
Monitoring of the Pre-Equilibrium Step in the Alkyne Hydration Reaction Catalyzed by Au(III) Complexes: A Computational Study Based on Experimental Evidences

 ,
,  ,
,  and
and
Abstract
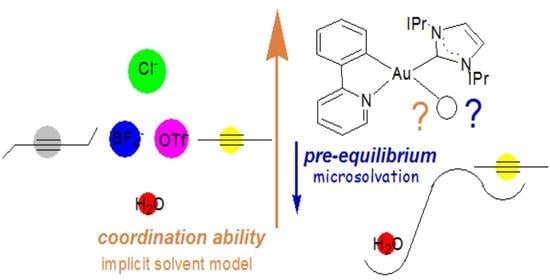
1. Introduction
2. Computational Details
3. Results and Discussion
3.1. Experimental Evidence on the Coordination Ability of [(ppy)Au(IPr)]2+
3.2. Coordination Ability of the [(ppy)Au(IPr)]2+ Fragment
3.3. Ligand Effect on the Coordination Ability: Alkynes vs. Water
3.4. Substitution of H2O With the Substrate: The Pre-Equilibrium Step
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Obradors, C.; Echavarren, A.M. Intriguing mechanistic labyrinths in gold(i) catalysis. Chem. Commun. 2014, 50, 16–28. [Google Scholar] [CrossRef]
- Wang, Y.-M.; Lackner, A.D.; Toste, F.D. Development of Catalysts and Ligands for Enantioselective Gold Catalysis. Acc. Chem. Res. 2014, 47, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Hashmi, A.S.K. Dual Gold Catalysis. Acc. Chem. Res. 2014, 47, 864–876. [Google Scholar] [CrossRef] [PubMed]
- Yeom, H.-S.; Shin, S. Catalytic Access to α-Oxo Gold Carbenes by N–O Bond Oxidants. Acc. Chem. Res. 2014, 47, 966. [Google Scholar] [CrossRef]
- Zhang, L. A Non-Diazo Approach to α-Oxo Gold Carbenes via Gold-Catalyzed Alkyne Oxidation. Acc. Chem. Res. 2014, 47, 877–888. [Google Scholar] [CrossRef] [PubMed]
- Qian, D.; Zhang, J. Gold-catalyzed cyclopropanation reactions using a carbenoid precursor toolbox. Chem. Soc. Rev. 2014, 44, 677–698. [Google Scholar] [CrossRef] [PubMed]
- Dorel, R.; Echavarren, A.M. Gold(I)-Catalyzed Activation of Alkynes for the Construction of Molecular Complexity. Chem. Rev. 2015, 115, 9028–9072. [Google Scholar] [CrossRef] [PubMed]
- Pflästerer, D.; Hashmi, A.S.K. Gold catalysis in total synthesis–recent achievements. Chem. Soc. Rev. 2016, 45, 1331–1367. [Google Scholar] [CrossRef]
- Alyabyev, S.B.; Beletskaya, I.P. Gold as a catalyst. Part I. Nucleophilic addition to the triple bond. Russ. Chem. Rev. 2017, 86, 689–749. [Google Scholar] [CrossRef]
- Alyabyev, S.B.; Beletskaya, I.P. Gold as a catalyst. Part II. Alkynes in the Reactions of Carbon–Carbon Bond Formation. Russ. Chem. Rev. 2018, 87, 948–1047. [Google Scholar] [CrossRef]
- Nijamudheen, A.; Datta, A. Gold-Catalyzed Cross-Coupling Reactions: An Overview of Design Strategies, Mechanistic Studies, and Applications. Chem. Eur. J. 2020, 26, 1442–1487. [Google Scholar] [CrossRef]
- Pažický, M.; Loos, A.; Ferreira, M.J.; Serra, D.; Vinokurov, N.; Rominger, F.; Jékel, C.; Hashmi, A.S.K.; Limbach, M. Synthesis, Reactivity, and Electrochemical Studies of Gold(I) and Gold(III) Complexes Supported byN-Heterocyclic Carbenes and Their Application in Catalysis. Organometallics 2010, 29, 4448–4458. [Google Scholar] [CrossRef]
- Wang, D.; Yang, Y.; Huang, R.; Wang, L.; Wan, H. Synthesis of Allenes through Triazole Gold(III) Catalyzed Rearrangement of Propargyl Vinyl Ethers. J. Chem. Res. 2016, 40, 645–647. [Google Scholar] [CrossRef]
- O’Neill, J.A.T.; Rosair, G.M.; Lee, A.-L. Gold(iii)–oxo complexes as catalysts in intramolecular hydroamination. Catal. Sci. Technol. 2012, 2, 1818–1821. [Google Scholar] [CrossRef]
- Wang, G.; Liu, X.; Chen, Y.; Yang, J.; Li, J.; Lin, L.; Feng, X. Diastereoselective and Enantioselective Alleno-aldol Reaction of Allenoates with Isatins to Synthesis of Carbinol Allenoates Catalyzed by Gold. ACS Catal. 2016, 6, 2482–2486. [Google Scholar] [CrossRef]
