
Drug Repurposing: A Review of Old and New Antibiotics for the Treatment of Malaria: Identifying Antibiotics with a Fast Onset of Antiplasmodial Action



Abstract
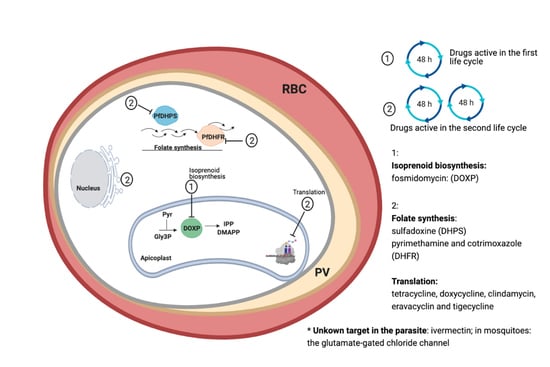
1. Introduction
1.1. Chemoprevention and Chemotherapy of Malaria
1.2. Drug Repurposing
1.3. Antibiotics for the Treatment of Malaria
2. Methods
3. Fast-Acting Antibiotics
3.1. Folate Synthesis Inhibitors
3.2. Tetracyclines
3.3. Fosmidomycin
3.4. Macrolides
4. Lincosamides
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Available online: https://www.who.int/publications/i/item/9789240015791 (accessed on 26 January 2021).
- Siciliano, G.; Alano, P. Enlightening the malaria parasite life cycle: Bioluminescent Plasmodium in fundamental and applied research. Front. Microbiol. 2015, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Van Biljon, R.; Niemand, J.; Van Wyk, R.; Clark, K.; Verlinden, B.; Abrie, C.; Von Grüning, H.; Smidt, W.; Smit, A.; Reader, J.; et al. Inducing controlled cell cycle arrest and re-entry during asexual proliferation of Plasmodium falciparum malaria parasites. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Campo, B.; Vandal, O.; Wesche, D.L.; Burrows, J.N. Killing the hypnozoite — drug discovery approaches to prevent relapse in Plasmodium vivax. Pathog. Glob. Health 2015, 109, 107–122. [Google Scholar] [CrossRef] [PubMed]
- Milner, D.A.; Whitten, R.O.; Kamiza, S.; Carr, R.; Liomba, G.; Dzamalala, C.; Seydel, K.B.; Molyneux, M.E.; Taylor, T.E. The systemic pathology of cerebral malaria in African children. Front. Cell Infect. Microbiol. 2014, 4, 1–13. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Available online: https://www.who.int/malaria/publications/atoz/rectal-artesunate-severe-malaria/en/ (accessed on 26 January 2021).
- Carvalho, L.P.; Kreidenweiss, A.; Held, J. The preclinical discovery and development of rectal artesunate for the treatment of malaria in young children: A review of the evidence. Expert. Opin. Drug Discov. 2021, 16, 13–22. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Available online: https://www.who.int/publications/i/item/9789241549127 (accessed on 26 January 2021).
- Markus, M.B. Killing of Plasmodium vivax by primaquine and tafenoquine. Trends Parasitol. 2019, 35, 857–859. [Google Scholar] [CrossRef] [PubMed]
- Hassett, M.R.; Roepe, P.D. Origin and spread of evolving artemisinin-resistant Plasmodium falciparum malarial parasites in Southeast Asia. Am. J. Trop. Med. Hyg. 2019, 101, 1204–1211. [Google Scholar] [CrossRef]
- Recht, J.; Ashley, E.A.; White, N.J. Use of primaquine and glucose-6-phosphate dehydrogenase deficiency testing: Divergent policies and practices in malaria endemic countries. PLoS Negl. Trop. Dis. 2018, 12, 1–27. [Google Scholar] [CrossRef]
- Hounkpatin, A.B.; Kreidenweiss, A.; Held, J. Clinical utility of tafenoquine in the prevention of relapse of Plasmodium vivax malaria: A review on the mode of action and emerging trial data. Infect. Drug Resist. 2019, 12, 553–570. [Google Scholar] [CrossRef]
- Van Eijk, A.M.; Larsen, D.A.; Kayentao, K.; Koshy, G.; Slaughter, D.E.C.; Roper, C.; Okell, L.C.; Desai, M.; Gutman, J.; Khairallah, C.; et al. Effect of Plasmodium falciparum sulfadoxine-pyrimethamine resistance on the effectiveness of intermittent preventive therapy for malaria in pregnancy in Africa: A systematic review and meta-analysis. Lancet Infect. Dis. 2019, 19, 546–556. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Available online: https://www.cdc.gov/malaria/travelers/drugs.html (accessed on 26 January 2021).
