveira Fariña, L.; da Silva de Lucca, R.A. Polyphenols as natural antioxidants in cosmetics applications. J. Cosmet. Dermatol. 2020, 19, 33–37. [Google Scholar] [CrossRef]
- Espinoza Vázquez, A.; Figueroa, I.A.; Gómez, F.J.R.; Vásquez, A.P.; Mata, R.; Ángeles Beltrán, D.; Miralrio, A.; Castro, M. (–)–Epicatechin gallate as a corrosion inhibitor for bronze in a saline medium and theoretical study. J. Mol. Struct. 2021, 1227, 129416. [Google Scholar] [CrossRef]
- Kurek-Górecka, A.; Górecki, M.; Rzepecka-Stojko, A.; Balwierz, R.; Stojko, J. Bee Products in Dermatology and Skin Care. Molecules 2020, 25, 556. [Google Scholar] [CrossRef] [Green Version]
- Maqsood, S.; Adiamo, O.; Ahmad, M.; Mudgil, P. Bioactive compounds from date fruit and seed as potential nutraceutical and functional food ingredients. Food Chem. 2020, 308, 125522. [Google Scholar] [CrossRef]
- Obaid, R.J.; Mughal, E.U.; Naeem, N.; Sadiq, A.; Alsantali, R.I.; Jassas, R.S.; Moussa, Z.; Ahmed, S.A. Natural and synthetic flavonoid derivatives as new potential tyrosinase inhibitors: A systematic review. RSC Adv. 2021, 11, 22159–22198. [Google Scholar] [CrossRef]
- Kumar, S.; Verma, N.; Kumar, N.; Patel, A.; Roy, P.; Pruthi, V.; Ahmed, N. Design, synthesis, molecular docking, and biological studies of novel phytoestrogen-tanaproget hybrids. Synth. Commun. 2016, 46, 460–474. [Google Scholar] [CrossRef]
- Sang, Z.; Wang, K.; Shi, J.; Cheng, X.; Zhu, G.; Wei, R.; Ma, Q.; Yu, L.; Zhao, Y.; Tan, Z.; et al. Apigenin-rivastigmine hybrids as multi-target-directed liagnds for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2020, 187, 111958. [Google Scholar] [CrossRef]
- Bozorov, K.; Zhao, J.; Aisa, H.A. 1,2,3-Triazole-containing hybrids as leads in medicinal chemistry: A recent overview. Bioorgan. Med. Chem. 2019, 27, 3511–3531. [Google Scholar] [CrossRef]
- Kumar, S.; Sharma, B.; Mehra, V.; Kumar, V. Recent accomplishments on the synthetic/biological facets of pharmacologically active 1H-1,2,3-triazoles. Eur. J. Med. Chem. 2021, 212, 113069. [Google Scholar] [CrossRef]
- Zhao, L.; Mao, L.; Hong, G.; Yang, X.; Liu, T. Design, synthesis and anticancer activity of matrine–1H-1,2,3-triazole–chalcone conjugates. Bioorg. Med. Chem. Lett. 2015, 25, 2540–2544. [Google Scholar] [CrossRef]
- Tome, S.M.; Tome, A.C.; Soengas, R.G.; Silva, A.M.S. Chalcones and Chromones in Copper-Catalyzed Azide–Alkyne Cycloadditions (CuAAC). Curr. Org. Chem. 2018, 22, 1307–1325. [Google Scholar] [CrossRef]
- Aly, M.R.E.S.; Saad, H.A.; Mohamed, M.A.M. Click reaction based synthesis, antimicrobial, and cytotoxic activities of new 1,2,3-triazoles. Bioorg. Med. Chem. Lett. 2015, 25, 2824–2830. [Google Scholar] [CrossRef]
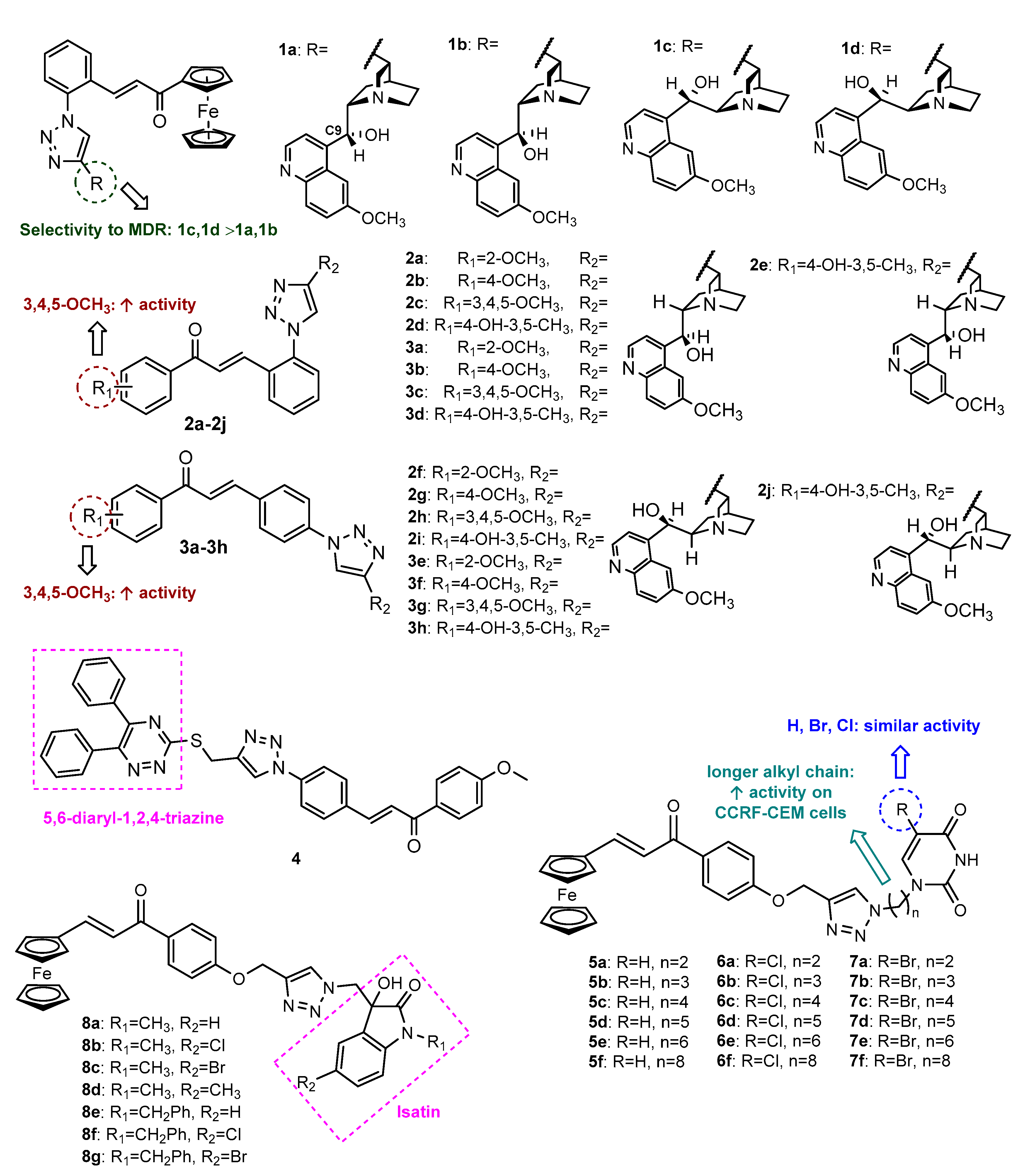
- Podolski-Renić, A.; Bősze, S.; Dinić, J.; Kocsis, L.; Hudecz, F.; Csámpai, A.; Pešić, M. Ferrocene–cinchona hybrids with triazolyl-chalcone linkers act as pro-oxidants and sensitize human cancer cell lines to paclitaxel. Metallomics 2017, 9, 1132–1141. [Google Scholar] [CrossRef]
