
On the Quantum Chemical Nature of Lead(II) “Lone Pair”


Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
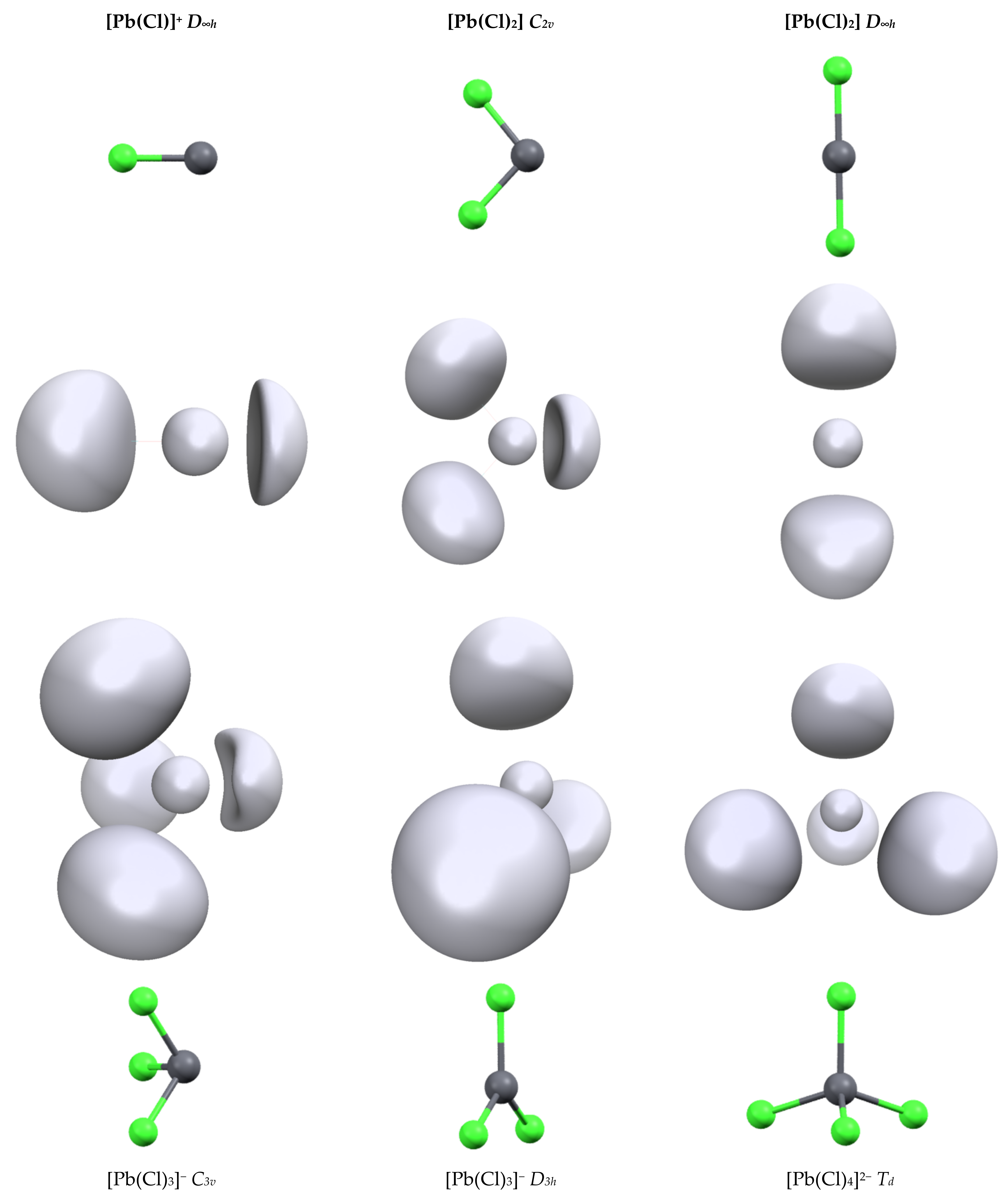
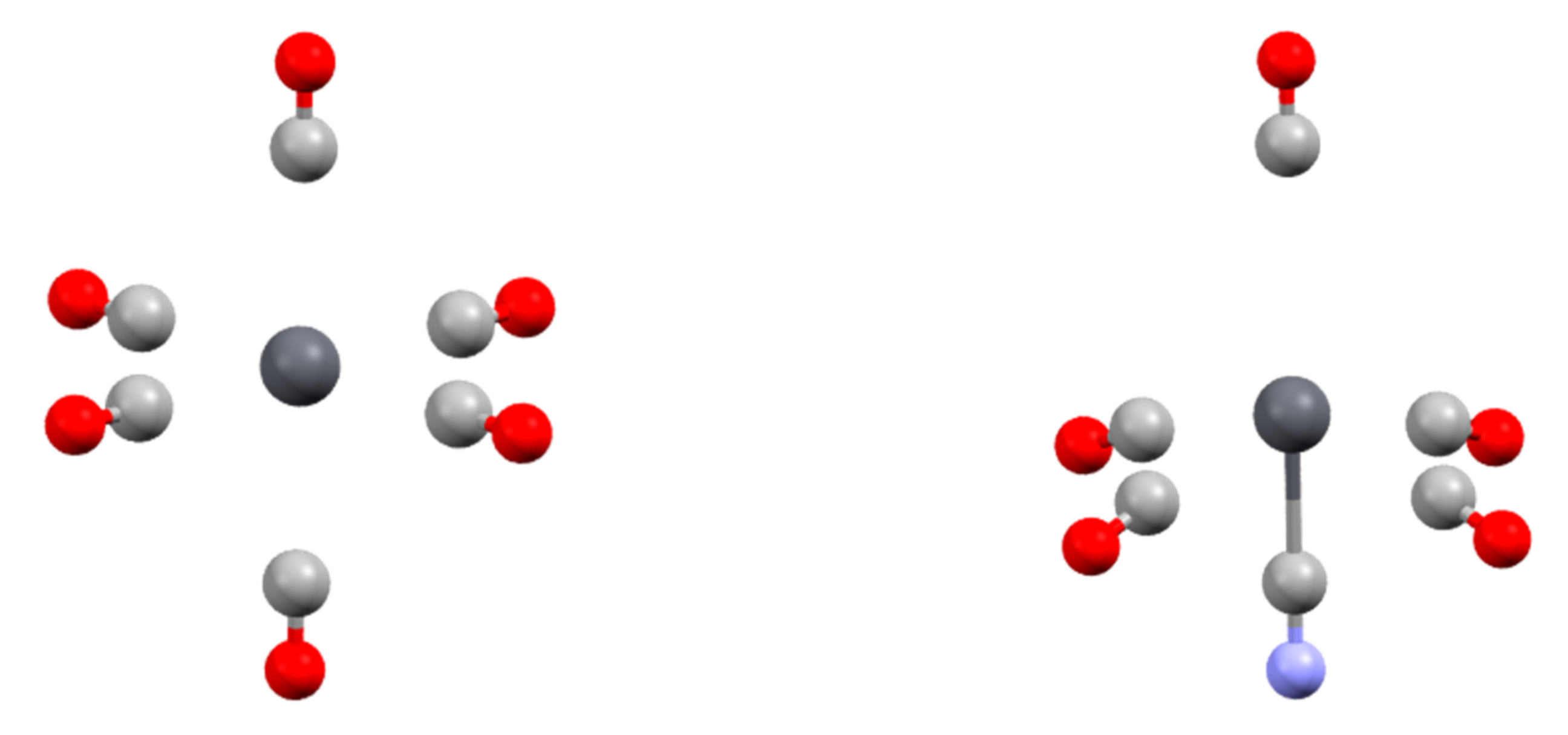
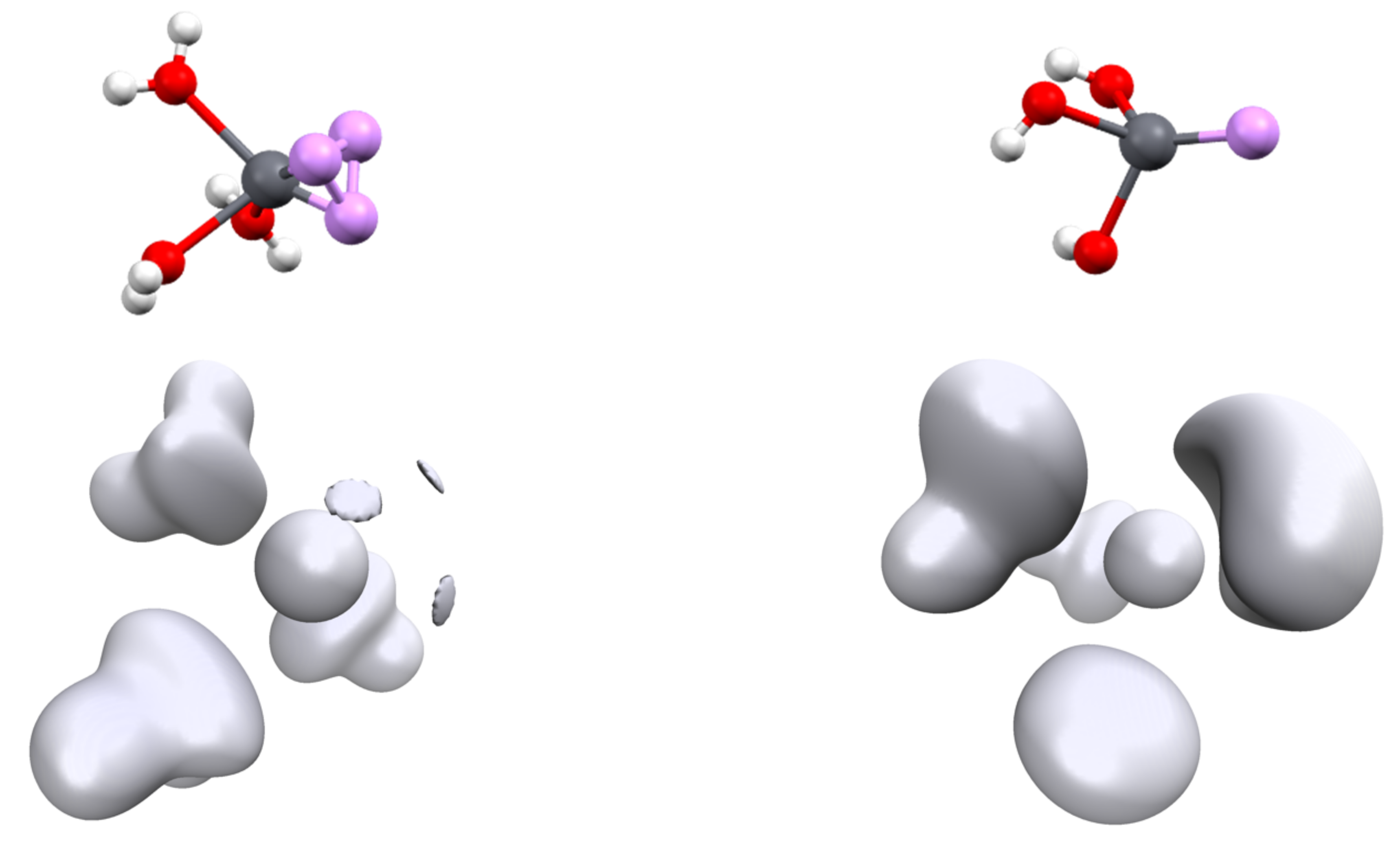
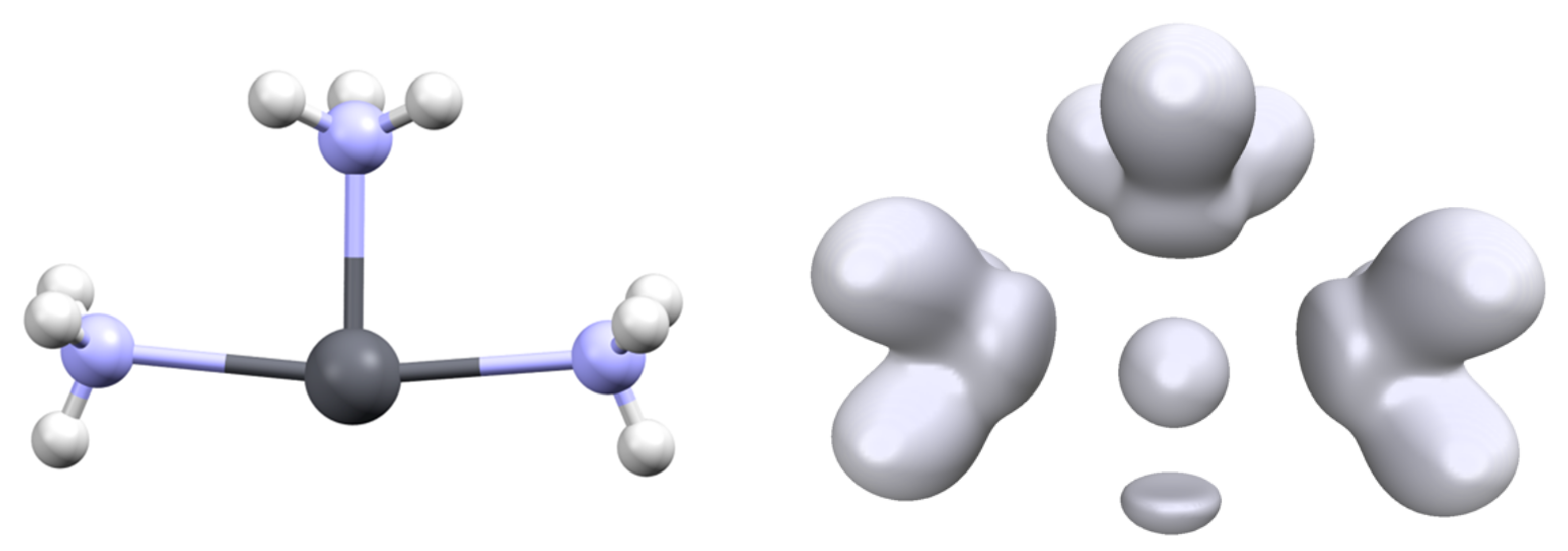
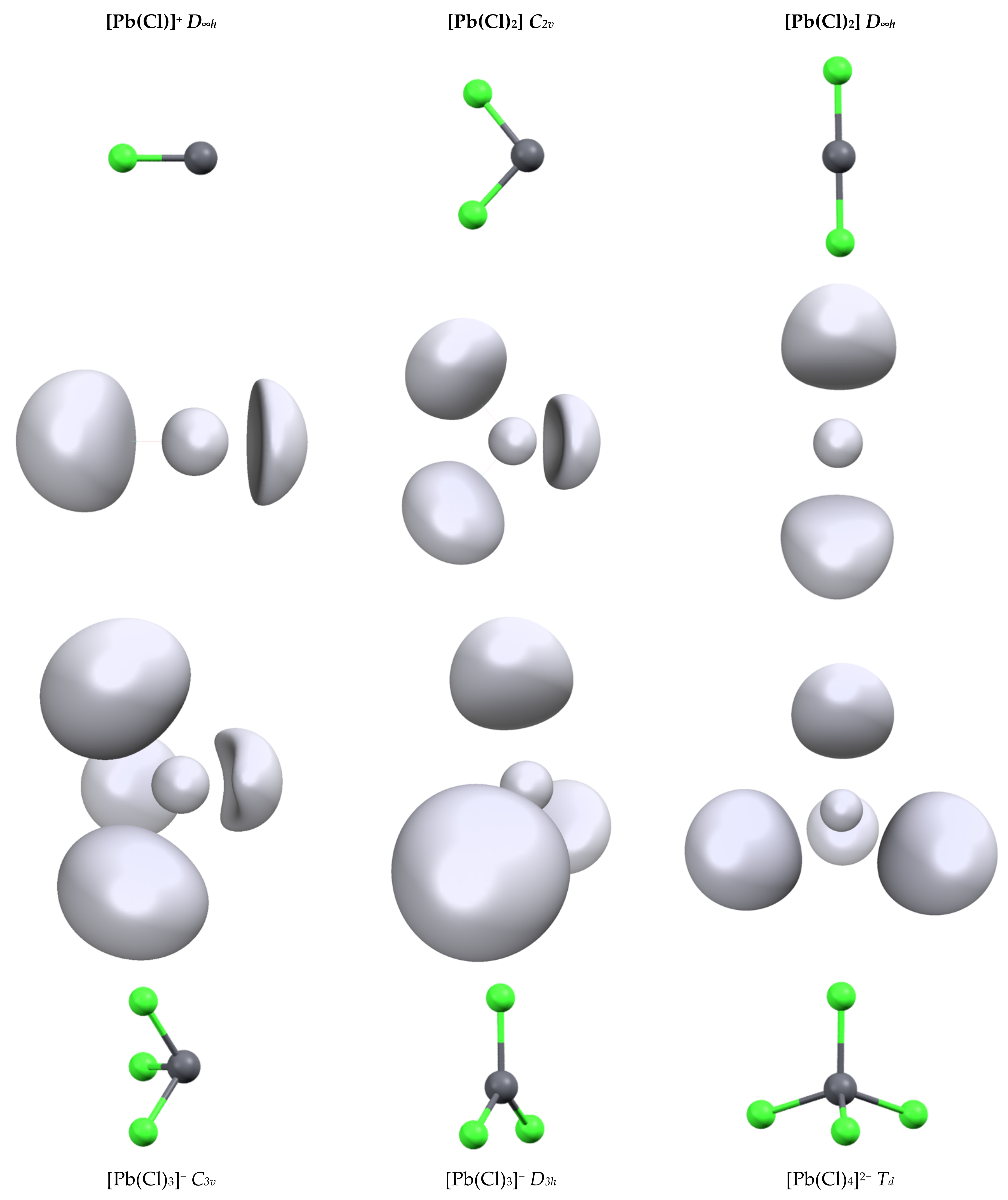
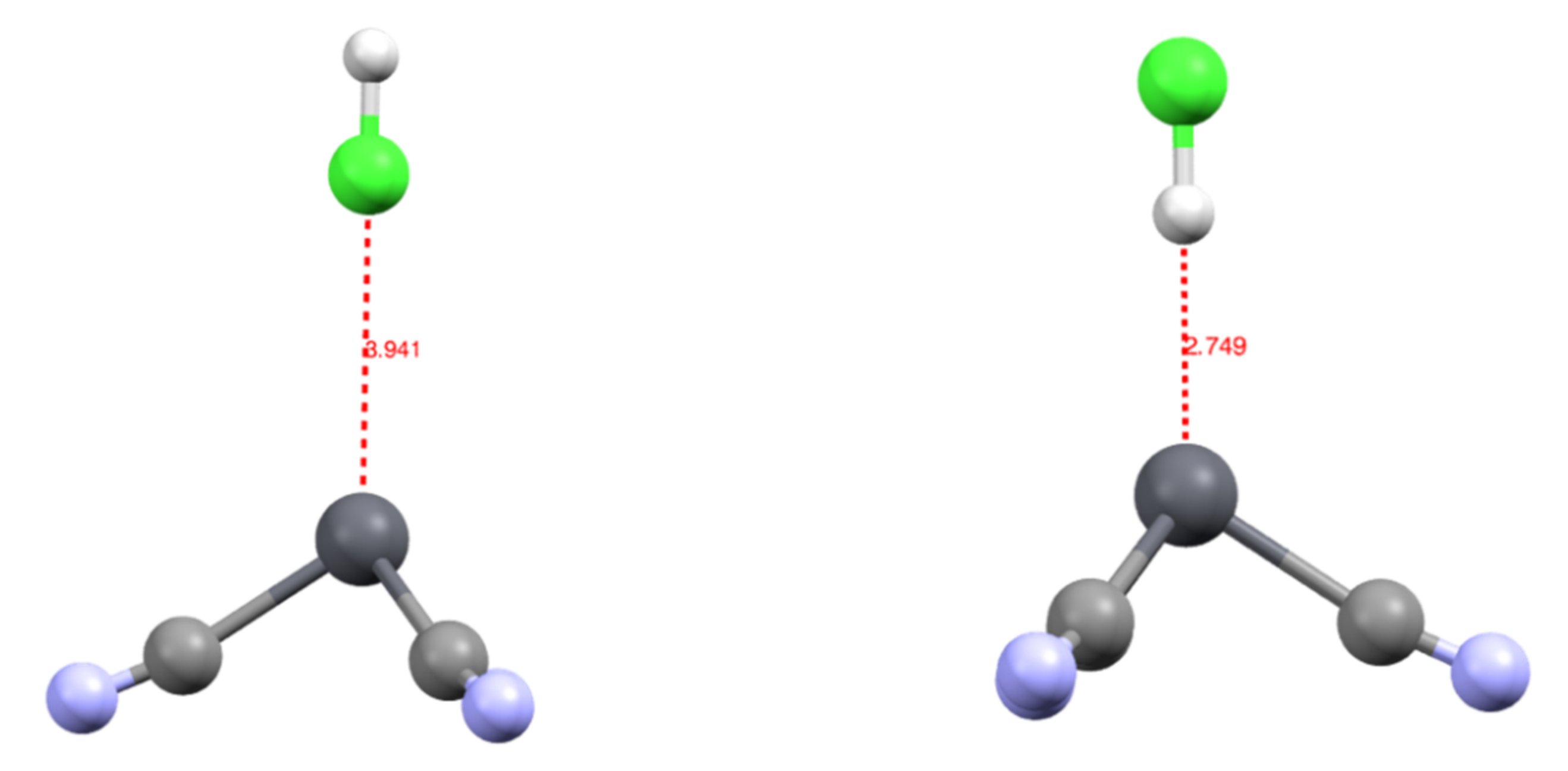
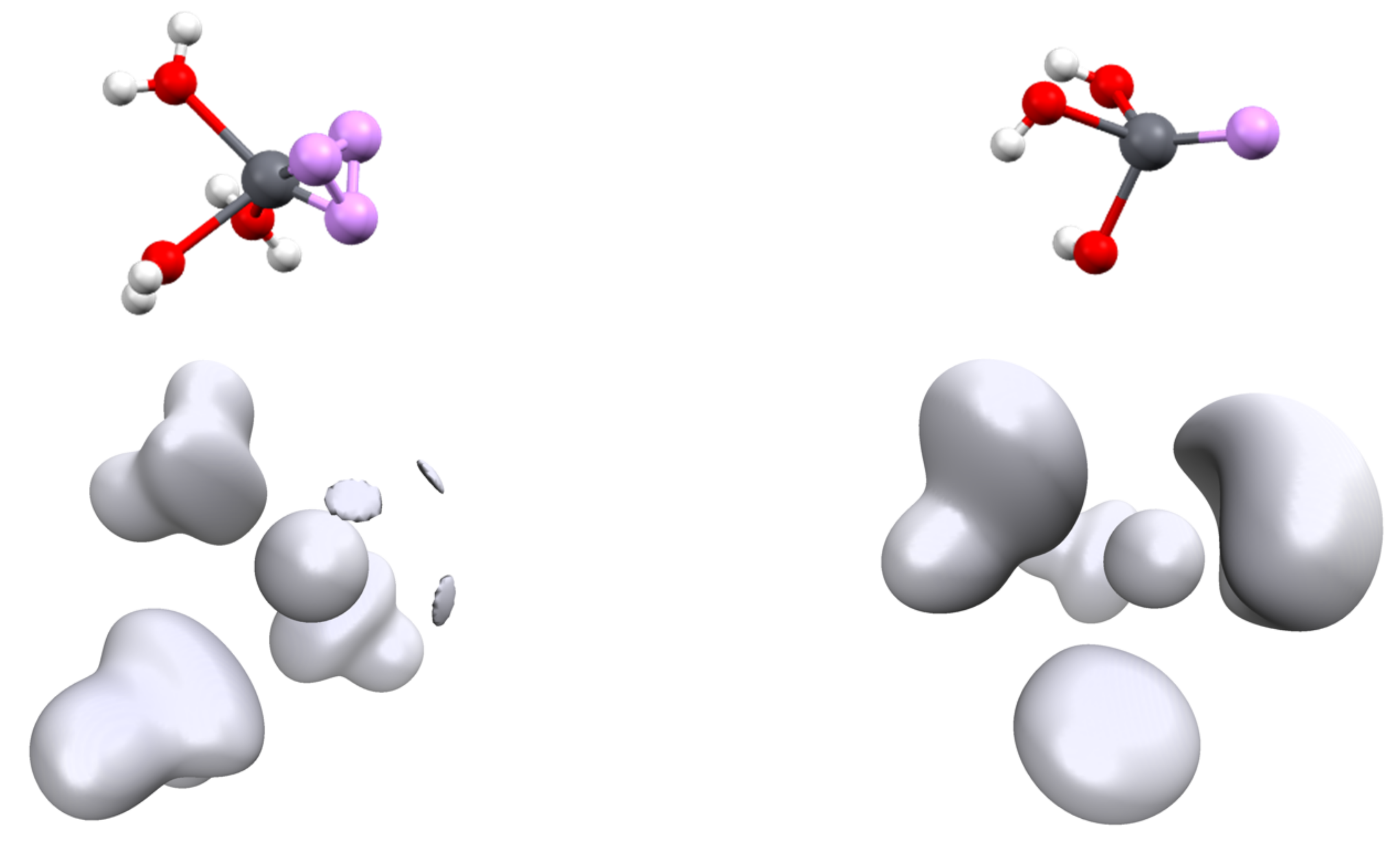
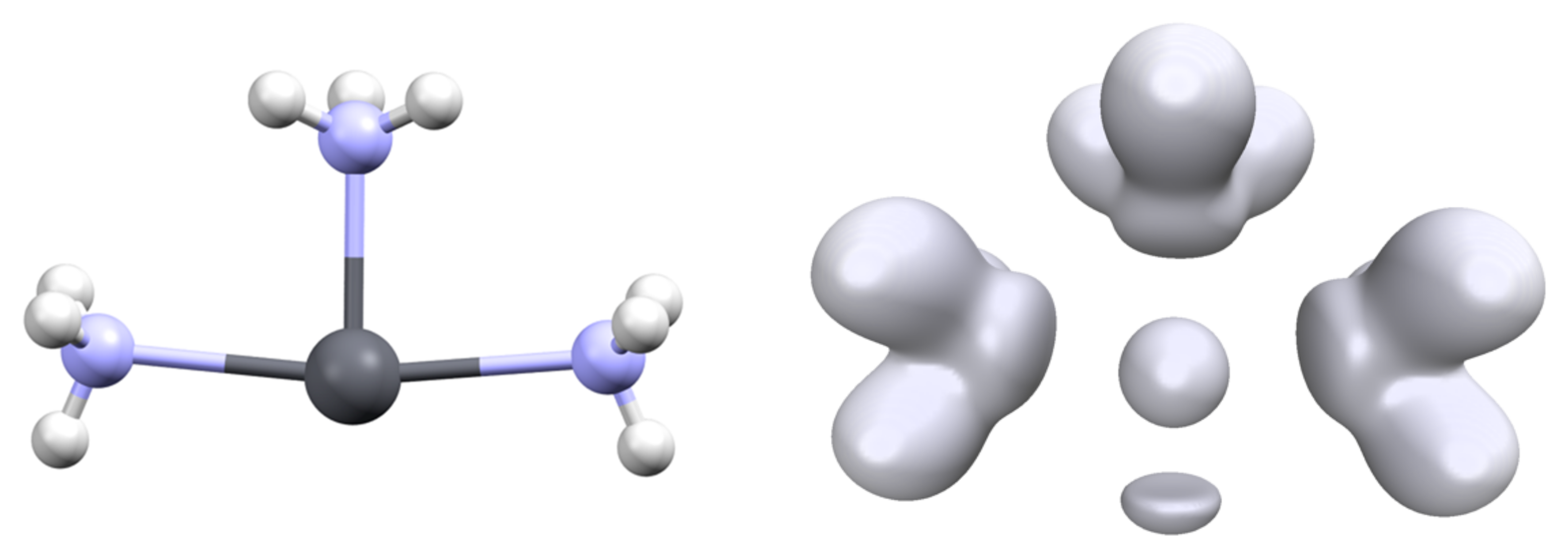
3.1. Structures and Properties of Lead Complexes
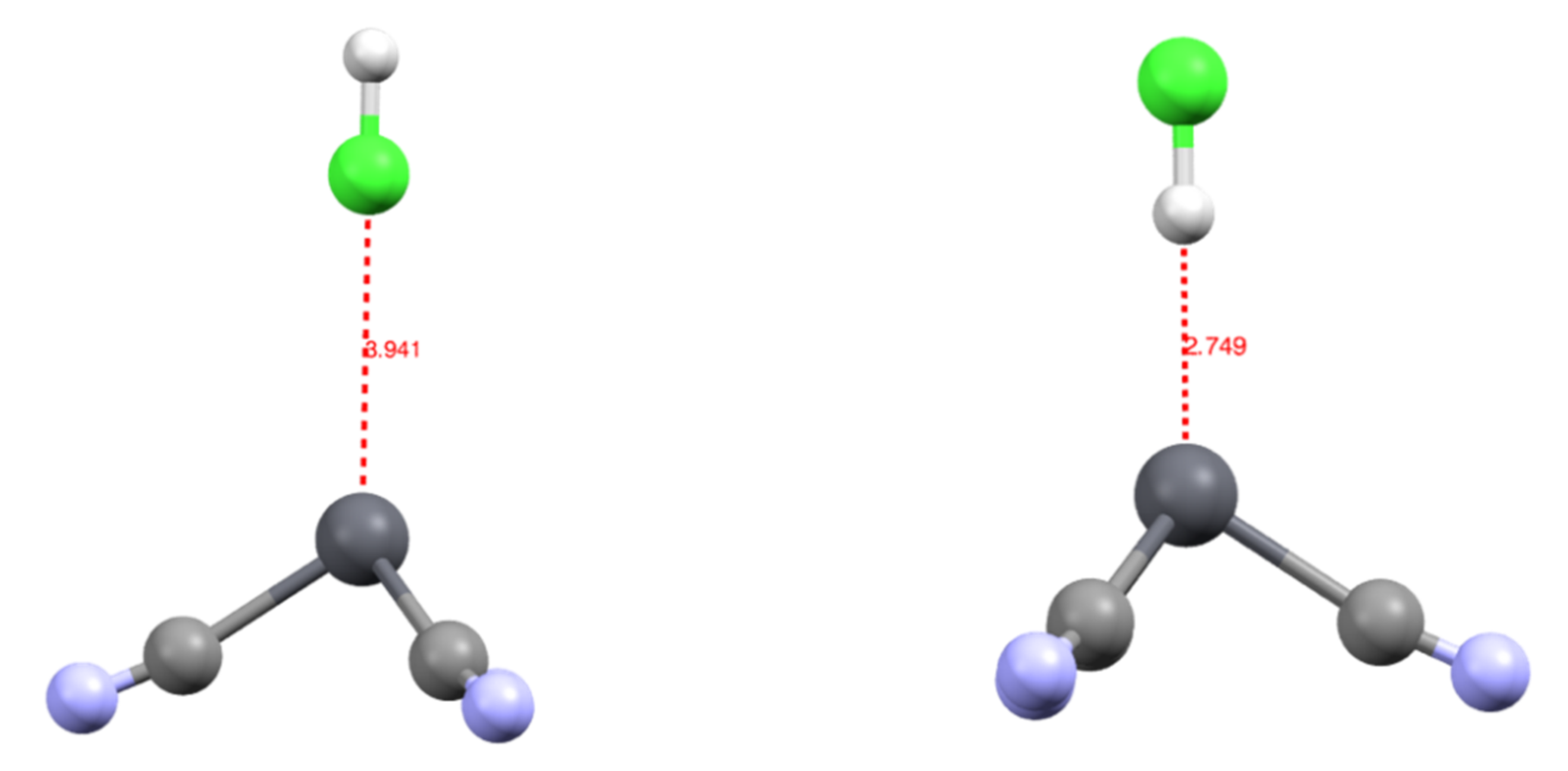
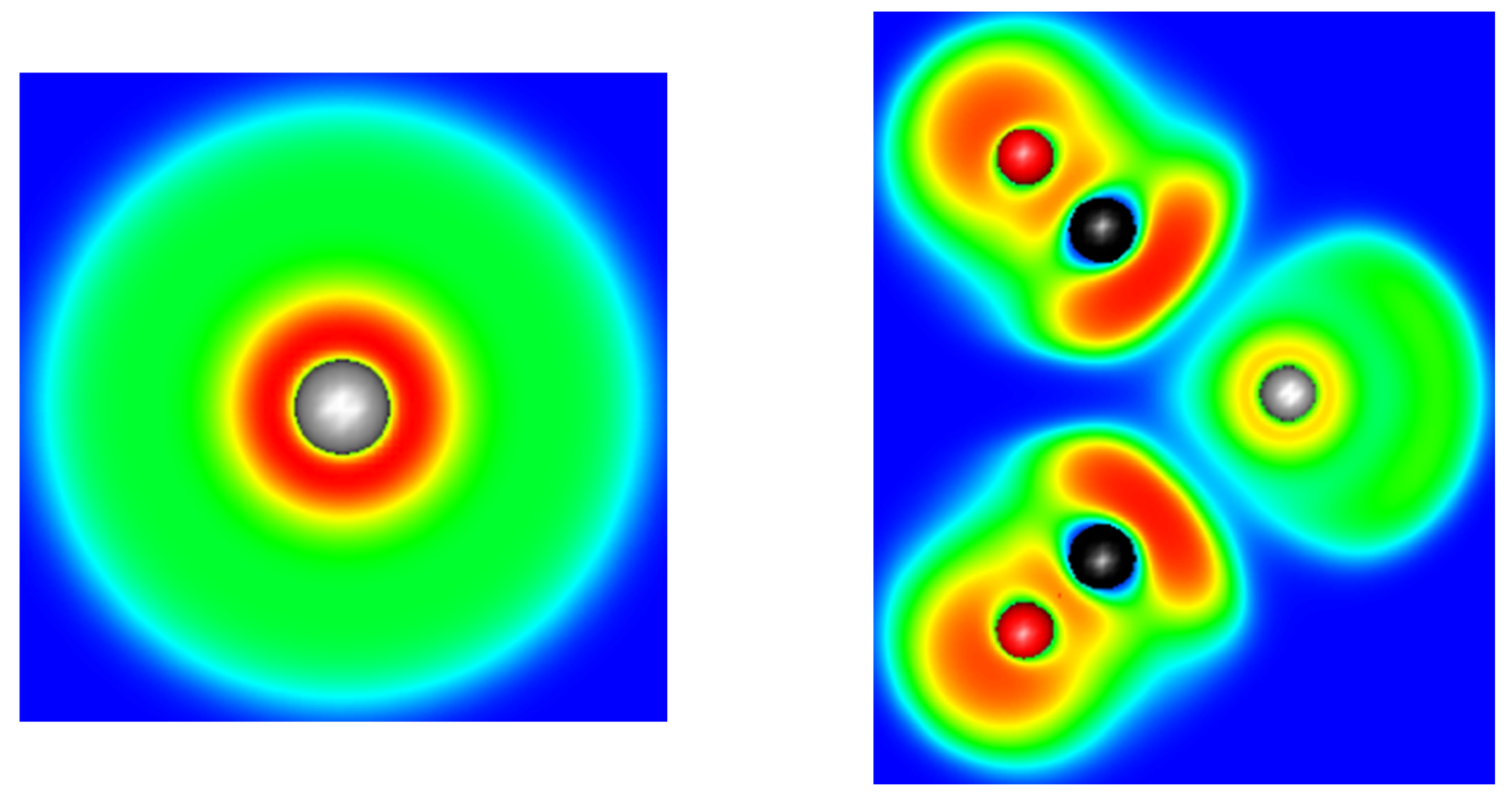
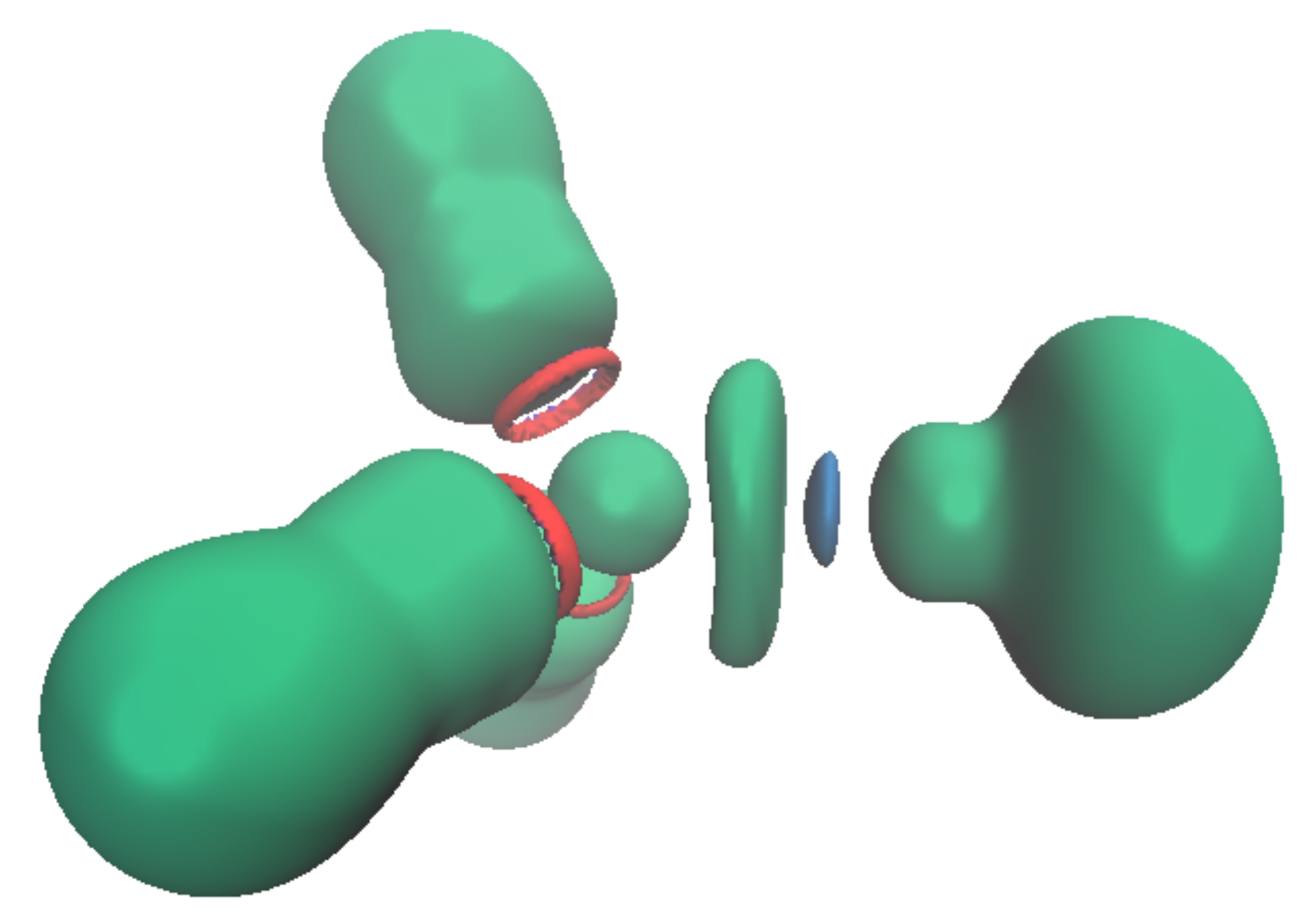
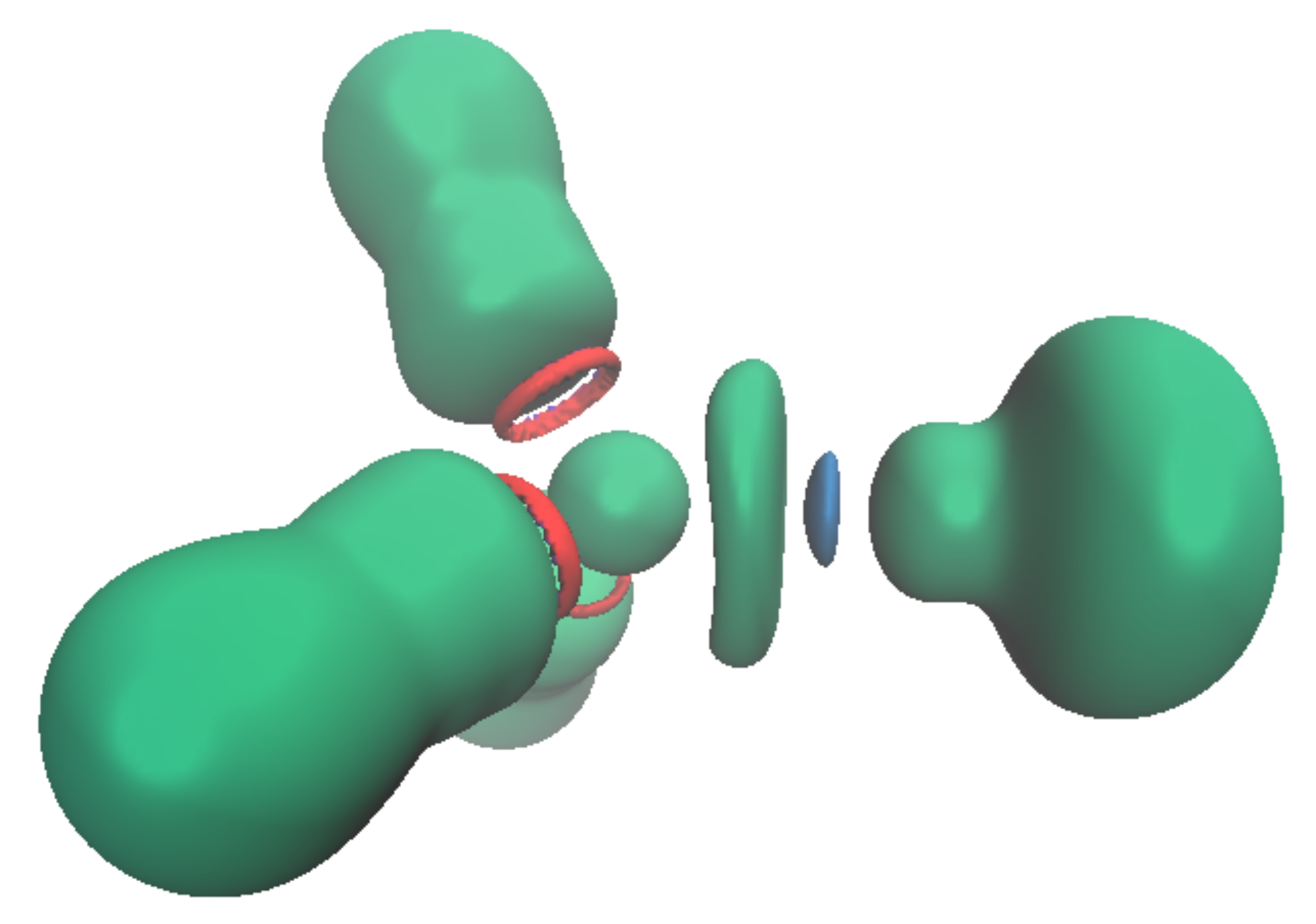
