
Metal-Promoted Heterocyclization: A Heterosynthetic Approach to Face a Pandemic Crisis



Abstract
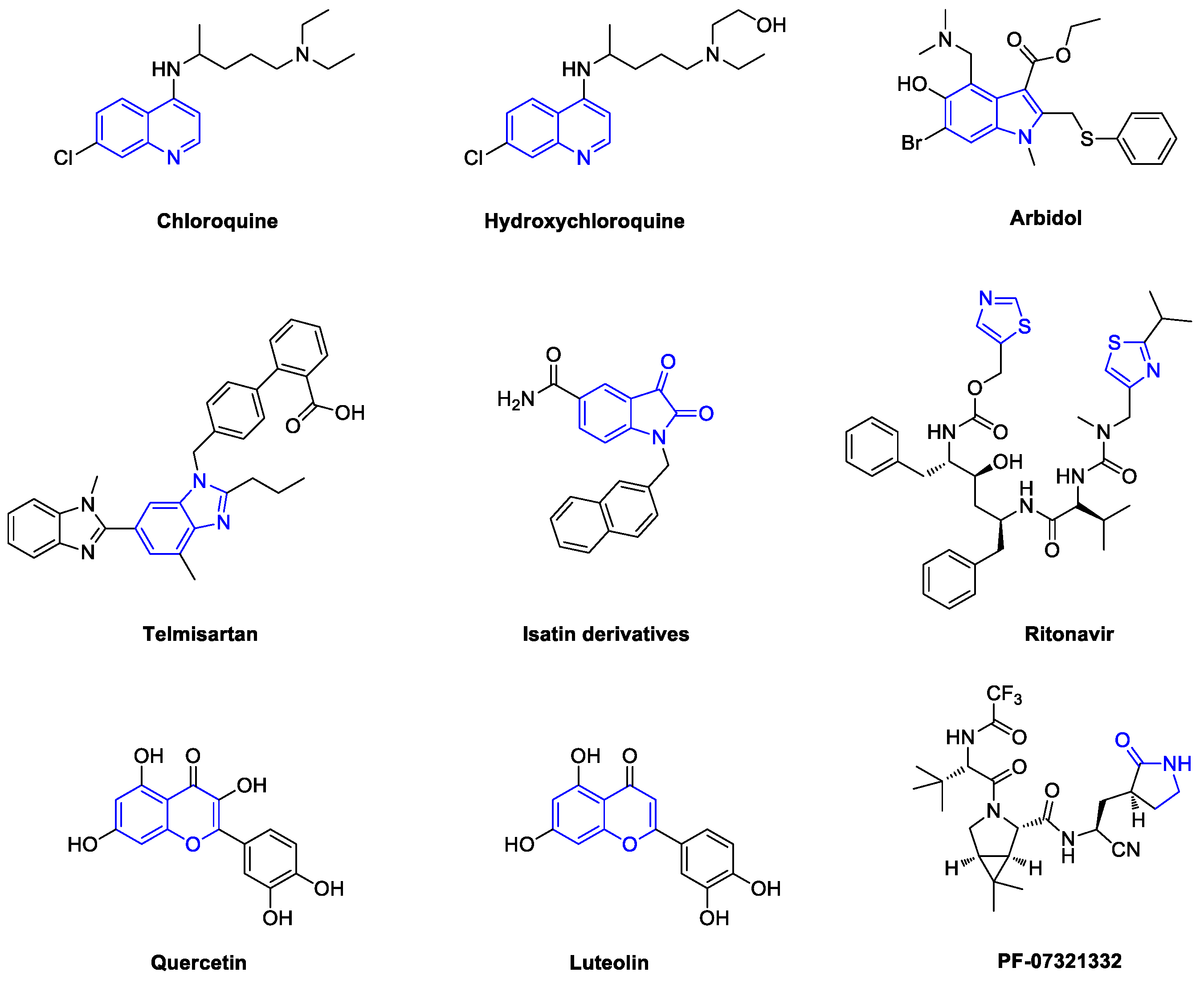
1. Introduction
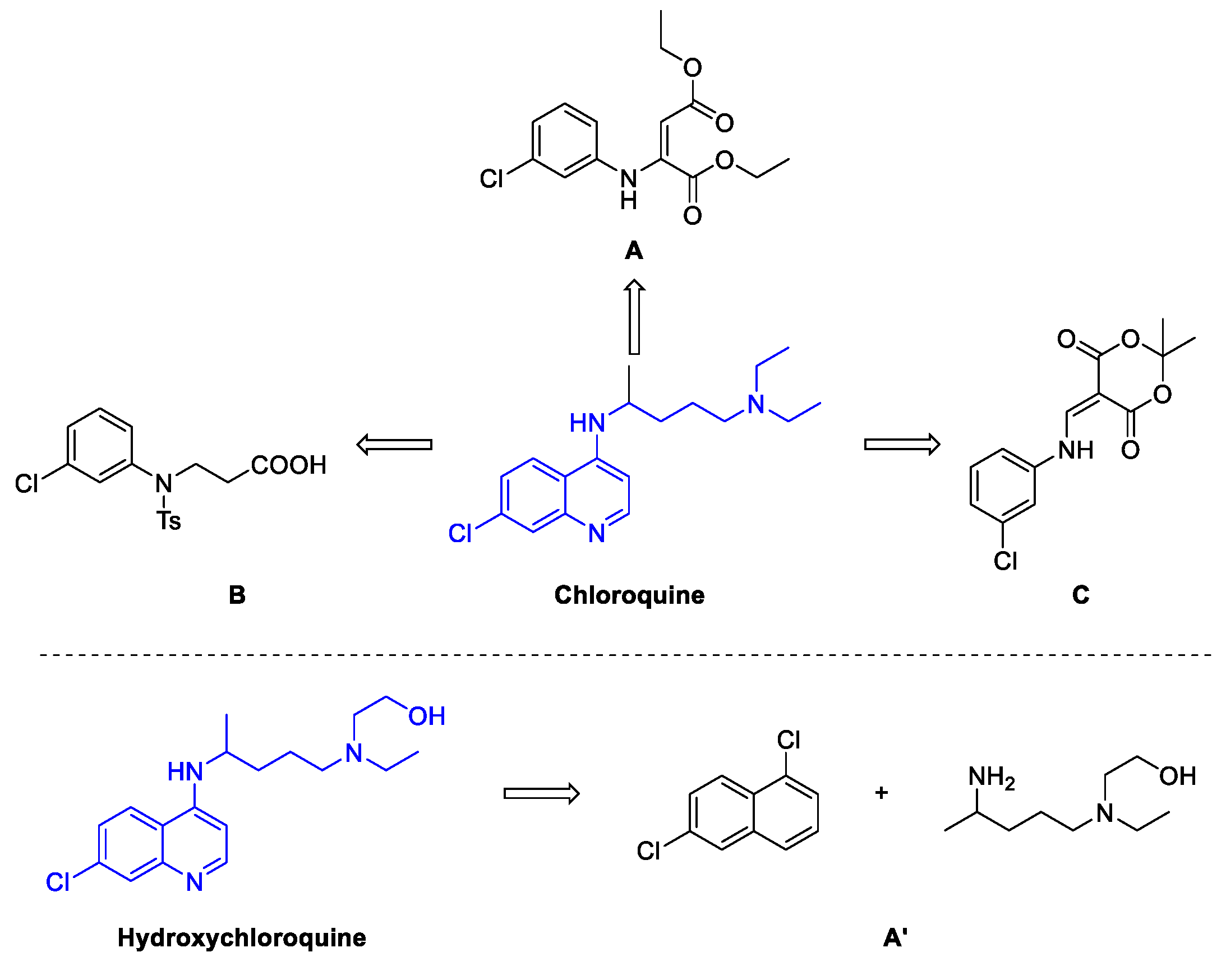
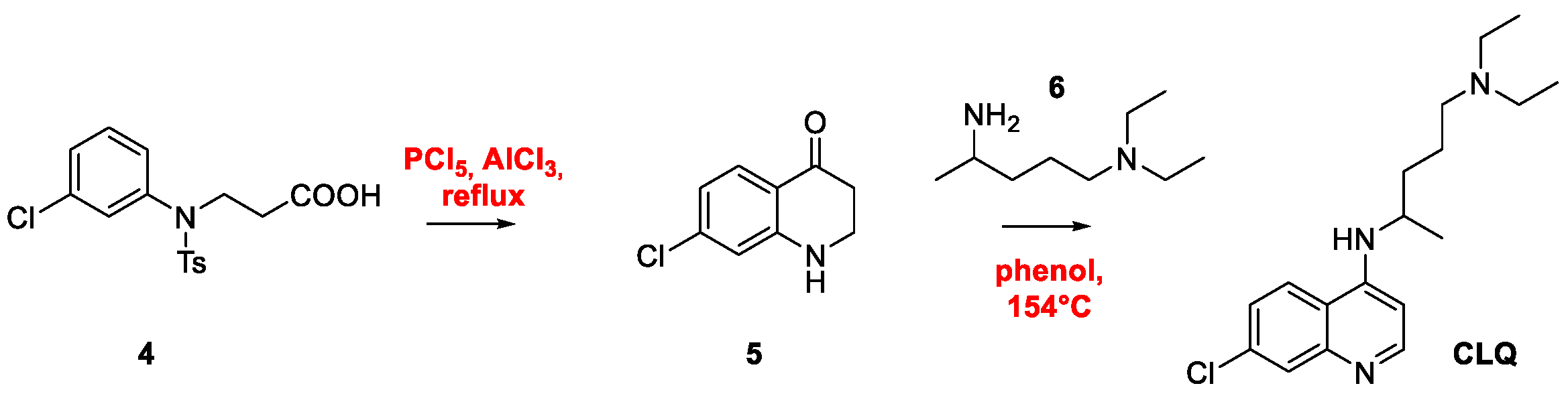
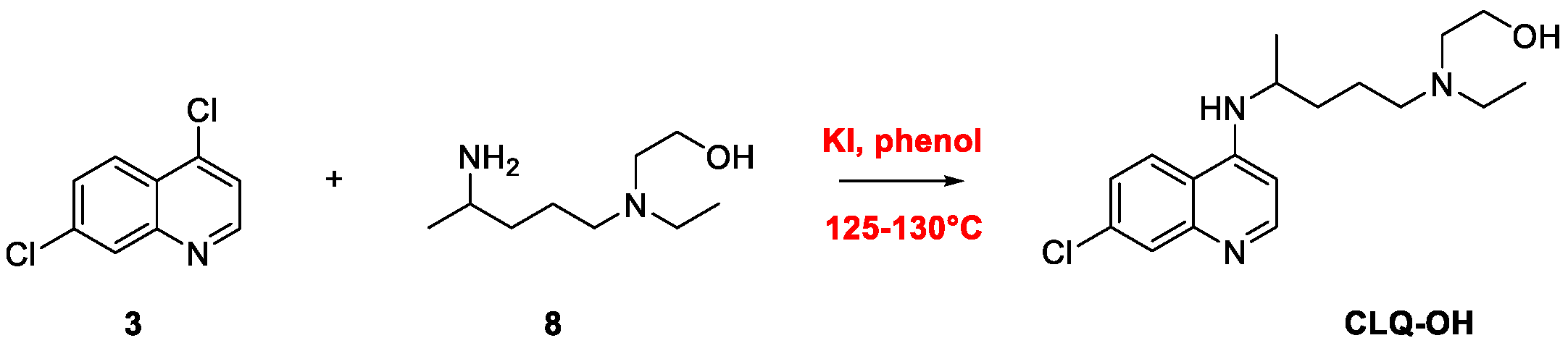
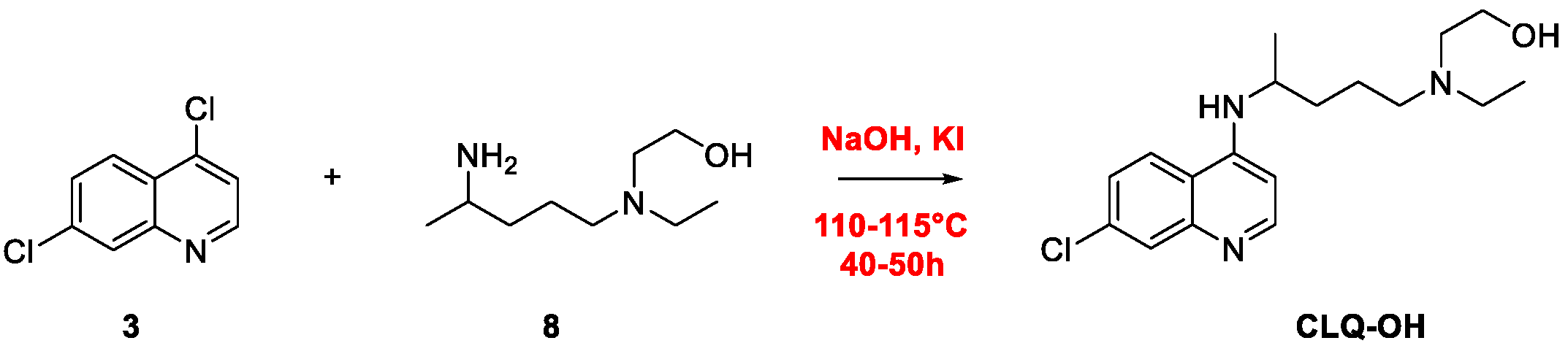
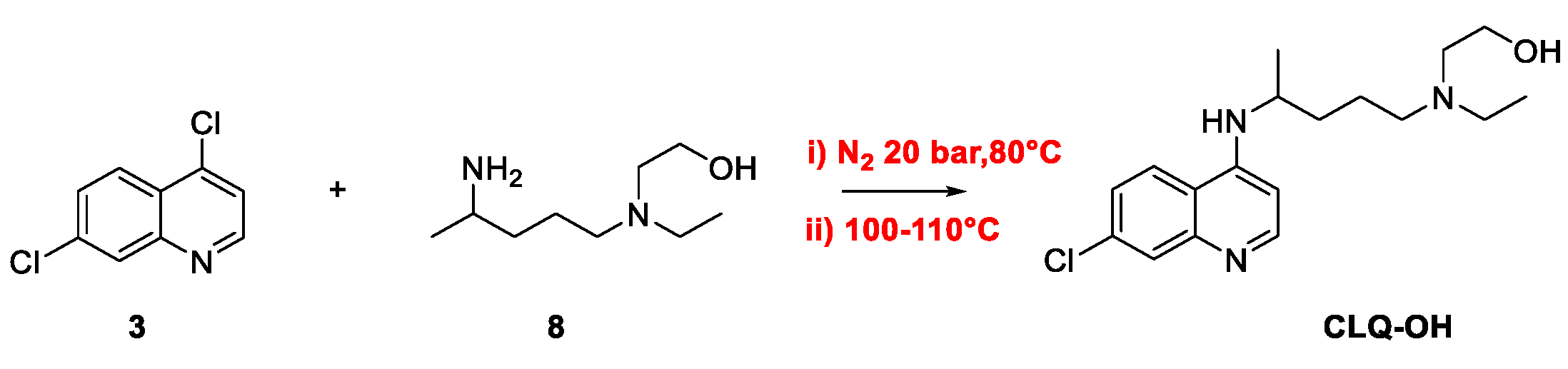
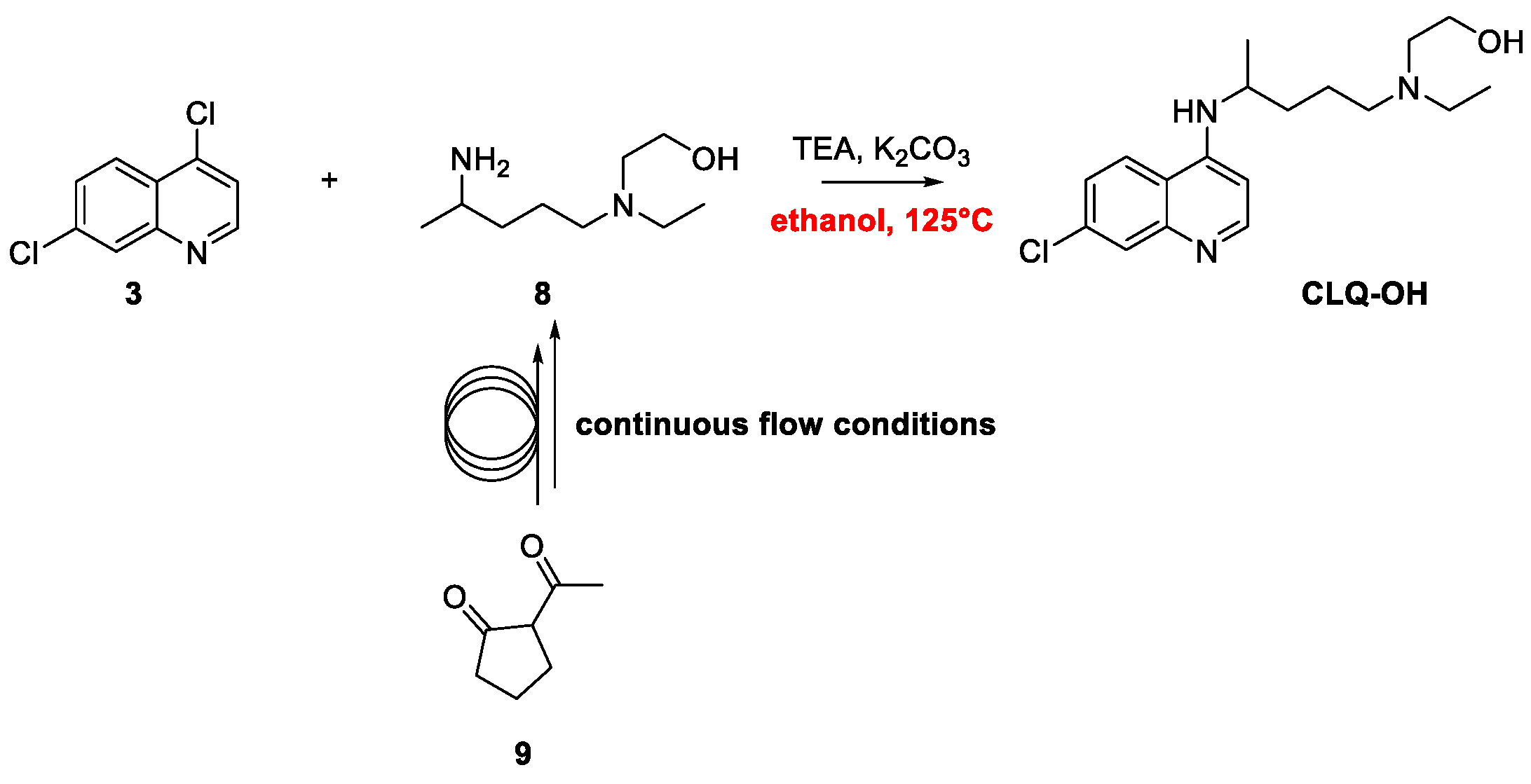
2. Chloroquine and Hydroxychloroquine
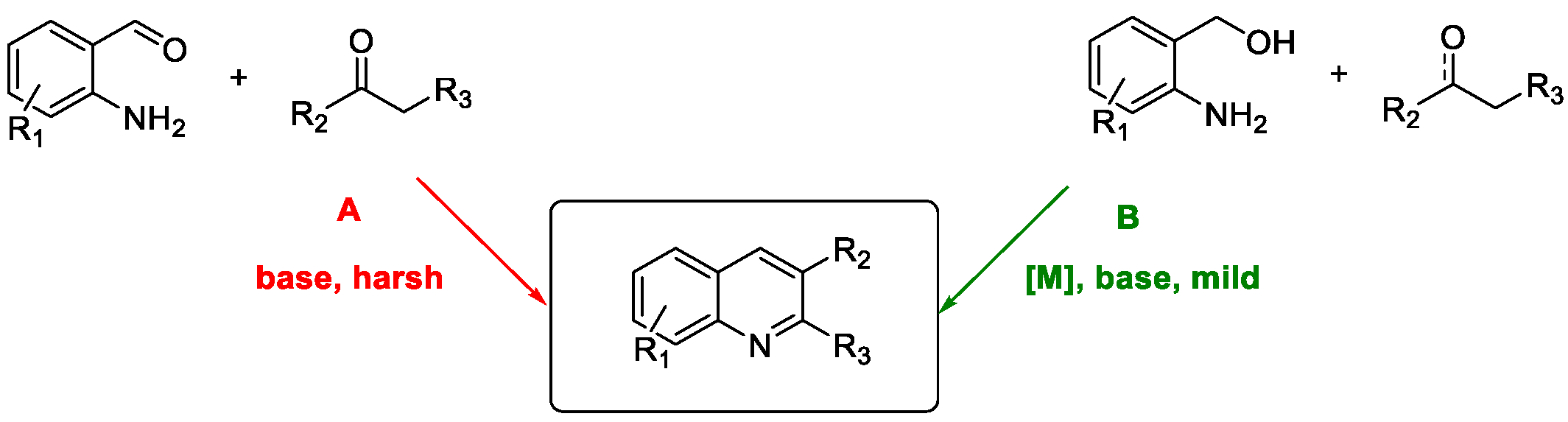
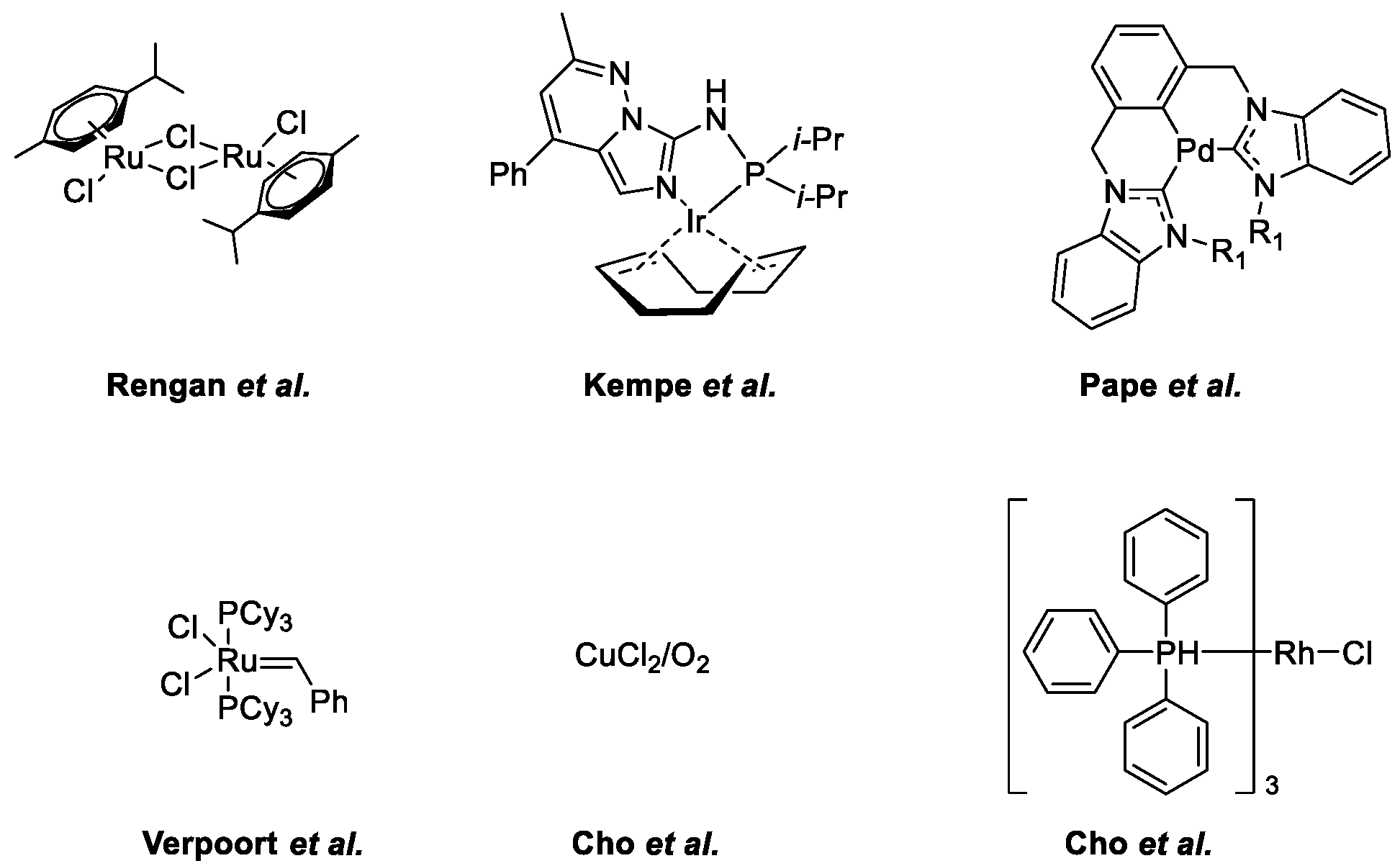
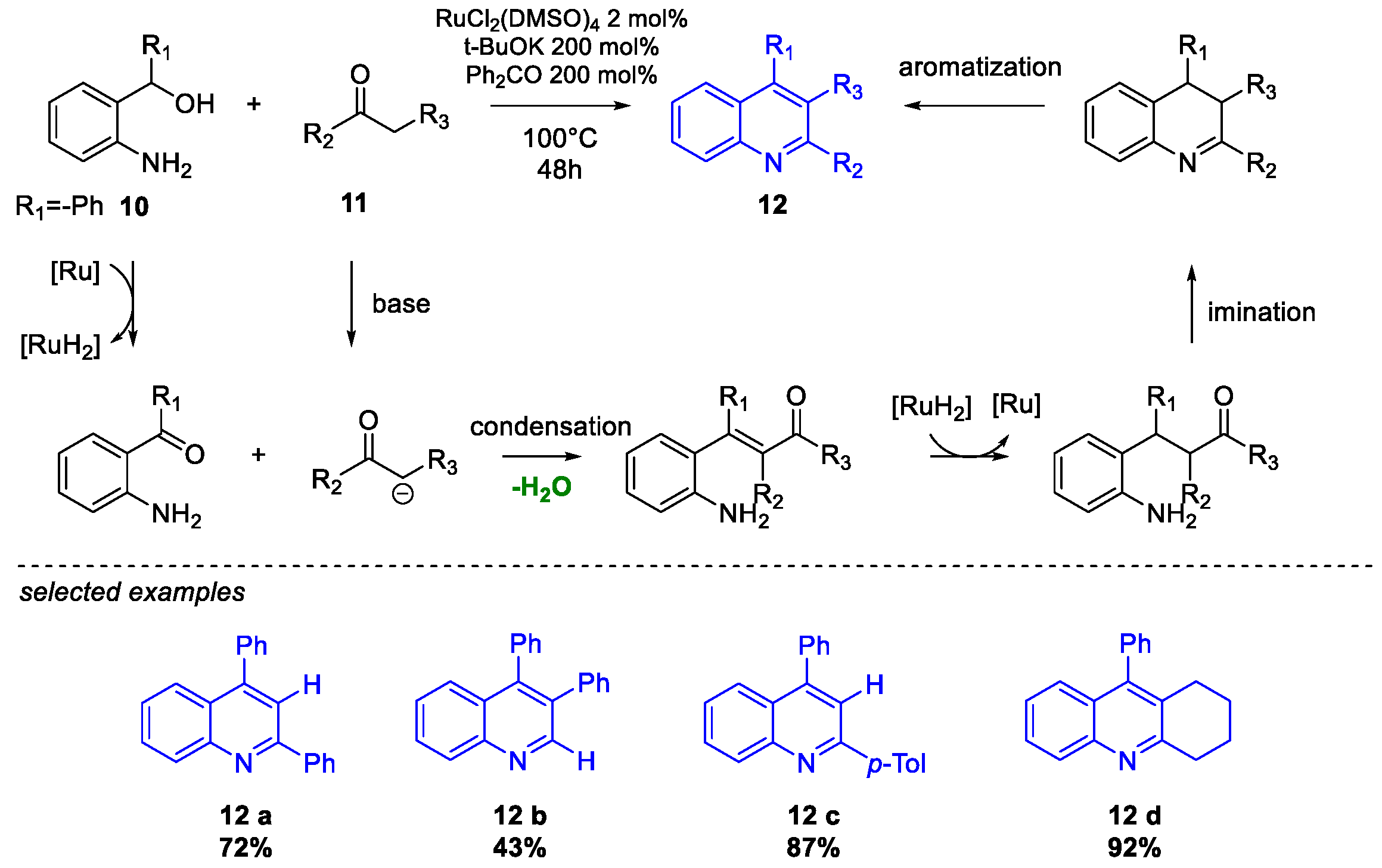
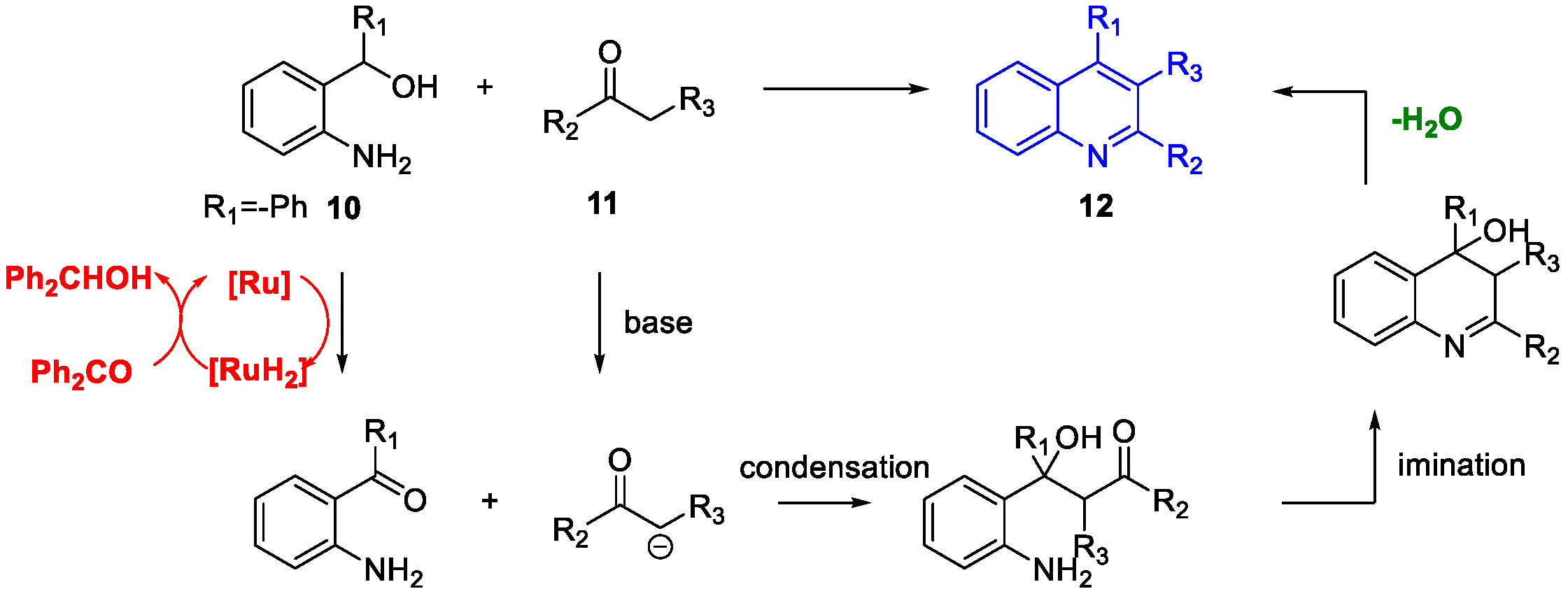
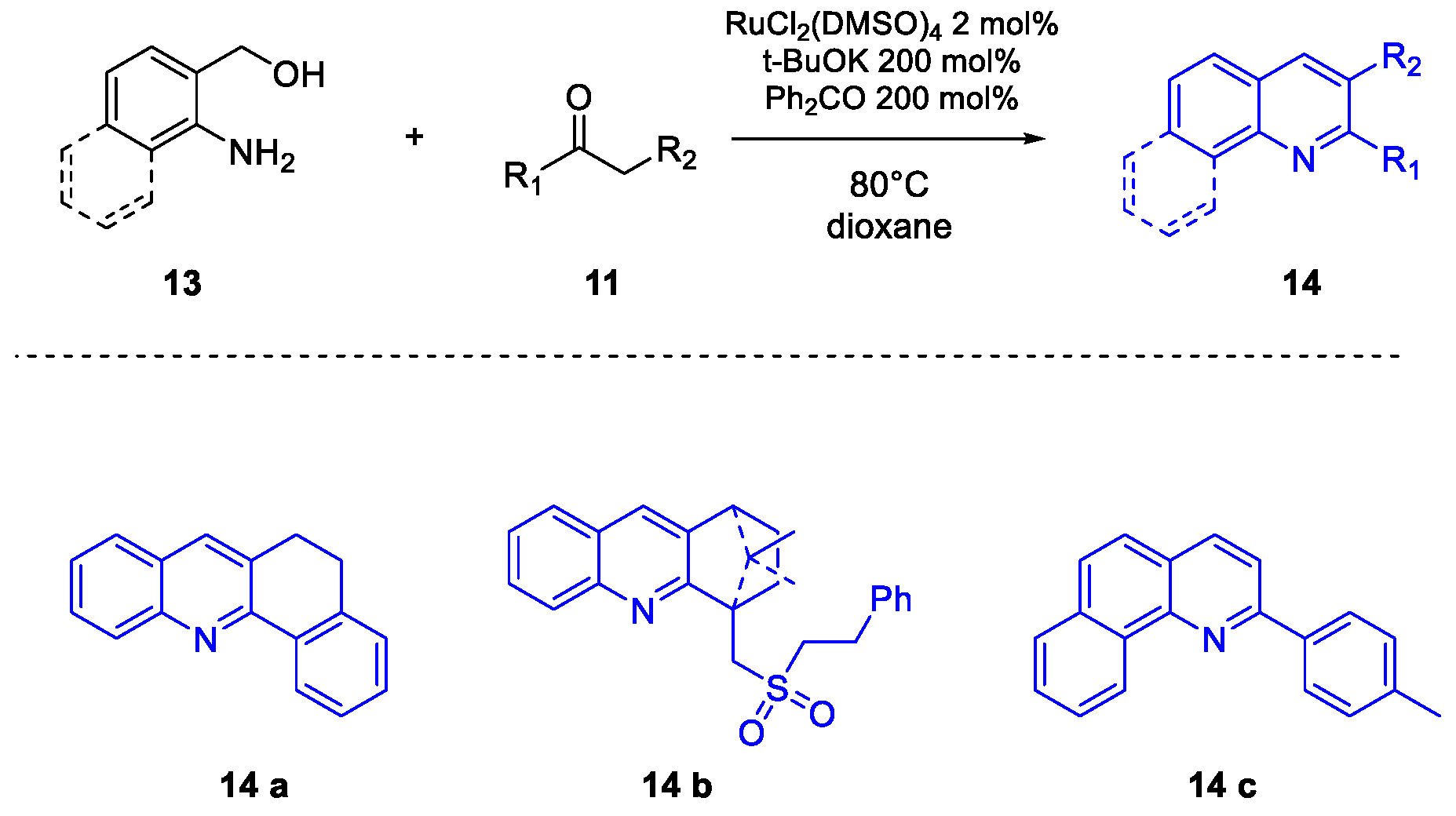
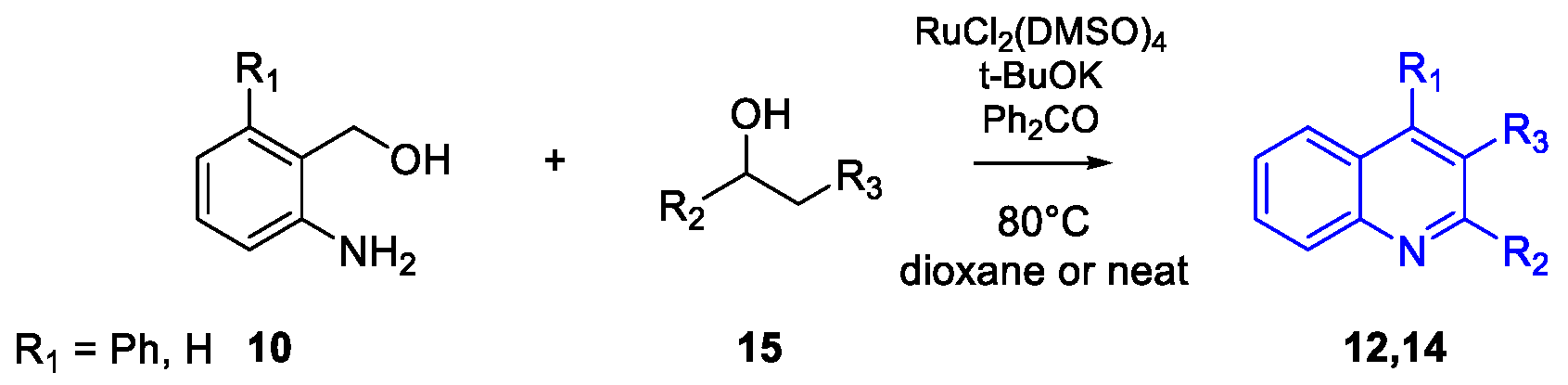
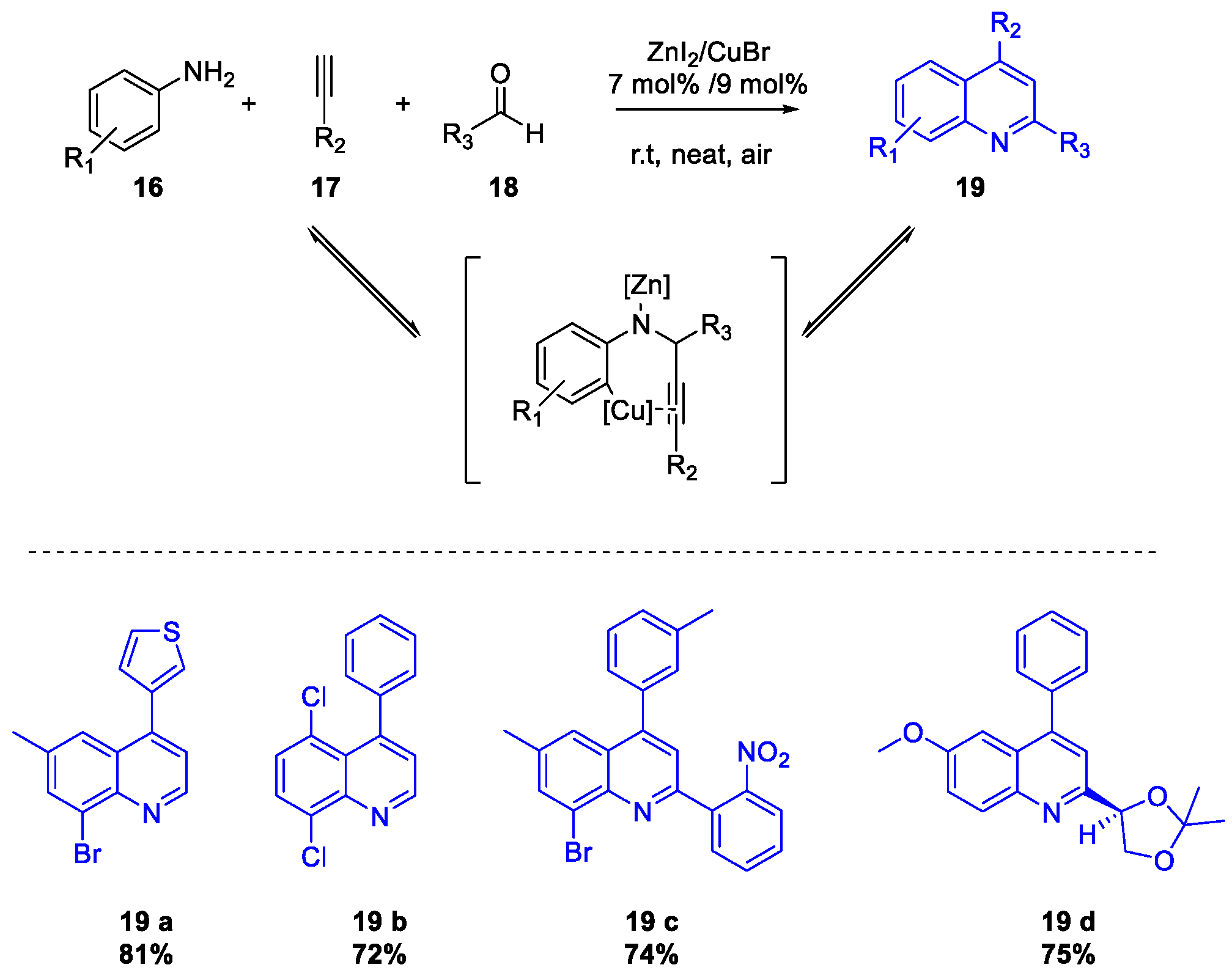
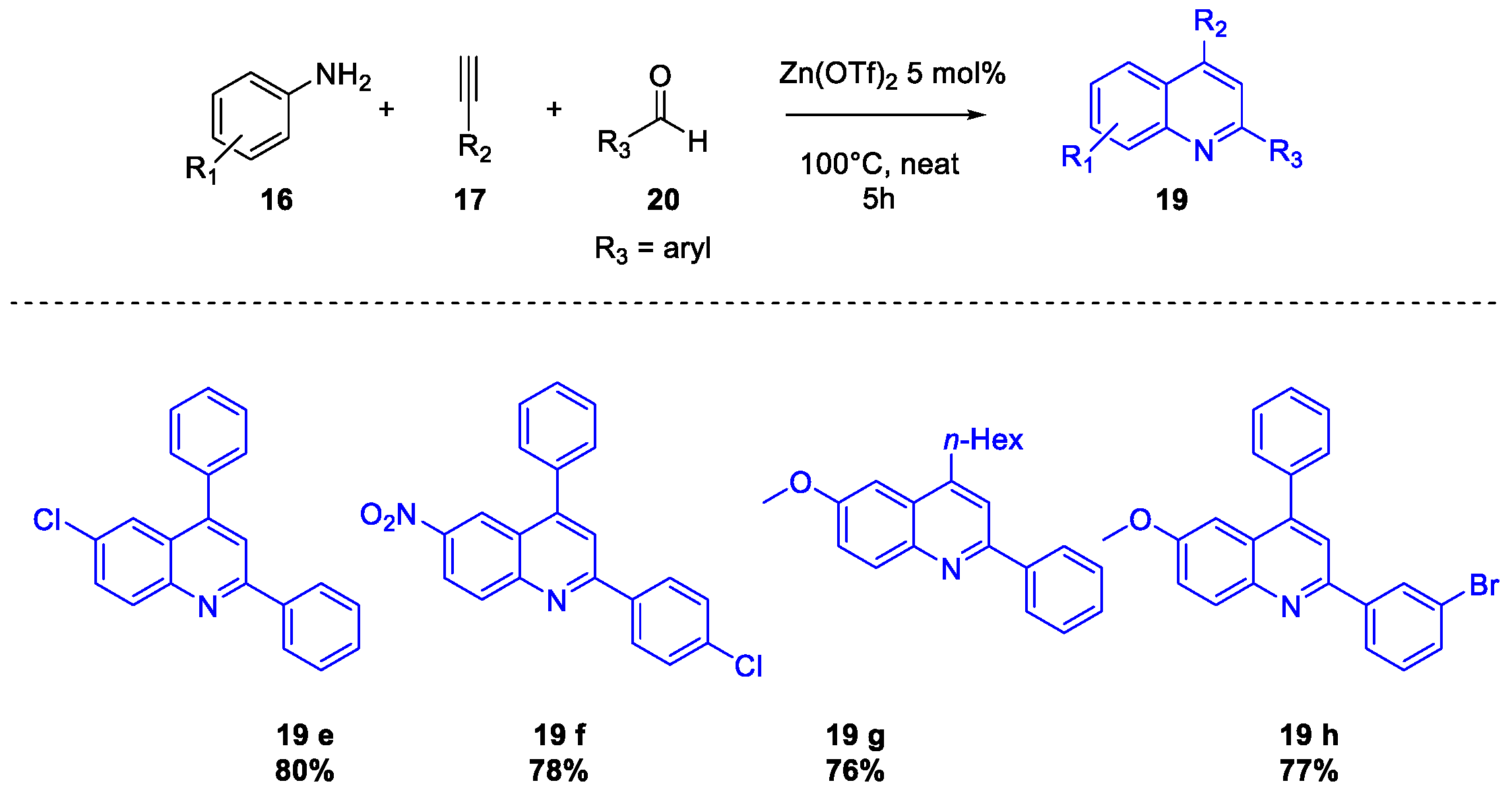
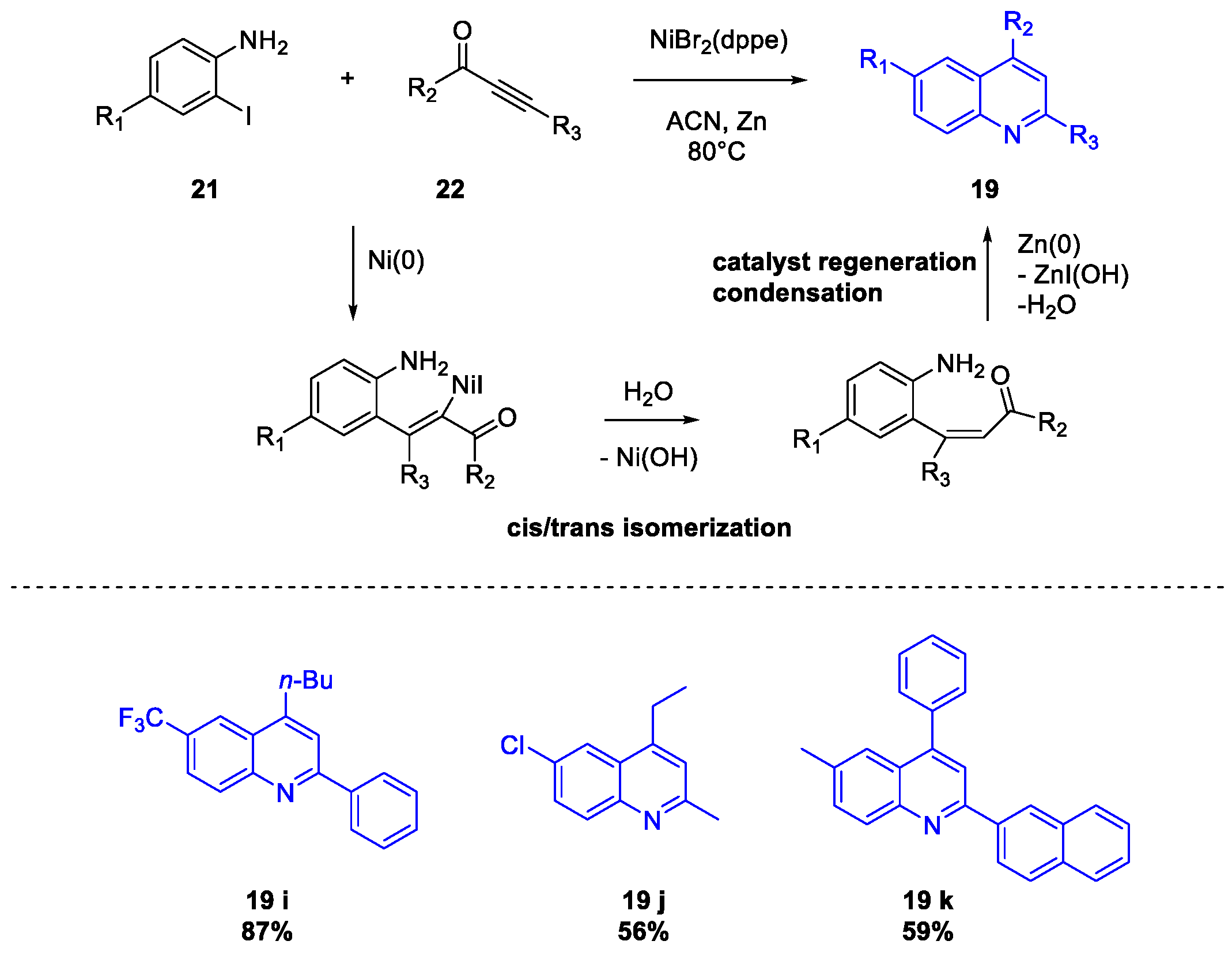
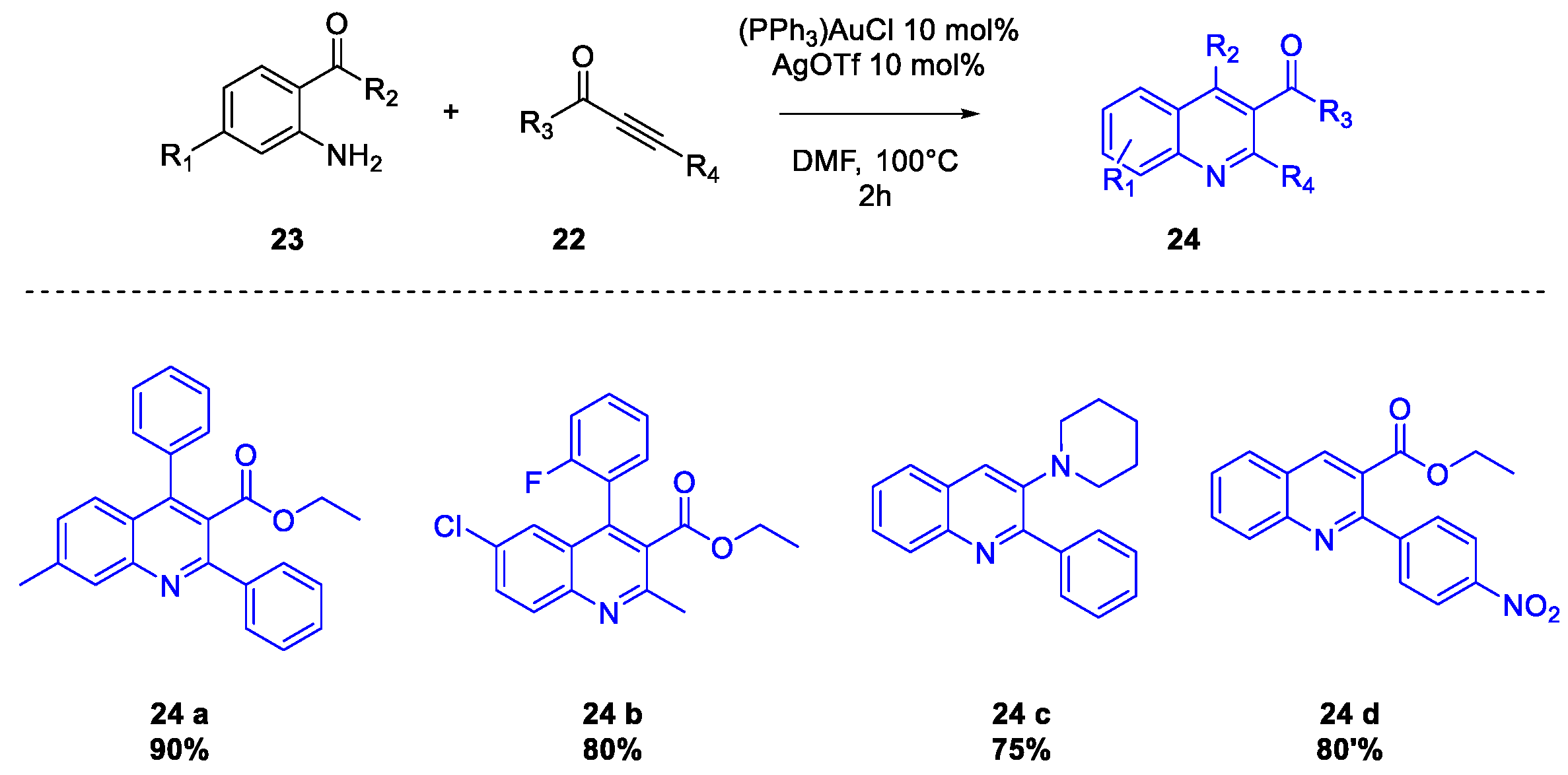
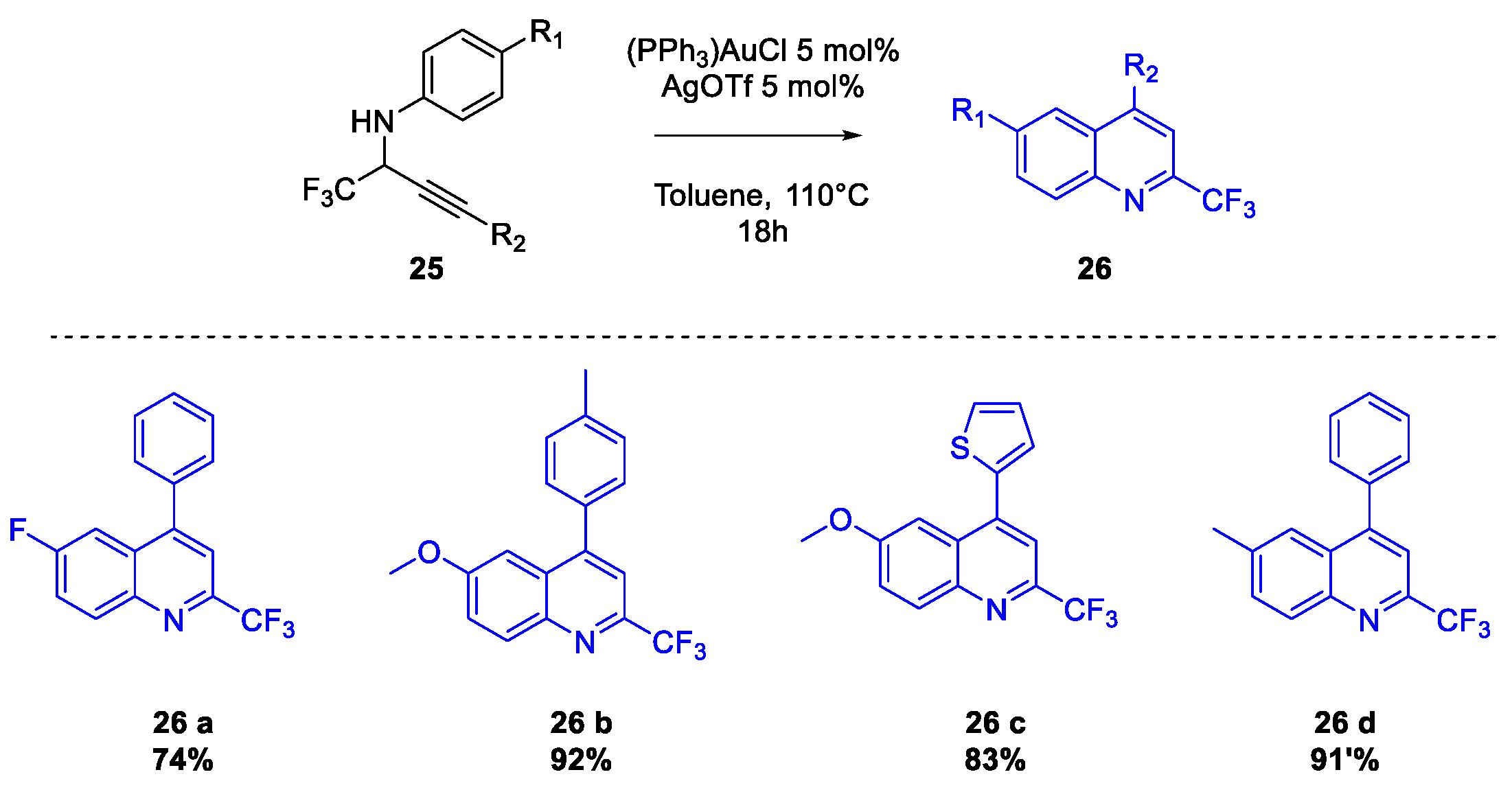
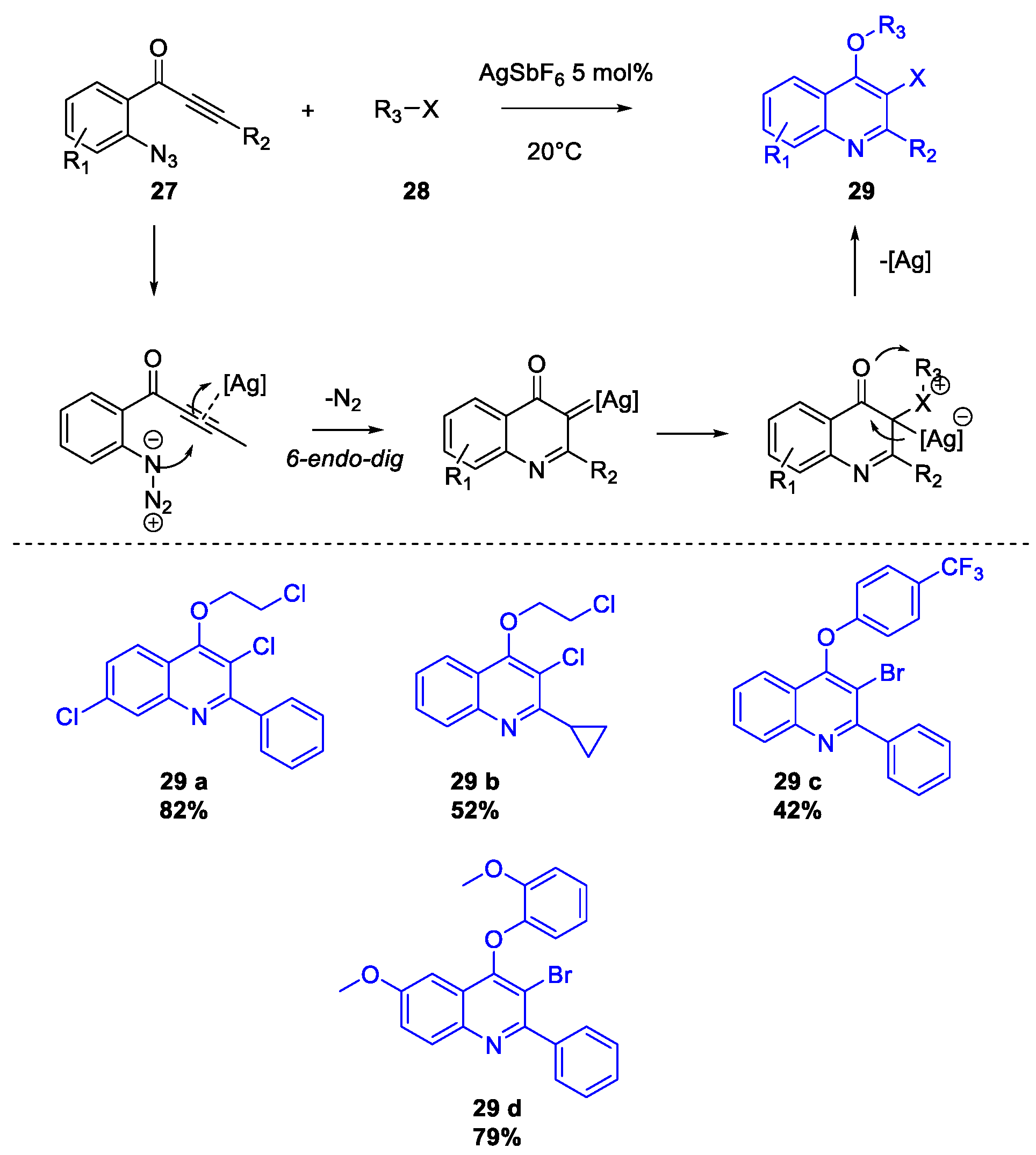
Quinoline Synthesis: Metal-Promoted Annulation
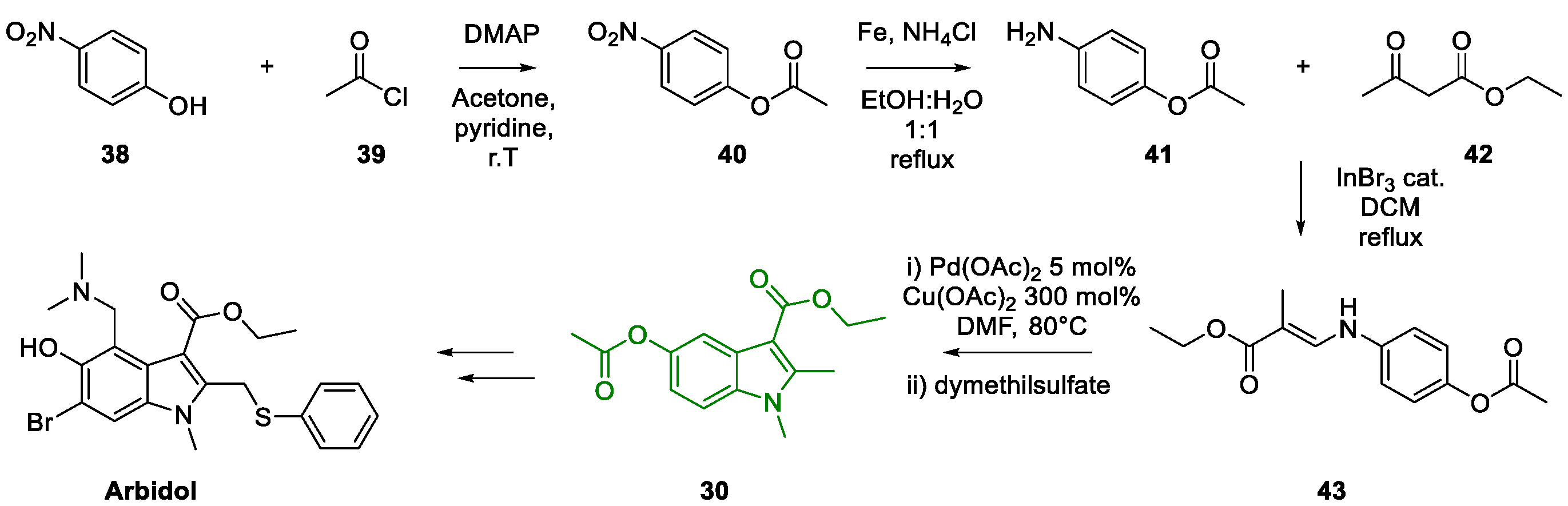
3. Arbidol
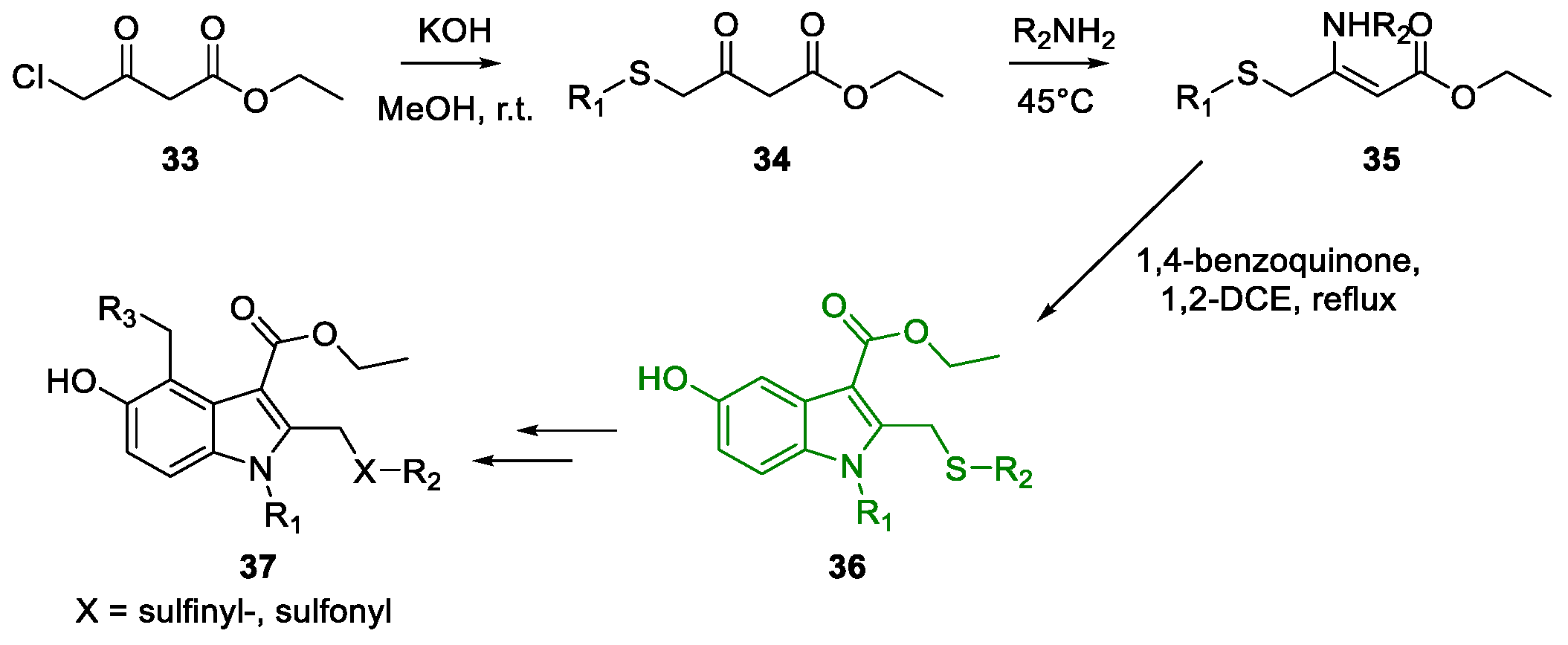
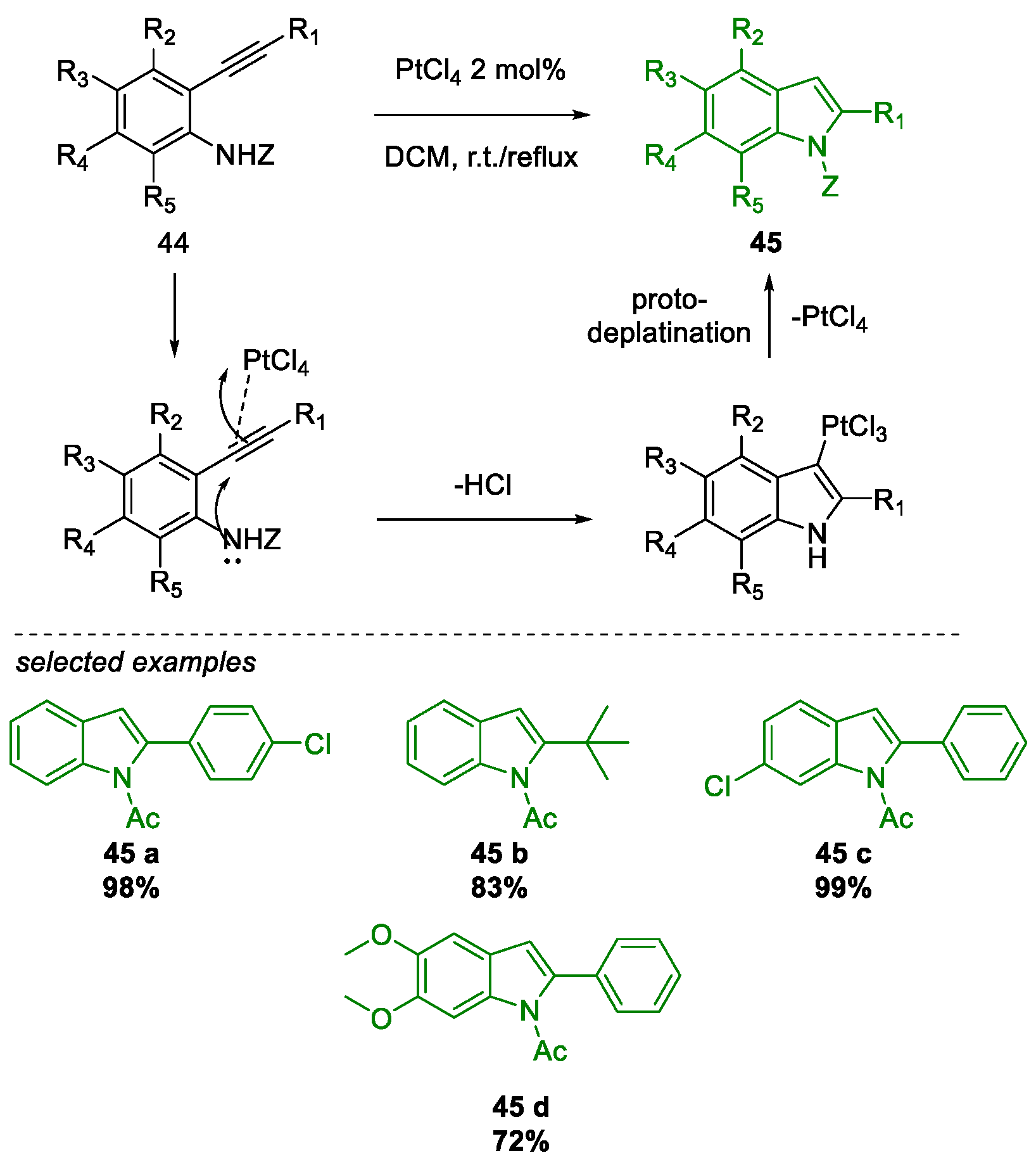
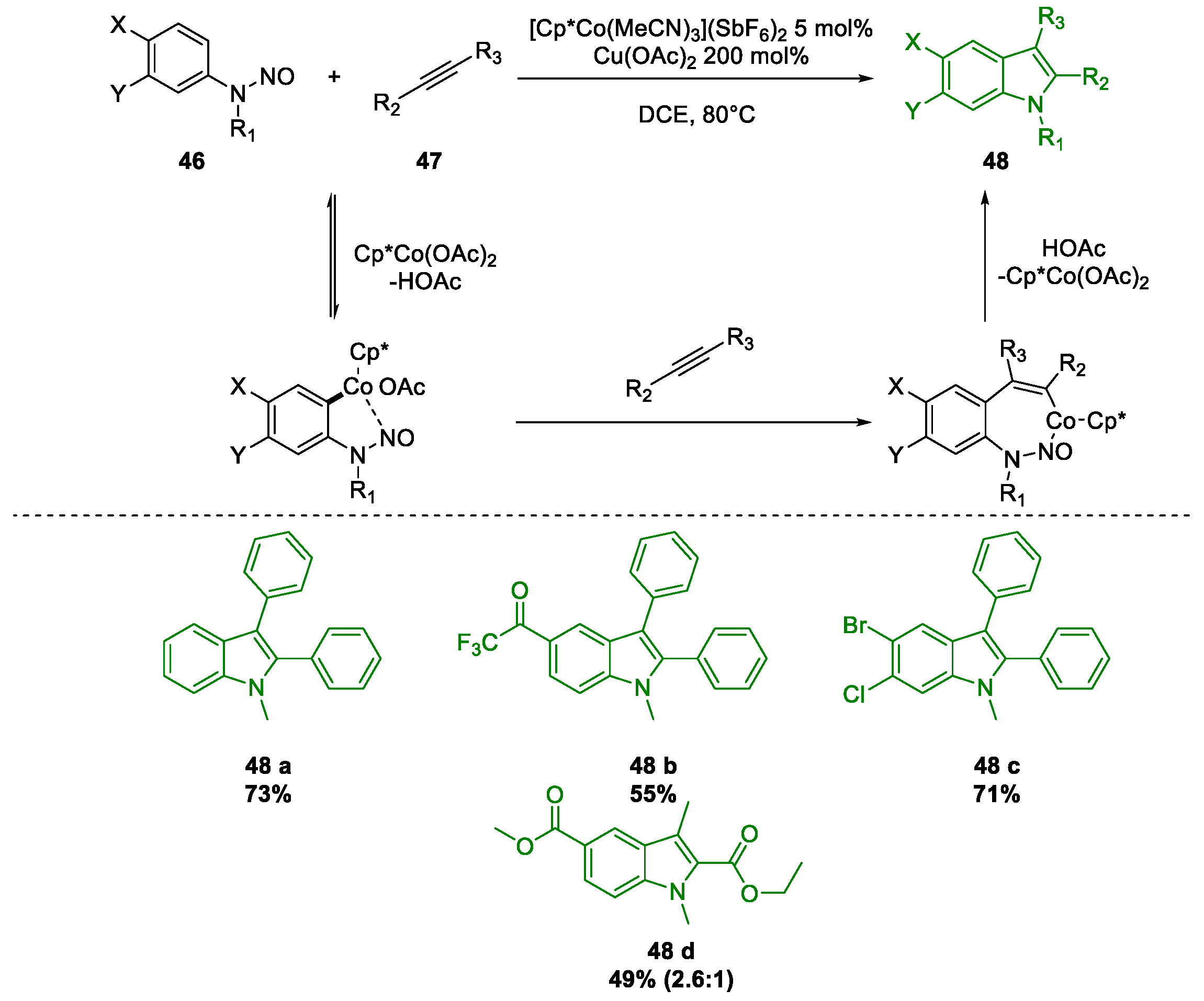
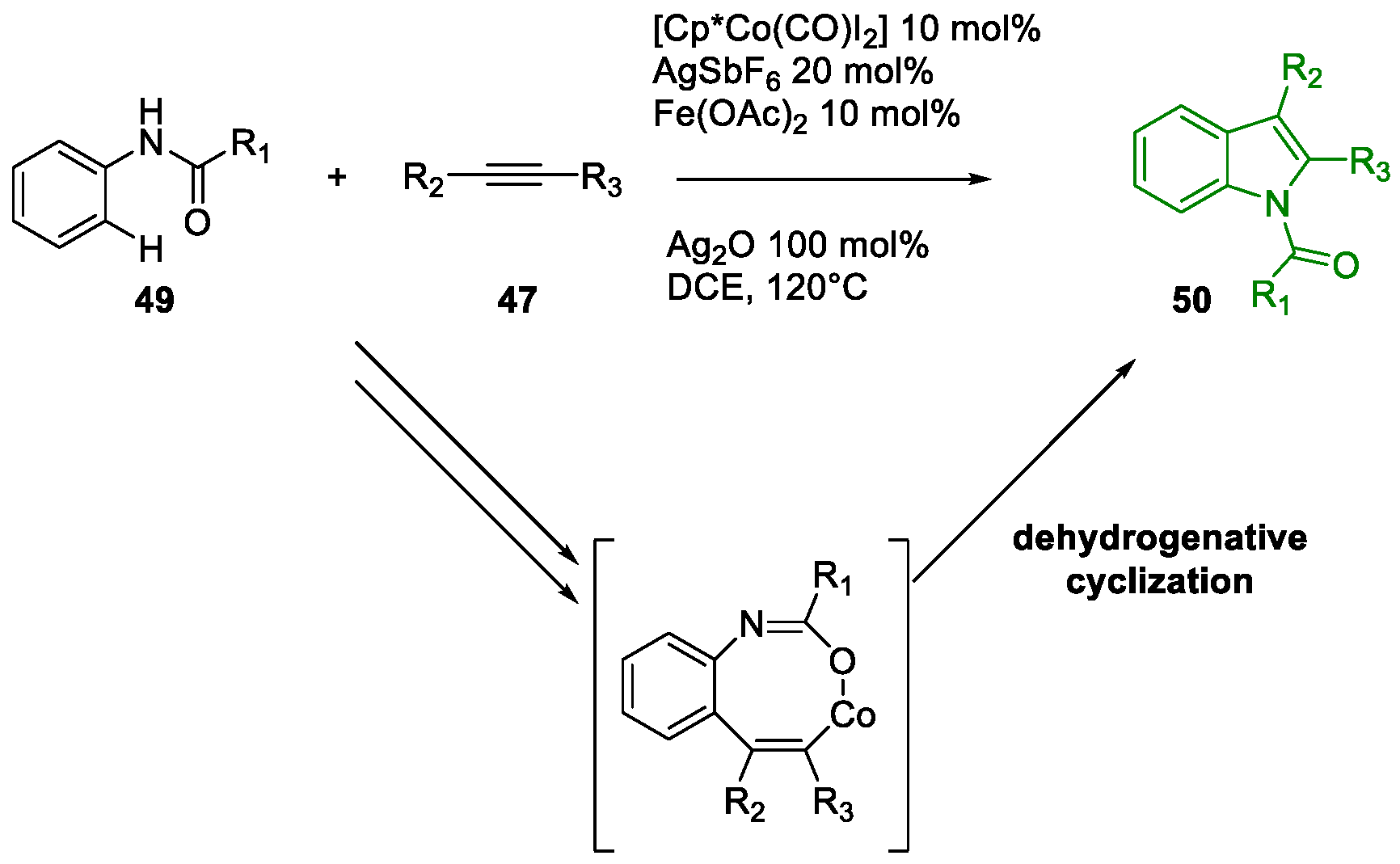
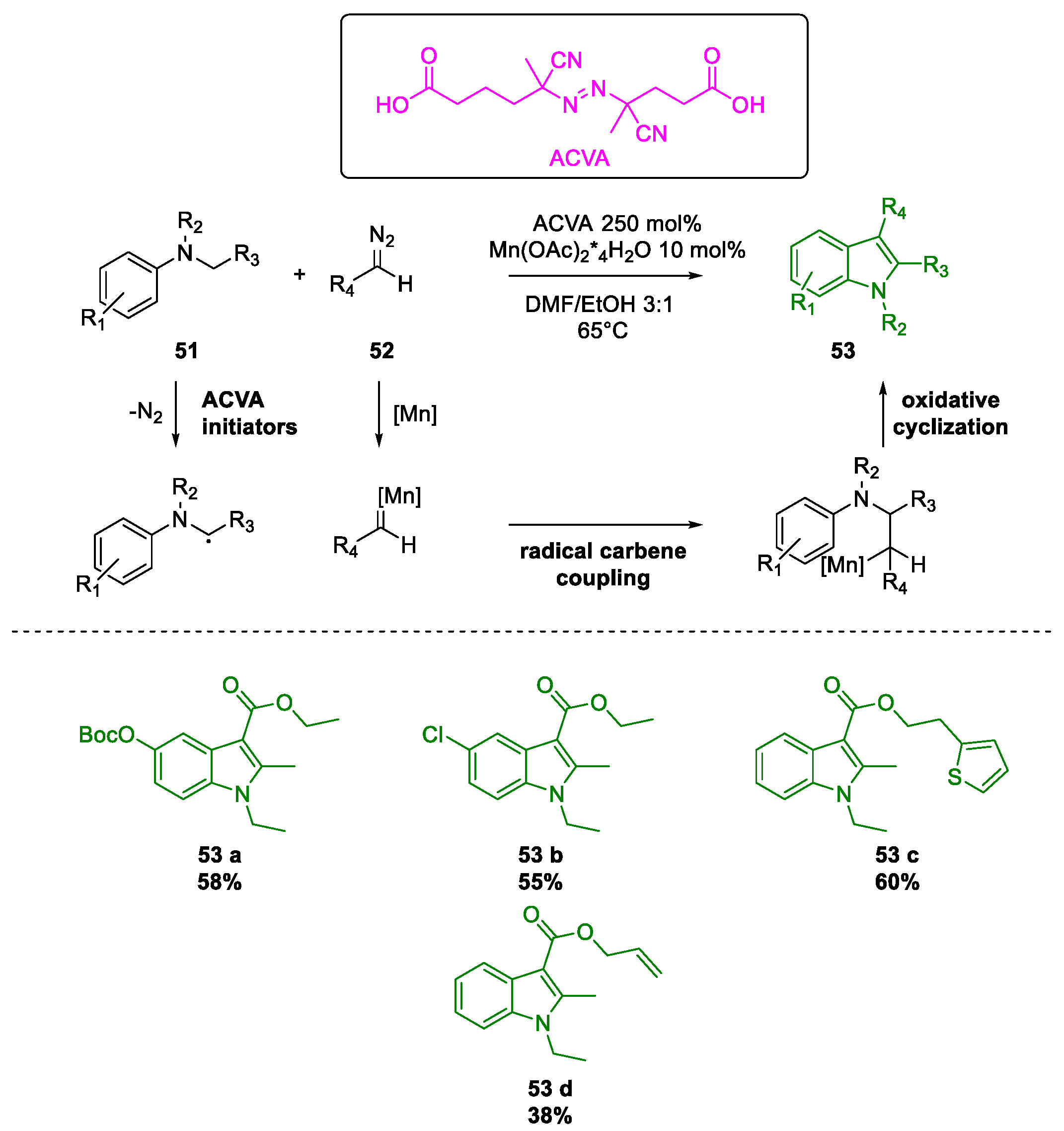
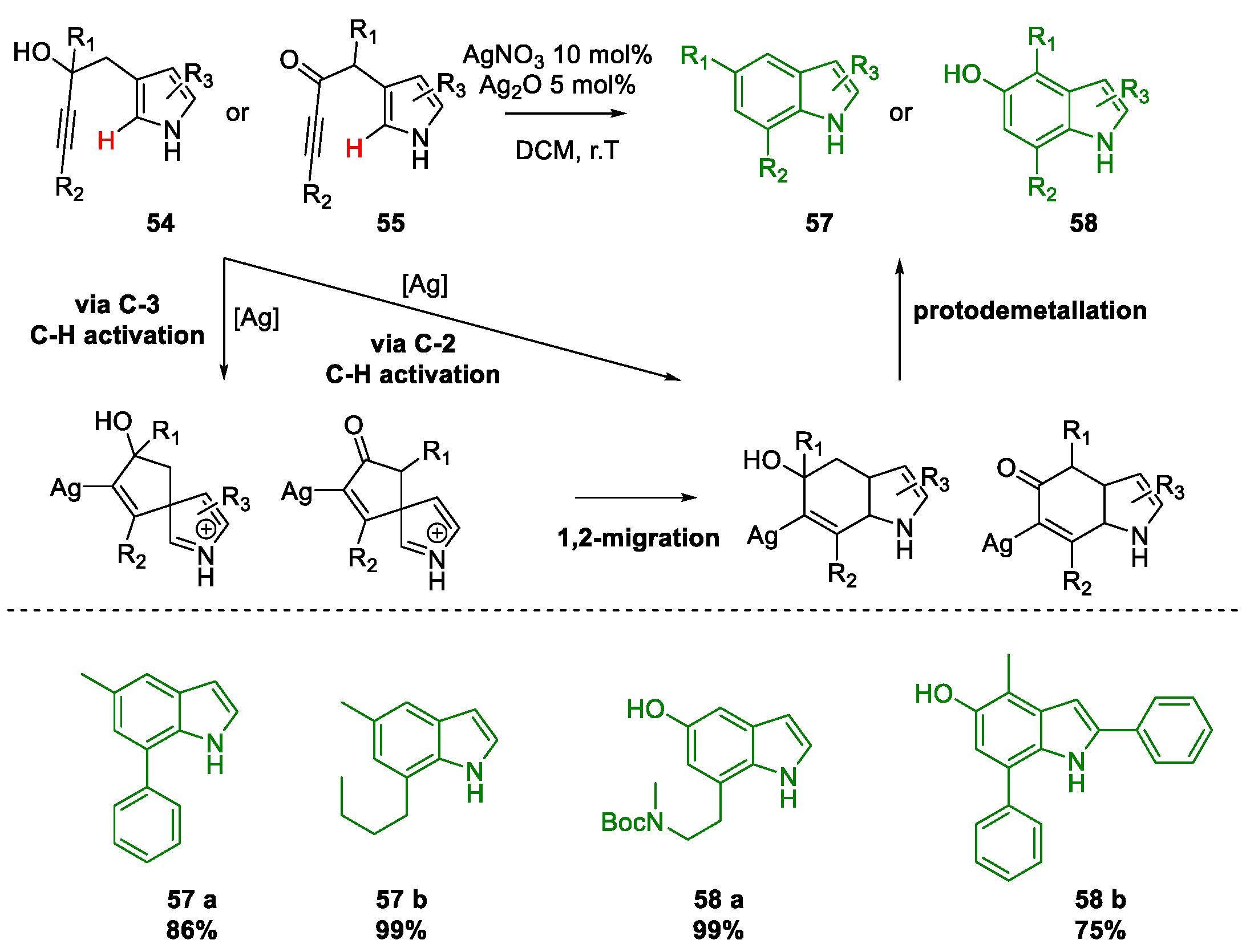
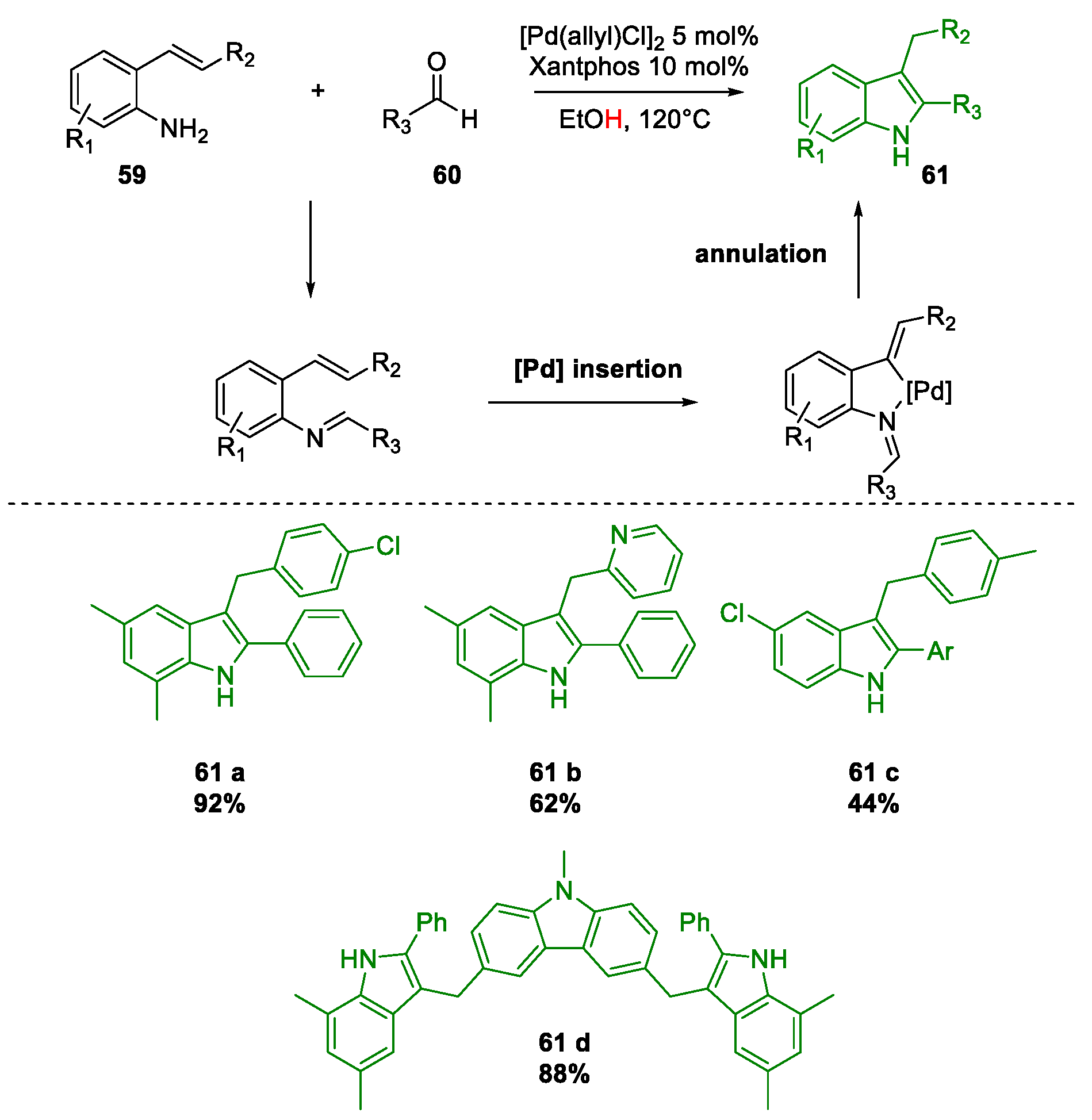
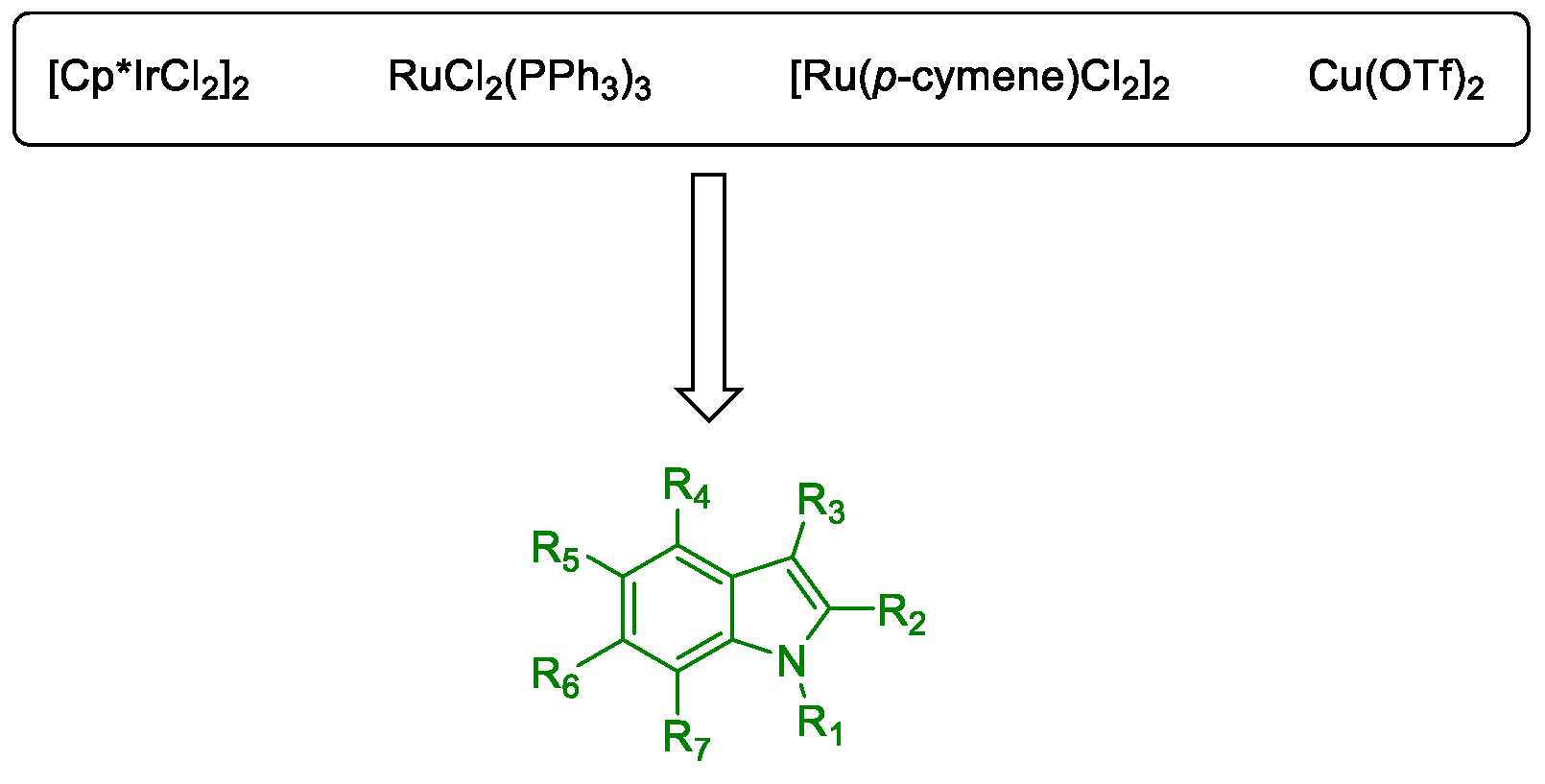
Metal-Promoted Heterocyclization to Achieve Polysubstituted Indoles
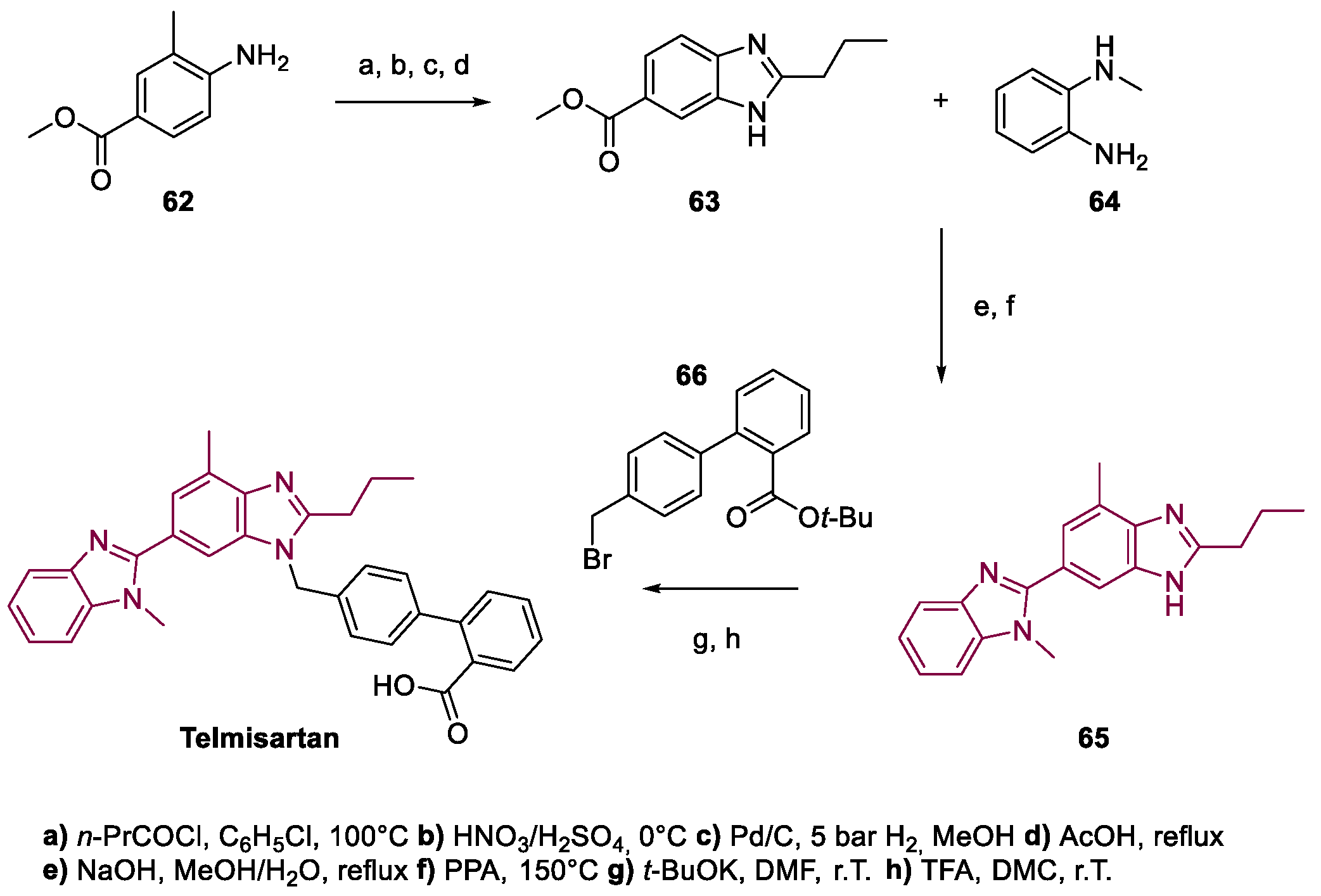
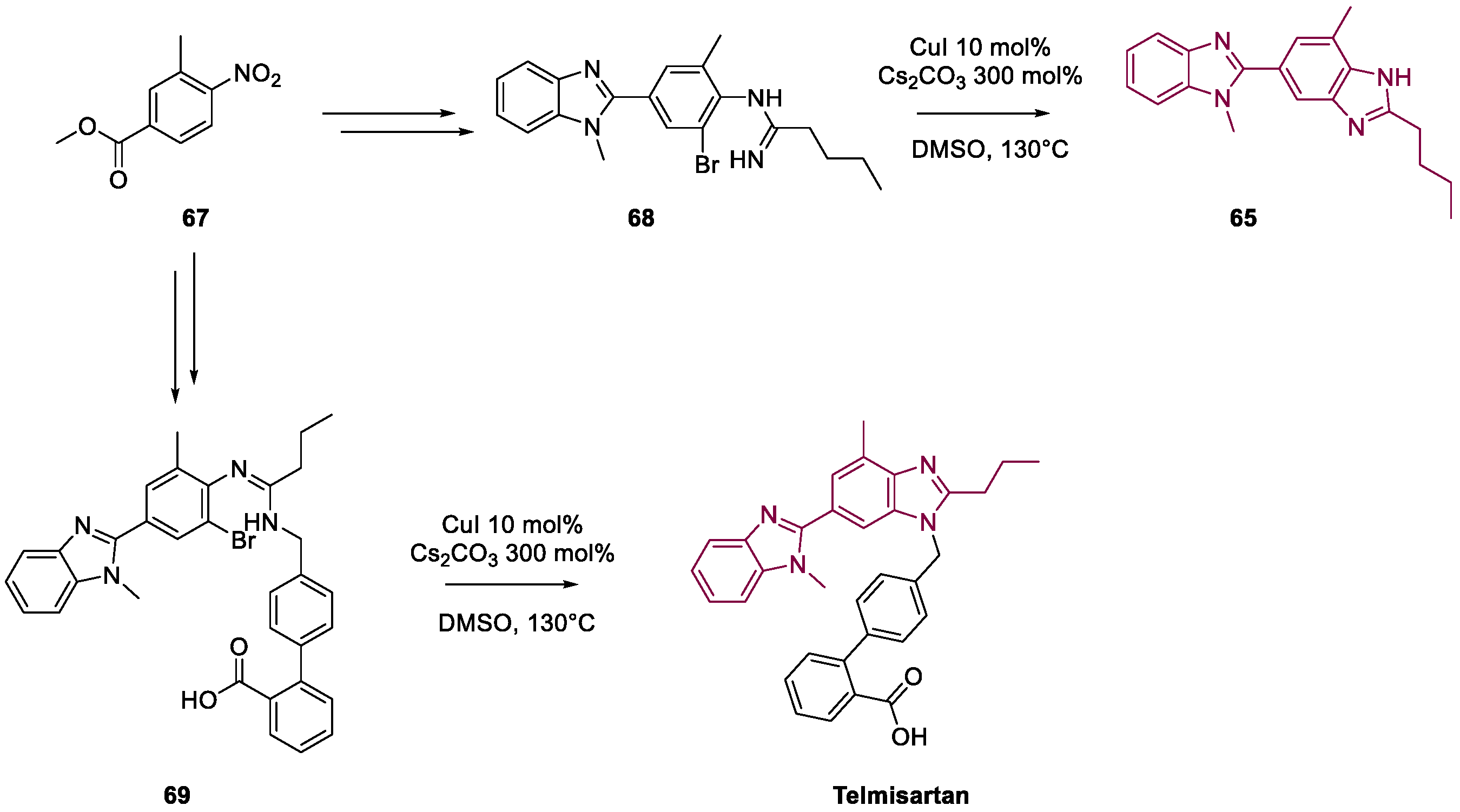
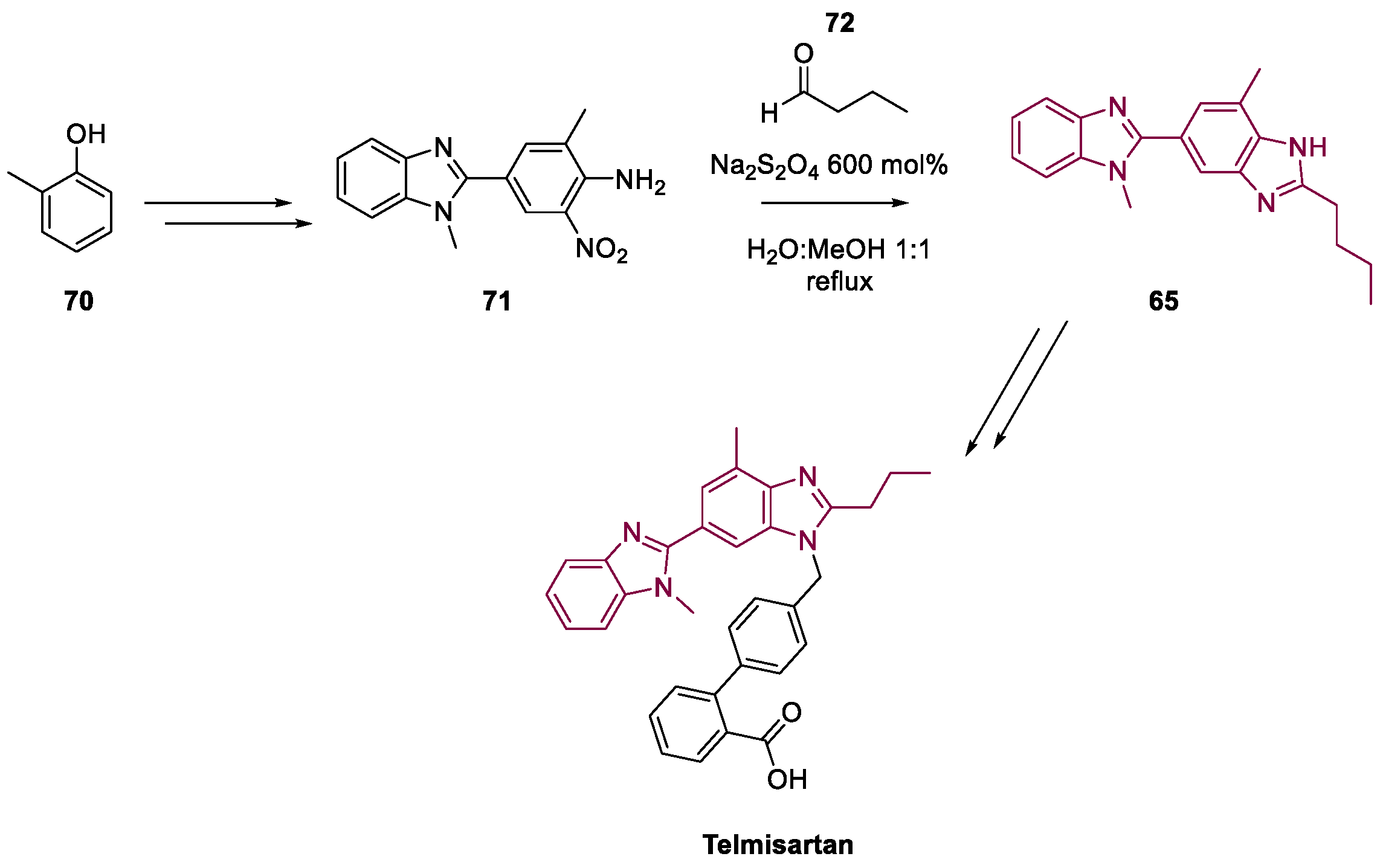
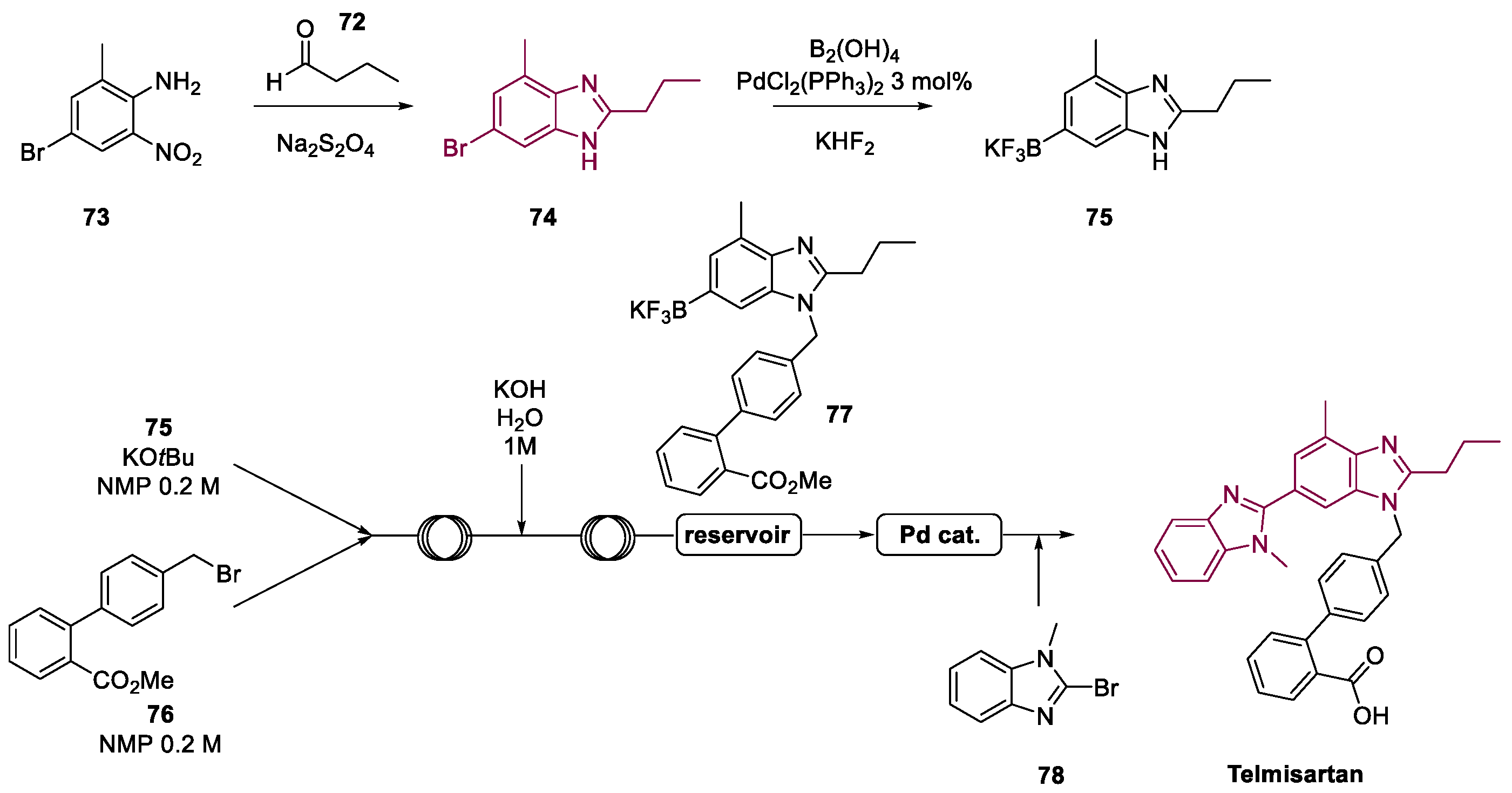
4. Telmisartan
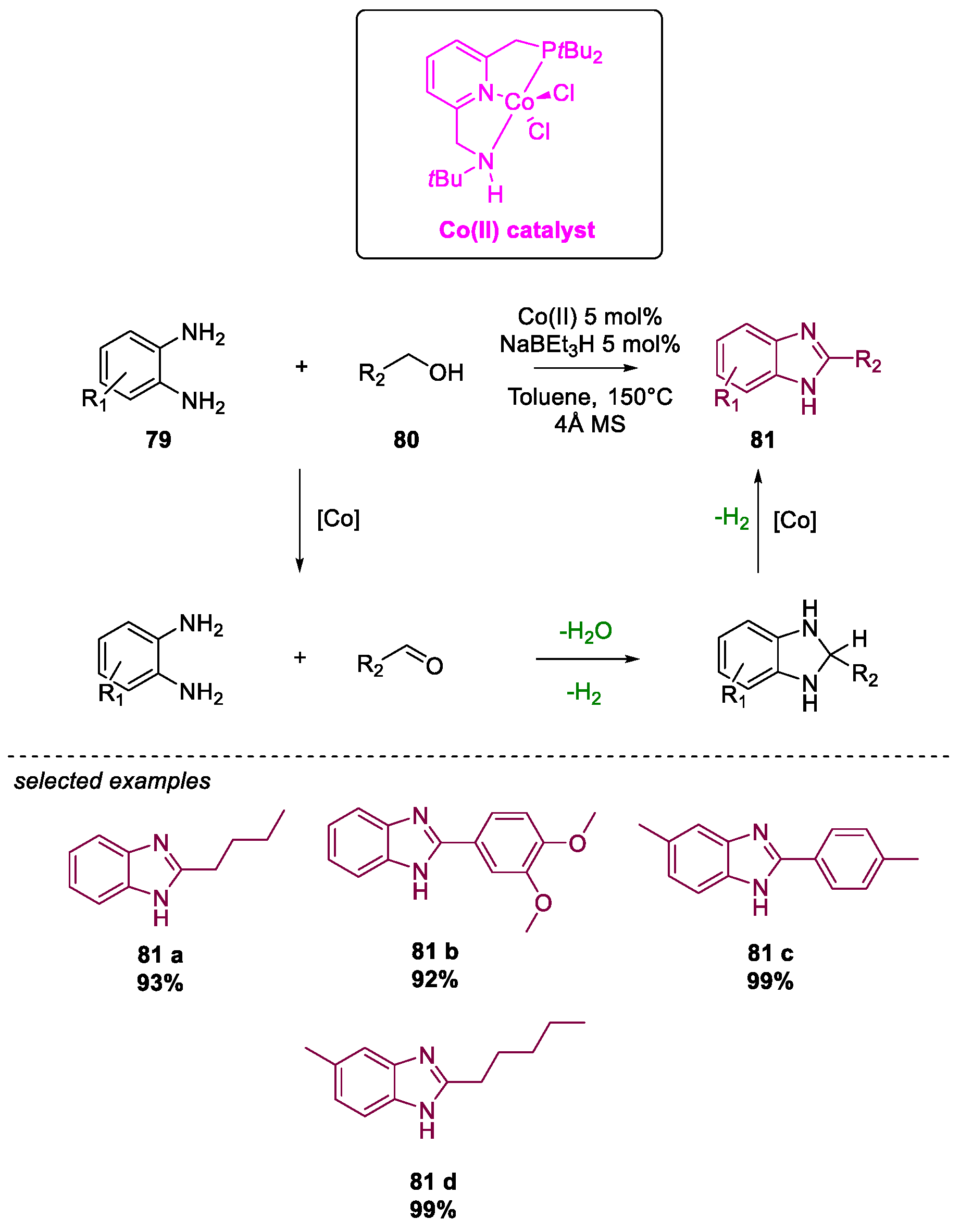
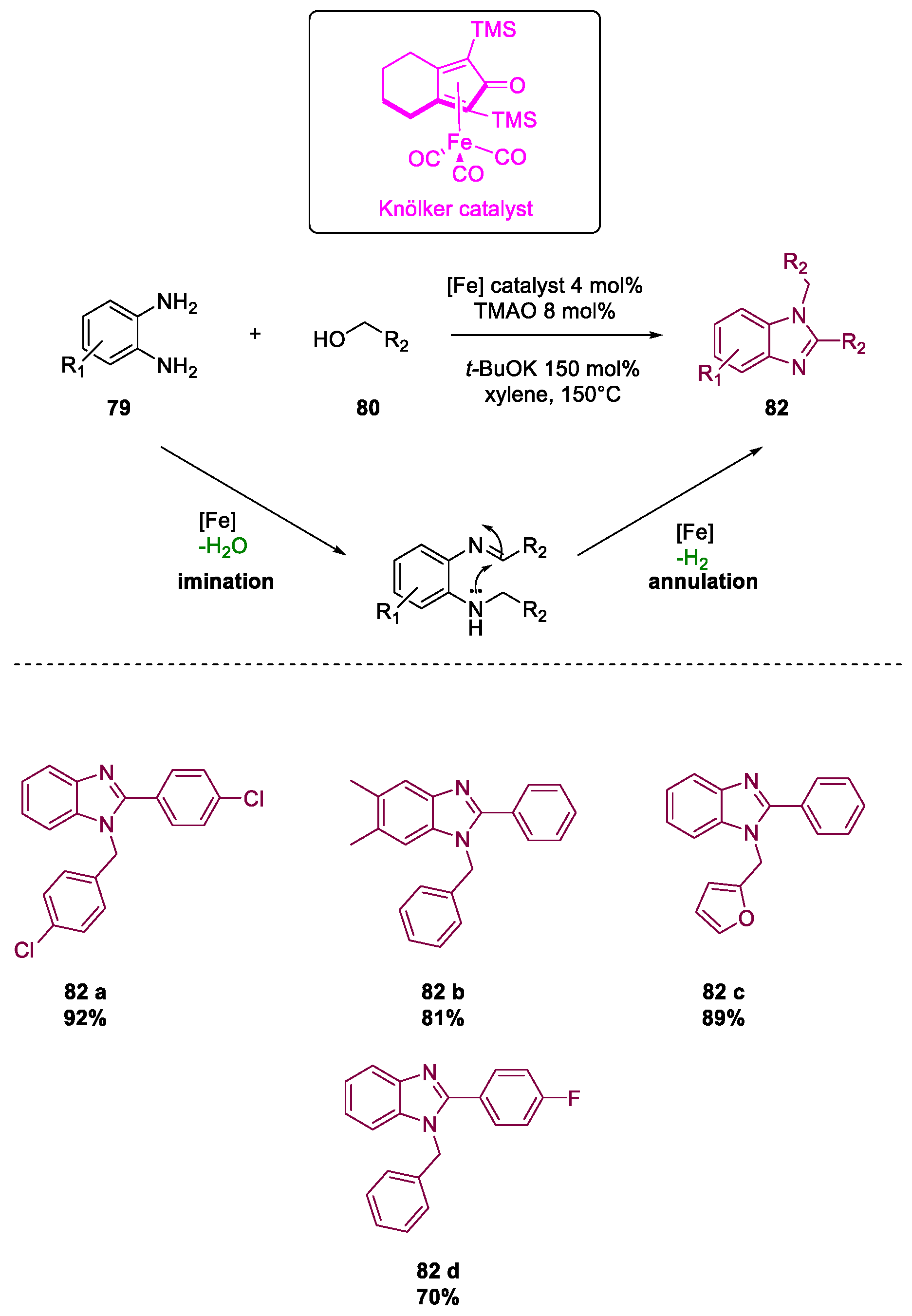
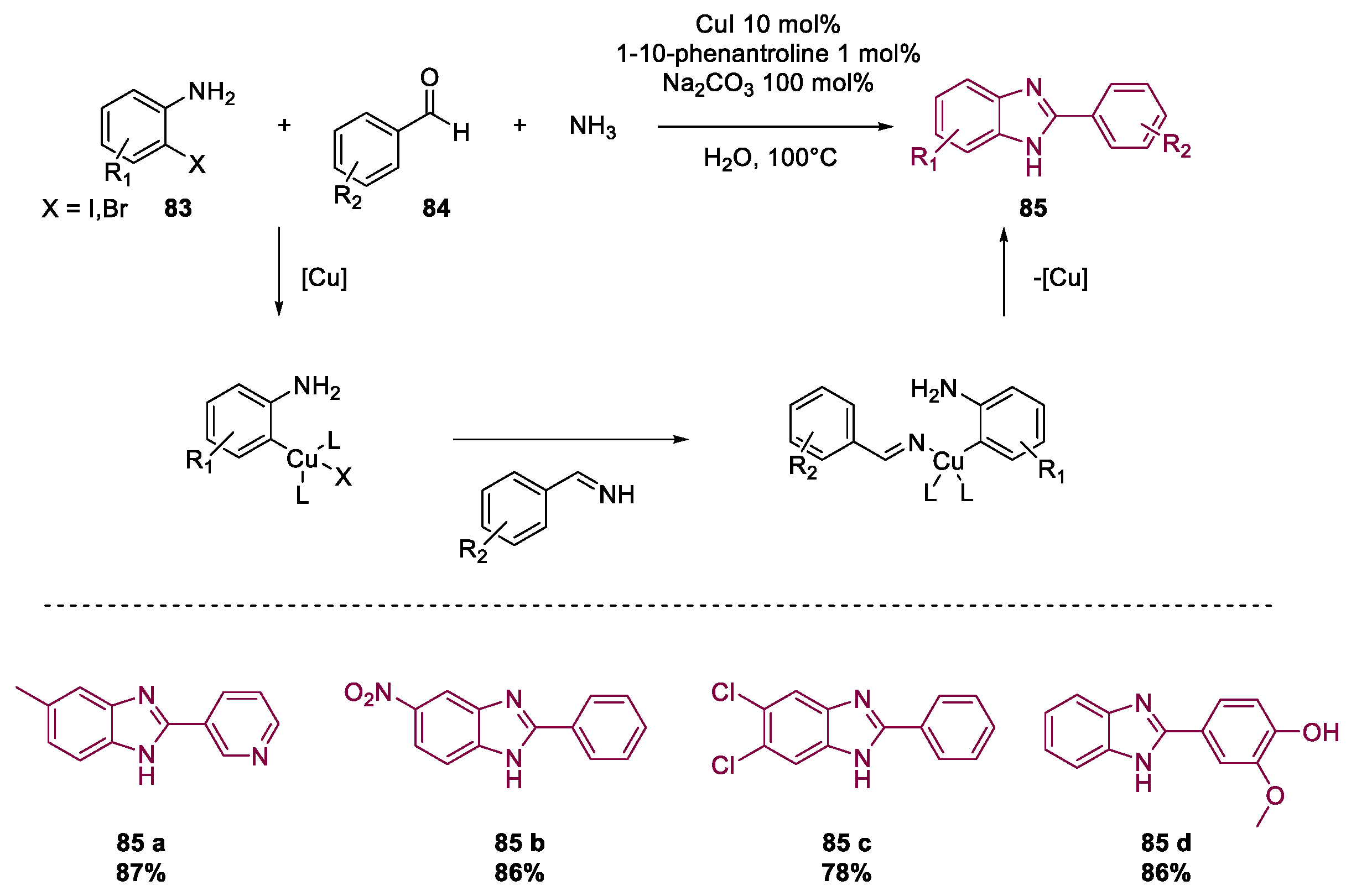
Metal-Catalyzed Annulation in Benzimidazole Synthesis
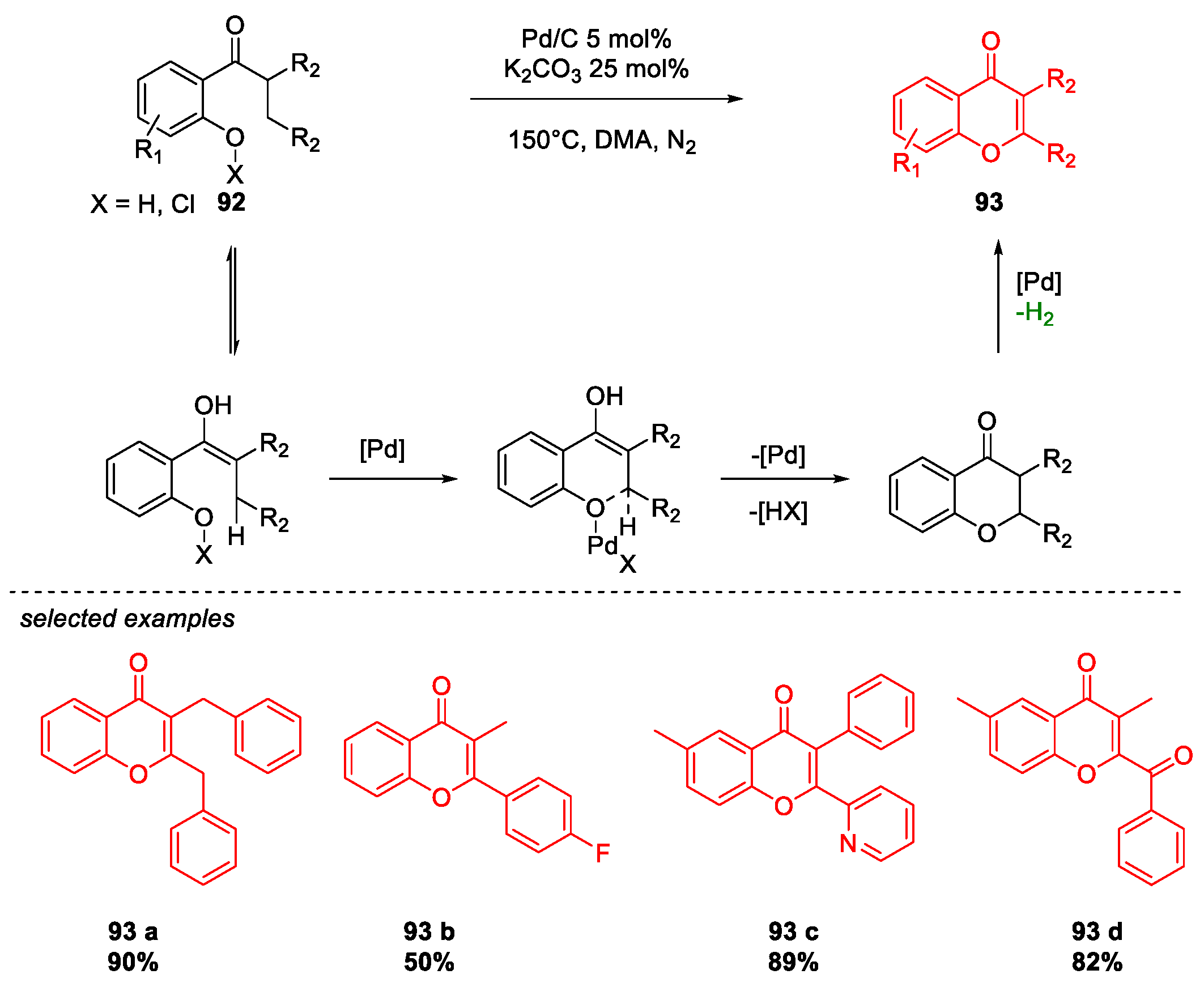
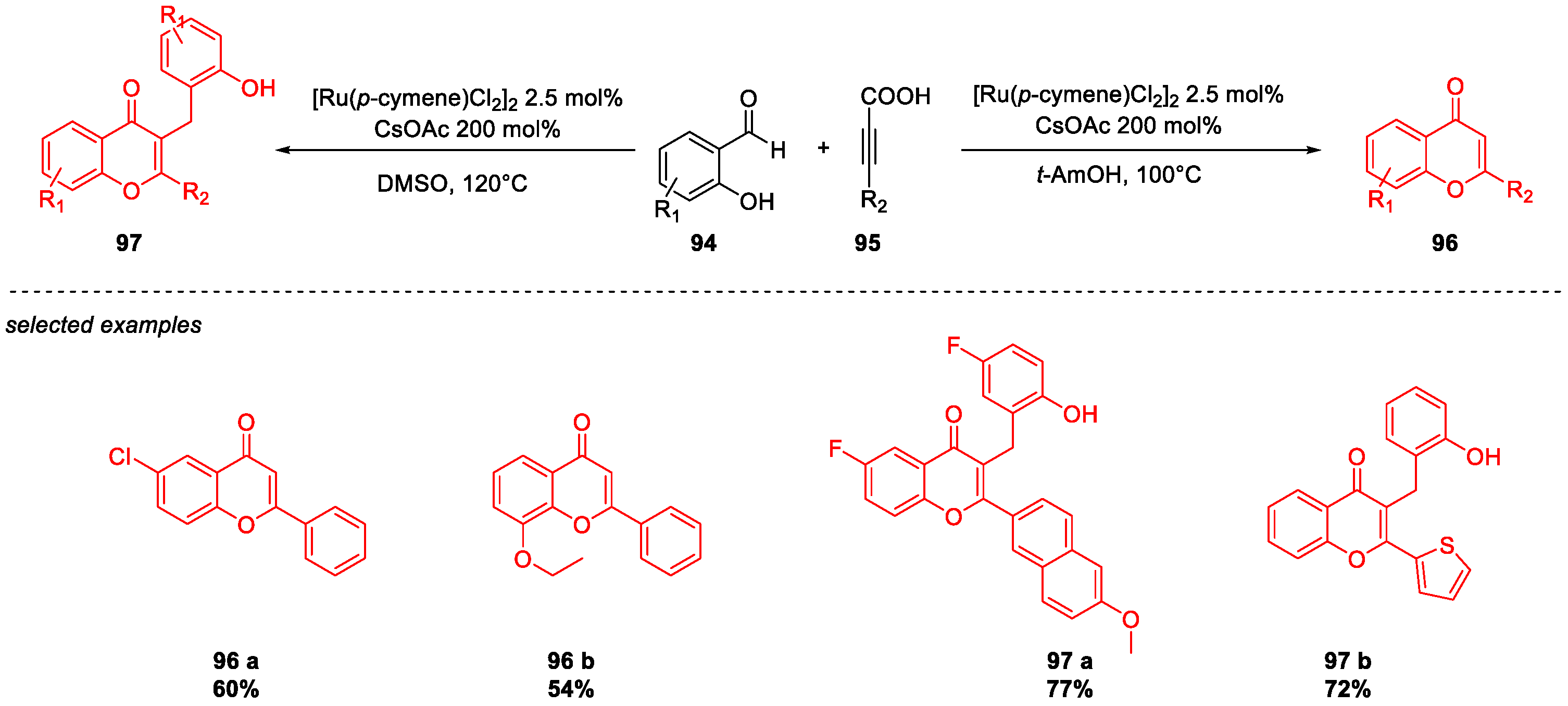
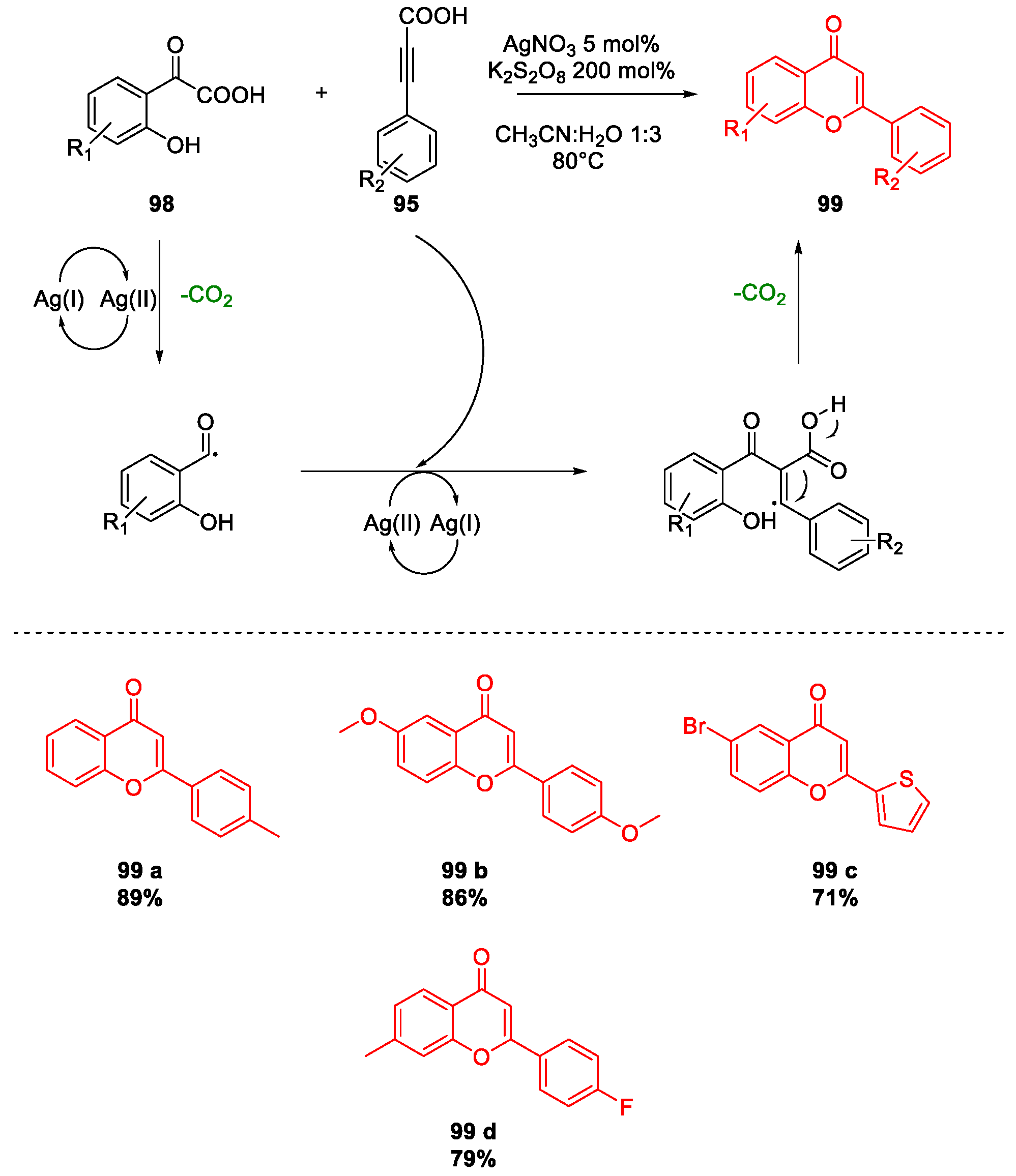
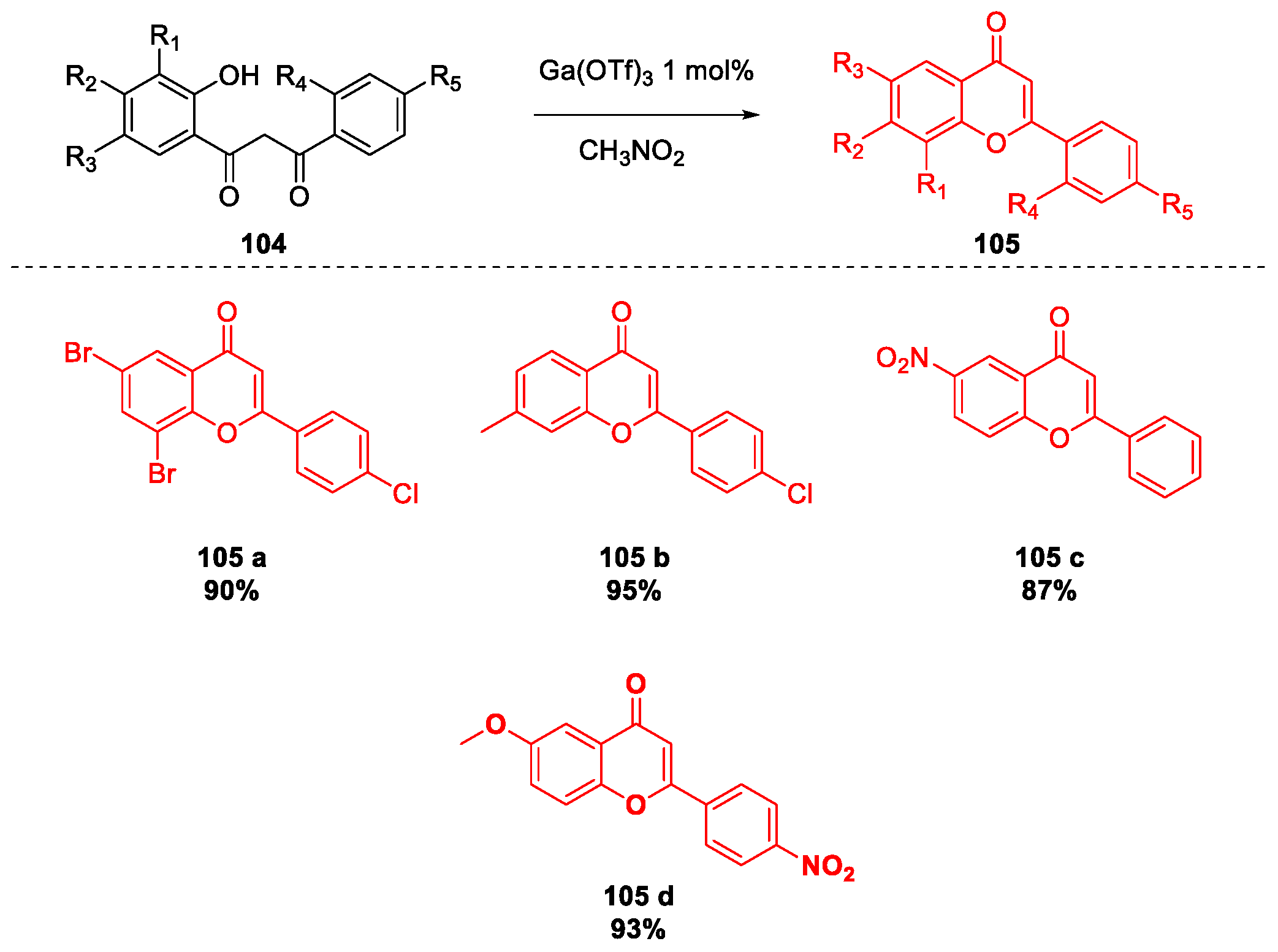
5. Quercetin and Luteolin
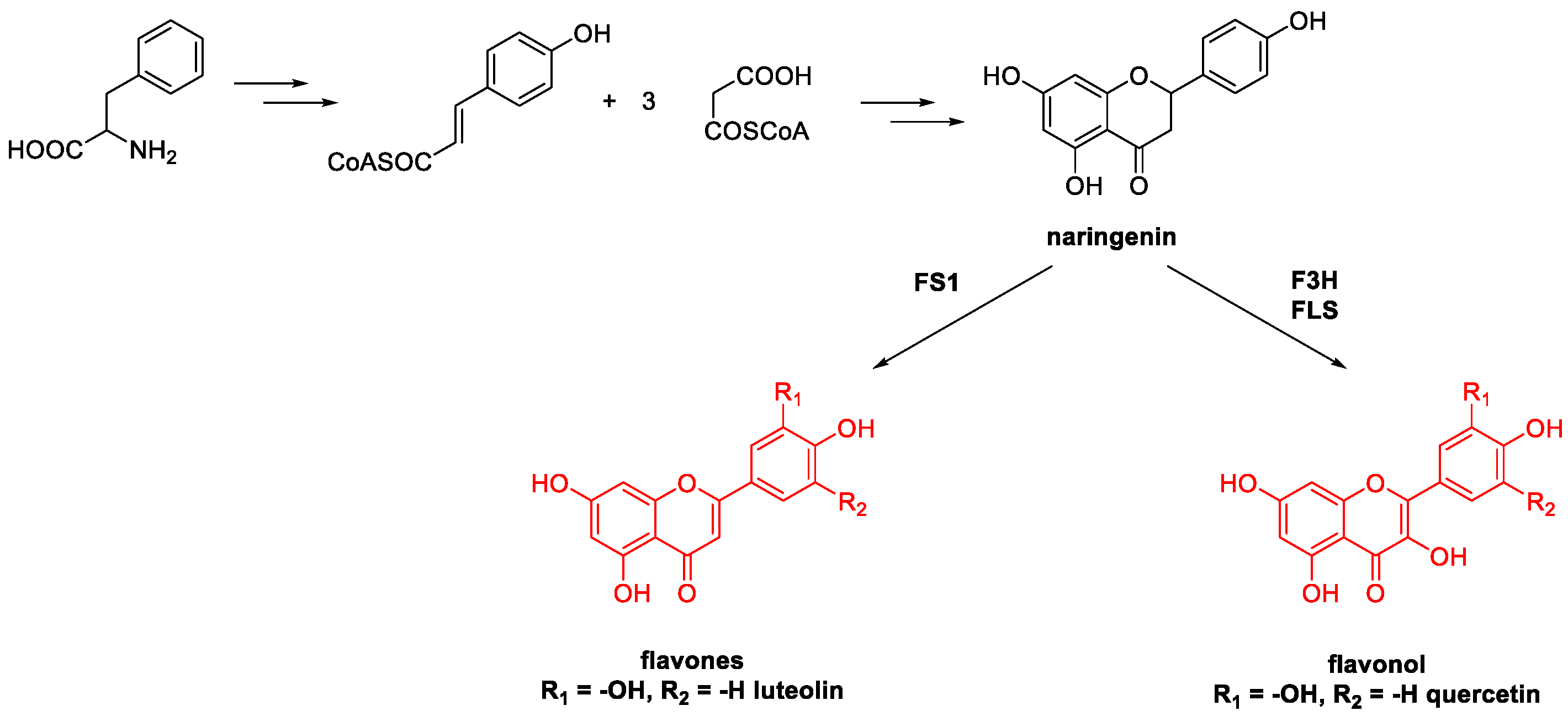
Metal-Catalyzed O-Heterocyclization to Flavonoids
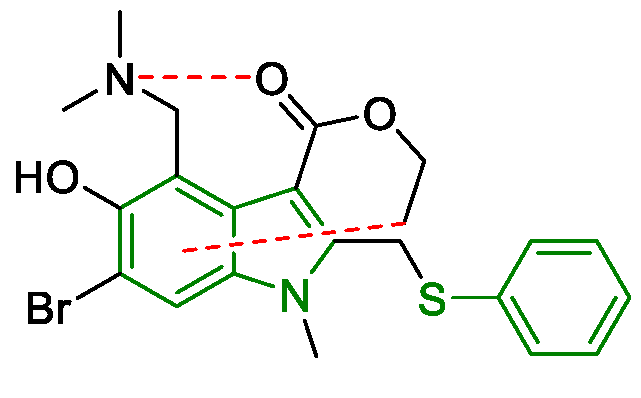
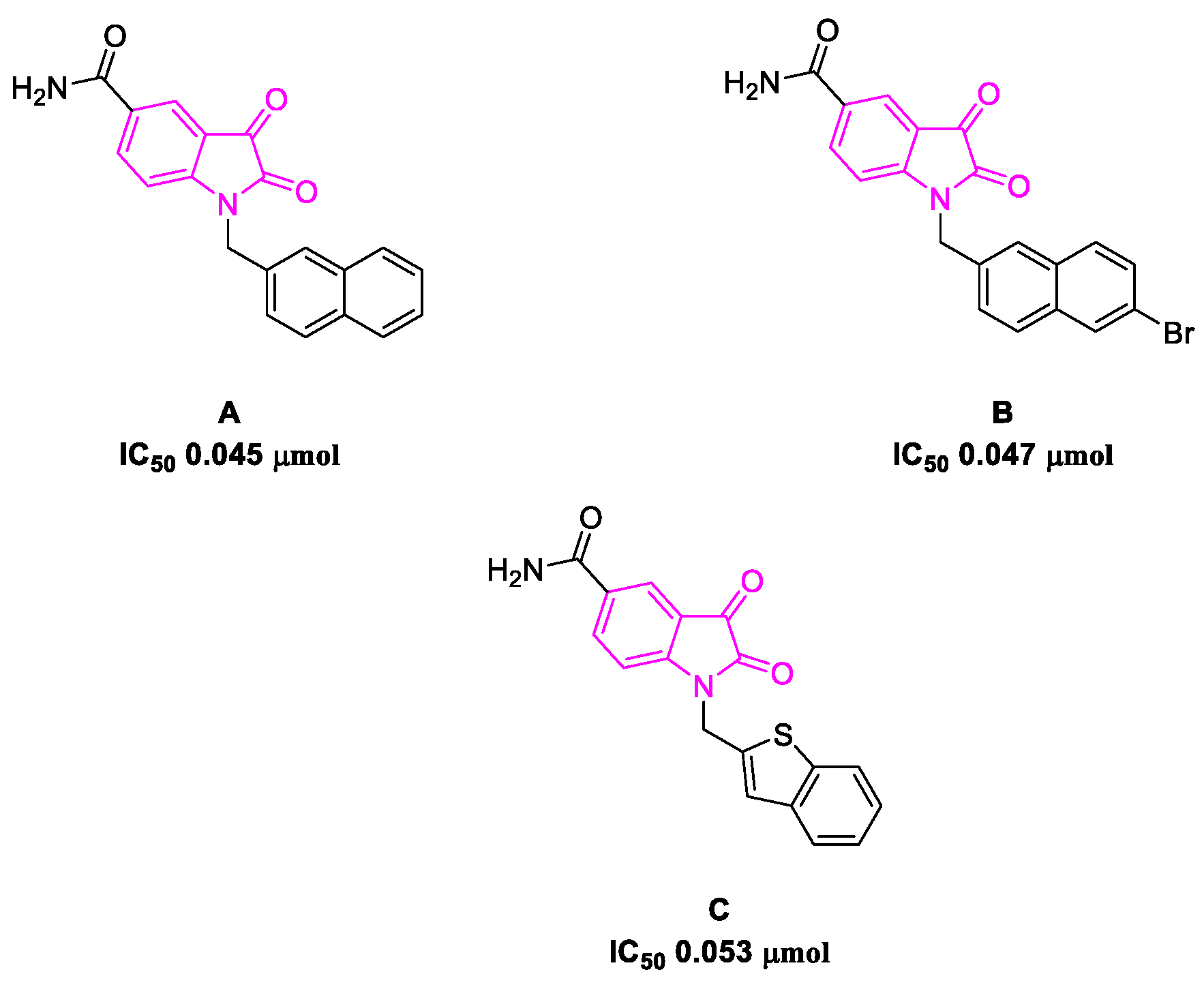
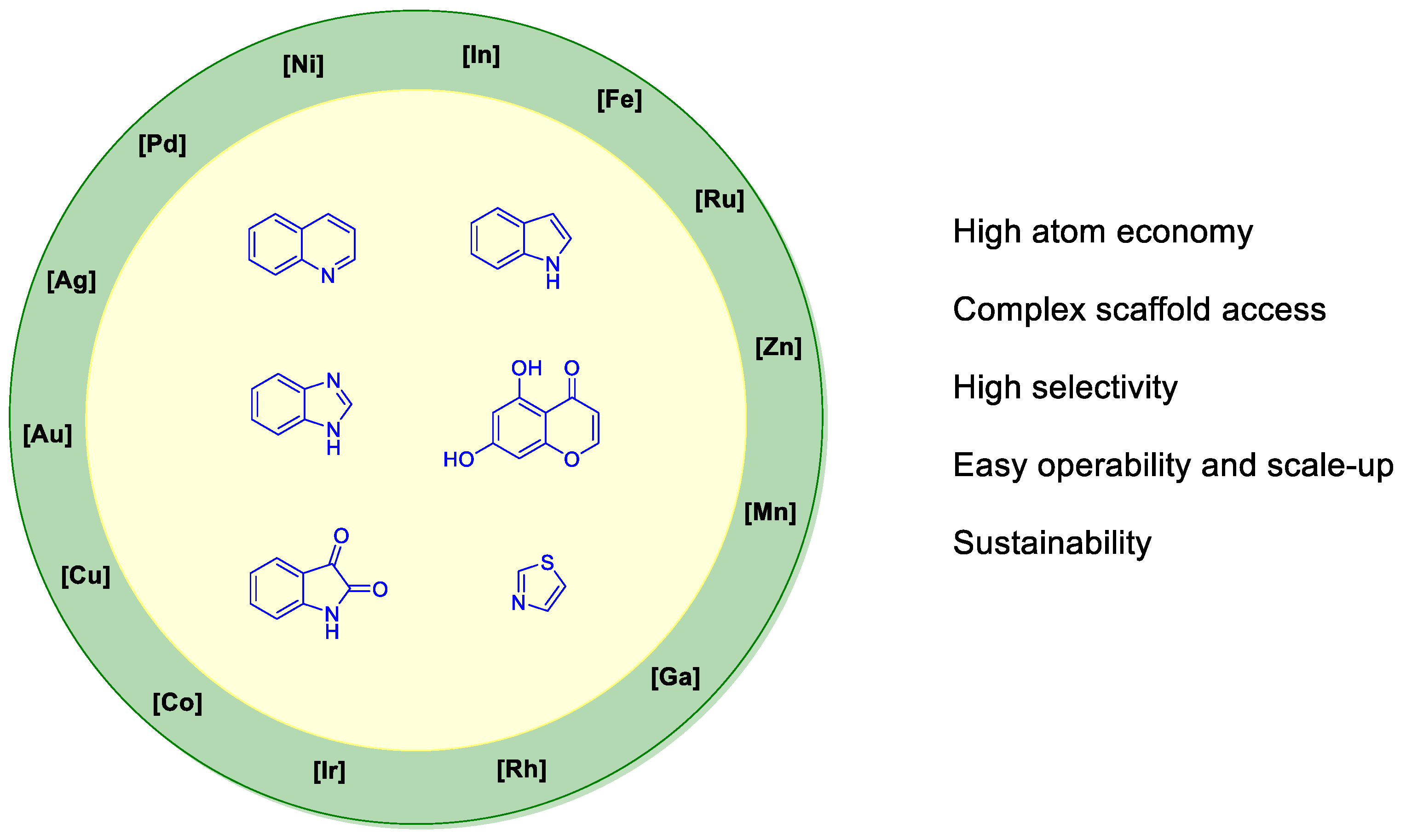
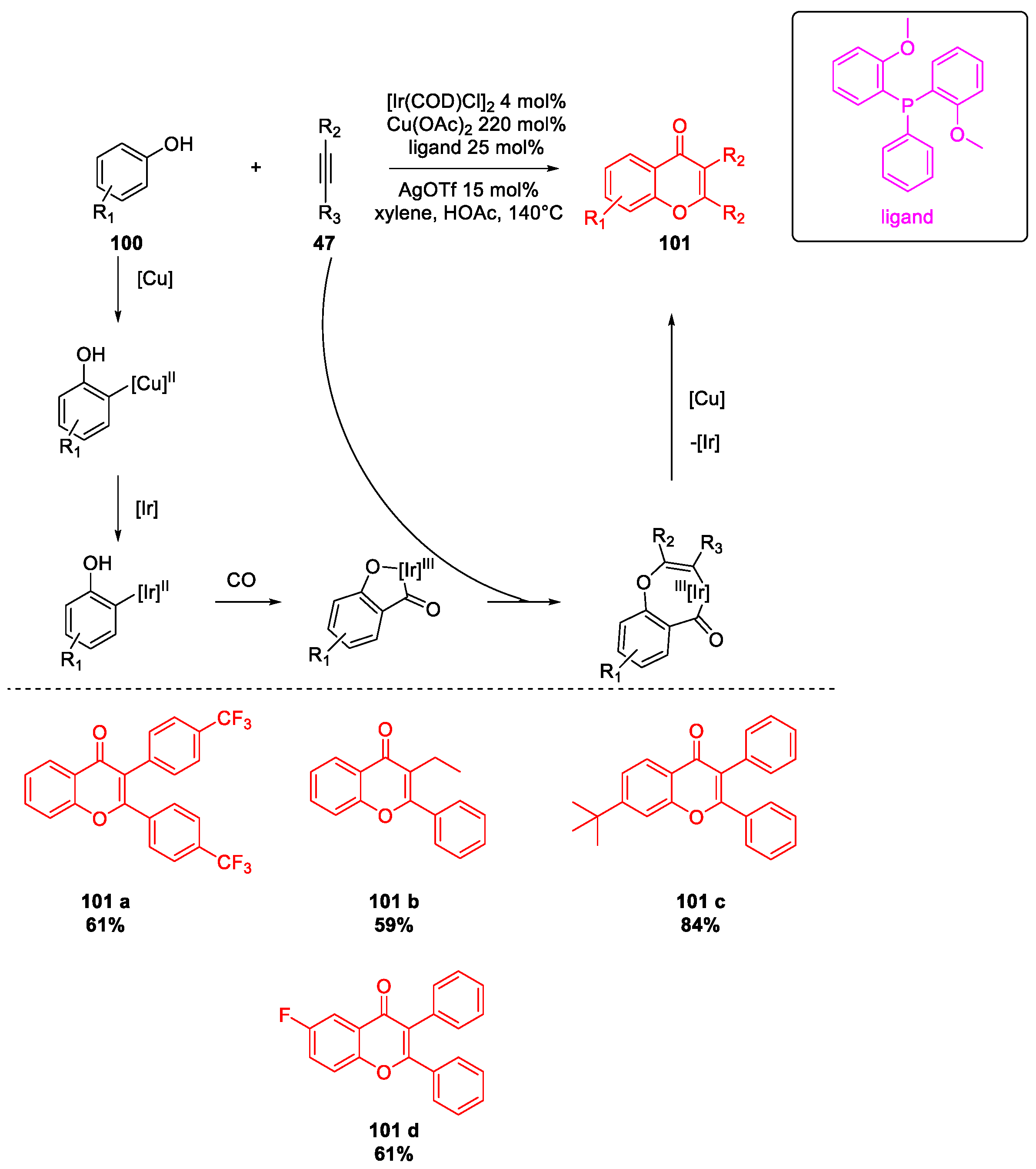
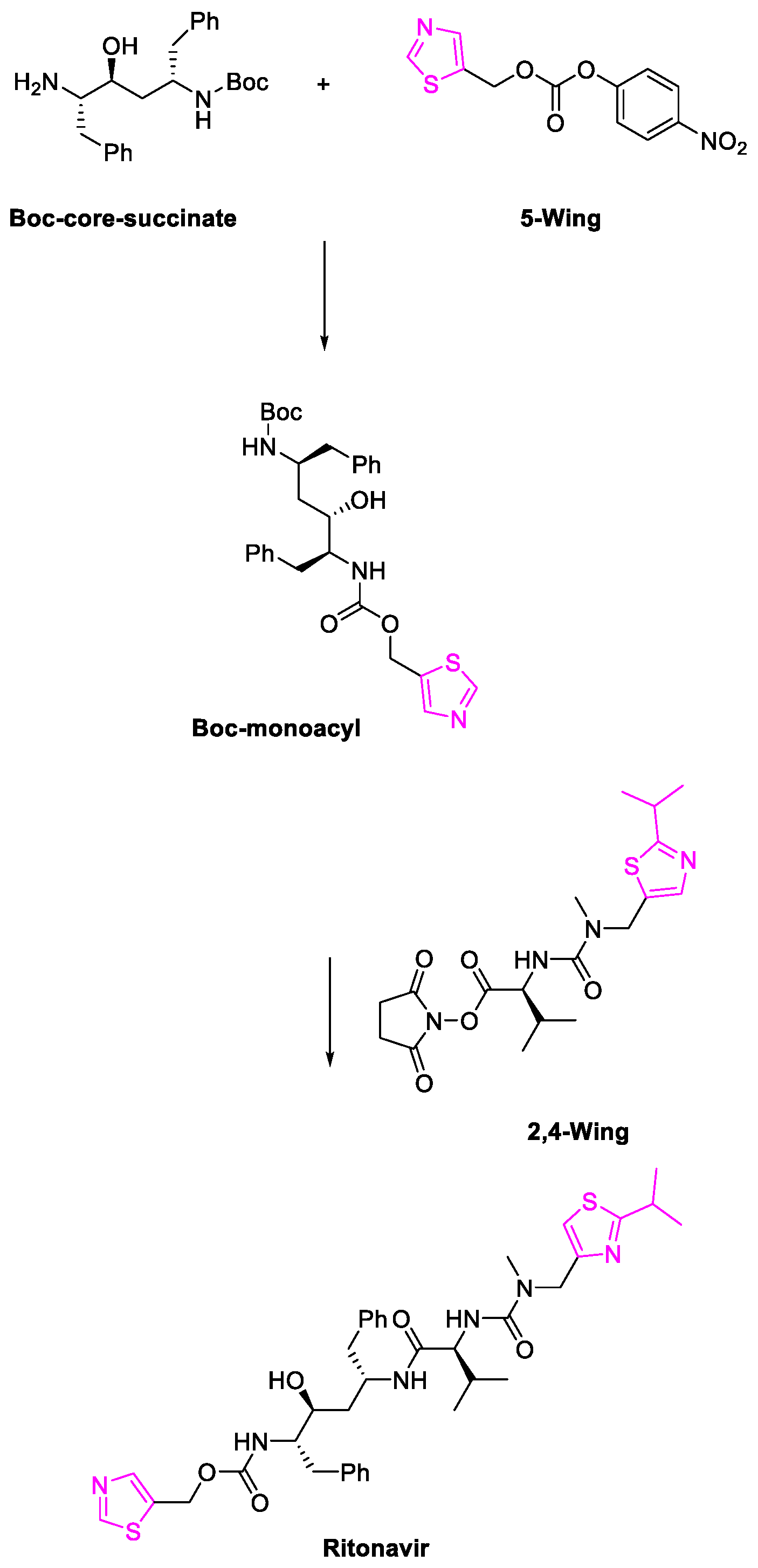
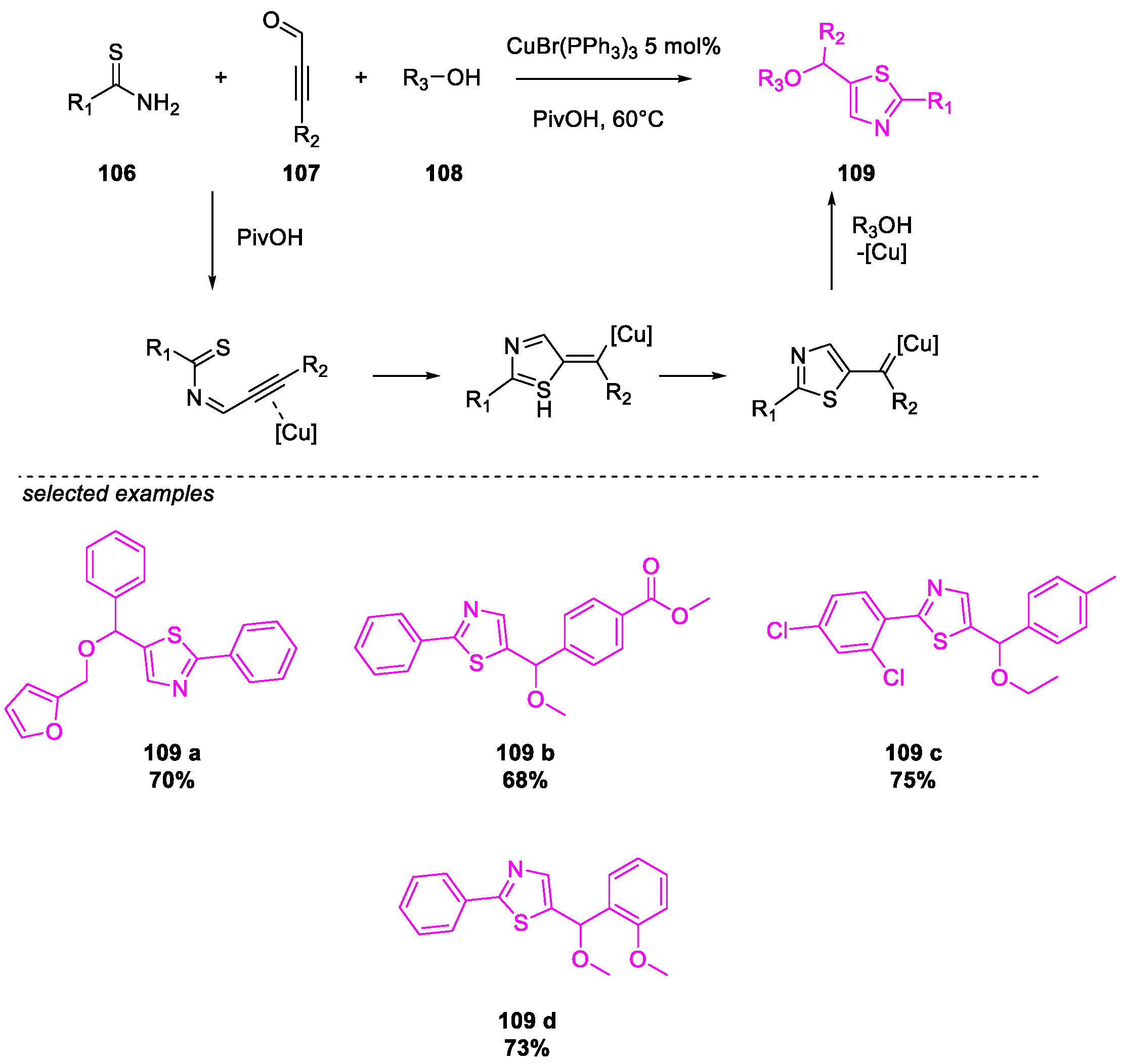
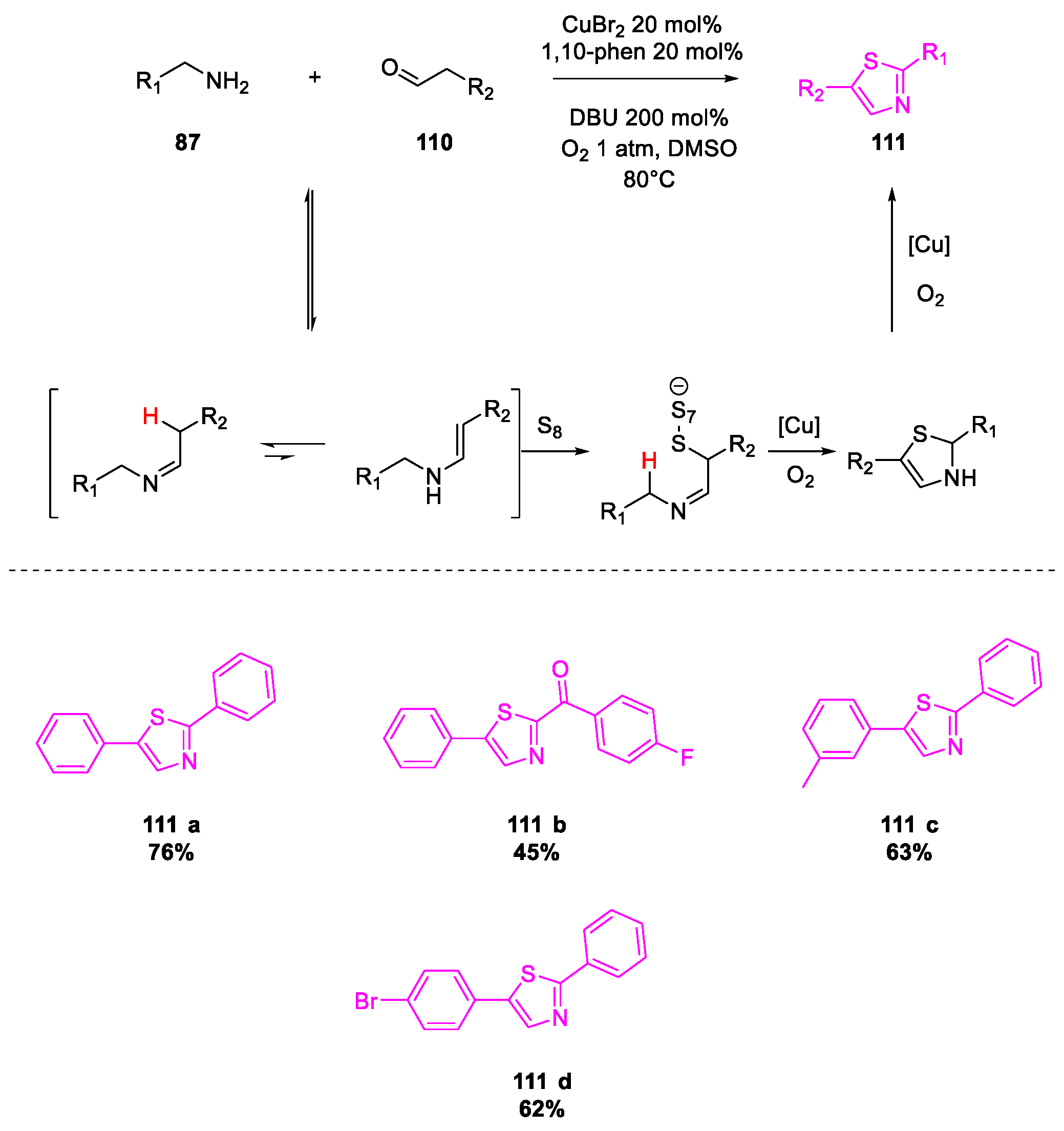
6. SARS-CoV-2 3CL Protease Target Drugs
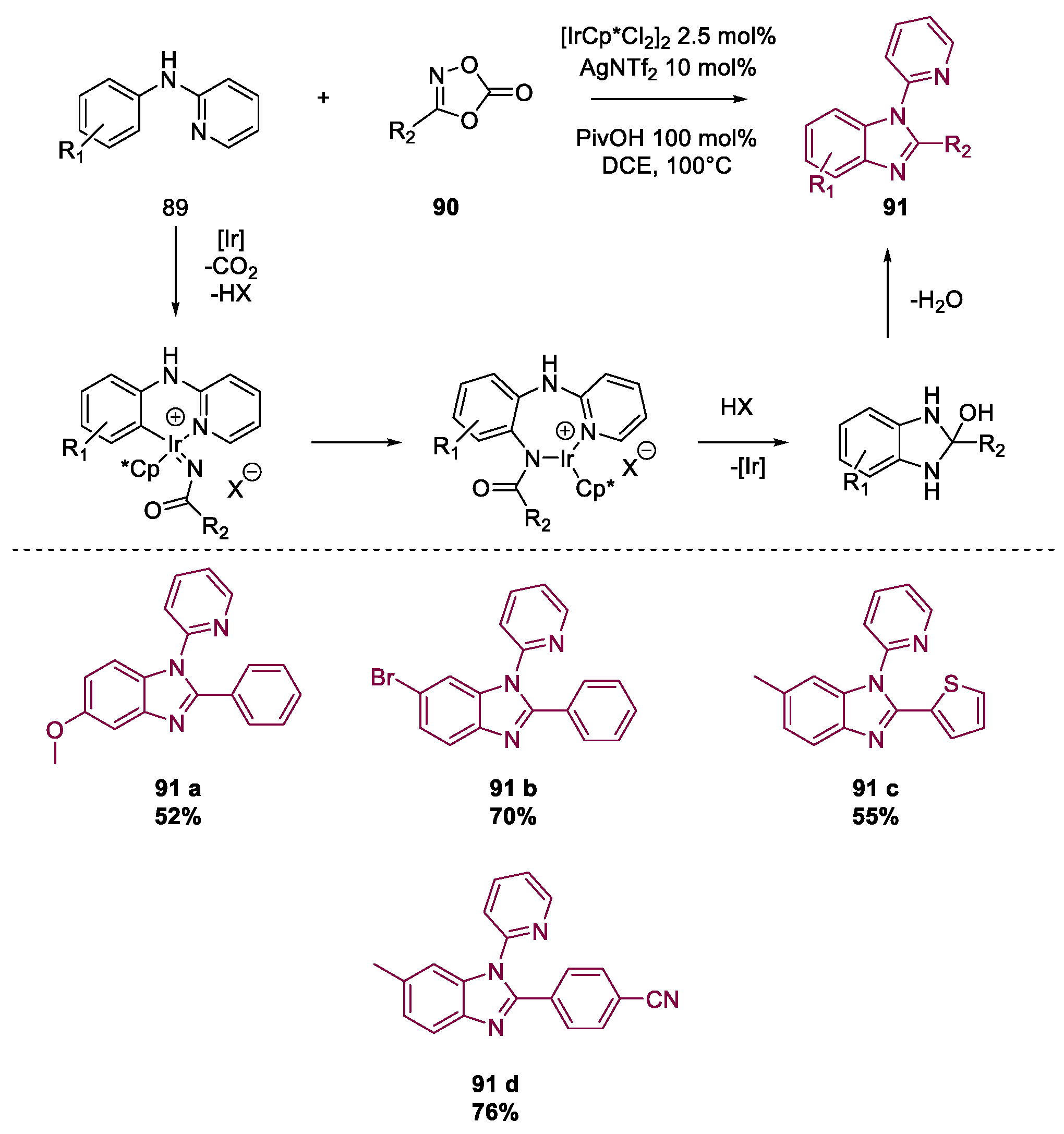
Miscellaneous Metal-Catalyzed Heterocycle Synthesis Approaches
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hardy, M.A.; Wright, B.A.; Bachman, J.L.; Boit, T.B.; Haley, H.M.S.; Knapp, R.R.; Lusi, R.F.; Okada, T.; Tona, V.; Garg, N.K.; et al. Treating a Global Health Crisis with a Dose of Synthetic Chemistry. ACS Cent. Sci. 2020, 6, 1017–1030. [Google Scholar] [CrossRef] [PubMed]
- European Centre for Disease Prevention and Control. Available online: https://www.ecdc.europa.eu/en/cases-2019-ncov-eueea (accessed on 25 February 2021).
- Mahase, E. Covid-19: Pfizer vaccine efficacy was 52% after first dose and 95% after second dose, paper shows. BMJ 2020, 371, m4826. [Google Scholar] [CrossRef] [PubMed]
- Wise, J. Covid-19: New data on Oxford AstraZeneca vaccine backs 12 week dosing interval. BMJ 2021, 372, n326. [Google Scholar] [CrossRef] [PubMed]
- Mahase, E. Covid-19: Vaccine candidate may be more than 90% effective, interim results indicate. BMJ 2020, 371, m4347. [Google Scholar] [CrossRef] [PubMed]
- Iacobucci, G. Covid-19: New UK variant may be linked to increased death rate, early data indicate. BMJ 2021, 372, n230. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhou, Q.; Li, Y.; Garner, L.V.; Watkins, S.P.; Carter, L.J.; Smoot, J.; Gregg, A.C.; Daniels, A.D.; Jervey, S.; et al. Research and Development on Therapeutic Agents and Vaccines for COVID-19 and Related Human Coronavirus Diseases. ACS Cent. Sci. 2020, 6, 315–331. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Chen, V.; Shannon, C.P.; Wei, X.-S.; Xiang, X.; Wang, X.; Wang, Z.-H.; Tebbutt, S.J.; Kollmann, T.R.; Fish, E.N. Interferon-α2b Treatment for COVID-19. Front. Immunol. 2020, 11, 1061. [Google Scholar] [CrossRef]