- Jernei, T.; Duro, C.; Dembo, A.; Lajko, E.; Takacs, A.; Kohidai, L.; Schlosser, G.; Csampai, A. Synthesis, Structure and In Vitro Cytotoxic Activity of Novel Cinchona-Chalcone Hybrids with 1,4-Disubstituted- and 1,5-Disubstituted 1,2,3-Triazole Linkers. Molecules 2019, 24, 4077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, D.-J.; Song, J.; Hou, Y.-H.; Zhao, R.-H.; Li, J.-H.; Mao, R.-W.; Yang, J.-J.; Li, P.; Zi, X.-L.; Li, Z.-H.; et al. Discovery of 5,6-diaryl-1,2,4-triazines hybrids as potential apoptosis inducers. Eur. J. Med. Chem. 2017, 138, 1076–1088. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Chen, H.; Yan, W.; Zheng, M.; Zhang, T.; Zhang, Y. Ferrocene-containing hybrids as potential anticancer agents: Current developments, mechanisms of action and structure-activity relationships. Eur. J. Med. Chem. 2020, 190, 112109. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Mehra, V.; Sadeghiani, N.; Mozaffari, S.; Parang, K.; Kumar, V. Ferrocenylchalcone–uracil conjugates: Synthesis and cytotoxic evaluation. Med. Chem. Res. 2018, 27, 1260–1268. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Saha, S.T.; Perumal, S.; Kaur, M.; Kumar, V. Azide–Alkyne Cycloaddition En Route to 1H-1,2,3-Triazole-Tethered Isatin–Ferrocene, Ferrocenylmethoxy–Isatin, and Isatin–Ferrocenylchalcone Conjugates: Synthesis and Antiproliferative Evaluation. ACS Omega 2018, 3, 1263–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
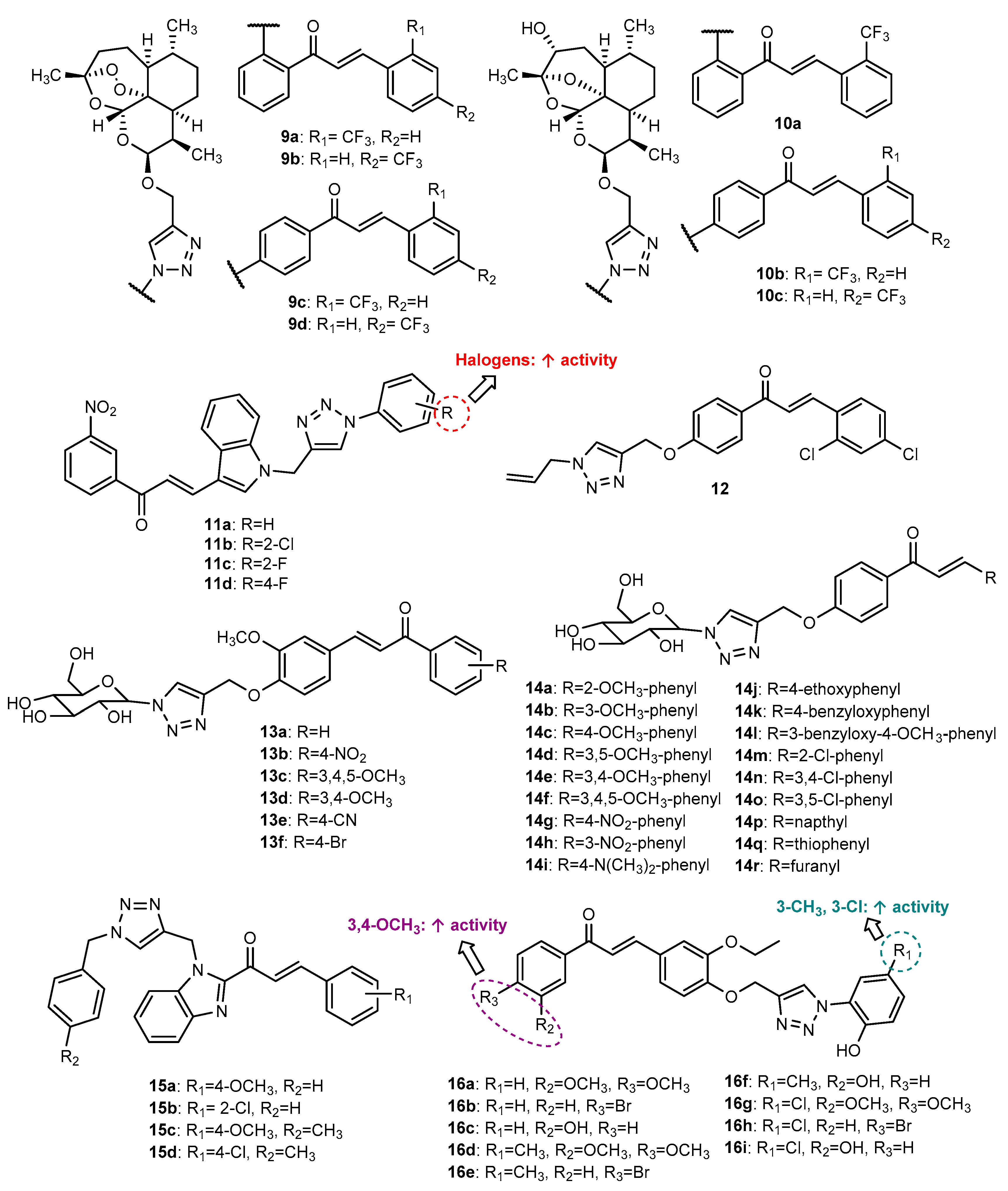
- Kapkoti, D.S.; Singh, S.; Luqman, S.; Bhakuni, R.S. Synthesis of novel 1,2,3-triazole based artemisinin derivatives and their antiproliferative activity. New J. Chem. 2018, 42, 5978–5995. [Google Scholar] [CrossRef]
- Aneja, B.; Arif, R.; Perwez, A.; Napoleon, J.V.; Hasan, P.; Rizvi, M.M.A.; Azam, A.; Rahisuddin; Abid, M. N-Substituted 1,2,3-Triazolyl-Appended Indole-Chalcone Hybrids as Potential DNA Intercalators Endowed with Antioxidant and Anticancer Properties. ChemistrySelect 2018, 3, 2638–2645. [Google Scholar] [CrossRef]
- Yan, W.; Xiangyu, C.; Ya, L.; Yu, W.; Feng, X. An orally antitumor chalcone hybrid inhibited HepG2 cells growth and migration as the tubulin binding agent. Investig. New Drugs 2019, 37, 784–790. [Google Scholar] [CrossRef]
- Manna, T.; Pal, K.; Jana, K.; Misra, A.K. Anti-cancer potential of novel glycosylated 1,4-substituted triazolylchalcone derivatives. Bioorg. Med. Chem. Lett. 2019, 29, 126615. [Google Scholar] [CrossRef]
- Djemoui, A.; Naouri, A.; Ouahrani, M.R.; Djemoui, D.; Lahcene, S.; Lahrech, M.B.; Boukenna, L.; Albuquerque, H.M.T.; Saher, L.; Rocha, D.H.A.; et al. A step-by-step synthesis of triazole-benzimidazole-chalcone hybrids: Anticancer activity in human cells+. J. Mol. Struct. 2020, 1204, 127487. [Google Scholar] [CrossRef]
- Gurrapu, N.; Praveen Kumar, E.; Kolluri, P.K.; Putta, S.; Sivan, S.K.; Subhashini, N.J.P. Synthesis, biological evaluation and molecular docking studies of novel 1,2,3-triazole tethered chalcone hybrids as potential anticancer agents. J. Mol. Struct. 2020, 1217, 128356. [Google Scholar] [CrossRef]
