
Molecular Regulation of Betulinic Acid on α3β4 Nicotinic Acetylcholine Receptors

 ,
,  ,
,
Abstract
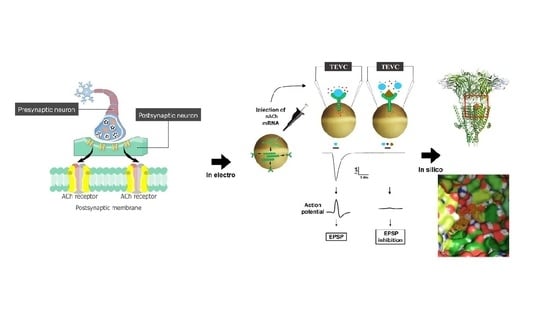
1. Introduction
2. Results
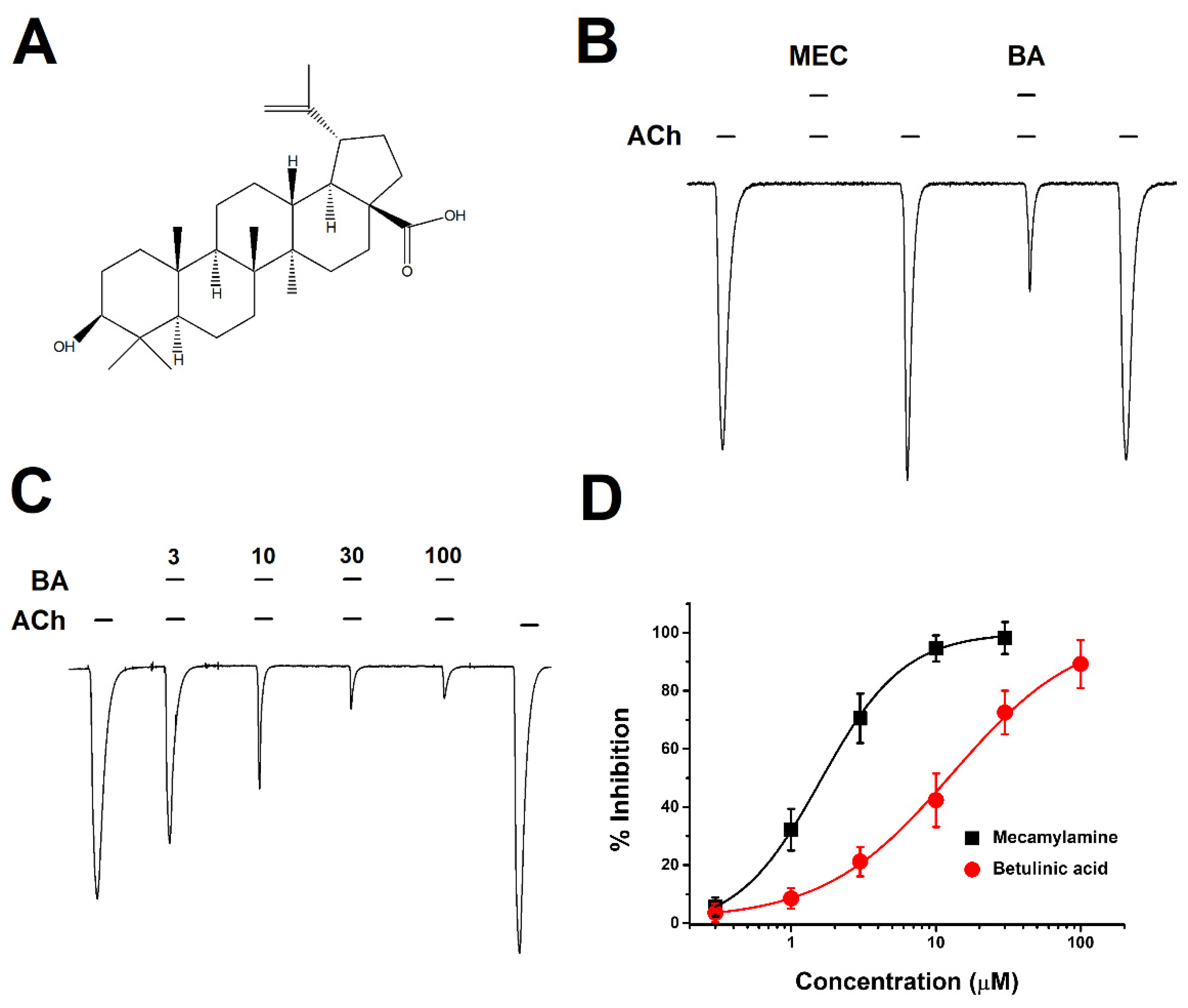
2.1. Concentration-Dependent Inhibitory Effect of BA on α3β4 nAChRs
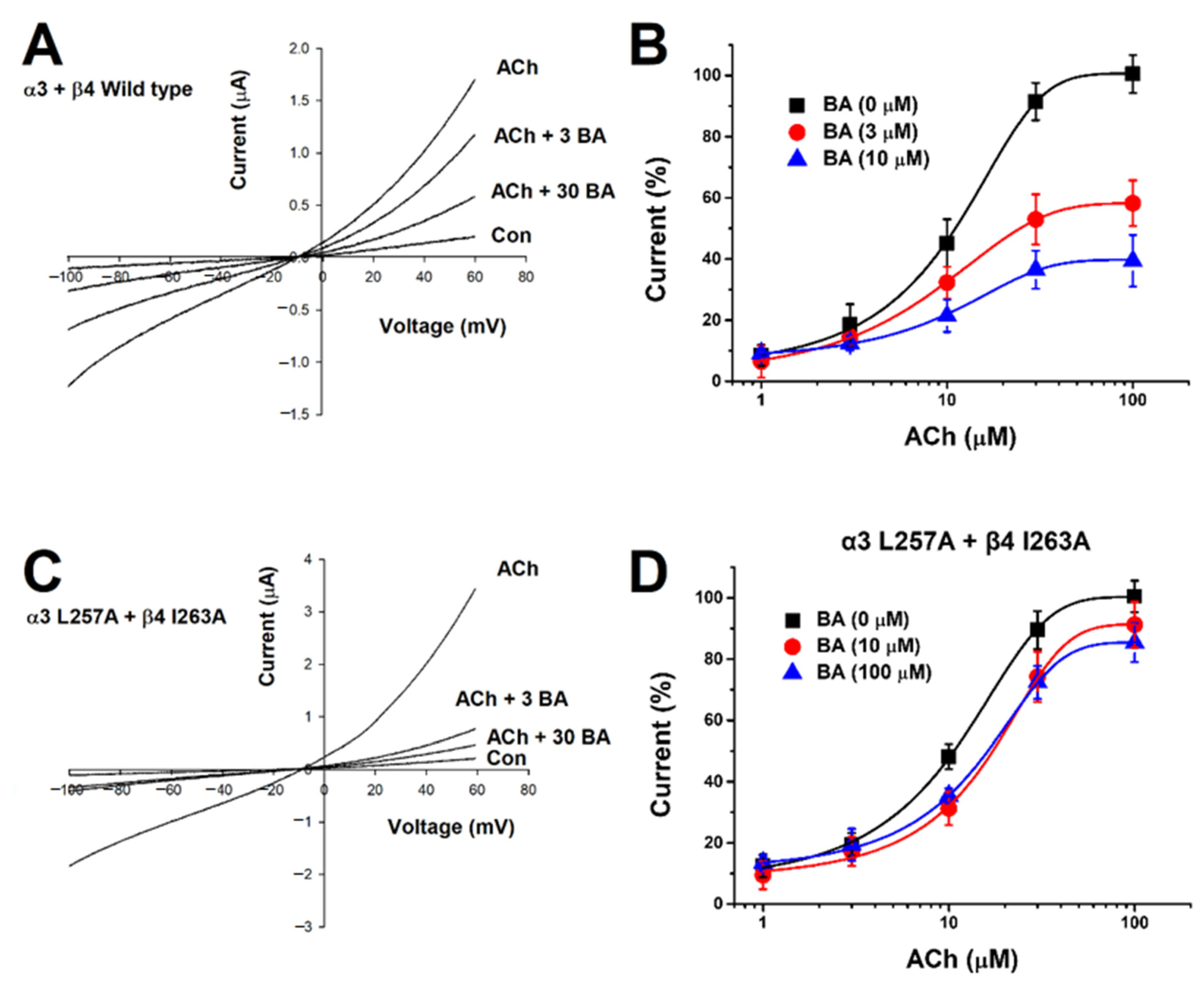
2.2. Current-Voltage Relationship of IACh and Non-Competitive Inhibition by Betulinic Acid
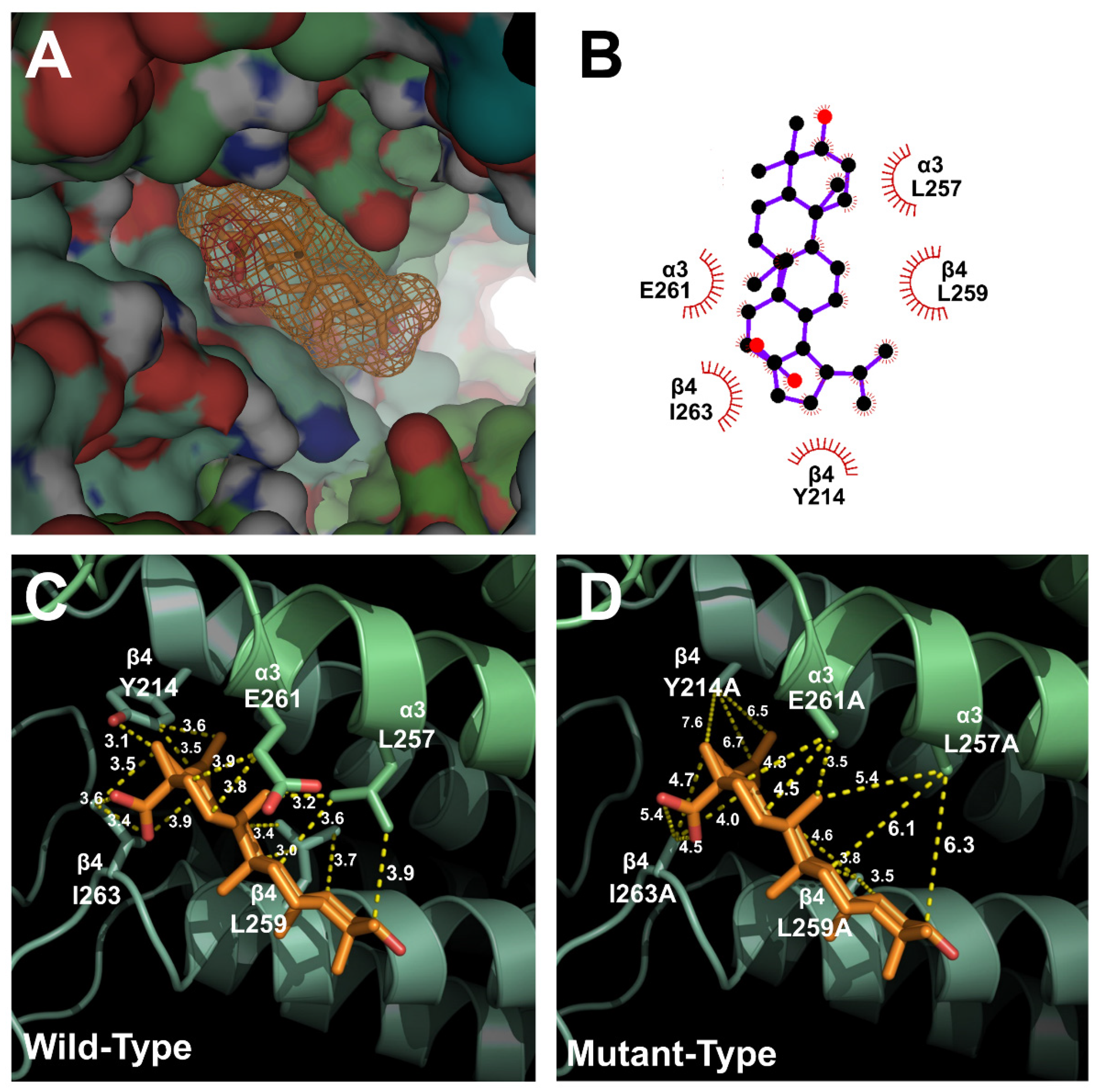
2.3. Docking Model of Betulinic Acid and Nicotinic Acetylcholine Receptor α3β4
2.4. Inhibitory Effect of Betulinic Acid on a Double-Mutant Nicotinic Acetylcholine Receptor α3β4
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Oocyte Preparation
4.3. In Vitro Transcription of mRNA and Injection into Oocytes
4.4. Mutagenesis
4.5. D Structure Modeling and Molecular-Docking Studies
4.6. Data Recording with Two-Electrode Voltage Clamp
4.7. Data Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Zhao, J.; Li, S.; Yang, F.; Li, P.; Wang, Y. Simultaneous determination of saponins and fatty acids in Ziziphus jujuba (Suanzaoren) by high performance liquid chromatography-evaporative light scattering detection and pressurized liquid extraction. J. Chromatogr. A 2006, 1108, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.-H.; Hsieh, M.-T.; Lee, Y.-S.; Lin, Y.-C.; Liao, J. Anxiolytic effect of seed of Ziziphus jujuba in mouse models of anxiety. J. Ethnopharmacol. 2000, 72, 435–441. [Google Scholar] [CrossRef]