- Montanel-Pérez, S.; Herrera, R.P.; Laguna, A.; Villacampa, M.D.; Gimeno, M.C. The fluxional amine gold(iii) complex as an excellent catalyst and precursor of biologically active acyclic carbenes. Dalton Trans. 2015, 44, 9052–9062. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-Y.; Horibe, T.; Jacobsen, C.B.; Toste, F.D. Stable gold(III) catalysts by oxidative addition of a carbon–carbon bond. Nat. Cell Biol. 2015, 517, 449–454. [Google Scholar] [CrossRef]
- Cui, J.-F.; Ko, H.-M.; Shing, K.-P.; Deng, J.-R.; Lai, N.C.-H.; Wong, M.-K. C,O-Chelated BINOL/Gold(III) Complexes: Synthesis and Catalysis with Tunable Product Profiles. Angew. Chem. Int. Ed. 2017, 56, 3074–3079. [Google Scholar] [CrossRef]
- Rekhroukh, F.; Blons, C.; Estevez, L.; Mallet-Ladeira, S.; Miqueu, K.; Amgoune, A.; Bourissou, D. Gold(III)-Arene Complexes by Insertion of Olefins into Gold-Aryl Bonds. Chem. Sci. 2017, 8, 4539–4545. [Google Scholar] [CrossRef] [PubMed]
- Teci, M.; Hueber, D.; Pale, P.; Toupet, L.; Blanc, A.; Brenner, E.; Matt, D. Metal Confinement through N -(9-Alkyl)fluorenyl-Substituted N-Heterocyclic Carbenes and Its Consequences in Gold-Catalysed Reactions Involving Enynes. Chem. A Eur. J. 2017, 23, 7809–7818. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.-F.; Deng, J.-R.; Wei, X.-Q.; Shao, S.-P.; Xiang, D.-P.; Wong, M.-K. Visual detection of formaldehyde by highly selective fluorophore labeling via gold(iii) complex-mediated three-component coupling reaction. Org. Biomol. Chem. 2015, 13, 7408–7411. [Google Scholar] [CrossRef]
- Hui, T.-W.; Cui, J.-F.; Wong, M.-K. Modular Synthesis of Propargylamine Modified Cyclodextrins by a Gold(iii)-Catalyzed Three-Component Coupling Reaction. RSC Adv. 2017, 7, 14477–14480. [Google Scholar] [CrossRef]
- Lee, J.S.; Kapustin, E.A.; Pei, X.; Llopis, S.; Yaghi, O.M.; Toste, F.D. Architectural Stabilization of a Gold(III) Catalyst in Metal-Organic Frameworks. Chem 2020, 6, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Song, L.; Farshadfar, K.; Rudolph, M.; Rominger, F.; Oeser, T.; Ariafard, A.; Hashmi, A.S.K. Acyl Migration versus Epoxidation in Gold Catalysis: Facile, Switchable, and Atom-Economic Synthesis of Acylindoles and Quinoline Derivatives. Angew. Chem. Int. Ed. 2020, 59, 471–478. [Google Scholar] [CrossRef]
- Jiang, J.-J.; Cui, J.-F.; Yang, B.; Ning, Y.; Lai, N.C.-H.; Wong, M.-K. Chiral Cyclometalated Oxazoline Gold(III) Complex-Catalyzed Asymmetric Carboalkoxylation of Alkynes. Org. Lett. 2019, 21, 6289–6294. [Google Scholar] [CrossRef]
- Lo, V.K.-Y.; Kung, K.K.-Y.; Wong, M.-K.; Che, C.-M. Gold(III) (C^N) complex-catalyzed synthesis of propargylamines via a three-component coupling reaction of aldehydes, amines and alkynes. J. Organomet. Chem. 2009, 694, 583–591. [Google Scholar] [CrossRef]
- Li, G.; Zhang, L. Gold-Catalyzed Intramolecular Redox Reaction of Sulfinyl Alkynes: Efficient Generation of α-Oxo Gold Carbenoids and Application in Insertion into R-CO Bonds. Angew. Chem. Int. Ed. 2007, 46, 5156–5159. [Google Scholar] [CrossRef]
- Shaw, M.; Thakur, R.; Kumar, A. Gold(III)-Catalyzed Glycosylation Using Phenylpropiolate Glycosides: Phenylpropiolic Ac-id, an Easily Separable and Reusable Leaving Group. J. Org. Chem. 2019, 84, 589–605. [Google Scholar] [CrossRef] [PubMed]
