
Cannabinoids in Breast Cancer: Differential Susceptibility According to Subtype

 and
and
Abstract
1. Cannabinoids: Overview
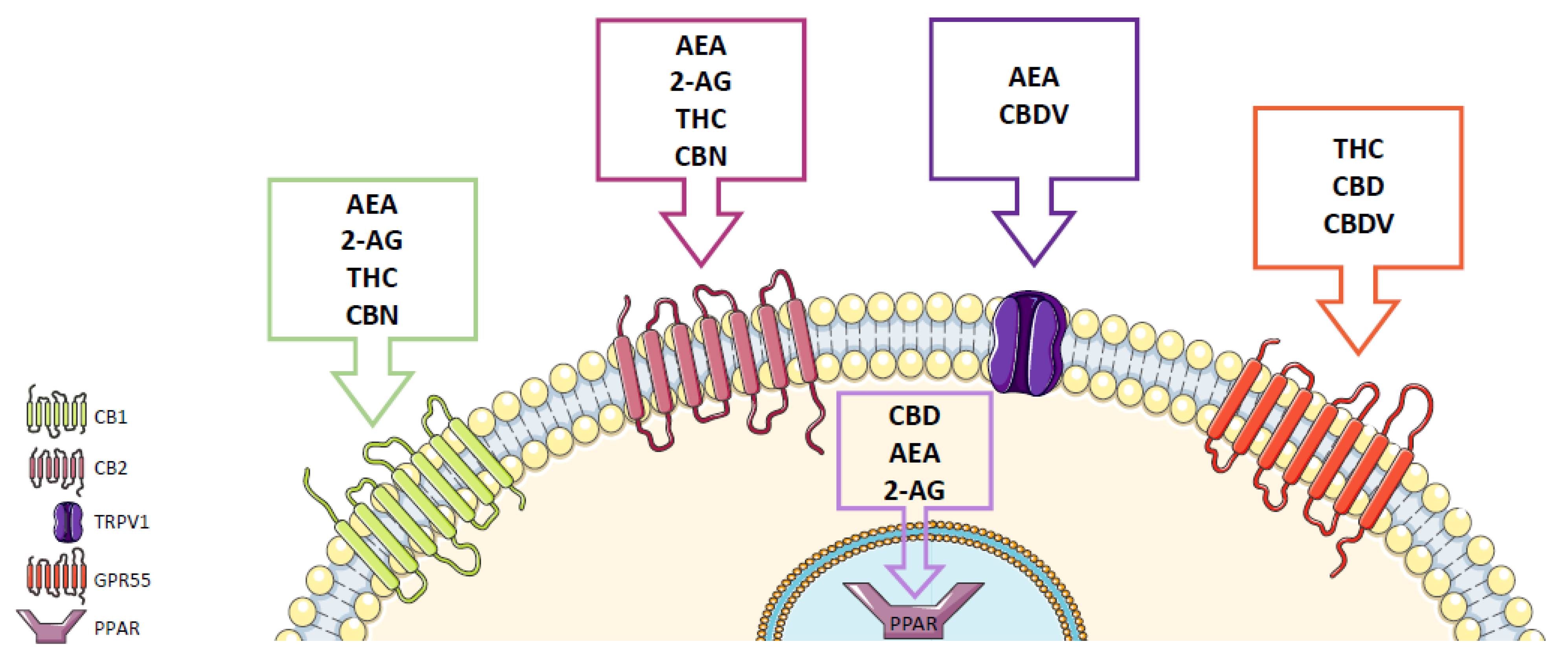
2. Cannabinoid Receptors
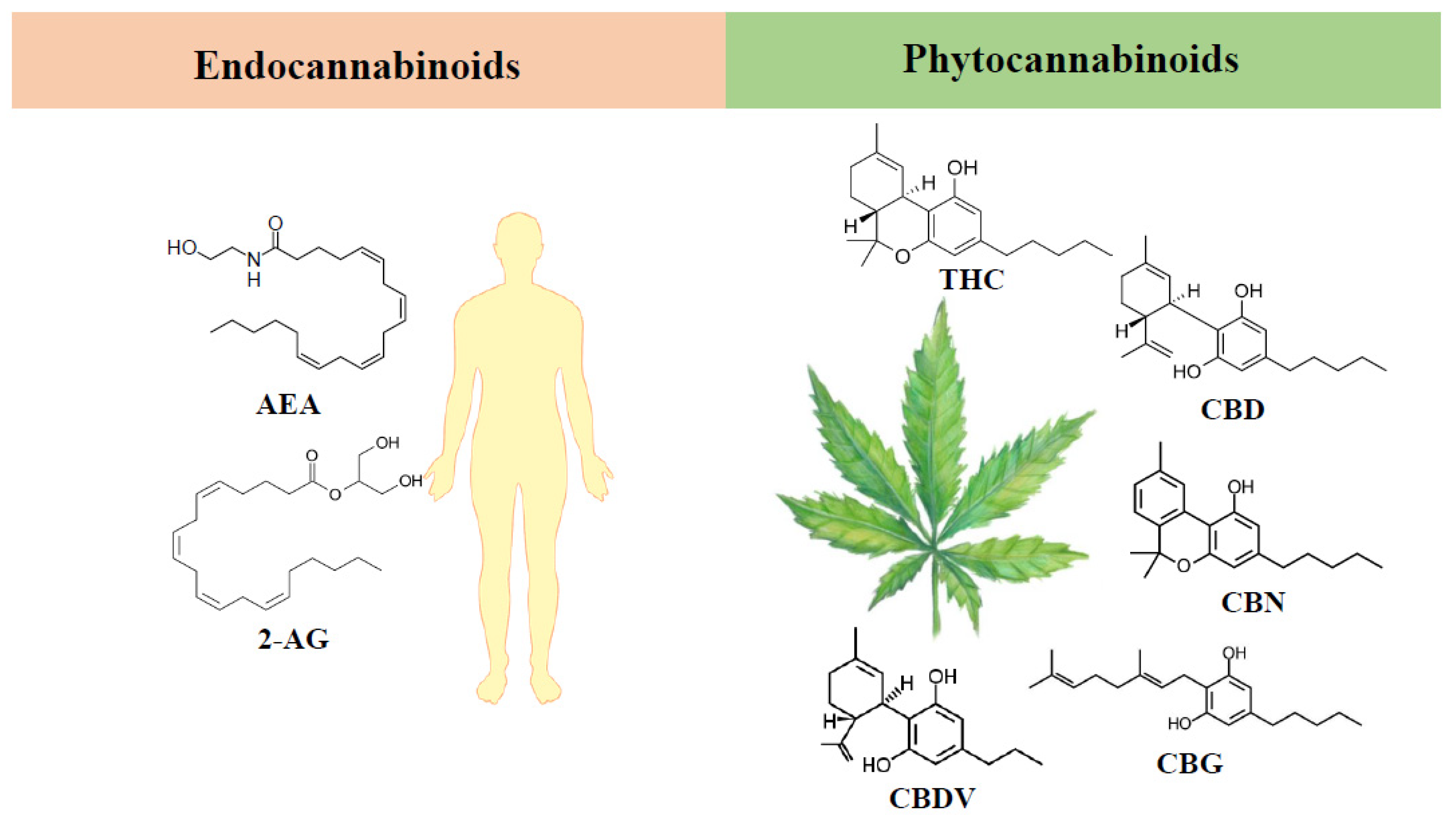
3. Endocannabinoid System
4. Phytocannabinoids
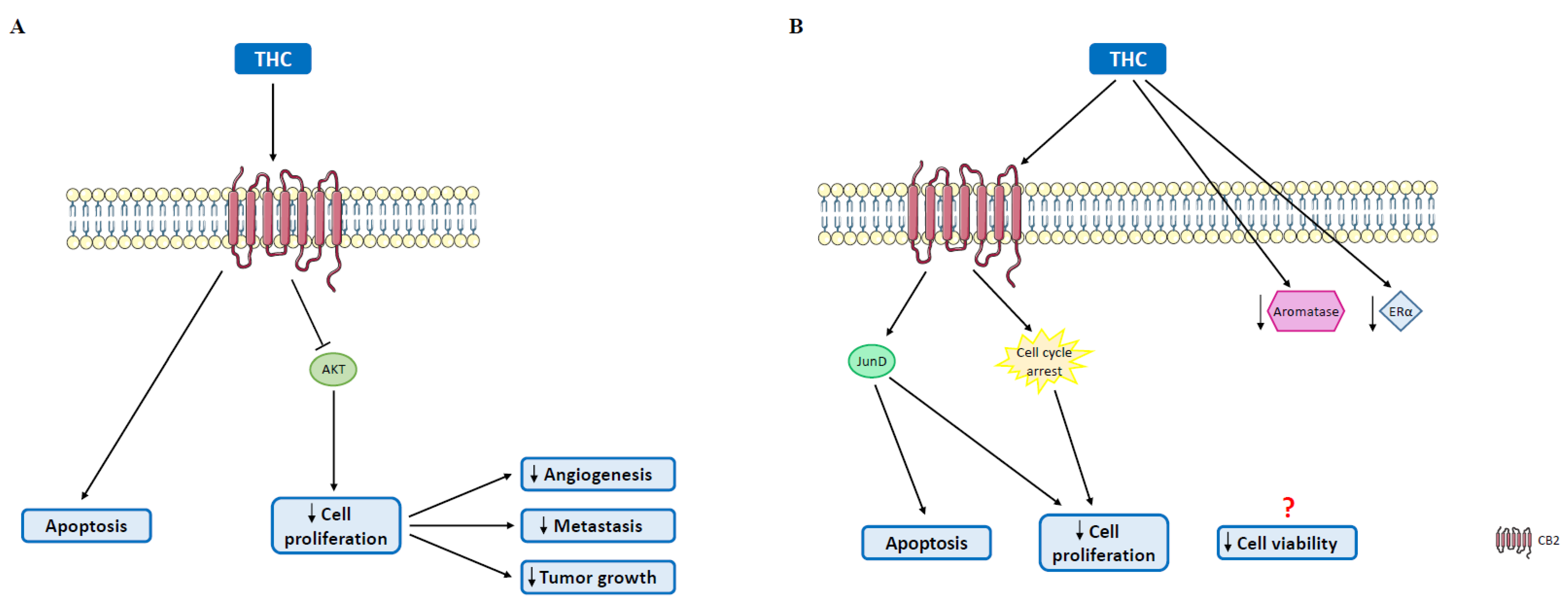
4.1. Δ9-Tetrahydrocannabinol
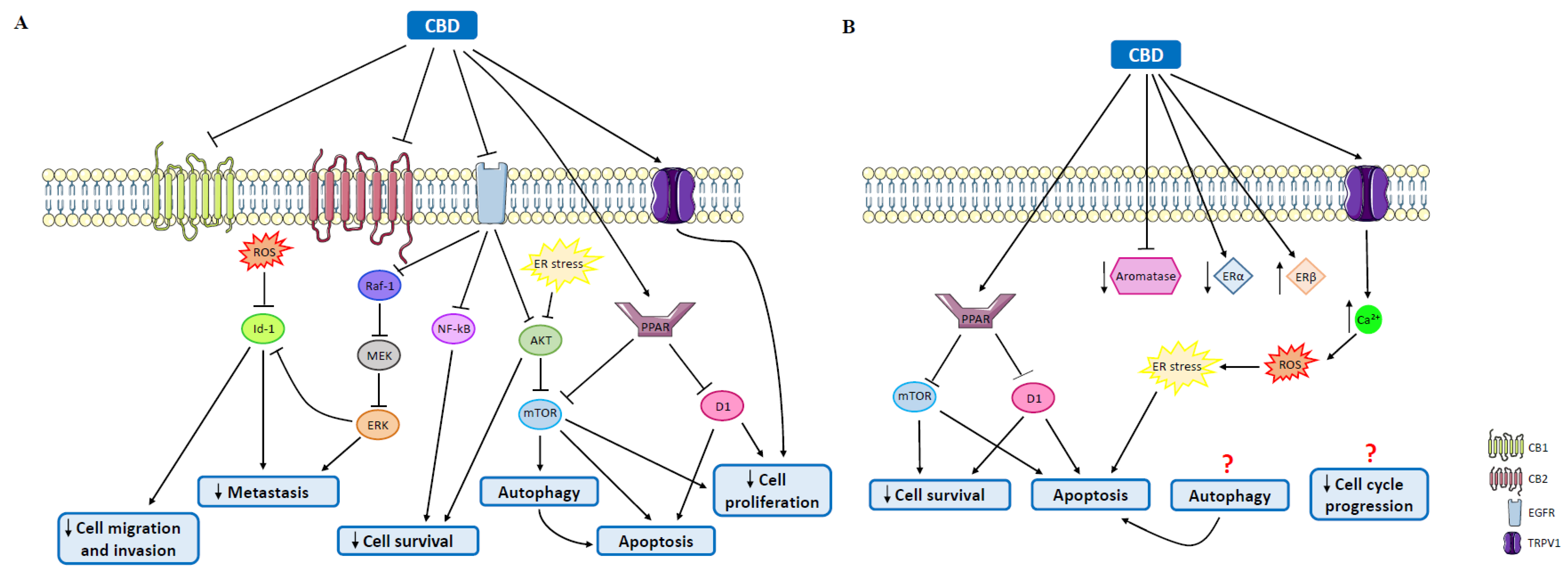
4.2. Cannabidiol
4.3. Minor Phytocannabinoids
5. Cannabinoids and Breast Cancer
5.1. Cannabinoids in Triple-Negative Breast Cancer
5.2. Cannabinoids in Human Epidermal Growth Factor Receptor 2-Positive Tumors
5.3. Cannabinoids in Luminal A Tumors
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 2-AG | 2-arachidonoylglycerol |
| 2-AGE | 2-arachidonoylglyceryl ether |
| ABHD6 | alpha beta hydrolase domain-6 |
| ABHD12 | alpha beta hydrolase domain-12 |
| AC | adenylyl cyclase |
| AEA | anandamide |
| AIs | aromatase inhibitors |
| AKT | protein kinase B |
| cAMP | cyclic adenosine monophosphate |
| CB1 | cannabinoid receptor 1 |
| CB2 | cannabinoid receptor 2 |
| CBs | cannabinoid receptors |
| CBD | cannabidiol |
| CBDA | cannabidiolic acid |
| CBDV | cannabidivarin |
| CBG | cannabigerol |
| CBN | cannabinol |
| CNS | central nervous system |
| COX-2 | cyclooxygenase-2 |
| D1 | cyclin D1 |
| DAGL | diacylglycerol lipase |
| ECS | endocannabinoid system |
| EGFR | epidermal growth factor receptor |
| EMT | endocannabinoid membrane transporter |
| ER | estrogen receptor |
| Erα | estrogen receptor α |
| Erβ | estrogen receptor β |
| ER stress | endoplasmic reticulum stress |
| ERK | extracellular signal-related kinase |
| FAAH | fatty acid amide hydrolase |
| FAK | focal adhesion kinase |
| GPR55 | G protein-coupled receptor 55 |
| HER2 | human epidermal growth factor receptor 2 |
| HER2+ | human epidermal growth factor receptor 2 positive |
| JunD | proto-oncogene JunD |
| LOXs | lipoxygenases |
| MAGL | monoacylglycerol lipase |
| MAPKs | mitogen-activated protein kinases |
| MEK | mitogen-activated protein kinase kinase |
| mTOR | mammalian target of rapamycin |
| NADA | N-arachidonoyldopamine |
| NAGly | N-arachidonoyl glycine |
| NAPE-PLD | N-acylphosphatidylethanolamine-phospholipase D |
| NF-kB | nuclear factor kB |
| NGF | nerve growth factor |
| ODA | oleamide |
| PKA | protein kinase A |
| PPARs | peroxisome proliferator-activated receptors |
| PR | progesterone receptor |
| Raf-1 | proto-oncogene Raf-1 |
| ROS | reactive oxygen species |
| THC | Δ9-tetrahydrocannabinol |
| THCA | Δ9-tetrahydrocannabinolic acid |
| TNBC | triple-negative breast cancer |
| TRP | transient receptor potential cation channels |
| TRPV1 | transient receptor potential vanilloid 1 |
| VEGF | vascular endothelial growth factor |
References
- Klumpers, L.; Thacker, D.L. A brief background on cannabis: From plant to medical indications. J. AOAC Int. 2019, 102, 412–420. [Google Scholar] [CrossRef]
- Pertwee, R.G. Cannabinoid pharmacology: The first 66 years. Br. J. Pharmacol. 2006, 147, S163–S171. [Google Scholar] [CrossRef]
- De Meijer, E.P.M.; Pertweem, R.G. Handbook of Cannabis. Handbooks in Psychopharmacology; Oxford University Press: Cambridge, UK, 2014. [Google Scholar]
- Robson, P.J. Therapeutic potential of cannabinoid medicines. Drug Test. Anal. 2014, 6, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, B.M.; Costa, M.; Almada, M.; Correia-Da-Silva, G.; Teixeira, N. Endogenous cannabinoids revisited: A biochemistry perspective. Prostaglandins Other Lipid Mediat. 2013, 102-103, 13–30. [Google Scholar] [CrossRef]
- Grimaldi, C.; Capasso, A. The endocannabinoid system in the cancer therapy: An overview. Curr. Med. Chem. 2011, 18, 1575–1583. [Google Scholar] [CrossRef]
- Bramness, J.G.; Dom, G.; Gual, A.; Mann, K.; Wurst, F.M. A survey on the medical use of cannabis in Europe: A position paper. Eur. Addict. Res. 2018, 24, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Chianese, G.; Taglialatela-Scafati, O. Cannabinoids: Occurrence and medicinal chemistry. Curr. Med. Chem. 2011, 18, 1085–1099. [Google Scholar] [CrossRef]
- Gould, J. The cannabis crop. Nat. Cell Biol. 2015, 525, S2–S3. [Google Scholar] [CrossRef]
- Alves, P.; Amaral, C.; Teixeira, N.; Correia-da-Silva, G. Cannabis sativa: Much more beyond Δ9-tetrahydrocannabinol. Pharmacol. Res. 2020, 157, 104822. [Google Scholar] [CrossRef]
- Maccarrone, M. Missing pieces to the endocannabinoid puzzle. Trends Mol. Med. 2020, 26, 263–272. [Google Scholar] [CrossRef]
- Turgeman, I.; Bar-Sela, G. Cannabis for cancer—Illusion or the tip of an iceberg: A review of the evidence for the use of Cannabis and synthetic cannabinoids in oncology. Expert Opin. Investig. Drugs 2019, 28, 285–296. [Google Scholar] [CrossRef]
- Fraguas-Sánchez, A.I.; Fernández-Carballido, A.; Torres-Suárez, A.I. Phyto-, endo- and synthetic cannabinoids: Promising chemotherapeutic agents in the treatment of breast and prostate carcinomas. Expert Opin. Investig. Drugs 2016, 25, 1311–1323. [Google Scholar] [CrossRef]
