
Prediction of Chromatography Conditions for Purification in Organic Synthesis Using Deep Learning



Abstract
1. Introduction
2. Related Work
3. Formal Definition of Tasks
- Task No. 1: Prediction of the solvent labels that make up the solvent system for chromatographic purification.
- Task No. 2: Prediction of the ratio between solvents if two solvents are used.
| Task No. 1 (Multiclass Classification) | Task No. 2 (Regression) | |
| Input | Let a chemical reaction be denoted as di and belong to a space of chemical reactions di ∈ D. Each di can be converted into a p-dimensional feature vector Xi = (xi,1, xi,2, …, xi,p) | |
| Output | Let Y = {y1, y2, … yN} be a label space of size N that represents possible solvents (class) labels | Let γi ⊆ [0, 1] be a continuous variable which represents the ratio between the solvents |
| Prediction function | Let η be a function that η(X) → Y correctly predicts | |
| a set of solvents labels | a ratio between solvents | |
| Method | Let Γ be a machine-learning algorithm that finds an approximation η′ (model) of a function η when given a learning dataset DL ⊂ D | |
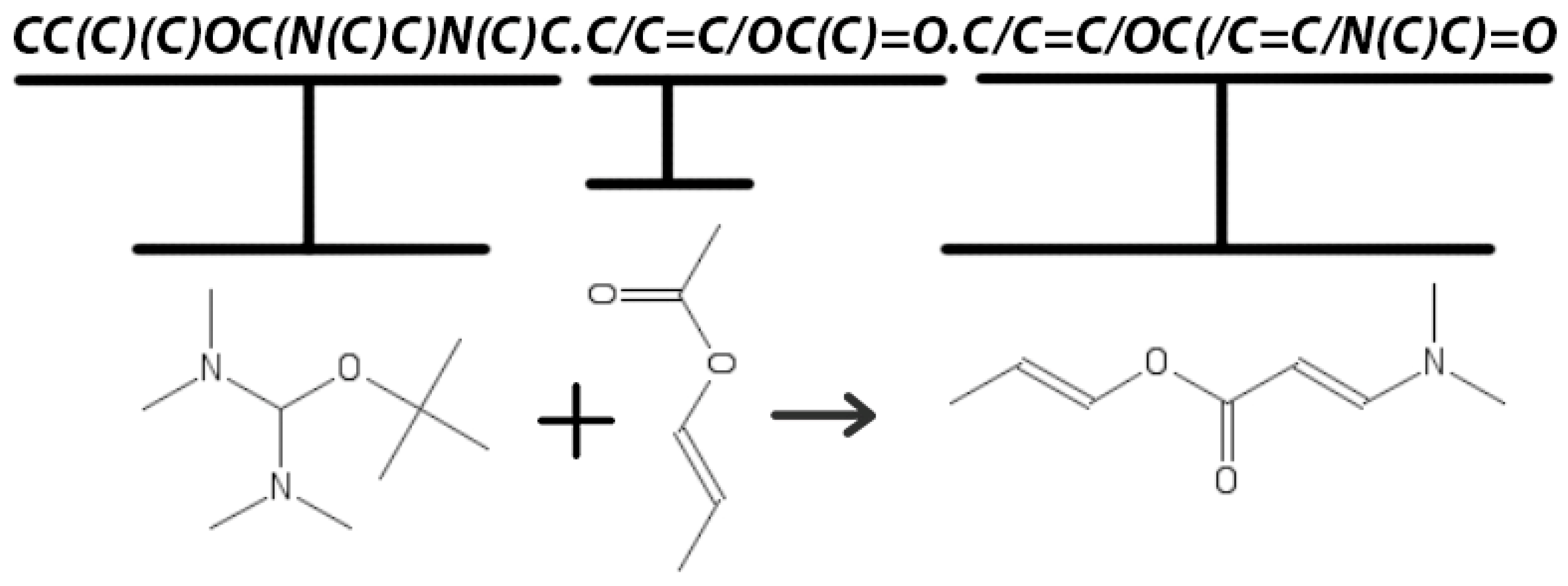
4. The Data
5. Materials and Methods
5.1. Vectorization
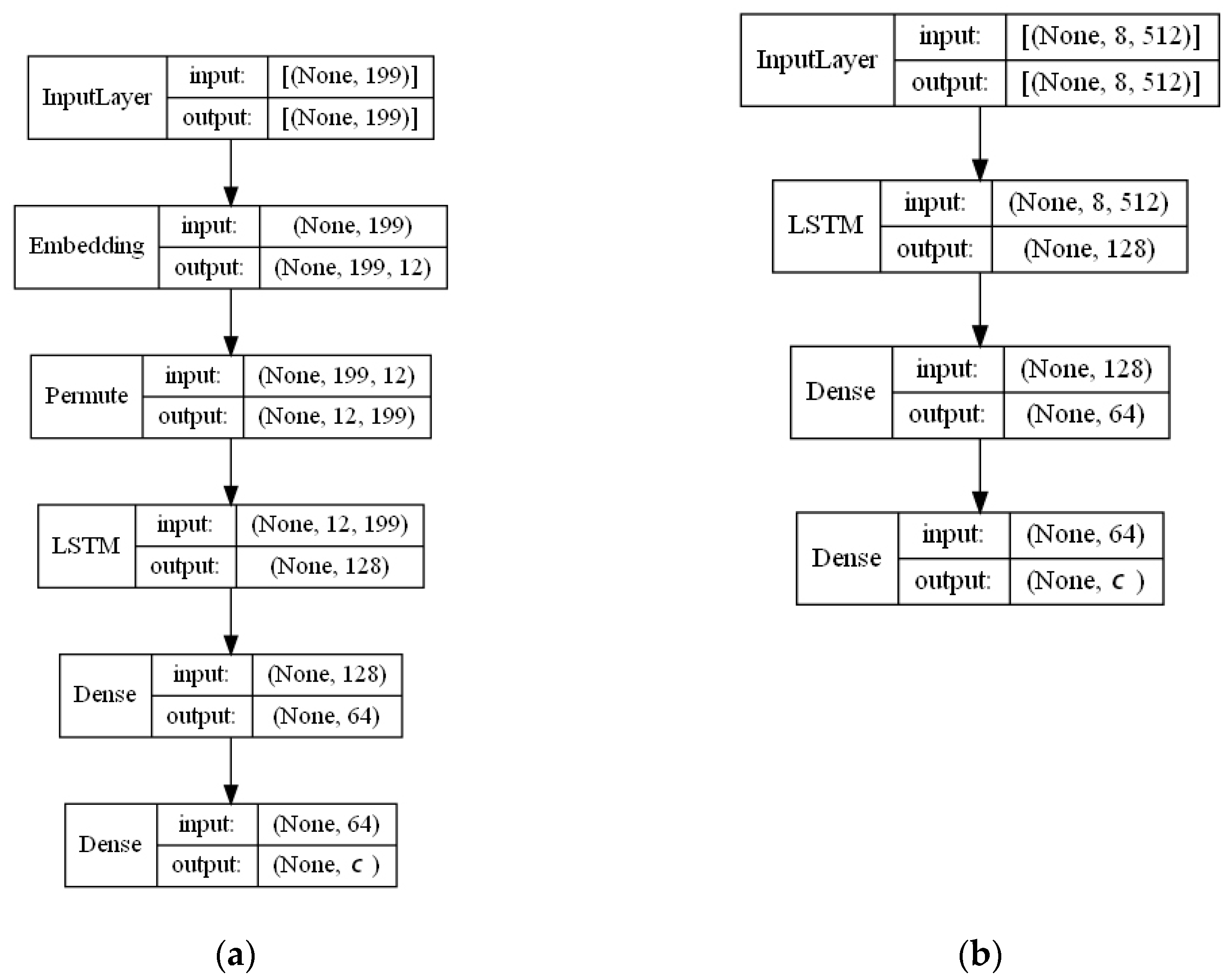
- Learned Embedding (LE). An object’s attributes—chemical structures in SMILES notation are concatenated into one line of symbols with a dot as their separator. The downstream supervised machine-learning task symbol representations (embeddings) can be learned jointly. 56 unique symbols are used in the SMILES representation and they all must be represented with N-dimensional vectors. N was set to 12 because larger values did not increase the accuracy in the preliminary experiments. Not to lose the input information, the maximum length of a symbol sequence was set to 200. Thus, the embedded 2D vector for any input sequence consisted of 200 rows (one row per symbol) of 12 vector values each. In short, each of 56 unique symbols is embedded with vectors of 12 dimensions that are trained the same way as weights are trained in a neural network. For a single instance, a matrix 200 × 12 is formed by looking up the symbol and its vector and later combining them into a matrix [41]. Some key advantages of a learned embedding type are the following: (1) encoding is fast, as it does not require any additional processing if compared to, e.g., fingerprints or descriptors; (2) embedding vectors are jointly learned with the training model, therefore complements each other well; (3) compared to one-hot encoding, learned embeddings provide a more uniform and less sparse vectorization which may extract more features of the molecular compounds.