- Jahanshahlu, L.; Rezaei, N. Monoclonal antibody as a potential anti-COVID-19. Biomed. Pharmacother. 2020, 129, 110337. [Google Scholar] [CrossRef]
- Li, D.; Hu, J.; Li, D.; Yang, W.; Yin, S.-F.; Qiu, R. Reviews on Biological Activity, Clinical Trial and Synthesis Progress of Small Molecules for the Treatment of COVID-19. Top. Curr. Chem. 2021, 379, 4. [Google Scholar] [CrossRef]
- McKee, D.L.; Sternberg, A.; Stange, U.; Laufer, S.; Naujokat, C. Candidate drugs against SARS-CoV-2 and COVID-19. Pharmacol. Res. 2020, 157, 104859. [Google Scholar] [CrossRef]
- Kouznetsov, V.V. COVID-19 treatment: Much research and testing, but far, few magic bullets against SARS-CoV-2 coronavirus. Eur. J. Med. Chem. 2020, 203, 112647. [Google Scholar] [CrossRef]
- Shagufta; Ahmad, I. The race to treat COVID-19: Potential therapeutic agents for the prevention and treatment of SARS-CoV-2. Eur. J. Med. Chem. 2021, 213, 113157. [Google Scholar] [CrossRef]
- Yao, X.; Ye, F.; Zhang, M.; Cui, C.; Huang, B.; Niu, P.; Liu, X.; Zhao, L.; Dong, E.; Song, C.; et al. In Vitro Antiviral Activity and Projection of Optimized Dosing Design of Hydroxychloroquine for the Treatment of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). Clin. Infect. Dis. 2020, 71, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; Li, Z.; Yuan, K.; Qu, X.; Chen, J.; Wang, G.; Zhang, H.; Luo, H.; Zhu, L.; Jiang, P.; et al. Small Molecules Blocking the Entry of Severe Acute Respiratory Syndrome Coronavirus into Host Cells. J. Virol. 2004, 78, 11334–11339. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Liu, H.; Sun, Q.; Liang, H.; Li, C.; Deng, X.; Liu, Y.; Lai, L. Potent inhibitors of SARS-CoV-2 3C-like protease derived from N-substituted isatin compounds. Eur. J. Med. Chem. 2020, 206, 112702. [Google Scholar] [CrossRef]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Vankadari, N. Arbidol: A potential antiviral drug for the treatment of SARS-CoV-2 by blocking trimerization of the spike glycoprotein. Int. J. Antimicrob. Agents 2020, 56, 105998. [Google Scholar] [CrossRef] [PubMed]
- Pathan, S.I.; Chundawat, N.S.; Chauhan, N.P.S.; Singh, G.P. A review on synthetic approaches of heterocycles via insertion-cyclization reaction. Synth. Commun. 2020, 50, 1251–1285. [Google Scholar] [CrossRef]
- Negi, M.; Chawla, P.A.; Faruk, A.; Chawla, V. Role of heterocyclic compounds in SARS and SARS CoV-2 pandemic. Bioorg. Chem. 2020, 104, 104315. [Google Scholar] [CrossRef] [PubMed]
- Hagar, M.; Ahmed, H.A.; Aljohani, G.; Alhaddad, O.A. Investigation of Some Antiviral N-Heterocycles as COVID 19 Drug: Molecular Docking and DFT Calculations. Int. J. Mol. Sci. 2020, 21, 3922. [Google Scholar] [CrossRef]
- Gomtsyan, A. Heterocycles in drugs and drug discovery. Chem. Heterocycl. Compd. 2012, 48, 7–10. [Google Scholar] [CrossRef]
- Das, R.R.; Jaiswal, N.; Dev, N.; Jaiswal, N.; Naik, S.S.; Sankar, J. Efficacy and Safety of Anti-malarial Drugs (Chloroquine and Hydroxy-Chloroquine) in Treatment of COVID-19 Infection: A Systematic Review and Meta-Analysis. Front. Med. 2020, 7. [Google Scholar] [CrossRef] [PubMed]
- Sang, P.; Tian, S.-H.; Meng, Z.-H.; Yang, L.-Q. Anti-HIV drug repurposing against SARS-CoV-2. RSC Adv. 2020, 10, 15775–15783. [Google Scholar] [CrossRef]
- Stebbing, J.; Phelan, A.; Griffin, I.; Tucker, C.; Oechsle, O.; Smith, D.; Richardson, P. COVID-19: Combining antiviral and anti-inflammatory treatments. Lancet Infect. Dis. 2020, 20, 400–402. [Google Scholar] [CrossRef]
