
Design, Synthesis, and Structure–Activity Relationship Study of Potent MAPK11 Inhibitors

 ,
,
Abstract
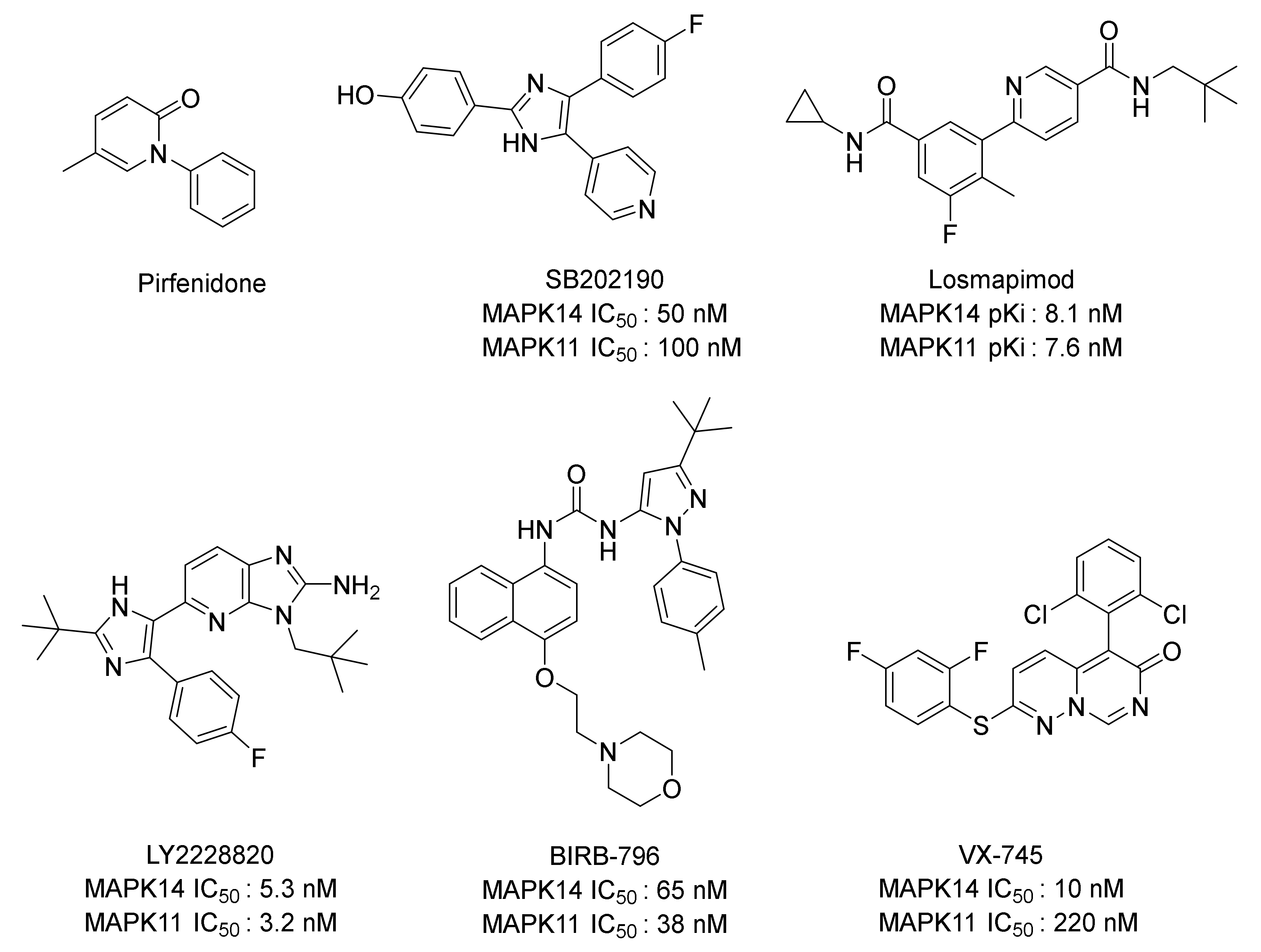
:1. Introduction
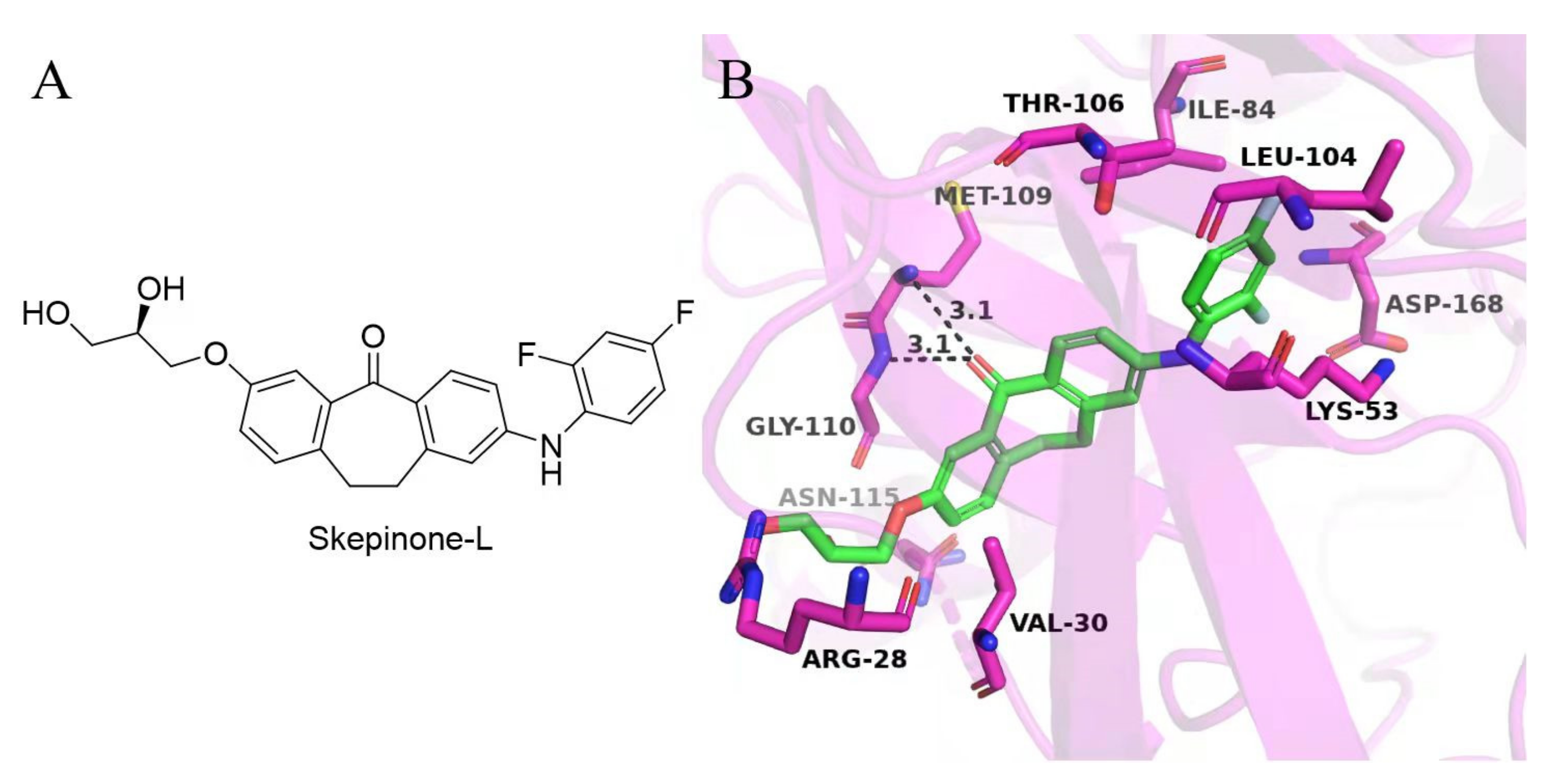
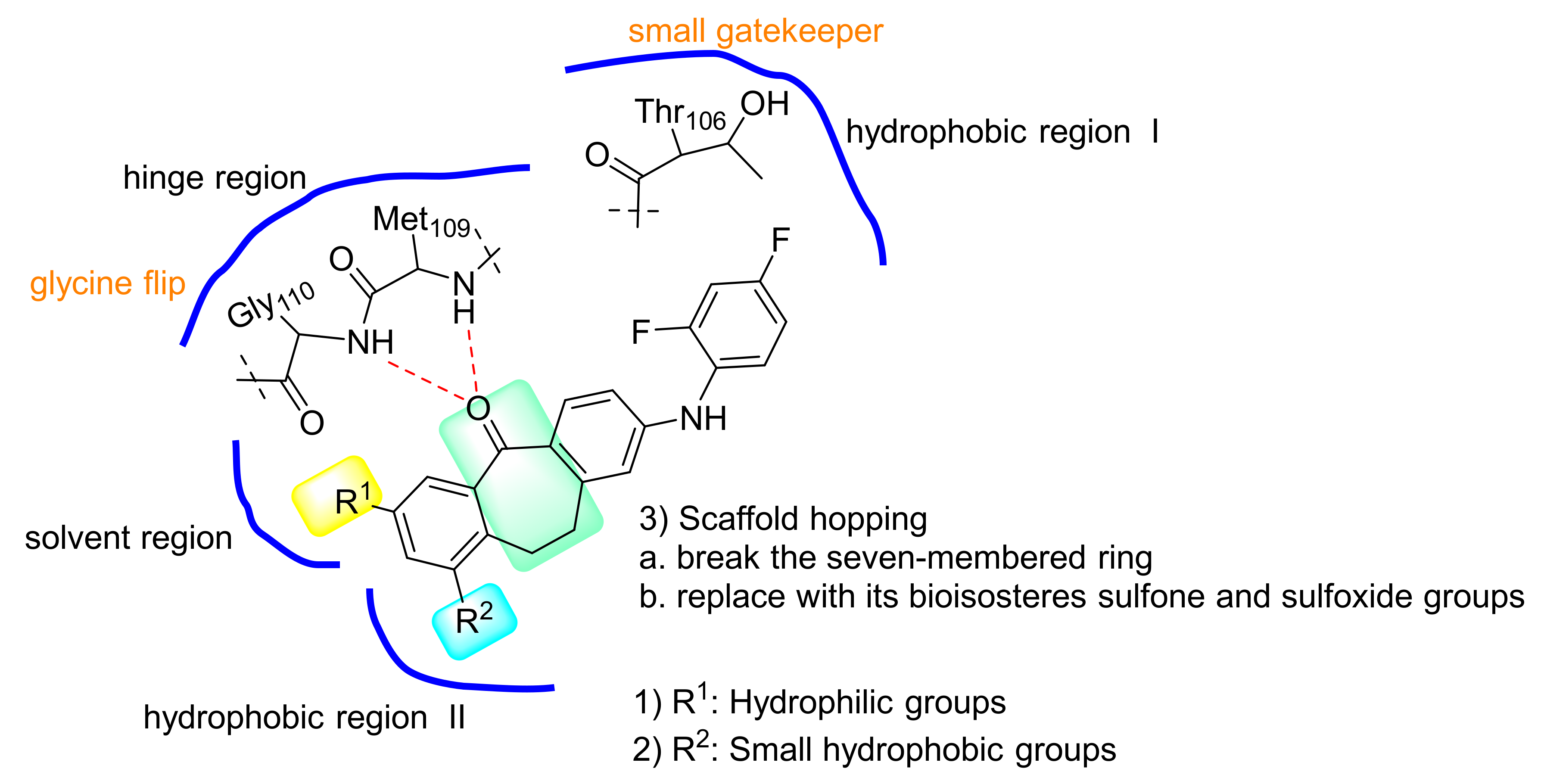
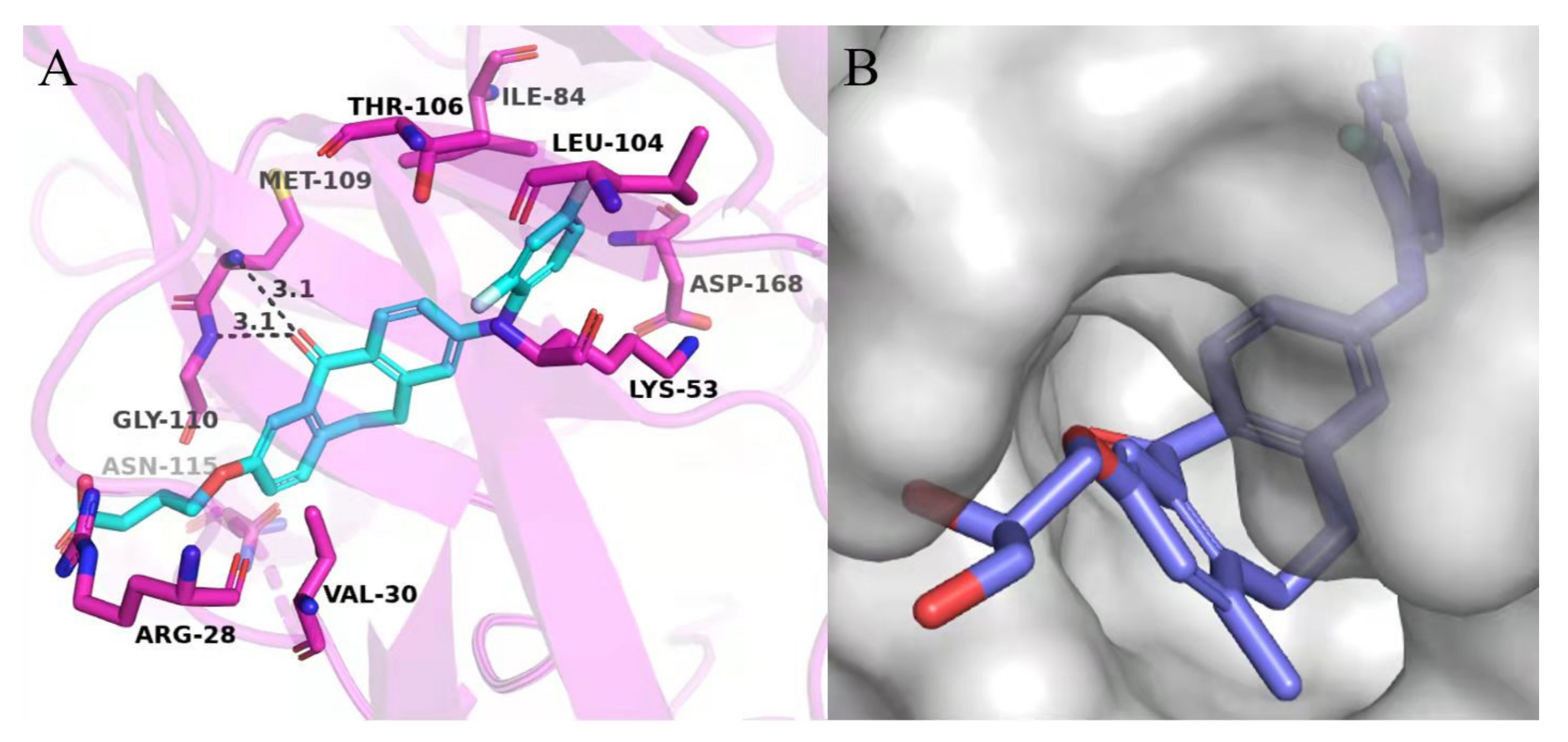
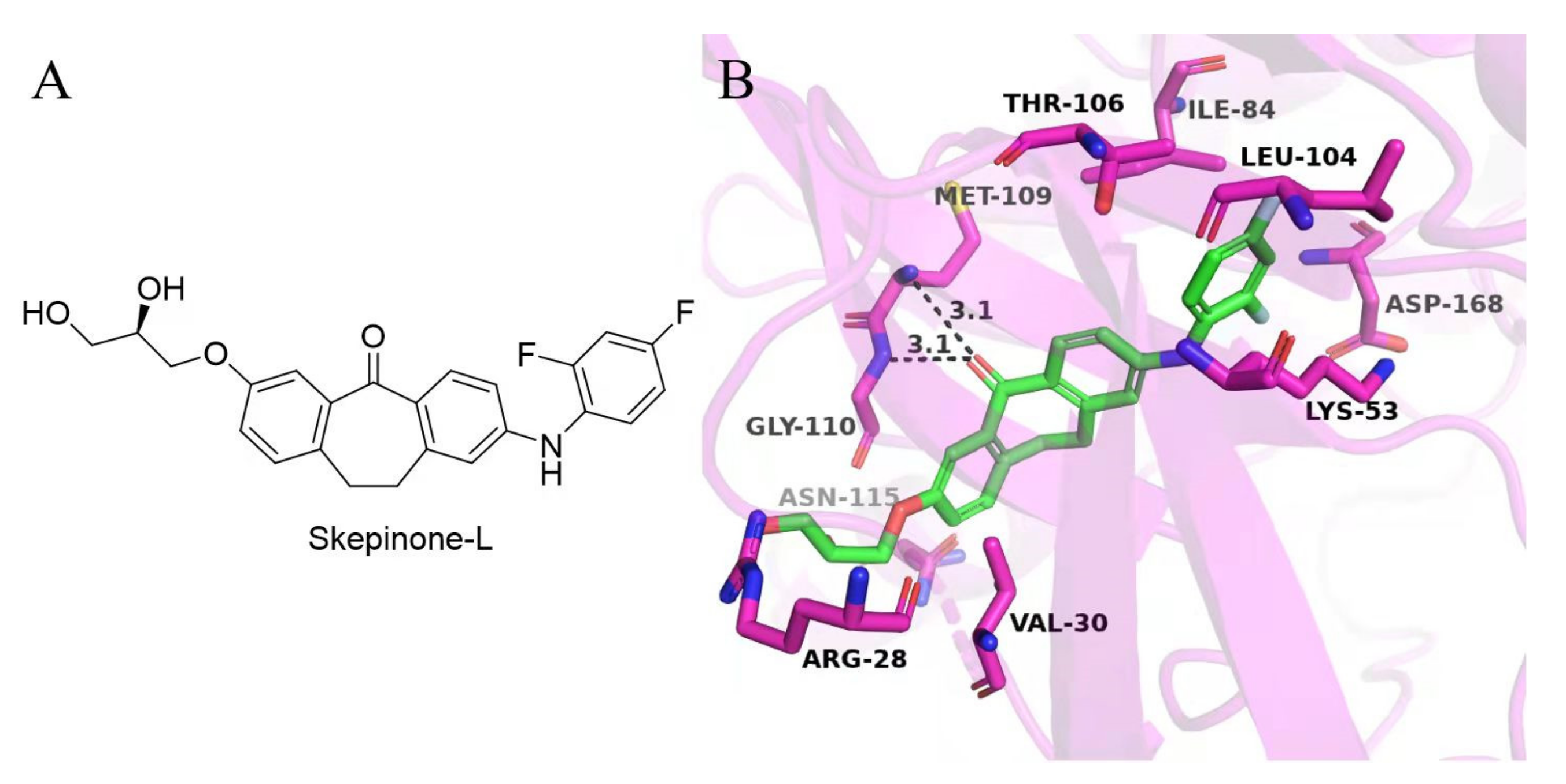
2. Results and Discussion
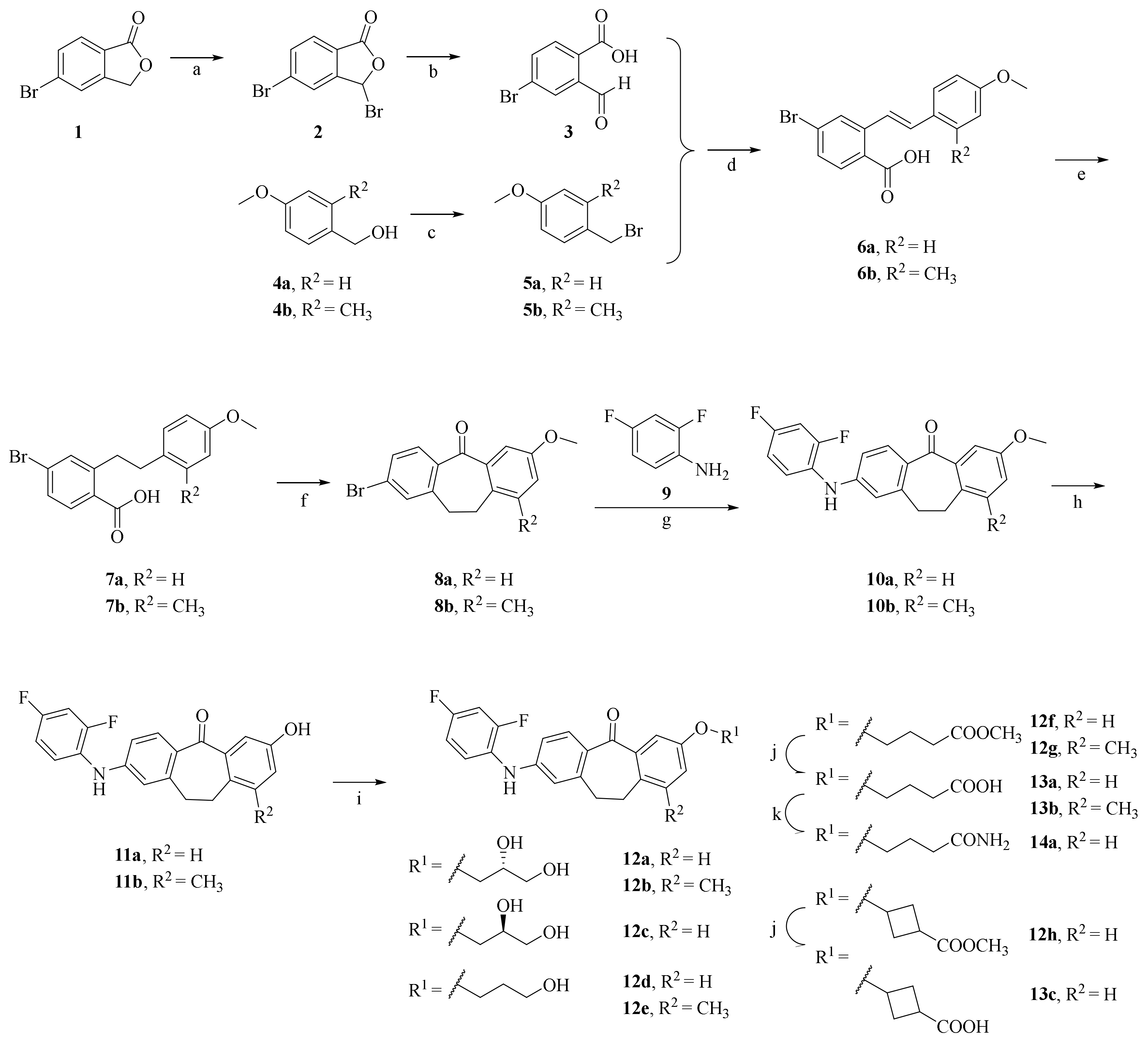
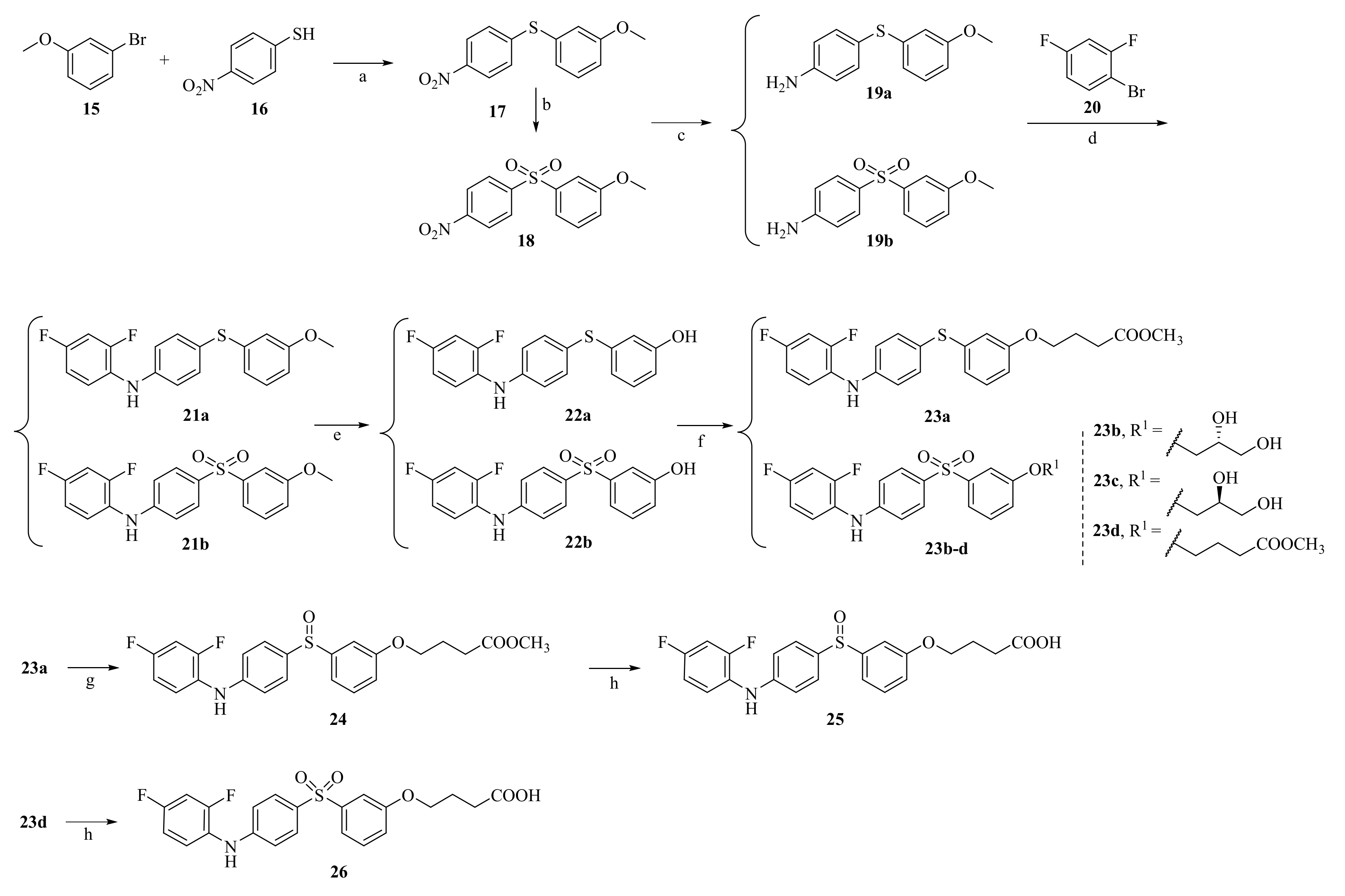
2.1. Synthesis
2.2. Structure–Activity Relationship
3. Materials and Methods
3.1. Chemistry
3.2. Enzyme Assay against MAPK11
3.3. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Sample Availability
References
- Ross, C.A.; Tabrizi, S.J. Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef]
- Wild, E.J.; Tabrizi, S.J. Therapies targeting DNA and RNA in Huntington’s disease. Lancet Neurol. 2017, 16, 837–847. [Google Scholar] [CrossRef] [Green Version]