- Extended-connectivity fingerprints (ECFP). Morgan fingerprints or extended-connectivity fingerprints (ECFP) [42] are representations of molecular structures explicitly created to capture chemical features. ECFPs are a variant of the Morgan algorithm that solves the molecular isomorphism problem, where two molecules numbered differently should produce the same fingerprint vector. ECFPs are very useful for the representation of topological structural information and have been used in a diverse set of applications, such as virtual screening [42], activity modeling [43], and machine learning [44,45,46]. Since the maximum number of molecules participating in a single reaction is 8, it results in an 8 × 512 matrix composed for every reaction. The main features of ECFPs are that they represent the presence of particular substructures by means of circular atom neighborhoods. ECFPs represent both the presence and absence of functionality, which are significant for extracting the molecule’s features. This results in a more informative vectorization method. Compared to the one-hot encoding method able to encode only characters or symbols, ECFPs encode fragments of the molecular structure.
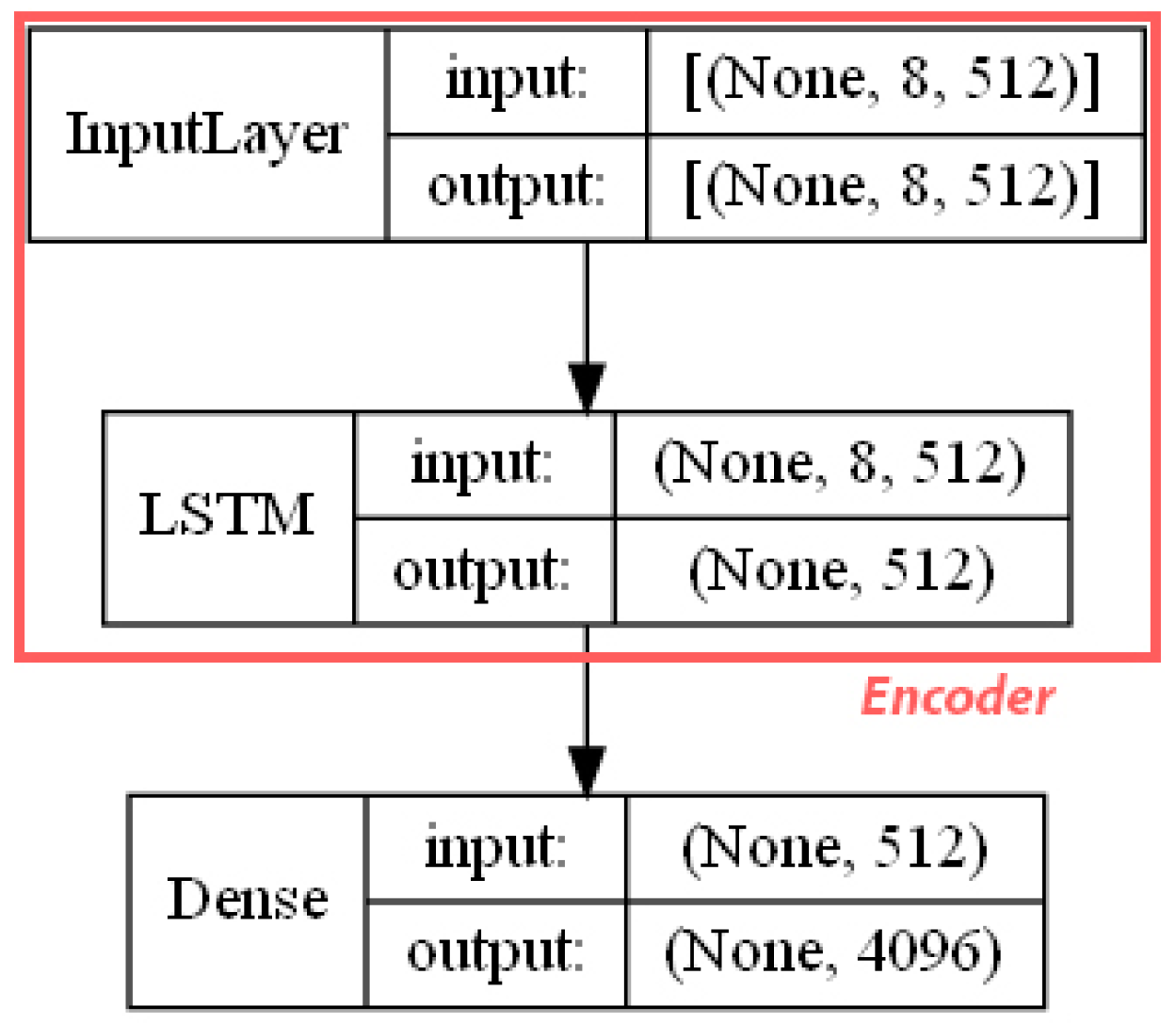
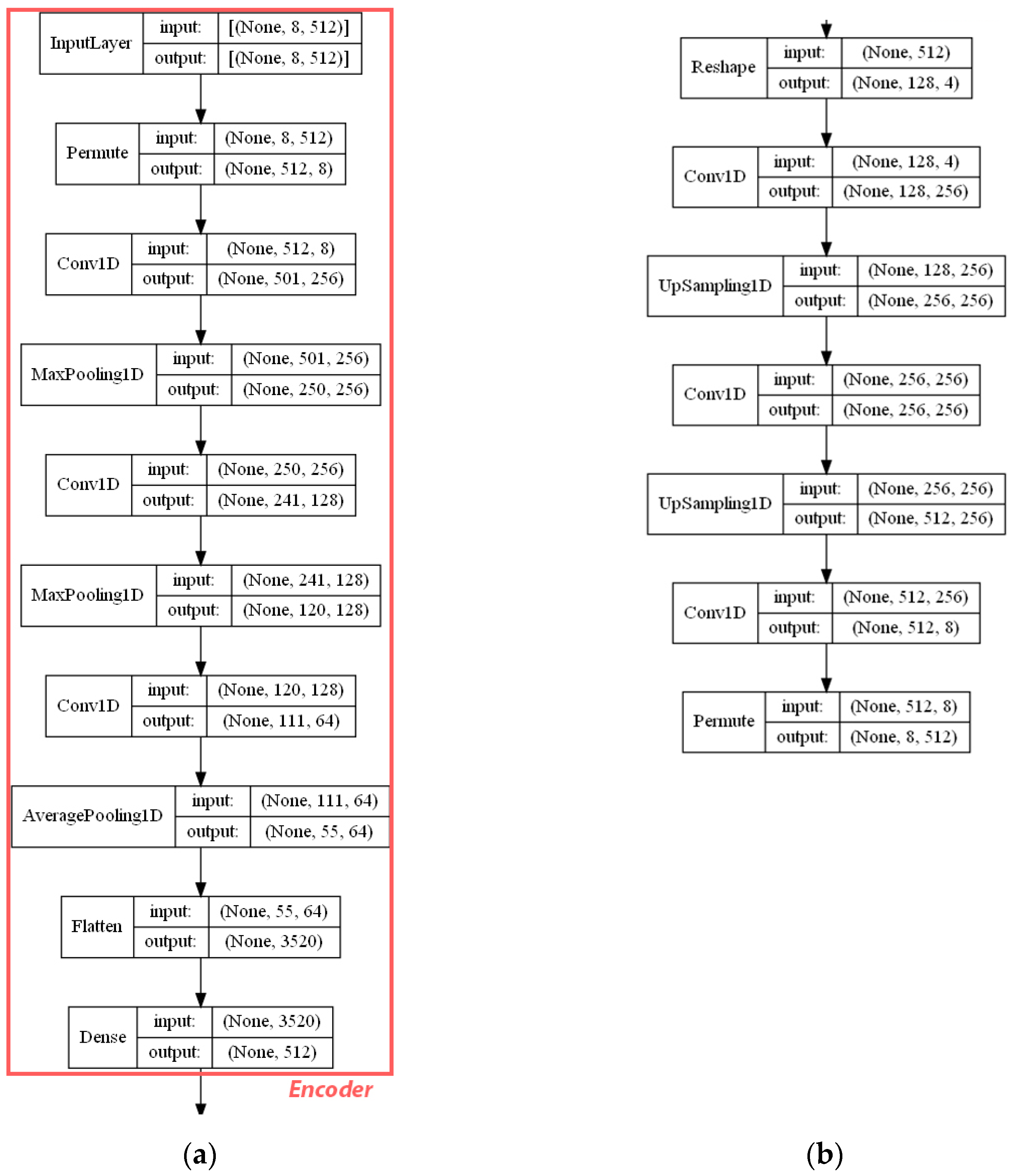
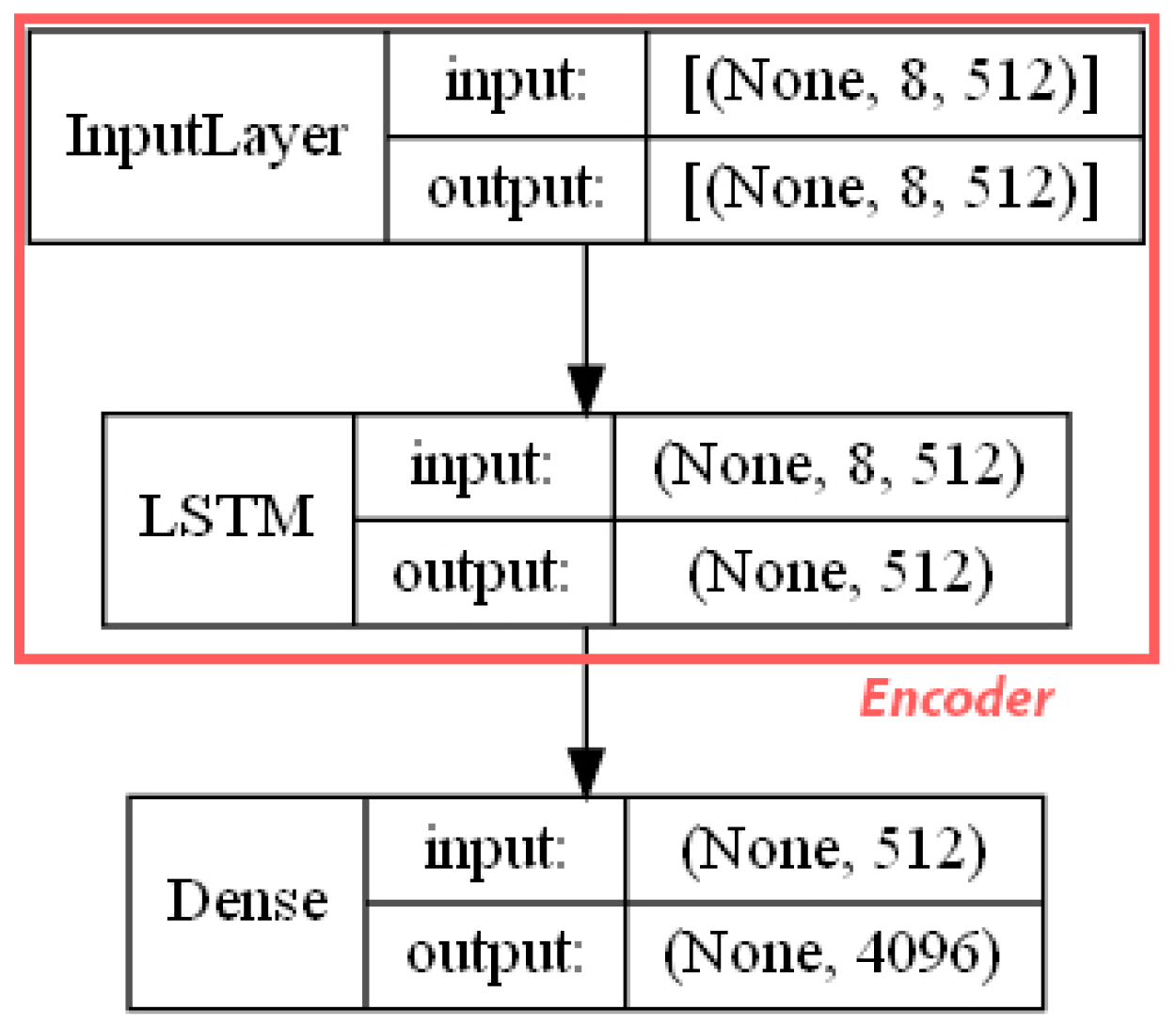
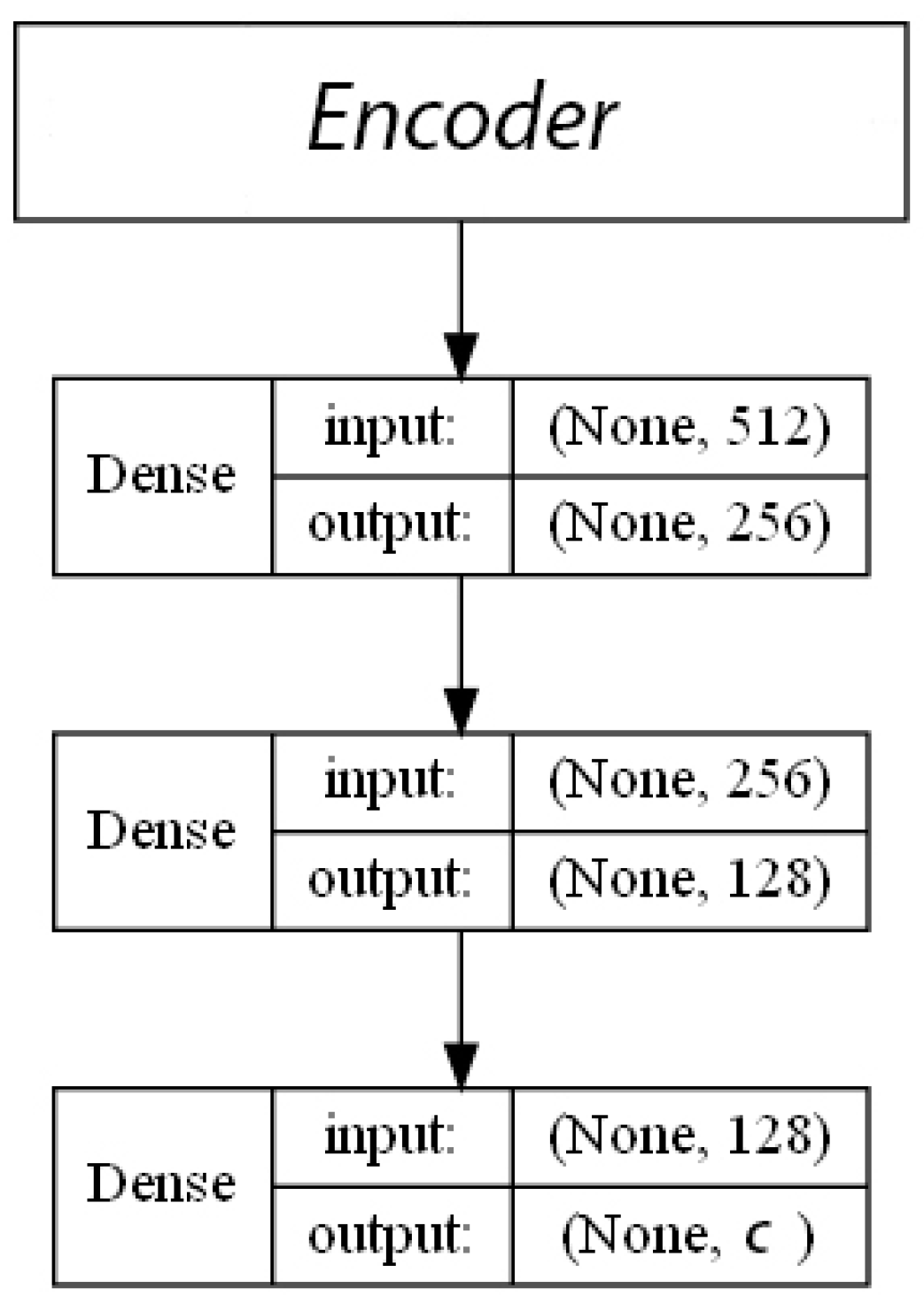
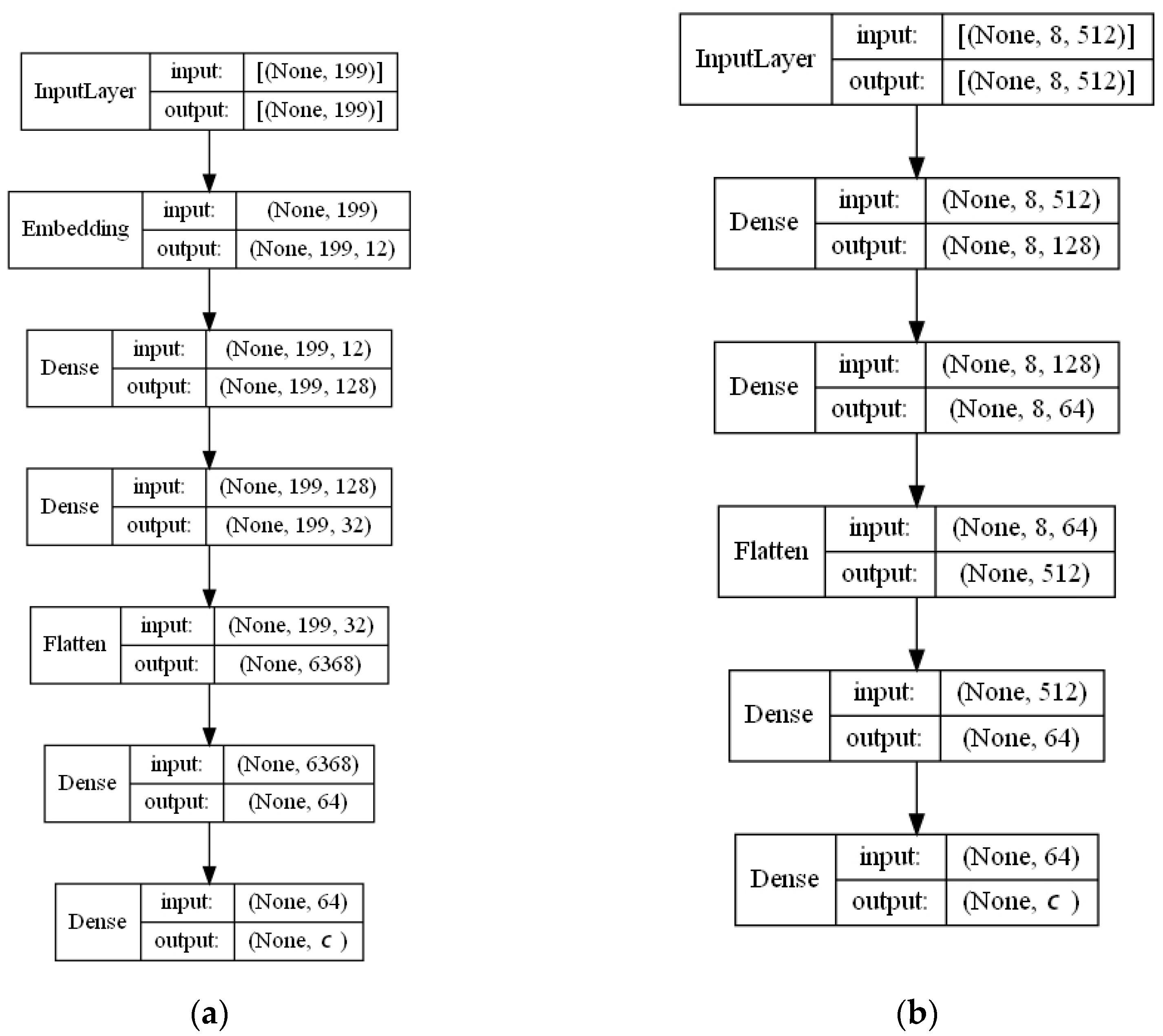
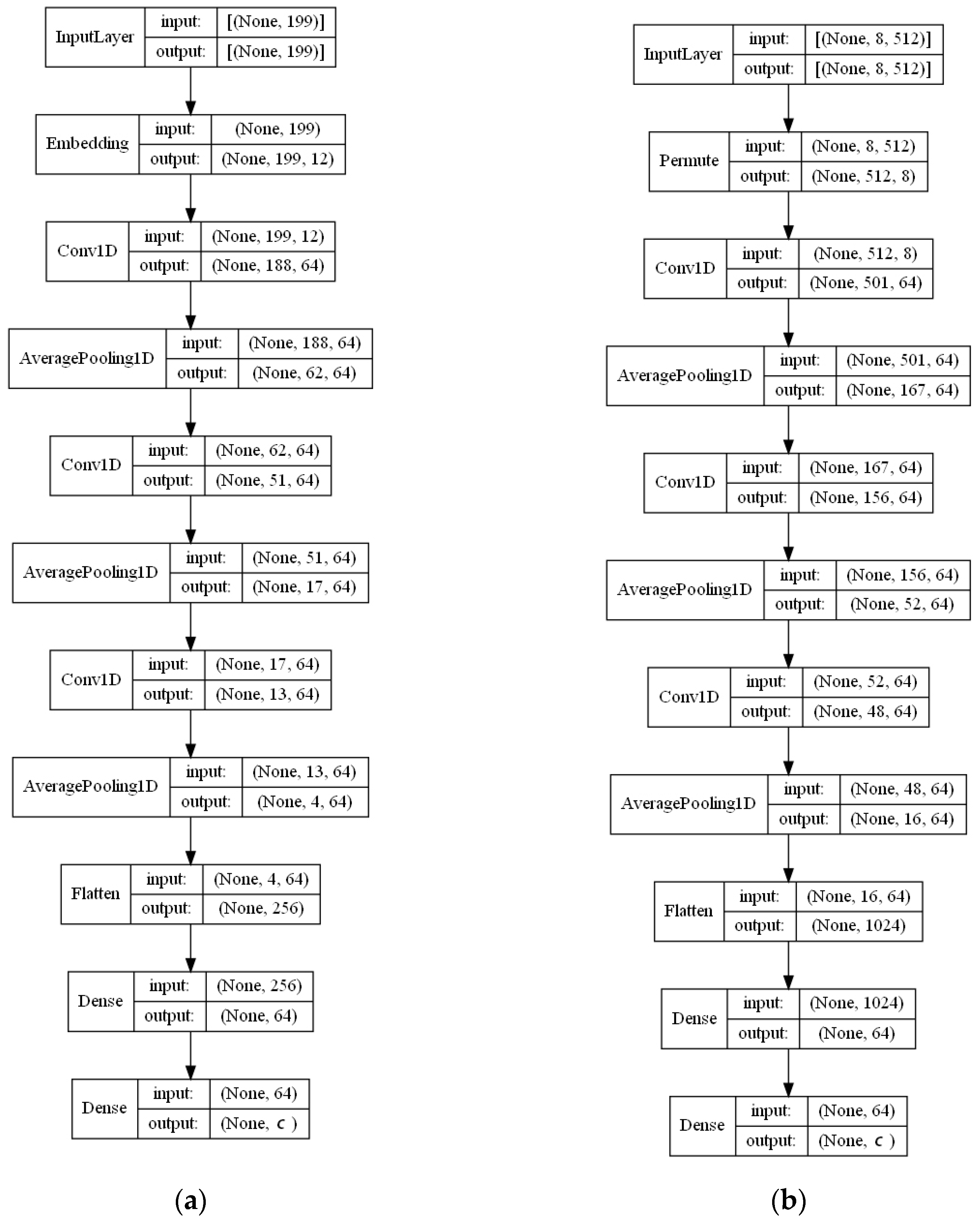
- ECFP auto-encoder (ECFP + E). Auto-encoders have been offered as the dimensionality reduction solution for rather large and sparse matrices of extended-connectivity fingerprints. The auto-encoder consists of two connected neural networks called encoder and decoder. Training of a neural network works by taking in ECFPs and trying to reproduce the same fingerprints using a bottleneck layer called a latent space. Later, encoder weights are transferred into a separate neural network. During the training, each instance passes the encoder layer and is compressed to the latent layer’s size. It is even hypothesized that an auto-encoder can denoise the data by learning only the key features representing a specific reaction. The main advantage of an encoder is that it learns how to map the larger multiple molecules input into smaller vectors during the training phase. This action forces the encoder to detect relevant ECFP parts and disregard irrelevant, which, in turn, may lead to higher accuracy. Auto-encoders were also proved to be successful for the dimensionality reduction in the QSAR (Quantitative structure-activity relationship) modeling [47].Furthermore, we present approaches (including encoder-decoder parts, topologies, and hyperparameter values) used to develop auto-encoders for vectorization.Three types of auto-encoders were constructed and investigated: (1) feed-forward, (3) 1DCNN, and (2) LSTM. Two types of input formats were used: (1) an unchanged ECFP with a size of 8 × 512 for LSTM and 1DCNN auto-encoders; (2) a flattened ECFP of size 1 × 4096 with a feed-forward auto-encoder for feed-forward. These input types were used to simplify encoders’ topology: i.e., the feed-forward