- Santhoshkumar, R.; Cheng, C. Reaching Green: Heterocycle Synthesis by Transition Metal-Catalyzed C−H Functionalization in Sustainable Medium. Chem. Eur. J. 2019, 25, 9366–9384. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, K.M.; Kapoor, A. Role of chloroquine and hydroxychloroquine in the treatment of COVID-19 infection—A systematic literature review. medRxiv 2020. [Google Scholar] [CrossRef]
- Meyerowitz, E.A.; Vannier, A.G.L.; Friesen, M.G.N.; Schoenfeld, S.; Gelfand, J.A.; Callahan, M.V.; Kim, A.Y.; Reeves, P.M.; Poznansky, M.C. Rethinking the role of hydroxychloroquine in the treatment of COVID-19. FASEB J. 2020, 34, 6027–6037. [Google Scholar] [CrossRef] [PubMed]
- Pagliano, P.; Piazza, O.; De Caro, F.; Ascione, T.; Filippelli, A. Is Hydroxychloroquine a Possible Postexposure Prophylaxis Drug to Limit the Transmission to Healthcare Workers Exposed to Coronavirus Disease 2019? Clin. Infect. Dis. 2020, 71, 887–888. [Google Scholar] [CrossRef]
- Patil, V.M.; Singhal, S.; Masand, N. A systematic review on use of aminoquinolines for the therapeutic management of COVID-19: Efficacy, safety and clinical trials. Life Sci. 2020, 254, 117775. [Google Scholar] [CrossRef] [PubMed]
- Surrey, A.R.; Hammer, H.F. Some 7-Substituted 4-Aminoquinoline Derivatives. J. Am. Chem. Soc. 1946, 68, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.S.; Buell, B.G. A New Synthesis of Chloroquine. J. Am. Chem. Soc. 1952, 74, 4513–4516. [Google Scholar] [CrossRef]
- Margolis, B.J.; Long, K.A.; Laird, D.L.T.; Ruble, J.C.; Pulley, S.R. Assembly of 4-Aminoquinolines via Palladium Catalysis: A Mild and Convenient Alternative to S N Ar Methodology. J. Org. Chem. 2007, 72, 2232–2235. [Google Scholar] [CrossRef]
- Surrey, A.R.; Hammer, H.F. The Preparation of 7-Chloro-4-(4-(N-ethyl-N-β-hydroxyethylamino)-1-methylbutylamino)-quinoline and Related Compounds. J. Am. Chem. Soc. 1950, 72, 1814–1815. [Google Scholar] [CrossRef]
- Kumar, A.V.; Vyas, K.D.; Singh, D.; Nanolavadekar, S.; Bhiae, S.; Jadhav, A. An Improved Process for the Preparation of 7-chloro-4-(5-N-Ethyl-N-2-Hydroxyethylamine)-2-pentyl Aminoquinoline and Its Intermediates. U.S. Patent WO 2005062723, 11 July 2005. [Google Scholar]
- Min, Y.S.; Cho, H.S.; Mo, K.W. New Preparation of Hydroxychloroquine. U.S. Patent WO 2010027150, 17 March 2010. [Google Scholar]
- Yu, E.; Mangunuru, H.P.R.; Telang, N.S.; Kong, C.J.; Verghese, J.; Gilliland III, S.E.; Ahmad, S.; Dominey, R.N.; Gupton, B.F. High-yielding continuous-flow synthesis of antimalarial drug hydroxychloroquine. Beilstein J. Org. Chem. 2018, 14, 583–592. [Google Scholar] [CrossRef]
- Chelucci, G.; Porcheddu, A. Synthesis of Quinolines via a Metal-Catalyzed Dehydrogenative N-Heterocyclization. Chem. Rec. 2017, 17, 200–216. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Kour, P.; Kumar, A. A review on transition-metal mediated synthesis of quinolines. J. Chem. Sci. 2018, 130, 73. [Google Scholar] [CrossRef]
- Eswaran, S.; Adhikari, A.V.; Chowdhury, I.H.; Pal, N.K.; Thomas, K.D. New quinoline derivatives: Synthesis and investigation of antibacterial and antituberculosis properties. Eur. J. Med. Chem. 2010, 45, 3374–3383. [Google Scholar] [CrossRef]
- Ramann, G.; Cowen, B. Recent Advances in Metal-Free Quinoline Synthesis. Molecules 2016, 21, 986. [Google Scholar] [CrossRef] [PubMed]
- Matada, B.S.; Yernale, N.G. The contemporary synthetic recipes to access versatile quinoline heterocycles. Synth. Commun. 2021, 51, 1133–1159. [Google Scholar] [CrossRef]
- Martínez, R.; Ramón, D.J.; Yus, M. RuCl2(dmso)4 Catalyzes the Solvent-Free Indirect Friedländer Synthesis of Polysubstituted Quinolines from Alcohols. European J. Org. Chem. 2007, 2007, 1599–1605. [Google Scholar] [CrossRef]
- Martínez, R.; Ramón, D.J.; Yus, M. Easy α-alkylation of ketones with alcohols through a hydrogen autotransfer process catalyzed by RuCl2(dmso)4. Tetrahedron 2006, 62, 8988–9001. [Google Scholar] [CrossRef]
