
Hybrid Drugs—A Strategy for Overcoming Anticancer Drug Resistance?


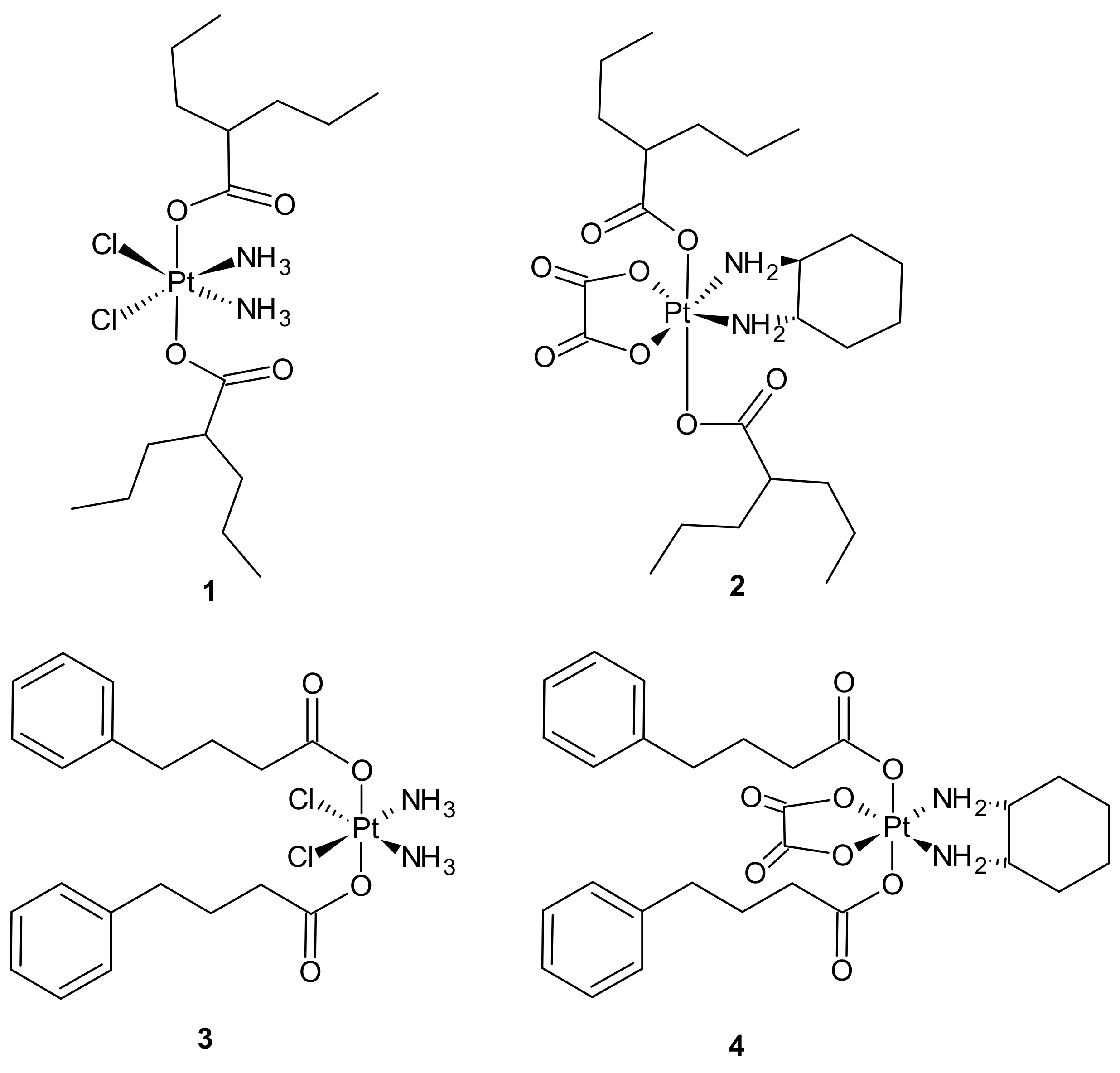{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
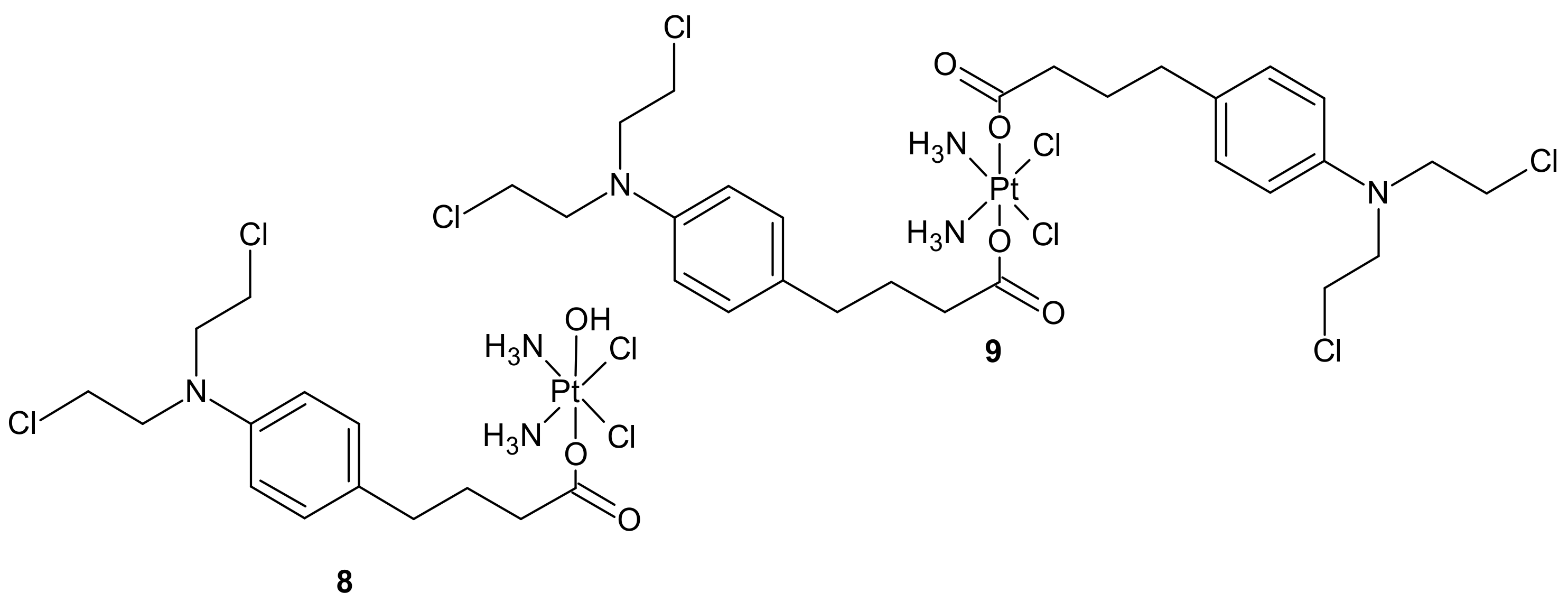
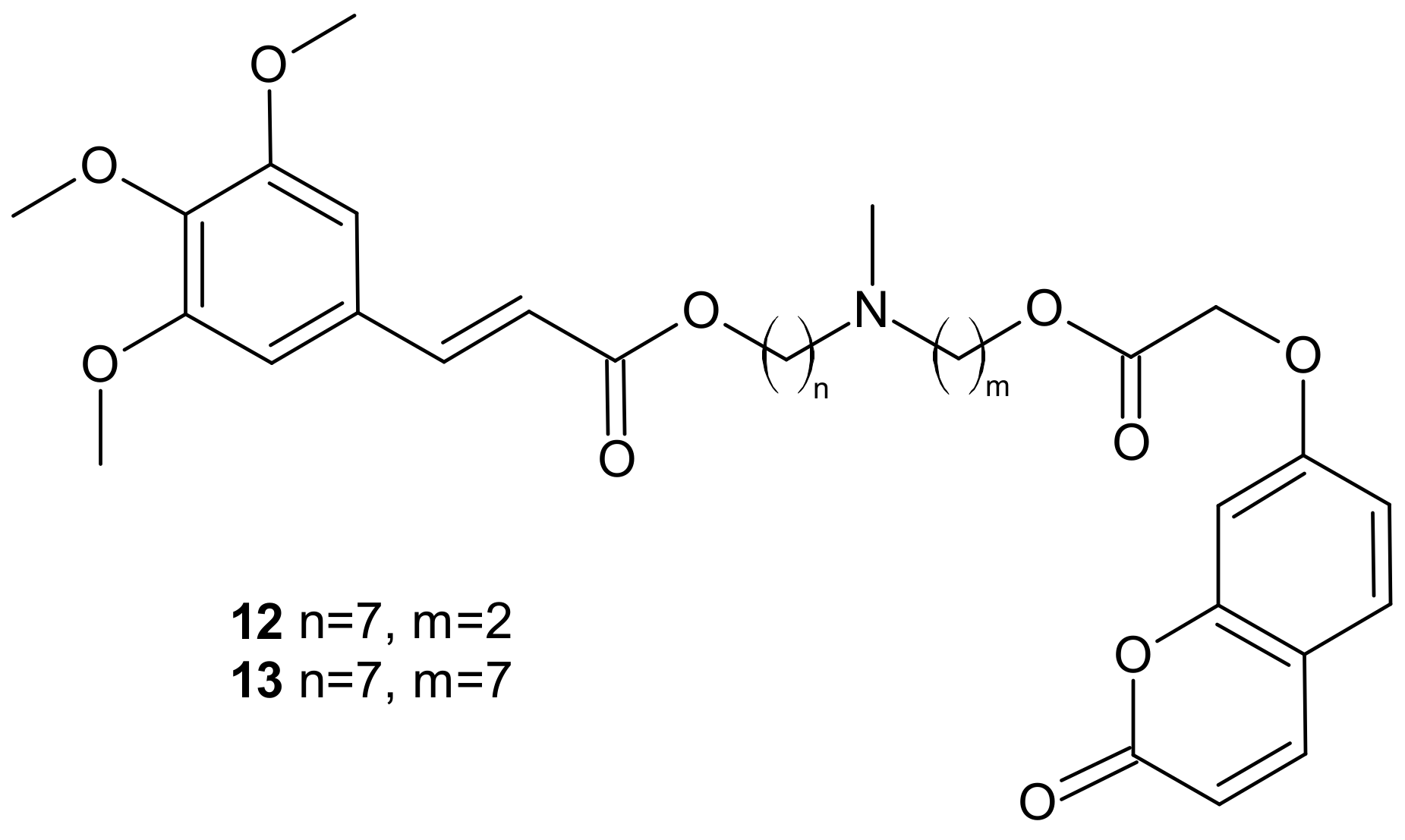{kind=link}
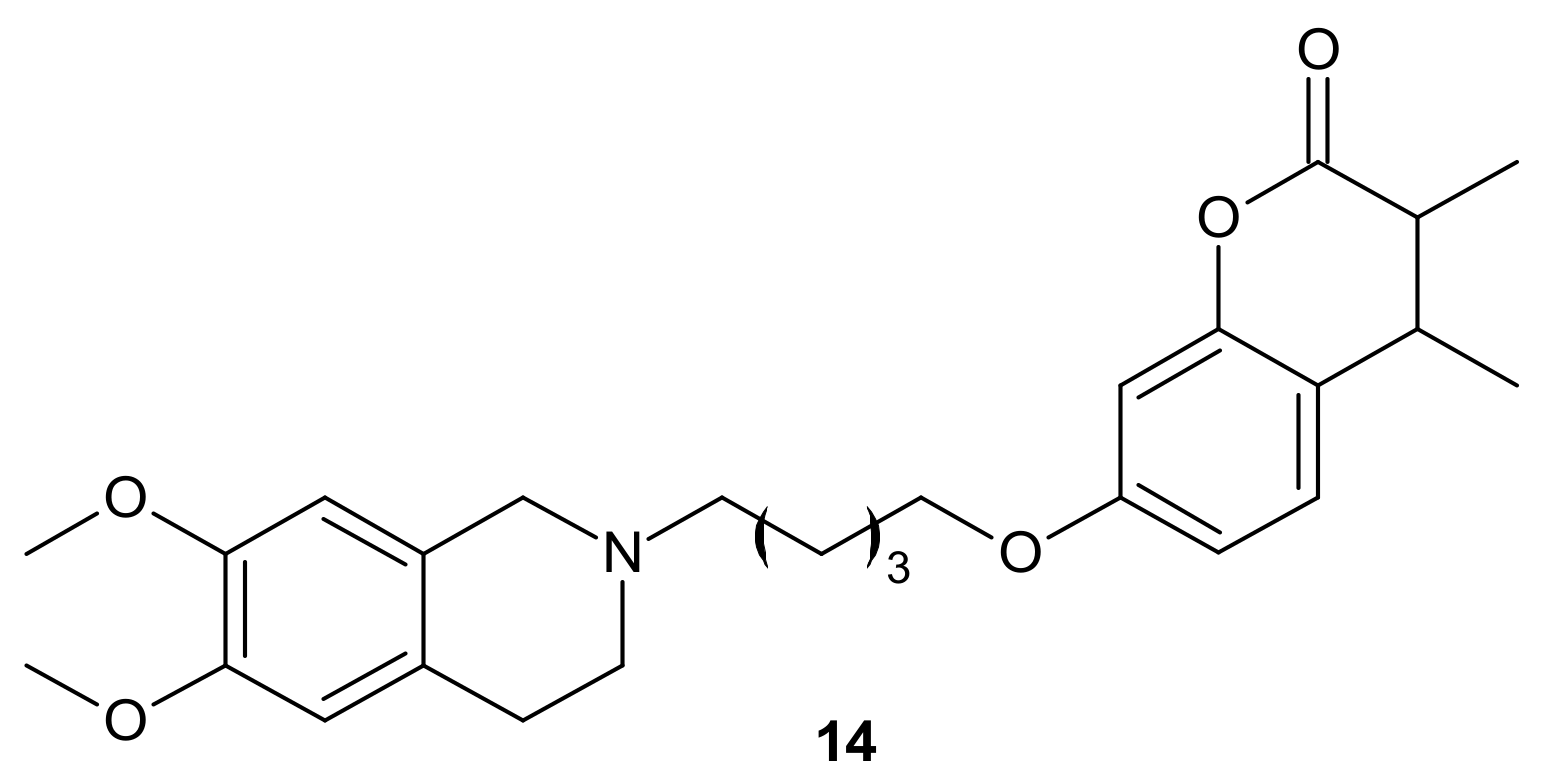{kind=link}