3.2. Topology and Nature of Interactions
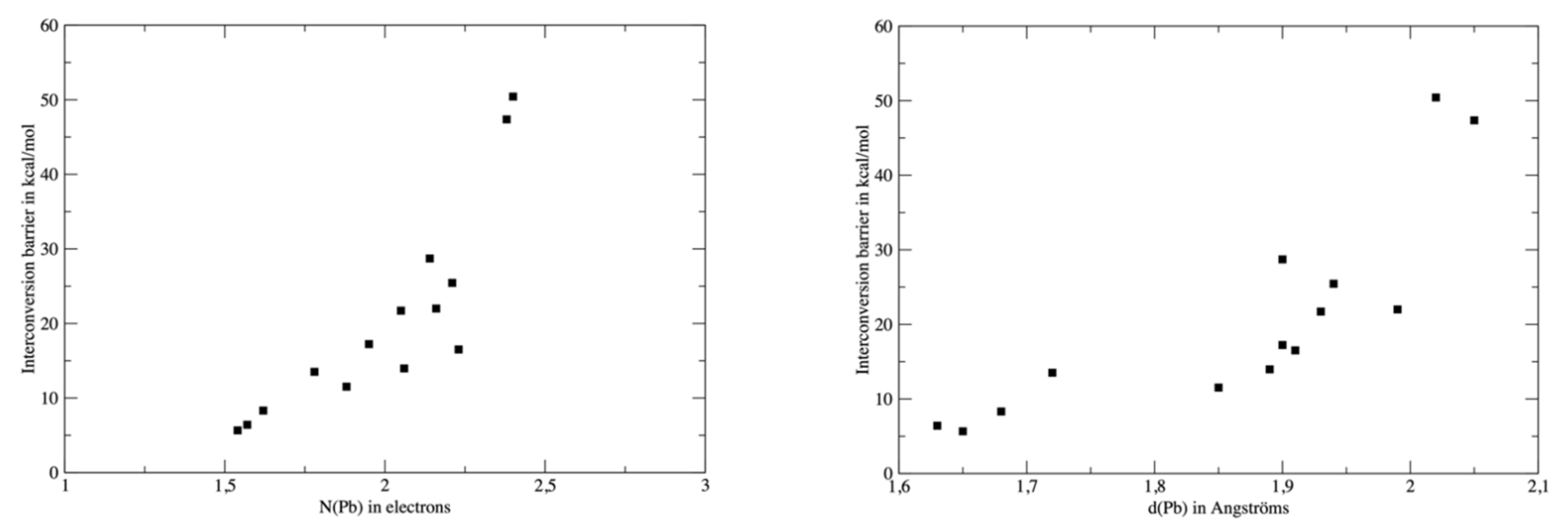
3.3. Interconversion Barriers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Casas, J.S.; Sordo, J. (Eds.) . Lead: Chemistry, Analytical Aspects, Environmental Impact and Health Effects; Elsevier: Amsterdam, The Netherlands, 2011. [Google Scholar]
- Gracia, R.C.; Snodgrass, W.R. Lead toxicity and chelation therapy. Am. J. Health Syst. Pharm. 2007, 64, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Magyar, J.S.; Weng, T.C.; Stern, C.M.; Dye, D.F.; Rous, B.W.; Payne, J.C.; Bridgewater, B.M.; Mijovilovich, A.; Parkin, G.; Zaleski, J.M.; et