- Castaneto, M.S.; Gorelick, D.A.; Desrosiers, N.A.; Hartman, R.L.; Pirard, S.; Huestis, M.A. Synthetic cannabinoids: Epidemiology, pharmacodynamics, and clinical implications. Drug Alcohol Depend. 2014, 144, 12–41. [Google Scholar] [CrossRef]
- ElSohly, M.A.; Radwan, M.M.; Gul, W.; Chandra, S.; Galal, A. Phytochemistry of Cannabis sativa L. In Phytocannabinoids. Progress in the Chemistry of Organic Natural Products; Kinghorn, A., Falk, H., Gibbons, S., Kobayashi, J., Eds.; Springer: Cham, Switzerland, 2017; Volume 103. [Google Scholar] [CrossRef]
- Fonseca, B.M.; Costa, M.A.; Almada, M.; Correia-da-Silva, G.; Teixeira, N.A. Determination and characterization of a cannabinoid receptor in rat brain. Mol. Pharmacol. 1988, 34, 605–613. [Google Scholar]
- Howlett, A.; Fleming, R.M. Cannabinoid inhibition of adenylate cyclase. Pharmacology of the response in neuroblastoma cell membranes. Mol. Pharmacol. 1984, 26, 532–538. [Google Scholar] [PubMed]
- Howlett, A. Cannabinoid inhibition of adenylate cyclase. Biochemistry of the response in neuroblastoma cell membranes. Mol. Pharmacol. 1985, 27, 429–436. [Google Scholar]
- Howlett, A.C. Inhibition of neuroblastoma adenylate cyclase by cannabinoid and nantradol compounds. Life Sci. 1984, 35, 1803–1810. [Google Scholar] [CrossRef]
- McPartland, J.M.; Glass, M. Functional mapping of cannabinoid receptor homologs in mammals, other vertebrates, and invertebrates. Gene 2003, 312, 297–303. [Google Scholar] [CrossRef]
- Sviženska, I.; Dubový, P.; Sulcova, A. Cannabinoid receptors 1 and 2 (CB1 and CB2), their distribution, ligands and functional involvement in nervous system structures—A short review. Pharmacol. Biochem. Behav. 2008, 90, 501–511. [Google Scholar] [CrossRef]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef]
- Caenazzo, L.; Hoehe, M.; Hsieh, W.-T.; Berrottini, W.; Bonner, T.; Gershon, E. HindIII identifies a two allele DNA polymorphism of the human cannabinoid receptor gene (CNR). Nucleic Acids Res. 1991, 19, 4798. [Google Scholar] [CrossRef][Green Version]
- Hoehe, M.R.; Caenazzo, L.; Martinez, M.M.; Hsieh, W.T.; Modi, W.S.; Gershon, E.S.; Bonner, T.I. Genetic and physical mapping of the human cannabinoid receptor gene to chromosome 6q14-q15. New Biol. 1991, 3, 880–885. [Google Scholar]
- Matsuda, L.A.; Lolait, S.J.; Brownstein, M.J.; Young, A.C.; Bonner, T.I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nat. Cell Biol. 1990, 346, 561–564. [Google Scholar] [CrossRef]
- Zou, S.; Kumar, U. Cannabinoid receptors and the endocannabinoid system: Signaling and function in the central nervous system. Int. J. Mol. Sci. 2018, 19, 833. [Google Scholar] [CrossRef]
- Gelfand, E.V.; Cannon, C.P. Rimonabant: A cannabinoid receptor type 1 blocker for management of multiple cardiometabolic risk factors. J. Am. Coll. Cardiol. 2006, 47, 1919–1926. [Google Scholar] [CrossRef] [PubMed]
- Pisanti, S.; Picardi, P.; D’Alessandro, A.; Laezza, C.; Bifulco, M. The endocannabinoid signaling system in cancer. Trends Pharmacol. Sci. 2013, 34, 273–282. [Google Scholar] [CrossRef]
- Pacher, P.; Kunos, G. Modulating the endocannabinoid system in human health and disease-successes and failures. FEBS J. 2013, 280, 1918–1943. [Google Scholar] [CrossRef]
- Pertwee, R.G. Cannabinoid receptors and pain. Prog. Neurobiol. 2001, 63, 569–611. [Google Scholar] [CrossRef]
- Shao, Z.; Yin, J.; Chapman, K.; Grzemska, M.; Clark, L.; Wang, J.; Rosenbaum, D.M. High-resolution crystal structure of the human CB1 cannabinoid receptor. Nat. Cell Biol. 2016, 540, 602–606. [Google Scholar] [CrossRef]
- Li, X.; Hua, T.; Vemuri, K.; Ho, J.-H.; Wu, Y.; Wu, L.; Popov, P.; Benchama, O.; Zvonok, N.; Locke, K.; et al. Crystal structure of the human cannabinoid receptor CB2. Cell 2019, 176, 459–467. [Google Scholar] [CrossRef]
- Sawzdargo, M.; Nguyen, T.; Lee, D.K.; Lynch, K.R.; Cheng, R.; Heng, H.H.Q.; George, S.R.; O’Dowd, B.F. Identification and cloning of three novel human G protein-coupled receptor genes GPR52, & Psi;GPR53 and GPR55: GPR55 is extensively expressed in human brain. Mol. Brain Res. 1999, 64, 193–198. [Google Scholar] [CrossRef]
- Devane, W.A.; Hanus, L.; Breuer, A.; Pertwee, R.G.; Stevenson, L.A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef] [PubMed]
- Mechoulam, R.; Ben-Shabat, S.; Hanus, L.; Ligumsky, M.; Kaminski, N.E.; Schatz, A.R.; Gopher, A.; Almog, S.; Martin, B.R.; Compton, D.R.; et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem. Pharmacol. 1995, 50, 83–90. [Google Scholar] [CrossRef]
- Sugiura, T.; Kondo, S.; Sukagawa, A.; Nakane, S.; Shinoda, A.; Itoh, K.; Yamashita, A.; Waku, K. 2-Arachidonoylgylcerol: A possible endogenous cannabinoid receptor ligand in brain. Biochem. Biophys. Res. Commun. 1995, 215, 89–97. [Google Scholar] [CrossRef]