- Nagaraju, R.; Gopichand, K.; Rao, N.N.; Ganai, A.M.; Kishan, E.; Rao, P.V. Synthesis and Anticancer Activity of a Novel Series of Tetrazolo[1,5-a]quinoline Based 1,2,3-Triazole Derivatives. Russ. J. Gen. Chem. 2020, 90, 314–318. [Google Scholar] [CrossRef]
- Raghavender, M.; Kumar, A.K.; Sunitha, V.; Vishnu, T.; Jalapathi, P. Synthesis and Cytotoxicity of Chalcone Based 1,2,3-Triazole Derivatives. Russ. J. Gen. Chem. 2020, 90, 697–702. [Google Scholar] [CrossRef]
- Sharma, B.; Gu, L.; Pillay, R.P.; Cele, N.; Awolade, P.; Singh, P.; Kaur, M.; Kumar, V. Design, synthesis, and anti-proliferative evaluation of 1H-1,2,3-triazole grafted tetrahydro-β-carboline-chalcone/ferrocenylchalcone conjugates in estrogen responsive and triple negative breast cancer cells. New J. Chem. 2020, 44, 11137–11147. [Google Scholar] [CrossRef]
- Latif, A.D.; Jernei, T.; Podolski-Renić, A.; Kuo, C.-Y.; Vágvölgyi, M.; Girst, G.; Zupkó, I.; Develi, S.; Ulukaya, E.; Wang, H.-C.; et al. Protoflavone-Chalcone Hybrids Exhibit Enhanced Antitumor Action through Modulating Redox Balance, Depolarizing the Mitochondrial Membrane, and Inhibiting ATR-Dependent Signaling. Antioxidants 2020, 9, 519. [Google Scholar] [CrossRef]
- Wong, I.L.K.; Zhu, X.; Chan, K.F.; Law, M.C.; Lo, A.M.Y.; Hu, X.; Chow, L.M.C.; Chan, T.H. Discovery of Novel Flavonoid Dimers To Reverse Multidrug Resistance Protein 1 (MRP1, ABCC1) Mediated Drug Resistance in Cancers Using a High Throughput Platform with “Click Chemistry”. J. Med. Chem. 2018, 61, 9931–9951. [Google Scholar] [CrossRef]
- Zhu, X.; Wong, I.L.K.; Chan, K.F.; Cui, J.; Law, M.C.; Chong, T.C.; Hu, X.; Chow, L.M.C.; Chan, T.H. Triazole Bridged Flavonoid Dimers as Potent, Nontoxic, and Highly Selective Breast Cancer Resistance Protein (BCRP/ABCG2) Inhibitors. J. Med. Chem. 2019, 62, 8578–8608. [Google Scholar] [CrossRef]
- McGown, A.; Ragazzon-Smith, A.; Hadfield, J.A.; Potgetier, H.; Ragazzon, P.A. Microwave-Assisted Synthesis of Novel Bis-Flavone Dimers as New Anticancer Agents. Lett. Org. Chem. 2019, 16, 66–75. [Google Scholar] [CrossRef]
- Mistry, B.; Patel, R.V.; Keum, Y.-S. Access to the substituted benzyl-1,2,3-triazolyl hesperetin derivatives expressing antioxidant and anticancer effects. Arab. J. Chem. 2017, 10, 157–166. [Google Scholar] [CrossRef] [Green Version]
- Gupta, N.; Qayum, A.; Raina, A.; Shankar, R.; Gairola, S.; Singh, S.; Sangwan, P.L. Synthesis and biological evaluation of novel bavachinin analogs as anticancer agents. Eur. J. Med. Chem. 2018, 145, 511–523. [Google Scholar] [CrossRef]
- Gutam, M.; Mokenapelli, S.; Yerrabelli, J.R.; Banerjee, S.; Roy, P.; Chitneni, P.R. Synthesis and cytotoxicity of novel (E)-2-phenylchroman-4-one-O-((1-substituted-1H-1,2,3-triazol-4-yl)methyl) oxime derivatives. Synth. Commun. 2020, 50, 1883–1891. [Google Scholar] [CrossRef]
- Ribeiro, R.; Eloy, M.A.; Francisco, C.S.; Javarini, C.L.; Ayusso, G.M.; Da Rocha Fonseca, V.; Romão, W.; Regasini, L.O.; Araujo, S.C.; Almeida, M.O.; et al. Flavonoid derivatives targeting BCR-ABL kinase: Semisynthesis, Molecular dynamic simulations and Enzymatic inhibition. Curr. Top. Med. Chem. 2021, 21, 1999–2017. [Google Scholar] [CrossRef] [PubMed]