auto-encoder does not require an additional flattening layer if the input is a one-dimensional vector.The latent dimension size, which is the key auto-encoder’s parameter, was set to 512. Out of tested latent dimensionality sizes (32, 64, 128, 256, 512) in the preliminary experiments, 512 produced the most accurate reconstructions and at the same time assured significant compression by 8 times compared to the vector size (4096) of the initial input. Binary cross-entropy loss function was used for the training of auto-encoders.Topologies of auto-encoders were selected experimentally based on the lowest loss scores on the testing dataset. These experiments helped us to end up with an exact parameter set for this particular vectorization type. The tuning of parameters was performed separately with each classification method. The optimization of parameters was performed by testing all values of the selected parameter, setting the best determined one, and then iteratively advancing to the optimization of the following parameter. Deep auto-encoders have been constructed by stacking feed-forward layers with different numbers of neurons (16, 32, 64, 128, 256, or 512). However, these deeper architectures underperformed shallow auto-encoders, therefore, were not selected for our further experiments. The investigation of the 1DCNN auto-encoder included the addition/removal of convolutional layers (with sizes of 8, 16, 32, 64, 128, 256), their kernels (with sizes from 2 to 20), and pooling layers (with sizes of 2, 3, 4, 5). The experiment investigating revealed the optimal topology containing three convolutional layers of sizes (256, 128, 64 in sequence with kernel sizes of 12, 10, 10) and pooling layers of sizes (2, 2, 2). Various LSTM auto-encoders were tested by stacking LSTM layers with different numbers of neurons (16, 32, 64, 128, 256, or 512). However, deeper architectures underperformed shallow auto-encoders.Figure A1, Figure A2 and Figure A3 (Appendix A) illustrate the topologies and sizes of layers of all three previously described auto-encoders (Feed-forward, LSTM, and 1D CNN). Figure A1 (Appendix A) represents feed-forward auto-encoder with a flattened input of 4096, one latent layer size of 512; Figure A2 (Appendix A) represents a 1D CNN auto-encoder. Kernel sizes of encoder are 12, 10, 10; kernel sizes of decoder are 1, 1, 1, 1; rectified linear units (ReLU) activation function used for convolutional layers. Figure A3 (Appendix A) represents the LSTM auto-encoder with one LSTM layer of size 512.Encoders were later combined with a simple feed-forward neural network (FFNN) illustrated in Figure A4 (Appendix A). All encoders output a 1 × 512 vector and therefore are compatible with the illustrated FFNN. Encoder’s weights were set to be untrainable except for the weights of the added neural network. In essence, different types of encoders take in an uncompressed ECFP encoding (with a size of 8 × 512) and produce (output) a compressed representation (with a size 1 × 512) which is an input of a second (i.e., FFNN) performing label classification (task No. 1) or prediction of the ratio between solvents (task No. 2). The only difference in the ratio prediction task is that the output layer size is set to 1 neuron (instead of 20) because it corresponds to a single numeric value; also, a linear activation function is used instead of a sigmoid. The encoders were kept stable in this stage, while different topologies of the FFNN were investigated. These investigations involved the addition/removal of fully connected layers with different sizes equal to 16, 32, 64, 128, 256, 512. The determined optimal topology (able to work well with all types of encoders) is illustrated in Figure A4 (Appendix A): it contains three fully connected layers.
5.2. Supervised Machine-Learning Approach
- Feed-Forward Neural Network (FFNN) is a simple DNN type in which information moves from input to output nodes without any loops. Despite it being much simpler than its successors, FFNNs have been successfully used to explore and visualize chemical space [48]. However, FFNNs are adjusted to learn the relationships between independent variables, which theoretically makes them unsuitable for solving tasks. Despite it, we will use FFNN in our experiments as the baseline approach to be able to compare the results with more sophisticated topologies.
- Convolutional neural networks (CNNs) [49] are a type of deep learning network initially developed for image recognition to detect recurring spatial patterns in the data. Recently CNNs are used in many applications to cope with the data, with grid patterns. Graph CNNs have been applied for predicting drug-target interactions [50]. Widely used CNN architectures VGG-19, ResNet512, AlexNet, DenseNet-201 [51,52,53,54] were used for the prediction of cytotoxicity for 8 cancer cell lines. In addition, CNNs have been proven to effectively identify steroids via deep learning retention time modeling when analyzed using gas chromatography [55]. It is hypothesized that molecules with similar chemical substructures or functional groups can behave similarly and form patterns recognized and processed by convolutional layers. It explains why CNNs are suitable for our solving tasks as well. Since we are dealing with sequences of symbols, we deal with models with 1D convolutions.
- Long short-term memory (LSTM) [56] neural networks are suitable for training on sequences due to a memory cell and feedback connections between cells. Contrary to their predecessors Simple Recurrent Neural Networks (RNNs), LSTMs do not suffer from the vanishing gradient problem and; therefore, learning longer sequential dependencies is not challenging anymore. Additional internal mechanisms (so-called gates) in LSTMs control information flow and carry relevant information forward while processing the sequence. Carrying information from earlier time moments in a sequence bears significant meaning on a conceptual level for the interpretation of chemical symbol sequences. For example, O and H symbols separately are less indicative than compared to (OH), which symbolizes a hydroxyl group or symbols c1 of aromaticity notation compared to an entire sequence cc1ccccc1 where c1 denotes a start and end of an aromatic ring. Thus, the sequential character of the input data explains the selection of LSTMs for our experiments.