- Martínez, R.; Ramón, D.J.; Yus, M. RuCl2(dmso)4 catalyzes the β-alkylation of secondary alcohols with primary alcohols through a hydrogen autotransfer process. Tetrahedron 2006, 62, 8982–8987. [Google Scholar] [CrossRef]
- Subramanian, M.; Sundar, S.; Rengan, R. Synthesis and structure of arene ruthenium(II) complexes: One-pot catalytic approach to synthesis of bioactive quinolines under mild conditions. Appl. Organomet. Chem. 2018, 32, e4582. [Google Scholar] [CrossRef]
- Ruch, S.; Irrgang, T.; Kempe, R. New Iridium Catalysts for the Selective Alkylation of Amines by Alcohols under Mild Conditions and for the Synthesis of Quinolines by Acceptor-less Dehydrogenative Condensation. Chem. A Eur. J. 2014, 20, 13279–13285. [Google Scholar] [CrossRef]
- Hahn, F.E.; Jahnke, M.C.; Pape, T. Synthesis of Pincer-Type Bis(benzimidazolin-2-ylidene) Palladium Complexes and Their Application in C−C Coupling Reactions. Organometallics 2007, 26, 150–154. [Google Scholar] [CrossRef]
- Vander Mierde, H.; Van Der Voort, P.; De Vos, D.; Verpoort, F. A Ruthenium-Catalyzed Approach to the Friedländer Quinoline Synthesis. European J. Org. Chem. 2008, 2008, 1625–1631. [Google Scholar] [CrossRef]
- Cho, C.S.; Ren, W.X.; Yoon, N.S. A recyclable copper catalysis in modified Friedländer quinoline synthesis. J. Mol. Catal. A Chem. 2009, 299, 117–120. [Google Scholar] [CrossRef]
- Cho, C.S.; Seok, H.J.; Shim, S.O. A rhodium-catalyzed route for oxidative coupling and cyclization of 2-aminobenzyl alcohol with ketones leading to quinolines. J. Heterocycl. Chem. 2005, 42, 1219–1222. [Google Scholar] [CrossRef]
- Mondal, R.R.; Khamarui, S.; Maiti, D.K. CuBr–ZnI2 Combo-Catalysis for Mild Cu I –Cu III Switching and sp 2 C–H Activated Rapid Cyclization to Quinolines and Their Sugar-Based Chiral Analogues: A UV–Vis and XPS Study. ACS Omega 2016, 1, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Sarode, P.B.; Bahekar, S.P.; Chandak, H.S. Zn(OTf)2-mediated C H activation: An expeditious and solvent-free synthesis of aryl/alkyl substituted quinolines. Tetrahedron Lett. 2016, 57, 5753–5756. [Google Scholar] [CrossRef]
- Korivi, R.P.; Cheng, C. Nickel-Catalyzed Cyclization of 2-Iodoanilines with Aroylalkynes: An Efficient Route for Quinoline Derivatives. J. Org. Chem. 2006, 71, 7079–7082. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Zeng, J.; Bai, Y.; Liu, X.-W. Access to Quinolines through Gold-Catalyzed Intermolecular Cycloaddition of 2-Aminoaryl Carbonyls and Internal Alkynes. J. Org. Chem. 2012, 77, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Fu, W.; Zou, G.; Xun, C.; Deng, D.; Ji, B. An efficient synthesis of 2-trifluoromethyl quinolines via gold-catalyzed cyclization of trifluoromethylated propargylamines. J. Fluor. Chem. 2012, 135, 195–199. [Google Scholar] [CrossRef]
- Xu, X.; Su, H.; Bao, M.; Huang, J.; Qiu, L. Silver-Catalyzed Carbocyclization of Azide-Tethered Alkynes: Expeditious Synthesis of Polysubstituted Quinolines. Adv. Synth. Catal. 2018, 361, adsc.201801425. [Google Scholar] [CrossRef]
- Wang, X.; Xie, P.; Sun, G.; Zhao, M.; Deng, Z.; Zhou, Y.; Bao, S. A systematic review and meta-analysis of the efficacy and safety of arbidol in the treatment of coronavirus disease 2019. Medicine (Baltimore) 2020, 99, e21402. [Google Scholar] [CrossRef]
- Proskurnina, E.V.; Izmailov, D.Y.; Sozarukova, M.M.; Zhuravleva, T.A.; Leneva, I.A.; Poromov, A.A. Antioxidant potential of antiviral drug umifenovir. Molecules 2020, 25, 1577. [Google Scholar] [CrossRef]
- Choudhary, S.; Silakari, O. Scaffold morphing of arbidol (umifenovir) in search of multi-targeting therapy halting the interaction of SARS-CoV-2 with ACE2 and other proteases involved in COVID-19. Virus Res. 2020, 289, 198146. [Google Scholar] [CrossRef]
- Kadam, R.U.; Wilson, I.A. Structural basis of influenza virus fusion inhibition by the antiviral drug Arbidol. Proc. Natl. Acad. Sci. USA 2017, 114, 206–214. [Google Scholar] [CrossRef]