- Sowjanya, T.; Jayaprakash Rao, Y.; Murthy, N.Y.S. Synthesis and antiproliferative activity of new 1,2,3-triazole/flavone hybrid heterocycles against human cancer cell lines. Russ. J. Gen. Chem. 2017, 87, 1864–1871. [Google Scholar] [CrossRef]
- Wang, G.; Yan, L.; Wang, Q. Synthesis and antiproliferative activity of flavonoid triazolyl glycosides. Heterocycl. Commun. 2018, 24, 119–124. [Google Scholar] [CrossRef]
- Rao, Y.J.; Sowjanya, T.; Thirupathi, G.; Murthy, N.Y.S.; Kotapalli, S.S. Synthesis and biological evaluation of novel flavone/triazole/benzimidazole hybrids and flavone/isoxazole-annulated heterocycles as antiproliferative and antimycobacterial agents. Mol. Divers. 2018, 22, 803–814. [Google Scholar] [CrossRef]
- Bian, J.; Ren, J.; Li, Y.; Wang, J.; Xu, X.; Feng, Y.; Tang, H.; Wang, Y.; Li, Z. Discovery of Wogonin-based PROTACs against CDK9 and capable of achieving antitumor activity. Bioorg. Chem. 2018, 81, 373–381. [Google Scholar] [CrossRef]
- Qi, Y.; Ding, Z.; Yao, Y.; Ma, D.; Ren, F.; Yang, H.; Chen, A. Novel triazole analogs of apigenin-7-methyl ether exhibit potent antitumor activity against ovarian carcinoma cells via the induction of mitochondrial-mediated apoptosis. Exp. Ther. Med. 2019, 17, 1670–1676. [Google Scholar] [CrossRef] [Green Version]
- Noole, V.; Krishna, T.; Godeshala, S.; Meraji, S.; Rege, K.; Reddy, C.K.; Kedika, B. Synthesis and Biological Evaluation of New 1,2,3-Triazole Derivatives of the Chrysin Flavonoid as Anticancer Agents. Anticancer Agents Med. Chem. 2021. [Google Scholar] [CrossRef]
- Ganesan, K.; Xu, B. Telomerase Inhibitors from Natural Products and Their Anticancer Potential. Int. J. Mol. Sci. 2018, 19, 13. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.F.; Ho, S.T.; Wen, R.; Fu, Y.; Zhang, L.; Wang, J.; Hu, C.; Shaw, P.C.; Liu, Y.; Cheng, M.S. Design, Synthesis and Molecular Docking Analysis of Flavonoid Derivatives as Potential Telomerase Inhibitors. Molecules 2019, 24, 3180. [Google Scholar] [CrossRef] [Green Version]
- Znati, M.; Horchani, M.; Latapie, L.; Ben Jannet, H.; Bouajila, J. New 1,2,3-triazole linked flavonoid conjugates: Microwave-assisted synthesis, cytotoxic activity and molecular docking studies. J. Mol. Struct. 2021, 1246, 131216. [Google Scholar] [CrossRef]
- Yerrabelly, J.R.; Gogula, T.; Erukala, Y.G.; Yerrabelly, H.; Gabriella, S. Synthesis and antiproliferative activity of Daidzein bridged bis-[1,2,3]-triazole derivatives: Double click strategy. Chem. Data Collect. 2020, 29, 100523. [Google Scholar] [CrossRef]
- Ashok, D.; Kavitha, R.; Gundu, S.; Rao, V.H. Microwave-assisted synthesis of new pyrazole derivatives bearing 1,2,3-triazole scaffold as potential antimicrobial agents. J. Serb. Chem. Soc. 2017, 82, 357–366. [Google Scholar] [CrossRef]
- Singh, A.; Gut, J.; Rosenthal, P.J.; Kumar, V. 4-Aminoquinoline-ferrocenyl-chalcone conjugates: Synthesis and anti-plasmodial evaluation. Eur. J. Med. Chem. 2017, 125, 269–277. [Google Scholar] [CrossRef]