- Chen, J.; Yan, A.L.; Lam, K.Y.; Lam, C.T.; Li, N.; Yao, P.; Xiong, A.; Dong, T.T.; Tsim, K.W. A chemically standardized extract of Ziziphus jujuba fruit (Jujube) stimulates expressions of neurotrophic factors and anti-oxidant enzymes in cultured astrocytes. Phytother. Res. 2014, 28, 1727–1730. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.J.; Baghdoyan, H.A.; Lydic, R. Neuropharmacology of sleep and wakefulness. Sleep Med. Clin. 2010, 5, 513–528. [Google Scholar] [CrossRef] [PubMed]
- Dani, J.A.; Bertrand, D. Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu. Rev. Pharm. Toxicol. 2007, 47, 699–729. [Google Scholar] [CrossRef] [PubMed]
- Karlin, A. Emerging structure of the nicotinic acetylcholine receptors. Nat. Rev. Neurosci. 2002, 3, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Reitstetter, R.; Lukas, R.J.; Gruener, R. Dependence of nicotinic acetylcholine receptor recovery from desensitization on the duration of agonist exposure. J. Pharmacol. Exp. Ther. 1999, 289, 656–660. [Google Scholar] [PubMed]
- Duga, S.; Soldà, G.; Asselta, R.; Bonati, M.T.; Dalprà, L.; Malcovati, M.; Tenchini, M.L. Characterization of the genomic structure of the human neuronal nicotinic acetylcholine receptor CHRNA5/A3/B4 gene cluster and identification of novel intragenic polymorphisms. J. Hum. Genet. 2001, 46, 640–648. [Google Scholar] [CrossRef][Green Version]
- Niwa, Y.; Kanda, G.N.; Yamada, R.G.; Shi, S.; Sunagawa, G.A.; Ukai-Tadenuma, M.; Fujishima, H.; Matsumoto, N.; Masumoto, K.-H.; Nagano, M. Muscarinic acetylcholine receptors Chrm1 and Chrm3 are essential for REM Sleep. Cell Rep. 2018, 24, 2231–2247.e7. [Google Scholar] [CrossRef] [PubMed]