- Pandey, M.; Rajamma, U. Huntington’s disease: The coming of age. J. Genet. 2018, 97, 649–664. [Google Scholar] [CrossRef] [PubMed]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harper, S.J.; Wilkie, N. MAPKs: New targets for neurodegeneration. Expert Opin. Ther. Targets 2003, 7, 187. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Fu, Y.; Liang, Y.; Song, H.; Yao, Y.; Wu, P.; Yao, Y.; Pan, Y.; Wen, X.; Ma, L.; et al. Suppression of MAPK11 or HIPK3 reduces mutant Huntingtin levels in Huntington’s disease models. Cell Res. 2017, 27, 1441–1465. [Google Scholar] [CrossRef]
- Jiang, Y.; Chen, C.; Li, Z.; Guo, W.; Gegner, J.A.; Lin, S.; Han, J. Characterization of the structure and function of a new mitogen-activated protein kinase (p38beta). J. Biol. Chem. 1996, 271, 17920–17926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, B.; Yang, M.X.; Young, D.B.; Janknecht, R.; Hunter, T.; Murray, B.W.; Barbosa, M.S. p38-2, a novel mitogen-activated protein kinase with distinct properties. J. Biol. Chem. 1997, 272, 19509–19517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, S.B.; Cameron, P.M.; O’Keefe, S.J.; Frantz-Wattley, B.; Thompson, J.; O’Neill, E.A.; Tennis, T.; Liu, L.; Becker, J.W.; Scapin, G. The three-dimensional structure of MAP kinase p38beta: Different features of the ATP-binding site in p38beta compared with p38alpha. Acta Crystallogr. D Biol. Crystallogr. 2009, 65 Pt 8, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Haller, V.; Nahidino, P.; Forster, M.; Laufer, S.A. An updated patent review of p38 MAP kinase inhibitors (2014–2019). Expert Opin. Ther. Pat. 2020, 30, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Muniyappa, H.; Das, K.C. Activation of c-Jun N-terminal kinase (JNK) by widely used specific p38 MAPK inhibitors SB202190 and SB203580: A MLK-3-MKK7-dependent mechanism. Cell. Signal. 