
Selective Synthesis of N-Acylnortropane Derivatives in Palladium-Catalysed Aminocarbonylation


Abstract
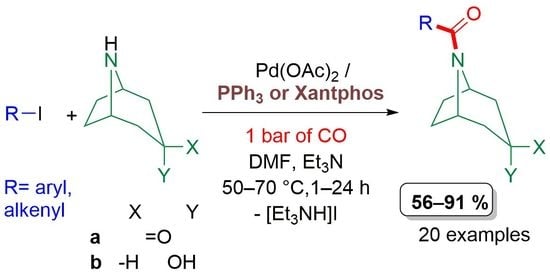
1. Introduction
2. Results
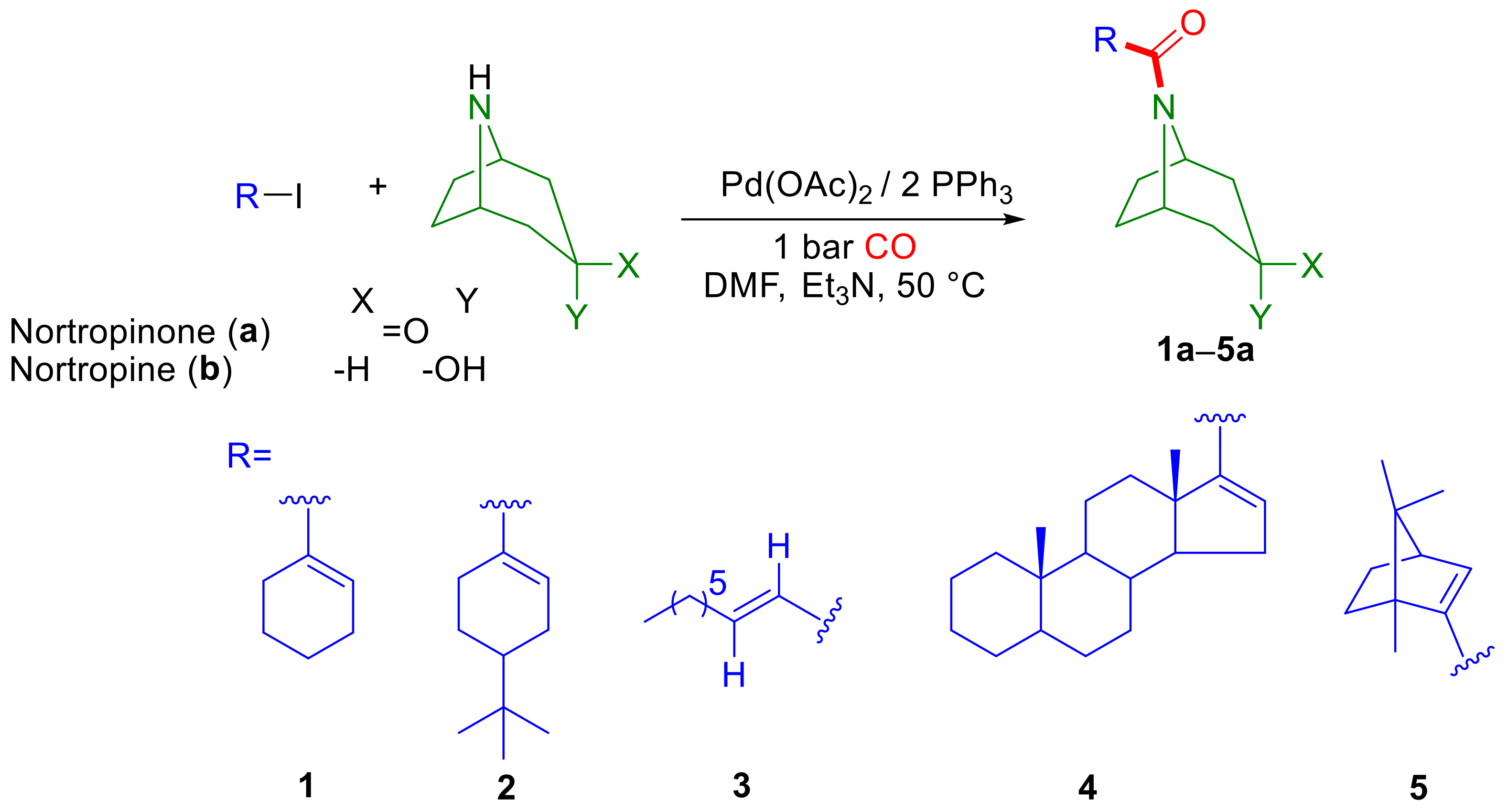
2.1. Aminocarbonylation of Iodoalkenes (1–5) in the Presence of Tropane-Based Nucleophiles (a, b)
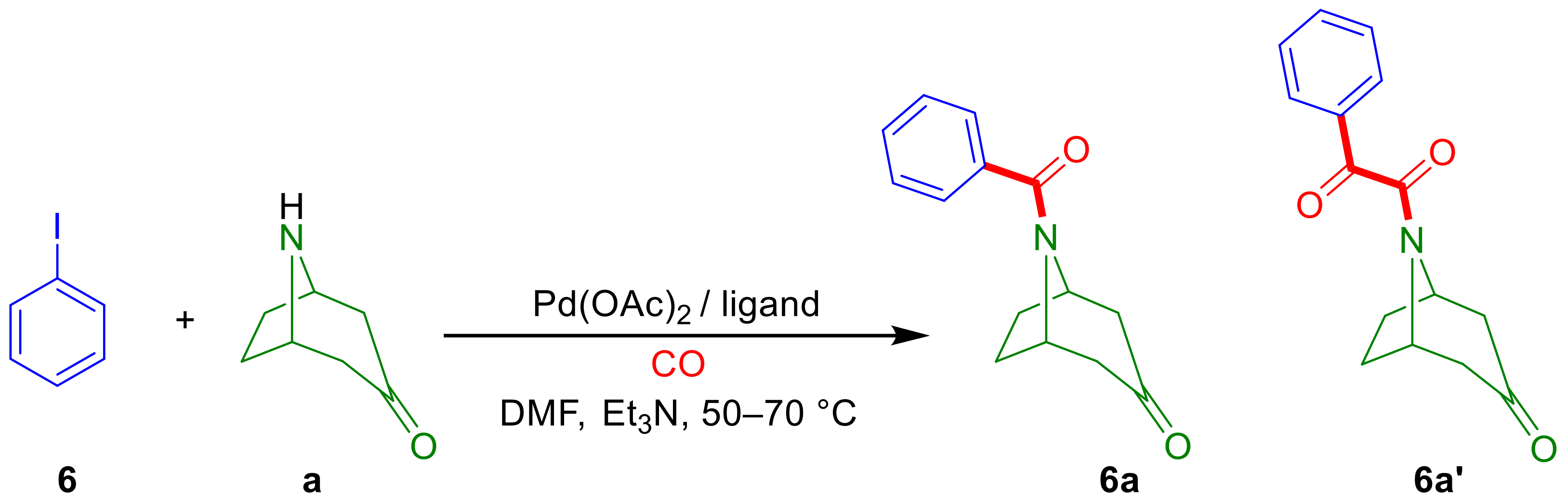
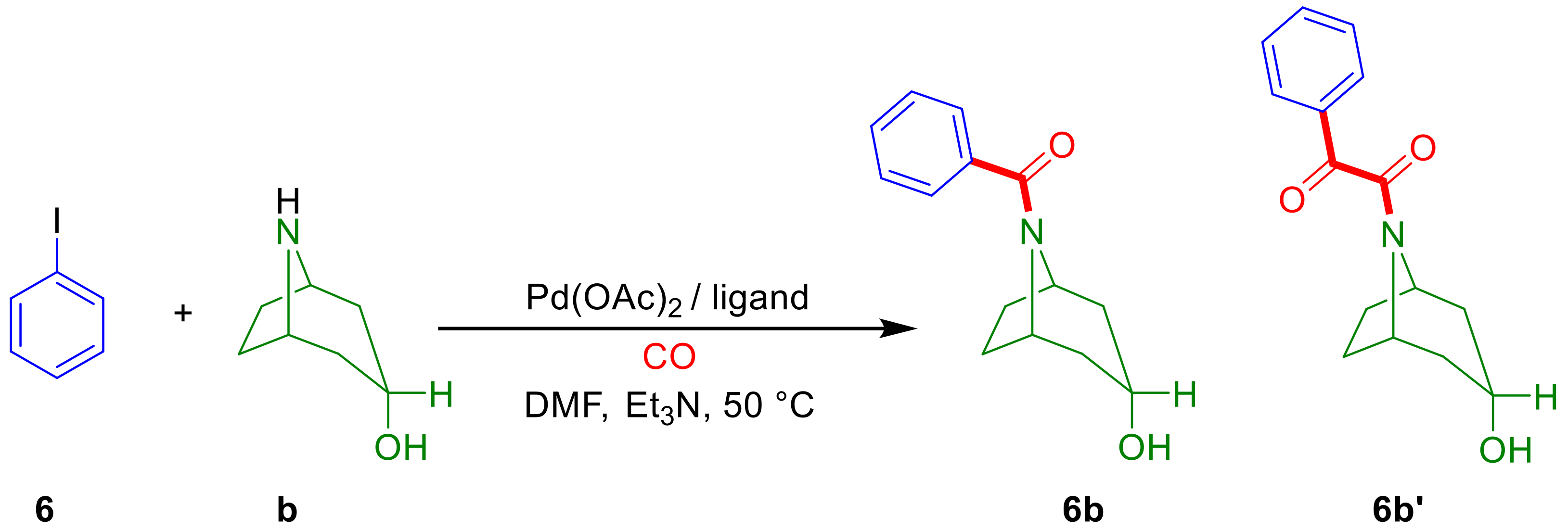
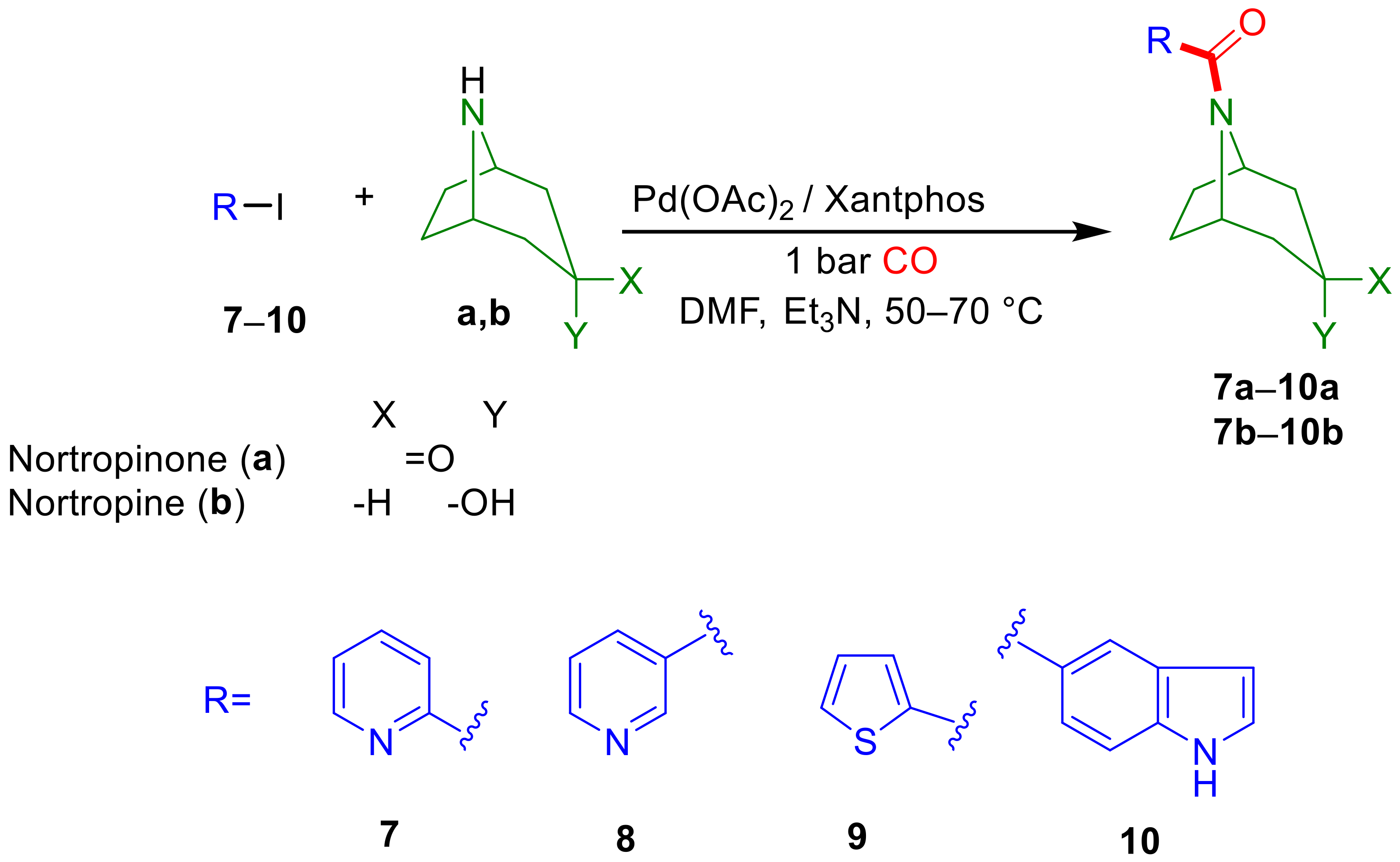
2.2. Aminocarbonylation of Iodoarenes (6–10) in the Presence of Tropane-Based Nucleophiles (a, b)
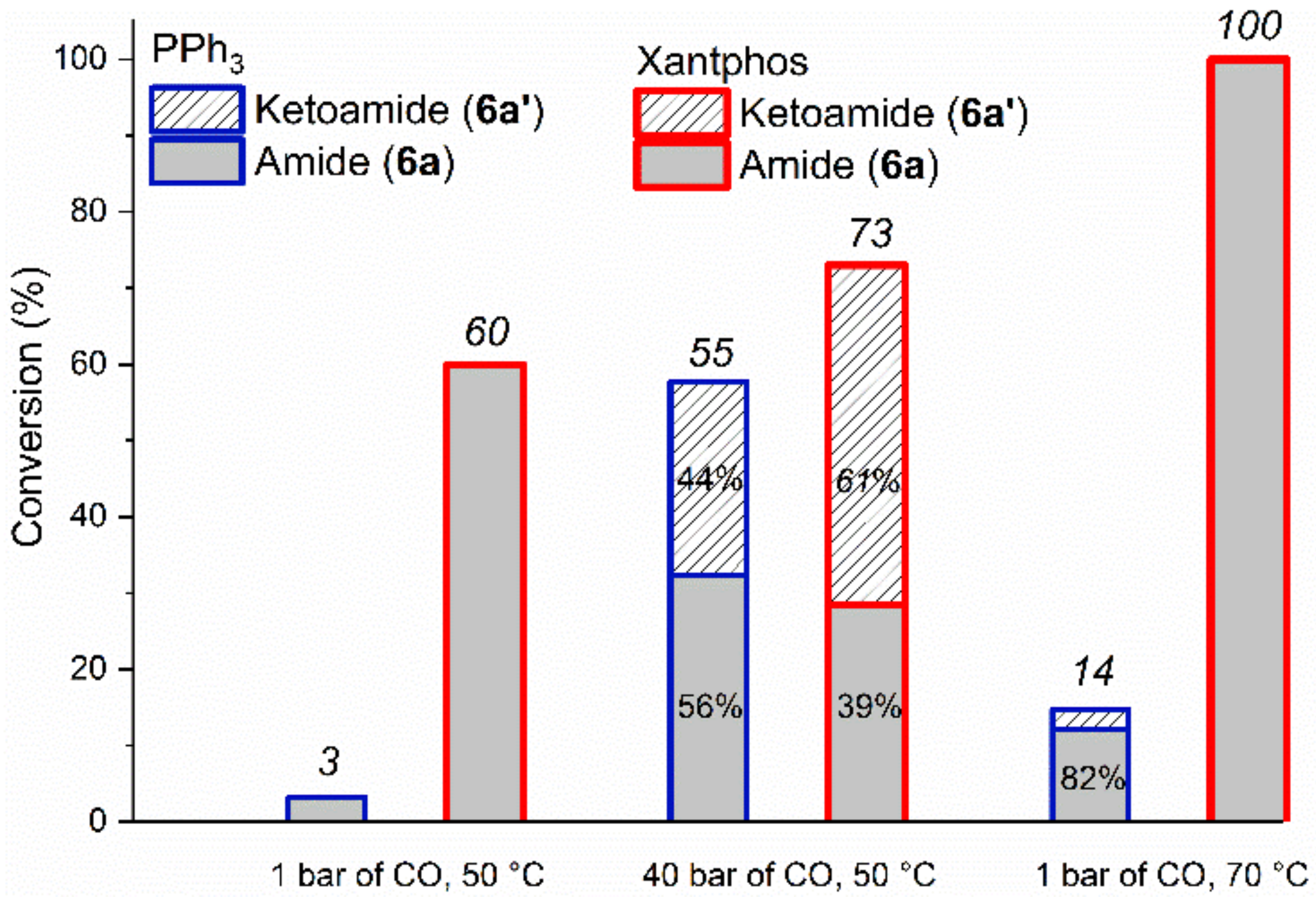
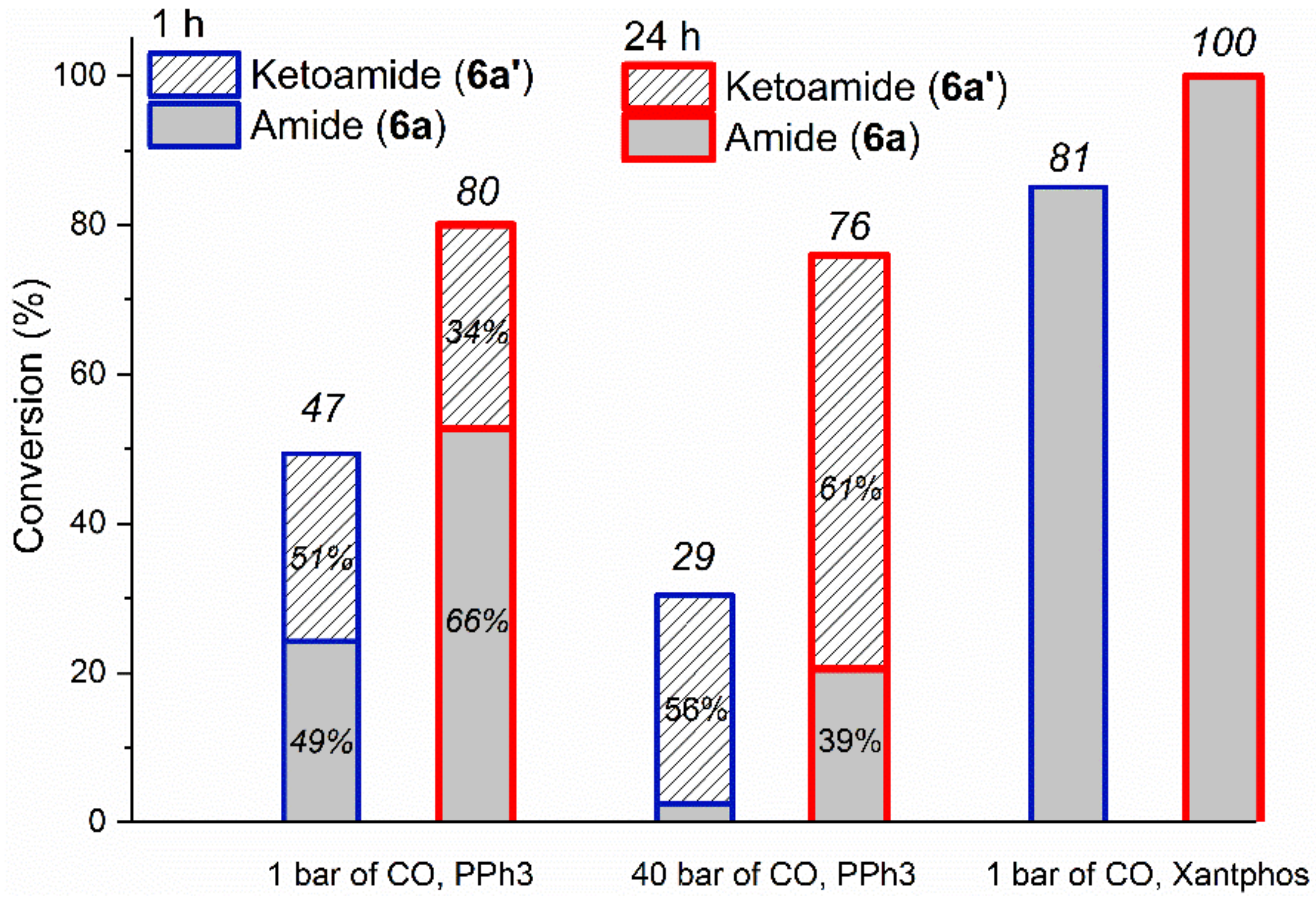
2.2.1. Optimization Study with Iodobenzene (6)
2.2.2. Extending the Scope of Iodoarene Substrates
3. Materials and Methods
3.1. General Procedures
3.2. Aminocarbonylation of Nortropane Derivatives (a, b) in the Presence of Iodoalkenes (1–5) and Iodoarenes (6–10) under Atmospheric Carbon Monoxide Pressure
3.3. Aminocarbonylation of Nortropane Derivatives (a, b) in the Presence of Iodoalkenes (1–5) and Iodoarenes (6–11) under High Carbon Monoxide Pressure
3.4. Characterization of the Products
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Pattabiraman, V.; Bode, J. Rethinking amide bond synthesis. Nature 2011, 480, 471–479. [Google Scholar] [CrossRef]
- Kaspar, A.; Reichert, J. Future directions for peptide therapeutics development. Drug Discov. Today 2013, 18, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.; Viswanadhan, V.; Wendoloski, J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- De Figueiredo, R.; Suppo, J.; Campagne, J. Nonclassical Routes for Amide Bond Formation. Chem. Rev. 2016, 116, 12029–12122. [Google Scholar] [CrossRef] [PubMed]
- Massolo, E.; Pirola, M.; Benaglia, M. Amide Bond Formation Strategies: Latest Advances on a Dateless Transformation. Eur. J. Org. Chem. 2020, 2020, 4641–4651. [Google Scholar] [CrossRef]
- Montalbetti, C.; Falque, V. Amide bond formation and peptide coupling. Tetrahedron 2005, 61, 10827–10852. [Google Scholar] [CrossRef]
- Challis, B.G.; Challis, J.A. PATAI’S Chemistry of Functional Groups. In Reactions of the Carboxamide Group; Zabicky, J., Ed.; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 1970. [Google Scholar]
- Klausner, Y.; Bodansky, M. The Azide Method in Peptide Synthesis: Its Scope and Limitations. Synthesis 1974, 549–559. [Google Scholar] [CrossRef]
