1. Introduction
Bacterial biofilms are aggregates of microorganisms anchored to a surface and embedded in a self-produced matrix of extracellular polymeric substances [
1]. Due to the properties of the matrix and to the intercellular interactions between the bacteria within, the biofilm develops, thus becoming increasingly sophisticated and offering the constituting bacteria several advantages over their planktonic counterparts [
2,
3]. These include an increased antibiotic resistance, elevated levels of lateral gene transfer, higher stress resistance and subversion of host defense mechanisms [
4,
5,
6]. Quorum sensing (QS), a process of cell-to-cell communication, is important for the control of several virulence factors and plays a key role during biofilm formation. In this process, cells communicate using auto-inducer signals. QS allows bacteria to synchronously adjust gene expression to alter their behavior in response to changes in population density and the surrounding bacterial community [
7].
Several clinically important pathogenic bacteria, such as cystic fibrosis-associated
Pseudomonas aeruginosa, surgical sites associated with
Staphylococcus aureus and many others, cause infection through biofilm formation [
8]. This can have devastating consequences. Microbes that reside in biofilms may not be eliminated by traditional antibiotics because of metabolic dormancy or molecular resistance mechanisms [
9]. The US National Institutes of Health estimate that 80% of all bacterial infections occurring in the human body are biofilm-related [
10]. Therefore, the overall burden of biofilm infections is significant, and it has been recognized as a serious threat to our society [
10]. Biofilm infections are not easily treated with existing antimicrobial approaches because the biofilm recalcitrance is a consequence of its complex physical and biological properties [
11]. QS signaling plays an important role in biofilm formation in such a way that specific QS signaling blockage is an effective way to prevent the biofilm formation of most pathogens. Additionally, QS inhibition does not affect the normal growth of the bacteria. Therefore, they do not create any evolutionary pressure for the emergence of multidrug-resistant bacteria. Consequently, QS inhibitors usually have a longer functional shelf life than modern antibiotics and are regarded as a promising therapeutic alternative in combined therapies [
12,
13].
Chromobacterium violaceum is a large, motile, Gram-negative bacillus, which lives on soil and water in tropical and subtropical regions, and it can act as an opportunistic pathogen for animals and humans. It enters through broken skin by contamination with soil or stagnant water [
14]. There have been reports of it causing localized skin and soft tissue infection and systemic or invasive infection. These include necrotizing fasciitis, visceral abscesses, osteomyelitis, and central nervous system disease [
15]. Infections due to
C. violaceum, albeit relatively rare, with less than 150 published clinical reports, are associated with high mortality [
16].
This bacterium is known for the production of a natural violet pigment with antibiotic properties, known as violacein, whose production is regulated via quorum-sensing [
17]. Since this QS-regulated trait is an easily observable and quantifiable trait,
C. violaceum is widely used as a model organism for QS research [
18]. The QS system in
C. violaceum is homologous of the LuxI/LuxR system found
in Vibrio fischeri, with the AHL synthase being CviI and the transcriptional activator being CviR [
19,
20].
Over the years, the usage of computer-aided drug design (CADD) as a preliminary stage of drug design has increased, finding application in the study of many problems, including in the search for quorum-sensing inhibitors [
21,
22,
23,
24,
25,
26,
27,
28,
29,
30,
31,
32,
33,
34,
35,
36]. This makes the entire process more cost-efficient and minimizes failures [
37]. Therefore, the aim of this study is to use CADD to understand the binding determinants and particularities of the CviR-binding pocket as a QS inhibition model target and to identify new promising molecules for blocking quorum-sensing and preventing biofilm formation. Different computational methods were combined in this study, including protein–ligand docking, virtual screening, MD simulations and MM/PB(GB)SA calculations.
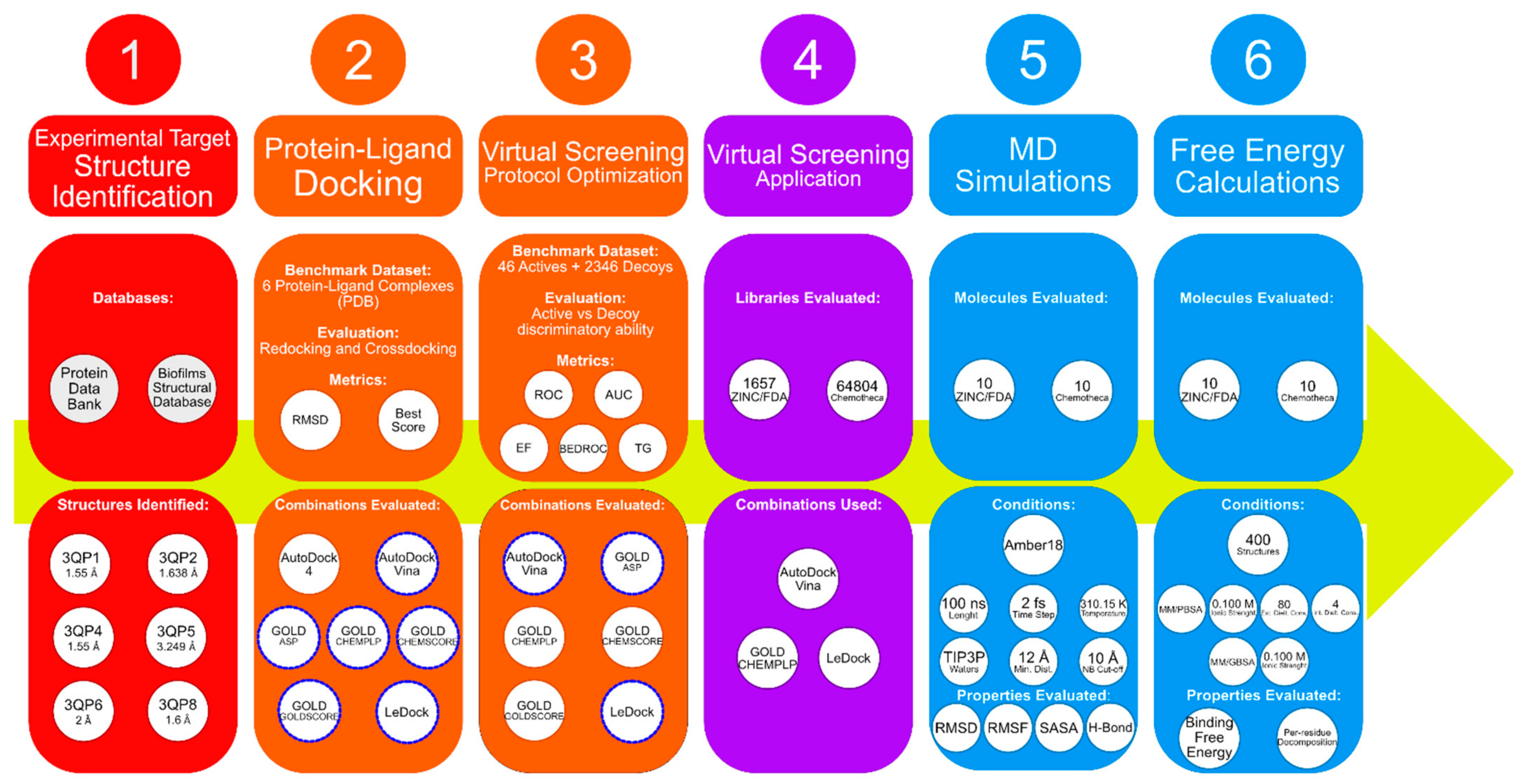
Figure 1 depicts the workflow of this work.