- Manjunatha, M.R.; Shandil, R.; Panda, M.; Sadler, C.; Ambady, A.; Panduga, V.; Kumar, N.; Mahadevaswamy, J.; Sreenivasaiah, M.; Narayan, A.; et al. Scaffold Morphing to Identify Novel DprE1 Inhibitors with Antimycobacterial Activity. ACS Med. Chem. Lett. 2019, 10, 1480–1485. [Google Scholar] [CrossRef]
- Trofimov, F.A.; Tsyshkova, N.G.; Zotova, S.A.; Grinev, A.N. Synthesis of a new antiviral agent, arbidole. Pharm. Chem. J. 1993, 27, 75–76. [Google Scholar] [CrossRef]
- Zhao, C.; Zhao, Y.; Chai, H.; Gong, P. Synthesis and in vitro anti-hepatitis B virus activities of some ethyl 5-hydroxy-1H-indole-3-carboxylates. Bioorg. Med. Chem. 2006, 14, 2552–2558. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Dong, J. Preparation Method of Arbidol Hydrochloride. CN Patent CN 102351778A, February 2012. [Google Scholar]
- Mancuso, R.; Dalpozzo, R. Recent Progress in the Transition Metal Catalyzed Synthesis of Indoles. Catalysts 2018, 8, 458. [Google Scholar] [CrossRef]
- Chaisan, N.; Kaewsri, W.; Thongsornkleeb, C.; Tummatorn, J.; Ruchirawat, S. PtCl 4 -catalyzed cyclization of N -acetyl-2-alkynylanilines: A mild and efficient synthesis of N -acetyl-2-substituted indoles. Tetrahedron Lett. 2018, 59, 675–680. [Google Scholar] [CrossRef]
- Liang, Y.; Jiao, N. Cationic Cobalt(III) Catalyzed Indole Synthesis: The Regioselective Intermolecular Cyclization of N-Nitrosoanilines and Alkynes. Angew. Chemie 2016, 128, 4103–4107. [Google Scholar] [CrossRef]
- Lu, Q.; Vásquez-Céspedes, S.; Gensch, T.; Glorius, F. Control over Organometallic Intermediate Enables Cp*Co(III) Catalyzed Switchable Cyclization to Quinolines and Indoles. ACS Catal. 2016, 6, 2352–2356. [Google Scholar] [CrossRef]
- Li, H.; Zhao, Y.; Ma, L.; Ma, M.; Jiang, J.; Wan, X. Radical-carbene coupling reaction: Mn-catalyzed synthesis of indoles from aromatic amines and diazo compounds. Chem. Commun. 2017, 53, 5993–5996. [Google Scholar] [CrossRef]
- Clarke, A.K.; Lynam, J.M.; Taylor, R.J.K.; Unsworth, W.P. “Back-to-Front” Indole Synthesis Using Silver(I) Catalysis: Unexpected C-3 Pyrrole Activation Mode Supported by DFT. ACS Catal. 2018, 8, 6844–6850. [Google Scholar] [CrossRef]
- San Jang, S.; Kim, Y.H.; Youn, S.W. Divergent Syntheses of Indoles and Quinolines Involving N1–C2–C3 Bond Formation through Two Distinct Pd Catalyses. Org. Lett. 2020, 22, 9151–9157. [Google Scholar] [CrossRef] [PubMed]
- Meesala, R.; Nagarajan, R. Synthesis of new heteroaryldi(diindolyl)methanes: Colorimetric detection of DNA by di(diindolylmethyl)carbazoles. J. Chem. Sci. 2009, 121, 183–187. [Google Scholar] [CrossRef]
- Chelucci, G. Metal-catalyzed dehydrogenative synthesis of pyrroles and indoles from alcohols. Coord. Chem. Rev. 2017, 331, 37–53. [Google Scholar] [CrossRef]
- Plosker, G.L. Telmisartan A Review of its Use in Hypertension. Drugs 2009, 69, 2477–2499. [Google Scholar] [CrossRef] [PubMed]
- Yadav, G.; Ganguly, S. Structure activity relationship (SAR) study of benzimidazole scaffold for different biological activities: A mini-review. Eur. J. Med. Chem. 2015, 97, 419–443. [Google Scholar] [CrossRef] [PubMed]
- Gurwitz, D. Angiotensin receptor blockers as tentative SARS-CoV-2 therapeutics. Drug Dev. Res. 2020, 81, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Rothlin, R.P.; Vetulli, H.M.; Duarte, M.; Pelorosso, F.G. Telmisartan as tentative angiotensin receptor blocker therapeutic for COVID-19. Drug Dev. Res. 2020, 81, 768–770. [Google Scholar] [CrossRef] [PubMed]
- Ries, U.J.; Mihm, G.; Narr, B.; Hasselbach, K.M.; Wittneben, H.; Entzeroth, M.; van Meel, J.C.A.; Wienen, W.; Hauel, N.H. 6-Substituted benzimidazoles as new nonpeptide angiotensin II receptor antagonists: Synthesis, biological activity, and structure-activity relationships. J. Med. Chem. 1993, 36, 4040–4051. [Google Scholar] [CrossRef]