- Ruivo, L.M.T.-G.; Baker, K.L.; Conway, M.W.; Kinsley, P.J.; Gilmour, G.; Phillips, K.G.; Isaac, J.T.; Lowry, J.P.; Mellor, J.R. Coordinated acetylcholine release in prefrontal cortex and hippocampus is associated with arousal and reward on distinct timescales. Cell Rep. 2017, 18, 905–917. [Google Scholar] [CrossRef]
- Scholey, A.; Benson, S.; Gibbs, A.; Perry, N.; Sarris, J.; Murray, G. Exploring the effect of Lactium™ and zizyphus complex on sleep quality: A double-blind, randomized placebo-controlled trial. Nutrients 2017, 9, 154. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Dong, J.-W.; Zhao, J.-H.; Tang, L.-N.; Zhang, J.-J. Herbal insomnia medications that target GABAergic systems: A review of the psychopharmacological evidence. Curr. Neuropharmacol. 2014, 12, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Yogeeswari, P.; Sriram, D. Betulinic acid and its derivatives: A review on their biological properties. Curr. Med. Chem. 2005, 12, 657–666. [Google Scholar] [CrossRef]
- Fulda, S. Betulinic acid for cancer treatment and prevention. Int. J. Mol. Sci. 2008, 9, 1096–1107. [Google Scholar] [CrossRef]
- Tamanoi, F.; Bathaie, S.Z. Natural Products and Cancer Signaling: Isoprenoids, Polyphenols and Flavonoids; Academic Press: Cambridge, MA, USA, 2014. [Google Scholar]
- Alqahtani, A.; Hamid, K.; Kam, A.; Wong, K.; Abdelhak, Z.; Razmovski-Naumovski, V.; Chan, K.; Li, K.M.; Groundwater, P.W.; Li, G.Q. The pentacyclic triterpenoids in herbal medicines and their pharmacological activities in diabetes and diabetic complications. Curr. Med. Chem. 2013, 20, 908–931. [Google Scholar] [PubMed]