- Okamoto, Y.; Morishita, J.; Tsuboi, K.; Tonai, T.; Ueda, N. Molecular characterization of a phospholipase D generating anandamide and Its congeners. J. Biol. Chem. 2004, 279, 5298–5305. [Google Scholar] [CrossRef]
- Battista, N.; Di Tommaso, M.; Bari, M.; Maccarrone, M. The endocannabinoid system: An overview. Front. Behav. Neurosci. 2012, 6, 9. [Google Scholar] [CrossRef] [PubMed]
- Glass, M.; Northup, J.K. Agonist selective regulation of G proteins by cannabinoid CB1and CB2Receptors. Mol. Pharmacol. 1999, 56, 1362–1369. [Google Scholar] [CrossRef]
- Pertwee, R.G.; Howlett, A.C.; Abood, M.E.; Alexander, S.P.H.; Di Marzo, V.; Elphick, M.R.; Greasley, P.J.; Hansen, H.S.; Kunos, K.; Mechoulam, R.; et al. International union of basic and clinical pharmacology. LXXIX. Cannabinoid receptors and their ligands: Beyond CB1 and CB2. Pharmacol. Rev. 2010, 62, 588–631. [Google Scholar] [CrossRef]
- Felder, C.C.; Joyce, K.E.; Briley, E.M.; Mansouri, J.; Mackie, K.; Blond, O.; Lai, Y.; Ma, A.L.; Mitchell, R.L. Comparison of the pharmacology and signal transduction of the human cannabinoid CB1 and CB2 receptors. Mol. Pharmacol. 1995, 48, 443–450. [Google Scholar]
- Zygmunt, P.M.; Petersson, J.; Andersson, D.; Chuang, H.-H.; Sørgård, M.; Di Marzo, V.; Julius, D.; Högestätt, E.D. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nat. Cell Biol. 1999, 400, 452–457. [Google Scholar] [CrossRef]
- O’Sullivan, S.E. Cannabinoids go nuclear: Evidence for activation of peroxisome proliferator-activated receptors. Br. J. Pharmacol. 2007, 152, 576–582. [Google Scholar] [CrossRef]
- Deutsch, D.G.; Chin, S.A. Enzymatic synthesis and degradation of anandamide, a cannabinoid receptor agonist. Biochem. Pharmacol. 1993, 46, 791–796. [Google Scholar] [CrossRef]
- Sugiura, T.; Kishimoto, S.; Oka, S.; Gokoh, M. Biochemistry, pharmacology and physiology of 2-arachidonoylglycerol, an endogenous cannabinoid receptor ligand. Prog. Lipid Res. 2006, 45, 405–446. [Google Scholar] [CrossRef]
- Prescott, S.M.; Majerus, P.W. Characterization of 1,2-diacylglycerol hydrolysis in human platelets. Demonstration of an arachidonoyl-monoacylglycerol intermediate. J. Biol. Chem. 1983, 258, 764–769. [Google Scholar] [CrossRef]
- Sugiura, T.; Waku, K. 2-Arachidonoylglycerol and the cannabinoid receptors. Chem. Phys. Lipids 2000, 108, 89–106. [Google Scholar] [CrossRef]
- Childers, S.R.; Breivogel, C.S. Cannabis and endogenous cannabinoid systems. Drug Alcohol Depend. 1998, 51, 173–187. [Google Scholar] [CrossRef]
- Bouaboula, M.; Hilairet, S.; Marchand, J.; Fajas, L.; Le Fur, G.; Casellas, P. Anandamide induced PPARγ transcriptional activation and 3T3-L1 preadipocyte differentiation. Eur. J. Pharmacol. 2005, 517, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Chen, X.; Zhang, J.; Chen, C. Inhibition of COX-2 expression by endocannabinoid 2-arachidonoylglycerol is mediated via PPAR-γ. Br. J. Pharmacol. 2011, 163, 1533–1549. [Google Scholar] [CrossRef]
- Maia, J.; Fonseca, B.M.; Cunha, S.C.; Braga, J.; Gonçalves, D.; Teixeira, N.; Correia-Da-Silva, G. Impact of tetrahydrocannabinol on the endocannabinoid 2-arachidonoylglycerol metabolism: ABHD6 and ABHD12 as novel players in human placenta. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2020, 1865, 158807. [Google Scholar] [CrossRef] [PubMed]
- Hanus, L.; Abu-Lafi, S.; Fride, E.; Breuer, A.; Vogel, Z.; Shalev, D.E.; Kustanovich, I.; Mechoulam, R. 2-Arachidonyl glyceryl ether, an endogenous agonist of the cannabinoid CB1 receptor. Proc. Natl. Acad. Sci. USA 2001, 98, 3662–3665. [Google Scholar] [CrossRef]
- Porter, A.C.; Sauer, J.-M.; Knierman, M.; Becker, G.W.; Berna, M.J.; Bao, J.; Nomikos, G.G.; Carter, P.; Bymaster, F.P.; Leese, A.B.; et al. Characterization of a Novel Endocannabinoid, Virodhamine, with Antagonist Activity at the CB1 Receptor. J. Pharmacol. Exp. Ther. 2002, 301, 1020–1024. [Google Scholar] [CrossRef]
- Huang, S.M.; Bisogno, T.; Trevisani, M.; Al-Hayani, A.; De Petrocellis, L.; Fezza, F.; Tognetto, M.; Petros, T.; Krey, J.F.; Chu, C.J.; et al. An endogenous capsaicin-like substance with high potency at recombinant and native vanilloid VR1 receptors. Proc. Natl. Acad. Sci. USA 2002, 99, 8400–8405. [Google Scholar] [CrossRef]
- Sheskin, T.; Hanuš, L.; Slager, J.; Vogel, A.Z.; Mechoulam, R. Structural requirements for binding of anandamide-type compounds to the brain cannabinoid receptor. J. Med. Chem. 1997, 40, 659–667. [Google Scholar] [CrossRef]
- Leggett, J.D.; Aspley, S.; Beckett, S.R.G.; D’Antona, A.M.; Kendall, D.A. Oleamide is a selective endogenous agonist of rat and human CB1 cannabinoid receptors. Br. J. Pharmacol. 2004, 141, 253–262. [Google Scholar] [CrossRef]
- Hillard, C.J. Circulating endocannabinoids: From whence do they Come and where are they going? Neuropsychopharmacology 2018, 43, 155–172. [Google Scholar] [CrossRef]
- Cecconi, S.; Rapino, C.; Di Nisio, V.; Rossi, G.; Maccarrone, M. The (endo)cannabinoid signaling in female reproduction: What are the latest advances? Prog. Lipid Res. 2019, 77, 101019. [Google Scholar] [CrossRef] [PubMed]