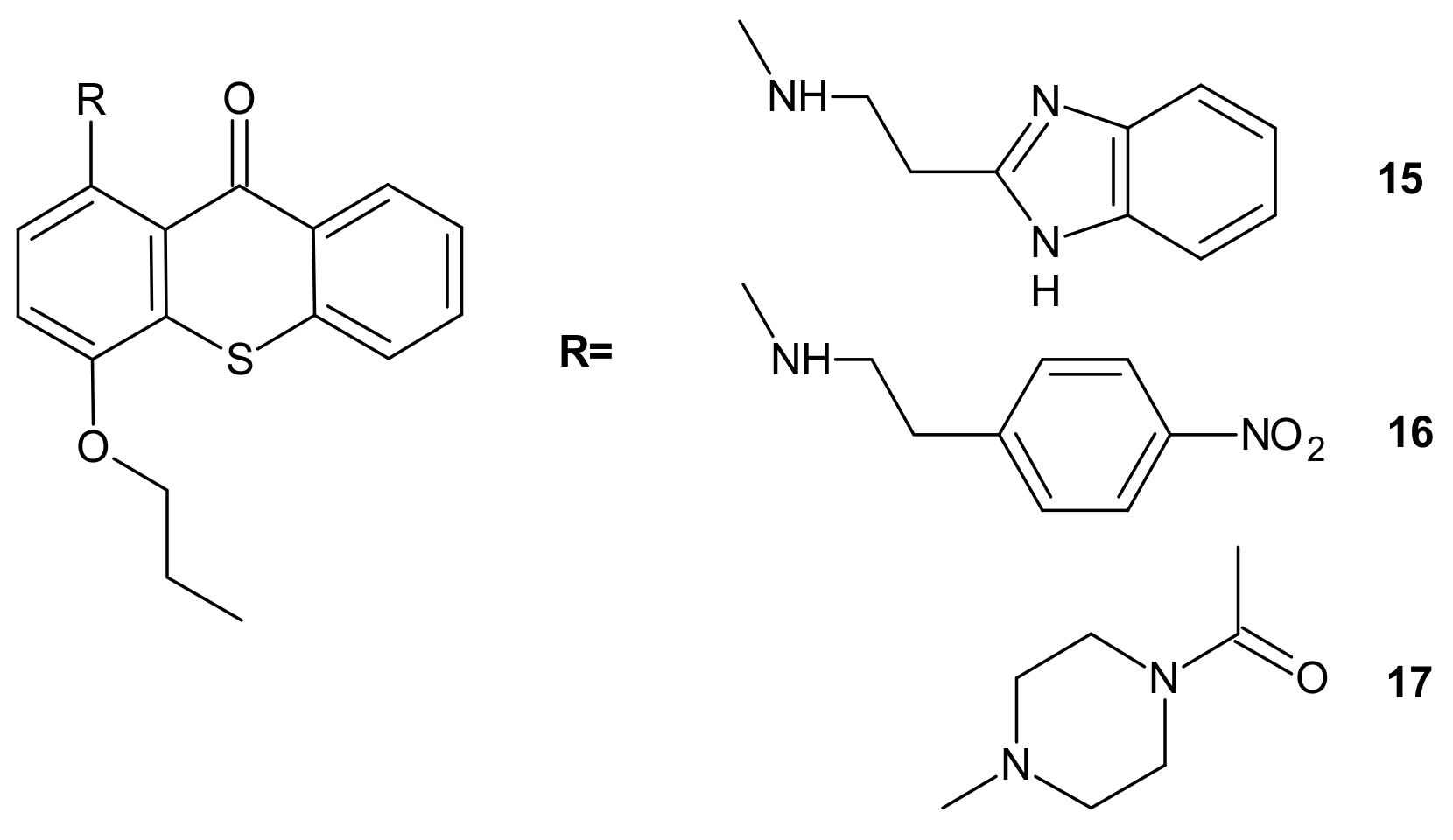{kind=link}
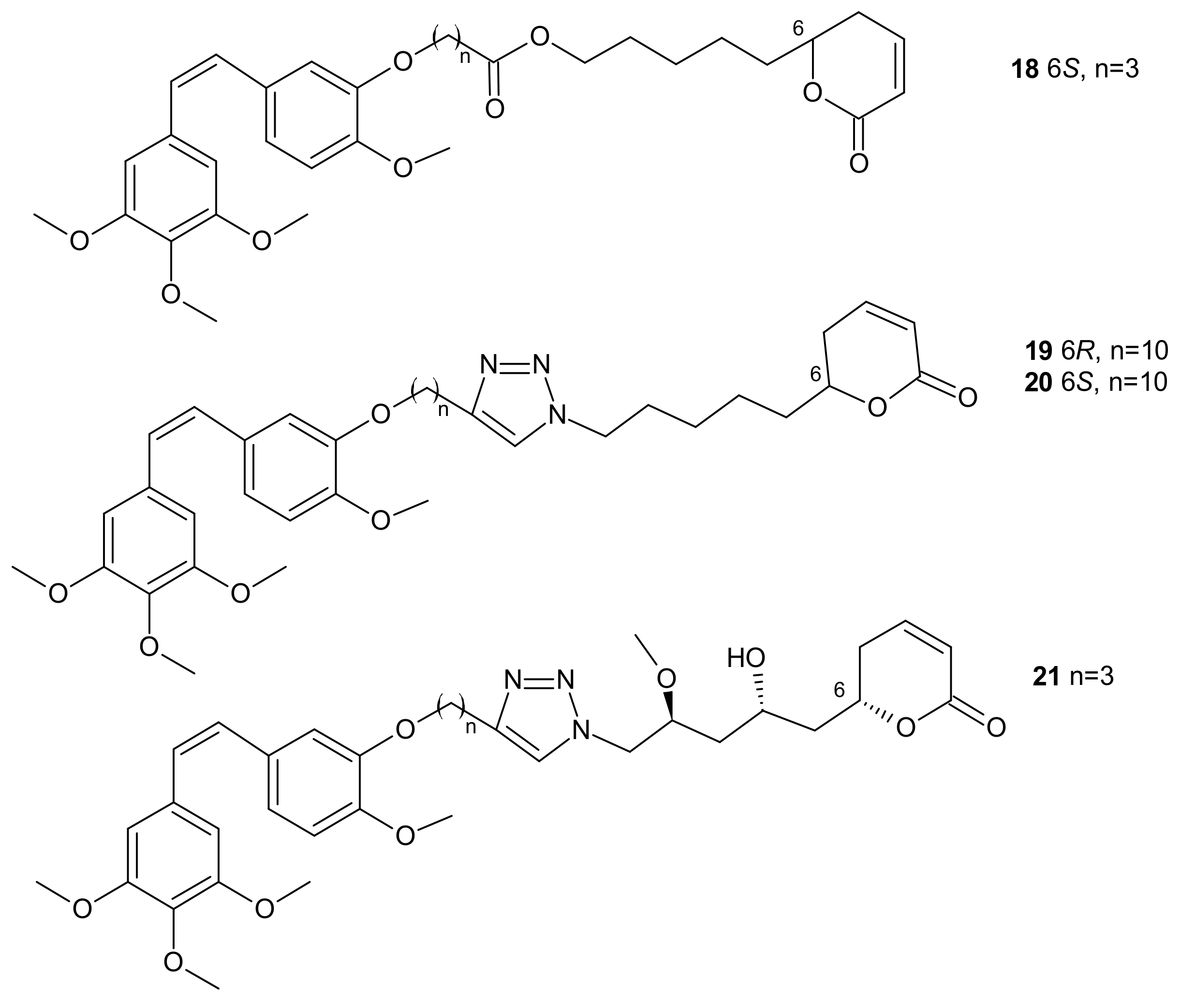{kind=link}
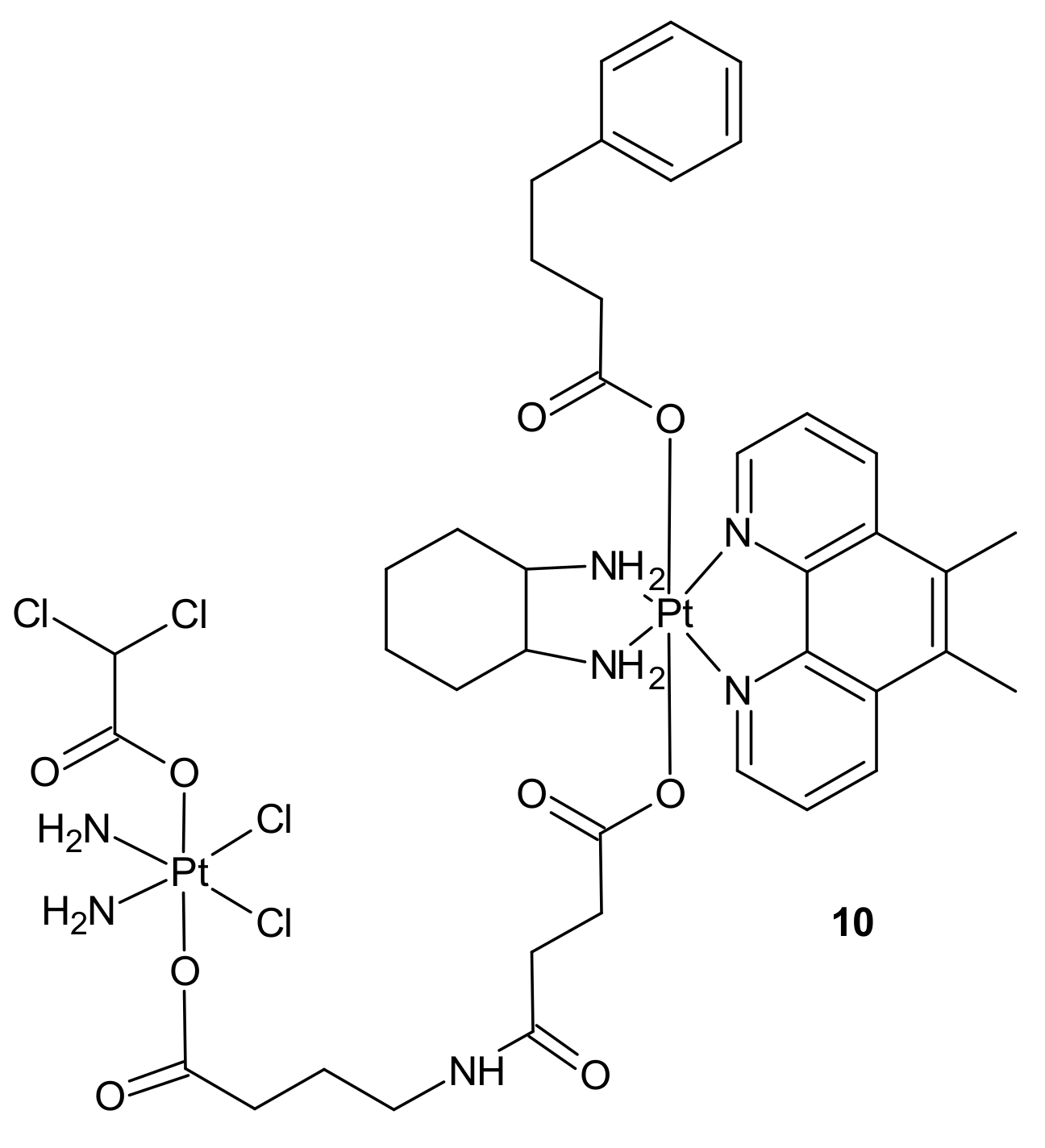
{kind=link}
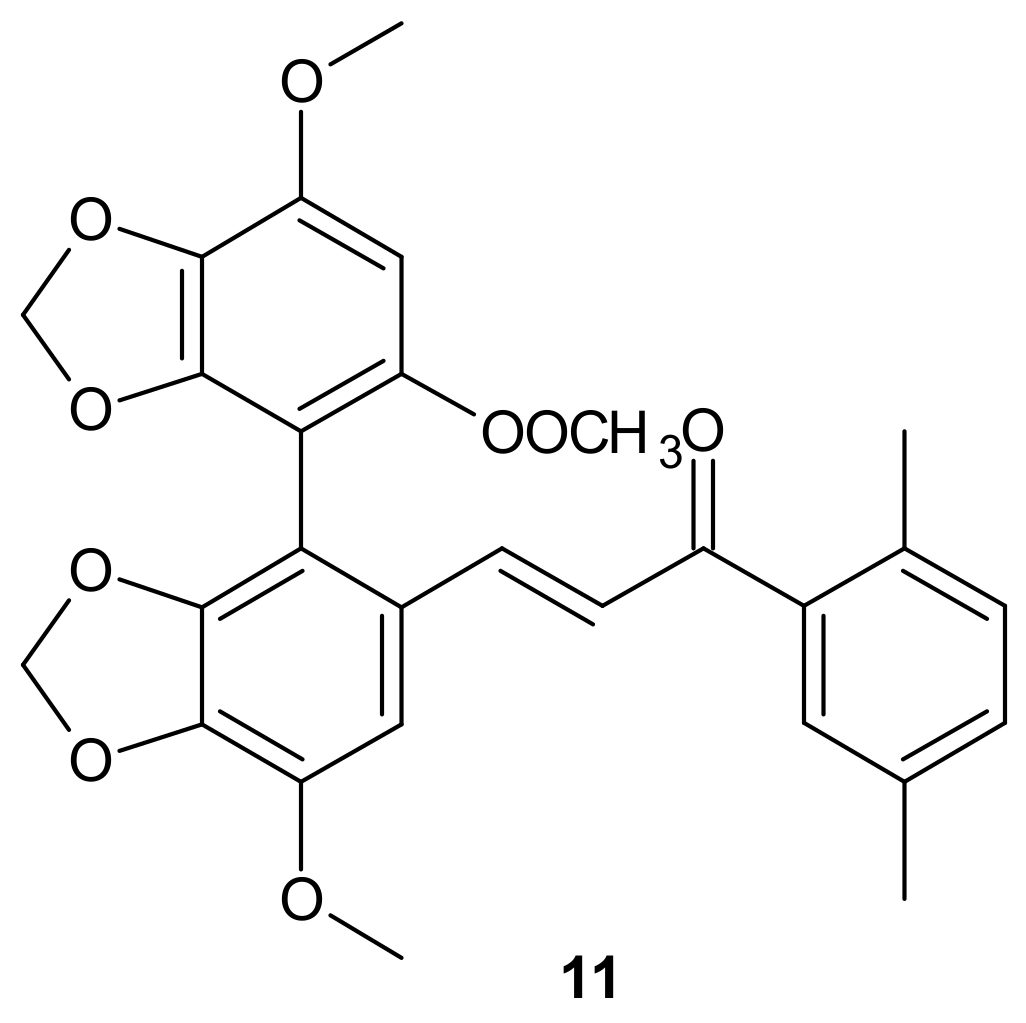
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Hybrid Drug—What Could It Be?