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Talevi, A.; Bellera, C.L. Challenges and opportunities with drug repurposing: Finding strategies to find alternative uses of therapeutics. Expert Opin. Drug Discov. 2020, 15, 397–401. [Google Scholar] [CrossRef]
- Jourdan, J.-P.; Bureau, R.; Rochais, C.; Dallemagne, P. Drug repositioning: A brief overview. J. Pharm. Pharmacol. 2020, 72, 1145–1151. [Google Scholar] [CrossRef]
- Andrews, K.T.; Fisher, G.; Skinner-Adams, T.S. Drug repurposing and human parasitic protozoan diseases. Int. J. Parasitol. Drugs Drug Resist. 2014, 4, 95–111. [Google Scholar] [CrossRef]
- Kapoor, G.; Saigal, S.; Elongavan, A. Action and resistance mechanisms of antibiotics: A guide for clinicians. J. Anaesthesiol. Clin. Pharmacol. 2017, 33, 300–305. [Google Scholar] [CrossRef]
- Gaillard, T.; Madamet, M.; Tsombeng, F.F.; Dormoi, J.; Pradines, B. Antibiotics in malaria therapy: Which antibiotics except tetracyclines and macrolides may be used against malaria? Malar. J. 2016, 15, 1–10. [Google Scholar] [CrossRef]
- Fontinha, D.; Moules, I.; Prudencio, M. Repurposing drugs to fight hepatic malaria parasites. Molecules 2020, 25, 3409. [Google Scholar] [CrossRef]
- Aung, N.M.; Nyein, P.P.; Kyi, M.M.; Hanson, J. Bacterial coinfection in adults with severe malaria. Clin. Infec. Dis. 2021, 72, 535–536. [Google Scholar] [CrossRef]
- Church, J.; Maitland, K. Invasive bacterial co-infection in African children with Plasmodium falciparum malaria: A systematic review. BMC Med. 2014, 12, 1–16. [Google Scholar] [CrossRef]
- Dahl, E.L.; Rosenthal, P.J. Multiple antibiotics exert delayed effects against the Plasmodium falciparum apicoplast. Antimicrob. Agents Chemother. 2007, 51, 3485–3490. [Google Scholar] [CrossRef]
- Pradel, G.; Schlitzer, M. Antibiotics in malaria therapy and their effect on the parasite apicoplast. Curr. Mol. Med. 2010, 10, 335–349. [Google Scholar] [CrossRef]
- Lim, L.; McFadden, G.I. The evolution, metabolism and functions of the apicoplast. Philos. Trans. R. Soc. B Biol. Sci. 2010, 365, 749–763. [Google Scholar] [CrossRef]
- Wilson, D.W.; Goodman, C.D.; Sleebs, B.E.; Weiss, G.E.; De Jong, N.W.; Angrisano, F.; Langer, C.; Baum, J.; Crabb, B.S.; Gilson, P.R.; et al. Macrolides rapidly inhibit red blood cell invasion by the human malaria parasite, Plasmodium falciparum. BMC Biol. 2015, 13, 1–19. [Google Scholar] [CrossRef]
- Ralph, S.A.; Van Dooren, G.G.; Waller, R.F.; Crawford, M.J.; Fraunholz, M.J.; Foth, B.J.; Tonkin, C.J.; Roos, D.S.; McFadden, G.I. Metabolic maps and functions of the Plasmodium falciparum apicoplast. Nat. Rev. Microbiol. 2004, 2, 203–216. [Google Scholar] [CrossRef]
- Seeber, F. Biogenesis of iron — sulphur clusters in amitochondriate and apicomplexan protists. Int. J. Parasitol. 2002, 32, 1207–1217. [Google Scholar] [CrossRef]
- Waller, R.F.; Keeling, P.J.; Donald, R.G.K.; Striepen, B.; Handman, E.; Lang-Unnasch, N.; Cowman, A.F.; Besra, G.S.; Roos, D.S.; McFadden, G.I. Nuclear-encoded proteins target to the plastid in Toxoplasma gondii and Plasmodium falciparum. Proc. Natl. Acad. Sci. USA 1998, 95, 12352–12357. [Google Scholar] [CrossRef]