- Velasco, G.; Sánchez, C.; Guzmán, M. Towards the use of cannabinoids as antitumour agents. Nat. Rev. Cancer 2012, 12, 436–444. [Google Scholar] [CrossRef]
- Gaoni, Y.; Mechoulam, R. Isolation, structure, and partial synthesis of an active constituent of hashish. J. Am. Chem. Soc. 1964, 86, 1646–1647. [Google Scholar] [CrossRef]
- Papaseit, E.; Pérez-Mañá, C.; Pérez-Acevedo, A.P.; Hladun, O.; Torres-Moreno, M.C.; Muga, R.; Torrens, M.; Farré, M. Cannabinoids: From pot to lab. Int. J. Med. Sci. 2018, 15, 1286–1295. [Google Scholar] [CrossRef]
- Pertwee, R.G. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: Δ9-tetrahydrocannabinol, cannabidiol and Δ9-tetrahydrocannabivarin. Br. J. Pharmacol. 2008, 153, 199–215. [Google Scholar] [CrossRef] [PubMed]
- Metna-Laurent, M.; Mondésir, M.; Grel, A.; Vallée, M.; Piazza, P.-V. Cannabinoid-induced tetrad in mice. Curr. Protoc. Neurosci. 2017, 80, 9–59. [Google Scholar] [CrossRef]
- Russo, E.B. Taming THC: Potential cannabis synergy and phytocannabinoid-terpenoid entourage effects. Br. J. Pharmacol. 2011, 163, 1344–1364. [Google Scholar] [CrossRef]
- Carroll, C.B.; Zeissler, M.L.; Hanemann, C.O.; Zajicek, J.P. Δ9-tetrahydrocannabinol (Δ9-THC) exerts a direct neuroprotective effect in a human cell culture model of Parkinson’s disease. Neuropathol. Appl. Neurobiol. 2012, 38, 535–547. [Google Scholar] [CrossRef]
- Shang, V.C.; Kendall, D.A.; Roberts, R.E. Δ9-Tetrahydrocannabinol reverses TNFα-induced increase in airway epithelial cell permeability through CB2 receptors. Biochem. Pharmacol. 2016, 120, 63–71. [Google Scholar] [CrossRef]
- Compton, W.M.; Grant, B.F.; Colliver, J.D.; Glantz, M.D.; Stinson, F.S. Prevalence of marijuana use disorders in the United States: 1991–1992 and 2001–2002. JAMA Psychiatry 2004, 291, 2114–2121. [Google Scholar] [CrossRef]
- Adams, R.; Pease, D.C.; Clark, J.H. Isolation of cannabinol, cannabidiol and quebrachitol from red oil of minnesota wild hemp. J. Am. Chem. Soc. 1940, 62, 2194–2196. [Google Scholar] [CrossRef]
- Mechoulam, R.; Shvo, Y. Hashish—I: The structure of cannabidiol. Tetrahedron 1963, 19, 2073–2078. [Google Scholar] [CrossRef]
- Černe, K. Toxicological properties of Δ9-tetrahydrocannabinol and cannabidiol. Arch. Ind. Hyg. Toxicol. 2020, 71, 1–11. [Google Scholar] [CrossRef]
- LaPrairie, R.B.; Bagher, A.M.; Kelly, M.E.M.; Denovanwright, E.M. Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. Br. J. Pharmacol. 2015, 172, 4790–4805. [Google Scholar] [CrossRef]
- Martinez-Pinilla, E.; Varani, K.; Reyes-Resina, I.; Angelats, E.; Vincenzi, F.; Ferreiro-Vera, C.; Oyarzabal, J.; Canela, E.I.; Lanciego, J.L.; Nadal, X.; et al. Binding and signaling studies disclose a potential allosteric site for cannabidiol in cannabinoid CB2 receptors. Front. Pharmacol. 2017, 8, 744. [Google Scholar] [CrossRef]
- Pisanti, S.; Malfitano, A.M.; Ciaglia, E.; Lamberti, A.; Ranieri, R.; Cuomo, G.; Abate, M.; Faggiana, G.; Proto, M.C.; Fiore, D.; et al. Cannabidiol: State of the art and new challenges for therapeutic applications. Pharmacol. Ther. 2017, 175, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Atalay, S.; Jarocka-Karpowicz, I.; Skrzydlewska, E. Antioxidative and anti-inflammatory properties of cannabidiol. Antioxidants 2019, 9, 21. [Google Scholar] [CrossRef]
- Izzo, A.A.; Borrelli, F.; Capasso, R.; Di Marzo, V.; Mechoulam, R. Non-psychotropic plant cannabinoids: New therapeutic opportunities from an ancient herb. Trends Pharmacol. Sci. 2009, 30, 515–527. [Google Scholar] [CrossRef]
- Gaoni, Y.; Mechoulam, R. The structure and synthesis of cannabigerol, a new hashish constituent. In Proceedings of the Chemical Society; RSC Publishing: London, UK, 1964; p. 82. [Google Scholar]
- Gaoni, Y.; Mechoulam, R. Isolation and structure of. DELTA.+-tetrahydrocannabinol and other neutral cannabinoids from hashish. J. Am. Chem. Soc. 1971, 93, 217–224. [Google Scholar] [CrossRef]
- Vollner, L.; Bieniek, D.; Korte, F. Hashish. XX. Cannabidivarin, a new hashish constituent. Tetrahedron Lett. 1969, 3, 145–147. [Google Scholar] [CrossRef]
- Hill, A.J.; Mercier, M.S.; Hill, T.D.M.; Glyn, S.E.; Jones, N.A.; Yamasaki, Y.; Futamura, T.; Duncan, M.; Stott, C.G.; Stephens, G.J.; et al. Cannabidivarin is anticonvulsant in mouse and rat. Br. J. Pharmacol. 2012, 167, 1629–1642. [Google Scholar] [CrossRef]
- Rosenthaler, S.; Pöhn, B.; Kolmanz, C.; Huu, C.N.; Krewenka, C.; Huber, A.; Kranner, B.; Rausch, W.-D.; Moldzio, R. Differences in receptor binding affinity of several phytocannabinoids do not explain their effects on neural cell cultures. Neurotoxicology Teratol. 2014, 46, 49–56. [Google Scholar] [CrossRef]
- Hill, T.D.M.; Cascio, M.-G.; Romano, B.; Duncan, M.; Pertwee, R.; Williams, C.; Whalley, B.J.; Hill, A.J. Cannabidivarin-rich cannabis extracts are anticonvulsant in mouse and rat via a CB1receptor-independent mechanism. Br. J. Pharmacol. 2013, 170, 679–692. [Google Scholar] [CrossRef]