- Mikolajczyk, M.; Kielbasinski, P. Recent Developments in the Carbodiimide Chemistry. Tetrahedron 1981, 37, 233–284. [Google Scholar] [CrossRef]
- Wittenberger, S.; McLaughlin, M. Preparation of endothelin antagonist ABT-627. Tetrahedron Lett. 1999, 40, 7175–7178. [Google Scholar] [CrossRef]
- Ishihara, K.; Ohara, S.; Yamamoto, H. 3,4,5-trifluorobenzeneboronic acid as an extremely active amidation catalyst. J. Org. Chem. 1996, 61, 4196–4197. [Google Scholar] [CrossRef] [PubMed]
- Gangwar, S.; Pauletti, G.; Siahaan, T.; Stella, V.; Borchardt, R. Synthesis of a novel esterase-sensitive cyclic prodrug of a hexapeptide using an (acyloxy)alkoxy promoiety. J. Org. Chem. 1997, 62, 1356–1362. [Google Scholar] [CrossRef]
- Rajappan, V.; Hosmane, R. Pentafluorophenol: A superior reagent for condensations in heterocyclic chemistry. Synth. Commun. 1998, 28, 753–764. [Google Scholar] [CrossRef]
- Sanchez-Sancho, F.; Mann, E.; Herradon, B. Efficient syntheses of polyannular heterocycles featuring microwave-accelerated Bischler-Napieralski reaction, stereoselective Heck cyclization, and Claisen rearrangement. Synlett 2000, 509–513. [Google Scholar] [CrossRef]
- Ragnarsson, U.; Grehn, L. Novel amine chemistry based on DMAP-catalyzed acylation. Acc. Chem. Res. 1998, 31, 494–501. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, X.; Wang, J.; Grinberg, N.; Krishnamurthy, D.; Senanayake, C. An improved method of amide synthesis using acyl chlorides. Tetrahedron Lett. 2009, 50, 2964–2966. [Google Scholar] [CrossRef]
- Engers, D.; Field, J.; Le, U.; Zhou, Y.; Bolinger, J.; Zamorano, R.; Blobaum, A.; Jones, C.; Jadhav, S.; Weaver, C.; et al. Discovery, Synthesis, and Structure-Activity Relationship Development of a Series of N-(4-Acetamido)phenylpicolinamides as Positive Allosteric Modulators of Metabotropic Glutamate Receptor 4 (mGlu(4)) with CNS Exposure in Rats. J. Med. Chem. 2011, 54, 1106–1110. [Google Scholar] [CrossRef]
- Bousfield, T.; Pearce, K.; Nyamini, S.; Angelis-Dimakis, A.; Camp, J. Synthesis of amides from acid chlorides and amines in the bio-based solvent Cyrene (TM). Green Chem. 2019, 21, 3675–3681. [Google Scholar] [CrossRef]
- Sayes, M.; Charette, A. Diphenylsilane as a coupling reagent for amide bond formation. Green Chem. 2017, 19, 5060–5064. [Google Scholar] [CrossRef]