- Salvador, J.A.; Leal, A.S.; Alho, D.P.; Gonçalves, B.M.; Valdeira, A.S.; Mendes, V.I.; Jing, Y. Highlights of pentacyclic triterpenoids in the cancer settings. In Studies in Natural Products Chemistry; Elsevier: Amsterdam, The Netherlands, 2014; Volume 41, pp. 33–73. [Google Scholar]
- Sahu, B.S.; Obbineni, J.M.; Sahu, G.; Singh, P.K.; Sonawane, P.J.; Sasi, B.K.; Allu, P.K.; Maji, S.K.; Bera, A.K.; Senapati, S. Molecular interactions of the physiological anti-hypertensive peptide catestatin with the neuronal nicotinic acetylcholine receptor. J. Cell Sci. 2012, 125, 2323–2337. [Google Scholar] [CrossRef] [PubMed]
- Patlolla, J.M.R.; Lubet, R.; Ely, M.; Zhang, Y.; Janakiram, N.B.; Madka, V.; Mohammed, A.; Steele, V.E.; Rao, C.V. Chemoprevention of Colon Cancer by DFMO, Sulindac and NO-Sulindac Administered Individually or in Combinations in Male F344 Rats; AACR: Philadelphia, PA, USA, 2011. [Google Scholar]
- Monti, J.M. The role of dorsal raphe nucleus serotonergic and non-serotonergic neurons, and of their receptors, in regulating waking and rapid eye movement (REM) sleep. Sleep Med. Rev. 2010, 14, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Zant, J.C.; Kim, T.; Prokai, L.; Szarka, S.; McNally, J.; McKenna, J.T.; Shukla, C.; Yang, C.; Kalinchuk, A.V.; McCarley, R.W. Cholinergic neurons in the basal forebrain promote wakefulness by actions on neighboring non-cholinergic neurons: An opto-dialysis study. J. Neurosci. 2016, 36, 2057–2067. [Google Scholar] [CrossRef]