- De Petrocellis, L.; Ligresti, A.; Moriello, A.S.; Allarà, M.; Bisogno, T.; Petrosino, S.; Stott, C.G.; Marzo, V.D. Effects of cannabinoids and cannabinoid-enriched Cannabis extracts on TRP channels and endocannabinoid metabolic enzymes. Br. J. Pharmacol. 2011, 163, 1479–1494. [Google Scholar] [CrossRef]
- Laun, A.S.; Shrader, S.; Brown, K.J.; Song, Z.-H. GPR3, GPR6, and GPR12 as novel molecular targets: Their biological functions and interaction with cannabidiol. Acta Pharmacol. Sin. 2019, 40, 300–308. [Google Scholar] [CrossRef]
- Anavi-Goffer, S.; Baillie, G.; Irving, A.J.; Gertsch, J.; Greig, I.R.; Pertwee, R.G.; Ross, R.A. Modulation of l-α-lysophosphatidylinositol/GPR55 mitogen-activated protein kinase (MAPK) signaling by cannabinoids. J. Biol. Chem. 2012, 287, 91–104. [Google Scholar] [CrossRef]
- Baker, D.; Pryce, G.; Giovannoni, G.; Thompson, A.J. The therapeutic potential of cannabis. Lancet Neurol. 2003, 2, 291–298. [Google Scholar] [CrossRef]
- Keating, G.M. Delta-9-tetrahydrocannabinol/cannabidiol oromucosal spray (Sativex®): A review in multiple sclerosis-related spasticity. Drugs 2017, 77, 563–574. [Google Scholar] [CrossRef]
- Urits, I.; Borchart, M.; Hasegawa, M.; Kochanski, J.; Orhurhu, V.; Viswanath, O. An update of current cannabis-based pharmaceuticals in pain medicine. Pain Ther. 2019, 8, 41–51. [Google Scholar] [CrossRef]
- Chen, J.W.; Borgelt, L.M.; Blackmer, A.B. Cannabidiol: A new hope for patients with dravet or Lennox-Gastaut syndromes. Ann. Pharmacother. 2019, 53, 603–611. [Google Scholar] [CrossRef]
- Malfitano, A.M.; Ciaglia, E.; Gangemi, G.; Gazzerro, P.; Laezza, C.; Bifulco, M. Update on the endocannabinoid system as an anticancer target. Expert Opin. Ther. Targets 2011, 15, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, B.M.; Teixeira, N.A.; Correia-Da-Silva, G. Cannabinoids as modulators of cell death: Clinical applications and future directions. Rev. Physiol. Biochem. Pharmacol. 2017, 173, 63–88. [Google Scholar] [CrossRef]
- Caffarel, M.M.; Andradas, C.; Perez-Gomez, E.; Guzmán, M.; Sanchez, C. Cannabinoids: A new hope for breast cancer therapy? Cancer Treat. Rev. 2012, 38, 911–918. [Google Scholar] [CrossRef]
- Munson, A.E.; Harris, L.S.; Friedman, M.A.; Dewey, W.L.; Carchman, R.A. Antineoplastic activity of cannabinoids2. J. Natl. Cancer Inst. 1975, 55, 597–602. [Google Scholar] [CrossRef]
- Lukong, K.E. Understanding breast cancer—The long and winding road. BBA Clin. 2017, 7, 64–77. [Google Scholar] [CrossRef]
- Wu, V.S.; Kanaya, N.; Lo, C.; Mortimer, J.; Chen, S. From bench to bedside: What do we know about hormone receptor-positive and human epidermal growth factor receptor 2-positive breast cancer? J. Steroid Biochem. Mol. Biol. 2015, 153, 45–53. [Google Scholar] [CrossRef]
- Goldhirsch, A.; Wood, W.C.; Coates, A.S.; Gelber, R.S.; Thürlimann, B.; Senn, H.-J. Strategies for subtypes—Dealing with the diversity of breast cancer: Highlights of the St. Gallen international expert consensus on the primary therapy of early breast cancer 2011. Ann. Oncol. 2011, 22, 1736–1747. [Google Scholar] [CrossRef]
- Goldhirsch, A.; Winer, E.P.; Coates, A.S.; Gelber, R.D.; Piccart-Gebhart, M.; Thürlimann, B.; Senn, H.J.; Panel members. personalizing the treatment of women with early breast cancer: Highlights of the St Gallen international expert consensus on the primary therapy of early breast cancer 2013. Ann. Oncol. 2013, 24, 2206–2223. [Google Scholar] [CrossRef]
- Cardoso, F.; Kyriakides, S.; Ohno, S.; Penault-Llorca, F.; Poortmans, P.; Rubio, I.T.; Zackrisson, S.; Senkus, E.; ESMO guidelines committee. Early breast cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2019, 30, 1194–1220. [Google Scholar] [CrossRef]
- Fragomeni, S.M.; Sciallis, A.; Jeruss, J.S. Molecular subtypes and local-regional control of breast cancer. Surg. Oncol. Clin. North Am. 2018, 27, 95–120. [Google Scholar] [CrossRef]
- Tsang, J.Y.S.; Tse, G.M. Molecular classification of breast cancer. Adv. Anat. Pathol. 2020, 27, 27–35. [Google Scholar] [CrossRef]
- Escrivá-De-Romaní, S.; Arumí, M.; Bellet, M.; Saura, C. HER2-positive breast cancer: Current and new therapeutic strategies. Breast 2018, 39, 80–88. [Google Scholar] [CrossRef]
- Society, A.C. Breast Cancer Facts & Figure 2019 and Figure 2020; American Cancer Society, Inc.: Atlanta, GA, USA, 2019. [Google Scholar]
- Johnson, K.S.; Conant, E.F.; Soo, M.S. Molecular subtypes of breast cancer: A review for breast radiologists. J. Breast Imaging 2021, 3, 12–24. [Google Scholar] [CrossRef]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef]
- Śledziński, P.; Zeyland, J.; Słomski, R.; Nowak, A. The current state and future perspectives of cannabinoids in cancer biology. Cancer Med. 2018, 7, 765–775. [Google Scholar] [CrossRef]
- Fraguas-Sánchez, A.I.; Torres-Suárez, A.I. Medical use of cannabinoids. Drugs 2018, 78, 1665–1703. [Google Scholar] [CrossRef] [PubMed]