2008, 20, 675–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newby, L.K.; Marber, M.S.; Melloni, C.; Sarov-Blat, L.; Aberle, L.H.; Aylward, P.E.; Cai, G.; de Winter, R.J.; Hamm, C.W.; Heitner, J.F.; et al. Losmapimod, a novel p38 mitogen-activated protein kinase inhibitor, in non-ST-segment elevation myocardial infarction: A randomised phase 2 trial. Lancet 2014, 384, 1187–1195. [Google Scholar] [CrossRef]
- Tate, C.M.; Blosser, W.; Wyss, L.; Evans, G.; Xue, Q.; Pan, Y.; Stancato, L. LY2228820 dimesylate, a selective inhibitor of p38 mitogen-activated protein kinase, reduces angiogenic endothelial cord formation in vitro and in vivo. J. Biol. Chem. 2013, 288, 6743–6753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruenbaum, L.M.; Schwartz, R.; Woska, J.R., Jr.; DeLeon, R.P.; Peet, G.W.; Warren, T.C.; Capolino, A.; Mara, L.; Morelock, M.M.; Shrutkowski, A.; et al. Inhibition of pro-inflammatory cytokine production by the dual p38/JNK2 inhibitor BIRB796 correlates with the inhibition of p38 signaling. Biochem. Pharmacol. 2009, 77, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Duffy, J.P.; Harrington, E.M.; Salituro, F.G.; Cochran, J.E.; Green, J.; Gao, H.; Bemis, G.W.; Evindar, G.; Galullo, V.P.; Ford, P.J.; et al. The Discovery of VX-745: A Novel and Selective p38alpha Kinase Inhibitor. ACS Med. Chem. Lett. 2011, 2, 758–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koeberle, S.C.; Romir, J.; Fischer, S.; Koeberle, A.; Schattel, V.; Albrecht, W.; Grutter, C.; Werz, O.; Rauh, D.; Stehle, T.; et al. Skepinone-L is a selective p38 mitogen-activated protein kinase inhibitor. Nat. Chem. Biol. 2011, 8, 141–143. [Google Scholar] [CrossRef] [PubMed]