3. Hybrid Drugs as an Answer to the Anticancer Drug Resistance Problem?
3.1. Overcoming Anticancer Drug Resistance—From the Drug Perspective
3.2. Overcoming Anticancer Drug Resistance—From the Mechanism of Development Perspective
3.2.1. Drug Transporters, Drug Efflux, Drug Uptake
3.2.2. Drug Inactivation (Enzymatic Detoxification)
3.2.3. DNA Damage Repair
3.2.4. Target Modifications
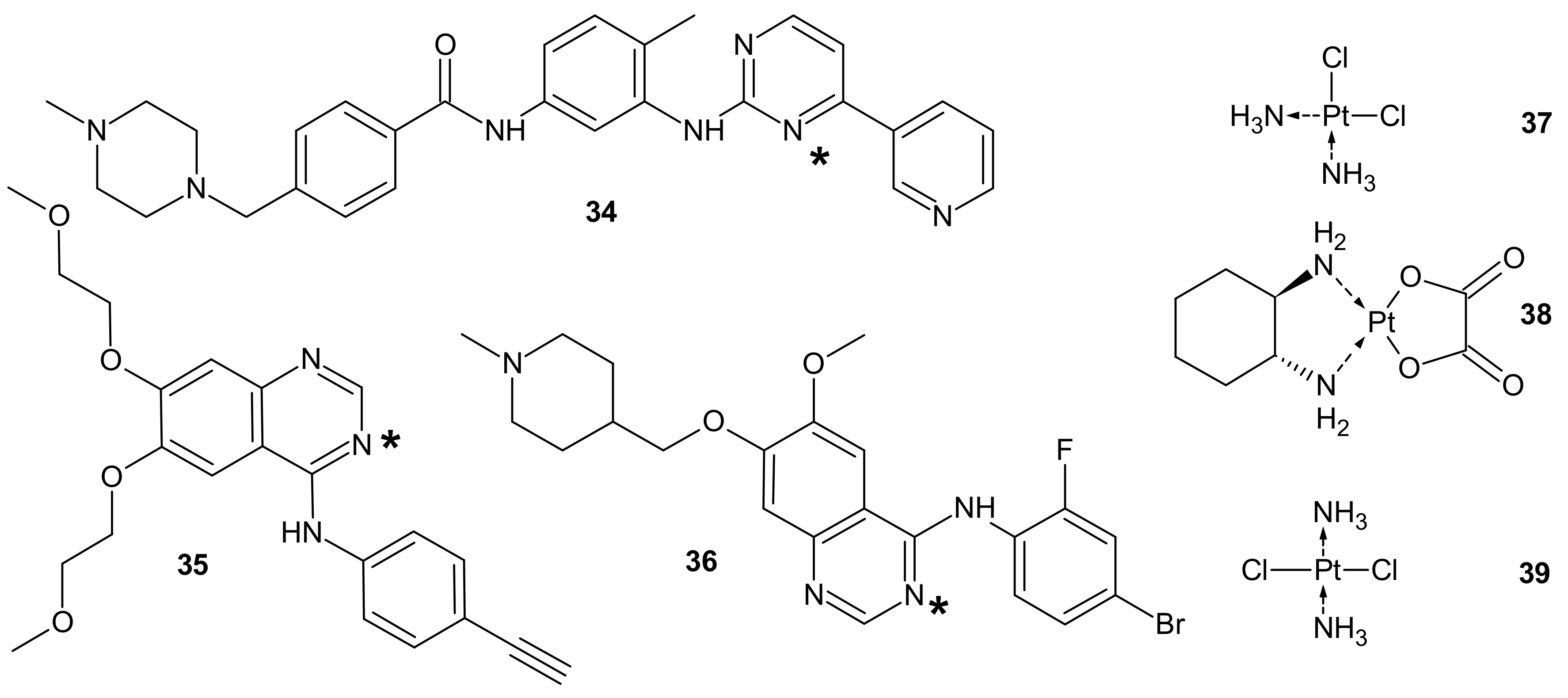
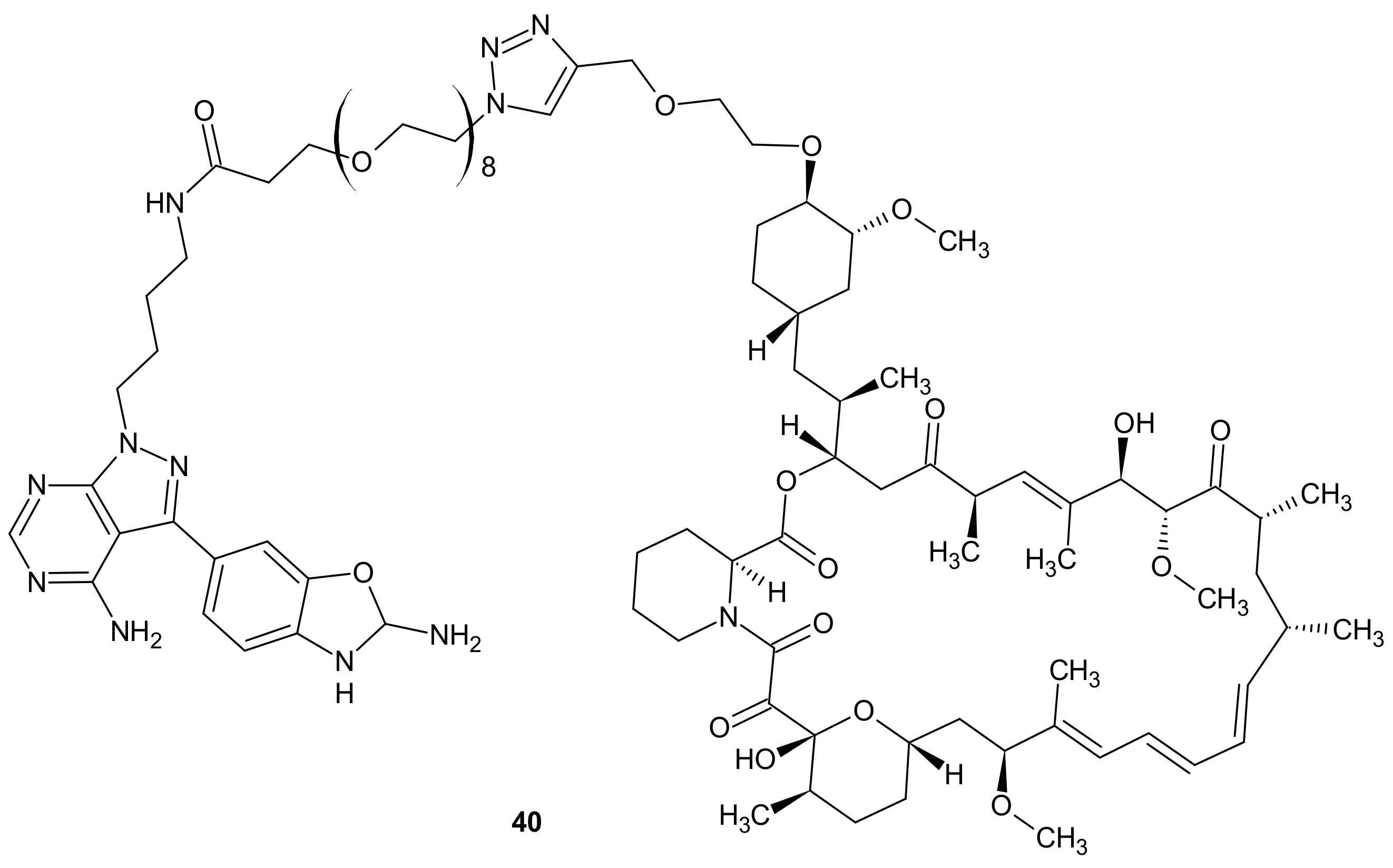
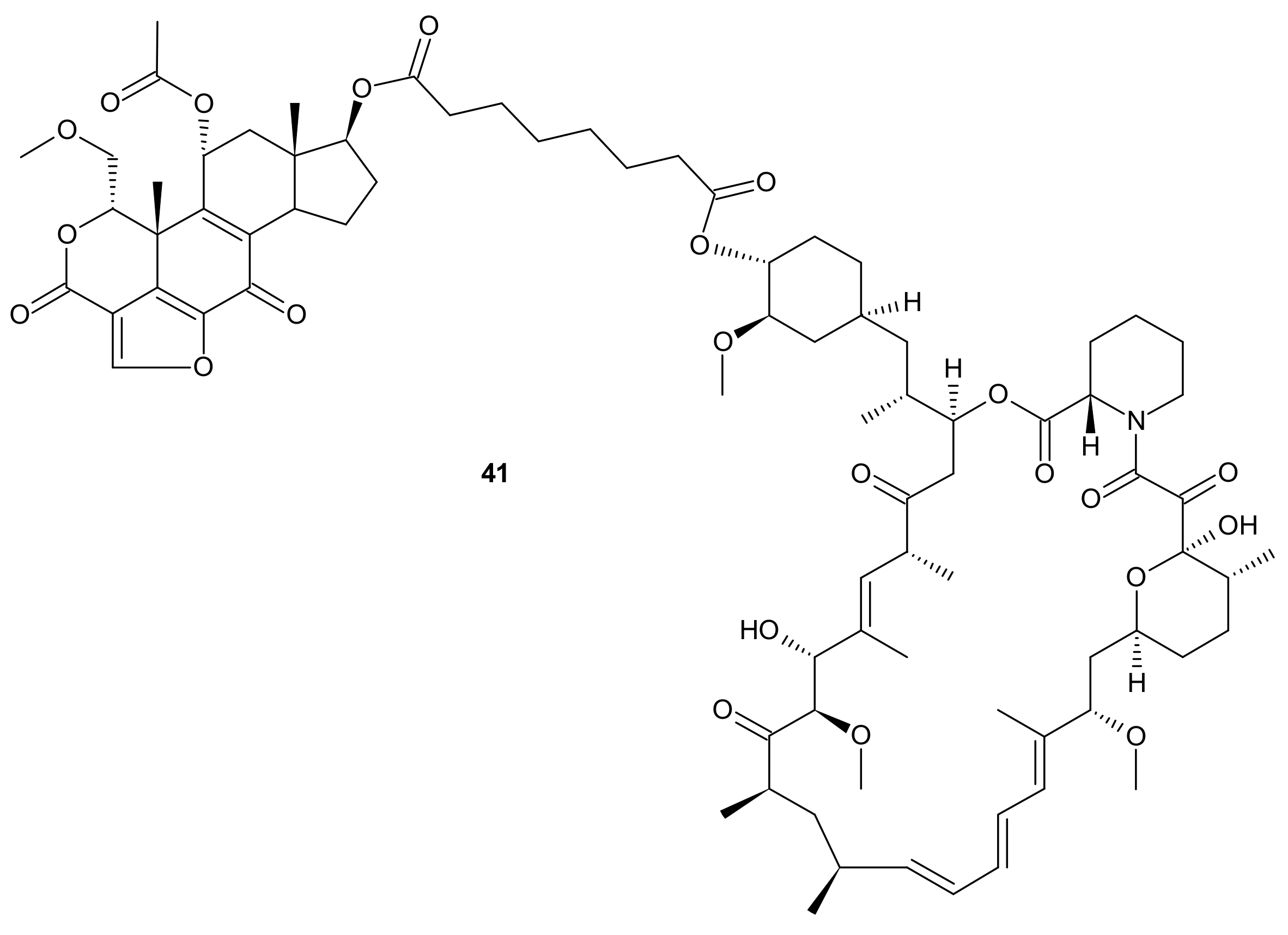
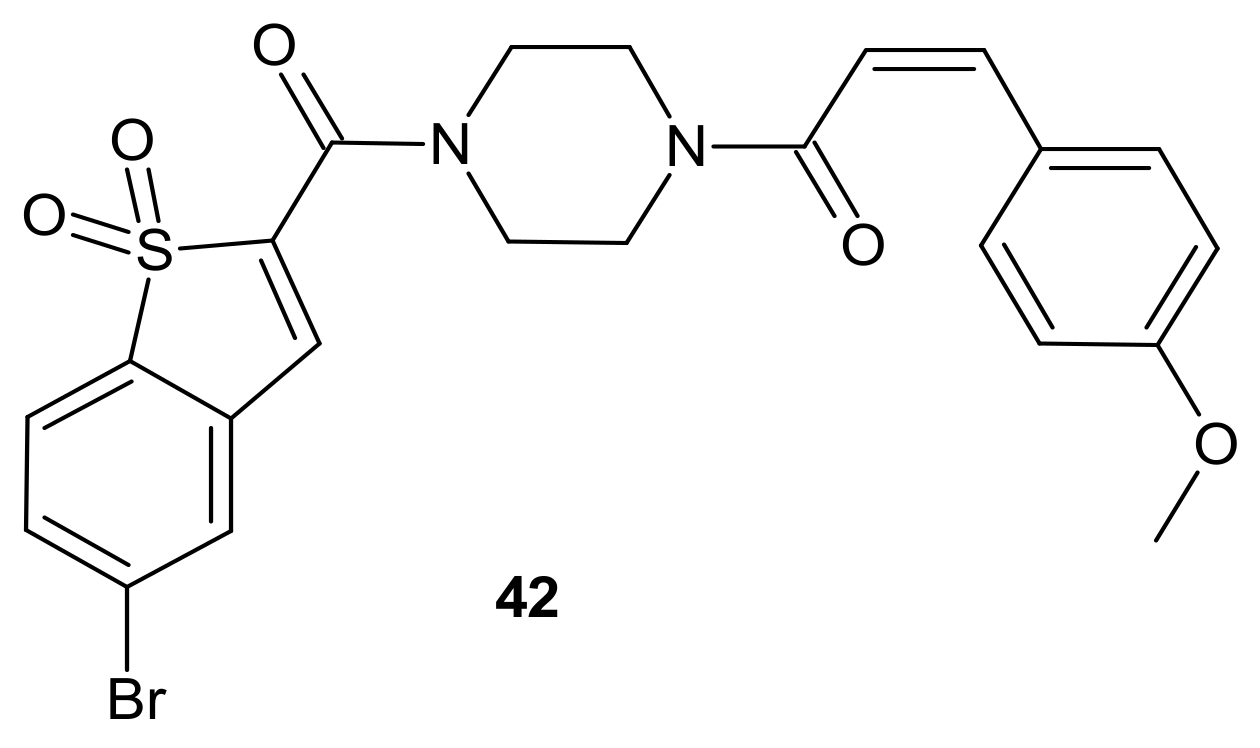
3.2.5. Alterations in Signaling Pathways
3.2.6. Epigenetic Alterations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef]
- Haider, T.; Pandey, V.; Banjare, N.; Gupta, P.N.; Soni, V. Drug resistance in cancer: Mechanisms and tackling strategies. Pharmacol. Rep. 2020, 72, 1125–1151. [Google Scholar] [CrossRef] [PubMed]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. 2019, 2, 141–160. [Google Scholar] [CrossRef]
- Delou, J.M.A.; Souza, A.S.O.; Souza, L.C.M.; Borges, H.L. Highlights in Resistance Mechanism Pathways for Combination Therapy. Cells 2019, 8. [Google Scholar] [CrossRef]
- Fojo, T.; Menefee, M. Mechanisms of multidrug resistance: The potential role of microtubule-stabilizing agents. Ann. Oncol. 2007, 18 (Suppl. 5), 3–8. [Google Scholar] [CrossRef]
- Cree, I.A.; Charlton, P. Molecular chess? Hallmarks of anti-cancer drug resistance. BMC Cancer 2017, 17, 10. [Google Scholar] [CrossRef] [PubMed]
- Falzone, L.; Salomone, S.; Libra, M. Evolution of Cancer Pharmacological Treatments at the Turn of the Third Millennium. Front. Pharmacol. 2018, 9, 1300. [Google Scholar] [CrossRef]
- Hopkins, A.L. Network pharmacology: The next paradigm in drug discovery. Nat. Chem. Biol. 2008, 4, 682–690. [Google Scholar] [CrossRef]
- Frei, E., 3rd; Karon, M.; Levin, R.H.; Freireich, E.J.; Taylor, R.J.; Hananian, J.; Selawry, O.; Holland, J.F.; Hoogstraten, B.; Wolman, I.J.; et al. The effectiveness of combinations of antileukemic agents in inducing and maintaining remission in children with acute leukemia. Blood 1965, 26, 642–656. [Google Scholar] [CrossRef]
- Bayat Mokhtari, R.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D. The contribution of Gianni Bonadonna to the history of chemotherapy. Cancer Chemother. Pharm. 2007, 60, 309–312. [Google Scholar] [CrossRef]
- Moxley, J.H., 3rd; De Vita, V.T.; Brace, K.; Frei, E., 3rd. Intensive combination chemotherapy and X-irradiation in Hodgkin’s disease. Cancer Res. 1967, 27, 1258–1263. [Google Scholar] [PubMed]
- Devita, V.T., Jr.; Serpick, A.A.; Carbone, P.P. Combination chemotherapy in the treatment of advanced Hodgkin’s disease. Ann. Intern. Med. 1970, 73, 881–895. [Google Scholar] [CrossRef]
- DeVita, V.T., Jr.; Lewis, B.J.; Rozencweig, M.; Muggia, F.M. The chemotherapy of Hodgkin’s disease: Past experiences and future directions. Cancer 1978, 42, 979–990. [Google Scholar] [CrossRef]
- Bonadonna, G.; Zucali, R.; Monfardini, S.; De Lena, M.; Uslenghi, C. Combination chemotherapy of Hodgkin’s disease with adriamycin, bleomycin, vinblastine, and imidazole carboxamide versus MOPP. Cancer 1975, 36, 252–259. [Google Scholar] [CrossRef]
- Santoro, A.; Bonadonna, G.; Valagussa, P.; Zucali, R.; Viviani, S.; Villani, F.; Pagnoni, A.M.; Bonfante, V.; Musumeci, R.; Crippa, F.; et al. Long-term results of combined chemotherapy-radiotherapy approach in Hodgkin’s disease: Superiority of ABVD plus radiotherapy versus MOPP plus radiotherapy. J. Clin. Oncol. 1987, 5, 27–37. [Google Scholar] [CrossRef]
- Canellos, G.P.; Devita, V.T.; Gold, G.L.; Chabner, B.A.; Schein, P.S.; Young, R.C. Cyclical combination chemotherapy for advanced breast carcinoma. Br. Med. J. 1974, 1, 218–220. [Google Scholar] [CrossRef][Green Version]
- Canellos, G.P.; Devita, V.T.; Gold, G.L.; Chabner, B.A.; Schein, P.S.; Young, R.C. Combination chemotherapy for advanced breast-cancer—response and effect on survival. Ann. Intern. Med. 1976, 84, 389–392. [Google Scholar] [CrossRef]
- Brambilla, C.; De Lena, M.; Rossi, A.; Valagussa, P.; Bonadonna, G. Response and survival in advanced breast cancer after two non-cross-resistant combinations. Br. Med. J. 1976, 1, 801–804. [Google Scholar] [CrossRef][Green Version]
- De Lena, M.; Brambilla, C.; Morabito, A.; Bonadonna, G. Adriamycin plus vincristine compared to and combined with cyclophosphamide, methotrexate, and 5-fluorouracil for advanced breast cancer. Cancer 1975, 35, 1108–1115. [Google Scholar] [CrossRef]
- Bonadonna, G.; Brusamolino, E.; Valagussa, P.; Rossi, A.; Brugnatelli, L.; Brambilla, C.; De Lena, M.; Tancini, G.; Bajetta, E.; Musumeci, R.; et al. Combination chemotherapy as an adjuvant treatment in operable breast cancer. N. Eng. Med. 1976, 294, 405–410. [Google Scholar] [CrossRef]
- Bonadonna, G.; Moliterni, A.; Zambetti, M.; Daidone, M.G.; Pilotti, S.; Gianni, L.; Valagussa, P. 30 years’ follow up of randomised studies of adjuvant CMF in operable breast cancer: Cohort study. Br. Med. J. 2005, 330, 217–220. [Google Scholar] [CrossRef]
- Hu, C.M.; Zhang, L. Nanoparticle-based combination therapy toward overcoming drug resistance in cancer. Biochem. Pharm. 2012, 83, 1104–1111. [Google Scholar] [CrossRef]
- Morphy, R.; Rankovic, Z. Designed multiple ligands. An emerging drug discovery paradigm. J. Med. Chem. 2005, 48, 6523–6543. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Williams, R.O. Polymeric nanomedicines for poorly soluble drugs in oral delivery systems: An update. J. Pharm. Investig. 2018, 48, 61–75. [Google Scholar] [CrossRef]
- Choi, Y.H.; Han, H.K. Nanomedicines: Current status and future perspectives in aspect of drug delivery and pharmacokinetics. J. Pharm. Investig. 2018, 48, 43–60. [Google Scholar] [CrossRef]
- Mohanty, A.; Uthaman, S.; Park, I.K. Utilization of Polymer-Lipid Hybrid Nanoparticles for Targeted Anti-Cancer Therapy. Molecules 2020, 25, 4377. [Google Scholar] [CrossRef]
- Dawidczyk, C.M.; Kim, C.; Park, J.H.; Russell, L.M.; Lee, K.H.; Pomper, M.G.; Searson, P.C. State-of-the-art in design rules for drug delivery platforms: Lessons learned from FDA-approved nanomedicines. J. Control. Release 2014, 187, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Onoue, S.; Yamada, S.; Chan, H.K. Nanodrugs: Pharmacokinetics and safety. Intj. Nanomed. 2014, 9, 1025–1037. [Google Scholar] [CrossRef]
- Guo, Z.; Sui, J.; Ma, M.; Hu, J.; Sun, Y.; Yang, L.; Fan, Y.; Zhang, X. pH-Responsive charge switchable PEGylated ε-poly-l-lysine polymeric nanoparticles-assisted combination therapy for improving breast cancer treatment. J. Control. Release 2020, 326, 350–364. [Google Scholar] [CrossRef]
- Miao, L.; Guo, S.; Zhang, J.; Kim, W.Y.; Huang, L. Nanoparticles with Precise Ratiometric Co-Loading and Co-Delivery of Gemcitabine Monophosphate and Cisplatin for Treatment of Bladder Cancer. Adv. Funct. Mater. 2014, 24, 6601–6611. [Google Scholar] [CrossRef]
- He, J.; Xiao, H.; Li, B.; Peng, Y.; Li, X.; Wang, Y.; Adamus, G.; Kowalczuk, M.; Shuai, X. The programmed site-specific delivery of the angiostatin sunitinib and chemotherapeutic paclitaxel for highly efficient tumor treatment. J. Mater. Chem. B 2019, 7, 4953–4962. [Google Scholar] [CrossRef]
- Zhang, L.; Radovic-Moreno, A.F.; Alexis, F.; Gu, F.X.; Basto, P.A.; Bagalkot, V.; Jon, S.; Langer, R.S.; Farokhzad, O.C. Co-delivery of hydrophobic and hydrophilic drugs from nanoparticle-aptamer bioconjugates. ChemMedChem 2007, 2, 1268–1271. [Google Scholar] [CrossRef]
- Kolishetti, N.; Dhar, S.; Valencia, P.M.; Lin, L.Q.; Karnik, R.; Lippard, S.J.; Langer, R.; Farokhzad, O.C. Engineering of self-assembled nanoparticle platform for precisely controlled combination drug therapy. Proc. Natl. Acad. Sci. USA 2010, 107, 17939. [Google Scholar] [CrossRef] [PubMed]
- Prasad, P.; Cheng, J.; Shuhendler, A.; Rauth, A.M.; Wu, X.Y. A novel nanoparticle formulation overcomes multiple types of membrane efflux pumps in human breast cancer cells. Drug Deliv. Transl. Res. 2012, 2, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Shuhendler, A.J.; Cheung, R.Y.; Manias, J.; Connor, A.; Rauth, A.M.; Wu, X.Y. A novel doxorubicin-mitomycin C co-encapsulated nanoparticle formulation exhibits anti-cancer synergy in multidrug resistant human breast cancer cells. Breast Cancer Res. Treat. 2010, 119, 255–269. [Google Scholar] [CrossRef]
- Shuhendler, A.J.; O’Brien, P.J.; Rauth, A.M.; Wu, X.Y. On the synergistic effect of doxorubicin and mitomycin C against breast cancer cells. Drug Metab. Drug Interact. 2007, 22, 201–233. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Lip, H.; He, C.; Cai, P.; Wang, Z.; Henderson, J.T.; Rauth, A.M.; Wu, X.Y. Multitargeted Nanoparticles Deliver Synergistic Drugs across the Blood-Brain Barrier to Brain Metastases of Triple Negative Breast Cancer Cells and Tumor-Associated Macrophages. Adv. Healthc Mater. 2019, 8, e1900543. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Wang, Z.; Li, C.; Duan, G.; Wang, K.; Li, Q.; Tao, T. RGD peptide-modified, paclitaxel prodrug-based, dual-drugs loaded, and redox-sensitive lipid-polymer nanoparticles for the enhanced lung cancer therapy. Biomed. Pharm. 2018, 106, 275–284. [Google Scholar] [CrossRef]