- Domino, E.; Yamamoto, K.; Dren, A. Role of cholinergic mechanisms in states of wakefulness and sleep. In Progress in Brain Research; Elsevier: Amsterdam, The Netherlands, 1968; Volume 28, pp. 113–133. [Google Scholar]
- Kodama, T.; Honda, Y. Acetylcholine and glutamate release during sleep–wakefulness in the pedunculopontine tegmental nucleus and norepinephrine changes regulated by nitric oxide. Psychiatry Clin. Neurosci. 1999, 53, 109–111. [Google Scholar] [CrossRef]
- Cuddapah, V.A.; Zhang, S.L.; Sehgal, A. Regulation of the blood–brain barrier by circadian rhythms and sleep. Trends Neurosci. 2019, 42, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Ballatore, C.; Huryn, D.M.; Smith III, A.B. Carboxylic acid (bio) isosteres in drug design. ChemMedChem 2013, 8, 385. [Google Scholar] [CrossRef]
- Crivori, P.; Cruciani, G.; Carrupt, P.-A.; Testa, B. Predicting blood− brain barrier permeation from three-dimensional molecular structure. J. Med. Chem. 2000, 43, 2204–2216. [Google Scholar] [CrossRef] [PubMed]
- Stępnik, K.; Kukula-Koch, W. In Silico Studies on Triterpenoid Saponins Permeation through the Blood–Brain Barrier Combined with Postmortem Research on the Brain Tissues of Mice Affected by Astragaloside IV Administration. Int. J. Mol. Sci. 2020, 21, 2534. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, C.; Legault, J.; Piochon-Gauthier, M.; Pichette, A. Advances in the synthesis and pharmacological activity of lupane-type triterpenoid saponins. Phytochem. Rev. 2011, 10, 521–544. [Google Scholar] [CrossRef]