- Singh, A.; Viljoen, A.; Kremer, L.; Kumar, V. Azide–alkyne cycloaddition en route to 4-aminoquinoline–ferrocenylchalcone conjugates: Synthesis and anti-TB evaluation. Future Med. Chem. 2017, 9, 1701–1708. [Google Scholar] [CrossRef]
- Kumar, S.; Saini, A.; Gut, J.; Rosenthal, P.J.; Raj, R.; Kumar, V. 4-Aminoquinoline-chalcone/-N-acetylpyrazoline conjugates: Synthesis and antiplasmodial evaluation. Eur. J. Med. Chem. 2017, 138, 993–1001. [Google Scholar] [CrossRef]
- Fadda, A.A.; Elattar, K.M. Reactivity of dehydroacetic acid in organic synthesis. Synth. Commun. 2016, 46, 1–30. [Google Scholar] [CrossRef]
- Lal, K.; Yadav, P.; Kumar, A.; Kumar, A.; Paul, A.K. Design, synthesis, characterization, antimicrobial evaluation and molecular modeling studies of some dehydroacetic acid-chalcone-1,2,3-triazole hybrids. Bioorg. Chem. 2018, 77, 236–244. [Google Scholar] [CrossRef]
- Yadav, P.; Lal, K.; Kumar, L.; Kumar, A.; Kumar, A.; Paul, A.K.; Kumar, R. Synthesis, crystal structure and antimicrobial potential of some fluorinated chalcone-1,2,3-triazole conjugates. Eur. J. Med. Chem. 2018, 155, 263–274. [Google Scholar] [CrossRef]
- Kiran, K.; Sarasija, M.; Ananda Rao, B.; Namratha, V.; Ashok, D.; Srinivasa Rao, A. Design, Synthesis, and Biological Activity of New Bis-1,2,3-triazole Derivatives Bearing Thiophene-Chalcone Moiety. Russ. J. Gen. Chem. 2019, 89, 1859–1866. [Google Scholar] [CrossRef]
- Pujari, V.K.; Vinnakota, S.; Kakarla, R.K.; Maroju, S.; Ganesh, A.; Pervaram, S. Microwave Assisted Synthesis and Antimicrobial Activity of (E)-1-{2/3/4-[(1-Aryl-1H-1,2,3-triazol-4-yl)methoxy]phenyl}-3-(2-morpholinoquinolin-3-yl)prop-2-en-1-ones. Russ. J. Gen. Chem. 2018, 88, 1502–1507. [Google Scholar] [CrossRef]
- Singh, G.; Arora, A.; Kalra, P.; Maurya, I.K.; Ruizc, C.E.; Estebanc, M.A.; Sinha, S.; Goyal, K.; Sehgal, R. A strategic approach to the synthesis of ferrocene appended chalcone linked triazole allied organosilatranes: Antibacterial, antifungal, antiparasitic and antioxidant studies. Bioorgan. Med. Chem. 2019, 27, 188–195. [Google Scholar] [CrossRef]
- Sunitha, V.; Kumar, A.K.; Jalapathi, P.; Lincoln, C.A. Synthesis and Antimicrobial Activity of Bis-1,2,3-triazole Based Chalcones. Russ. J. Gen. Chem. 2020, 90, 154–159. [Google Scholar] [CrossRef]
- Nagaraja, M.; Kalluraya, B.; Asma; Shreekanth, T. K.; Kumar, M.S. Synthesis of chalcone precursor via Cu(I) catalyzed 1,3-dipolar reaction of functionalized acetylene and pyrazole embedded dipole. J. Heterocycl. Chem. 2020, 57, 3642–3652. [Google Scholar] [CrossRef]
- Yadav, P.; Lal, K.; Kumar, A. Antimicrobial Screening, in Silico Studies and QSAR of Chalcone-based 1,4-disubstituted 1,2,3-triazole Hybrids. Drug Res. 2021, 71, 149–156. [Google Scholar] [CrossRef]
- Vishnuvardhan, M.; Pradeep, M.; Gangadhar, T. An efficient microwave assisted synthesis and antimicrobial activity of novel p-Tolyloxyquinoline-Triazole hybrid derivatives. Chem. Data Collect. 2021, 31, 100612. [Google Scholar] [CrossRef]