- Galvez, A.; Schaack, C.; Noda, H.; Bode, J. Chemoselective Acylation of Primary Amines and Amides with Potassium Acyltrifluoroborates under Acidic Conditions. J. Am. Chem. Soc. 2017, 139, 1826–1829. [Google Scholar] [CrossRef]
- Braddock, D.; Lickiss, P.; Rowley, B.; Pugh, D.; Purnomo, T.; Santhakumar, G.; Fussell, S. Tetramethyl Orthosilicate (TMOS) as a Reagent for Direct Amidation of Carboxylic Acids. Org. Lett. 2018, 20, 950–953. [Google Scholar] [CrossRef] [PubMed]
- Sabatini, M.; Boulton, L.; Sheppard, T. Borate esters: Simple catalysts for the sustainable synthesis of complex amides. Sci. Adv. 2017, 3. [Google Scholar] [CrossRef]
- Schoenberg, A.; Bartoletti, I.; Heck, R.F. Palladium-catalyzed carboalcoxilation of aryl, benzyl and vinylic halides. J. Org. Chem. 1974, 39, 3318–3326. [Google Scholar] [CrossRef]
- Schoenberg, A.; Heck, R.F. Palladium-catalyzed amidation of aryl, heterocyclic, and vinylic halides. J. Org. Chem. 1974, 39, 3327–3331. [Google Scholar] [CrossRef]
- Schoenberg, A.; Heck, R.F. Palladium-catalyzed formylation of aryl, heterocyclic, and vinylic halides. J. Am. Chem. Soc. 1974, 96, 7761–7765. [Google Scholar] [CrossRef]
- Wu, X.; Neumann, H.; Beller, M. Synthesis of Heterocycles via Palladium-Catalyzed Carbonylations. Chem. Rev. 2013, 113, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Gribble, G. Metal-catalyzed amidation. Tetrahedron 2012, 68, 9867–9923. [Google Scholar] [CrossRef]
- Wu, X.; Neumann, H.; Beller, M. Palladium-catalyzed carbonylative coupling reactions between Ar-X and carbon nucleophiles. Chem. Soc. Rev. 2011, 40, 4986–5009. [Google Scholar] [CrossRef] [PubMed]
- Magano, J.; Dunetz, J. Large-Scale Applications of Transition Metal-Catalyzed Couplings for the Synthesis of Pharmaceuticals. Chem. Rev. 2011, 111, 2177–2250. [Google Scholar] [CrossRef] [PubMed]
- Grigg, R.; Mutton, S. Pd-catalysed carbonylations: Versatile technology for discovery and process chemists. Tetrahedron 2010, 66, 5515–5548. [Google Scholar] [CrossRef]
- Brennfuhrer, A.; Neumann, H.; Beller, M. Palladium-Catalyzed Carbonylation Reactions of Aryl Halides and Related Compounds. Angew. Chem. Int. Ed. 2009, 48, 4114–4133. [Google Scholar] [CrossRef] [PubMed]
- Barnard, C. Palladium-Catalyzed Carbonylation—A Reaction Come of Age. Organometallics 2008, 27, 5402–5422. [Google Scholar] [CrossRef]
- Skoda-Foldes, R.; Kollar, L. Synthetic applications of palladium catalysed carbonylation of organic halides. Curr. Org. Chem. 2002, 6, 1097–1119. [Google Scholar] [CrossRef]