- Alfayez, M.; Kantarjian, H.; Kadia, T.; Ravandi-Kashani, F.; Daver, N. CPX-351 (vyxeos) in AML. Leuk. Lymphoma 2020, 61, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Musso, L.; Dallavalle, S.; Zunino, F. Perspectives in the development of hybrid bifunctional antitumour agents. Biochem. Pharmacol. 2015, 96, 297–305. [Google Scholar] [CrossRef]
- Zheng, W.; Zhao, Y.; Luo, Q.; Zhang, Y.; Wu, K.; Wang, F.Y. Multi-Targeted Anticancer Agents. Curr. Top. Med. Chem. 2017, 17, 3084–3098. [Google Scholar] [CrossRef] [PubMed]
- Nepali, K.; Sharma, S.; Sharma, M.; Bedi, P.M.S.; Dhar, K.L. Rational approaches, design strategies, structure activity relationship and mechanistic insights for anticancer hybrids. Eur. J. Med. Chem. 2014, 77, 422–487. [Google Scholar] [CrossRef] [PubMed]
- Fortin, S.; Bérubé, G. Advances in the development of hybrid anticancer drugs. Expert Opin. Drug Discov. 2013, 8, 1029–1047. [Google Scholar] [CrossRef]
- Fu, R.-g.; Sun, Y.; Sheng, W.-b.; Liao, D.-f. Designing multi-targeted agents: An emerging anticancer drug discovery paradigm. Eur. J. Med. Chem. 2017, 136, 195–211. [Google Scholar] [CrossRef]
- Viegas-Junior, C.; Danuello, A.; Bolzani, V.D.; Barreir, E.J.; Fraga, C.A.M. Molecular hybridization: A useful tool in the design of new drug prototypes. Curr. Med. Chem. 2007, 14, 1829–1852. [Google Scholar] [CrossRef] [PubMed]
- Ai, T.; Cui, H.; Chen, L. Multi-targeted histone deacetylase inhibitors in cancer therapy. Curr. Med. Chem. 2012, 19, 475–487. [Google Scholar] [CrossRef] [PubMed]
- De Lera, A.R.; Ganesan, A. Epigenetic polypharmacology: From combination therapy to multitargeted drugs. Clin. Epigenetics 2016, 8, 105. [Google Scholar] [CrossRef]
- Wermuth, C.G.; Ganellin, C.R.; Lindberg, P.; Mitscher, L.A. Glossary of terms used in medicinal chemistry (IUPAC Recommendations 1998). Pure Appl. Chem. 1998, 70, 1129–1143. [Google Scholar] [CrossRef]
- Morphy, R.; Kay, C.; Rankovic, Z. From magic bullets to designed multiple ligands. Drug Discov. Today 2004, 9, 641–651. [Google Scholar] [CrossRef]
- Pedrosa, M.D.; da Cruz, R.M.D.; Viana, J.D.; de Moura, R.O.; Ishiki, H.M.; Barbosa, J.M.; Diniz, M.; Scotti, M.T.; Scotti, L.; Mendonca, F.J.B. Hybrid Compounds as Direct Multitarget Ligands: A Review. Curr. Top. Med. Chem. 2017, 17, 1044–1079. [Google Scholar] [CrossRef]
- Gediya, L.K.; Njar, V.C.O. Promise and challenges in drug discovery and development of hybrid anticancer drugs. Expert Opin. Drug Discov. 2009, 4, 1099–1111. [Google Scholar] [CrossRef] [PubMed]
- Luan, Y.; Li, J.; Bernatchez, J.A.; Li, R. Kinase and Histone Deacetylase Hybrid Inhibitors for Cancer Therapy. J. Med. Chem 2019, 62, 3171–3183. [Google Scholar] [CrossRef] [PubMed]
- Decker, M. 1—Introduction. In Design of Hybrid Molecules for Drug Development; Decker, M., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 1–3. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Passarella, D.; Comi, D.; Vanossi, A.; Paganini, G.; Colombo, F.; Ferrante, L.; Zuco, V.; Danieli, B.; Zunino, F. Histone deacetylase and microtubules as targets for the synthesis of releasable conjugate compounds. Bioorganic Med. Chem. Lett. 2009, 19, 6358–6363. [Google Scholar] [CrossRef]
- Meunier, B. Hybrid Molecules with a Dual Mode of Action: Dream or Reality? Acc. Chem. Res. 2008, 41, 69–77. [Google Scholar] [CrossRef]
- Dallavalle, S.; Dobričić, V.; Lazzarato, L.; Gazzano, E.; Machuqueiro, M.; Pajeva, I.; Tsakovska, I.; Zidar, N.; Fruttero, R. Improvement of conventional anti-cancer drugs as new tools against multidrug resistant tumors. Drug Resist. Updates 2020, 50. [Google Scholar] [CrossRef] [PubMed]
- Khoury, A.; Deo, K.M.; Aldrich-Wright, J.R. Recent advances in platinum-based chemotherapeutics that exhibit inhibitory and targeted mechanisms of action. J. Inorg. Biochem. 2020, 207. [Google Scholar] [CrossRef] [PubMed]
- Dilruba, S.; Kalayda, G.V. Platinum-based drugs: Past, present and future. Cancer Chemother. Pharmacol. 2016, 77, 1103–1124. [Google Scholar] [CrossRef] [PubMed]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Al-Taweel, N.; Varghese, E.; Florea, A.M.; Busselberg, D. Cisplatin (CDDP) triggers cell death of MCF-7 cells following disruption of intracellular calcium (Ca2+ (i)) homeostasis. J. Toxicol. Sci. 2014, 39, 765–774. [Google Scholar] [CrossRef]
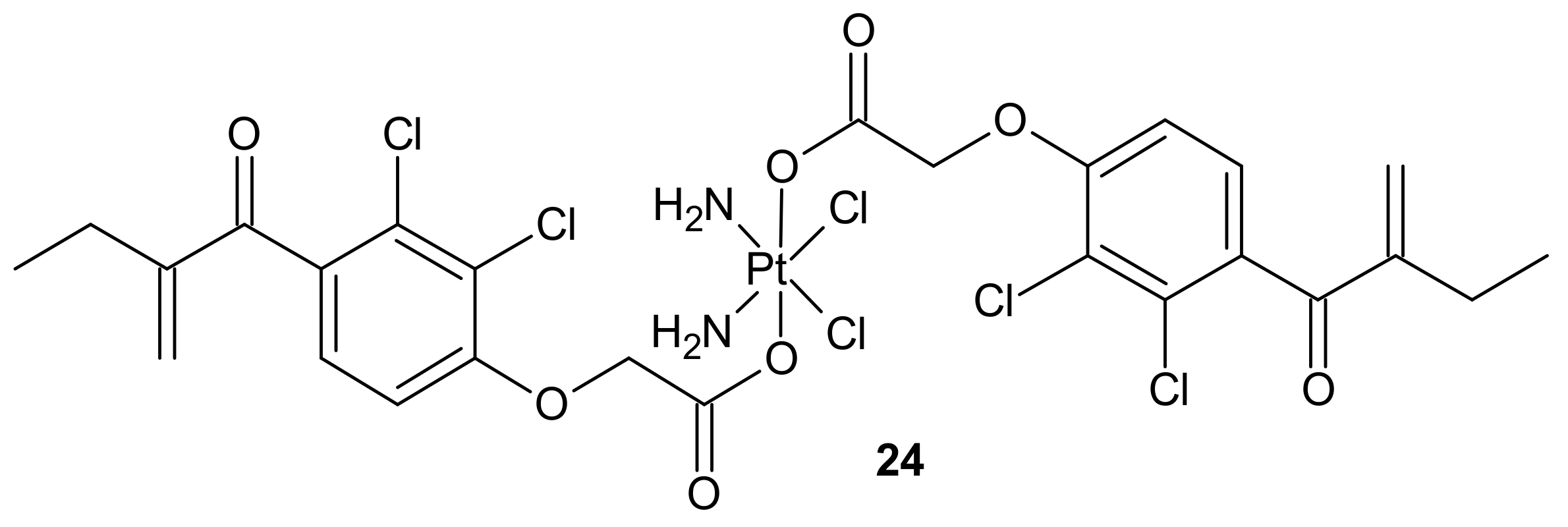
- Li, X.; Liu, Y.; Tian, H. Current Developments in Pt(IV) Prodrugs Conjugated with Bioactive Ligands. Bioinorg. Chem. Appl. 2018, 2018, 8276139. [Google Scholar] [CrossRef]
- Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. The Next Generation of Platinum Drugs: Targeted Pt(II) Agents, Nanoparticle Delivery, and Pt(IV) Prodrugs. Chem. Rev. 2016, 116, 3436–3486. [Google Scholar] [CrossRef]
- Huang, X.C.; Huang, R.Z.; Gou, S.H.; Wang, Z.M.; Liao, Z.X.; Wang, H.S. Combretastatin A-4 Analogue: A Dual-Targeting and Tubulin Inhibitor Containing Antitumor Pt(IV) Moiety with a Unique Mode of Action. Bioconjugate Chem. 2016, 27, 2132–2148. [Google Scholar] [CrossRef]
- Novohradsky, V.; Zerzankova, L.; Stepankova, J.; Vrana, O.; Raveendran, R.; Gibson, D.; Kasparkova, J.; Brabec, V. New insights into the molecular and epigenetic effects of antitumor Pt(IV)-valproic acid conjugates in human ovarian cancer cells. Biochem. Pharm. 2015, 95, 133–144. [Google Scholar] [CrossRef]
- Raveendran, R.; Braude, J.P.; Wexselblatt, E.; Novohradsky, V.; Stuchlikova, O.; Brabec, V.; Gandin, V.; Gibson, D. Pt(IV) derivatives of cisplatin and oxaliplatin with phenylbutyrate axial ligands are potent cytotoxic agents that act by several mechanisms of action. Chem. Sci. 2016, 7, 2381–2391. [Google Scholar] [CrossRef]
- Huang, X.C.; Huang, R.Z.; Gou, S.H.; Wang, Z.M.; Liao, Z.X.; Wang, H.S. Platinum(IV) complexes conjugated with phenstatin analogue as inhibitors of microtubule polymerization and reverser of multidrug resistance. Bioorganic Med. Chem. 2017, 25, 4686–4700. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Hua, S.; Huang, R.; Liu, Z.; Gou, S.; Wang, Z.; Liao, Z.; Wang, H. Dual-targeting antitumor hybrids derived from Pt(IV) species and millepachine analogues. Eur. J. Med. Chem. 2018, 148, 1–25. [Google Scholar] [CrossRef]
- Ma, Z.Y.; Wang, D.B.; Song, X.Q.; Wu, Y.G.; Chen, Q.; Zhao, C.L.; Li, J.Y.; Cheng, S.H.; Xu, J.Y. Chlorambucil-conjugated platinum(IV) prodrugs to treat triple-negative breast cancer in vitro and in vivo. Eur. J. Med. Chem. 2018, 157, 1292–1299. [Google Scholar] [CrossRef]
- Petruzzella, E.; Braude, J.P.; Aldrich-Wright, J.R.; Gandin, V.; Gibson, D. A Quadruple-Action Platinum(IV) Prodrug with Anticancer Activity Against KRAS Mutated Cancer Cell Lines. Angew. Chem.-Int. Ed. 2017, 56, 11539–11544. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Pastan, I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu. Rev. Biochem. 1993, 62, 385–427. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP–dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef]
- Vaidyanathan, A.; Sawers, L.; Gannon, A.L.; Chakravarty, P.; Scott, A.L.; Bray, S.E.; Ferguson, M.J.; Smith, G. ABCB1 (MDR1) induction defines a common resistance mechanism in paclitaxel- and olaparib-resistant ovarian cancer cells. Br. J. Cancer 2016, 115, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Q. Sunitinib reverse multidrug resistance in gastric cancer cells by modulating Stat3 and inhibiting P-gp function. Cell Biochem. Biophys. 2013, 67, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Szakács, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.P.; Hsieh, C.H.; Wu, Y.S. The emergence of drug transporter-mediated multidrug resistance to cancer chemotherapy. Mol. Pharm. 2011, 8, 1996–2011. [Google Scholar] [CrossRef]
- Lagas, J.S.; Fan, L.; Wagenaar, E.; Vlaming, M.L.; van Tellingen, O.; Beijnen, J.H.; Schinkel, A.H. P-glycoprotein (P-gp/Abcb1), Abcc2, and Abcc3 determine the pharmacokinetics of etoposide. Clin. Cancer Res. 2010, 16, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Stanković, T.; Dinić, J.; Podolski-Renić, A.; Musso, L.; Burić, S.S.; Dallavalle, S.; Pešić, M. Dual Inhibitors as a New Challenge for Cancer Multidrug Resistance Treatment. Curr. Med. Chem. 2019, 26, 6074–6106. [Google Scholar] [CrossRef]
- Mohammad, I.S.; He, W.; Yin, L. Understanding of human ATP binding cassette superfamily and novel multidrug resistance modulators to overcome MDR. Biomed. Pharm. 2018, 100, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Waghray, D.; Zhang, Q. Inhibit or Evade Multidrug Resistance P-Glycoprotein in Cancer Treatment. J. Med. Chem. 2018, 61, 5108–5121. [Google Scholar] [CrossRef]
- Wu, C.P.; Calcagno, A.M.; Ambudkar, S.V. Reversal of ABC drug transporter-mediated multidrug resistance in cancer cells: Evaluation of current strategies. Curr. Mol. Pharm. 2008, 1, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.K.; Ren, Z.G.; Tang, X.B.; Peng, H.; Ma, Y.F.; Lai, Y.S.; Peng, S.X.; Zhang, Y.H. Synthesis and biological evaluation of bifendate-chalcone hybrids as a new class of potential P-glycoprotein inhibitors. Bioorganic Med. Chem. 2012, 20, 2540–2548. [Google Scholar] [CrossRef]
- Teodori, E.; Braconi, L.; Bua, S.; Lapucci, A.; Bartolucci, G.; Manetti, D.; Romanelli, M.N.; Dei, S.; Supuran, C.T.; Coronnello, M. Dual P-Glycoprotein and CA XII Inhibitors: A New Strategy to Reverse the P-gp Mediated Multidrug Resistance (MDR) in Cancer Cells. Molecules 2020, 25, 1748. [Google Scholar] [CrossRef]