3. Results and Discussion
Table 2 compares the best redocking performance obtained with Autodock4, Vina, CHEMPLP, CHEMSCORE, GOLDSCORE, ASP and LeDock in terms of RMSD between the predicted pose and the X-ray crystallographic pose. One important detail is that 3QP2 was separated into two different files, one for each conformation of the important residue Met89. 3QP2a has the intermediate conformation, while 3QP2b has a similar conformation to 3QP1.
The RMSD values show that the best docking program for reproducing experimental poses of the ligand is LeDock, with an average RMSD of 0.74 Å. All scoring functions from GOLD also have a good performance. On the other hand, the programs, which performed worse were Vina and AutoDock 4, with an average RMSD of 1.44 Å and 1.13 Å, respectively. Vina has the worse overall RMSD value for the redocking of 3QP6. In this case, the program placed the molecule the opposite way when compared to the crystallographic structure. However, this could be due to the fact that a smaller molecule is being docked in a binding pocket previously accommodating a larger ligand. In reality, when C6-HSL binds to this structure (in this strain, C6-HSL functions as a partial antagonist), there is a shift in the DNA-binding domain, which will help fill the binding pocket [
20]. Nevertheless, most RMSD values are below 2 Å, which indicates a good overall performance from all the different molecular docking software/scoring functions. This good performance is mainly seen with the lactone head group, with most programs struggling with the acyl chain. The scores from all redocking procedures are available in
Table S1 in the supplementary materials.
The cross-docking studies (details on
Tables S2 and S3 in the supplementary materials) show that, on average, the best results are seen on structures 3QP6 and 3QP8, both from strain 12472. From strain 31532, the structure, which generates the best scores, is 3QP4. In contrast, the worst results in all ligands and software are seen on structure 3QP5. This may be due to the worse resolution of this structure. Similar to the redocking studies, C6-HSL shows less affinity when compared to all other ligands, while C10-HSL and CL show the best results. This behavior is shown in most structures and software. One major difference from the redocking and cross-docking studies is the behavior of 3QP6. As expected, the bad performance during the redocking studies was due to the ligand, C6-HSL. When docking other ligands, 3QP6 is frequently the best structure that leads to the best results.
Globally, the redocking and cross-docking studies suggest that most combinations of docking program/scoring function, when optimized, perform similarly. For these structures, the poorest results were obtained with AutoDock 4 that is also the most time-consuming program.