- Activation functions. All layers except for the output one use rectified linear units (ReLU) activation function, and it is done on purpose. This activation function has statistically outperformed sigmoid and Tanh activation functions in similar bioactivity modeling tasks [57]. Additionally, ReLU is faster to compute compared to non-linear activation functions. Moreover, it generalizes better and converges faster [58]. In our preliminary tests, the ReLU activation function led to more stable training and convergence; moreover, it outperformed the other tested functions: i.e., Tanh, sigmoid, SoftMax, SeLU.The last layer’s activation function type fully depends on the solving task. Prediction of solvents is a multilabel prediction problem where the target output contains multiple independent and binary variables. We have used a binary cross-entropy loss function that works best with a sigmoid activation function. For task No. 1, we have chosen the sigmoid activation function because (1) it is the only compatible function with binary cross-entropy loss function used for loss calculation (2) sigmoid function’s returns values from the range [0–1]. Therefore, it is easier for the network to classify each binary label. As for task No. 2 (which is used to predict the ratio), the linear activation function is selected. This function is the most common activation function used for regression problems because it returns an unbounded numerical value.
- Optimizers. Optimizer Adam is a popular algorithm due to its ability to adjust learning rate according to circumstances in contrast to classical algorithms such as stochastic gradient descent (SGD), maintaining a single learning rate during the whole training process. In our experiments, Adam was selected due to these reasons: (1) the convergence of the model is significantly faster compared to classical optimizers; (2) the learning rate is controlled and does not lead to volatile training, especially at the end.
- Batch size. The batch size is one more important hyperparameter that impacts training stability and speed. Since the datasets (in Section 4) we are using in our research cover chemical experimental data collected in a long period of time, the factor of the noise in the data cannot completely be ruled out. Typically, to avoid a volatile learning process, a larger batch size is chosen to smooth over noisy instances. On the other hand, larger batch sizes require more V-RAM and slow down the learning process. Out of several options, 256 was selected to be a good compromise for our case. as the most effective.
- Epochs. During one epoch, the entire training dataset is passed through the network once. Since the learning process in DNNs is iterative, updating weights with only one pass is not enough. To avoid the negative effect of overfitting, the training dataset was split (90% training and 10% validation), and models were trained on 40 epochs: this number allowed to equalize the performance of training and validation loss functions during the model training process.