- Hikawa, H.; Matsumoto, M.; Tawara, S.; Kikkawa, S.; Azumaya, I. Gold(III)/Sodium Diphenylphosphinobenzene-3-sulfonate (TPPMS) Catalyzed Dehydrative N-Benzylation of Electron-Deficient Anilines in Water. Synthesis 2019, 51, 2729–2736. [Google Scholar] [CrossRef]
- Xie, J.; Li, H.; Zhou, J.; Cheng, Y.; Zhu, C. A Highly Efficient Gold-Catalyzed Oxidative C-C Coupling from C-H Bonds Using Air as Oxidant. Angew. Chem. Int. Ed. Engl. 2012, 51, 1252–1255. [Google Scholar] [CrossRef]
- Chipman, A.; Gouranourimi, A.; Farshadfar, K.; Olding, A.; Yates, B.F.; Ariafard, A. A Computational Mechanistic Investi-gation into Reduction of Gold(III) Complexes by Amino Acid Glycine: A New Variant for Amine Oxidation. Chem. Eur. J. 2018, 24, 8361–8368. [Google Scholar] [CrossRef]
- Aguilar, D.; Contel, M.; Urriolabeitia, E.P. Mechanistic Insights into the One-Pot Synthesis of Propargylamines from Terminal Alkynes and Amines in Chlorinated Solvents Catalyzed by Gold Compounds and Nanoparticles. Chem. Eur. J. 2010, 16, 9287–9296. [Google Scholar] [CrossRef] [PubMed]
- Oliver-Meseguer, J.; Cabrero-Antonino, J.R.; Domínguez, I.; Leyva-Pérez, A.; Corma, A. Small Gold Clusters Formed in Solution Give Reaction Turnover Numbers of 10 at Room Temperature. Science 2012, 338, 1452. [Google Scholar] [CrossRef]
- Durović, M.D.; Bugarčić, Ž.D.; van Eldik, R. Stability and reactivity of gold compounds—From fundamental aspects to ap-plications. Coord. Chem. Rev. 2017, 338, 186–206. [Google Scholar] [CrossRef]
- Gaillard, S.; Slawin, A.M.Z.; Bonura, A.T.; Stevens, E.D.; Nolan, S.P. Synthetic and structural studies of [AuCl3(NHC)] complexes. Organometallics 2010, 29, 394–402. [Google Scholar] [CrossRef]
- de Frémont, P.; Singh, R.; Stevens, E.D.; Petersen, J.L.; Nolan, S.P. Synthesis, Characterization and Reactivity of N-Heterocyclic Carbene Gold(III) Complexes. Organometallics 2007, 26, 1376–1385. [Google Scholar] [CrossRef]
- Hashmi, A.S.K.; Blanco, M.C.; Fischer, D.; Bats, J.W. Gold Catalysis: Evidence for the In-situ Reduction of Gold(III) During the Cyclization of Allenyl Carbinols. Eur. J. Org. Chem. 2006, 2006, 1387–1389. [Google Scholar] [CrossRef]
- Kumar, R.; Nevado, C. Cyclometalated Gold(III) Complexes: Synthesis, Reactivity, and Physicochemical Properties. Angew. Chem. Int. Ed. 2017, 56, 1994–2015. [Google Scholar] [CrossRef] [PubMed]
- Gregori, L.; Sorbelli, D.; Belpassi, L.; Tarantelli, F.; Belanzoni, P. Alkyne Activation with Gold(III) Complexes: A Quantitative Assessment of the Ligand Effect by Charge-Displacement Analysis. Inorg. Chem. 2019, 58, 3115–3129. [Google Scholar] [CrossRef] [PubMed]
- Segato, J.; Del Zotto, A.; Belpassi, L.; Belanzoni, P.; Zuccaccia, D. Hydration of alkynes catalyzed by [Au(X)(L)(ppy)]X in the green solvent γ-valerolactone under acid-free conditions: The importance of the pre-equilibrium step. Catal. Sci. Technol. 2020, 10, 7757–7767. [Google Scholar] [CrossRef]
- Hintermann, L.; Labonne, A. Catalytic hydration of alkynes and its application in synthesis. ChemInform 2007, 8, 1121–1150. [Google Scholar] [CrossRef]
- Alonso, F.; Beletskaya, I.P.; Yus, M. Transition-Metal-Catalyzed Addition of Heteroatom−Hydrogen Bonds to Alkynes. Chem. Rev. 2004, 104, 3079–3160. [Google Scholar] [CrossRef]
- Ciancaleoni, G.; Belpassi, L.; Zuccaccia, D.; Tarantelli, F.; Belanzoni, P. Counterion Effect in the Reaction Mechanism of NHC Gold(I)-Catalyzed Alkoxylation of Alkynes: Computational Insight into Experiment. ACS Catal. 2015, 5, 803–814. [Google Scholar] [CrossRef]