- Kopecka, J.; Campia, I.; Jacobs, A.; Frei, A.P.; Ghigo, D.; Wollscheid, B.; Riganti, C. Carbonic anhydrase XII is a new therapeutic target to overcome chemoresistance in cancer cells. Oncotarget 2015, 6, 9. [Google Scholar] [CrossRef] [PubMed]
- Kopecka, J.; Rankin, G.M.; Salaroglio, I.C.; Poulsen, S.-A.; Riganti, C. P-glycoprotein-mediated chemoresistance is reversed by carbonic anhydrase XII inhibitors. Oncotarget 2016, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Rullo, M.; Niso, M.; Pisani, L.; Carrieri, A.; Colabufo, N.A.; Cellamare, S.; Altomare, C.D. 1,2,3,4-Tetrahydroisoquinoline/2H-chromen-2-one conjugates as nanomolar P-glycoprotein inhibitors: Molecular determinants for affinity and selectivity over multidrug resistance associated protein 1. Eur. J. Med. Chem. 2019, 161, 433–444. [Google Scholar] [CrossRef]
- Palmeira, A.; Vasconcelos, M.H.; Paiva, A.; Fernandes, M.X.; Pinto, M.; Sousa, E. Dual inhibitors of P-glycoprotein and tumor cell growth: (Re)discovering thioxanthones. Biochem. Pharmacol. 2012, 83, 57–68. [Google Scholar] [CrossRef]
- Torijano-Gutiérrez, S.; Vilanova, C.; Díaz-Oltra, S.; Murga, J.; Falomir, E.; Carda, M.; Redondo-Horcajo, M.; Díaz, J.F.; Barasoain, I.; Marco, J.A. The Mechanism of the Interactions of Pironetin Analog/Combretastatin A-4 Hybrids with Tubulin. Arch. Pharm. 2015, 348, 541–547. [Google Scholar] [CrossRef]
- Cirla, A.; Mann, J. Combretastatins: From natural products to drug discovery. Nat. Prod. Rep. 2003, 20, 558–564. [Google Scholar] [CrossRef]
- Shan, Y.; Zhang, J.; Liu, Z.; Wang, M.; Dong, Y. Developments of combretastatin A-4 derivatives as anticancer agents. Curr. Med. Chem. 2011, 18, 523–538. [Google Scholar] [CrossRef] [PubMed]
- Tron, G.C.; Pirali, T.; Sorba, G.; Pagliai, F.; Busacca, S.; Genazzani, A.A. Medicinal Chemistry of Combretastatin A4: Present and Future Directions. J. Med. Chem. 2006, 49, 3033–3044. [Google Scholar] [CrossRef] [PubMed]
- Usui, T.; Watanabe, H.; Nakayama, H.; Tada, Y.; Kanoh, N.; Kondoh, M.; Asao, T.; Takio, K.; Nishikawa, K.; Kitahara, T.; et al. The anticancer natural product pironetin selectively targets Lys352 of alpha-tubulin. Chem. Biol. 2004, 11, 799–806. [Google Scholar] [CrossRef]
- Coulup, S.K.; Georg, G.I. Revisiting microtubule targeting agents: α-Tubulin and the pironetin binding site as unexplored targets for cancer therapeutics. Bioorg. Med. Chem. Lett. 2019, 29, 1865–1873. [Google Scholar] [CrossRef]
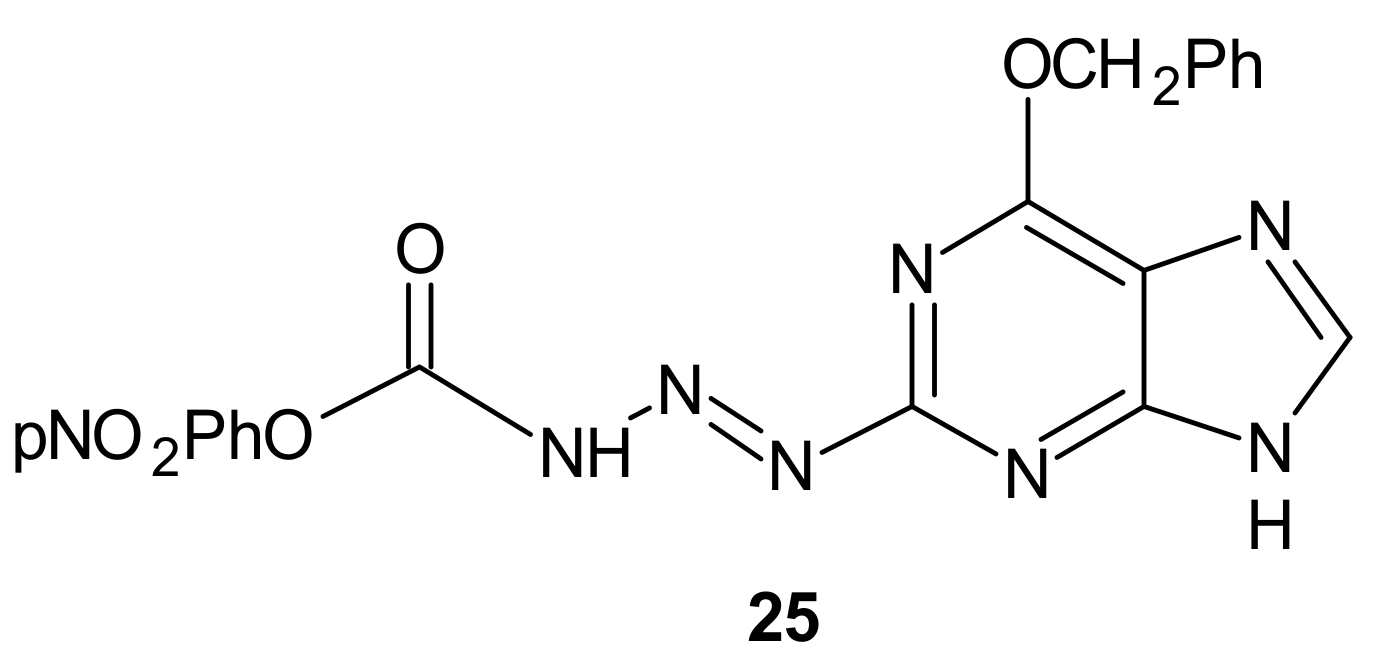
- Brzezińska, A.; Wińska, P.; Balińska, M. Cellular aspects of folate and antifolate membrane transport. Acta Biochim. Pol. 2000, 47, 735–749. [Google Scholar] [CrossRef] [PubMed]
- Jansen, G.; Mauritz, R.; Drori, S.; Sprecher, H.; Kathmann, I.; Bunni, M.; Priest, D.G.; Noordhuis, P.; Schornagel, J.H.; Pinedo, H.M.; et al. A Structurally Altered Human Reduced Folate Carrier with Increased Folic Acid Transport Mediates a Novel Mechanism of Antifolate Resistance. J. Biol. Chem. 1998, 273, 30189–30198. [Google Scholar] [CrossRef]
- Cai, B.; Liao, A.; Lee, K.K.; Ban, J.S.; Yang, H.S.; Im, Y.J.; Chun, C. Design, synthesis of methotrexate-diosgenin conjugates and biological evaluation of their effect on methotrexate transport-resistant cells. Steroids 2016, 116, 45–51. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Tian, Y.; Zhang, X.; Bing, B.; Zhang, L.; Wang, H.; Zhao, W. Anti-tumour and immunomodulating activities of diosgenin, a naturally occurring steroidal saponin. Nat. Prod. Res. 2012, 26, 2243–2246. [Google Scholar] [CrossRef] [PubMed]
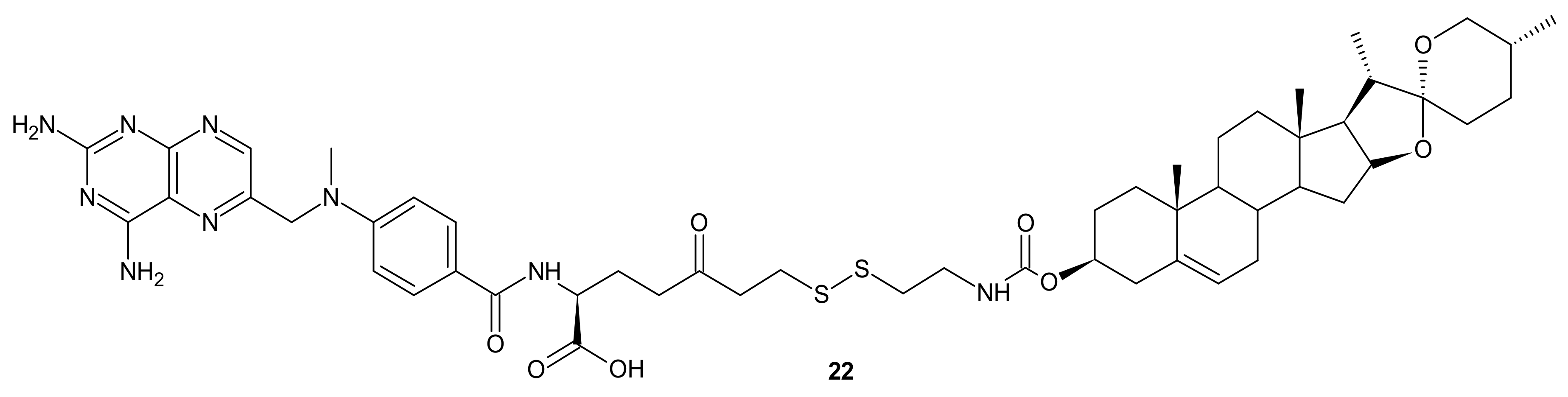
- Xue, X.; Liang, X.-J. Overcoming drug efflux-based multidrug resistance in cancer with nanotechnology. Chin. J. Cancer 2012, 31, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, R.B.; Choe, Y.H.; McGuire, J.; Conover, C.D. Effective drug delivery by PEGylated drug conjugates. Adv. Drug Deliv. Rev. 2003, 55, 217–250. [Google Scholar] [CrossRef]
- Elvira, C.; Gallardo, A.; San Roman, J.; Cifuentes, A. Covalent polymer-drug conjugates. Molecules 2005, 10, 114–125. [Google Scholar] [CrossRef]
- Huang, P.; Wang, D.L.; Su, Y.; Huang, W.; Zhou, Y.F.; Cui, D.X.; Zhu, X.Y.; Yan, D.Y. Combination of Small Molecule Prodrug and Nanodrug Delivery: Amphiphilic Drug-Drug Conjugate for Cancer Therapy. J. Am. Chem. Soc. 2014, 136, 11748–11756. [Google Scholar] [CrossRef]
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. Glutathione transferases. Annu Rev. Pharm. Toxicol. 2005, 45, 51–88. [Google Scholar] [CrossRef]
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003, 22, 7369–7375. [Google Scholar] [CrossRef]
- Hayes, J.D.; Pulford, D.J. The glutathione S-transferase supergene family: Regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 445–600. [Google Scholar] [CrossRef] [PubMed]
- Lien, S.; Larsson, A.K.; Mannervik, B. The polymorphic human glutathione transferase T1-1, the most efficient glutathione transferase in the denitrosation and inactivation of the anticancer drug 1,3-bis(2-chloroethyl)-1-nitrosourea. Biochem. Pharm. 2002, 63, 191–197. [Google Scholar] [CrossRef]
- Paumi, C.M.; Ledford, B.G.; Smitherman, P.K.; Townsend, A.J.; Morrow, C.S. Role of multidrug resistance protein 1 (MRP1) and glutathione S-transferase A1-1 in alkylating agent resistance. Kinetics of glutathione conjugate formation and efflux govern differential cellular sensitivity to chlorambucil versus melphalan toxicity. J. Biol. Chem. 2001, 276, 7952–7956. [Google Scholar] [CrossRef] [PubMed]
- Meijer, C.; Mulder, N.H.; Timmer-Bosscha, H.; Sluiter, W.J.; Meersma, G.J.; de Vries, E.G. Relationship of cellular glutathione to the cytotoxicity and resistance of seven platinum compounds. Cancer Res. 1992, 52, 6885–6889. [Google Scholar]
- Parker, L.J.; Italiano, L.C.; Morton, C.J.; Hancock, N.C.; Ascher, D.B.; Aitken, J.B.; Harris, H.H.; Campomanes, P.; Rothlisberger, U.; De Luca, A.; et al. Studies of glutathione transferase P1-1 bound to a platinum(IV)-based anticancer compound reveal the molecular basis of its activation. Chemistry 2011, 17, 7806–7816. [Google Scholar] [CrossRef]
- Ang, W.H.; Khalaila, I.; Allardyce, C.S.; Juillerat-Jeanneret, L.; Dyson, P.J. Rational Design of Platinum(IV) Compounds to Overcome Glutathione-S-Transferase Mediated Drug Resistance. J. Am. Chem. Soc. 2005, 127, 1382–1383. [Google Scholar] [CrossRef]
- Zanellato, I.; Bonarrigo, I.; Sardi, M.; Alessio, M.; Gabano, E.; Ravera, M.; Osella, D. Evaluation of Platinum–Ethacrynic Acid Conjugates in the Treatment of Mesothelioma. ChemMedChem 2011, 6, 2287–2293. [Google Scholar] [CrossRef] [PubMed]
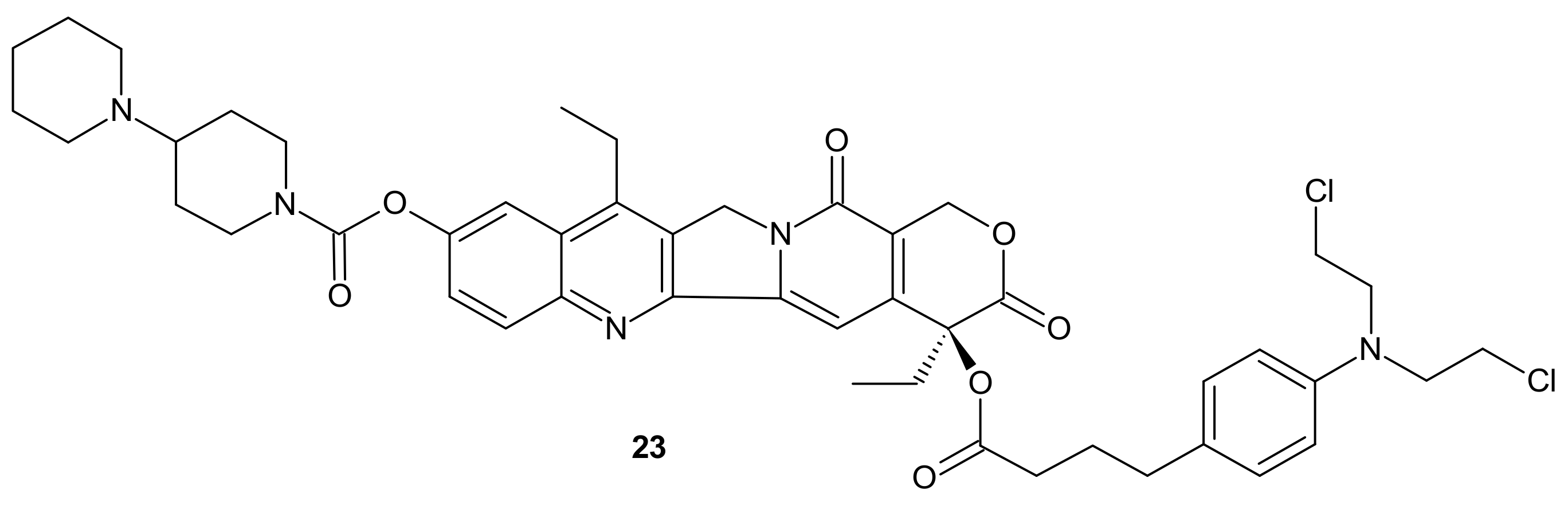
- Sun, G.H.; Fan, T.J.; Zhao, L.J.; Zhou, Y.; Zhong, R.G. The potential of combi-molecules with DNA-damaging function as anticancer agents. Future Med. Chem. 2017, 9, 403–435. [Google Scholar] [CrossRef]
- Bouwman, P.; Jonkers, J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat. Rev. Cancer 2012, 12, 587–598. [Google Scholar] [CrossRef]
- Kaina, B.; Margison, G.P.; Christmann, M. Targeting O⁶-methylguanine-DNA methyltransferase with specific inhibitors as a strategy in cancer therapy. Cell Mol. Life Sci. 2010, 67, 3663–3681. [Google Scholar] [CrossRef] [PubMed]
- Sarkaria, J.N.; Kitange, G.J.; James, C.D.; Plummer, R.; Calvert, H.; Weller, M.; Wick, W. Mechanisms of chemoresistance to alkylating agents in malignant glioma. Clin. Cancer Res. 2008, 14, 2900–2908. [Google Scholar] [CrossRef] [PubMed]
- Wanner, M.J.; Koch, M.; Koomen, G.-J. Synthesis and Antitumor Activity of Methyltriazene Prodrugs Simultaneously Releasing DNA-Methylating Agents and the Antiresistance Drug O6-Benzylguanine. J. Med. Chem. 2004, 47, 6875–6883. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.; Baumann, R.P.; Patridge, E.; Penketh, P.G.; Shyam, K.; Ishiguro, K.; Sartorelli, A.C. Chloroethylating and methylating dual function antineoplastic agents display superior cytotoxicity against repair proficient tumor cells. Bioorg. Med. Chem. Lett. 2013, 23, 1853–1859. [Google Scholar] [CrossRef][Green Version]
- Sun, G.; Zhang, N.; Zhao, L.; Fan, T.; Zhang, S.; Zhong, R. Synthesis and antitumor activity evaluation of a novel combi-nitrosourea prodrug: Designed to release a DNA cross-linking agent and an inhibitor of O(6)-alkylguanine-DNA alkyltransferase. Bioorg. Med. Chem. 2016, 24, 2097–2107. [Google Scholar] [CrossRef]
- Ye, Q.; Liu, K.; Shen, Q.; Li, Q.; Hao, J.; Han, F.; Jiang, R.-W. Reversal of Multidrug Resistance in Cancer by Multi-Functional Flavonoids. Front. Oncol. 2019, 9, 487. [Google Scholar] [CrossRef] [PubMed]
- Hurtado, M.; Sankpal, U.T.; Ranjan, A.; Maram, R.; Vishwanatha, J.K.; Nagaraju, G.P.; El-Rayes, B.F.; Basha, R. Investigational agents to enhance the efficacy of chemotherapy or radiation in pancreatic cancer. Crit. Rev. Oncol. Hematol. 2018, 126, 201–207. [Google Scholar] [CrossRef]
- Wang, X.; Li, L.Y.; Pei, S.N.; Zhu, Q.; Chen, F.H. Disruption of SSBs repair to combat platinum resistance via the JWA-targeted Pt(IV) prodrug conjugated with a wogonin derivative. Pharmazie 2020, 75, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Guerra, B.; Issinger, O.G. Protein kinase CK2 in human diseases. Curr. Med. Chem. 2008, 15, 1870–1886. [Google Scholar] [CrossRef]