- Zhang, J.; Li, R.; Zhu, F.; Sun, C.; Shen, J. An improved synthesis of telmisartan via the copper-catalyzed cyclization of o -haloarylamidines. RSC Adv. 2020, 10, 13717–13721. [Google Scholar] [CrossRef]
- Wang, P.; Zheng, G.; Wang, Y.; Wang, X.; Wei, H.; Xiang, W. Highly practical and cost-efficient synthesis of telmisartan: An antihypertensive drug. Tetrahedron 2012, 68, 2509–2512. [Google Scholar] [CrossRef]
- Martin, A.D.; Siamaki, A.R.; Belecki, K.; Gupton, B.F. A flow-based synthesis of telmisartan. J. Flow Chem. 2015, 5, 145–147. [Google Scholar] [CrossRef]
- Horton, D.A.; Bourne, G.T.; Smythe, M.L. The Combinatorial Synthesis of Bicyclic Privileged Structures or Privileged Substructures. Chem. Rev. 2003, 103, 893–930. [Google Scholar] [CrossRef]
- Preston, P.N. Synthesis, reactions, and spectroscopic properties of benzimidazoles. Chem. Rev. 1974, 74, 279–314. [Google Scholar] [CrossRef]
- Daw, P.; Ben-David, Y.; Milstein, D. Direct Synthesis of Benzimidazoles by Dehydrogenative Coupling of Aromatic Diamines and Alcohols Catalyzed by Cobalt. ACS Catal. 2017, 7, 7456–7460. [Google Scholar] [CrossRef]
- Putta, R.R.; Chun, S.; Lee, S.B.; Oh, D.-C.; Hong, S. Iron-Catalyzed Acceptorless Dehydrogenative Coupling of Alcohols with Aromatic Diamines: Selective Synthesis of 1,2-Disubstituted Benzimidazoles. Front. Chem. 2020, 8. [Google Scholar] [CrossRef]
- Cimarelli, C.; Di Nicola, M.; Diomedi, S.; Giovannini, R.; Hamprecht, D.; Properzi, R.; Sorana, F.; Marcantoni, E. An efficient one-pot two catalyst system in the construction of 2-substituted benzimidazoles: Synthesis of benzimidazo[1,2-c]quinazolines. Org. Biomol. Chem. 2015, 13, 11687–11695. [Google Scholar] [CrossRef] [PubMed]
- Ke, F.; Zhang, P.; Lin, C.; Lin, X.; Xu, J.; Zhou, X. Synthesis of benzimidazoles by CuI-catalyzed three-component reaction of 2-haloaniline, ammonia and aldehyde in water. Org. Biomol. Chem. 2018, 16, 8090–8094. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Tan, Z.; Zhao, H.; Chen, X.; Jiang, H.; Zhang, M. Aerobic Copper-Catalyzed Synthesis of Benzimidazoles from Diaryl- and Alkylamines via Tandem Triple C–H Aminations. ACS Catal. 2018, 8, 2242–2246. [Google Scholar] [CrossRef]
- Xia, J.; Yang, X.; Li, Y.; Li, X. Iridium(III)-Catalyzed Synthesis of Benzimidazoles via C–H Activation and Amidation of Aniline Derivatives. Org. Lett. 2017, 19, 3243–3246. [Google Scholar] [CrossRef]
- Gurung, A.B.; Ali, M.A.; Lee, J.; Farah, M.A.; Al-Anazi, K.M. Unravelling lead antiviral phytochemicals for the inhibition of SARS-CoV-2 Mpro enzyme through in silico approach. Life Sci. 2020, 255, 117831. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Last 25 Years. J. Nat. Prod. 2007, 70, 461–477. [Google Scholar] [CrossRef]
- Colunga Biancatelli, R.M.L.; Berrill, M.; Catravas, J.D.; Marik, P.E. Quercetin and Vitamin C: An Experimental, Synergistic Therapy for the Prevention and Treatment of SARS-CoV-2 Related Disease (COVID-19). Front. Immunol. 2020, 11, 1451. [Google Scholar] [CrossRef]
- Ansari, W.A.; Ahamad, T.; Khan, M.A.; Khan, Z.A.; Khan, M.F. Luteolin: A Dietary Molecule as Potential Anti-COVID-19 Agent. Researchsquare 2020, 1–10. [Google Scholar] [CrossRef]
- Theoharides, T.C. COVID-19, pulmonary mast cells, cytokine storms, and beneficial actions of luteolin. BioFactors 2020, 46, 306–308. [Google Scholar] [CrossRef]
- Derosa, G.; Maffioli, P.; D’Angelo, A.; Di Pierro, F. A role for quercetin in coronavirus disease 2019 (COVID-19). Phyther. Res. 2021, 35, 1230–1236. [Google Scholar] [CrossRef]
- Winkel-Shirley, B. Flavonoid Biosynthesis. A Colorful Model for Genetics, Biochemistry, Cell Biology, and Biotechnology. Plant Physiol. 2001, 126, 485–493. [Google Scholar] [CrossRef]
- Shawan, M.M.A.K.; Halder, S.K.; Hasan, M.A. Luteolin and abyssinone II as potential inhibitors of SARS-CoV-2: An in silico molecular modeling approach in battling the COVID-19 outbreak. Bull. Natl. Res. Cent. 2021, 45, 27. [Google Scholar] [CrossRef]