- Shears, M.J.; Botté, C.Y.; Mcfadden, G.I. Fatty acid metabolism in the Plasmodium apicoplast: Drugs, doubts and knockouts. Mol. Biochem. Parasitol. 2015, 199, 34–50. [Google Scholar] [CrossRef]
- Ke, H.; Sigala, P.A.; Miura, K.; Morrisey, J.M.; Mather, M.W.; Crowley, J.R.; Henderson, J.P.; Goldberg, D.E.; Long, C.A.; Vaidya, A.B. The heme biosynthesis pathway is essential for Plasmodium falciparum development in mosquito stage but not in blood stages. J. Biol. Chem. 2014, 289, 1–17. [Google Scholar] [CrossRef]
- Uddin, T.; McFadden, G.I.; Goodman, C.D. Validation of putative apicoplast-targeting drugs using a chemical supplementation assay in cultured human malaria. Antimicrob. Agents Chemother. 2018, 62, 34827–34837. [Google Scholar] [CrossRef]
- Yeh, E.; DeRisi, J.L. Chemical rescue of malaria parasites lacking an apicoplast defines organelle function in blood-stage Plasmodium falciparum. PLoS Biol. 2011, 9, 1–10. [Google Scholar] [CrossRef]
- Kennedy, K.; Crisafulli, E.M.; Ralph, S.A. Delayed death by plastid inhibition in apicomplexan parasites. Trends Parasitol. 2019, 35, 747–759. [Google Scholar] [CrossRef]
- Njau, J.D.; Goodman, C.A.; Kachur, S.P.; Mulligan, J.; Munkondya, J.S.; Mchomvu, N.; Abdulla, S.; Bloland, P.; Mills, A. The costs of introducing artemisinin-based combination therapy: Evidence from district-wide implementation in rural Tanzania. Malar. J. 2008, 7, 1–14. [Google Scholar] [CrossRef]
- Triglia, T.; Cowman, A.F. The mechanism of resistance to sulfa drugs in Plasmodium falciparum. Drug Resist. Updat. 1999, 2, 15–19. [Google Scholar] [CrossRef]
- Bzik, D.J.; Li, W.B.; Horii, T.; Inselburg, J. Molecular cloning and sequence analysis of the Plasmodium falciparum dihydrofolate reductase-thymidylate synthase gene. Proc. Natl. Acad. Sci. USA 1987, 84, 8360–8364. [Google Scholar] [CrossRef]
- Cowman, A.F.; Foote, S.J. Chemotherapy and drug resistance in malaria. Int. J. Parasitol. 1990, 20, 503–513. [Google Scholar] [CrossRef]
- Peterson, D.S.; Walliker, D.; Wellems, T.E. Evidence that a point mutation in dihydrofolate reductase-thymidylate synthase confers resistance to pyrimethamine in falciparum malaria. Proc. Natl. Acad. Sci. USA 1988, 85, 9114–9118. [Google Scholar] [CrossRef]
- Triglia, T.; Wang, P.; Sims, P.F.; Hyde, J.E.; Cowman, A.F. Allelic exchange at the endogenous genomic locus in Plasmodium falciparum proves the role of dihydropteroate synthase in sulfadoxine-resistant malaria. EMBO J. 1998, 17, 3807–3815. [Google Scholar] [CrossRef]
- Wang, P.; Read, M.; Sims, P.F.G.; Hyde, J.E. Sulfadoxine resistance in the human malaria parasite Plasmodium falciparum is determined by mutations in dihydropteroate synthetase and an additional factor associated with folate utilization. Mol. Microbiol. 1997, 23, 979–986. [Google Scholar] [CrossRef]
- World Health Organization. Available online: https://www.who.int/publications/i/item/9789240012813 (accessed on 26 January 2021).