- Zhang, H.; Zheng, X.; Li, J.; Liu, Q.; Huang, X.-X.; Ding, H.; Suzuki, R.; Muramatsu, M.; Song, S.-J. Flavonoid-triazolyl hybrids as potential anti-hepatitis C virus agents: Synthesis and biological evaluation. Eur. J. Med. Chem. 2021, 218, 113395. [Google Scholar] [CrossRef]
- Zhang, C.; Li, N.; Niu, F. Baicalein triazole prevents respiratory tract infection by RSV through suppression of oxidative damage. Microb. Pathog. 2019, 131, 227–233. [Google Scholar] [CrossRef]
- Bhowmik, S.; Anand, P.; Das, R.; Sen, T.; Akhter, Y.; Das, M.C.; De, U.C. Synthesis of new chrysin derivatives with substantial antibiofilm activity. Mol. Divers. 2021. [Google Scholar] [CrossRef]
- Yerrabelly, J.R.; Mallepaka, P. Facile Synthesis of Novel Isoflavone/1,2,3-Triazole HybridHeterocycles as Potential Antimicrobial Agents. Russ. J. Gen. Chem. 2020, 90, 911–916. [Google Scholar] [CrossRef]
- Bansal, R.; Singh, R. Exploring the potential of natural and synthetic neuroprotective steroids against neurodegenerative disorders: A literature review. Med. Res. Rev. 2018, 38, 1126–1158. [Google Scholar] [CrossRef] [PubMed]
- Ayaz, M.; Sadiq, A.; Junaid, M.; Ullah, F.; Ovais, M.; Ullah, I.; Ahmed, J.; Shahid, M. Flavonoids as Prospective Neuroprotectants and Their Therapeutic Propensity in Aging Associated Neurological Disorders. Front. Aging Neurosci. 2019, 11, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lima, L.K.F.; Pereira, S.K.S.; Junior, R.; Santos, F.; Nascimento, A.S.; Feitosa, C.M.; Figuerêdo, J.S.; Cavalcante, A.D.N.; Araújo, E.; Rai, M. A Brief Review on the Neuroprotective Mechanisms of Vitexin. Biomed Res. Int. 2018, 2018, 4785089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sooknual, P.; Pingaew, R.; Phopin, K.; Ruankham, W.; Prachayasittikul, S.; Ruchirawat, S.; Prachayasittikul, V. Synthesis and neuroprotective effects of novel chalcone-triazole hybrids. Bioorg. Chem. 2020, 105, 104384. [Google Scholar] [CrossRef]
- Abeysinghe, A.A.D.T.; Deshapriya, R.D.U.S.; Udawatte, C. Alzheimer’s disease; a review of the pathophysiological basis and therapeutic interventions. Life Sci. 2020, 256, 117996. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, A.; Ekavali. A review on Alzheimer’s disease pathophysiology and its management: An update. Pharmacol. Rep. 2015, 67, 195–203. [Google Scholar] [CrossRef]
- Shi, S.; Wang, H.; Wang, J.; Wang, Y.; Xue, X.; Hou, Z.; Yao, G.-D.; Huang, X.-X.; Zhao, H.; Liu, Q.; et al. Semi-synthesis and biological evaluation of flavone hybrids as multifunctional agents for the potential treatment of Alzheimer’s disease. Bioorg. Chem. 2020, 100, 103917. [Google Scholar] [CrossRef]
- Çelik, F.; Türkan, F.; Aras, A.; Atalar, M.N.; Karaman, H.S.; Ünver, Y.; Kahriman, N. Synthesis of novel 1,2,3 triazole derivatives and assessment of their potential cholinesterases, glutathione S-transferase enzymes inhibitory properties: An in vitro and in silico study. Bioorg. Chem. 2021, 107, 104606. [Google Scholar] [CrossRef]