- Tawfic, S.; Yu, S.; Wang, H.; Faust, R.; Davis, A.; Ahmed, K. Protein kinase CK2 signal in neoplasia. Histol. Histopathol. 2001, 16, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Miyata, Y. Protein kinase CK2 in health and disease: CK2: The kinase controlling the Hsp90 chaperone machinery. Cell Mol. Life Sci. 2009, 66, 1840–1849. [Google Scholar] [CrossRef]
- Mottet, D.; Ruys, S.P.; Demazy, C.; Raes, M.; Michiels, C. Role for casein kinase 2 in the regulation of HIF-1 activity. Intj. Cancer 2005, 117, 764–774. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.S.; Turowec, J.P.; Duncan, K.E.; Vilk, G.; Wu, C.; Lüscher, B.; Li, S.S.; Gloor, G.B.; Litchfield, D.W. A peptide-based target screen implicates the protein kinase CK2 in the global regulation of caspase signaling. Sci. Signal. 2011, 4, 30. [Google Scholar] [CrossRef]
- Piazza, F.A.; Ruzzene, M.; Gurrieri, C.; Montini, B.; Bonanni, L.; Chioetto, G.; Di Maira, G.; Barbon, F.; Cabrelle, A.; Zambello, R.; et al. Multiple myeloma cell survival relies on high activity of protein kinase CK2. Blood 2006, 108, 1698–1707. [Google Scholar] [CrossRef]
- Becherel, O.J.; Jakob, B.; Cherry, A.L.; Gueven, N.; Fusser, M.; Kijas, A.W.; Peng, C.; Katyal, S.; McKinnon, P.J.; Chen, J.; et al. CK2 phosphorylation-dependent interaction between aprataxin and MDC1 in the DNA damage response. Nucleic Acids Res. 2010, 38, 1489–1503. [Google Scholar] [CrossRef]
- Chon, H.J.; Bae, K.J.; Lee, Y.; Kim, J. The casein kinase 2 inhibitor, CX-4945, as an anti-cancer drug in treatment of human hematological malignancies. Front. Pharm. 2015, 6, 70. [Google Scholar] [CrossRef]
- Richter, A.; Roolf, C.; Hamed, M.; Gladbach, Y.S.; Sender, S.; Konkolefski, C.; Knübel, G.; Sekora, A.; Fuellen, G.; Vollmar, B.; et al. Combined Casein Kinase II inhibition and epigenetic modulation in acute B-lymphoblastic leukemia. BMC Cancer 2019, 19, 202. [Google Scholar] [CrossRef]
- Siddiqui-Jain, A.; Bliesath, J.; Macalino, D.; Omori, M.; Huser, N.; Streiner, N.; Ho, C.B.; Anderes, K.; Proffitt, C.; O’Brien, S.E.; et al. CK2 inhibitor CX-4945 suppresses DNA repair response triggered by DNA-targeted anticancer drugs and augments efficacy: Mechanistic rationale for drug combination therapy. Mol. Cancer 2012, 11, 994–1005. [Google Scholar] [CrossRef]
- Gomes, A.M.; Soares, M.V.; Ribeiro, P.; Caldas, J.; Póvoa, V.; Martins, L.R.; Melão, A.; Serra-Caetano, A.; de Sousa, A.B.; Lacerda, J.F.; et al. Adult B-cell acute lymphoblastic leukemia cells display decreased PTEN activity and constitutive hyperactivation of PI3K/Akt pathway despite high PTEN protein levels. Haematologica 2014, 99, 1062–1068. [Google Scholar] [CrossRef]
- Kim, H.; Choi, K.; Kang, H.; Lee, S.-Y.; Chi, S.-W.; Lee, M.-S.; Song, J.; Im, D.; Choi, Y.; Cho, S. Identification of a novel function of CX-4945 as a splicing regulator. PLoS ONE 2014, 9, e94978. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui-Jain, A.; Drygin, D.; Streiner, N.; Chua, P.; Pierre, F.; O’Brien, S.E.; Bliesath, J.; Omori, M.; Huser, N.; Ho, C.; et al. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010, 70, 10288–10298. [Google Scholar] [CrossRef] [PubMed]
- Buontempo, F.; Orsini, E.; Martins, L.R.; Antunes, I.; Lonetti, A.; Chiarini, F.; Tabellini, G.; Evangelisti, C.; Melchionda, F.; Pession, A.; et al. Cytotoxic activity of the casein kinase 2 inhibitor CX-4945 against T-cell acute lymphoblastic leukemia: Targeting the unfolded protein response signaling. Leukemia 2014, 28, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Huang, X.; Wu, M.; Gou, S.; Hu, W. A CK2-targeted Pt(IV) prodrug to disrupt DNA damage response. Cancer Lett. 2017, 385, 168–178. [Google Scholar] [CrossRef]
- Chen, Y.-f.; Fu, L.-w. Mechanisms of acquired resistance to tyrosine kinase inhibitors. Acta Pharm. Sin. B 2011, 1, 197–207. [Google Scholar] [CrossRef]
- Dumontet, C.; Sikic, B.I. Mechanisms of Action of and Resistance to Antitubulin Agents: Microtubule Dynamics, Drug Transport, and Cell Death. J. Clin. Oncol. 1999, 17, 1061. [Google Scholar] [CrossRef] [PubMed]
- Krause, W. Resistance to anti-tubulin agents: From vinca alkaloids to epothilones. Cancer Drug Resist. 2019, 2, 82–106. [Google Scholar] [CrossRef]
- Leonard, G.D.; Fojo, T.; Bates, S.E. The role of ABC transporters in clinical practice. Oncologist 2003, 8, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Ohishi, Y.; Oda, Y.; Basaki, Y.; Kobayashi, H.; Wake, N.; Kuwano, M.; Tsuneyoshi, M. Expression of beta-tubulin isotypes in human primary ovarian carcinoma. Gynecol. Oncol. 2007, 105, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Izutsu, N.; Maesawa, C.; Shibazaki, M.; Oikawa, H.; Shoji, T.; Sugiyama, T.; Masuda, T. Epigenetic modification is involved in aberrant expression of class III beta-tubulin, TUBB3, in ovarian cancer cells. Intj. Oncol. 2008, 32, 1227–1235. [Google Scholar] [CrossRef][Green Version]
- De Donato, M.; Mariani, M.; Petrella, L.; Martinelli, E.; Zannoni, G.F.; Vellone, V.; Ferrandina, G.; Shahabi, S.; Scambia, G.; Ferlini, C. Class III β-tubulin and the cytoskeletal gateway for drug resistance in ovarian cancer. J. Cell Physiol. 2012, 227, 1034–1041. [Google Scholar] [CrossRef]
- Roque, D.M.; Buza, N.; Glasgow, M.; Bellone, S.; Bortolomai, I.; Gasparrini, S.; Cocco, E.; Ratner, E.; Silasi, D.A.; Azodi, M.; et al. Class III β-tubulin overexpression within the tumor microenvironment is a prognostic biomarker for poor overall survival in ovarian cancer patients treated with neoadjuvant carboplatin/paclitaxel. Clin. Exp. Metastasis 2014, 31, 101–110. [Google Scholar] [CrossRef]
- Parker, A.L.; Turner, N.; McCarroll, J.A.; Kavallaris, M. βIII-Tubulin alters glucose metabolism and stress response signaling to promote cell survival and proliferation in glucose-starved non-small cell lung cancer cells. Carcinogenesis 2016, 37, 787–798. [Google Scholar] [CrossRef]
- Parker, A.L.; Teo, W.S.; McCarroll, J.A.; Kavallaris, M. An Emerging Role for Tubulin Isotypes in Modulating Cancer Biology and Chemotherapy Resistance. Int. J. Mol. Sci. 2017, 18, 1434. [Google Scholar] [CrossRef]
- Stengel, C.; Newman, S.P.; Leese, M.P.; Potter, B.V.; Reed, M.J.; Purohit, A. Class III beta-tubulin expression and in vitro resistance to microtubule targeting agents. Br. J. Cancer 2010, 102, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Raghavan, S.; Ihnat, M.; Thorpe, J.E.; Disch, B.C.; Bastian, A.; Bailey-Downs, L.C.; Dybdal-Hargreaves, N.F.; Rohena, C.C.; Hamel, E.; et al. The design and discovery of water soluble 4-substituted-2,6-dimethylfuro[2,3-d]pyrimidines as multitargeted receptor tyrosine kinase inhibitors and microtubule targeting antitumor agents. Bioorganic Med. Chem. 2014, 22, 3753–3772. [Google Scholar] [CrossRef]
- Gangjee, A.; Zeng, Y.; Ihnat, M.; Warnke, L.A.; Green, D.W.; Kisliuk, R.L.; Lin, F.T. Novel 5-substituted, 2,4-diaminofuro[2,3-d]pyrimidines as multireceptor tyrosine kinase and dihydrofolate reductase inhibitors with antiangiogenic and antitumor activity. Bioorg. Med. Chem. 2005, 13, 5475–5491. [Google Scholar] [CrossRef] [PubMed]
- Gangjee, A.; Li, W.; Lin, L.; Zeng, Y.; Ihnat, M.; Warnke, L.A.; Green, D.W.; Cody, V.; Pace, J.; Queener, S.F. Design, synthesis, and X-ray crystal structures of 2,4-diaminofuro[2,3-d]pyrimidines as multireceptor tyrosine kinase and dihydrofolate reductase inhibitors. Bioorg. Med. Chem. 2009, 17, 7324–7336. [Google Scholar] [CrossRef] [PubMed]
- Gangjee, A.; Zhao, Y.; Lin, L.; Raghavan, S.; Roberts, E.G.; Risinger, A.L.; Hamel, E.; Mooberry, S.L. Synthesis and discovery of water-soluble microtubule targeting agents that bind to the colchicine site on tubulin and circumvent Pgp mediated resistance. J. Med. Chem. 2010, 53, 8116–8128. [Google Scholar] [CrossRef] [PubMed]
- Gangjee, A.; Zhao, Y.; Hamel, E.; Westbrook, C.; Mooberry, S.L. Synthesis and biological activities of (R)- and (S)-N-(4-Methoxyphenyl)-N,2,6-trimethyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-aminium chloride as potent cytotoxic antitubulin agents. J. Med. Chem. 2011, 54, 6151–6155. [Google Scholar] [CrossRef] [PubMed]
- Gangjee, A.; Zhao, Y.; Raghavan, S.; Rohena, C.C.; Mooberry, S.L.; Hamel, E. Structure-activity relationship and in vitro and in vivo evaluation of the potent cytotoxic anti-microtubule agent N-(4-methoxyphenyl)-N,2,6-trimethyl-6,7-dihydro-5H-cyclopenta[d]pyrimidin-4-aminium chloride and its analogues as antitumor agents. J. Med. Chem. 2013, 56, 6829–6844. [Google Scholar] [CrossRef] [PubMed]
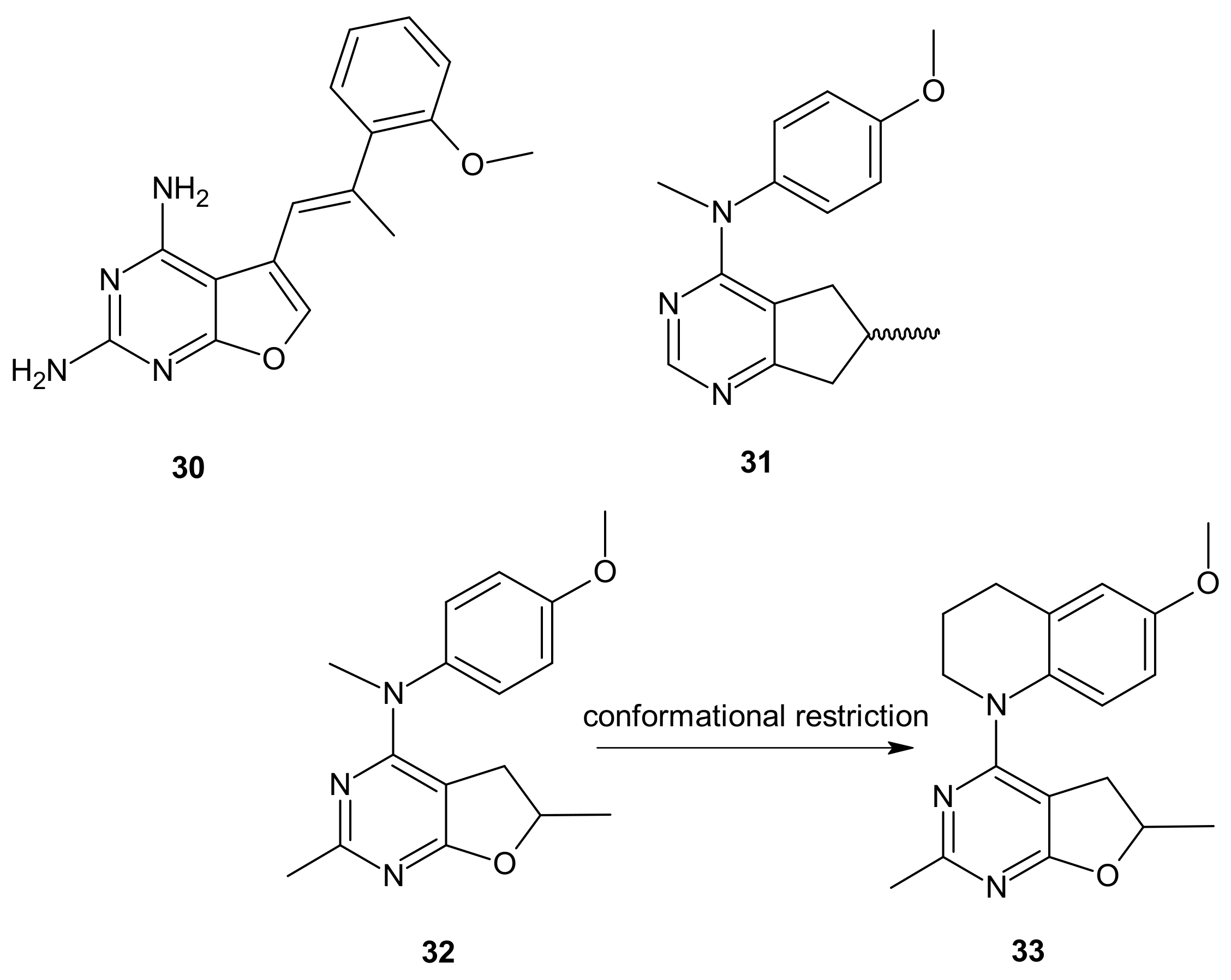
- Zhang, X.; Raghavan, S.; Ihnat, M.; Hamel, E.; Zammiello, C.; Bastian, A.; Mooberry, S.L.; Gangjee, A. The design, synthesis and biological evaluation of conformationally restricted 4-substituted-2,6-dimethylfuro[2,3-d]pyrimidines as multi-targeted receptor tyrosine kinase and microtubule inhibitors as potential antitumor agents. Bioorganic Med. Chem. 2015, 23, 2408–2423. [Google Scholar] [CrossRef]
- Jiao, Q.; Bi, L.; Ren, Y.; Song, S.; Wang, Q.; Wang, Y.-s. Advances in studies of tyrosine kinase inhibitors and their acquired resistance. Mol. Cancer 2018, 17, 36. [Google Scholar] [CrossRef]
- Iqbal, N.; Iqbal, N. Imatinib: A breakthrough of targeted therapy in cancer. Chemother. Res. Pract. 2014, 2014, 357027. [Google Scholar] [CrossRef]
- Tomaselli, D.; Lucidi, A.; Rotili, D.; Mai, A. Epigenetic polypharmacology: A new frontier for epi-drug discovery. Med. Res. Rev. 2020, 40, 190–244. [Google Scholar] [CrossRef]