- Wu, X.; Neumann, H.; Beller, M. Selective Palladium-Catalyzed Aminocarbonylation of Aryl Halides with CO and Ammonia. Chem. Eur. J. 2010, 16, 9750–9753. [Google Scholar] [CrossRef] [PubMed]
- Gadge, S.; Bhanage, B. Recent developments in palladium catalysed carbonylation reactions. RSC Adv. 2014, 4, 10367–10389. [Google Scholar] [CrossRef]
- Wu, X. Palladium-catalyzed carbonylative transformation of aryl chlorides and aryl tosylates. RSC Adv. 2016, 6, 83831–83837. [Google Scholar] [CrossRef]
- Peng, J.; Qi, X.; Wu, X. Recent Achievements in Carbonylation Reactions: A Personal Account. Synlett 2017, 28, 175–194. [Google Scholar] [CrossRef]
- Marosvolgyi-Hasko, D.; Lengyel, B.; Tukacs, J.; Kollar, L.; Mika, L. Application of -Valerolactone as an Alternative Biomass-Based Medium for Aminocarbonylation Reactions. ChemPlusChem 2016, 81, 1224–1229. [Google Scholar] [CrossRef]
- Marosvolgyi-Hasko, D.; Kegl, T.; Kollar, L. Substituent effects in aminocarbonylation of para-substituted iodobenzenes. Tetrahedron 2016, 72, 7509–7516. [Google Scholar] [CrossRef]
- Nagymihaly, Z.; Caturello, N.; Takatsy, A.; Aragay, G.; Kollar, L.; Albuquerque, R.; Csok, Z. Palladium-Mediated Catalysis Leads to Intramolecular Narcissistic Self-Sorting on a Cavitand Platform. J. Org. Chem. 2017, 82, 390–396. [Google Scholar] [CrossRef]
- Gergely, M.; Kollar, L. Aminocarbonylation (hydrazinocarbonylation) of iodoalkenes and iodoarenes. Tetrahedron 2017, 73, 838–844. [Google Scholar] [CrossRef]
- Gergely, M.; Boros, B.; Kollar, L. High-yielding synthesis of N-triazolyl carboxamides via palladium-catalysed aminocarbonylation. Tetrahedron 2017, 73, 6736–6741. [Google Scholar] [CrossRef]
- Mikle, G.; Boros, B.; Kollar, L. Asymmetric aminocarbonylation of iodoalkenes in the presence of alpha-phenylethylamine as an N-nucleophile. Tetrahedron Asymmetry 2017, 28, 1733–1738. [Google Scholar] [CrossRef]
- Nagymihaly, Z.; Csok, Z.; Kollar, L. Influence of base additives on the selectivity of palladium-catalysed aminocarbonylation: Highly selective functionalization of a cavitand scaffold. Mol. Catal. 2018, 444, 70–75. [Google Scholar] [CrossRef]
- Kollar, L.; Varga, M.; Dornyei, A.; Takacs, A. Functionalisation of the uracil ring via palladium-catalysed aminocarbonylation. Tetrahedron 2019, 75, 4632–4639. [Google Scholar] [CrossRef]