- Vazquez, J.; Baghdoyan, H.A. Basal forebrain acetylcholine release during REM sleep is significantly greater than during waking. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 280, R598–R601. [Google Scholar] [CrossRef] [PubMed]
- Mowry, K.L. Using the Xenopus Oocyte Toolbox. Cold Spring Harb. Protoc. 2020, 2020, pdb–top095844. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subunit Mutants | Imax | IC50 | nH |
|---|---|---|---|
| Wild α3 + Wild β4 | 101.5 ± 5.9 | 12.8 ± 2.0 | 0.9 ± 0.1 |
| α3 (L257A) + Wild β4 | 48.9 ± 2.7 | 18.6 ± 1.7 | 1.6 ± 0.1 |
| Wild α3 + β4 (I263A) | 33.1 ± 3.3 | 13.4 ± 1.9 | 2.7 ± 1.0 |
| α3 (L257A) + β4 (I263A) | 7.4 | 26.2 | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.; Jung, W.; Eom, S.; Yeom, H.D.; Park, H.-D.; Lee, J.H. Molecular Regulation of Betulinic Acid on α3β4 Nicotinic Acetylcholine Receptors. Molecules 2021, 26, 2659. https://doi.org/10.3390/molecules26092659
Lee S, Jung W, Eom S, Yeom HD, Park H-D, Lee JH. Molecular Regulation of Betulinic Acid on α3β4 Nicotinic Acetylcholine Receptors. Molecules. 2021; 26(9):2659. https://doi.org/10.3390/molecules26092659
Chicago/Turabian StyleLee, Shinhui, Woog Jung, Sanung Eom, Hye Duck Yeom, Heui-Dong Park, and Junho H. Lee. 2021. "Molecular Regulation of Betulinic Acid on α3β4 Nicotinic Acetylcholine Receptors" Molecules 26, no. 9: 2659. https://doi.org/10.3390/molecules26092659
APA StyleLee, S., Jung, W., Eom, S., Yeom, H. D., Park, H.-D., & Lee, J. H. (2021). Molecular Regulation of Betulinic Acid on α3β4 Nicotinic Acetylcholine Receptors. Molecules, 26(9), 2659. https://doi.org/10.3390/molecules26092659