al. Reexamination of lead (II) coordination preferences in sulfur-rich sites: Implications for a critical mechanism of lead poisoning. J. Am. Chem. Soc. 2005, 127, 9495–9505. [Google Scholar] [CrossRef] [Green Version]
- Khatik, S.K.; Thakur, R.S.; Lead, G.D. The heavy metal in soil water and plant environment. J. Ind. Pollut. Control. 2006, 22, 233. [Google Scholar]
- Mudring, A.-V. Stereochemical activity of lone pairs in heavier main-group element compounds. In Inorganic Chemistry in Focus; Meyer, G., Naumann, D., Wesemann, L., Eds.; Wiley: Hoboken, NJ, USA, 2006; Volume III, p. 15. [Google Scholar]
- Walsh, A.; Watson, G.W. The origin of the stereochemically active Pb (II) lone pair: DFT calculations on PbO and PbS. J. Solid State Chem. 2005, 178, 1422–1428. [Google Scholar] [CrossRef]
- Harrison, P.G. Silicon, Germanium, Tin and Lead. In Comprehensive Coordination Chemistry; Pergamon Press: Oxford, UK, 1987; Volume 3, p. 183. [Google Scholar]
- Parr, J. Germanium, Tin, and Lead. In Comprehensive Coordination Chemistry II.; Parkin, G.F., Ed.; Elsevier: Amsterdam, The Netherlands, 2003; Volume 3, p. 545. [Google Scholar]
- Claudio, E.S.; Goldwin, H.A.; Magyar, J.S. Fundamental coordination chemistry, environmental chemistry, and biochemistry of lead (II). Progr. Inorg. Chem. 2003, 51, 1. [Google Scholar] [CrossRef]