6. Results
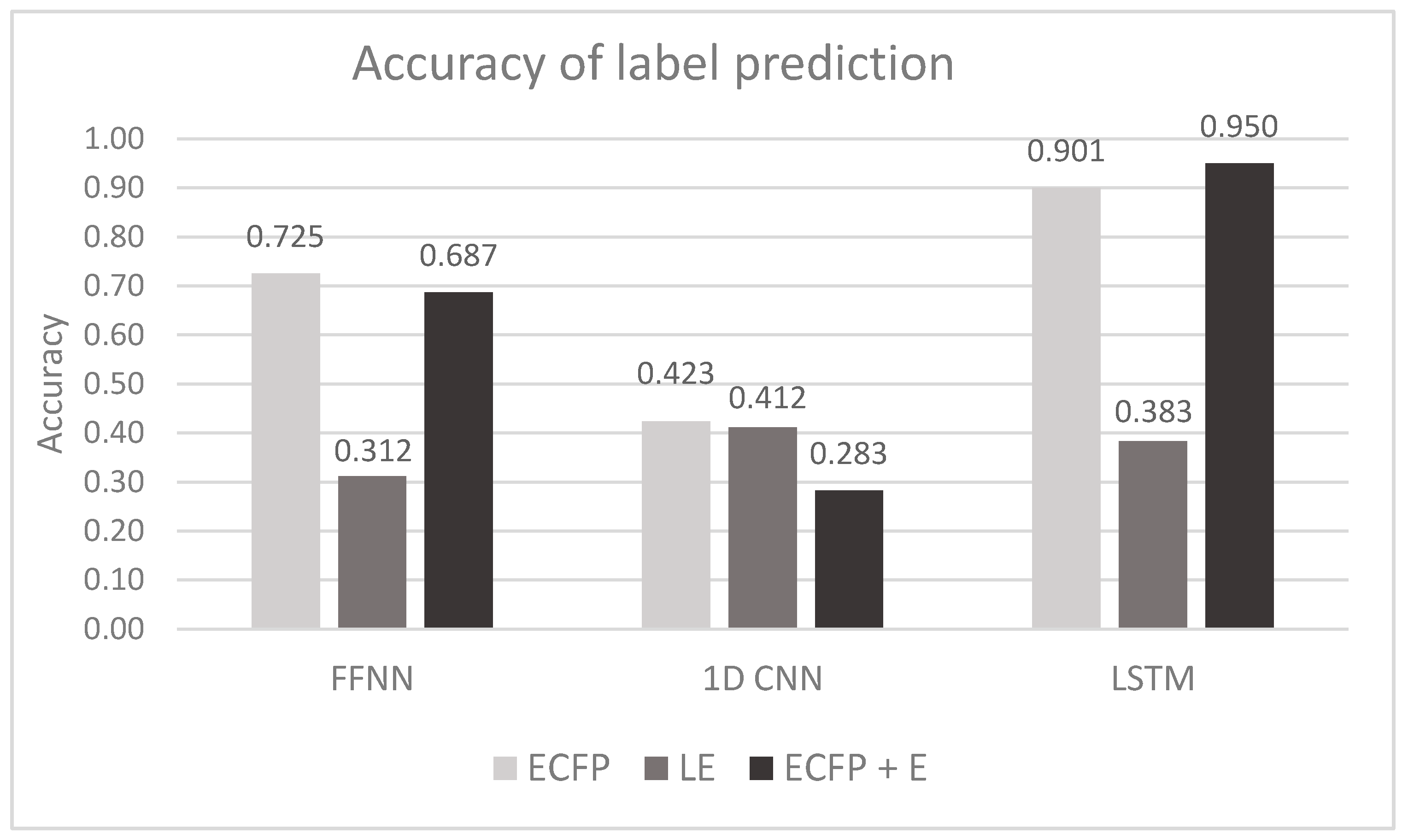
6.1. Task No. 1. Prediction of Solvents Used in Chromatographic Purification (Section 3)
6.2. Task No. 2: Prediction of Ratio between Solvents, When Their Number Is Two
7. Discussion
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A







References
- Ojima, I. Great Challenges in Organic Chemistry. Front. Chem. 2017, 5, 52. [Google Scholar] [CrossRef]
- Virshup, A.M.; Contreras-García, J.; Wipf, P.; Yang, W.; Beratan, D.N. Stochastic Voyages into Uncharted Chemical Space Produce a Representative Library of All Possible Drug-Like Compounds. J. Am. Chem. Soc. 2013, 135, 7296–7303. [Google Scholar] [CrossRef]
- Reymond, J.-L. The Chemical Space Project. Acc. Chem. Res. 2015, 48, 722–730. [Google Scholar] [CrossRef]
- Duch, W.; Setiono, R.; Zurada, J.M. Computational Intelligence Methods for Rule-Based Data Understanding. Proc. IEEE 2004, 92, 771–805. [Google Scholar] [CrossRef]
- Gani, R.; Jiménez-González, C.; Constable, D.J.C. Method for Selection of Solvents for Promotion of Organic Reactions. Comput. Chem. Eng. 2005, 29, 1661–1676. [Google Scholar] [CrossRef]
- Peiretti, F.; Brunel, J.M. Artificial Intelligence: The Future for Organic Chemistry? ACS Omega 2018, 3, 13263–13266. [Google Scholar] [CrossRef] [PubMed]
- Korovina, K.; Xu, S.; Kandasamy, K.; Neiswanger, W.; Poczos, B.; Schneider, J.; Xing, E. ChemBO: Bayesian Optimization of Small Organic Molecules with Synthesizable Recommendations. In Proceedings of the Twenty Third International Conference on Artificial Intelligence and Statistics (PMLR), Online. 26–28 August 2020; Volume 108, pp. 3393–3403. [Google Scholar]
- Genheden, S.; Thakkar, A.; Chadimová, V.; Reymond, J.-L.; Engkvist, O.; Bjerrum, E. AiZynthFinder: A Fast, Robust and Flexible Open-Source Software for Retrosynthetic Planning. J. Cheminform. 2020, 12, 70. [Google Scholar] [CrossRef] [PubMed]
- Tetko, I.V.; Karpov, P.; Van Deursen, R.; Godin, G. State-of-the-Art Augmented NLP Transformer Models for Direct and Single-Step Retrosynthesis. Nat. Commun. 2020, 11, 5575. [Google Scholar] [CrossRef]
- Brown, N.; Ertl, P.; Lewis, R.; Luksch, T.; Reker, D.; Schneider, N. Artificial Intelligence in Chemistry and Drug Design. J. Comput.-Aided Mol. Des. 2020, 34, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Grygorenko, O.O.; Volochnyuk, D.M.; Ryabukhin, S.V.; Judd, D.B. The Symbiotic Relationship between Drug Discovery and Organic Chemistry. Chem. Eur. J. 2019, 26, 1196–1237. [Google Scholar] [CrossRef] [PubMed]
- Patel, L.; Shukla, T.; Huang, X.; Ussery, D.W.; Wang, S. Machine Learning Methods in Drug Discovery. Molecules 2020, 25, 5277. [Google Scholar] [CrossRef]
- Ma, C.; Ren, Y.; Yang, J.; Ren, Z.; Yang, H.; Liu, S. Improved Peptide Retention Time Prediction in Liquid Chromatography through Deep Learning. Anal. Chem. 2018, 90, 10881–10888. [Google Scholar] [CrossRef] [PubMed]
- Moruz, L.; Käll, L. Peptide Retention Time Prediction. Mass Spec. Rev. 2016, 36, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Wang, L.; Wang, N.; Guo, F.; Liu, J.; Chen, Y.; Liu, J.; Zhao, Y.; Jiang, L. Separation of Organic Liquid Mixture by Flexible Nanofibrous Membranes with Precisely Tunable Wettability. NPG Asia Mater. 2016, 8, e334. [Google Scholar] [CrossRef]
- Coskun, O. Separation Tecniques: CHROMATOGRAPHY. North Clin. Istanbul. 2016, 3, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Chai, C.; Armarego, W.L.F. Purification of Laboratory Chemicals, 5th ed.; Butterworth-Heinemann Press: Woburn, MA, USA, 2014; ISBN 9780080515465. [Google Scholar]
- Bade, R.; Bijlsma, L.; Sancho, J.V.; Hernández, F. Critical Evaluation of a Simple Retention Time Predictor Based on LogKow as a Complementary Tool in the Identification of Emerging Contaminants in Water. Talanta 2015, 139, 143–149. [Google Scholar] [CrossRef]
- D’Archivio, A.A. Artificial Neural Network Prediction of Retention of Amino Acids in Reversed-Phase HPLC under Application of Linear Organic Modifier Gradients and/or PH Gradients. Molecules 2019, 24, 632. [Google Scholar] [CrossRef]
- Randazzo, G.M.; Tonoli, D.; Hambye, S.; Guillarme, D.; Jeanneret, F.; Nurisso, A.; Goracci, L.; Boccard, J.; Rudaz, S. Prediction of Retention Time in Reversed-Phase Liquid Chromatography as a Tool for Steroid Identification. Anal. Chim. Acta 2016, 916, 8–16. [Google Scholar] [CrossRef]
- Zhang, X.; Li, J.; Wang, C.; Song, D.; Hu, C. Identification of Impurities in Macrolides by Liquid Chromatography–Mass Spectrometric Detection and Prediction of Retention Times of Impurities by Constructing Quantitative Structure–Retention Relationship (QSRR). J. Pharm. Biomed. Anal. 2017, 145, 262–272. [Google Scholar] [CrossRef]
- Komsta, Ł.; Skibiński, R.; Berecka, A.; Gumieniczek, A.; Radkiewicz, B.; Radoń, M. Revisiting Thin-Layer Chromatography as a Lipophilicity Determination Tool—A Comparative Study on Several Techniques with a Model Solute Set. J. Pharm. Biomed. Anal. 2010, 53, 911–918. [Google Scholar] [CrossRef]
- Aalizadeh, R.; Thomaidis, N.S.; Bletsou, A.A.; Gago-Ferrero, P. Quantitative Structure–Retention Relationship Models To Support Nontarget High-Resolution Mass Spectrometric Screening of Emerging Contaminants in Environmental Samples. J. Chem. Inf. Model. 2016, 56, 1384–1398. [Google Scholar] [CrossRef]
- Haddad, P.R.; Taraji, M.; Szücs, R. Prediction of Analyte Retention Time in Liquid Chromatography. Anal. Chem. 2020, 93, 228–256. [Google Scholar] [CrossRef] [PubMed]
- Marlot, L.; Batteau, M.; Faure, K. Classification of Biphasic Solvent Systems According to Abraham Descriptors for Countercurrent Chromatography. J. Chromatogr. A 2020, 1617, 460820. [Google Scholar] [CrossRef]
- Winter, R.; Montanari, F.; Noé, F.; Clevert, D.-A. Learning Continuous and Data-Driven Molecular Descriptors by Translating Equivalent Chemical Representations. Chem. Sci. 2019, 10, 1692–1701. [Google Scholar] [CrossRef]
- Chakravarti, S.K. Distributed Representation of Chemical Fragments. ACS Omega 2018, 3, 2825–2836. [Google Scholar] [CrossRef]
- Su, Y.; Wang, Z.; Jin, S.; Shen, W.; Ren, J.; Eden, M.R. An Architecture of Deep Learning in QSPR Modeling for the Prediction of Critical Properties Using Molecular Signatures. AIChE J. 2019, 65, e16678. [Google Scholar] [CrossRef]