- Szoke, G.; Takacs, A.; Kollar, L. Synthesis of Pyridazine Dicarboxamides via Highly Selective Palladium-catalyzed Aminocarbonylation. J. Heterocycl. Chem. 2016, 53, 2020–2024. [Google Scholar] [CrossRef]
- Takacs, A.; Marosvolgyi-Hasko, D.; Kabak-Solt, Z.; Damas, L.; Rodrigues, F.; Carrilho, R.; Pineiro, M.; Pereira, M.; Kollar, L. Functionalization of indole at C-5 or C-7 via palladium-catalysed double carbonylation. A facile synthesis of indole ketocarboxamides and carboxamide dimers. Tetrahedron 2016, 72, 247–256. [Google Scholar] [CrossRef]
- Szoke, G.; Takacs, A.; Berente, Z.; Petz, A.; Kollar, L. Synthesis of amino-substituted pyridylglyoxylamides via palladium-catalysed aminocarbonylation. Tetrahedron 2016, 72, 3063–3067. [Google Scholar] [CrossRef]
- Takacs, A.; Varga, G.; Kardos, J.; Kollar, L. Palladium-catalysed aminocarbonylation of diiodopyridines. Tetrahedron 2017, 73, 2131–2138. [Google Scholar] [CrossRef]
- Cox, R.; Ritson, D.; Dane, T.; Berge, J.; Charmant, J.; Kantacha, A. Room temperature palladium catalysed coupling of acyl chlorides with terminal alkynes. Chem. Commun. 2005, 1037–1039. [Google Scholar] [CrossRef]
- Cox, R.J.; Evitt, A.S. Acyl palladium species in synthesis: Single-step synthesis of alpha, beta-unsaturated ketones from acid chlorides. Org. Biomol. Chem. 2007, 5, 229–232. [Google Scholar] [CrossRef]
- Gao, B.; Zhang, G.; Zhou, X.; Huang, H. Palladium-catalyzed regiodivergent hydroaminocarbonylation of alkenes to primary amides with ammonium chloride. Chem. Sci. 2018, 9, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Willand, N.; Folleas, B.; Boutillon, C.; Verbraeken, L.; Gesquiere, J.; Tartar, A.; Deprez, B. Efficient, two-step synthesis of N-substituted nortropinone derivatives. Tetrahedron Lett. 2007, 48, 5007–5011. [Google Scholar] [CrossRef]
- Neufeldt, S.; Jimenez-Oses, G.; Comins, D.; Houk, K. A Twist on Facial Selectivity of Hydride Reductions of Cyclic Ketones: Twist-Boat Conformers in Cyclohexanone, Piperidone, and Tropinone Reactions. J. Org. Chem. 2014, 79, 11609–11618. [Google Scholar] [CrossRef]
- Affolter, O.; Baro, A.; Laschat, S.; Fischer, P. Acylation of tropane alkaloids displaying reversed diastereoselectivities under enzymatic versus chemical conditions. Zeitschr. Naturforsch. Sect. B J. Chem. Sci. 2007, 62, 82–92. [Google Scholar] [CrossRef]