- Fraguas-Sánchez, A.I.; Martín-Sabroso, C.; Torres-Suárez, A.I. Insights into the effects of the endocannabinoid system in cancer: A review. Br. J. Pharmacol. 2018, 175, 2566–2580. [Google Scholar] [CrossRef] [PubMed]
- Guindon, J.; Hohmann, A.G. The endocannabinoid system and cancer: Therapeutic implication. Br. J. Pharmacol. 2011, 163, 1447–1463. [Google Scholar] [CrossRef]
- Laezza, C.; Pagano, C.; Navarra, G.; Pastorino, O.; Proto, M.C.; Fiore, D.; Piscopo, C.; Gazzerro, P.; Bifulco, M. The endocannabinoid system: A target for cancer treatment. Int. J. Mol. Sci. 2020, 21, 747. [Google Scholar] [CrossRef]
- Bisogno, T.; Hanuš, L.; De Petrocellis, L.; Tchilibon, S.; Ponde, D.E.; Brandi, I.; Moriello, A.S.; Davis, J.B.; Mechoulam, R.; Di Marzo, V. Molecular targets for cannabidiol and its synthetic analogues: Effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br. J. Pharmacol. 2001, 134, 845–852. [Google Scholar] [CrossRef]
- Ligresti, A.; Moriello, A.S.; Starowicz, K.; Matias, I.; Pisanti, S.; De Petrocellis, L.; Laezza, C.; Portella, G.; Bifulco, M.; Di Marzo, V. Antitumor activity of plant cannabinoids with emphasis on the effect of cannabidiol on human breast carcinoma. J. Pharmacol. Exp. Ther. 2006, 318, 1375–1387. [Google Scholar] [CrossRef]
- Shrivastava, A.; Kuzontkoski, P.M.; Groopman, J.E.; Prasad, A. Cannabidiol induces programmed cell death in breast cancer cells by coordinating the cross-talk between apoptosis and autophagy. Mol. Cancer Ther. 2011, 10, 1161–1172. [Google Scholar] [CrossRef]
- Sultan, A.S.; Marie, M.; Sheweita, S.A. Novel mechanism of cannabidiol-induced apoptosis in breast cancer cell lines. Breast 2018, 41, 34–41. [Google Scholar] [CrossRef]
- McKallip, R.J.; Nagarkatti, M.; Nagarkatti, P.S. Delta-9-tetrahydrocannabinol enhances breast cancer growth and metastasis by suppression of the antitumor immune response. J. Immunol. 2005, 174, 3281–3289. [Google Scholar] [CrossRef]
- Elbaz, M.; Nasser, M.W.; Ravi, J.; Wani, N.A.; Ahirwar, D.K.; Zhao, H.; Oghumu, S.; Satoskar, A.R.; Shilo, K.; Carson, W.E.; et al. Modulation of the tumor microenvironment and inhibition of EGF/EGFR pathway: Novel anti-tumor mechanisms of Cannabidiol in breast cancer. Mol. Oncol. 2015, 9, 906–919. [Google Scholar] [CrossRef]
- McAllister, S.D.; Murase, R.; Christian, R.T.; Lau, D.; Zielinski, A.J.; Allison, J.; Almanza, C.; Pakdel, A.; Lee, J.; Limbad, C.; et al. Pathways mediating the effects of cannabidiol on the reduction of breast cancer cell proliferation, invasion, and metastasis. Breast Cancer Res. Treat. 2011, 129, 37–47. [Google Scholar] [CrossRef]
- McAllister, S.D.; Christian, R.T.; Horowitz, M.P.; Garcia, A.; Desprez, P.-Y. Cannabidiol as a novel inhibitor of Id-1 gene expression in aggressive breast cancer cells. Mol. Cancer Ther. 2007, 6, 2921–2927. [Google Scholar] [CrossRef]
- Sarmiento-Salinas, F.L.; Delgado-Magallón, A.; Montes-Alvarado, J.B.; Ramírez-Ramírez, D.; Flores-Alonso, J.C.; Cortés-Hernández, P.; Reyes-Leyva, J.; Herrera-Camacho, I.; Anaya-Ruiz, M.; Pelayo, R.; et al. Breast cancer subtypes present a differential production of reactive oxygen species (ROS) and susceptibility to antioxidant treatment. Front. Oncol. 2019, 9, 480. [Google Scholar] [CrossRef] [PubMed]
- Jezierska-Drutel, A.; Rosenzweig, S.A.; Neumann, C.A. Role of oxidative stress and the microenvironment in breast cancer development and progression. Adv. Cancer Res. 2013, 119, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Lu, P.; Beeraka, N.M.; Sukocheva, O.A.; Madhunapantula, S.V.; Liu, J.; Sinelnikov, M.Y.; Nikolenko, V.N.; Bulygin, K.V.; Mikhaleva, L.M.; et al. Mitochondrial mutations and mitoepigenetics: Focus on regulation of oxidative stress-induced responses in breast cancers. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef]
- Schoeman, R.; Beukes, N.; Frost, C. Cannabinoid combination induces cytoplasmic vacuolation in MCF-7 breast cancer cells. Molecules 2020, 25, 4682. [Google Scholar] [CrossRef]
- Qamri, Z.; Preet, A.; Nasser, M.W.; Bass, C.E.; Leone, G.; Barsky, S.H.; Ganju, R.K. Synthetic cannabinoid receptor agonists inhibit tumor growth and metastasis of breast cancer. Mol. Cancer Ther. 2009, 8, 3117–3129. [Google Scholar] [CrossRef]
- Grimaldi, C.; Pisanti, S.; Laezza, C.; Malfitano, A.M.; Santoro, A.; Vitale, M.; Caruso, M.G.; Notarnicola, M.; Iacuzzo, I.; Portella, G.; et al. Anandamide inhibits adhesion and migration of breast cancer cells. Exp. Cell Res. 2006, 312, 363–373. [Google Scholar] [CrossRef]
- Laezza, C.; Pisanti, S.; Malfitano, A.M.; Bifulco, M. The anandamide analog, Met-F-AEA, controls human breast cancer cell migration via the RHOA/RHO kinase signaling pathway. Endocr-Relat. Cancer 2008, 15, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Nasser, M.W.; Qamri, Z.; Deol, Y.S.; Smith, D.; Shilo, K.; Zou, X.; Ganju, R.K. Crosstalk between chemokine receptor CXCR4 and cannabinoid receptor CB2 in modulating breast cancer growth and invasion. PLoS ONE 2011, 6, e23901. [Google Scholar] [CrossRef]