- Kotsias, P.-C.; Arús-Pous, J.; Chen, H.; Engkvist, O.; Tyrchan, C.; Bjerrum, E.J. Direct Steering of de Novo Molecular Generation with Descriptor Conditional Recurrent Neural Networks. Nat. Mach. Intell. 2020, 2, 254–265. [Google Scholar] [CrossRef]
- Xue, Y.; Li, Z.R.; Yap, C.W.; Sun, L.Z.; Chen, X.; Chen, Y.Z. Effect of Molecular Descriptor Feature Selection in Support Vector Machine Classification of Pharmacokinetic and Toxicological Properties of Chemical Agents. J. Chem. Inf. Comput. Sci. 2004, 44, 1630–1638. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Bombarelli, R.; Wei, J.N.; Duvenaud, D.; Hernández-Lobato, J.M.; Sánchez-Lengeling, B.; Sheberla, D.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Adams, R.P.; Aspuru-Guzik, A. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent. Sci. 2018, 4, 268–276. [Google Scholar] [CrossRef]
- Samanta, S.; O’Hagan, S.; Swainston, N.; Roberts, T.J.; Kell, D.B. VAE-Sim: A Novel Molecular Similarity Measure Based on a Variational Autoencoder. Molecules 2020, 25, 3446. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Ryu, S.; Kim, J.W.; Kim, W.Y. Molecular Generative Model Based on Conditional Variational Autoencoder for de Novo Molecular Design. J. Cheminform. 2018, 10, 31. [Google Scholar] [CrossRef]
- Xiong, Z.; Wang, D.; Liu, X.; Zhong, F.; Wan, X.; Li, X.; Li, Z.; Luo, X.; Chen, K.; Jiang, H.; et al. Pushing the Boundaries of Molecular Representation for Drug Discovery with the Graph Attention Mechanism. J. Med. Chem. 2019, 63, 8749–8760. [Google Scholar] [CrossRef]
- Rogers, D.; Hahn, M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Coley, C.W.; Barzilay, R.; Jaakkola, T.S.; Green, W.H.; Jensen, K.F. Prediction of Organic Reaction Outcomes Using Machine Learning. ACS Cent. Sci. 2017, 3, 434–443. [Google Scholar] [CrossRef]
- Feng, F.; Lai, L.; Pei, J. Computational Chemical Synthesis Analysis and Pathway Design. Front. Chem. 2018, 6, 199. [Google Scholar] [CrossRef]
- Sun, W.; Li, M.; Li, Y.; Wu, Z.; Sun, Y.; Lu, S.; Xiao, Z.; Zhao, B.; Sun, K. The Use of Deep Learning to Fast Evaluate Organic Photovoltaic Materials. Adv. Theory Simul. 2018, 2, 1800116. [Google Scholar] [CrossRef]
- Lowe, D. Chemical Reactions from US Patents (1976-Sep2016), 2017. [CrossRef]
- Weininger, D. SMILES, a Chemical Language and Information System. 1. Introduction to Methodology and Encoding Rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Keras Embedding Layer. Available online: https://keras.io/api/layers/core_layers/embedding/ (accessed on 10 December 2020).
- Gimeno, A.; Ojeda-Montes, M.; Tomás-Hernández, S.; Cereto-Massagué, A.; Beltrán-Debón, R.; Mulero, M.; Pujadas, G.; Garcia-Vallvé, S. The Light and Dark Sides of Virtual Screening: What Is There to Know? IJMS 2019, 20, 1375. [Google Scholar] [CrossRef]
- Wójcikowski, M.; Kukiełka, M.; Stepniewska-Dziubinska, M.M.; Siedlecki, P. Development of a Protein–Ligand Extended Connectivity (PLEC) Fingerprint and Its Application for Binding Affinity Predictions. Bioinformatics 2018, 35, 1334–1341. [Google Scholar] [CrossRef]
- Minami, T.; Okuno, Y. Number Density Descriptor on Extended-Connectivity Fingerprints Combined with Machine Learning Approaches for Predicting Polymer Properties. MRS Adv. 2018, 3, 2975–2980. [Google Scholar] [CrossRef]
- Ponting, D.J.; van Deursen, R.; Ott, M.A. Machine Learning Predicts Degree of Aromaticity from Structural Fingerprints. J. Chem. Inf. Model. 2020, 60, 4560–4568. [Google Scholar] [CrossRef] [PubMed]
- Friederich, P.; Krenn, M.; Tamblyn, I.; Aspuru-Guzik, A. Scientific intuition inspired by machine learning generated hypotheses. Mach. Learn. Sci. Technol. 2021, 2, 025027. [Google Scholar] [CrossRef]
- Alsenan, S.; Al-Turaiki, I.; Hafez, A. Autoencoder-Based Dimensionality Reduction for QSAR Modeling. In Proceedings of the 2020 3rd International Conference on Computer Applications & Information Security (ICCAIS), Riyadh, Saudi, 19–21 March 2020; 2020; pp. 1–4. [Google Scholar] [CrossRef]
- Karlov, D.S.; Sosnin, S.; Tetko, I.V.; Fedorov, M.V. Chemical Space Exploration Guided by Deep Neural Networks. RSC Adv. 2019, 9, 5151–5157. [Google Scholar] [CrossRef]
- Indolia, S.; Goswami, A.K.; Mishra, S.P.; Asopa, P. Conceptual Understanding of Convolutional Neural Network-A Deep Learning Approach. Procedia Comput. Sci. 2018, 132, 679–688. [Google Scholar] [CrossRef]
- Torng, W.; Altman, R.B. Graph Convolutional Neural Networks for Predicting Drug-Target Interactions. J. Chem. Inf. Model. 2019, 59, 4131–4149. [Google Scholar] [CrossRef]
- Guerra, E.; de Lara, J.; Malizia, A.; Díaz, P. Supporting User-Oriented Analysis for Multi-View Domain-Specific Visual Languages. Inf. Softw. Technol. 2009, 51, 769–784. [Google Scholar] [CrossRef]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Deep residual learning for image recognition. arXiv 2015, arXiv:1512.03385. [Google Scholar]
- Krizhevsky, A.; Sutskever, I.; Hinton, G.E. ImageNet Classification with Deep Convolutional Neural Networks. Commun. ACM 2017, 60, 84–90. [Google Scholar] [CrossRef]
- Huang, G.; Liu, Z.; van der Maaten, L.; Weinberger, K.Q. Densely connected convolutional networks. arXiv 2016, arXiv:1608.06993. [Google Scholar]
- Randazzo, G.M.; Bileck, A.; Danani, A.; Vogt, B.; Groessl, M. Steroid Identification via Deep Learning Retention Time Predictions and Two-Dimensional Gas Chromatography-High Resolution Mass Spectrometry. J. Chromatogr. A 2020, 1612, 460661. [Google Scholar] [CrossRef]
- Hochreiter, S.; Schmidhuber, J. Long Short-Term Memory. Neural Comput. 1997, 9, 1735–1780. [Google Scholar] [CrossRef] [PubMed]
- Koutsoukas, A.; Monaghan, K.J.; Li, X.; Huan, J. Deep-Learning: Investigating Deep Neural Networks Hyper-Parameters and Comparison of Performance to Shallow Methods for Modeling Bioactivity Data. J. Cheminform. 2017, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Zeiler, M.D.; Ranzato, M.; Monga, R.; Mao, M.; Yang, K.; Le, Q.V.; Nguyen, P.; Senior, A.; Vanhoucke, V.; Dean, J.; et al. On Rectified Linear Units for Speech Processing. In Proceedings of the 2013 IEEE International Conference on Acoustics, Speech and Signal Processing, Vancouver, BC, Canada, 26–31 May 2013; IEEE: Vancouver, CB, Canada, 2013. [Google Scholar] [CrossRef]
- Tensorflow. Available online: https://www.tensorflow.org (accessed on 10 December 2020).