- Wang, Z.; Hou, Z.; Yao, S.; Lin, M.; Song, H. A new and recyclable system based on tropin ionic liquids for resolution of several racemic amino acids. Anal. Chim. Acta. 2017, 960, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Lyapkalo, I.; Hogermeier, J.; Reissig, H. Synthesis of new tropinone derivatives by palladium-catalyzed couplings of 8-azabicyclo[3.2.1]oct-2-enyl nonatlates. Tetrahedron 2004, 60, 7721–7729. [Google Scholar] [CrossRef]
- Cheng, J.; Moore, Z.; Stevens, E.; Trudell, M. Stereoselective syntheses of the three isomers of ethylene glycol bis(tropane-3-carboxylate). J. Org. Chem. 2002, 67, 5433–5436. [Google Scholar] [CrossRef]
- Ghosh, S.; Kinney, W.; Gauthier, D.; Lawson, E.; Hudlicky, T.; Maryanoff, B. Convenient preparation of aryl-substituted nortropanes by Suzuki-Miyaura methodology. Can. J. Chem. 2006, 84, 555–560. [Google Scholar] [CrossRef]
- Horvath, L.; Berente, Z.; Kollar, L. High-yielding aminocarbonylation of 3-iodo-2-tropene by using amino acid esters as N-nucleophiles. Lett. Org. Chem. 2007, 4, 236–238. [Google Scholar] [CrossRef]
- Nocquet, P.; Opatz, T. Total Synthesis of (+/-)-Scopolamine: Challenges of the Tropane Ring. Eur. J. Org. Chem. 2016, 2016, 1156–1164. [Google Scholar] [CrossRef]
- Fodor, G.; Nádor, K. The stereochemistry of tropane alkaloids. Part I. The configuration of tropine and ψ-tropine. J. Chem. Soc. 1953, 721–723. [Google Scholar] [CrossRef]
- Bollinger, S.; Hubner, H.; Heinenmann, F.; Meyer, K.; Gmeiner, P. Novel Pyridylmethylamines as Highly Selective 5-HT1A Superagonists. J. Med. Chem. 2010, 53, 7167–7179. [Google Scholar] [CrossRef]
- Kohnen-Johannsen, K.; Kayser, O. Tropane Alkaloids: Chemistry, Pharmacology, Biosynthesis and Production. Molecules 2019, 24, 796. [Google Scholar] [CrossRef]
- Skoda-Foldes, R.; Kollar, L. Palladium-Catalyzed Aminocarbonylation of Iodoalkenes and Iodoarenes. Lett. Org. Chem. 2010, 7, 621–633. [Google Scholar] [CrossRef]
- Kiss, M.; Takacs, A.; Kollar, L. Highly Selective Synthesis of Carboxamides via Transition Metal Catalysed Aminocarbonylation. Curr. Green Chem. 2015, 2, 319–338. [Google Scholar] [CrossRef]
- Amatore, C.; Jutand, A.; Mbarki, M. Evidence of the formation of zerovalentpalladium from Pd(OAc)2 and triphenylphosphine. Organometallics 1992, 11, 3009–3013. [Google Scholar] [CrossRef]