- Gatto, M.; Belanzoni, P.; Belpassi, L.; Biasiolo, L.; Del Zotto, A.; Tarantelli, F.; Zuccaccia, D. Solvent-, Silver-, and Acid-Free NHC-Au-X Catalyzed Hydration of Alkynes. The Pivotal Role of the Counterion. ACS Catal. 2016, 6, 7363–7376. [Google Scholar] [CrossRef]
- D’Amore, L.; Ciancaleoni, G.; Belpassi, L.; Tarantelli, F.; Zuccaccia, D.; Belanzoni, P. Unraveling the Anion/Ligand Interplay in the Reaction Mechanism of Gold(I)-Catalyzed Alkoxylation of Alkynes. Organometallics 2017, 36, 2364–2376. [Google Scholar] [CrossRef]
- Gatto, M.; Baratta, W.; Belanzoni, P.; Belpassi, L.; Del Zotto, A.; Tarantelli, F.; Zuccaccia, D. Hydration and alkoxylation of alkynes catalyzed by NHC-Au-OTf. Green Chem. 2018, 20, 2125–2134. [Google Scholar] [CrossRef]
- Biasiolo, L.; Trinchillo, M.; Belanzoni, P.; Belpassi, L.; Busico, V.; Ciancaleoni, G.; D’Amora, A.; Macchioni, A.; Tarantelli, F.; Zuccaccia, D. Unexpected Anion Effect in the Alkoxylation of Alkynes Catalyzed by N-Heterocyclic Carbene (NHC) Cationic Gold Complexes. Chem. Eur. J. 2014, 20, 14594–14598. [Google Scholar] [CrossRef] [PubMed]
- Tolbatov, I.; Coletti, C.; Marrone, M.; Re, N. Insight into the substitution mechanism of antitumor Au(I) N-heterocyclic car-bene complexes by cysteine and selenocysteine. Inorg. Chem. 2020, 59, 3312–3320. [Google Scholar] [CrossRef] [PubMed]
- SCM. Theoretical Chemistry, ADF User’s Guide; Release 2016; Vrije Universiteit: Amsterdam, The Netherlands, 2016; Available online: http://www.scm.com (accessed on 30 March 2021).
- Fonseca Guerra, C.; Snijders, J.G.; te Velde, G.; Baerends, E.J. Towards an order-N DFT method. Theor. Chem. Acc. 1998, 99, 391–403. [Google Scholar] [CrossRef]
- Velde, G.T.; Bickelhaupt, F.M.; Baerends, E.J.; Guerra, C.F.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Swart, M.; Bickelhaupt, F.M. QUILD: QUantum-regions interconnected by local descriptions. J. Comput. Chem. 2007, 29, 724–734. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Klamt, A.; Schüürmann, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 1993, 2, 799–805. [Google Scholar] [CrossRef]
- Klamt, A. Conductor-like screening model for real solvents: A new approach to the quantitative calculation of salvation phe-nomena. J. Phys. Chem. 1995, 99, 2224–2235. [Google Scholar] [CrossRef]
- Klamt, A.; Jonas, V. Treatment of the outlying charge in continuum solvation models. J. Chem. Phys. 1996, 105, 9972–9981. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic regular two-component Hamiltonians. J. Chem. Phys. 1993, 99, 4597–4610. [Google Scholar] [CrossRef]
- van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic total energy using regular approximations. J. Chem. Phys. 1994, 101, 9783–9792. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Ehlers, A.; Baerends, E.J. Geometry optimizations in the zero order regular approximation for relativistic ef-fects. J. Chem. Phys. 1999, 110, 8943–8953. [Google Scholar] [CrossRef]
- Tlahuext-Aca, A.; Hopkinson, M.N.; Daniliuc, C.G.; Glorius, F. Oxidative Addition to Gold(I) by Photoredox Catalysis: Straightforward Access to Diverse (C,N)-Cyclometalated Gold(III) Complexes. Chem. Eur. J. 2016, 22, 11587–11592. [Google Scholar] [CrossRef]
- Yepes, D.; Neese, F.; List, B.; Bistoni, G. Unveiling the Delicate Balance of Steric and Dispersion Interactions in Organocatalysis Using High-Level Computational Methods. J. Am. Chem. Soc. 2020, 142, 3613–3625. [Google Scholar] [CrossRef]