- Caffarel, M.M.; Andradas, C.; Mira, E.; Pérez-Gómez, E.; Cerutti, C.; Moreno-Bueno, G.; Flores, J.M.; García-Real, I.; Palacios, J.; Mañes, S.; et al. Cannabinoids reduce ErbB2-driven breast cancer progression through Akt inhibition. Mol. Cancer 2010, 9, 196. [Google Scholar] [CrossRef]
- De Petrocellis, L.; Melck, D.; Palmisano, A.; Bisogno, T.; Laezza, C.; Bifulco, M.; Di Marzo, V. The endogenous cannabinoid anandamide inhibits human breast cancer cell proliferation. Proc. Natl. Acad. Sci. USA 1998, 95, 8375–8380. [Google Scholar] [CrossRef]
- Melck, D.; De Petrocellis, L.; Orlando, P.; Bisogno, T.; Laezza, C.; Bifulco, M.; Marzot, V.D. Suppression of nerve growth factor Trk receptors and prolactin receptors by endocannabinoids leads to inhibition of human breast and prostate cancer cell proliferation. Endocrinology 2000, 141, 118–126. [Google Scholar] [CrossRef]
- Melck, D.; Rueda, D.; Galve-Roperh, I.; De Petrocellis, L.; Guzmán, M.; Marzot, V.D. Involvement of the cAMP/protein kinase a pathway and of mitogen-activated protein kinase in the anti-proliferative effects of anandamide in human breast cancer cells. FEBS Lett. 1999, 463, 235–240. [Google Scholar] [CrossRef]
- Caffarel, M.M.; Moreno-Bueno, G.; Cerutti, C.; Palacios, J.; Guzman, M.; Mechta-Grigoriou, F.; Sanchez, C. JunD is involved in the antiproliferative effect of Delta9-tetrahydrocannabinol on human breast cancer cells. Oncogene 2008, 27, 5033–5044. [Google Scholar] [CrossRef]
- Caffarel, M.M.; Sarrió, D.; Palacios, J.; Guzmán, M.; Sánchez, C. Delta9-tetrahydrocannabinol inhibits cell cycle progression in human breast cancer cells through Cdc2 regulation. Cancer Res. 2006, 66, 6615–6621. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Yamaori, S.; Motoya, E.; Matsunaga, T.; Kimura, T.; Yamamoto, I.; Watanabe, K. Delta(9)-Tetrahydrocannabinol enhances MCF-7 cell proliferation via cannabinoid receptor-independent signaling. Toxicology 2008, 245, 141–146. [Google Scholar] [CrossRef]
- Watanabe, K.; Motoya, E.; Matsuzawa, N.; Funahashi, T.; Kimura, T.; Matsunaga, T.; Arizono, K.; Yamamoto, I. Marijuana extracts possess the effects like the endocrine disrupting chemicals. Toxicology 2005, 206, 471–478. [Google Scholar] [CrossRef]
- Takeda, S.; Yamamoto, I.; Watanabe, K. Modulation of Delta9-tetrahydrocannabinol-induced MCF-7 breast cancer cell growth by cyclooxygenase and aromatase. Toxicology 2009, 259, 25–32. [Google Scholar] [CrossRef]
- De la Harpe, A.; Beukes, N.; Frost, C.L. CBD activation of TRPV1 induces oxidative signaling and subsequent ER stress in breast cancer cell lines. Biotechnol. Appl. Biochem. 2021. [Google Scholar] [CrossRef]
- Amaral, C.; Trouille, F.M.; Almeida, C.; Correia-Da-Silva, G.; Teixeira, N. Unveiling the mechanism of action behind the anti-cancer properties of cannabinoids in ER+ breast cancer cells: Impact on aromatase and steroid receptors. J. Steroid Biochem. Mol. Biol. 2021, 210, 105876. [Google Scholar] [CrossRef]
- Bhattacharjee, A.; Hossain, M.U.; Chowdhury, Z.M.; Rahman, S.A.; Bhuyan, Z.A.; Salimullah, M.; Keya, C.A. Insight of druggable cannabinoids against estrogen receptor β in breast cancer. J. Biomol. Struct. Dyn. 2021, 39, 1688–1697. [Google Scholar] [CrossRef]
- Von Bueren, A.O.; Schlumpf, M.; Lichtensteiger, W. Delta(9)-tetrahydrocannabinol inhibits 17beta-estradiol-induced proliferation and fails to activate androgen and estrogen receptors in MCF7 human breast cancer cells. Anticancer. Res. 2008, 28, 85–89. [Google Scholar]
- Almada, M.; Oliveira, A.; Amaral, C.; Fernandes, P.; Ramos, M.J.; Fonseca, B.; Correia-Da-Silva, G.; Teixeira, N. Anandamide targets aromatase: A breakthrough on human decidualization. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2019, 1864, 158512. [Google Scholar] [CrossRef]
- Almada, M.; Amaral, C.; Oliveira, A.; Fernandes, P.; Ramos, M.J.; Fonseca, B.M.; Correia-Da-Silva, G.; Teixeira, N. Cannabidiol (CBD) but not tetrahydrocannabinol (THC) dysregulate in vitro decidualization of human endometrial stromal cells by disruption of estrogen signaling. Reprod. Toxicol. 2020, 93, 75–82. [Google Scholar] [CrossRef]
- Benito, S.B.; Seijo-Vila, M.; Caro-Villalobos, M.; Tundidor, I.; Andradas, C.; García-Taboada, E.; Wade, J.; Smith, S.; Guzmán, M.; Pérez-Gómez, E.; et al. Appraising the “entourage effect”: Antitumor action of a pure cannabinoid versus a botanical drug preparation in preclinical models of breast cancer. Biochem. Pharmacol. 2018, 157, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Prather, P.L.; Francis Devaraj, F.; Dates, C.R.; Greer, A.K.; Bratton, S.M.; Ford, B.M.; Franks, L.N.; Radominska-Pandya, A. CB1 and CB2 receptors are novel molecular targets for Tamoxifen and 4OH-Tamoxifen. Biochem. Biophys. Res. Commun. 2013, 441, 339–343. [Google Scholar] [CrossRef]