- Wang, M.; Fang, L.; Liu, T.; Chen, X.; Zheng, Y.; Zhang, Y.; Chen, S.; Li, Z. Discovery of 7-O-1, 2, 3-triazole hesperetin derivatives as multi-target-directed ligands against Alzheimer’s disease. Chem. Biol. Interact. 2021, 342, 109489. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhang, Y.-L.; Li, Z.; Shi, W.; Ji, Y.-R.; Guo, Y.-H.; Huang, C.; Sun, G.-P.; Li, J. Design and synthesis of 7-O-1,2,3-triazole hesperetin derivatives to relieve inflammation of acute liver injury in mice. Eur. J. Med. Chem. 2021, 213, 113162. [Google Scholar] [CrossRef]
- Rani, A.; Singh, A.; Kaur, J.; Singh, G.; Bhatti, R.; Gumede, N.; Kisten, P.; Singh, P.; Sumanjit; Kumar, V. 1H-1,2,3-triazole grafted tacrine-chalcone conjugates as potential cholinesterase inhibitors with the evaluation of their behavioral tests and oxidative stress in mice brain cells. Bioorg. Chem. 2021, 114, 105053. [Google Scholar] [CrossRef]
- Asadipour, A.; Noushini, S.; Moghimi, S.; Mahdavi, M.; Nadri, H.; Moradi, A.; Shabani, S.; Firoozpour, L.; Foroumadi, A. Synthesis and biological evaluation of chalcone-triazole hybrid derivatives as 15-LOX inhibitors. Z. Naturforsch. B 2018, 73, 77–83. [Google Scholar] [CrossRef]
- Boshra, A.N.; Abdu-Allah, H.H.M.; Mohammed, A.F.; Hayallah, A.M. Click chemistry synthesis, biological evaluation and docking study of some novel 2′-hydroxychalcone-triazole hybrids as potent anti-inflammatory agents. Bioorg. Chem. 2020, 95, 103505. [Google Scholar] [CrossRef]
- Mengheres, G.; Olajide, O.; Hemming, K. The synthesis and anti-inflammatory evaluation of 1,2,3-triazole linked isoflavone benzodiazepine hybrids. Arkivoc 2020, 2020, 306–321. [Google Scholar] [CrossRef]
- Linzembold, I.; Czett, D.; Boddi, K.; Kurtan, T.; Kiraly, S.B.; Gulyas-Fekete, G.; Takatsy, A.; Lorand, T.; Deli, J.; Agocs, A.; et al. Study on the Synthesis, Antioxidant Properties, and Self-Assembly of Carotenoid-Flavonoid Conjugates. Molecules 2020, 25, 636. [Google Scholar] [CrossRef] [Green Version]
- Ngameni, B.; Erdoğan, M.; Kuete, V.; Dalkılıç, E.; Ngadjui, B.T.; Daştan, A. Synthesis and structural characterization of novel O-substituted phenolic and chalcone derivatives with antioxidant activity. J. Chem. Res. 2020, 45, 159–165. [Google Scholar] [CrossRef]
- Singh, G.; Sushma; Singh, A.; Priyanka; Chowdhary, K.; Singh, J.; Esteban, M.A.; Espinosa-Ruíz, C.; González-Silvera, D. Designing of chalcone functionalized 1,2,3-triazole allied bis-organosilanes as potent antioxidants and optical sensor for recognition of Sn2+ and Hg2+ ions. J. Organomet. Chem. 2021, 953, 122049. [Google Scholar] [CrossRef]
- Lomelino, C.; McKenna, R. Carbonic anhydrase inhibitors: A review on the progress of patent literature (2011–2016). Expert Opin. Ther. Pat. 2016, 26, 947–956. [Google Scholar] [CrossRef]
- Singh, P.; Swain, B.; Thacker, P.S.; Sigalapalli, D.K.; Purnachander Yadav, P.; Angeli, A.; Supuran, C.T.; Arifuddin, M. Synthesis and carbonic anhydrase inhibition studies of sulfonamide based indole-1,2,3-triazole chalcone hybrids. Bioorg. Chem. 2020, 99, 103839. [Google Scholar] [CrossRef]