- Davidovich, R.L.; Stavila, V.; Marinin, D.V.; Voit, E.I.; Whitmire, K.H. Stereochemistry of lead(II) complexes with oxygen donor ligands. Coord. Chem. Rev. 2009, 253, 1316–1352. [Google Scholar]
- Shimoni-Livny, L.; Glusker, J.P.; Bock, C.W. Lone Pair Functionality in Divalent Lead Compounds. Inorg. Chem. 1998, 37, 1853–1867. [Google Scholar] [CrossRef]
- Esteban-Gomez, D.; Platas-Iglesias, C.; Enriquez-Perez, T.; Avecilla, F.; de Blas, A.; Rodriguez-Blas, T. Lone-Pair Activity in Lead(II) Complexes with Unsymmetrical Lariat Ethers. Inorg. Chem. 2006, 45, 5407–5416. [Google Scholar] [CrossRef] [PubMed]
- Gourlaouen, C.; Parisel, O. Is an electronic shield at the molecular origin of lead poisoning? A computational modeling experiment. Angew. Chem. Int. Ed. 2007, 46, 553–556. [Google Scholar] [CrossRef]
- Van Severen, M.-C.; Gourlaouen, C.; Parisel, O. Application of the topological analysis of the electronic localization function to archetypical [Pb(II)Ln]p complexes: The bonding of Pb2+ revisited. J. Comput. Chem. 2009, 31, 185–194. [Google Scholar] [CrossRef] [PubMed]
- TURBOMOLE V7.0 2015. A Development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, Since 2007. Available online: http://www.turbomole.com (accessed on 20 December 2021).
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Reed, A.E.; Curtis, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Glendening, E.D.; Reed, A.E.; Carpenter, J.E.; Weinhold, F. NBO Version 3.1.; Gaussian, Inc.: Pittsburgh, PA, USA, 2003. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Noury, S.; Krokidis, X.; Fuster, F.; Silvi, B. TopMod Package, 1997. This Package is Available on the Web Site of the Laboratoire de Chimie Théorique, Université Pierre et Marie Curie (UMR 7616, CNRS-UPMC). Available online: https://www.lct.jussieu.fr/pagesperso/fuster/TOPMOD/topmod-manual.pdf (accessed on 20 December 2021).