- Ross, J.A.; Kasum, C.M. Dietary Flavonoids: Bioavailability, Metabolic Effects, and Safety. Annu. Rev. Nutr. 2002, 22, 19–34. [Google Scholar] [CrossRef]
- Zhao, X.; Zhou, J.; Lin, S.; Jin, X.; Liu, R. C–H Functionalization via Remote Hydride Elimination: Palladium Catalyzed Dehydrogenation of ortho-Acyl Phenols to Flavonoids. Org. Lett. 2017, 19, 976–979. [Google Scholar] [CrossRef] [PubMed]
- Raja, G.C.E.; Ryu, J.Y.; Lee, J.; Lee, S. Ruthenium-Catalyzed C–H Activation of Salicylaldehyde and Decarboxylative Coupling of Alkynoic Acids for the Selective Synthesis of Homoisoflavonoids and Flavones. Org. Lett. 2017, 19, 6606–6609. [Google Scholar] [CrossRef]
- Meng, M.; Wang, G.; Yang, L.; Cheng, K.; Qi, C. Silver-catalyzed Double Decarboxylative Radical Alkynylation/Annulation of Arylpropiolic Acids with α-keto Acids: Access to Ynones and Flavones under Mild Conditions. Adv. Synth. Catal. 2018, 360, 1218–1231. [Google Scholar] [CrossRef]
- Zhu, F.; Wang, Z.; Li, Y.; Wu, X.-F. Iridium-Catalyzed and Ligand-Controlled Carbonylative Synthesis of Flavones from Simple Phenols and Internal Alkynes. Chem. A Eur. J. 2017, 23, 3276–3279. [Google Scholar] [CrossRef]
- Properzi, R.; Marcantoni, E. Construction of heterocyclic structures by trivalent cerium salts promoted bond forming reactions. Chem. Soc. Rev. 2014, 43, 779–791. [Google Scholar] [CrossRef]
- Ahmed, N.; Ali, H.; van Lier, J.E. Silica gel supported InBr3 and InCl3: New catalysts for the facile and rapid oxidation of 2′-hydroxychalcones and flavanones to their corresponding flavones under solvent free conditions. Tetrahedron Lett. 2005, 46, 253–256. [Google Scholar] [CrossRef]
- Jin, C.; He, F.; Wu, H.; Chen, J.; Su, W. A facile synthesis of flavones catalysed by gallium(III) triflate. J. Chem. Res. 2009, 2009, 27–29. [Google Scholar] [CrossRef]
- Hui, D.S.; Azhar, E.I.; Madani, T.A.; Ntoumi, F.; Kock, R.; Dar, O.; Ippolito, G.; Mchugh, T.D.; Memish, Z.A.; Drosten, C.; et al. The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health—The latest 2019 novel coronavirus outbreak in Wuhan, China. Int. J. Infect. Dis. 2020, 91, 264–266. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Nutho, B.; Mahalapbutr, P.; Hengphasatporn, K.; Pattaranggoon, N.C.; Simanon, N.; Shigeta, Y.; Hannongbua, S.; Rungrotmongkol, T. Why Are Lopinavir and Ritonavir Effective against the Newly Emerged Coronavirus 2019? Atomistic Insights into the Inhibitory Mechanisms. Biochemistry 2020, 59, 1769–1779. [Google Scholar] [CrossRef] [PubMed]
- Horby, P.W.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Emberson, J.; Palfreeman, A.; Raw, J.; Elmahi, E.; Prudon, B.; et al. Lopinavir-ritonavir in patients admitted to hospital with COVID-19 (RECOVERY): A randomised, controlled, open-label, platform trial. Lancet 2020, 396, 1345–1352. [Google Scholar] [CrossRef]
- Uzunova, K.; Filipova, E.; Pavlova, V.; Vekov, T. Insights into antiviral mechanisms of remdesivir, lopinavir/ritonavir and chloroquine/hydroxychloroquine affecting the new SARS-CoV-2. Biomed. Pharmacother. 2020, 131, 110668. [Google Scholar] [CrossRef]
- Kappelhoff, B.S.; Huitema, A.D.R.; Crommentuyn, K.M.L.; Mulder, J.W.; Meenhorst, P.L.; van Gorp, E.C.M.; Mairuhu, A.T.A.; Beijnen, J.H. Development and validation of a population pharmacokinetic model for ritonavir used as a booster or as an antiviral agent in HIV-1-infected patients. Br. J. Clin. Pharmacol. 2005, 59, 174–182. [Google Scholar] [CrossRef][Green Version]
- Kempf, D.J.; Marsh, K.C.; Kumar, G.; Rodrigues, A.D.; Denissen, J.F.; McDonald, E.; Kukulka, M.J.; Hsu, A.; Granneman, G.R.; Baroldi, P.A.; et al. Pharmacokinetic enhancement of inhibitors of the human immunodeficiency virus protease by coadministration with ritonavir. Antimicrob. Agents Chemother. 1997, 41, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Sevrioukova, I.; Poulos, T. Ritonavir Analogues as a Probe for Deciphering the Cytochrome P450 3A4 Inhibitory Mechanism. Curr. Top. Med. Chem. 2014, 14, 1348–1355. [Google Scholar] [CrossRef]