- Son, N.T.; Suenaga, M.; Matsunaga, Y.; Van Chinh, L.; Kubo, M.; Harada, K.; Cuong, N.M.; Fukuyama, Y. Serine protease inhibitors and activators from Dalbergia tonkinensis species. J. Nat. Med. 2020, 74, 257–263. [Google Scholar] [CrossRef]
- Allocati, N.; Masulli, M.; Di Ilio, C.; Federici, L. Glutathione transferases: Substrates, inihibitors and pro-drugs in cancer and neurodegenerative diseases. Oncogenesis 2018, 7, 8. [Google Scholar] [CrossRef]
- Dong, S.C.; Sha, H.H.; Xu, X.Y.; Hu, T.M.; Lou, R.; Li, H.; Wu, J.Z.; Dan, C.; Feng, J. Glutathione S-transferase π: A potential role in antitumor therapy. Drug Des. Dev. Ther. 2018, 12, 3535–3547. [Google Scholar] [CrossRef] [Green Version]
- Nie, J.-P.; Qu, Z.-N.; Chen, Y.; Chen, J.-H.; Jiang, Y.; Jin, M.-N.; Yu, Y.; Niu, W.-Y.; Duan, H.-Q.; Qin, N. Discovery and anti-diabetic effects of novel isoxazole based flavonoid derivatives. Fitoterapia 2020, 142, 104499. [Google Scholar] [CrossRef]
- Gorantla, J.N.; Maniganda, S.; Pengthaisong, S.; Ngiwsara, L.; Sawangareetrakul, P.; Chokchaisiri, S.; Kittakoop, P.; Svasti, J.; Ketudat Cairns, J.R. Chemoenzymatic and Protecting-Group-Free Synthesis of 1,4-Substituted 1,2,3-Triazole-α-d-glucosides with Potent Inhibitory Activity toward Lysosomal α-Glucosidase. ACS Omega 2021, 6, 25710–25719. [Google Scholar] [CrossRef]
- Almeida, J.R.; Palmeira, A.; Campos, A.; Cunha, I.; Freitas, M.; Felpeto, A.B.; Turkina, M.V.; Vasconcelos, V.; Pinto, M.; Correia-da-Silva, M.; et al. Structure-Antifouling Activity Relationship and Molecular Targets of Bio-Inspired(thio)xanthones. Biomolecules 2020, 10, 1126. [Google Scholar] [CrossRef]
- Pereira, D.; Gonçalves, C.; Martins, B.T.; Palmeira, A.; Vasconcelos, V.; Pinto, M.; Almeida, J.R.; Correia-da-Silva, M.; Cidade, H. Flavonoid Glycosides with a Triazole Moiety for Marine Antifouling Applications: Synthesis and Biological Activity Evaluation. Mar. Drugs 2021, 19, 5. [Google Scholar] [CrossRef]



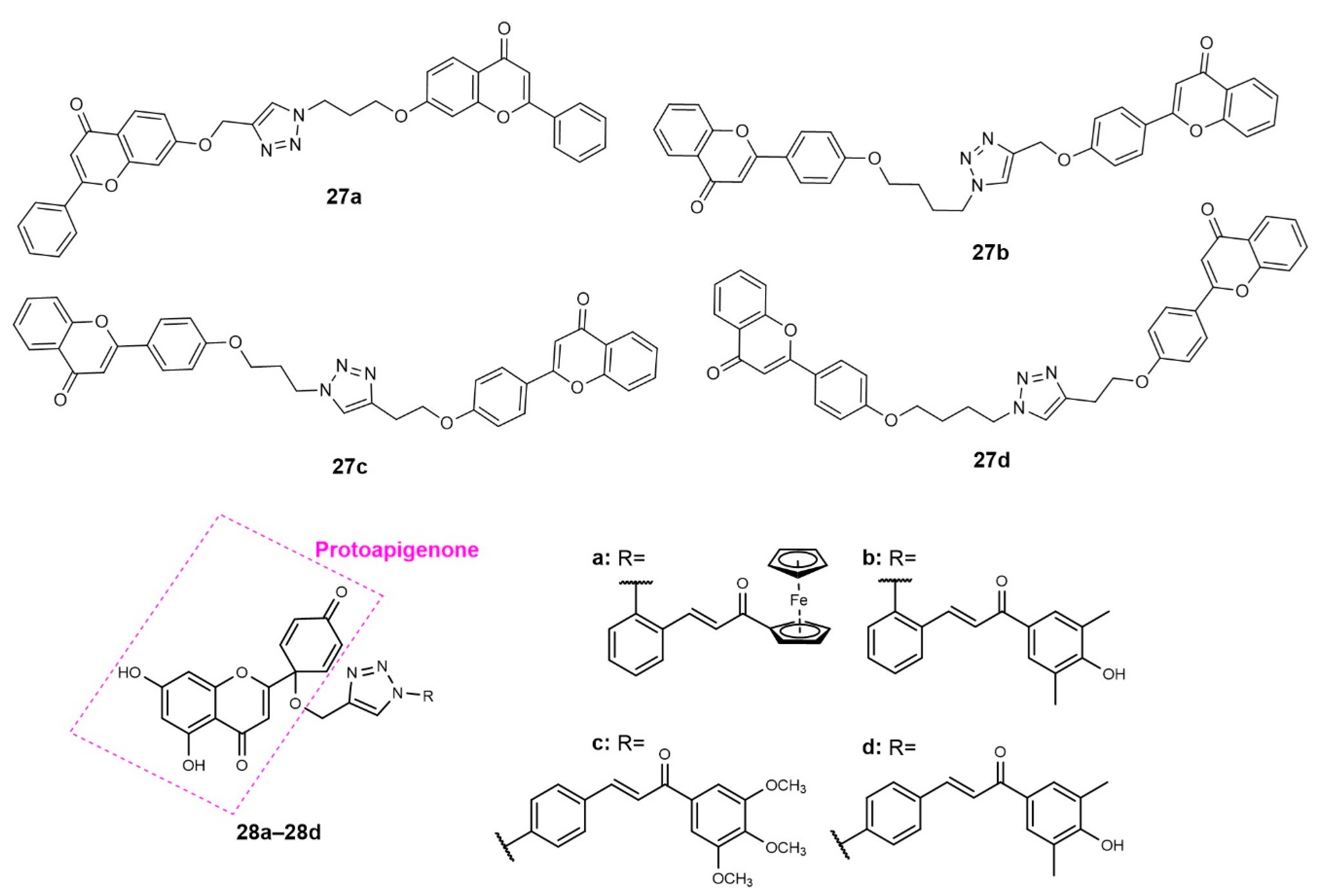
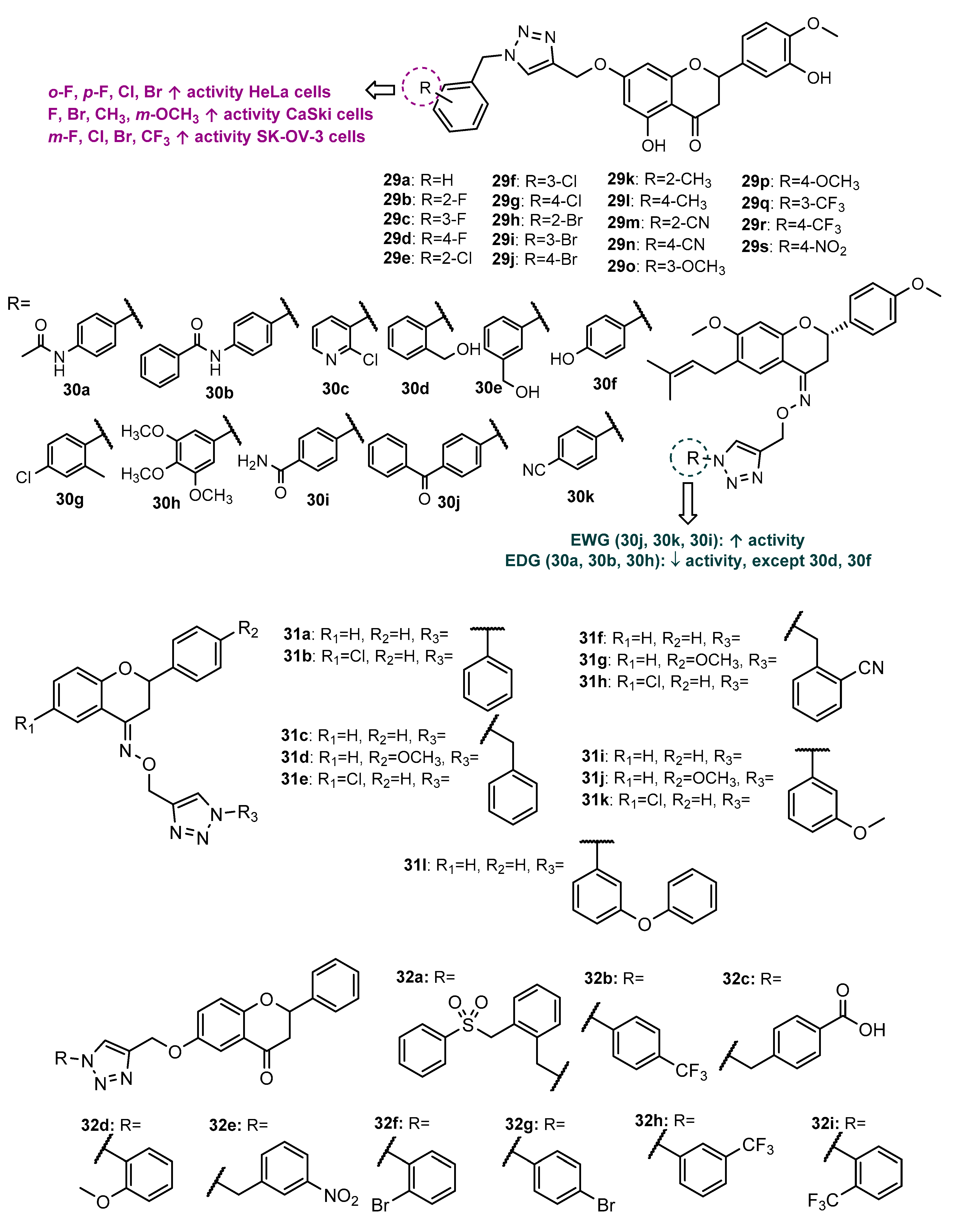
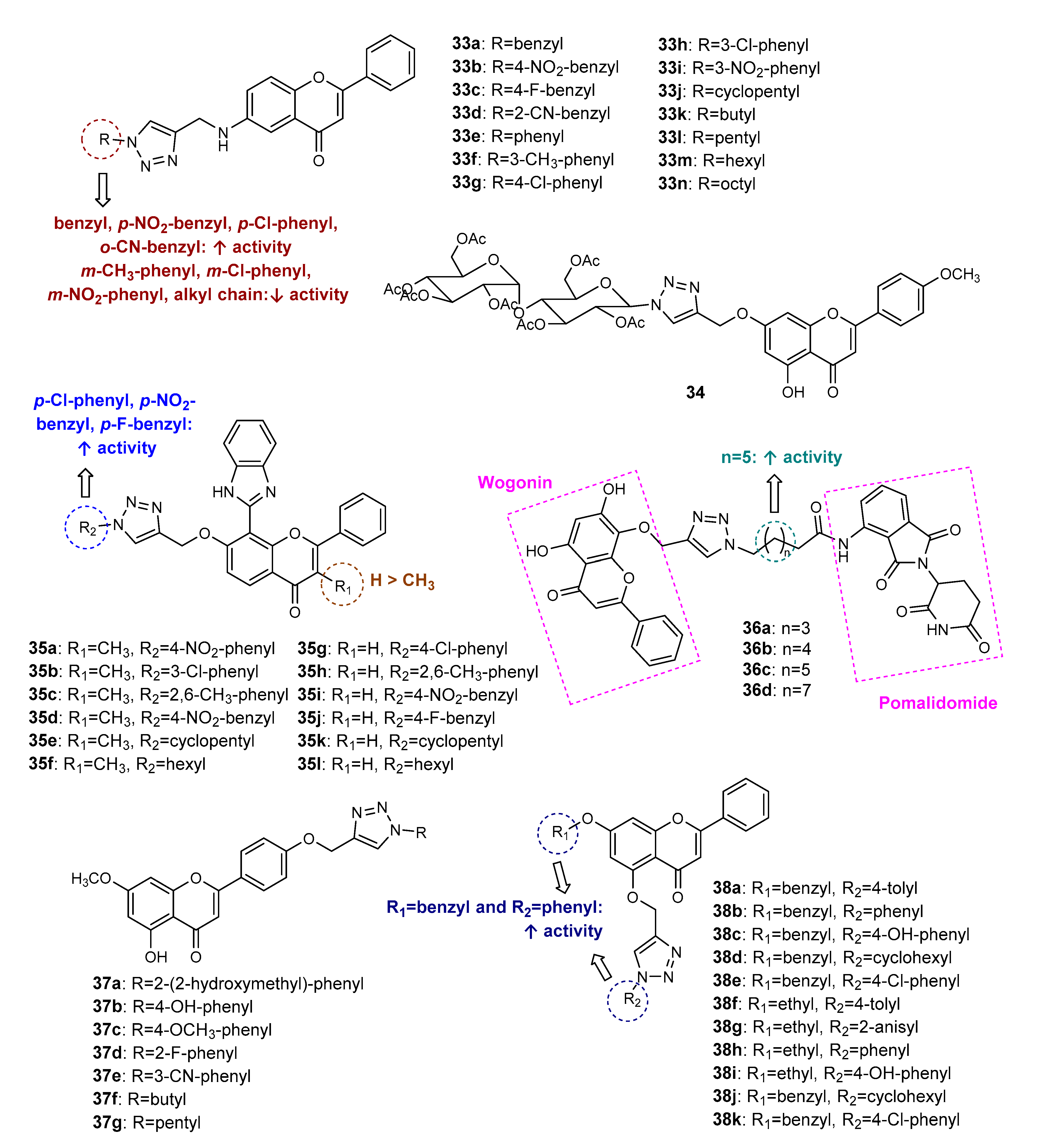
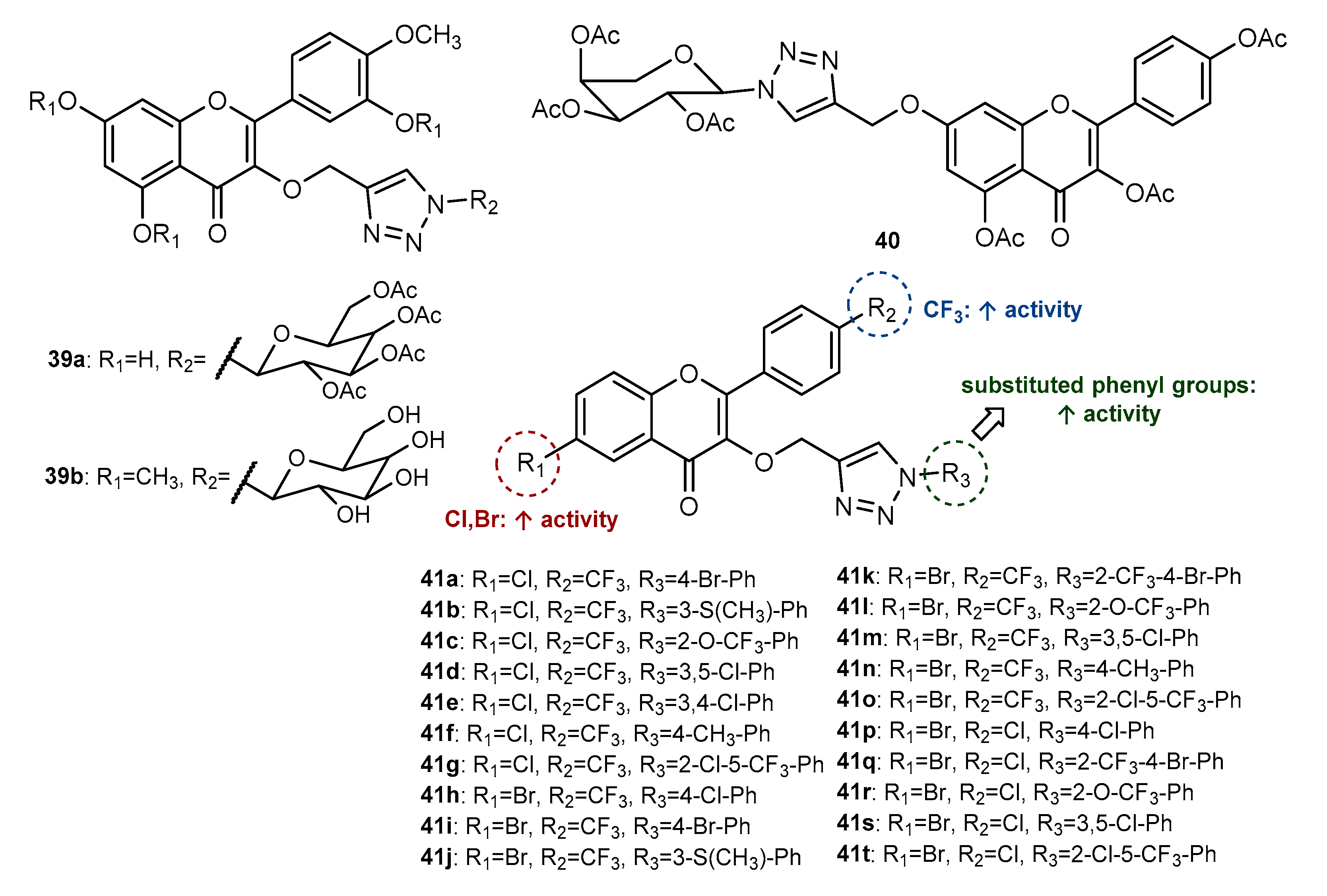


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pereira, D.; Pinto, M.; Correia-da-Silva, M.; Cidade, H. Recent Advances in Bioactive Flavonoid Hybrids Linked by 1,2,3-Triazole Ring Obtained by Click Chemistry. Molecules 2022, 27, 230. https://doi.org/10.3390/molecules27010230
Pereira D, Pinto M, Correia-da-Silva M, Cidade H. Recent Advances in Bioactive Flavonoid Hybrids Linked by 1,2,3-Triazole Ring Obtained by Click Chemistry. Molecules. 2022; 27(1):230. https://doi.org/10.3390/molecules27010230
Chicago/Turabian StylePereira, Daniela, Madalena Pinto, Marta Correia-da-Silva, and Honorina Cidade. 2022. "Recent Advances in Bioactive Flavonoid Hybrids Linked by 1,2,3-Triazole Ring Obtained by Click Chemistry" Molecules 27, no. 1: 230. https://doi.org/10.3390/molecules27010230
APA StylePereira, D., Pinto, M., Correia-da-Silva, M., & Cidade, H. (2022). Recent Advances in Bioactive Flavonoid Hybrids Linked by 1,2,3-Triazole Ring Obtained by Click Chemistry. Molecules, 27(1), 230. https://doi.org/10.3390/molecules27010230