Regarding the structures, the main conclusions emerging from these results are that both structures from strain 12472 (3QP6 and 3QP8) displayed the highest scores with all scoring functions, from which 3QP6 is the structure that generated the best results. From strain 31532, 3QP4 displayed the higher and most consistent scores for all scoring functions, while 3QP1, 3QP2a and 3QP2b presented more variable scores. Finally, 3QP5 was consistently the worst performing of all available structures of CviR.
After evaluating and optimizing the docking of known CviR ligands with well-characterized and experimentally defined binding poses, the next step was to evaluate the ability of this protocol in distinguishing between known CviR binders and non-binders.
Table 3 and
Table 4 compare the ability of Vina, CHEMPLP, CHEMSCORE, GOLDSCORE, ASP, LeDock, as well as all the different PDB structures, in distinguishing experimentally confirmed CviR binders (actives) and non-binders (decoys) through the usage of several different evaluation metrics.
The best-performing programs were consistent across most of the metrics, with Vina and GOLD with the CHEMPLP scoring function (GOLD/CHEMPLP) being more capable of distinguishing the experimentally confirmed binders from the decoys, generally followed by LeDock. With this in mind, the programs used in the virtual screening stage of this work were Vina, GOLD/CHEMPLP and LeDock (see
Figure 1).
Finally, it is important to select the receptor structures. Across nearly all metrics, the best discrimination ability between binders and non-binders was obtained when using 3QP6 and 3QP8. However, since they are both from strain 12472, the decision was made to use the best performing structure from each strain. The structure from strain 31532 that generally generated the best results was 3QP4. Therefore, the two CviR structures used in the virtual screening step of this work were 3QP4 and 3QP6.
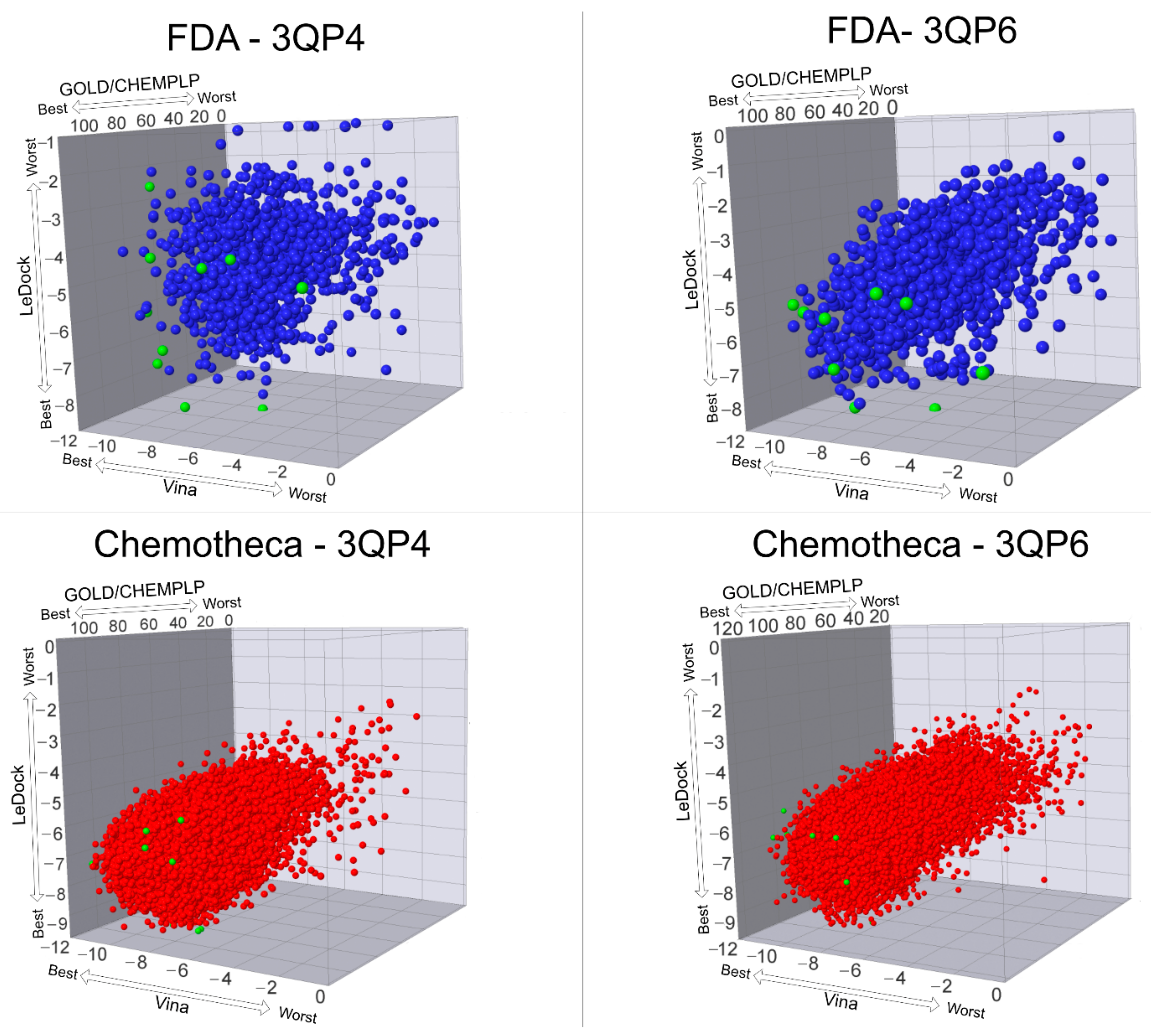
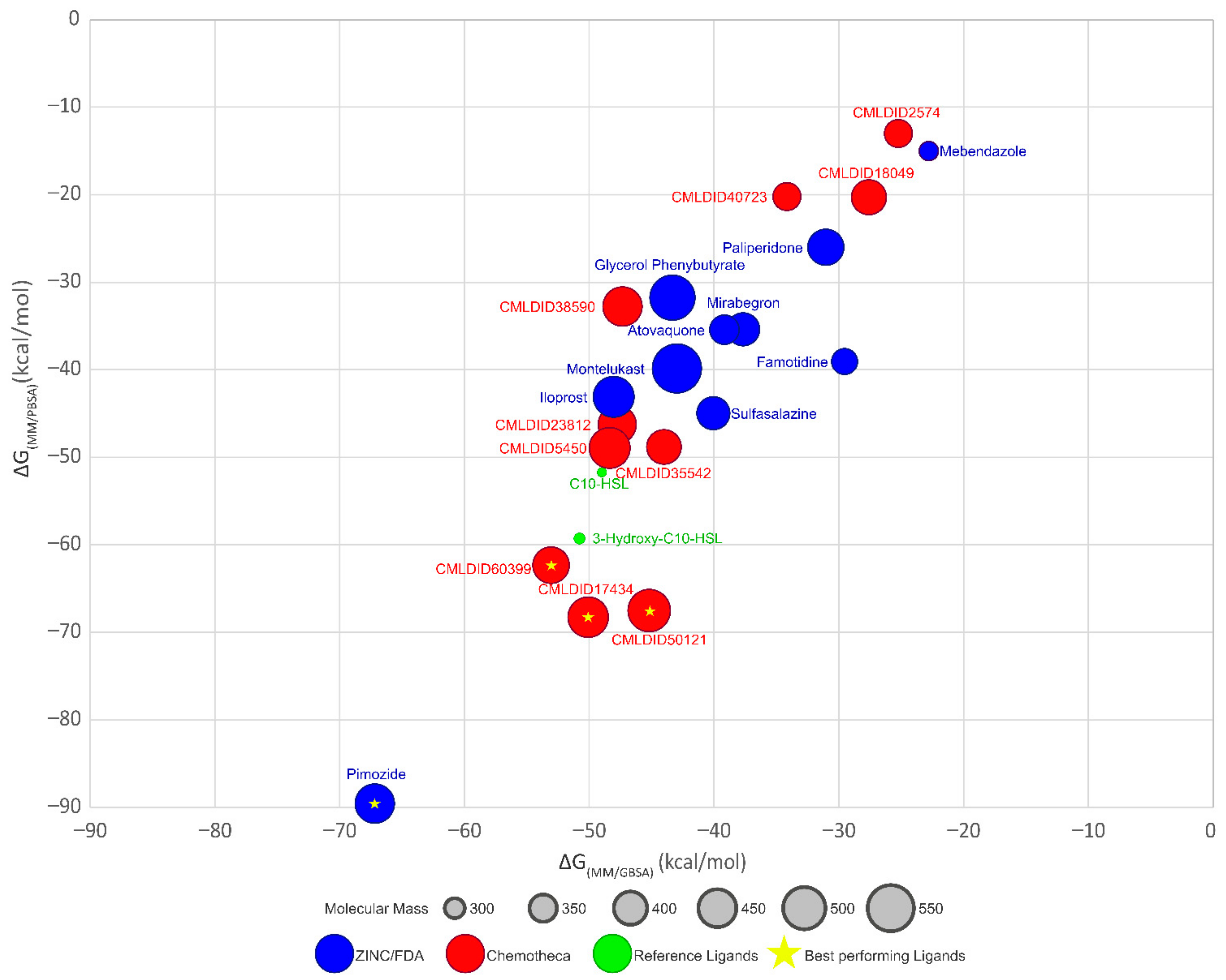
The optimized protein–ligand docking and virtual screening protocols were then applied to two libraries of compounds: the US Food and Drug Administration (USFDA) approved database available on ZINC (ZINC/FDA) to consider possible drug repurposing alternatives, and the more general Mu.Ta.Lig. Virtual Chemotheca. The results of all virtual screenings are in
Figure 2.
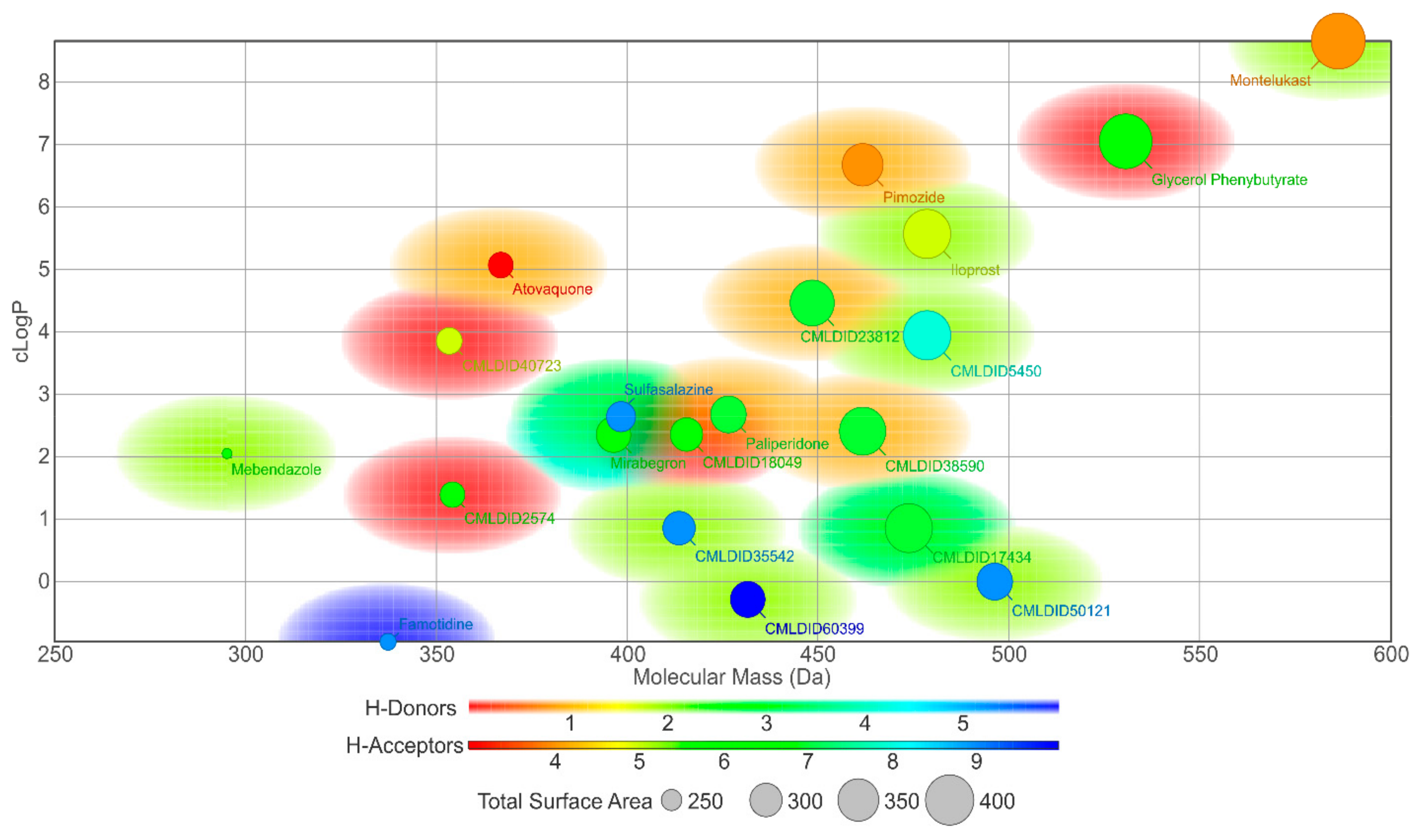
In this study, different molecular docking programs and different PDB structures were used in a consensus approach. This approach can explore many potential ligands with greater accuracy, contributing to a more rigorous selection of the molecules for testing. From the top solutions in the ranked lists obtained from the VS, 10 molecules from each library were selected for the MD simulations. For each library, the selection was based on the position of the molecules on the different VS protocols. The choice took into consideration the following criteria: (i) molecules among the top 25 solutions for the two structures considered in the VS; (ii) molecules among the top 25 solutions with more than one scoring function; (iii) molecular diversity taking into consideration the physical-chemical properties (
Figure 3).