- Chemburkar, S.R.; Bauer, J.; Deming, K.; Spiwek, H.; Patel, K.; Morris, J.; Henry, R.; Spanton, S.; Dziki, W.; Porter, W.; et al. Dealing with the Impact of Ritonavir Polymorphs on the Late Stages of Bulk Drug Process Development. Org. Process Res. Dev. 2000, 4, 413–417. [Google Scholar] [CrossRef]
- Badavath, V.N.; Kumar, A.; Samanta, P.K.; Maji, S.; Das, A.; Blum, G.; Jha, A.; Sen, A. Determination of potential inhibitors based on isatin derivatives against SARS-CoV-2 main protease (mpro): A molecular docking, molecular dynamics and structure-activity relationship studies. J. Biomol. Struct. Dyn. 2020, 1–19. [Google Scholar] [CrossRef]
- Tomassetti, M.; Lupidi, G.; Piermattei, P.; Rossi, F.V.; Lillini, S.; Bianchini, G.; Aramini, A.; Ciufolini, M.A.; Marcantoni, E. Catalyst-Free Synthesis of Polysubstituted 5-Acylamino-1,3-Thiazoles via Hantzsch Cyclization of α-Chloroglycinates. Molecules 2019, 24, 3846. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, X.; Zhu, B.; Guo, P.; Pei, Y.; He, Q.; Cao, H. Cu(I)-Catalyzed Three-Component Cyclization for the Construction of Functionalized Thiazoles. J. Org. Chem. 2020, 85, 10118–10124. [Google Scholar] [CrossRef]
- Wang, X.; Qiu, X.; Wei, J.; Liu, J.; Song, S.; Wang, W.; Jiao, N. Cu-Catalyzed Aerobic Oxidative Sulfuration/Annulation Approach to Thiazoles via Multiple Csp 3–H Bond Cleavage. Org. Lett. 2018, 20, 2632–2636. [Google Scholar] [CrossRef]
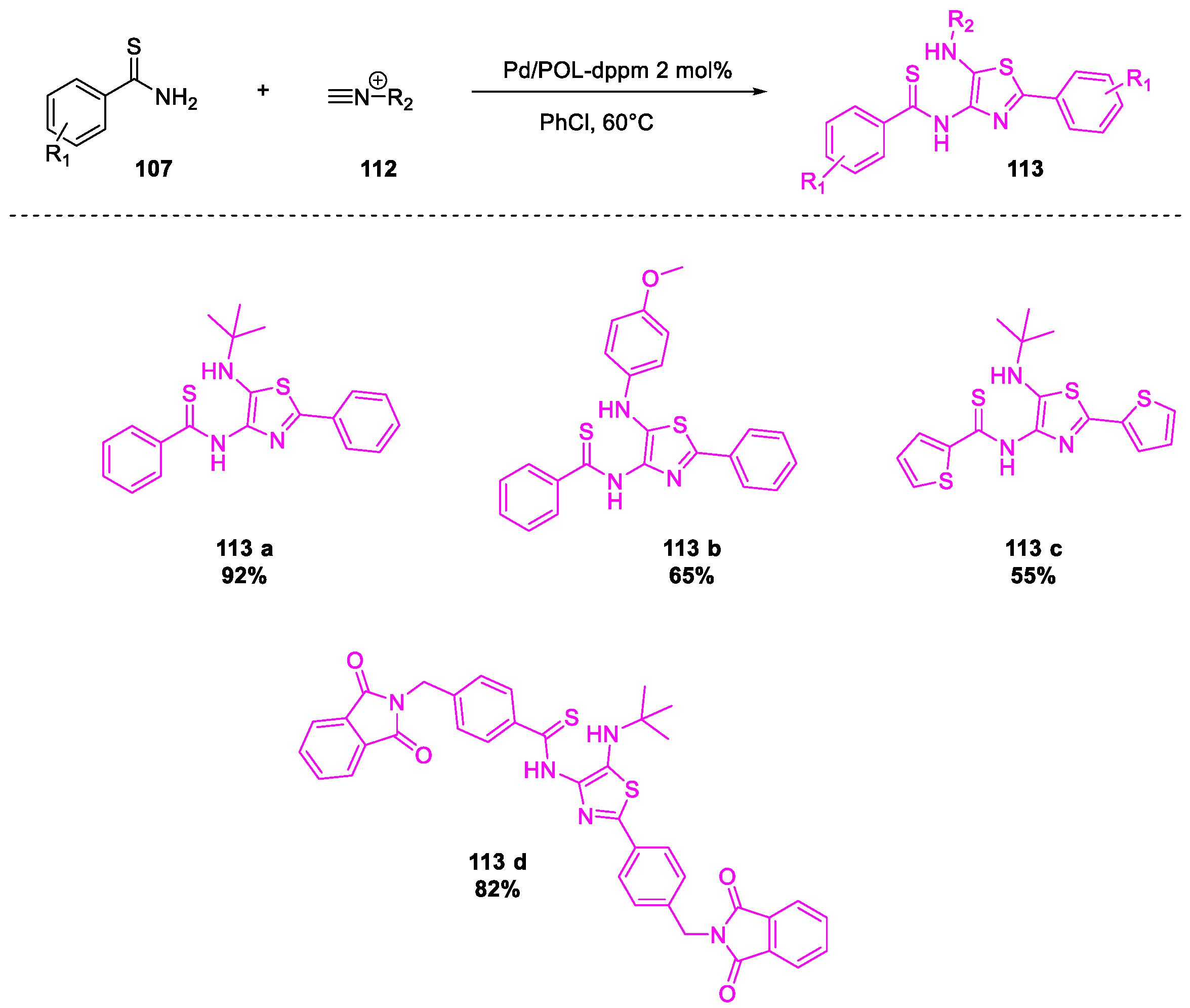
- Tong, W.; Li, W.-H.; He, Y.; Mo, Z.-Y.; Tang, H.-T.; Wang, H.-S.; Pan, Y.-M. Palladium-Metalated Porous Organic Polymers as Recyclable Catalysts for the Chemioselective Synthesis of Thiazoles from Thiobenzamides and Isonitriles. Org. Lett. 2018, 20, 2494–2498. [Google Scholar] [CrossRef]
- Liu, Y.-C.; Zhang, R.; Wu, Q.-Y.; Chen, Q.; Yang, G.-F. Recent Developments in the Synthesis and Applications of Isatins. Org. Prep. Proced. Int. 2014, 46, 317–362. [Google Scholar] [CrossRef]
- Salvanna, N.; Ramesh, P.; Santosh Kumar, K.; Das, B. Copper-catalyzed aerobic oxidative intramolecular amidation of 2-aminophenylacetylenes: A domino process for the synthesis of isatin. New J. Chem. 2017, 41, 13754–13759. [Google Scholar] [CrossRef]
- Zheng, Y.; Li, J.; Yu, X.; Lv, S.; Hai, L.; Wu, Y. Ferric(III) chloride catalyzed intramolecular cyclization of N-alkyl-2-oxo-acetanilides: A facile access to isatins. Tetrahedron Lett. 2016, 57, 39–42. [Google Scholar] [CrossRef]
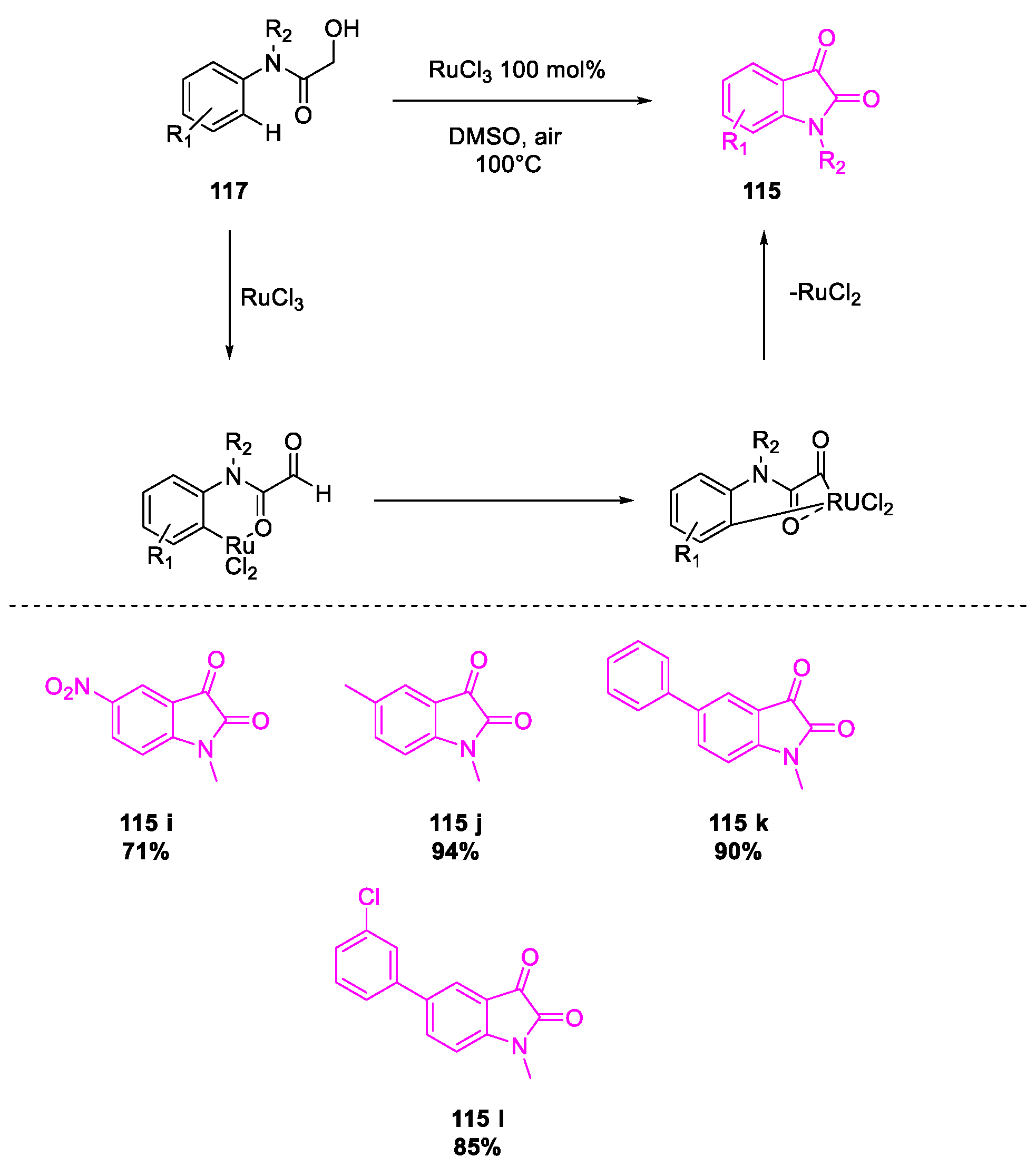
- Wang, Y.; Li, W.; Cheng, X.; Zhan, Z.; Ma, X.; Guo, L.; Jin, H.; Wu, Y. Ru(III)-mediated intramolecular ortho-C(sp2)–H activation/oxidative acylation: One-pot synthesis of isatins from α-hydroxy amides. Tetrahedron 2016, 72, 3193–3197. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


























































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossi, F.V.; Gentili, D.; Marcantoni, E. Metal-Promoted Heterocyclization: A Heterosynthetic Approach to Face a Pandemic Crisis. Molecules 2021, 26, 2620. https://doi.org/10.3390/molecules26092620
Rossi FV, Gentili D, Marcantoni E. Metal-Promoted Heterocyclization: A Heterosynthetic Approach to Face a Pandemic Crisis. Molecules. 2021; 26(9):2620. https://doi.org/10.3390/molecules26092620
Chicago/Turabian StyleRossi, Federico Vittorio, Dario Gentili, and Enrico Marcantoni. 2021. "Metal-Promoted Heterocyclization: A Heterosynthetic Approach to Face a Pandemic Crisis" Molecules 26, no. 9: 2620. https://doi.org/10.3390/molecules26092620
APA StyleRossi, F. V., Gentili, D., & Marcantoni, E. (2021). Metal-Promoted Heterocyclization: A Heterosynthetic Approach to Face a Pandemic Crisis. Molecules, 26(9), 2620. https://doi.org/10.3390/molecules26092620