- Wei, Y.; Poon, D.C.; Fei, R.; Lam, A.S.M.; Au-Yeung, S.C.F.; To, K.K.W. A platinum-based hybrid drug design approach to circumvent acquired resistance to molecular targeted tyrosine kinase inhibitors. Sci. Rep. 2016, 6, 25363. [Google Scholar] [CrossRef]
- Meng, L.-h.; Zheng, X.F.S. Toward rapamycin analog (rapalog)-based precision cancer therapy. Acta Pharmacol. Sin. 2015, 36, 1163–1169. [Google Scholar] [CrossRef]
- Rodrik-Outmezguine, V.S.; Okaniwa, M.; Yao, Z.; Novotny, C.J.; McWhirter, C.; Banaji, A.; Won, H.; Wong, W.; Berger, M.; de Stanchina, E.; et al. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature 2016, 534, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Kuroshima, K.; Yoshino, H.; Okamura, S.; Tsuruda, M.; Osako, Y.; Sakaguchi, T.; Sugita, S.; Tatarano, S.; Nakagawa, M.; Enokida, H. Potential new therapy of Rapalink-1, a new generation mammalian target of rapamycin inhibitor, against sunitinib-resistant renal cell carcinoma. Cancer Sci. 2020, 111, 1607–1618. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Yu, S.; Zhao, W.; Qin, S.; Chu, Q.; Wu, K. EGFR-TKIs resistance via EGFR-independent signaling pathways. Mol. Cancer 2018, 17, 53. [Google Scholar] [CrossRef]
- Ayral-Kaloustian, S.; Gu, J.; Lucas, J.; Cinque, M.; Gaydos, C.; Zask, A.; Chaudhary, I.; Wang, J.; Di, L.; Young, M.; et al. Hybrid Inhibitors of Phosphatidylinositol 3-Kinase (PI3K) and the Mammalian Target of Rapamycin (mTOR): Design, Synthesis, and Superior Antitumor Activity of Novel Wortmannin−Rapamycin Conjugates. J. Med. Chem. 2010, 53, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Meric-Bernstam, F.; Gonzalez-Angulo, A.M. Targeting the mTOR signaling network for cancer therapy. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 2278–2287. [Google Scholar] [CrossRef] [PubMed]
- Guertin, D.A.; Sabatini, D.M. The pharmacology of mTOR inhibition. Sci. Signal. 2009, 2, pe24. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Pandolfi, P.P. The PTEN–PI3K pathway: Of feedbacks and cross-talks. Oncogene 2008, 27, 5527–5541. [Google Scholar] [CrossRef]
- Sun, S.Y.; Rosenberg, L.M.; Wang, X.; Zhou, Z.; Yue, P.; Fu, H.; Khuri, F.R. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res. 2005, 65, 7052–7058. [Google Scholar] [CrossRef]
- Shi, Y.; Yan, H.; Frost, P.; Gera, J.; Lichtenstein, A. Mammalian target of rapamycin inhibitors activate the AKT kinase in multiple myeloma cells by up-regulating the insulin-like growth factor receptor/insulin receptor substrate-1/phosphatidylinositol 3-kinase cascade. Mol. Cancer 2005, 4, 1533–1540. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yue, P.; Kim, Y.A.; Fu, H.; Khuri, F.R.; Sun, S.Y. Enhancing mammalian target of rapamycin (mTOR)-targeted cancer therapy by preventing mTOR/raptor inhibition-initiated, mTOR/rictor-independent Akt activation. Cancer Res. 2008, 68, 7409–7418. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhou, X. Research progress of mTOR inhibitors. Eur. J. Med. Chem. 2020, 208, 112820. [Google Scholar] [CrossRef]
- Chiu, H.C.; Chou, D.L.; Huang, C.T.; Lin, W.H.; Lien, T.W.; Yen, K.J.; Hsu, J.T. Suppression of Stat3 activity sensitizes gefitinib-resistant non small cell lung cancer cells. Biochem Pharm. 2011, 81, 1263–1270. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Zhuang, G.; Cao, Y.; Du, P.; Kim, H.J.; Settleman, J. Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell 2014, 26, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Ji, T.; Gong, D.; Han, Z.; Wei, X.; Yan, Y.; Ye, F.; Ding, W.; Wang, J.; Xia, X.; Li, F.; et al. Abrogation of constitutive Stat3 activity circumvents cisplatin resistant ovarian cancer. Cancer Lett. 2013, 341, 231–239. [Google Scholar] [CrossRef]
- Zhang, W.; Guo, J.; Li, S.; Ma, T.; Xu, D.; Han, C.; Liu, F.; Yu, W.; Kong, L. Discovery of monocarbonyl curcumin-BTP hybrids as STAT3 inhibitors for drug-sensitive and drug-resistant breast cancer therapy. Sci. Rep. 2017, 7, 46352. [Google Scholar] [CrossRef]
- Ai, Y.; Zhu, B.; Ren, C.; Kang, F.; Li, J.; Huang, Z.; Lai, Y.; Peng, S.; Ding, K.; Tian, J.; et al. Discovery of New Monocarbonyl Ligustrazine-Curcumin Hybrids for Intervention of Drug-Sensitive and Drug-Resistant Lung Cancer. J. Med. Chem. 2016, 59, 1747–1760. [Google Scholar] [CrossRef]
- Chen, Q.H. Curcumin-based anti-prostate cancer agents. Anticancer Agents Med. Chem. 2015, 15, 138–156. [Google Scholar] [CrossRef]
- Prakobwong, S.; Gupta, S.C.; Kim, J.H.; Sung, B.; Pinlaor, P.; Hiraku, Y.; Wongkham, S.; Sripa, B.; Pinlaor, S.; Aggarwal, B.B. Curcumin suppresses proliferation and induces apoptosis in human biliary cancer cells through modulation of multiple cell signaling pathways. Carcinogenesis 2011, 32, 1372–1380. [Google Scholar] [CrossRef]
- Wei, C.C.; Ball, S.; Lin, L.; Liu, A.; Fuchs, J.R.; Li, P.K.; Li, C.; Lin, J. Two small molecule compounds, LLL12 and FLLL32, exhibit potent inhibitory activity on STAT3 in human rhabdomyosarcoma cells. Intj. Oncol. 2011, 38, 279–285. [Google Scholar]
- Bill, M.A.; Fuchs, J.R.; Li, C.; Yui, J.; Bakan, C.; Benson, D.M., Jr.; Schwartz, E.B.; Abdelhamid, D.; Lin, J.; Hoyt, D.G.; et al. The small molecule curcumin analog FLLL32 induces apoptosis in melanoma cells via STAT3 inhibition and retains the cellular response to cytokines with anti-tumor activity. Mol. Cancer 2010, 9, 165. [Google Scholar] [CrossRef] [PubMed]
- Fossey, S.L.; Bear, M.D.; Lin, J.; Li, C.; Schwartz, E.B.; Li, P.K.; Fuchs, J.R.; Fenger, J.; Kisseberth, W.C.; London, C.A. The novel curcumin analog FLLL32 decreases STAT3 DNA binding activity and expression, and induces apoptosis in osteosarcoma cell lines. BMC Cancer 2011, 11, 112. [Google Scholar] [CrossRef]
- Chen, H.; Yang, Z.; Ding, C.; Xiong, A.; Wild, C.; Wang, L.; Ye, N.; Cai, G.; Flores, R.M.; Ding, Y.; et al. Discovery of potent anticancer agent HJC0416, an orally bioavailable small molecule inhibitor of signal transducer and activator of transcription 3 (STAT3). Eurj. Med. Chem. 2014, 82, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Yang, Z.; Ding, C.; Chu, L.; Zhang, Y.; Terry, K.; Liu, H.; Shen, Q.; Zhou, J. Fragment-based drug design and identification of HJC0123, a novel orally bioavailable STAT3 inhibitor for cancer therapy. Eur. J. Med. Chem. 2013, 62, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Damaskos, C.; Valsami, S.; Kontos, M.; Spartalis, E.; Kalampokas, T.; Kalampokas, E.; Athanasiou, A.; Moris, D.; Daskalopoulou, A.; Davakis, S.; et al. Histone Deacetylase Inhibitors: An Attractive Therapeutic Strategy Against Breast Cancer. Anticancer Res. 2017, 37, 35. [Google Scholar] [CrossRef]
- Yang, X.J.; Seto, E. HATs and HDACs: From structure, function and regulation to novel strategies for therapy and prevention. Oncogene 2007, 26, 5310–5318. [Google Scholar] [CrossRef]
- Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene 2007, 26, 5541–5552. [Google Scholar] [CrossRef]
- Bass, A.K.A.; El-Zoghbi, M.S.; Nageeb, E.S.M.; Mohamed, M.F.A.; Badr, M.; Abuo-Rahma, G. Comprehensive review for anticancer hybridized multitargeting HDAC inhibitors. Eur. J. Med. Chem. 2021, 209, 65. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Wan, Y.; Xiao, Y.; Xia, C.; Duan, G. Dual-Target Inhibitors Based on HDACs: Novel Antitumor Agents for Cancer Therapy. J. Med. Chem. 2020, 63, 8977–9002. [Google Scholar] [CrossRef]
- Tanimoto, A.; Takeuchi, S.; Arai, S.; Fukuda, K.; Yamada, T.; Roca, X.; Ong, S.T.; Yano, S. Histone Deacetylase 3 Inhibition Overcomes BIM Deletion Polymorphism-Mediated Osimertinib Resistance in EGFR-Mutant Lung Cancer. Clin. Cancer Res. 2017, 23, 3139–3149. [Google Scholar] [CrossRef]
- Kim, M.J.; Kim, D.E.; Jeong, I.G.; Choi, J.; Jang, S.; Lee, J.H.; Ro, S.; Hwang, J.J.; Kim, C.S. HDAC Inhibitors Synergize Antiproliferative Effect of Sorafenib in Renal Cell Carcinoma Cells. Anticancer Res. 2012, 32, 3161–3168. [Google Scholar] [PubMed]
- Nakagawa, T.; Takeuchi, S.; Yamada, T.; Ebi, H.; Sano, T.; Nanjo, S.; Ishikawa, D.; Sato, M.; Hasegawa, Y.; Sekido, Y.; et al. EGFR-TKI resistance due to BIM polymorphism can be circumvented in combination with HDAC inhibition. Cancer Res. 2013, 73, 2428–2434. [Google Scholar] [CrossRef]
- Mahboobi, S.; Pilsl, B.; Sellmer, A. Generation and Assessment of Fusions between HDACi and TKi. Methods Mol. Biol. 2017, 1510, 405–412. [Google Scholar] [CrossRef]
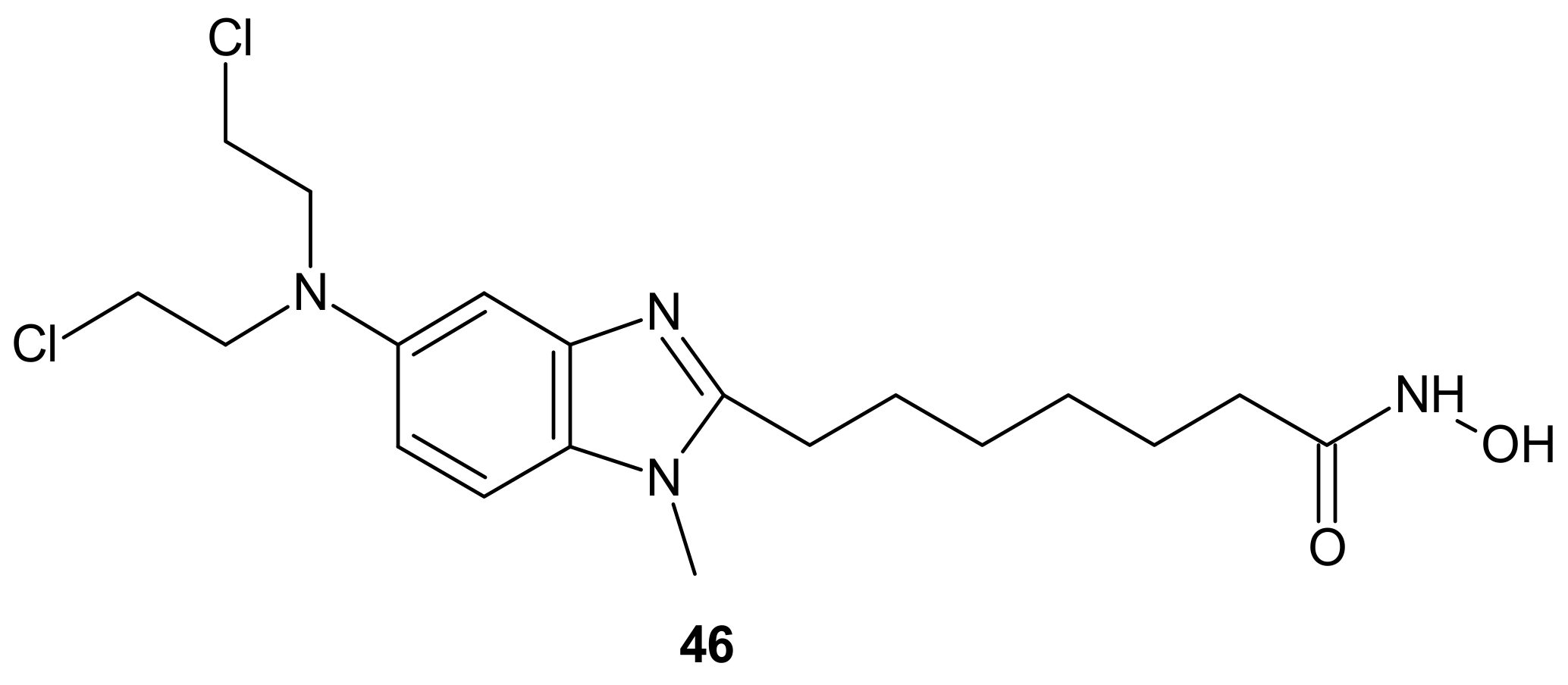
- Mahboobi, S.; Dove, S.; Sellmer, A.; Winkler, M.; Eichhorn, E.; Pongratz, H.; Ciossek, T.; Baer, T.; Maier, T.; Beckers, T. Design of chimeric histone deacetylase- and tyrosine kinase-inhibitors: A series of imatinib hybrides as potent inhibitors of wild-type and mutant BCR-ABL, PDGF-Rbeta, and histone deacetylases. J. Med. Chem. 2009, 52, 2265–2279. [Google Scholar] [CrossRef]
- Mahboobi, S.; Sellmer, A.; Winkler, M.; Eichhorn, E.; Pongratz, H.; Ciossek, T.; Baer, T.; Maier, T.; Beckers, T. Novel chimeric histone deacetylase inhibitors: A series of lapatinib hybrides as potent inhibitors of epidermal growth factor receptor (EGFR), human epidermal growth factor receptor 2 (HER2), and histone deacetylase activity. J. Med. Chem. 2010, 53, 8546–8555. [Google Scholar] [CrossRef] [PubMed]
- Beckers, T.; Mahboobi, S.; Sellmer, A.; Winkler, M.; Eichhorn, E.; Pongratz, H.; Maier, T.; Ciossek, T.; Baer, T.; Kelter, G.; et al. Chimerically designed HDAC- and tyrosine kinase inhibitors. A series of erlotinib hybrids as dual-selective inhibitors of EGFR, HER2 and histone deacetylases. MedChemComm 2012, 3, 829–835. [Google Scholar] [CrossRef]