- Bistoni, G.; Auer, A.A.; Neese, F. Understanding the Role of Dispersion in Frustrated Lewis Pairs and Classical Lewis Adducts: A Domain-Based Local Pair Natural Orbital Coupled Cluster Study. Chem. Eur. J. 2017, 23, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Sromek, A.W.; Rubina, M.; Gevorgyan, V. 1,2-halogen migration in haloallenyl ketones: Regiodivergent synthesis of halofurans. J. Am. Chem. Soc. 2005, 127, 10500–10501. [Google Scholar] [CrossRef]
- Morita, N.; Yasuda, A.; Shibata, M.; Ban, S.; Hashimoto, Y.; Okamoto, I.; Tamura, O. Gold(I)/(III)-catalyzed synthesis of cyclic ethers; valency-controlled cyclization modes. Org. Lett. 2015, 17, 2668–2671. [Google Scholar] [CrossRef]
- Crabtree, R.T. (Ed.) The Organometallic Chemistry of the Transition Metals; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014. [Google Scholar]
- Chambrier, I.; Rocchigiani, L.; Hughes, D.L.; Budzelaar, P.M.H.; Bochmann, M. Thermally Stable Gold(III) Alkene and Alkyne Complexes: Synthesis, Structures, and Assessment of the Trans-Influence on Gold-Ligand Bond Enthalpies. Chem. Eur. J. 2018, 24, 11467–11474. [Google Scholar] [CrossRef] [PubMed]
- Rocchigiani, L.; Fernandez-Cestau, J.; Agonigi, G.; Chambrier, I.; Budzelaar, P.H.M.; Bochmann, M. Gold(III) alkyne complexes: Bonding and reaction pathways. Angew. Chem. Int. Ed. Engl. 2017, 56, 13861–13865. [Google Scholar] [CrossRef]
- Sorbelli, D.; Belpassi, L.; Tarantelli, F.; Belanzoni, P. Ligand Effect on Bonding in Gold(III) Carbonyl Complexes. Inorg. Chem. 2018, 57, 6161–6175. [Google Scholar] [CrossRef] [PubMed]
- Macchioni, A. Ion Pairing in Transition-Metal Organometallic Chemistry. Chem. Rev. 2005, 105, 2039–2074. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cl− | OTf− | BF4− | 3-hexyne | 2-butyne | H2O | |
|---|---|---|---|---|---|---|
| ΔE | −54.3 | −34.3 | −24.7 | −31.4 | −27.4 | −15.2 |
| ΔH | −51.5 | −32.4 | −21.6 | −27.5 | −23.8 | −13.4 |
| ΔG | −44.0 | −17.5 | −12.5 | −15.9 | −12.6 | −0.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sabatelli, F.; Segato, J.; Belpassi, L.; Del Zotto, A.; Zuccaccia, D.; Belanzoni, P. Monitoring of the Pre-Equilibrium Step in the Alkyne Hydration Reaction Catalyzed by Au(III) Complexes: A Computational Study Based on Experimental Evidences. Molecules 2021, 26, 2445. https://doi.org/10.3390/molecules26092445
Sabatelli F, Segato J, Belpassi L, Del Zotto A, Zuccaccia D, Belanzoni P. Monitoring of the Pre-Equilibrium Step in the Alkyne Hydration Reaction Catalyzed by Au(III) Complexes: A Computational Study Based on Experimental Evidences. Molecules. 2021; 26(9):2445. https://doi.org/10.3390/molecules26092445
Chicago/Turabian StyleSabatelli, Flavio, Jacopo Segato, Leonardo Belpassi, Alessandro Del Zotto, Daniele Zuccaccia, and Paola Belanzoni. 2021. "Monitoring of the Pre-Equilibrium Step in the Alkyne Hydration Reaction Catalyzed by Au(III) Complexes: A Computational Study Based on Experimental Evidences" Molecules 26, no. 9: 2445. https://doi.org/10.3390/molecules26092445
APA StyleSabatelli, F., Segato, J., Belpassi, L., Del Zotto, A., Zuccaccia, D., & Belanzoni, P. (2021). Monitoring of the Pre-Equilibrium Step in the Alkyne Hydration Reaction Catalyzed by Au(III) Complexes: A Computational Study Based on Experimental Evidences. Molecules, 26(9), 2445. https://doi.org/10.3390/molecules26092445