- RDKit. Available online: http://www.rdkit.org (accessed on 9 October 2020).
- Le, N.Q.K.; Do, D.T.; Chiu, F.-Y.; Yapp, E.K.Y.; Yeh, H.-Y.; Chen, C.-Y. XGBoost Improves Classification of MGMT Promoter Methylation Status in IDH1 Wildtype Glioblastoma. J. Pers. Med. 2020, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- Le, N.Q.K.; Do, D.T.; Hung, T.N.K.; Lam, L.H.T.; Huynh, T.-T.; Nguyen, N.T.K. A Computational Framework Based on Ensemble Deep Neural Networks for Essential Genes Identification. Int. J. Mol. Sci. 2020, 21, 9070. [Google Scholar] [CrossRef] [PubMed]
- Duvenaud, D.; Maclaurin, D.; Aguilera-Iparraguirre, J.; Gómez-Bombarelli, R.; Hirzel, T.; Aspuru-Guzik, A.; Adams, R.P. Convolutional Networks on Graphs for Learning Molecular Fingerprints. arXiv 2015, arXiv:1512.03385. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Raw Text | Solvents | Ratio |
|---|---|---|
| the residue is chromatographed on silica gel with hexane/ethyl acetate 9:1 | Hexane, ethyl acetate | 9:1 |
| the residue was purified by column chromatography on silicagel with ethylacetate/methanol (1:1) | Ethyl acetate, methanol | 1:1 |
| the residue was chromatographed rapidly on 300 g of silica gel 60 eluting with ethyl acetate/hexane (2:3 parts by volume) | Hexane, ethyl acetate | 2:3 |
| Dataset 1 (DS1) | |
|---|---|
| Input | Output (Labels) |
| OCc(cc1)cc2c1OCO2.BrCC = CCBr.[Na].BrCC = CCOCc(cc1)c1OCO2 | Hexane, ethyl acetate |
| Dataset 2 (DS2) | |
| Input | Output (Ratio) |
| OCc(cc1)cc2c1OCO2.BrCC = CCBr.[Na].BrCC = CCOCc(cc1)c1OCO2 | 2:3 |
| Class Label | Training Subset (Number of Instances) | Testing Subset (Number of Instances) | Total (Number of Instances) |
|---|---|---|---|
| Ethyl acetate | 255,251 | 28,361 | 283,612 |
| Hexane | 155,858 | 17,318 | 173,175 |
| Dichloromethane | 85,643 | 9516 | 95,159 |
| Methanol | 84,626 | 9403 | 94,029 |
| Chloroform | 31,582 | 3509 | 35,091 |
| Petroleum ether | 16,964 | 1885 | 18,849 |
| Diethyl ether | 7943 | 883 | 8825 |
| Toluene | 7636 | 848 | 8484 |
| Acetone | 6139 | 682 | 6821 |
| Ethanol | 3424 | 380 | 3804 |
| Dataset 2 | |||
|---|---|---|---|
| Training Subset | Testing Subset | Total | |
| Numb. of instances | 245,096 | 27,233 | 272,329 |
| Mean | 0.356 | 0.355 | 0.356 |
| Standard deviation | 0.306 | 0.304 | 0.306 |
| Minimum value | 0.001 | 0.001 | 0.001 |
| 25% quartile | 0.100 | 0.100 | 0.100 |
| 50% quartile | 0.250 | 0.250 | 0.250 |
| 75% quartile | 0.500 | 0.500 | 0.500 |
| Maximum value | 0.998 | 0.998 | 0.998 |
| Vectorization | Training Dataset | FFNN | 1D CNN | LSTM |
|---|---|---|---|---|
| ECFP | Accuracy Precision Recall F1-score | 0.725 ± 0.002 | 0.423 ± 0.002 | 0.901 ± 0.001 |
| 0.899 ± 0.002 | 0.686 ± 0.004 | 0.960 ± 0.002 | ||
| 0.862 ± 0.003 | 0.709 ± 0.004 | 0.962 ± 0.001 | ||
| 0.697 ± 0.003 | 0.880 ± 0.004 | 0.961 ± 0.002 | ||
| LE | Accuracy Precision Recall F1-score | 0.312 ± 0.005 | 0.412 ± 0.012 | 0.383 ± 0.019 |
| 0.605 ± 0.004 | 0.687 ± 0.004 | 0.628 ± 0.007 | ||
| 0.603 ± 0.006 | 0.708 ± 0.003 | 0.683 ± 0.004 | ||
| 0.654 ± 0.005 | 0.604 ± 0.004 | 0.654 ± 0.005 | ||
| ECFP + E | Accuracy Precision Recall F1-score | 0.687 ± 0.002 | 0.283 ± 0.002 | 0.950 ± 0.001 |
| 0.845 ± 0.004 | 0.558 ± 0.004 | 0.977 ± 0.002 | ||
| 0.807 ± 0.006 | 0.591 ± 0.011 | 0.979 ± 0.002 | ||
| 0.574 ± 0.005 | 0.825 ± 0.007 | 0.978 ± 0.002 |
| Vectorization | Training Dataset | FFNN | 1D CNN | LSTM |
|---|---|---|---|---|
| ECFP | R-squared Pearson R MSE | 0.890 ± 0.001 | 0.568 ± 0.002 | 0.885 ± 0.001 |
| 0.941 ± 0.005 | 0.691 ± 0.001 | 0.941 ± 0.001 | ||
| 0.013 ± 0.001 | 0.040 ± 0.001 | 0.010 ± 0.001 | ||
| LE | R-squared Pearson R MSE | 0.200 ± 0.002 | 0.226 ± 0.004 | 0.143 ± 0.003 |
| 0.459 ± 0.001 | 0.475 ± 0.004 | 0.372 ± 0.002 | ||
| 0.080 ± 0.001 | 0.072 ± 0.001 | 0.084 ± 0.003 | ||
| ECFP + E | R-squared Pearson R MSE | 0.701 ± 0.003 | 0.847 ± 0.002 | 0.982 ± 0.001 |
| 0.831 ± 0.003 | 0.896 ± 0.007 | 0.984 ± 0.001 | ||
| 0.026 ± 0.002 | 0.013 ± 0.001 | 0.002 ± 0.001 |
| Class Label | Precision | Ratio | F1-Score | Number of Examples |
|---|---|---|---|---|
| Chloroform | 0.976 | 0.955 | 0.966 | 3424 |
| Dichloromethane | 0.980 | 0.956 | 0.968 | 9626 |
| Ethyl acetate | 0.983 | 0.990 | 0.986 | 28,472 |
| Diethyl ether | 0.921 | 0.951 | 0.936 | 834 |
| Hexane | 0.969 | 0.979 | 0.974 | 17,216 |
| Petroleum ether | 0.956 | 0.947 | 0.952 | 1836 |
| Acetone | 0.964 | 0.939 | 0.951 | 680 |
| Toluene | 0.974 | 0.935 | 0.954 | 817 |
| Methanol | 0.979 | 0.964 | 0.972 | 9558 |
| Ethanol | 0.949 | 0.967 | 0.958 | 354 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vaškevičius, M.; Kapočiūtė-Dzikienė, J.; Šlepikas, L. Prediction of Chromatography Conditions for Purification in Organic Synthesis Using Deep Learning. Molecules 2021, 26, 2474. https://doi.org/10.3390/molecules26092474
Vaškevičius M, Kapočiūtė-Dzikienė J, Šlepikas L. Prediction of Chromatography Conditions for Purification in Organic Synthesis Using Deep Learning. Molecules. 2021; 26(9):2474. https://doi.org/10.3390/molecules26092474
Chicago/Turabian StyleVaškevičius, Mantas, Jurgita Kapočiūtė-Dzikienė, and Liudas Šlepikas. 2021. "Prediction of Chromatography Conditions for Purification in Organic Synthesis Using Deep Learning" Molecules 26, no. 9: 2474. https://doi.org/10.3390/molecules26092474
APA StyleVaškevičius, M., Kapočiūtė-Dzikienė, J., & Šlepikas, L. (2021). Prediction of Chromatography Conditions for Purification in Organic Synthesis Using Deep Learning. Molecules, 26(9), 2474. https://doi.org/10.3390/molecules26092474