- Amatore, C.; Carre, E.; Jutand, A.; Mbarki, M.; Meyer, G. Evidence for the Ligation of Palladium(0) Complexes by Acetate Ions: Consequences on the Mechanism of Their Oxidative Addition with Phenyl Iodide and PhPd(OAc)(PPh3)2 as Intermediate in the Heck Reaction. Organometallics 1995, 14, 5605–5614. [Google Scholar] [CrossRef]
- Csakai, Z.; Skoda-Foldes, R.; Kollar, L. NMR investigation of Pd(II)-Pd(0) reduction in the presence of mono- and ditertiary phosphines. Inorg. Chim. Acta 1999, 286, 93–97. [Google Scholar] [CrossRef]
- Takacs, A.; Farkas, R.; Kollar, L. High-yielding synthesis of 2-arylacrylamides via homogeneous catalytic aminocarbonylation of α-iodostyrene and α,α’-diiodo-1,4-divinylbenzene. Tetrahedron 2008, 64, 61–66. [Google Scholar] [CrossRef]
- Takacs, A.; Acs, P.; Berente, Z.; Wolfling, J.; Schneider, G.; Kollar, L. Novel 13 beta- and 13 alpha-D-homo steroids: 17a-carboxamido-D-homoestra-1,3,5(10),17-tetraene derivatives via palladium-catalyzed aminocarbonylations. Steroids 2010, 75, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Kollar, L.; Takacs, A. Novel synthesis of 3-carboxamidolactam derivatives via palladium-catalysed aminocarbonylation. Tetrahedron 2018, 74, 6116–6128. [Google Scholar] [CrossRef]
- Muller, E.; Peczely, G.; Skoda-Foldes, R.; Takacs, E.; Kokotos, G.; Bellis, E.; Kollar, L. Homogeneous catalytic aminocarbonylation of iodoalkenes and iodobenzene with amino acid esters under conventional conditions and in ionic liquids. Tetrahedron 2005, 61, 797–802. [Google Scholar] [CrossRef]
- Kiss, M.; Maho, S.; Boddi, K.; Boros, B.; Kollar, L. Palladium-catalyzed diaminocarbonylation: Synthesis of androstene dimers containing 3,3′- or 17,17′-dicarboxamide spacers. Monatsh. Chem. 2015, 146, 357–364. [Google Scholar] [CrossRef]
- Barton, D.; O’Brien, R.; Sternhell, S. A new reaction of hydrazones. J. Chem. Soc. 1962, 470–476. [Google Scholar] [CrossRef]
- Barton, D.; Bashiardes, G.; Fourrey, J. An Improved Preparation of Vinyl Iodides. Tetrahedron Lett. 1983, 24, 1605–1608. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry | Substrate | Amine | R. Time [h] | Conv. b [18] | Isolated Yield c [18] |
| 1 | 1-iodocyclohexene (1) | a | 4 | 3 | n.i. |
| 2 | 1-iodocyclohexene (1) | a | 24 | 42 | n.i. |
| 3 | 1-iodocyclohexene (1) | a | 48 | 88 | n.i. |
| 4 | 1-iodocyclohexene (1) d | a | 4 | 70 | n.i. |
| 5 | 1-iodocyclohexene (1) d | a | 24 | 100 | 80 (1a) |
| 6 | 4-tert-butyl-1-iodocyclohexene (2) | a | 4 | 100 | 86 (2a) |
| 7 | trans-1-iodo-1-octene (3) | a | 4 | 100 | 81 (3a) |