- Huovinen, P. Increases in rates of resistance to trimethoprim. Clin. Infect. Dis. 1997, 24, 63–66. [Google Scholar] [CrossRef]
- Brown, G.M. The biosynthesis of folic acid. J. Biol. Chem. 1963, 249, 536–540. [Google Scholar]
- Manyando, C.; Njunju, E.M.; D’Alessandro, U.; Van Geertruyden, J.-P. Safety and efficacy of co-trimoxazole for treatment and prevention of Plasmodium falciparum malaria: Asystematic review. PLoS ONE 2013, 8, e56916. [Google Scholar] [CrossRef]
- Kamya, M.R.; Gasasira, A.F.; Achan, J.; Mebrahtu, T.; Ruel, T.; Kekitiinwa, A.; Charlebois, E.D.; Rosenthal, P.J.; Havlir, D.; Dorsey, G. Effects of trimethoprim-sulfamethoxazole and insecticide-treated bednets on malaria among HIV-infected Ugandan children. AIDS 2007, 21, 2059–2066. [Google Scholar] [CrossRef]
- Sandison, T.G.; Homsy, J.; Arinaitwe, E.; Wanzira, H.; Kakuru, A.; Bigira, V.; Kalamya, J.; Vora, N.; Kublin, J.; Kamya, M.R.; et al. Protective efficacy of co-trimoxazole prophylaxis against malaria in HIV exposed children in rural Uganda: A randomised clinical trial. BMJ 2011, 342, 1–10. [Google Scholar] [CrossRef]
- Mermin, J.; Ekwaru, J.P.; Liechty, C.A.; Were, W.; Downing, R.; Ransom, R.; Weidle, P.; Lule, J.; Coutinho, A.; Solberg, P. Effect of co-trimoxazole prophylaxis, antiretroviral therapy, and insecticide-treated bednets on the frequency of malaria in HIV-1-infected adults in Uganda: A prospective cohort study. Lancet 2006, 367, 1256–1261. [Google Scholar] [CrossRef]
- Fasan, P.O. Trimethoprim plus sulphamethoxazole compared with chloroquine in the treatment and suppression of malaria in African schoolchildren. Ann. Trop Med. Parasitol. 1971, 65, 117–121. [Google Scholar] [CrossRef]
- Sowunmi, A.; Fateye, B.A.; Adedeji, A.A.; Fehintola, F.A.; Bamgboye, A.E.; Babalola, C.P.; Happi, T.C.; Gbotosho, G.O. Effects of antifolates — co-trimoxazole and pyrimethamine- sulfadoxine—on gametocytes in children with acute, symptomatic, uncomplicated, Plasmodium falciparum malaria. Mem. Inst. Oswaldo Cruz 2005, 100, 451–455. [Google Scholar] [CrossRef]
- Hamel, M.J.; Kublin, J.; Mkandala, C.; Chizani, N.; Steketee, R.; Holtz, T.; Bloland, P.; Kaimila, N.; Kazembe, P. Efficacy of trimethroprim-sulfamethoxazole compared with sulfadoxine-pyrimethamine plus erythromycin for the treatment of uncomplicated malaria in children with integrated management of childhood illness dual classifications of malaria and pneumonia. Am. J. Trop. Med. Hyg. 2005, 73, 609–615. [Google Scholar] [CrossRef]
- Thera, M.A.; Sehdev, P.S.; Coulibaly, D.; Traore, K.; Garba, M.N.; Cissoko, Y.; Kone, A.; Guindo, A.; Dicko, A.; Beavogui, A.B.; et al. Impact of trimethoprim-sulfamethoxazole prophylaxis on falciparum malaria infection and disease. J. Infect. Dis. 2005, 192, 1823–1829. [Google Scholar] [CrossRef]
- Mbeye, N.M.; Ter Kuile, F.O.; Davies, M.-A.; Phiri, K.S.; Egger, M.; Wandeler, G.; Africa, I.-S. Cotrimoxazole prophylactic treatment prevents malaria in children in sub-Saharan Africa: Systematic review and meta-analysis. Trop. Med. Int. Health 2015, 19, 1057–1067. [Google Scholar] [CrossRef]
- Daniels, B.; Coutsoudis, A.; Moodley-Govender, E.; Mulol, H.; Spooner, E.; Kiepiela, P.; Reddy, S.; Zako, L.; Ho, N.T.; Kuhn, L.; et al. Effect of co-trimoxazole prophylaxis on morbidity and mortality of HIV-exposed, HIV-uninfected infants in South Africa: A randomised controlled, non-inferiority trial. Lancet Glob. Health 2019, 7, 1717–1727. [Google Scholar] [CrossRef]
- Van Geertruyden, J.-P.; Menten, J.; Colebunders, R.; Korenromp, E.; D’Alessandro, U. The impact of HIV-1 on the malaria parasite biomass in adults in sub-Saharan Africa contributes to the emergence of antimalarial drug resistance. Malar. J. 2008, 13, 1–13. [Google Scholar] [CrossRef]
- Juma, D.W.; Muiruri, P.; Yuhas, K.; John-Stewart, G.; Ottichilo, R.; Waitumbi, J.; Singa, B.; Polyak, C.; Kamau, E. The prevalence and antifolate drug resistance profiles of Plasmodium falciparum in study participants randomized to discontinue or continue cotrimoxazole prophylaxis. PLoS Negl. Trop. Dis. 2019, 13, e0007223. [Google Scholar] [CrossRef]
- MMV-Supported Projects. Available online: https://www.mmv.org/research-development/mmv-supported-projects (accessed on 29 January 2021).
- Chopra, I.; Roberts, M. Tetracycline antibiotics: Mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol. Mol. Biol. Rev. 2001, 65, 232–260. [Google Scholar] [CrossRef]
- Andrei, S.; Droc, G.; Stefan, G. FDA approved antibacterial drugs: 2018-2019. Discoveries 2019, 7, e102. [Google Scholar] [CrossRef]
- Maxwell, I.H. Partial removal of bound transfer RNA from polysomes engaged in protein synthesis in vitro after addition of tetracycline. Biochim. Biophys. Acta (BBA) Nucleic Acids Protein Synth. 1967, 138, 337–346. [Google Scholar] [CrossRef]
- Pioletti, M.; Schlünzen, F.; Harms, J.; Zarivach, R.; Glühmann, M.; Avila, H.; Bashan, A.; Bartels, H.; Auerbach, T.; Jacobi, C.; et al. Crystal structures of complexes of the small ribosomal subunit with tetracycline, edeine and IF3. EMBO J. 2001, 20, 1829–1839. [Google Scholar] [CrossRef]
- Sánchez, A.R.; Rogers, R.S.; Sheridan, P.J. Tetracycline and other tetracycline-derivative staining of the teeth and oral cavity. Int J. Dermatol. 2004, 43, 709–715. [Google Scholar] [CrossRef]
- Imboden, C.A., Jr.; Cooper, W.C.; Coatney, G.R.; Jeffery, G.M. Studies in human malaria. XXIX. Trials of aureomycin, chloramphenicol, penicillin, and dihydrostreptomycin against the Chesson strain of Plasmodium vivax. J. Natl. Malar. Soc. 1950, 9, 377–380. [Google Scholar]
- Sanchez, F.R.; Casillas, J.; Paredes, M.; Velazquez, J.; Riebeling, Q.B. Terramycin in malaria therapy. Pan. Am. Med. Womans. J. 1952, 59, 10–15. [Google Scholar]
- Grande, E.N.; Sanchez, A.R.; Sanchez, F.R. The treatment of malaria with tetracycline. Antibiot. Med. Clin. Ther. 1956, 3, 193196. [Google Scholar] [CrossRef]
- Tan, K.R.; Magill, A.J.; Arguin, P.M.; Parise, M.E.; Prevention, C.F.D.C.A. Doxycycline for malaria chemoprophylaxis and treatment: Report from the CDC expert meeting on malaria chemoprophylaxis. Am. J. Trop. Med. Hyg. 2011, 84, 517–531. [Google Scholar] [CrossRef]
- Starzengruber, P.; Thriemer, K.; Haque, R.; Khan, W.A.; Fuehrer, H.P.; Siedl, A.; Hofecker, V.; Ley, B.; Wernsdorfer, W.H.; Noedl, H. Antimalarial activity of tigecycline, a novel glycylcycline antibiotic. Antimicrob. Agents Chemother. 2009, 53, 4040–4042. [Google Scholar] [CrossRef] [PubMed]
- Ribatski-Silva, D.; Bassi, C.L.; Martin, T.O.G.; Alves-Junior, E.; Gomes, L.T.; Fontes, C.J.F. In vitro antimalarial activity of tigecycline against Plasmodium falciparum culture-adapted reference strains and clinical isolates from the Brazilian Amazon. Rev. Soc. Bras. Med. Trop. 2014, 47, 110–112. [Google Scholar] [CrossRef] [PubMed]
- Held, J.; Zanger, P.; Issifou, S.; Kremsner, P.G.; Mordmüller, B. In vitro activity of tigecycline in Plasmodium falciparum culture-adapted strains and clinical isolates from Gabon. Int. J. Antimicrob. Agents 2010, 35, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Held, J.; Zanger, P.; Issifou, S.; Kremsner, P.G.; Mordmüller, B. Functional, biophysical, and structural bases for antibacterial activity of tigecycline. Antimicrob. Agents Chemother. 2006, 50, 2156–2166. [Google Scholar] [CrossRef]
- Sahu, R.; Walker, L.A.; Tekwani, B.L. In vitro and in vivo anti-malarial activity of tigecycline, a glycylcycline antibiotic, in combination with chloroquine. Malar. J. 2014, 414, 1–7. [Google Scholar] [CrossRef]
- Koehne, E.; Kreidenweiss, A.; Adegbite, B.R.; Manego, R.Z.; McCall, M.B.; Mombo-Ngoma, G.; Adegnika, A.A.; Agnandji, S.T.; Mordmüller, B.; Held, J. In vitro activity of eravacycline, a novel synthetic halogenated tetracycline, against the malaria parasite Plasmodium falciparum. J. Glob. Antimicrob. Resist. 2020, 24, 93–97. [Google Scholar] [CrossRef]
- Parkinson, E.I.; Erb, A.; Eliot, A.C.; Ju, K.-S.; Metcalf, W.W. Fosmidomycin biosynthesis diverges from related phosphonate natural products. Nat. Chem. Biol. 2019, 15, 1049–1056. [Google Scholar] [CrossRef]
- Davey, M.S.; Tyrrell, J.M.; Howe, R.A.; Walsh, T.R.; Moser, B.; