
Redox Speciation of Vanadium in Estuarine Waters Using Improved Methodology Based on Anion Exchange Chromatography Coupled to HR ICP-MS System



Abstract
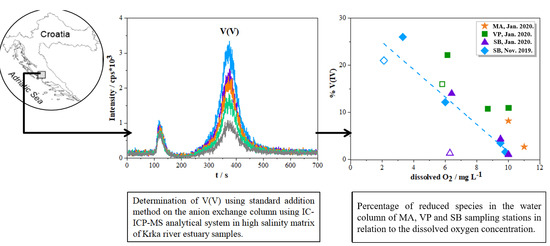
{kind=link}
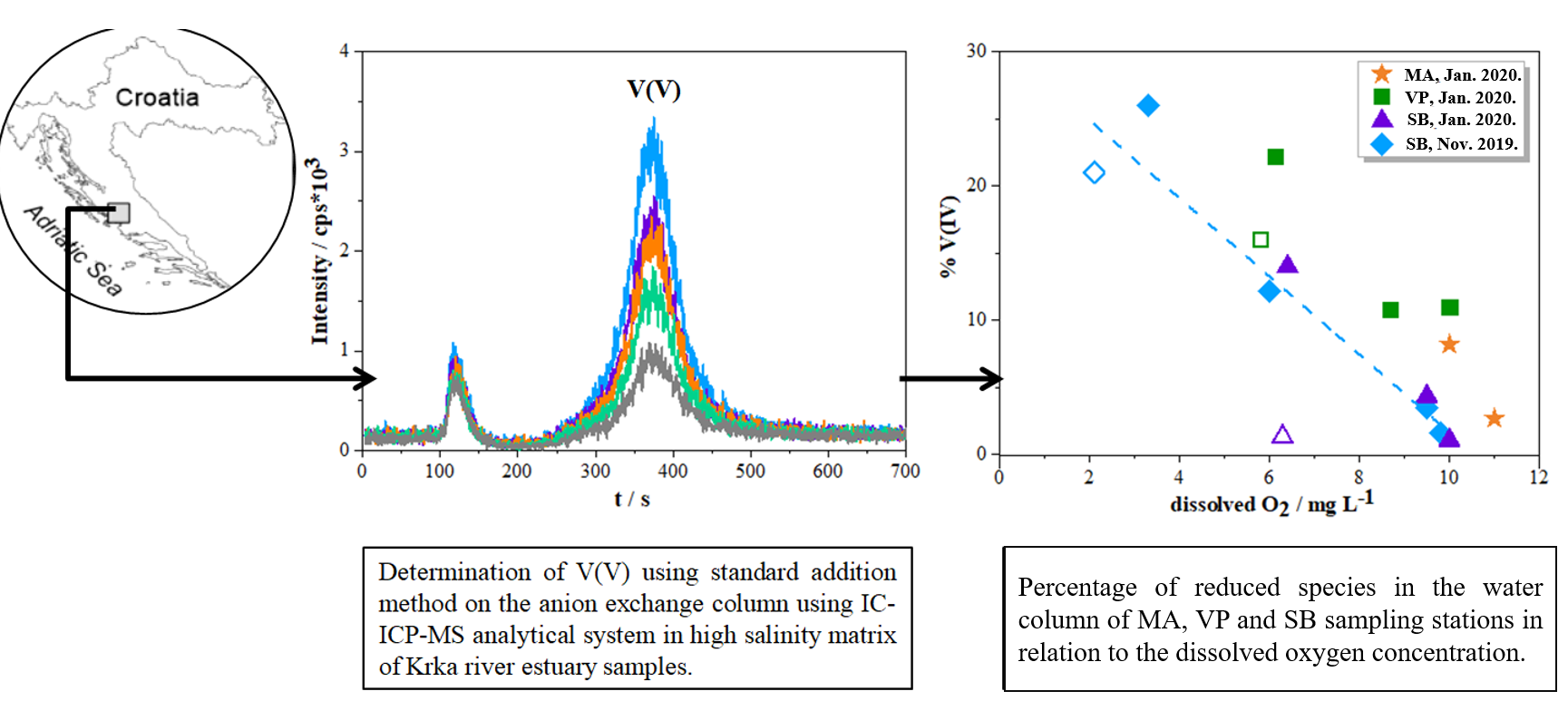
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Methodology Improvements
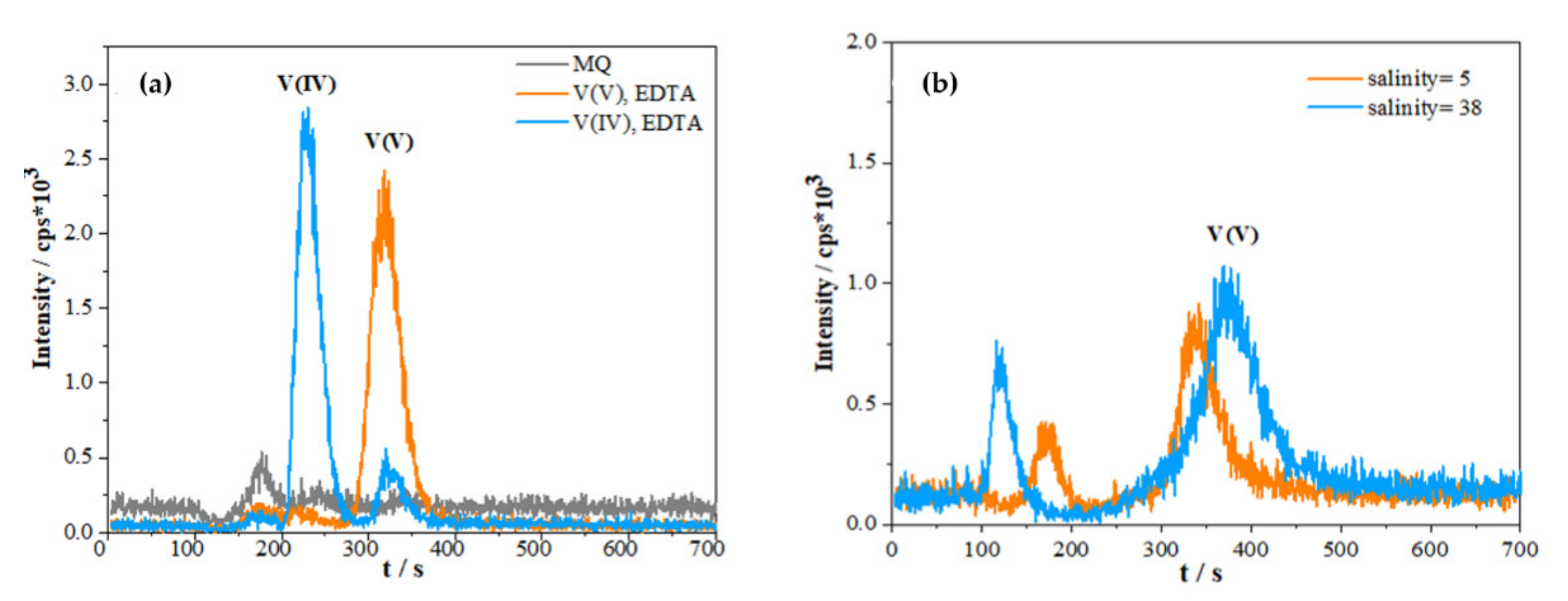
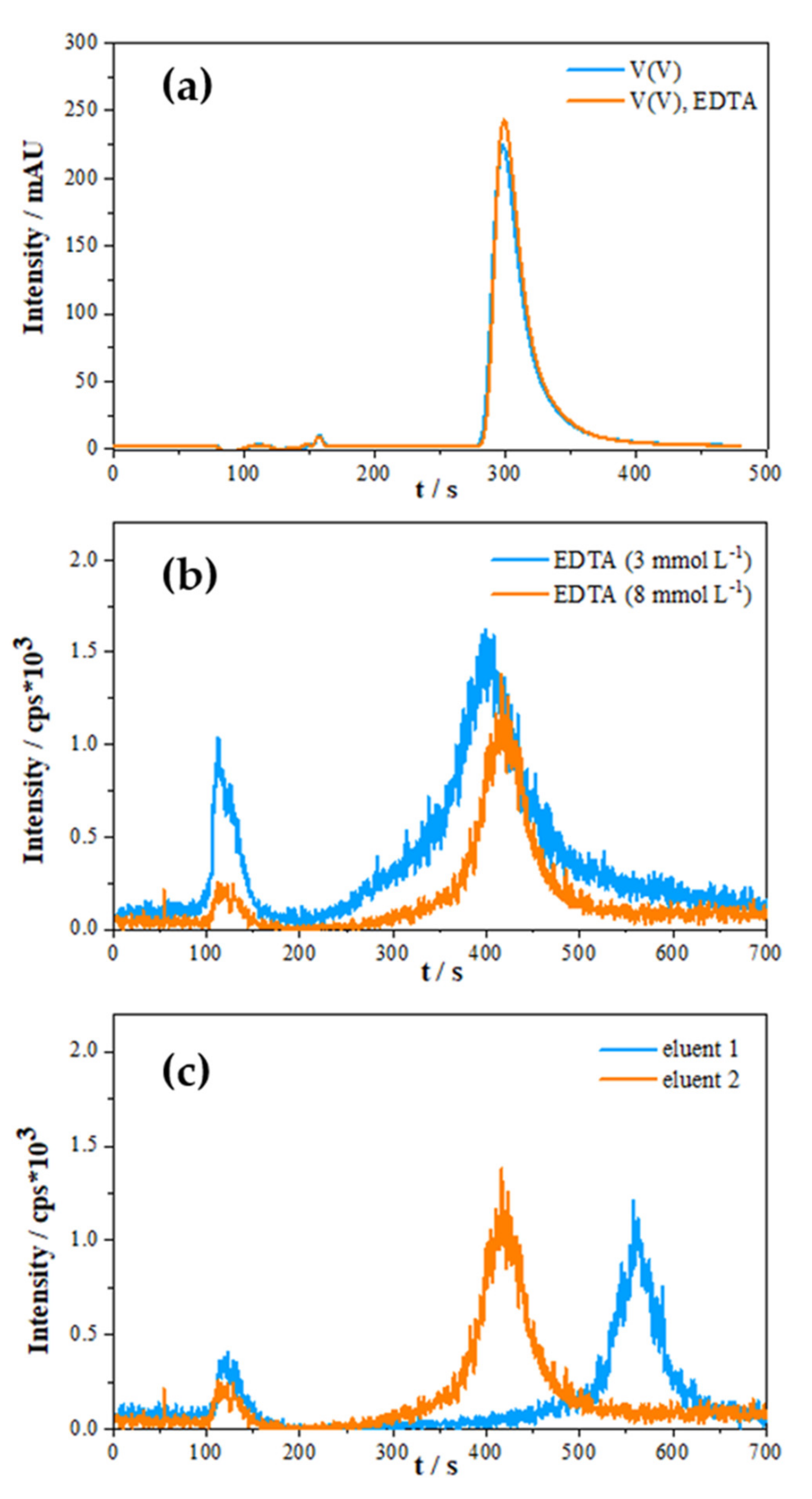
2.1.1. Effect of Sample Matrix
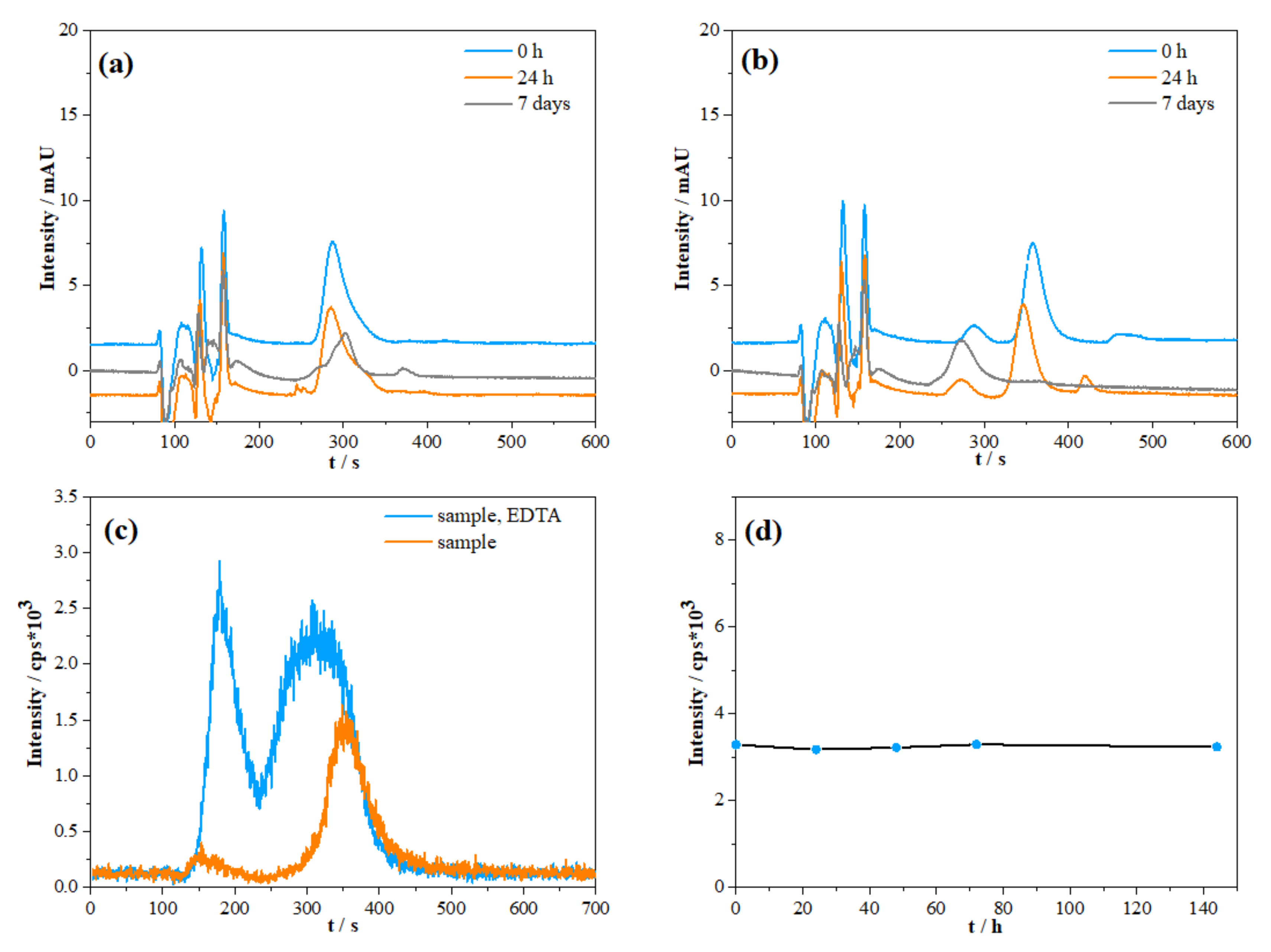
2.1.2. Species Preservation and Sample Pre-Treatment
2.1.3. Optimisation of Eluent Composition
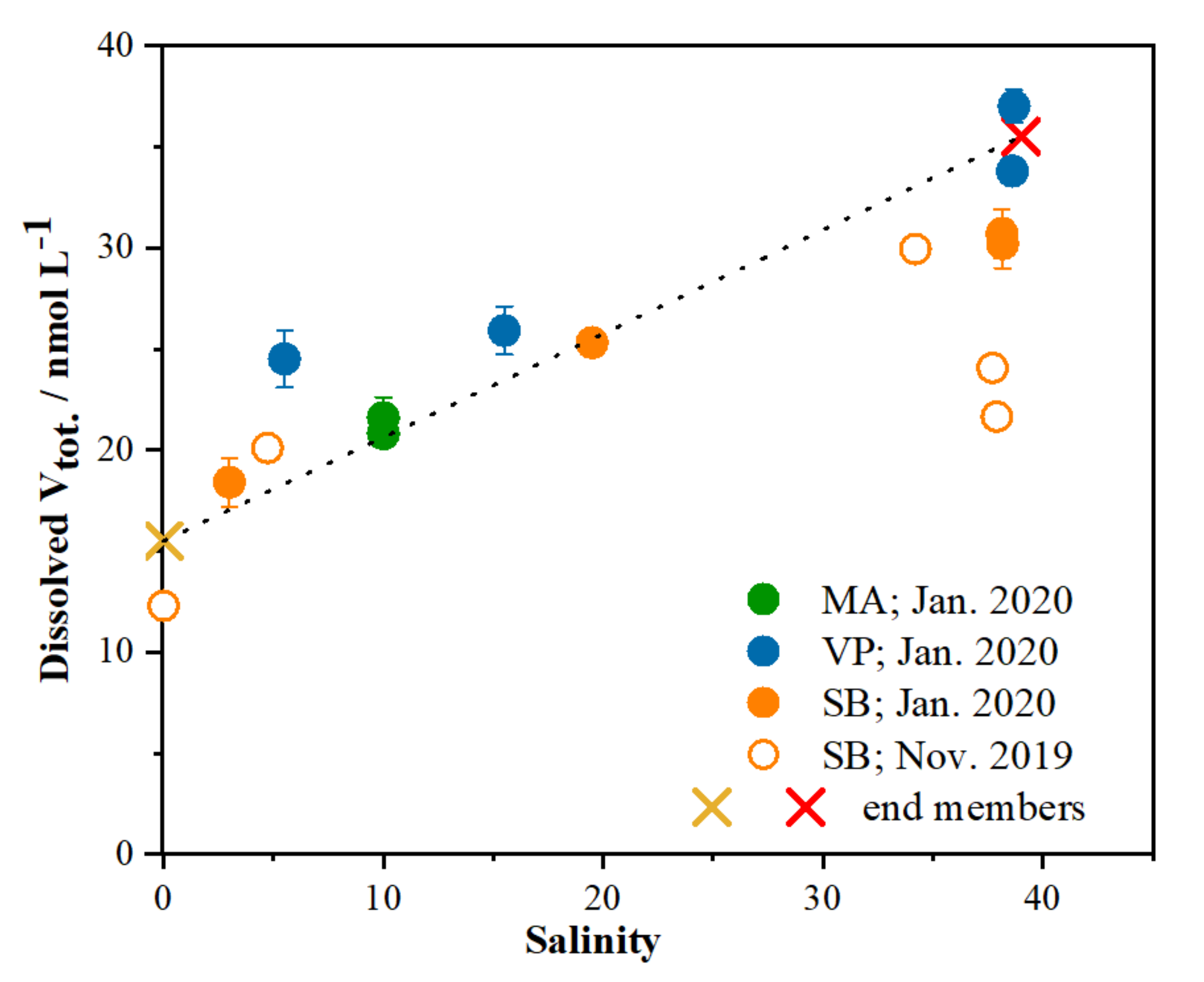
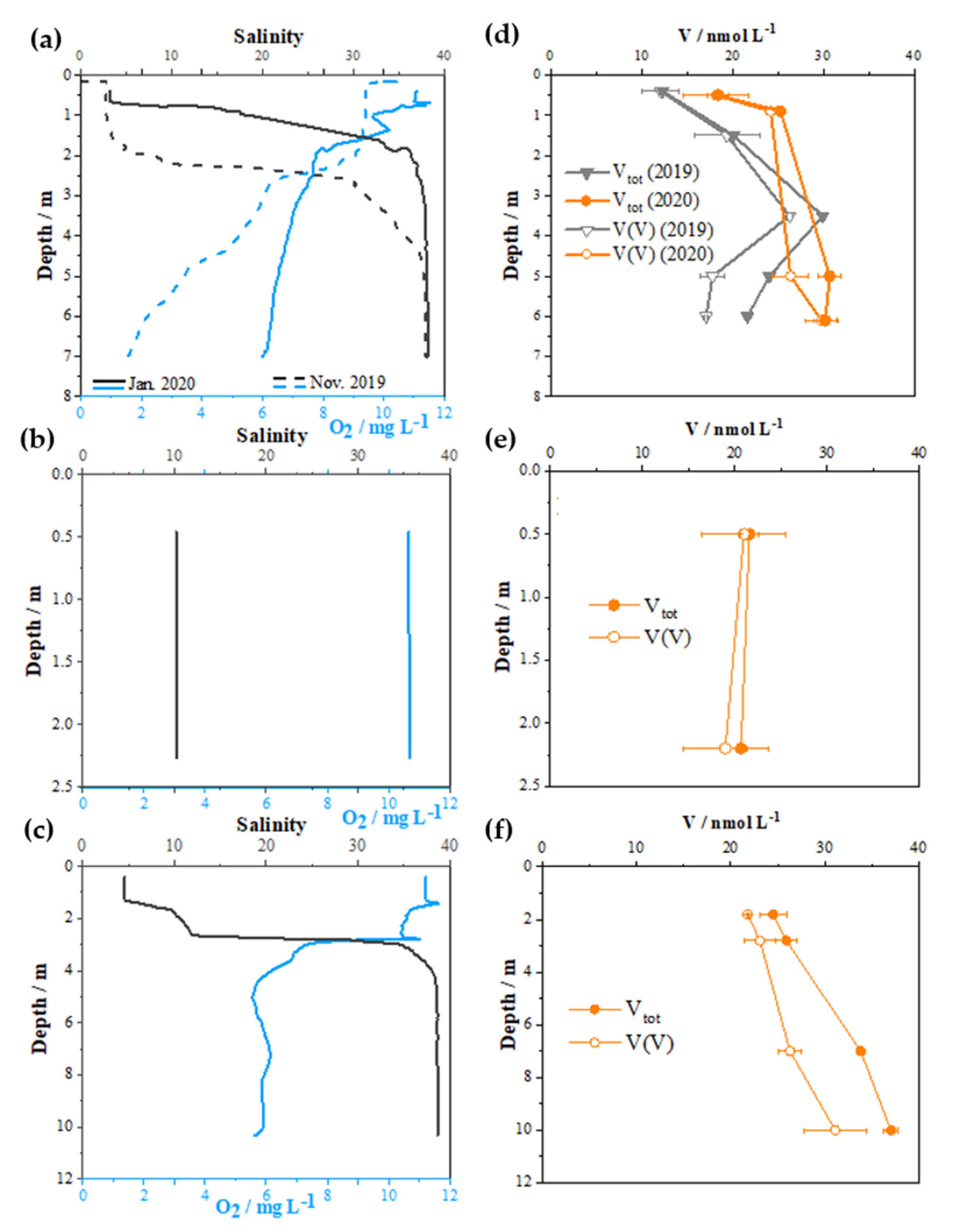
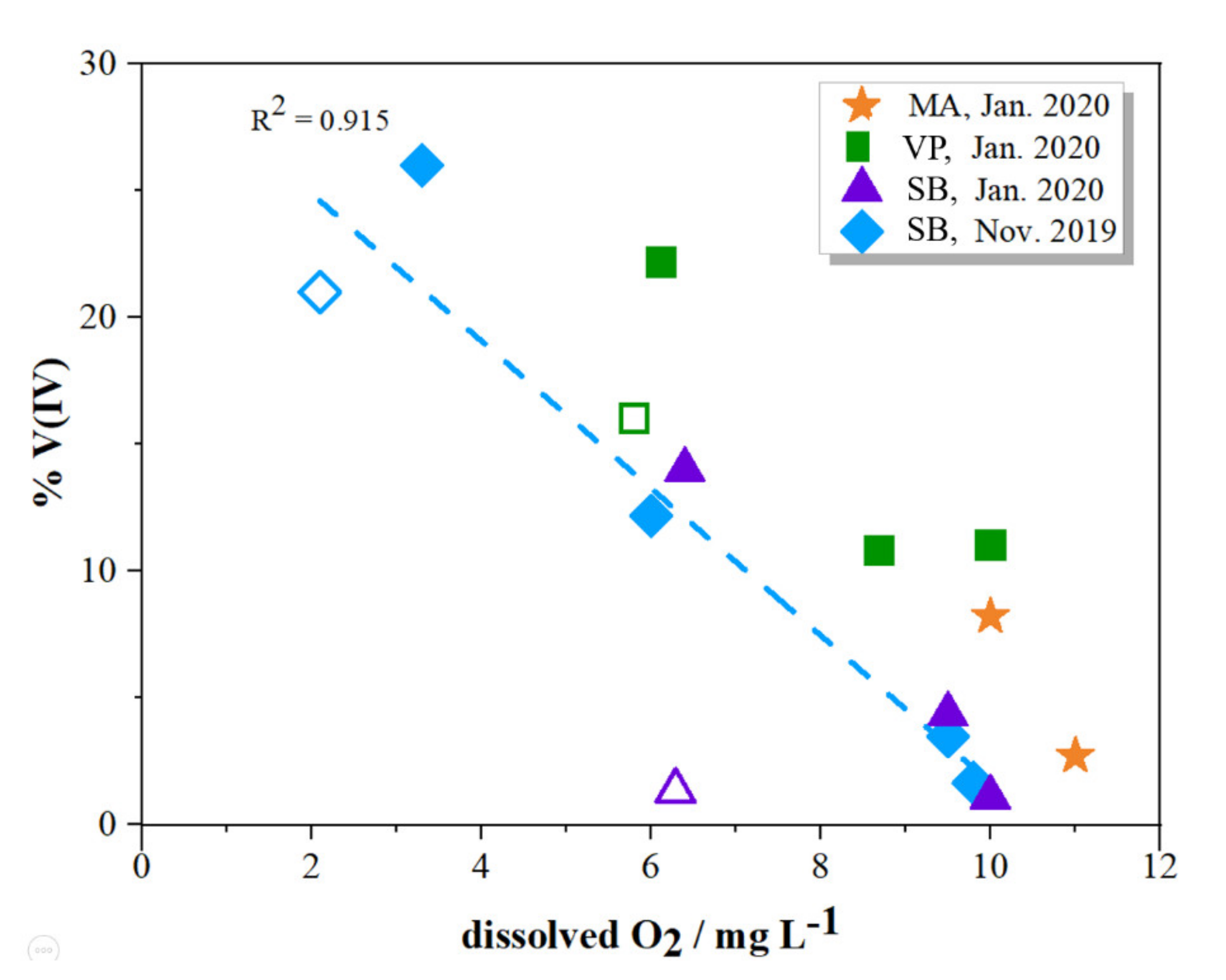
2.2. Distribution of V Redox Speciation in Krka River Estuary Samples
3. Materials and Methods
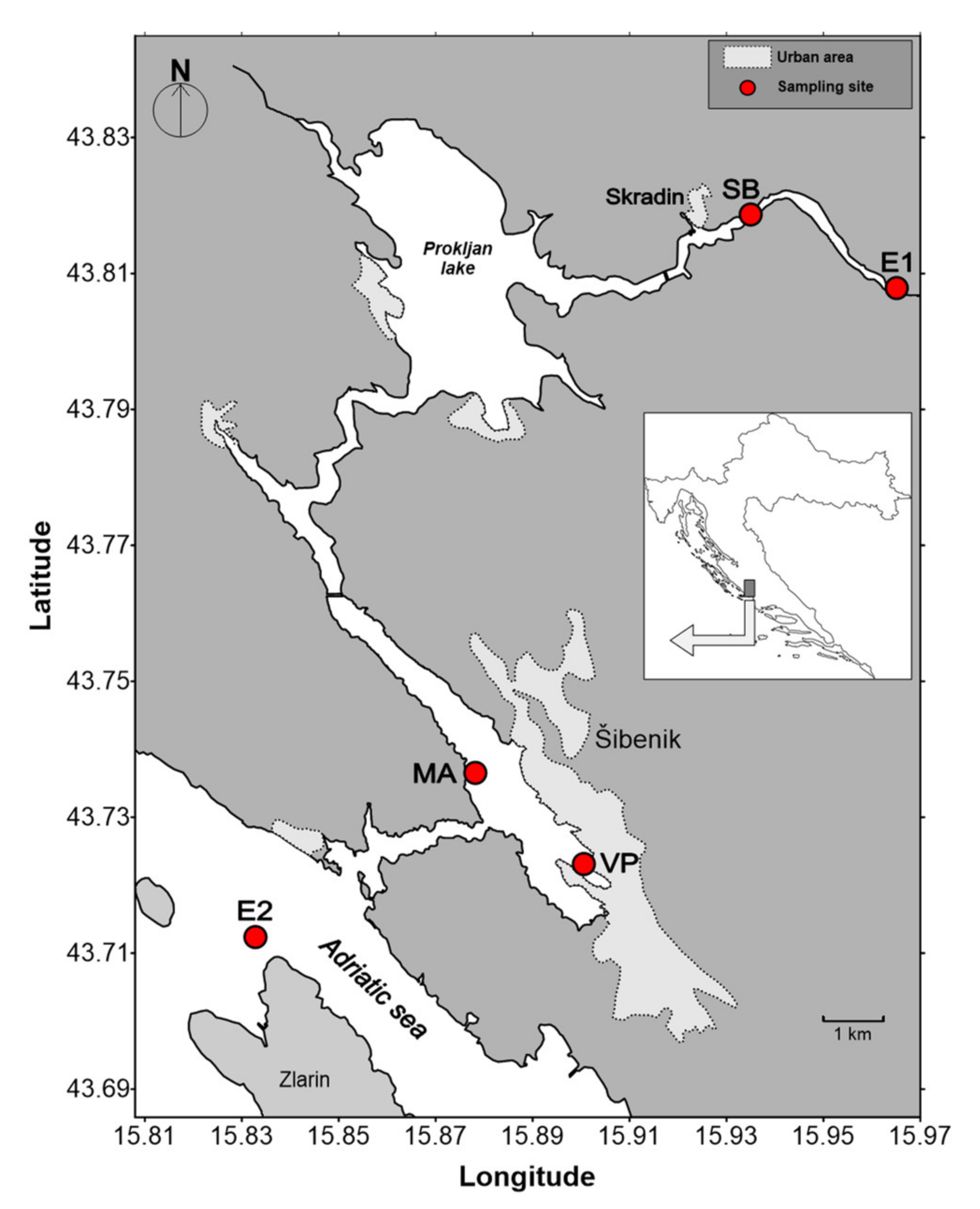
3.1. Study Site
3.2. Equipment and Chemicals
3.3. Determination of Vtot and V Redox Species
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Sample Availability
References
- Huang, J.H.; Huang, F.; Evans, L.; Glasauer, S. Vanadium: Global (bio)geochemistry. Chem. Geol. 2015, 417, 68–89. [Google Scholar] [CrossRef]
- Gustafsson, J.P. Vanadium geochemistry in the biogeosphere –speciation, solid-solution interactions, and ecotoxicity. Appl. Geochem. 2019, 102, 1–25. [Google Scholar] [CrossRef]
- Bernárdez, P.; Ospina-Alvarez, N.; Caetano, M.; Prego, R. Fluvial contributions of nutrient salts, dissolved trace elements and organic carbon to the sea by pristine temperate rivers (SW Europe). Environ. Chem. 2013, 10, 42–53. [Google Scholar] [CrossRef]
- Wang, D.; Sañudo Wilhelmy, S.A. Vanadium speciation and cycling in coastal waters. Mar. Chem. 2009, 117, 52–58. [Google Scholar] [CrossRef]
- Collier, R.W. Collier Particulate and dissolved vanadium in North Pacific Ocean. Nature 1984, 309, 441–444. [Google Scholar] [CrossRef]
- Pyrzyńska, K. Selected problems in speciation analysis of vanadium in water samples. Chem. Anal. 2006, 51, 339–350. [Google Scholar]
- Costa Pessoa, J. Thirty years through vanadium chemistry. J. Inorg. Biochem. 2015, 147, 4–24. [Google Scholar] [CrossRef]
- Xiong, C.; Qin, Y.; Hu, B. On-line separation/preconcentration of V(IV)/V(V) in environmental water samples with CTAB-modified alkyl silica microcolumn and their determination by inductively coupled plasma-optical emission spectrometry. J. Hazard. Mater. 2010, 178, 164–170. [Google Scholar] [CrossRef]
- Shi, Y.X.; Mangal, V.; Guéguen, C. Influence of dissolved organic matter on dissolved vanadium speciation in the Churchill River estuary (Manitoba, Canada). Chemosphere 2016, 154, 367–374. [Google Scholar] [CrossRef]
- Strady, E.; Blanc, G.; Schäfer, J.; Coynel, A.; Dabrin, A. Dissolved uranium, vanadium and molybdenum behaviours during contrasting freshwater discharges in the Gironde Estuary (SW France). Estuar. Coast. Shelf Sci. 2009, 83, 550–560. [Google Scholar] [CrossRef]
- Fiket, Ž.; Ivanić, M.; Turk, M.F.; Mikac, N.; Kniewald, G. Distribution of trace elements in waters of the zrmanja river estuary (eastern adriatic coast, croatia). Croat. Chem. Acta 2018, 91, 29–41. [Google Scholar] [CrossRef]
- Cindrić, A.M.; Garnier, C.; Oursel, B.; Pižeta, I.; Omanović, D. Evidencing the natural and anthropogenic processes controlling trace metals dynamic in a highly stratified estuary: The Krka River estuary (Adriatic, Croatia). Mar. Pollut. Bull. 2015, 94, 199–216. [Google Scholar] [CrossRef]
- Chester, R. Marine Geochemistry; Department of Earth Sciences, Ed.; Academic Division Publishing Unwin Hyman Ltd.: Liverpool, NY, USA, 1990; ISBN 9789401094900. [Google Scholar]
- Shiller, A.M.; Mao, L. Dissolved vanadium in rivers: Effects of silicate weathering. Chem. Geol. 2000, 165, 13–22. [Google Scholar] [CrossRef]
- De Arruda Xaviera, D.; dos Santos, V.F.; de Miranda, A.G.O.; Berrêdo, J.F. Determination of background geochemistry of an Amazon estuary: The Cuñaní Estuary–Amapá. Mar. Pollut. Bull. 2020, 155, 111144. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.; Couto, C.; Ribeiro, A.R.; Maia, A.S.; Santos, M.; Tiritan, M.E.; Pinto, E.; Almeida, A.A. Distribution and environmental assessment of trace elements contamination of water, sediments and flora from Douro River estuary, Portugal. Sci. Total Environ. 2018, 639, 1381–1393. [Google Scholar] [CrossRef]
- Knežević, L.; Cukrov, N.; Bura-Nakić, E. Ion-exchange chromatography as a tool for investigating vanadium speciation in sediments: Preliminary studies. J. Soils Sediments. 2020, 20, 2733–2740. [Google Scholar] [CrossRef]
- Li, X.S.; Le, X.C. Speciation of vanadium in oilsand coke and bacterial culture by high performance liquid chromatography inductively coupled plasma mass spectrometry. Anal. Chim. Acta. 2007, 602, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, K.; Kageyama, S.; Unohara, N. Mutual separation and preconcentration of vanadium(V) and vanadium(IV) in natural waters with chelating functional group immobilized silica gels followed by determination of vanadium by inductively coupled plasma atomic emission spectrometry. Analyst 1992, 117, 13–17. [Google Scholar] [CrossRef]
- Colina, M.; Gardiner, P.H.E.; Rivas, Z.; Troncone, F. Determination of vanadium species in sediment, mussel and fish muscle tissue samples by liquid chromatography-inductively coupled plasma-mass spectrometry. Anal. Chim. Acta. 2005, 538, 107–115. [Google Scholar] [CrossRef]
- Wuilloud, R.G.; Wuilloud, J.C.; Olsina, R.A.; Martinez, L.D. Speciation and preconcentration of vanadium(V) and vanadium(IV) in water samples by flow injection-inductively coupled plasma optical emission spectrometry and ultrasonic nebulization. Analyst 2001, 126, 715–719. [Google Scholar] [CrossRef]
- Pyrzyńska, K.; Wierzbicki, T. Determination of vanadium species in environmental samples. Talanta 2004, 64, 823–829. [Google Scholar] [CrossRef]
- Chen, Z.L.; Owens, G. Trends in speciation analysis of vanadium in environmental samples and biological fluids—A review. Anal. Chim. Acta. 2008, 607, 1–14. [Google Scholar] [CrossRef]
- Cornelis, R.; Caruso, J.A.; Crews, H.; Heumann, K.G. Handbook of Elemental Speciation. In Handbook of Elemental Speciation II: Species in the Environment, Food, Medicine and Occupational Health; John Wiley& Sons, LTD: Chichester, England, 2005; Volume 3, pp. 464–488. [Google Scholar]
- Shijo, Y.; Sato, H.; Uehara, N.; Aratake, S. Simultaneous Determination of Trace Amounts of Copper, Nickel and Vanadium in Sea-water by High-performance Liquid Chromatography After Extraction and Back-extraction. Analyst 1996, 121, 325–328. [Google Scholar] [CrossRef]
- Agrawal, Y.K.; Menon, S.K.; Jain, V.K. Liquid-liquid extraction, separation and preconcentration, membrane transportation and ICP-AES determination of vanadium with dibenzo-18-crown-6. Indian J. Chem. 2003, 42, 1008–1012. [Google Scholar]
- Filik, H.; Berker, K.I.; Balkis, N.; Apak, R. Simultaneous preconcentration of vanadium(V/IV) species with palmitoyl quinolin-8-ol bonded to amberlite XAD 2 and their separate spectrophotometric determination with 4-(2-pyridylazo)-resorcinol using CDTA as masking agent. Anal. Chim. Acta 2004, 518, 173–179. [Google Scholar] [CrossRef]
- Seubert, A. On-line coupling of ion chromatography with ICP-AES and ICP-MS. TrAC Trends. Anal. Chem. 2001, 20, 274–287. [Google Scholar] [CrossRef]
- Gros, N. Ion chromatographic analyses of sea waters, brines and related samples. Water 2013, 5, 659–676. [Google Scholar] [CrossRef]
- Singh, R.P.; Abbas, N.M.; Smesko, S.A. Suppressed ion chromatographic analysis of anions in environmental waters containing high salt concentrations. J. Chromatogr. A 1996, 733, 73–91. [Google Scholar] [CrossRef]
- Lu, H.; Mou, S.; Riviello, J.M. Effects of high concentration of chloride on the determination of trace anions by ion chromatography. J. Liq. Chromatogr. Relat. Technol. 2000, 23, 2181–2193. [Google Scholar] [CrossRef]
- Wann, C.C.; Jiang, S.J. Determination of vanadium species in water samples by liquid chromatography-inductively coupled plasma mass spectrometry. Anal. Chim. Acta 1997, 357, 211–218. [Google Scholar] [CrossRef]
- Aureli, F.; Ciardullo, S.; Pagano, M.; Raggi, A.; Cubadda, F. Speciation of vanadium(iv) and (v) in mineral water by anion exchange liquid chromatography-inductively coupled plasma mass spectrometry after EDTA complexation. J. Anal. At. Spectrom. 2008, 23, 1009–1016. [Google Scholar] [CrossRef]
- Chen, Z.L.; Owens, G.; Naidu, R. Confirmation of vanadium complex formation using electrospray mass spectrometry and determination of vanadium speciation by sample stacking capillary electrophoresis. Anal. Chim. Acta 2007, 585, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Lohmayer, R.; Reithmaier, G.M.S.; Bura-Nakić, E.; Planer-Friedrich, B. Ion-Pair Chromatography Coupled to Inductively Coupled Plasma-Mass Spectrometry (IPC-ICP-MS) as a Method for Thiomolybdate Speciation in Natural Waters. Anal. Chem. 2015, 87, 3388–3395. [Google Scholar] [CrossRef] [PubMed]
- Krachler, M.; Emons, H. Urinary antimony speciation by HPLC-ICP-MS. J. Anal. At. Spectrom. 2001, 16, 20–25. [Google Scholar] [CrossRef][Green Version]
- Wallschläger, D.; London, J. Determination of inorganic selenium species in rain and sea waters by anion exchange chromatography-hydride generation-inductively-coupled plasma-dynamic reaction cell-mass spectrometry (AEC-HG-ICP-DRC-MS). J. Anal. At. Spectrom. 2004, 19, 1119–1127. [Google Scholar] [CrossRef]
- Novič, M.; Divjak, B.; Pihlar, B.; Hudnik, V. Influence of the sample matrix composition on the accuracy of the ion chromatographic determination of anions. J. Chromatogr. A 1996, 739, 35–42. [Google Scholar] [CrossRef]
- Agatemor, C.; Beauchemin, D. Matrix effects in inductively coupled plasma mass spectrometry: A review. Anal. Chim. Acta 2011, 706, 66–83. [Google Scholar] [CrossRef]
- Jen, J.F.; Yang, S.M. Simultaneous speciation determination of vanadium(IV) and vanadium(V) as EDTA complexes by liquid chromatography with UV detection. Anal. Chim. Acta 1994, 289, 97–104. [Google Scholar] [CrossRef]
- Kanamori, K.; Sakurai, M.; Kinoshita, T.; Uyama, T.; Ueki, T.; Michibata, H. Direct reduction from vanadium(V) to vanadium(IV) by NADPH in the presence of EDTA. A consideration of the reduction and accumulation of vanadium in the ascidian blood cells. J. Inorg. Biochem. 1999, 77, 157–161. [Google Scholar] [CrossRef]
- Kanamori, K.; Kinebuchi, Y.; Michibata, H. Reduction of vanadium(IV) to vanadium(III) by cysteine methyl ester in water in the presence of amino polycarboxylates. Chem. Lett. 1997, 26, 423–424. [Google Scholar] [CrossRef]
- Kilibarda, N.; Afton, S.E.; Harrington, J.M.; Yan, F.; Levine, K.E. Rapid speciation and determination of vanadium compounds using ion-pair reversed-phase ultra-high-performance liquid chromatography inductively coupled plasma-sector field mass spectrometry. J. Chromatogr. A 2013, 1304, 121–126. [Google Scholar] [CrossRef]
- Mishra, A.P.; Khan, R.; Pandey, R.R. Kinetic studies on effects of EDTA and surfactants on Reduction of vanadium(V) to vanadium(IV) in sulphuric acid medium. Indian J. Chem. 2009, 48, 1228–1234. [Google Scholar]
- Wang, D.; Sañudo-Wilhelmy, S.A. Development of an analytical protocol for the determination of V (IV) and V (V) in seawater: Application to coastal environments. Mar. Chem. 2008, 112, 72–80. [Google Scholar] [CrossRef]
- Baran, E.J. Oxovanadium(IV) and oxovanadium(V) complexes relevant to biological systems. J. Inorg. Biochem. 2000, 80, 1–10. [Google Scholar] [CrossRef]
- Reuter, J.H.; Perdue, E.M. Importance of heavy metal-organic matter interactions in natural waters. Geochim. Cosmochim. Acta 1977, 41, 325–334. [Google Scholar] [CrossRef]
- Chen, Z.; Mahmudur Rahman, M.; Naidu, R. Speciation of vanadium by anion-exchange chromatography with inductively coupled plasma mass spectrometry and confirmation of vanadium complex formation using electrospray mass spectrometry. J. Anal. At. Spectrom. 2007, 22, 811–816. [Google Scholar] [CrossRef]
- LeGras, C.A.A. Simultaneous determination of anions and divalent cations using ion chromatography with EDTA as eluent. Analyst 1993, 118, 1035–1041. [Google Scholar] [CrossRef]
- Legović, T.; Petricioli, D.; Ẑutić, V. Hypoxia in a pristine stratified estuary (Krka, Adriatic Sea). Mar. Chem. 1991, 32, 347–359. [Google Scholar] [CrossRef]
- Cukrov, N.; Doumandji, N.; Garnier, C.; Tucaković, I.; Dang, D.H.; Omanović, D.; Cukrov, N. Anthropogenic mercury contamination in sediments of Krka River estuary (Croatia). Environ. Sci. Pollut. Res. 2020, 27, 7628–7638. [Google Scholar] [CrossRef] [PubMed]
- Cukrov, N.; Frančišković-Bilinski, S.; Mikac, N.; Roje, V. Natural and anthropogenic influences recorded in sediments from the Krka river estuary (Eastern Adriatic coast), evaluated by statistical methods. Fresenius Environ. Bull. 2008, 17, 855–863. [Google Scholar]
- Louis, Y.; Garnier, C.; Lenoble, V.; Mounier, S.; Cukrov, N.; Omanović, D.; Pižeta, I. Kinetic and equilibrium studies of copper-dissolved organic matter complexation in water column of the stratified Krka River estuary (Croatia). Mar. Chem. 2009, 114, 110–119. [Google Scholar] [CrossRef]
- Legović, T. Exchange of water in a stratified estuary with an application to Krka (Adriatic Sea). Mar. Chem. 1991, 32, 121–135. [Google Scholar] [CrossRef]
- Pađan, J.; Marcinek, S.; Cindrić, A.M.; Layglon, N.; Lenoble, V.; Salaün, P.; Garnier, C.; Omanović, D. Improved voltammetric methodology for chromium redox speciation in estuarine waters. Anal. Chim. Acta 2019, 1089, 40–47. [Google Scholar] [CrossRef]
- Pađan, J.; Marcinek, S.; Cindrić, A.M.; Layglon, N.; Garnier, C.; Salaün, P.; Cobelo-García, A.; Omanović, D. Determination of sub-picomolar levels of platinum in the pristine Krka River estuary (Croatia) using improved voltammetric methodology. Environ. Chem. 2020, 17, 77–84. [Google Scholar] [CrossRef]
- Marcinek, S.; Santinelli, C.; Cindrić, A.-M.; Evangelista, V.; Gonnelli, M.; Layglon, N.; Mounier, S.; Lenoble, V.; Omanović, D. Dissolved organic matter dynamics in the pristine Krka River estuary (Croatia). Mar. Chem. 2020, 225, 103848. [Google Scholar] [CrossRef]
- Cukrov, N.; Cmuk, P.; Mlakar, M.; Omanović, D. Spatial distribution of trace metals in the Krka River, Croatia: An example of the self-purification. Chemosphere 2008, 72, 1559–1566. [Google Scholar] [CrossRef] [PubMed]
- Grẑetić, Z.; Precali, R.; Degobbis, D.; Škrivanić, A. Nutrient enrichment and phytoplankton response in an Adriatic karstic estuary. Mar. Chem. 1991, 32, 313–331. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Knežević, L.; Omanović, D.; Bačić, N.; Mandić, J.; Bura-Nakić, E. Redox Speciation of Vanadium in Estuarine Waters Using Improved Methodology Based on Anion Exchange Chromatography Coupled to HR ICP-MS System. Molecules 2021, 26, 2436. https://doi.org/10.3390/molecules26092436
Knežević L, Omanović D, Bačić N, Mandić J, Bura-Nakić E. Redox Speciation of Vanadium in Estuarine Waters Using Improved Methodology Based on Anion Exchange Chromatography Coupled to HR ICP-MS System. Molecules. 2021; 26(9):2436. https://doi.org/10.3390/molecules26092436
Chicago/Turabian StyleKnežević, Lucija, Dario Omanović, Niko Bačić, Jelena Mandić, and Elvira Bura-Nakić. 2021. "Redox Speciation of Vanadium in Estuarine Waters Using Improved Methodology Based on Anion Exchange Chromatography Coupled to HR ICP-MS System" Molecules 26, no. 9: 2436. https://doi.org/10.3390/molecules26092436
APA StyleKnežević, L., Omanović, D., Bačić, N., Mandić, J., & Bura-Nakić, E. (2021). Redox Speciation of Vanadium in Estuarine Waters Using Improved Methodology Based on Anion Exchange Chromatography Coupled to HR ICP-MS System. Molecules, 26(9), 2436. https://doi.org/10.3390/molecules26092436