- Koeberle, S.C.; Fischer, S.; Schollmeyer, D.; Schattel, V.; Grutter, C.; Rauh, D.; Laufer, S.A. Design, synthesis, and biological evaluation of novel disubstituted dibenzosuberones as highly potent and selective inhibitors of p38 mitogen activated protein kinase. J. Med. Chem. 2012, 55, 5868–5877. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
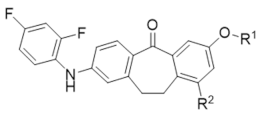 | |||
| Compounds | R1 | R2 | MAPK11 Enzyme Inhibitory Activity IC50/nM |
| 12a (Skepinone-L) | 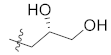 | H | 19.2 ± 0.9 |
| 12c | 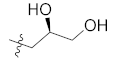 | H | 32.0 ± 0.6 |
| 12d | 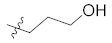 | H | 25.9 ± 0.7 |
| 12f | 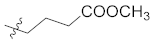 | H | 61.0 ± 2.1 |
| 13a |  | H | 6.4 ± 0.6 |
| 13c | 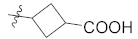 | H | 14.3 ± 0.6 |
| 14a |  | H | 8.4 ± 0.1 |
| 12b | 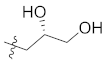 | CH3 | 14.5 ± 0.3 |
| 12e | 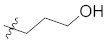 | CH3 | 16.4 ± 0.6 |
| 13b |  | CH3 | 4.2 ± 0.1 |
| SB202190 b | 34.2 ± 0.8 | ||
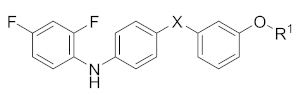 | |||
| Compounds | X | R1 | MAPK11 Enzyme Inhibitory Activity IC50/nM |
| 23b | 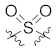 |  | >10,000 |
| 23c | 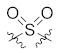 | 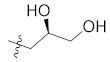 | >10,000 |
| 26 | 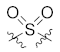 | 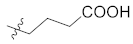 | >10,000 |
| 25 | 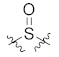 |  | 2547.3 ± 682.1 |
| 12a (Skepinone-L) | 19.2 ± 0.9 | ||
| SB202190 b | 34.2 ± 0.8 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gong, M.; Tu, M.; Sun, H.; Li, L.; Zhu, L.; Li, H.; Zhao, Z.; Li, S. Design, Synthesis, and Structure–Activity Relationship Study of Potent MAPK11 Inhibitors. Molecules 2022, 27, 203. https://doi.org/10.3390/molecules27010203
Gong M, Tu M, Sun H, Li L, Zhu L, Li H, Zhao Z, Li S. Design, Synthesis, and Structure–Activity Relationship Study of Potent MAPK11 Inhibitors. Molecules. 2022; 27(1):203. https://doi.org/10.3390/molecules27010203
Chicago/Turabian StyleGong, Mengdie, Mingyan Tu, Hongxia Sun, Lu Li, Lili Zhu, Honglin Li, Zhenjiang Zhao, and Shiliang Li. 2022. "Design, Synthesis, and Structure–Activity Relationship Study of Potent MAPK11 Inhibitors" Molecules 27, no. 1: 203. https://doi.org/10.3390/molecules27010203
APA StyleGong, M., Tu, M., Sun, H., Li, L., Zhu, L., Li, H., Zhao, Z., & Li, S. (2022). Design, Synthesis, and Structure–Activity Relationship Study of Potent MAPK11 Inhibitors. Molecules, 27(1), 203. https://doi.org/10.3390/molecules27010203