- Takeda, S.; Yoshida, K.; Nishimura, H.; Harada, M.; Okajima, S.; Miyoshi, H.; Okamoto, Y.; Amamoto, T.; Watanabe, K.; Omiecinski, C.J.; et al. Δ9-Tetrahydrocannabinol disrupts estrogen-signaling through up-regulation of estrogen receptor β (ERβ). Chem. Res. Toxicol. 2013, 26, 1073–1079. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Breast Cancer Subtype | HER2 Expression | ER Expression | PR Expression | Ki67 Expression | Prevalence |
|---|---|---|---|---|---|
| Luminal A | Negative | Positive | High | Low | 73% |
| Luminal B | Positive or Negative | Positive | Low or Any | High or Any | 11% |
| HER2+ | Positive | Negative | Negative | - | 12–20% |
| TNBC | Negative | Negative | Negative | - | 15–20% |
| Model | Cannabinoid | Biological Effect | Mechanism of Action | Reference |
|---|---|---|---|---|
| Xenograft-Based and PyMT Genetically Engineered Models | JWH-133 | Tumor growth reduction; angiogenesis inhibition | COX-2/PGE2 axis inhibition through CB2 | [121] |
| MDA-MB-231 Cells | Met-F-AEA | Cell migration impairment; cell cycle arrest | Inhibition of (FAK)/Src and RhoA-ROCK pathways through CB1 | [122,123] |
| MDA-MB-231 Cells | JWH-133 | Cell migration impairment; cell cycle arrest | COX-2/PGE2 axis inhibition through CB2 | [121] |
| MDA-MB-231 Cells | JWH-015 | Cell migration impairment | Inhibition of ERK and cytoskeletal focal adhesion and stress fiber formation through CB2 | [124] |
| MDA-MB-231 Cells | WIN 55,212-2 | Cell migration impairment; cell cycle arrest | COX-2/PGE2 axis inhibition through CB2 | [124] |
| MDA-MB-231 Cells | CBD | Cell proliferation reduction; apoptosis; autophagy | Endoplasmic reticulum stress and AKT/mTOR inhibition | [111] |
| MDA-MB-231 Cells | CBD | Cell proliferation reduction | TRPV1 receptors and uncharacterized CBD targets | [109,110] |
| MDA-MB-231 Cells | CBD | Cell proliferation reduction | Increased ROS production | [110,115,116] |
| MDA-MB-231 Xenografts in Immune-Deficient Mice and Orthotopic Xenografts from 4T1 Cells in Syngeneic BALB/c Mice | CBD | Cell proliferation reduction | Downregulation of Id-1 | [110,115] |
| MDA-MB-231 and 4T1 Cells | CBD | Cell proliferation reduction | Downregulation of Id-1 | [115,116] |
| SUM159, 4T1.2 and SPC2 Cells | CBD | Cell proliferation reduction; impairment of cell migration; invasion | Inhibition of EGF/EGFR signaling | [114] |
| MDA-MB-231 Cells | CBD | Apoptosis | Interplay among PPARy, mTOR and cyclin D1 | [112] |
| MDA-MB-231 and MDA-MB436 Cells | CBG | Cell viability reduction; impairment of cell migration | Decreased Id-1 expression | [116] |
| MDA-MB-231 and MDA-MB436 Cells | CBN | Cell viability reduction; impairment of cell migration | Decreased Id-1 expression | [116] |
| Model | Cannabinoid | Biological effect | Mechanism of action | Reference |
|---|---|---|---|---|
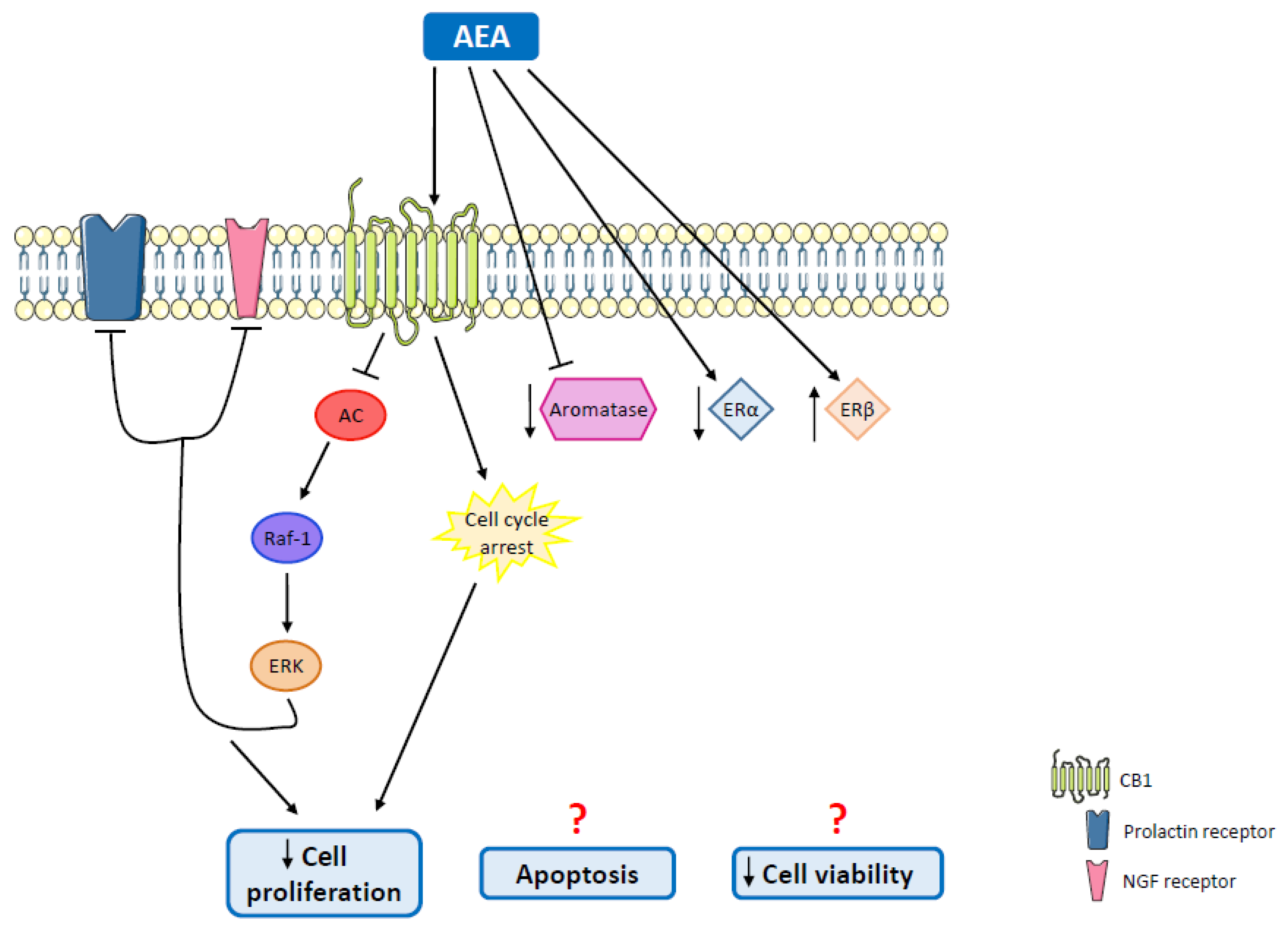
| MCF-7 and EFM-19 cells | AEA | Cell proliferation inhibition | Cell cycle arrest; CB1 activation; Raf-1/ERK/MAPK pathway activation | [126] |
| MCF-7 cells | AEA | Decreased cell proliferation | CB1 activation | [127,128] |
| MCF-7aro cells | AEA | Apoptosis; reduction in aromatase and ERα protein levels; upregulation of ERβ; aromatase inhibition | Cell cycle arrest | [135] |
| T-47D cells | CBD | Cell survival impairment; apoptosis | Interplay among PPARy, mTOR and cyclin D1 | [112] |
| MCF-7aro cells | CBD | Apoptosis; autophagy; reduction in aromatase and ERα protein levels; upregulation of ERβ; aromatase inhibition | Cell cycle arrest | [135] |
| MCF-7 cells | CBD | Apoptosis | Endoplasmic reticulum stress; disruption of protein folding | [134] |
| EVSA-T cells | THC | Apoptosis | Cell cycle arrest mediated by CB2 | [129,130] |
| MCF-7 | THC | Decreased cell proliferation | [132] | |
| MCF-7aro cells | THC | Apoptosis; reduction in aromatase and ERα protein levels | Cell cycle arrest | [135] |
| MCF-7 | CBG | Decreased cell proliferation | [110] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almeida, C.F.; Teixeira, N.; Correia-da-Silva, G.; Amaral, C. Cannabinoids in Breast Cancer: Differential Susceptibility According to Subtype. Molecules 2022, 27, 156. https://doi.org/10.3390/molecules27010156
Almeida CF, Teixeira N, Correia-da-Silva G, Amaral C. Cannabinoids in Breast Cancer: Differential Susceptibility According to Subtype. Molecules. 2022; 27(1):156. https://doi.org/10.3390/molecules27010156
Chicago/Turabian StyleAlmeida, Cristina Ferreira, Natércia Teixeira, Georgina Correia-da-Silva, and Cristina Amaral. 2022. "Cannabinoids in Breast Cancer: Differential Susceptibility According to Subtype" Molecules 27, no. 1: 156. https://doi.org/10.3390/molecules27010156
APA StyleAlmeida, C. F., Teixeira, N., Correia-da-Silva, G., & Amaral, C. (2022). Cannabinoids in Breast Cancer: Differential Susceptibility According to Subtype. Molecules, 27(1), 156. https://doi.org/10.3390/molecules27010156