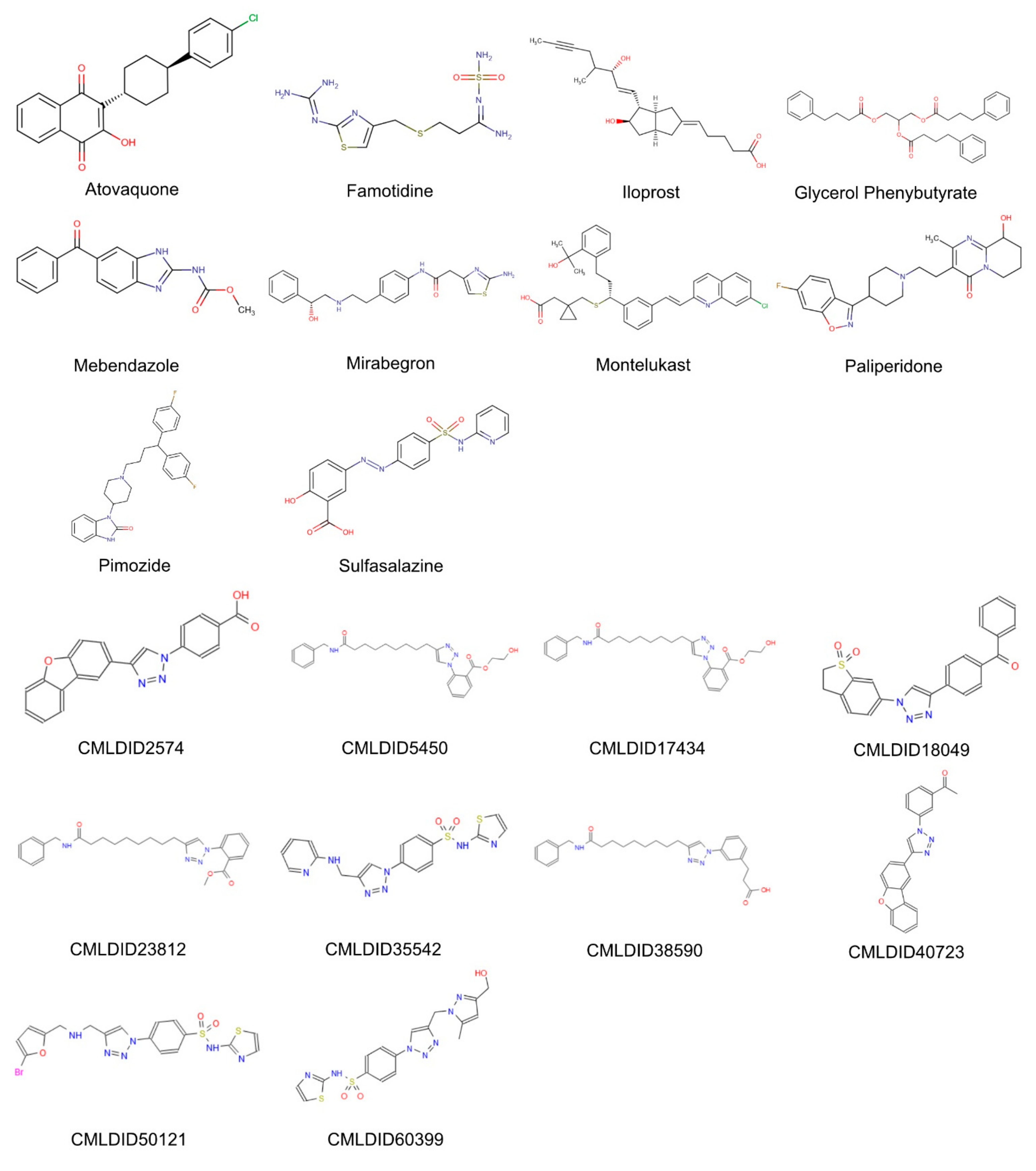
The molecules from the ZINC/FDA database were atovaquone, famotidine, iloprost, mebendazole, mirabegron, montelukast, paliperidone, pimozide, glycerol phenylbutyrate, and sulfasalazine (
Figure 4). The molecules from Chemotheca were CMLDID2574, CMLDID5450, CMLDID17434, CMLDID18049, CMLDID23812, CMLDID35542, CMLDID38590, CMLDID40723, CMLDID50121 and CMLDID60399 (
Figure 4). As a display of the variability of the selected molecules, multiple important properties (cLogP, molecular mass, total surface area, hydrogen donors and acceptors) of the 20 selected molecules are displayed in
Figure 3. These properties and graphs were calculated using the data visualization and analysis program, DataWarrior [
76]. Comparisons between the score of the 20 selected molecules and the scores obtained by the known active molecules is available in
Figures S2–S7 in the supplementary materials.
Based on the EF 1% for the actives/decoys training set, we estimated the minimum number of actives expected to be found on the 25 first positions of each VS (
Table 5).
The next step of this work involved 100 ns of molecular dynamics simulation for each of the above-mentioned molecules in complex with 3QP6. This was done to validate the protein–ligand docking results, to evaluate the structural stability of the protein–ligand complex and to carry out the MM/PB(GB)SA calculation. For reference, MD simulations of 3-hydroxy-C10-HSL and C10-HSL in complex with the protein were also performed. While the native ligand of the strain 12472 of
C. violaceum is 3-hydroxy-C10-HSL, C10-HSL is an agonist for this protein [
20,
77]. Additionally, this ligand was the agonist that generated the highest scores on the cross-docking studies with 3QP6. Consequently, C10-HSL, together with 3-hydroxy-C10-HSL, was selected as the reference ligands. Since these MD simulations were used for the refinement of the virtual screening results, only the ligand-binding domain was considered. In the future, we intend to perform further studies for a better understanding of how these ligands could affect the DNA-binding domain.
To assess the structural stability of the complexes, RMSD calculations were performed for the Cα atoms of each complex and for the ligands.
Considering the selected ligands from the ZINC/FDA database and from the Chemotheca database, all complexes exhibit low RMSD values through the simulation. Most ligands also display low RMSD values than the docking prediction (
Table 6). However, multiple molecules display higher values, suggesting an induced-fit adjustment to the CviR-binding pocket during the MD simulation. The low standard deviation confirms that after the initial 40 ns of simulation, the ligand positions are well stabilized. This is further confirmed by the solvent-accessible surface area analysis. An increase in SASA from the initial pose predicted from docking would imply that the ligand was exiting the binding pocket. Fortunately, all ligands display a stable SASA value along the simulation. This, together with the RMSD results, confirms that all the selected ligands form stable complexes with CviR. Further information on the MD simulations is available in the
supplementary materials (Figures S8–S11 and Table S4).
The final step of this study was to perform a MM/PB(GB)SA analysis for each ligand–receptor complex to determine the binding free energy of each ligand. For these calculations, only the last 40 ns of MD simulation were considered. Results are presented in
Table 7 and
Figure 5.