| 8 | 17-iodoandrost-16-ene (4) | a | 24 | 100 | 80 (4a) |
| 9 | 2-iodobornene (5) | a | 4 | 75 | n.i. |
| 10 | 2-iodobornene (5) | a | 24 | 100 | 77 (5a) |
| 11 | 1-iodocyclohexene (1) | b | 1 | 100 | 89 (1b) |
| 12 | 4-tert-butyl-1-iodocyclohexene (2) | b | 1 | 100 | 79 (2b) |
| 13 | trans-1-iodo-1-octene (3) | b | 1 | 100 | 65 (3b) |
| 14 | 17-iodoandrost-16-ene (4) | b | 1 | 80 | n.i. |
| 15 | 17-iodoandrost-16-ene (4) | b | 6 | 100 | 55 (4b) |
| 16 | 2-iodobornene (5) | b | 1 | 100 | 63 (5b) |
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | Amine | R. Time [h] | Temp [°C] | Conv. b [18] | Isolated Yield c [18] |
| 1 | 2-Iodopyridine (7) | a | 6 | 70 | 100 | 90 (7a) |
| 2 | 3-Iodopyridine (8) | a | 6 d | 70 | 66 | n.i. |
| 3 | 3-Iodopyridine (8) | a | 24 | 70 | 100 | 82 (8a) |
| 4 | 2-Iodothiophene (9) | a | 6 d | 70 | 82 | n.i. |
| 5 | 2-Iodothiophene (9) | a | 24 | 70 | 100 | 82 (9a) |
| 6 | 5-Iodoindole (10) | a | 6 d | 70 | 47 | n.i. |
| 7 | 5-Iodoindole (10) | a | 24 d | 70 | 68 | n.i. |
| 8 | 5-Iodoindole (10) | a | 48 | 70 | 86 | 68 (10a) |
| 9 | 2-Iodopyridine (7) | b | 2 | 50 | 100 | 73 (7b) |
| 10 | 3-Iodopyridine (8) | b | 2 | 50 | 100 | 62 (8b) |
| 11 | 2-Iodothiophene (9) | b | 2 d | 50 | 85 | n.i. |
| 12 | 2-Iodothiophene (9) | b | 6 | 50 | 100 | 92 (9b) |
| 13 | 5-Iodoindole (10) | b | 2 d | 50 | 90 | n.i. |
| 14 | 5-Iodoindole (10) | b | 4 | 50 | 100 | 51 (10b) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kollár, L.; Erdélyi, Á.; Rasheed, H.; Takács, A. Selective Synthesis of N-Acylnortropane Derivatives in Palladium-Catalysed Aminocarbonylation. Molecules 2021, 26, 1813. https://doi.org/10.3390/molecules26061813
Kollár L, Erdélyi Á, Rasheed H, Takács A. Selective Synthesis of N-Acylnortropane Derivatives in Palladium-Catalysed Aminocarbonylation. Molecules. 2021; 26(6):1813. https://doi.org/10.3390/molecules26061813
Chicago/Turabian StyleKollár, László, Ádám Erdélyi, Haroon Rasheed, and Attila Takács. 2021. "Selective Synthesis of N-Acylnortropane Derivatives in Palladium-Catalysed Aminocarbonylation" Molecules 26, no. 6: 1813. https://doi.org/10.3390/molecules26061813
APA StyleKollár, L., Erdélyi, Á., Rasheed, H., & Takács, A. (2021). Selective Synthesis of N-Acylnortropane Derivatives in Palladium-Catalysed Aminocarbonylation. Molecules, 26(6), 1813. https://doi.org/10.3390/molecules26061813