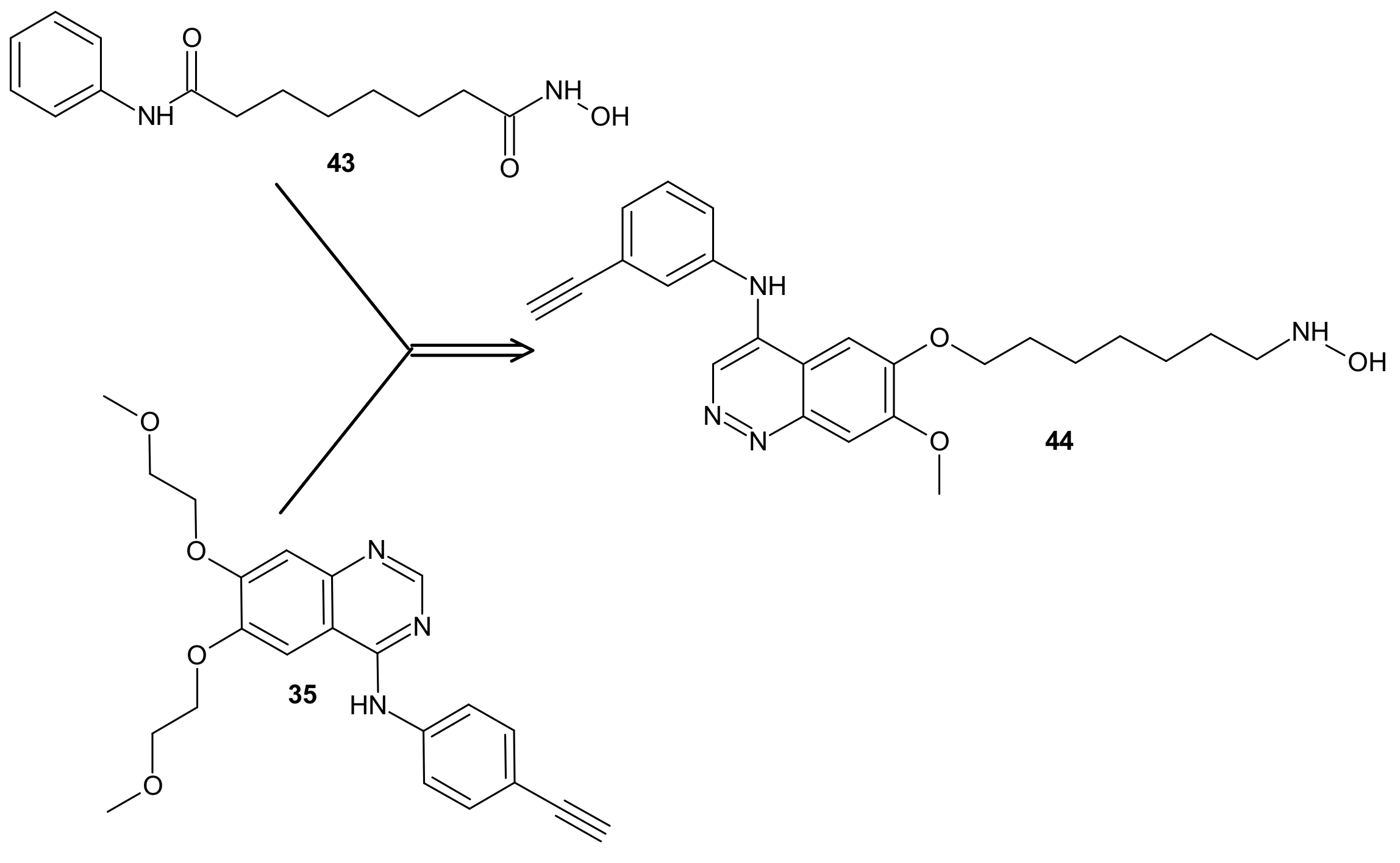
- Cai, X.; Zhai, H.-X.; Wang, J.; Forrester, J.; Qu, H.; Yin, L.; Lai, C.-J.; Bao, R.; Qian, C. Discovery of 7-(4-(3-Ethynylphenylamino)-7-methoxyquinazolin-6-yloxy)-N-hydroxyheptanamide (CUDC-101) as a Potent Multi-Acting HDAC, EGFR, and HER2 Inhibitor for the Treatment of Cancer. J. Med. Chem. 2010, 53, 2000–2009. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Pursell, N.W.; Samson, M.E.S.; Atoyan, R.; Ma, A.W.; Selmi, A.; Xu, W.L.; Cai, X.; Voi, M.; Savagner, P.; et al. Potential Advantages of CUDC-101, a Multitargeted HDAC, EGFR, and HER2 Inhibitor, in Treating Drug Resistance and Preventing Cancer Cell Migration and Invasion. Mol. Cancer Ther. 2013, 12, 925–936. [Google Scholar] [CrossRef]
- Lai, C.-J.; Bao, R.; Tao, X.; Wang, J.; Atoyan, R.; Qu, H.; Wang, D.-G.; Yin, L.; Samson, M.; Forrester, J.; et al. CUDC-101, a Multitargeted Inhibitor of Histone Deacetylase, Epidermal Growth Factor Receptor, and Human Epidermal Growth Factor Receptor 2, Exerts Potent Anticancer Activity. Cancer Res. 2010, 70, 3647–3656. [Google Scholar] [CrossRef]
- Shimizu, T.; LoRusso, P.M.; Papadopoulos, K.P.; Patnaik, A.; Beeram, M.; Smith, L.S.; Rasco, D.W.; Mays, T.A.; Chambers, G.; Ma, A.; et al. Phase I First-in-Human Study of CUDC-101, a Multitargeted Inhibitor of HDACs, EGFR, and HER2 in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2014, 20, 5032–5040. [Google Scholar] [CrossRef]
- Galloway, T.J.; Wirth, L.J.; Colevas, A.D.; Gilbert, J.; Bauman, J.E.; Saba, N.F.; Raben, D.; Mehra, R.; Ma, A.W.; Atoyan, R.; et al. A Phase I Study of CUDC-101, a Multitarget Inhibitor of HDACs, EGFR, and HER2, in Combination with Chemoradiation in Patients with Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2015, 21, 1566–1573. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Y.; Mehta, A.; Boufraqech, M.; Davis, S.; Wang, J.; Tian, Z.; Yu, Z.; Boxer, M.B.; Kiefer, J.A.; et al. Dual inhibition of HDAC and EGFR signaling with CUDC-101 induces potent suppression of tumor growth and metastasis in anaplastic thyroid cancer. Oncotarget 2015, 6, 9073–9085. [Google Scholar] [CrossRef]
- De Leo, S.; Trevisan, M.; Fugazzola, L. Recent advances in the management of anaplastic thyroid cancer. Thyroid Res. 2020, 13, 17. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, J. CUDC-101 Overcomes Arsenic Trioxide Resistance Via Caspase-Dependent PML-Rarα Degradation in Acute Promyelocytic Leukemia. Blood 2019, 134, 5054. [Google Scholar] [CrossRef]
- Zhang, T.Z.; Ma, D.; Wei, D.N.; Lu, T.T.; Yu, K.L.; Zhang, Z.Y.; Wang, W.L.; Fang, Q.; Wang, J.S. CUDC-101 overcomes arsenic trioxide resistance via caspase-dependent promyelocytic leukemia-retinoic acid receptor alpha degradation in acute promyelocytic leukemia. Anti-Cancer Drugs 2020, 31, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.; Lai, C.J.; Bao, R.; Wang, D.G.; Wang, J.; Xu, G.X.; Atoyan, R.; Qu, H.; Yin, L.; Samson, M.; et al. Cancer network disruption by a single molecule inhibitor targeting both histone deacetylase activity and phosphatidylinositol 3-kinase signaling. Clin. Cancer Res. 2012, 18, 4104–4113. [Google Scholar] [CrossRef] [PubMed]
- Ali, D.; Alshammari, H.; Vishnubalaji, R.; Chalisserry, E.P.; Hamam, R.; Alfayez, M.; Kassem, M.; Aldahmash, A.; Alajez, N.M. CUDC-907 Promotes Bone Marrow Adipocytic Differentiation Through Inhibition of Histone Deacetylase and Regulation of Cell Cycle. Stem Cells Dev. 2017, 26, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Atoyan, R.; Borek, M.A.; Dellarocca, S.; Samson, M.E.; Ma, A.W.; Xu, G.X.; Patterson, T.; Tuck, D.P.; Viner, J.L.; et al. Dual HDAC and PI3K Inhibitor CUDC-907 Downregulates MYC and Suppresses Growth of MYC-dependent Cancers. Mol. Cancer 2017, 16, 285–299. [Google Scholar] [CrossRef]
- Kotian, S.; Zhang, L.; Boufraqech, M.; Gaskins, K.; Gara, S.K.; Quezado, M.; Nilubol, N.; Kebebew, E. Dual Inhibition of HDAC and Tyrosine Kinase Signaling Pathways with CUDC-907 Inhibits Thyroid Cancer Growth and Metastases. Clin. Cancer Res. 2017, 23, 5044–5054. [Google Scholar] [CrossRef] [PubMed]
- Mondello, P.; Derenzini, E.; Asgari, Z.; Philip, J.; Brea, E.J.; Seshan, V.; Hendrickson, R.C.; de Stanchina, E.; Scheinberg, D.A.; Younes, A. Dual inhibition of histone deacetylases and phosphoinositide 3-kinase enhances therapeutic activity against B cell lymphoma. Oncotarget 2017, 8, 14017–14028. [Google Scholar] [CrossRef][Green Version]
- Li, X.; Su, Y.; Madlambayan, G.; Edwards, H.; Polin, L.; Kushner, J.; Dzinic, S.H.; White, K.; Ma, J.; Knight, T.; et al. Antileukemic activity and mechanism of action of the novel PI3K and histone deacetylase dual inhibitor CUDC-907 in acute myeloid leukemia. Haematologica 2019, 104, 2225–2240. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Xia, H.Y.; Bai, S.S.; Zhao, J.L.; Edwards, H.; Li, X.Y.; Yang, Y.R.; Lyu, J.; Wang, G.; Zhan, Y.; et al. CUDC-907, a novel dual PI3K and HDAC inhibitor, in prostate cancer: Antitumour activity and molecular mechanism of action. J. Cell. Mol. Med. 2020, 24, 7239–7253. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Bian, X.; Lin, W. The dual HDAC-PI3K inhibitor CUDC-907 displays single-agent activity and synergizes with PARP inhibitor olaparib in small cell lung cancer. J. Exp. Clin. Cancer Res. 2020, 39, 219. [Google Scholar] [CrossRef]
- Oki, Y.; Kelly, K.R.; Flinn, I.; Patel, M.R.; Gharavi, R.; Ma, A.; Parker, J.; Hafeez, A.; Tuck, D.; Younes, A. CUDC-907 in relapsed/refractory diffuse large B-cell lymphoma, including patients with MYC-alterations: Results from an expanded phase I trial. Haematologica 2017, 102, 1923–1930. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Berdeja, J.G.; Patel, M.R.; Flinn, I.; Gerecitano, J.F.; Neelapu, S.S.; Kelly, K.R.; Copeland, A.R.; Akins, A.; Clancy, M.S.; et al. Safety, tolerability, and preliminary activity of CUDC-907, a first-in-class, oral, dual inhibitor of HDAC and PI3K, in patients with relapsed or refractory lymphoma or multiple myeloma: An open-label, dose-escalation, phase 1 trial. Lancet Oncol. 2016, 17, 622–631. [Google Scholar] [CrossRef]
- Shulman, D.S.; Carlowicz, C.; Gustafson, C.; Vo, K.T.; Fox, E.; Muscal, J.A.; Supko, J.G.; Place, A.E.; Chi, S.N.; Shusterman, S.; et al. Phase I multicenter trial of CUDC-907 in children and young adults with relapsed/refractory solid tumors, CNS tumors, and lymphomas. J. Clin. Oncol. 2018, 36, 10542. [Google Scholar] [CrossRef]
- Available online: www.clinicaltrials.gov (accessed on 30 March 2021).
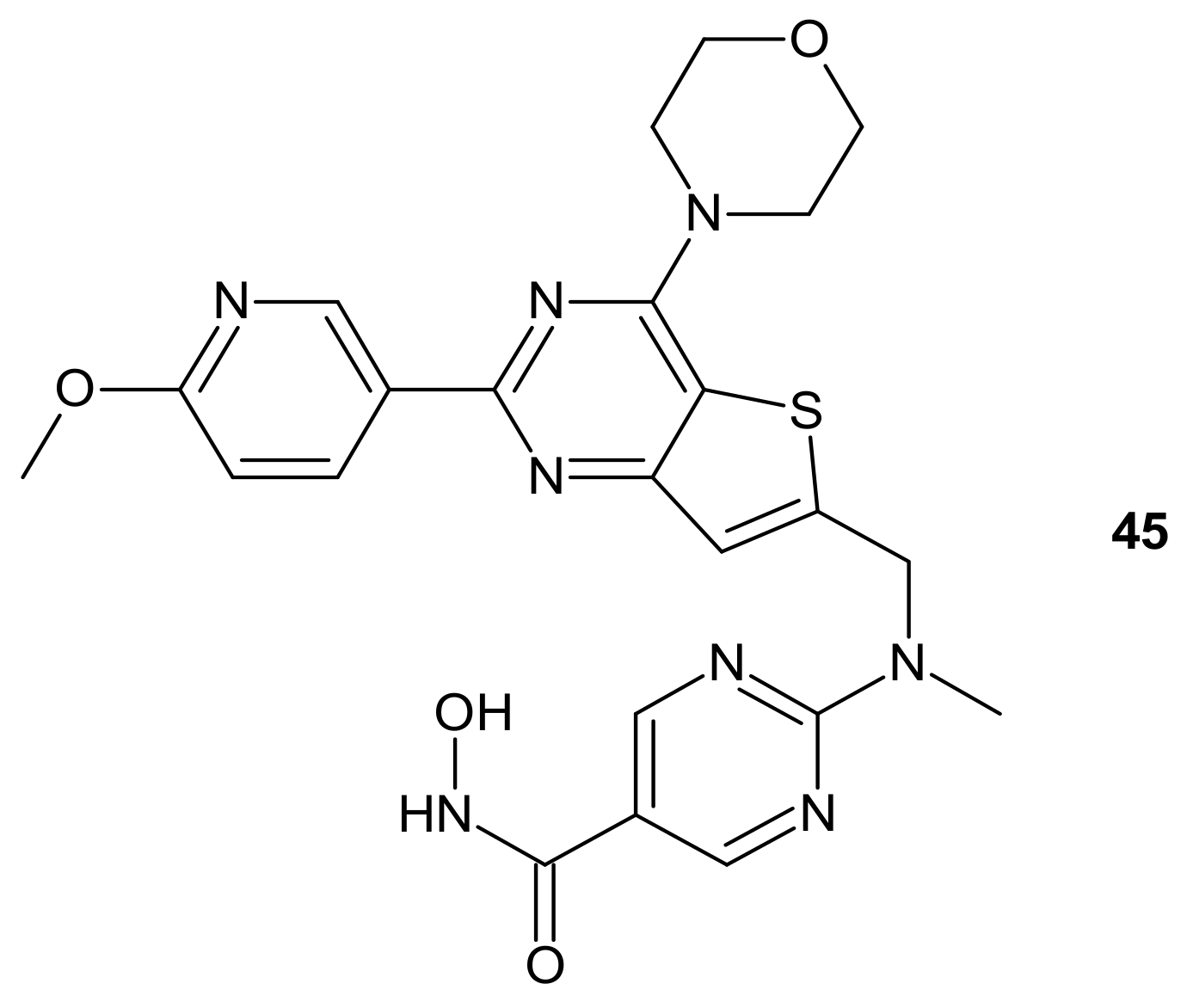
- Mehrling, T.; Chen, Y. The Alkylating-HDAC Inhibition Fusion Principle: Taking Chemotherapy to the Next Level with the First in Class Molecule EDO-S101. Anti-Cancer Agents Med. Chem. 2016, 16, 20–28. [Google Scholar] [CrossRef] [PubMed]
- López-Iglesias, A.A.; San-Segundo, L.; González-Méndez, L.; Hernández-García, S.; Primo, D.; Garayoa, M.; Hernández, A.B.; Paíno, T.; Mateos, M.-V.; Chen, Y.; et al. The Alkylating Histone Deacetylase Inhibitor Fusion Molecule Edo-S101 Displays Full Bi-Functional Properties in Preclinical Models of Hematological Malignancies. Blood 2014, 124, 2100. [Google Scholar] [CrossRef]
- Chesi, M.; Garbitt, V.; Bergsagel, P.L. Identification of Novel Therapeutic Targets in the Clinically Predictive Vk*MYC Mouse Model of Multiple Myeloma. Blood 2014, 124, 415. [Google Scholar] [CrossRef]
- Besse, L.; Kraus, M.; Besse, A.; Bader, J.; Silzle, T.; Mehrling, T.; Driessen, C. The first-in-class alkylating HDAC inhibitor EDO-S101 is highly synergistic with proteasome inhibition against multiple myeloma through activation of multiple pathways. Blood Cancer J. 2017, 7. [Google Scholar] [CrossRef]
- Festuccia, C.; Mancini, A.; Colapietro, A.; Gravina, G.L.; Vitale, F.; Marampon, F.; Delle Monache, S.; Pompili, S.; Cristiano, L.; Vetuschi, A.; et al. The first-in-class alkylating deacetylase inhibitor molecule tinostamustine shows antitumor effects and is synergistic with radiotherapy in preclinical models of glioblastoma. J. Hematol. Oncol. 2018, 11, s13045-018. [Google Scholar]
- U.S. Food & Drug Administration. Designating an Orphan Product: Drugs and Biological Products. Available online: https://www.accessdata.fda.gov/scripts/opdlisting/oopd/listResult.cfm (accessed on 30 March 2021).

























Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szumilak, M.; Wiktorowska-Owczarek, A.; Stanczak, A. Hybrid Drugs—A Strategy for Overcoming Anticancer Drug Resistance? Molecules 2021, 26, 2601. https://doi.org/10.3390/molecules26092601
Szumilak M, Wiktorowska-Owczarek A, Stanczak A. Hybrid Drugs—A Strategy for Overcoming Anticancer Drug Resistance? Molecules. 2021; 26(9):2601. https://doi.org/10.3390/molecules26092601
Chicago/Turabian StyleSzumilak, Marta, Anna Wiktorowska-Owczarek, and Andrzej Stanczak. 2021. "Hybrid Drugs—A Strategy for Overcoming Anticancer Drug Resistance?" Molecules 26, no. 9: 2601. https://doi.org/10.3390/molecules26092601
APA StyleSzumilak, M., Wiktorowska-Owczarek, A., & Stanczak, A. (2021). Hybrid Drugs—A Strategy for Overcoming Anticancer Drug Resistance? Molecules, 26(9), 2601. https://doi.org/10.3390/molecules26092601