The results demonstrate that most ligands present a lower affinity towards CviR than the reference ligands (MM/GBSA predicted values between −22.7 ± 0.1 and −67.2 ± 0.2, against −49.0 ± 0.2 and −51.0 ± 0.2 kcal/mol and MM/PBSA predicted values between −13.0 ± 0.1 and −89.6 ± 0.2, against −51.7 ± 0.2 and 59.6 ± 0.2 kcal/mol) The only ligand from the ZINC/FDA database, which displays higher affinity towards CviR is pimozide (−67.2 ± 0.2 and −89.7 ± 0.2 kcal/mol, respectively). Pimozide is used as an antipsychotic agent and for the suppression of vocal and motor tics in patients with Tourette syndrome. Although its exact mechanism of action is unknown, it is thought that it inhibits the dopamine D2 receptor [
78].
For the molecules from Chemotheca, only CMLDID17434 (−68.1 ± 0.2 kcal/mol for MM/PBSA) and CMLDID60399 (−53.0 ± 0.2 kcal/mol for MM/GBSA and −61.8 ± 0.2 kcal/mol for MM/PBSA) with both methods, and CMLDID50121 using MM/PBSA (−67.4 ± 0.2 kcal/mol) display higher affinity towards CviR than C10-HSL and 3-hydroxy-C10-HSL.
To provide additional confirmation of these results, and to better assess if the best performing ligands are indeed true binders, replicate MD runs were performed. For each of the four best-performing ligands, three additional MD simulations were performed, and the average RMSD, RMSF, SASA and number of H bonds were calculated. These results are shown in
Table 8.
Across all evaluated metrics, the replicas display similar results to the original runs. Furthermore, the percentage of SASA buried on all replicas gives additional strength to the prediction that these ligands are true binders and remain tightly bound.
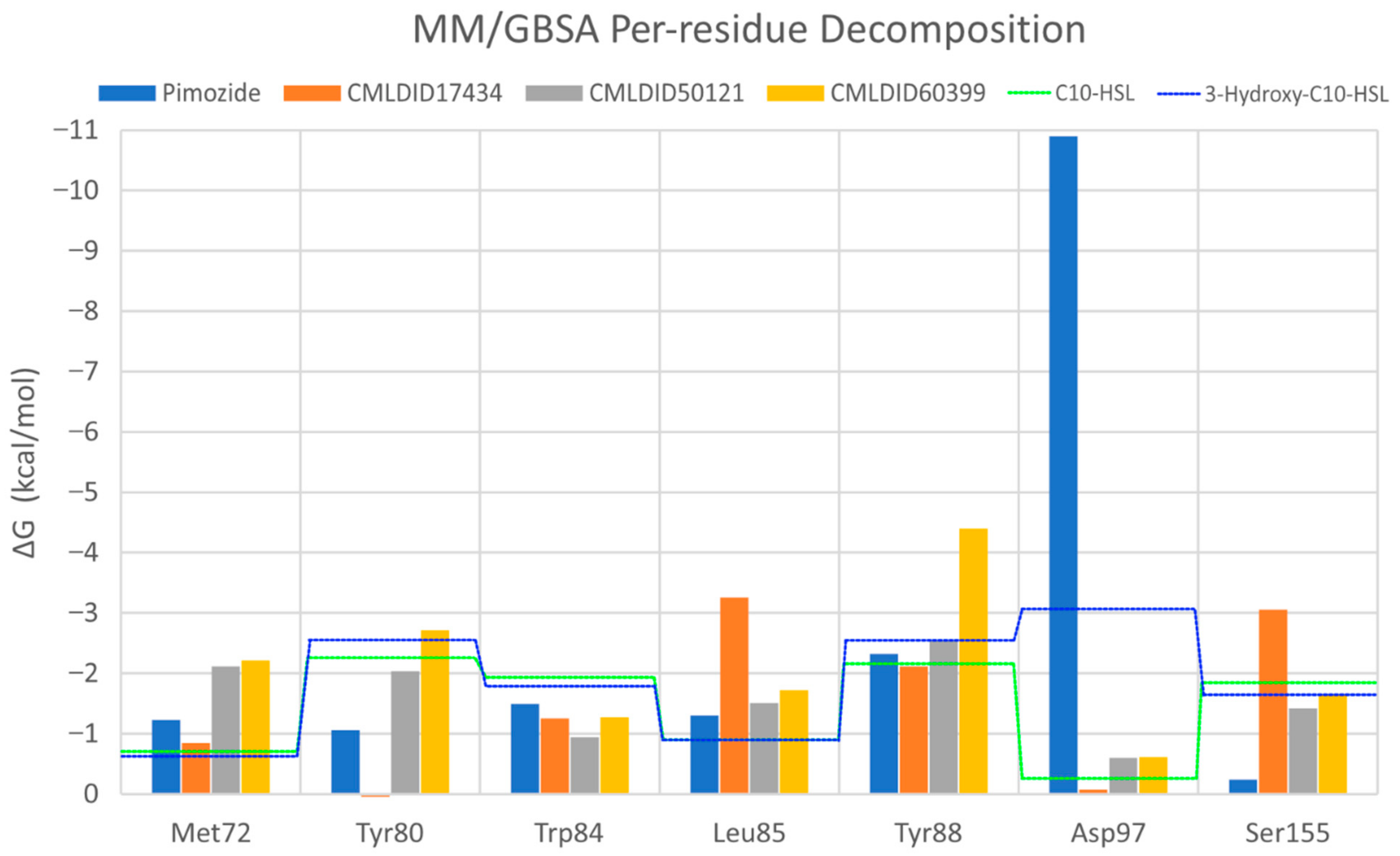
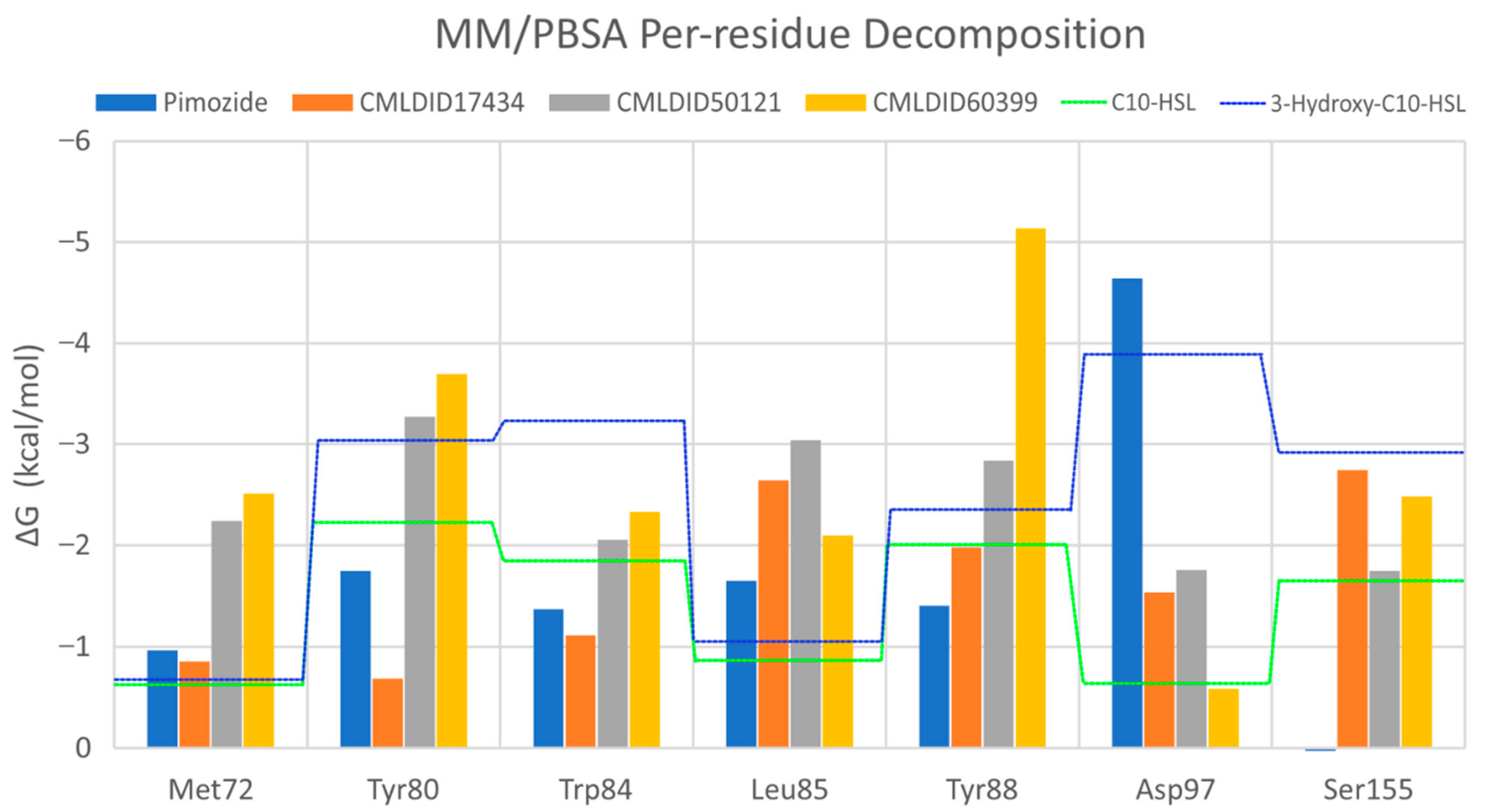
To further analyze the affinity between the ligands and the receptor, the overall Gibbs energy of association was decomposed into the contribution associated with each residue (
Figure 6 and
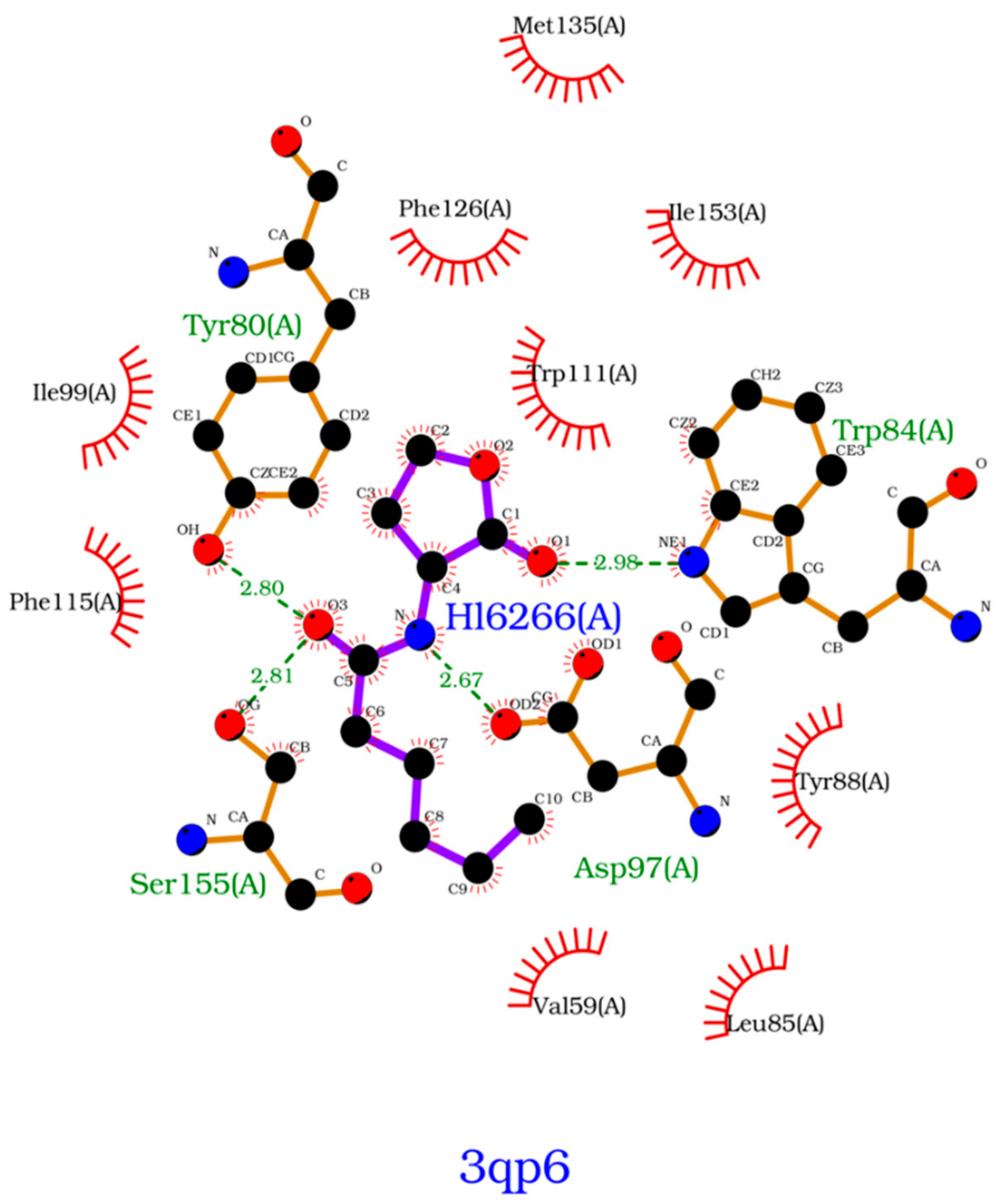
Figure 7). On 3-hydroxy-C10-HSL, the residues with the higher contribution to the Gibbs energy of association are Tyr80, Trp84, Tyr88, Asp97 and Ser155. As for C10-HSL, using both methods, the amino acids that have a bigger impact are Tyr80, Trp84, Tyr88 and Ser155. When comparing with the already known interactions map obtained with LigPlot+ [
79] (
Figure 8), these results show great similarity. The only difference is the contribution of Asp97 in the interaction with C10-HSL. While this residue is considered to have an important role in the binding of ligand to CviR, the calculations show a less favorable result for Asp97.
Overall, the residues with the greatest contribution for the affinity of the ligands towards CviR are Met72, Tyr80, Trp84, Leu85, Tyr88 and Ser155. For pimozide, the bigger contribution is obtained from Tyr80 (MM/PBSA), Tyr88 (MM/GBSA), and Asp97. Whereas on most ligands, the calculations show a less favorable contribution from this residue, with pimozide, it is the major contribution for the affinity between the ligand and the protein. For CMLDID17434, the residues with the highest contribution to the overall result using both methods are Leu85, Tyr88 and Ser155. In the case of CMLDID50121, Tyr80, Leu85 and Tyr88 are the residues with the most impact in the predicted affinity towards CviR. Lastly, for CMLDID60399, the amino acids with the greater impact are Met72, Tyr80, Tyr88.
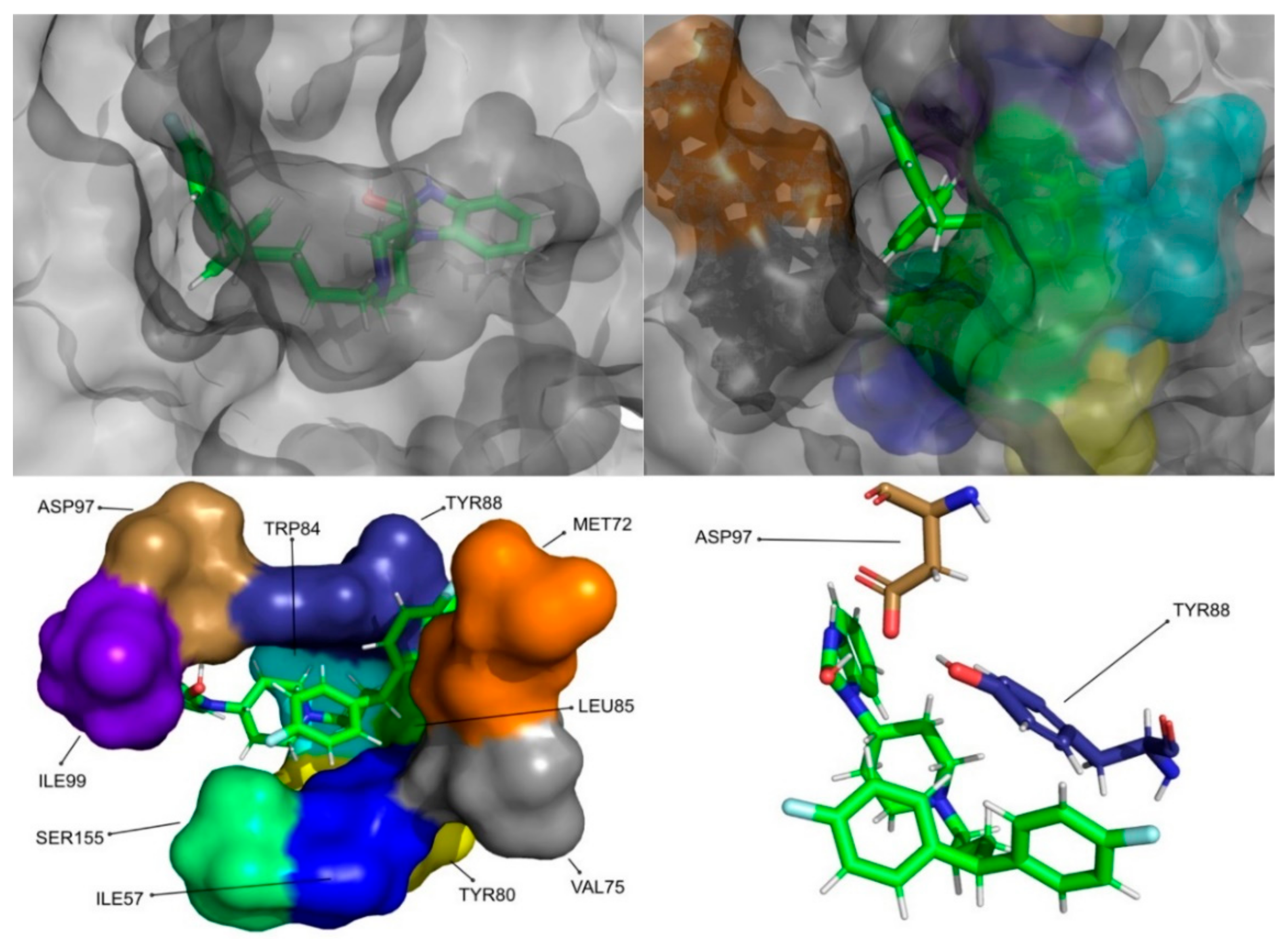
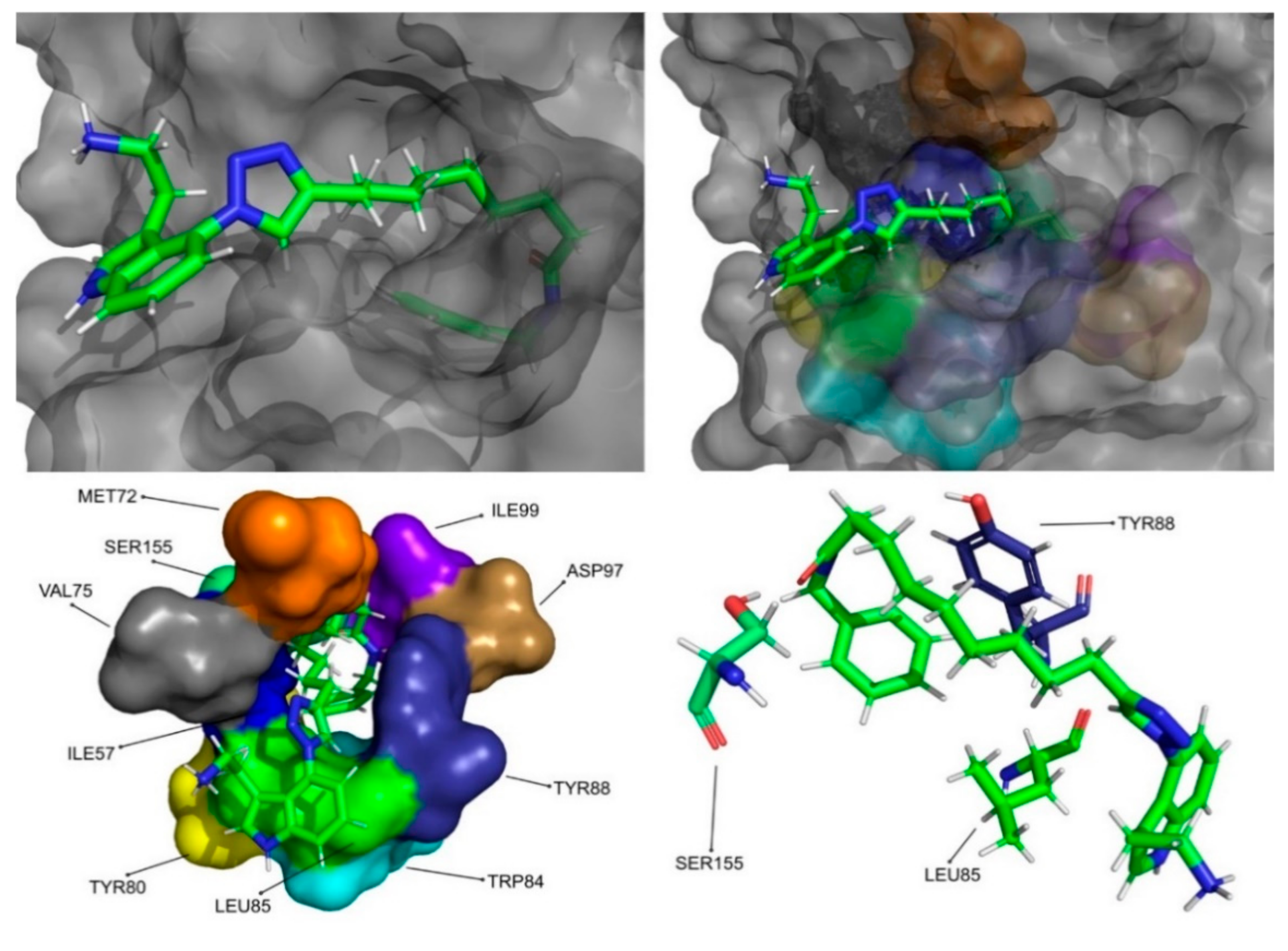
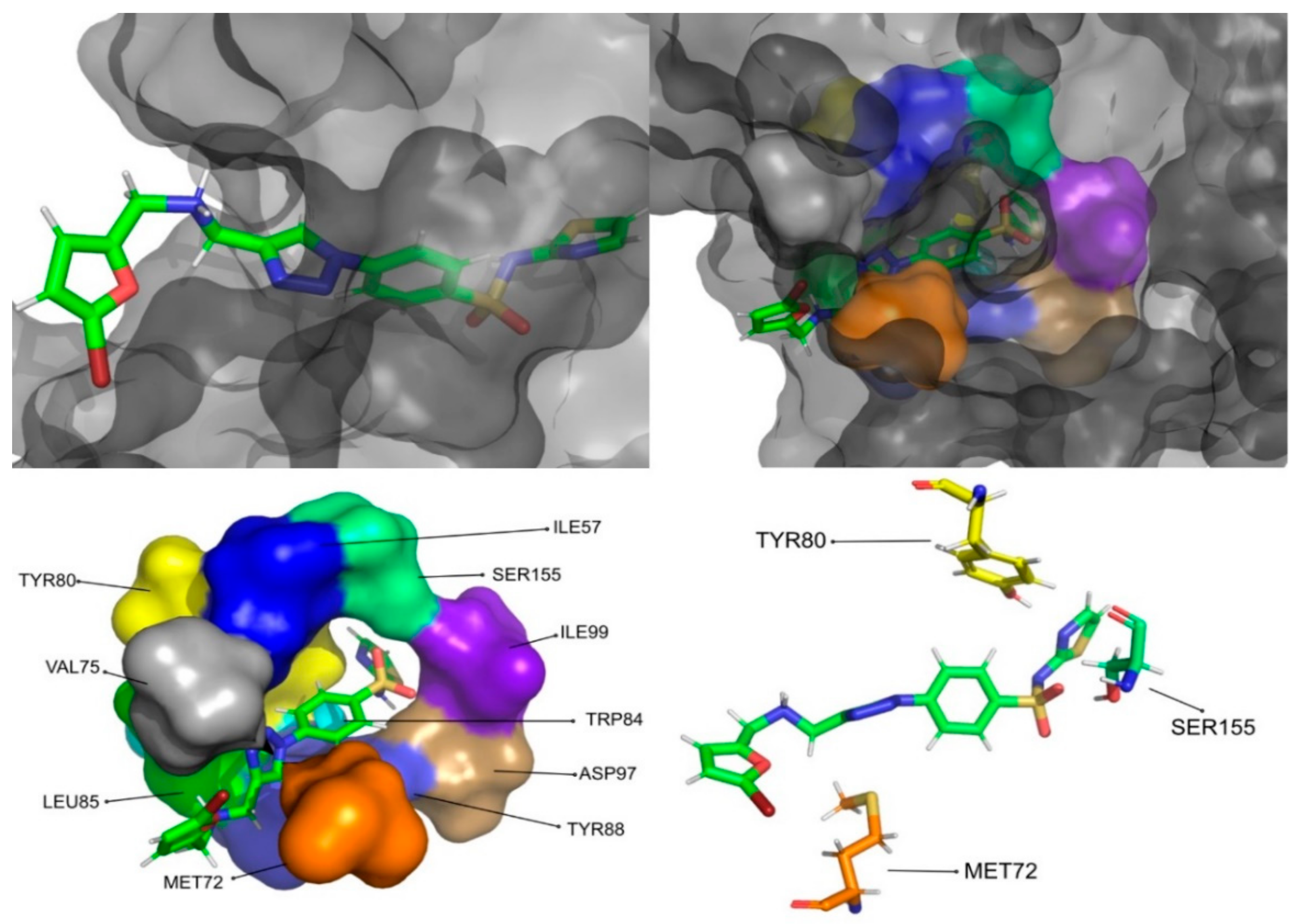
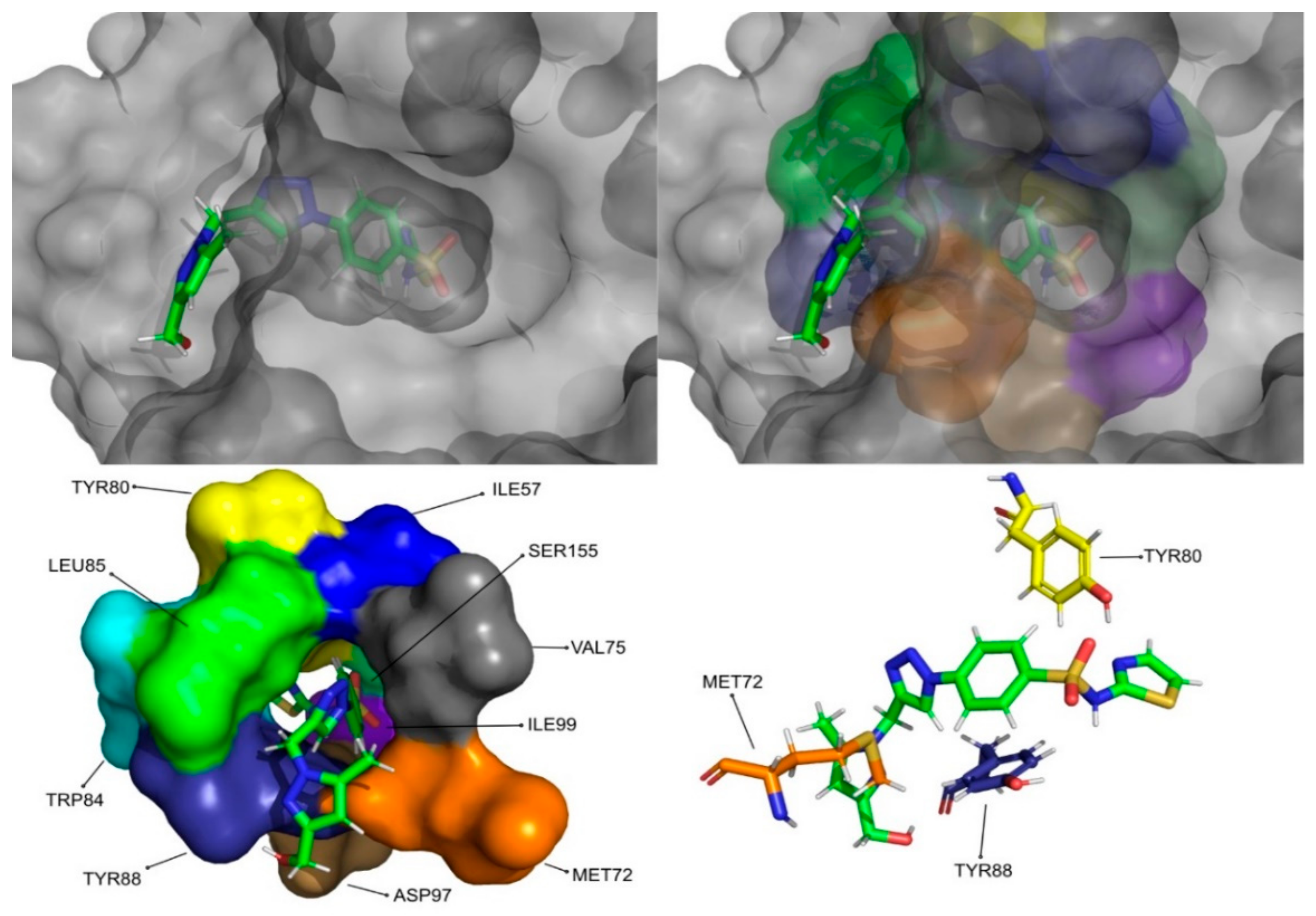
The molecular dynamics simulations and the MM/PB(GB)SA resulted in four molecules with higher or comparable binding affinities to the reference ligands, 3-hydroxy-C10-HSL and C10-HSL. These molecules were pimozide (
Figure 9) from the ZINC/FDA database and CMLDID17434 (
Figure 10), CMLDID50121 (
Figure 11) and CMLDID60399 (
Figure 12) from Chemotheca.

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}