- Noury, S.; Krokidis, X.; Fuster, F.; Silvi, B. Computational tools for the electron localization function topological analysis. Comput. Chem. 1999, 23, 597–604. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Savin, A.; Nesper, R.; Wengert, S.; Fässler, T.F. ELF: The Electron Localization Function. Ang. Chem. Int. Ed. 1997, 36, 1808–1832. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Savin, A.; Silvi, B.; Colonna, F. Topological analysis of the electron localization function applied to delocalized bonds. Can. J. Chem. 1996, 74, 1088–1096. [Google Scholar] [CrossRef]
- Poater, J.; Duran, M.; Solà, M.; Silvi, B. Theoretical Evaluation of Electron Delocalization in Aromatic Molecules by Means of Atoms in Molecules (AIM) and Electron Localization Function (ELF) Topological Approaches. Chem. Rev. 2005, 105, 3911–3947. [Google Scholar] [CrossRef]
- Contreras-Garcia, J.; Martin Pendas, A.; Recio, J.M.; Silvi, B. Computation of Local and Global Properties of the Electron Localization Function Topology in Crystals. J. Chem. Theory Comput. 2009, 5, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Kohout, M.; Savin, A. Influence of core–valence separation of electron localization function. J. Comput. Chem. 1997, 18, 1431–1439. [Google Scholar] [CrossRef]
- Joubert, L.; Silvi, B.; Picard, G. Topological approach in the structural and bonding characterization of lanthanide trihalide molecules. Theor. Chem. Acc. 2000, 104, 109–115. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contreras-Garcia, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.; Yang, W. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theory. Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef]
- Desclaux, J.P. Relativistic Dirac-Fock expectation values for atoms with Z = 1 to Z = 120. Atom. Data Nucl. Tables 1973, 12, 311–406. [Google Scholar] [CrossRef]
- Crabtree, R.H. The Organometallic Chemistry of the Transition Metals, 6th ed.; Wiley: Hoboken, NJ, USA, 2014. [Google Scholar]
- Gourlaouen, C.; Gérard, H.; Piquemal, J.-P.; Parisel, O. Understanding Lead Chemistry from Topological Insights: The Transition between Holo- and Hemidirected Structures within the [Pb(CO)n]2+ Model Series. Chem. Eur. J. 2008, 14, 2730–2743. [Google Scholar] [CrossRef] [PubMed]
- de Courcy, B.; Pedersen, L.G.; Parisel, O.; Gresh, N.; Silvi, B.; Pilmé, J.; Piquemal, J.-P. Understanding selectivity of hard and soft metal cations within biological systems using the subvalence concept. I. Application to blood coagulation: Direct cation-protein electronic effects vs. indirect interactions through water networks. J. Chem. Theory. Comput. 2010, 6, 1048–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hancock, R.; Reibenspies, J.; Maumela, H. Structural Effects of the Lone Pair on Lead(II), and Parallels with the Coordination Geometry of Mercury(II). Does the Lone Pair on Lead(II) Form H-Bonds? Structures of the Lead(II) and Mercury(II) Complexes of the Pendant-Donor Macrocycle DOTAM (1,4,7,10-Tetrakis(carbamoylmethyl)-1,4,7,10-tetraazacyclododecane). Inorg. Chem. 2004, 43, 2981–2987. [Google Scholar]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pb-C | C-Pb-C | 6s | 6p | V(Pb) | |||
|---|---|---|---|---|---|---|---|
| ω(Pb) | N(Pb) | d(Pb) | |||||
| [Pb(CO)]2+ | 2.612 | NC | 1.94 | 0.17 | 165.6 | 1.25 | 1.583 |
| [Pb(CO)2]2+ | 2.627 | 83.1 | 1.93 | 0.35 | 160.6 | 1.42 | 1.587 |
| [Pb(CO)3]2+ | 2.635 | 82.1 | 1.91 | 0.58 | 156.0 | 1.52 | 1.620 |
| [Pb(CO)4]2+ | 2.635 2.777 | 81.5 150.0 | 1.95 | 0.59 | 135.3 | 1.44 | 1.616 |
| [Pb(CO)5]2+ | 2.635 2.796 | 76.3 86.6 152.5 | 1.95 | 0.71 | 106.5 | 1.27 | 1.618 |
| [Pb(CO)6]2+ | 2.845 | 90.0 | 1.98 | 0.77 | |||
| Pb-Cl | Cl-Pb-Cl | 6s | 6p | V(Pb) | |||
|---|---|---|---|---|---|---|---|
| ω(Pb) | N(Pb) | d(Pb) | |||||
| [Pb(Cl)]+ | 2.348 | NC | 1.91 | 0.55 | 216.4 | 1.78 | 1.707 |
| [Pb(Cl2] (C2v) | 2.449 | 99.3 | 1.87 | 0.89 | 213.2 | 1.95 | 1.787 |
| [Pb(Cl2] (D∞h) | 2.566 | 180 | 1.99 | 0.39 | 182.7 | 1.32 | 1.401 |
| [Pb(Cl)3]− (C3v) | 2.560 | 99.8 | 1.84 | 0.96 | 186.6 | 1.94 | 1.837 |
| [Pb(Cl)3]− (D3h) | 2.660 | 120.0 | 1.93 | 0.72 | 79.48 | 0.64 | 1.439 |
| [Pb(Cl)4]2− (Td) | 2.781 | 109.5 | 1.93 | 0.74 | 56.9 | 0.62 | 1.225 |
| Pb-L | L-Pb-L | 6s | 6p | V(Pb) | |||
|---|---|---|---|---|---|---|---|
| ω(Pb) | N(Pb) | d(Pb) | |||||
| [Pb(H)3]− | 1.848 | 91.2 | 1.66 | 2.45 | 286.5 | 2.51 | 1.966 |
| [Pb(Me)3]− | 2.328 | 90.8 | 1.68 | 1.80 | 282.9 | 2.48 | 1.988 |
| [Pb(F)3]− | 2.115 | 97.1 | 1.78 | 0.61 | 210.9 | 2.01 | 1.898 |
| [Pb(Cl)3]− | 2.560 | 99.8 | 1.84 | 0.96 | 186.6 | 1.94 | 1.837 |
| [Pb(Br)3]− | 2.719 | 100.6 | 1.87 | 1.15 | 183.5 | 1.96 | 1.810 |
| [Pb(I)3]− | 2.911 | 100.8 | 1.88 | 1.34 | 182.6 | 2.04 | 1.787 |
| [Pb(CN)3]− | 2.306 | 91.9 | 1.73 | 1.50 | 212.5 | 2.19 | 1.882 |
| [Pb(OH)3]− | 2.176 | 91.8 | 1.78 | 0.96 | 236.7 | 2.13 | 1.935 |
| [Pb(SH)3]− | 2.636 | 89.9 | 1.80 | 1.34 | 214.4 | 2.17 | 1.859 |
| [Pb(HCN)3]2+ | 2.415 | 84.7 | 1.89 | 0.43 | 161.8 | 1.65 | 1.682 |
| [Pb(CO)3]2+ | 2.635 | 82.1 | 1.91 | 0.58 | 156.0 | 1.52 | 1.620 |
| [Pb(OH2)3]2+ | 2.374 | 83.8 | 1.91 | 0.31 | 158.9 | 1.58 | 1.638 |
| [Pb(NH3)3]2+ | 2.451 | 90.0 | 1.89 | 0.61 | 172.9 | 1.82 | 1.690 |
| Complex | Structure | ∆G (B3LYP) | ∆G (ωB97XD) |
|---|---|---|---|
| [Pb(H)3]− | D3h | 53.1 | 50.4 |
| [Pb(Me)3]− | T shape | 44.8 | 47.4 |
| [Pb(CN)3]− | T shape | 27.0 | 28.7 |
| [Pb(OH)3]− | T shape | 23.6 | 25.4 |
| [Pb(NH2)3]− | T shape | 20.7 | 22.0 |
| [Pb(OMe)3]− | T shape | 19.6 | 21.7 |
| [Pb(F)3]− | D3h | 16.0 | 17.2 |
| [Pb(SMe)3]− | T shape | 15.2 | 16.5 |
| [Pb(SH)3]− | T shape | 14.4 | 14.0 |
| [Pb(NH3)3]2+ | T shape | 12.2 | 13.5 |
| [Pb(Cl)3]− | D3h | 10.7 | 11.5 |
| [Pb(HCN)3]2+ | T shape | 7.9 | 8.3 |
| [Pb(CO)3]2+ | T shape | 6.4 | 6.4 |
| [Pb(OH2)3]2+ | T shape | 5.2 | 5.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gourlaouen, C.; Piquemal, J.-P. On the Quantum Chemical Nature of Lead(II) “Lone Pair”. Molecules 2022, 27, 27. https://doi.org/10.3390/molecules27010027
Gourlaouen C, Piquemal J-P. On the Quantum Chemical Nature of Lead(II) “Lone Pair”. Molecules. 2022; 27(1):27. https://doi.org/10.3390/molecules27010027
Chicago/Turabian StyleGourlaouen, Christophe, and Jean-Philip Piquemal. 2022. "On the Quantum Chemical Nature of Lead(II) “Lone Pair”" Molecules 27, no. 1: 27. https://doi.org/10.3390/molecules27010027
APA StyleGourlaouen, C., & Piquemal, J.-P. (2022). On the Quantum Chemical Nature of Lead(II) “Lone Pair”. Molecules, 27(1), 27. https://doi.org/10.3390/molecules27010027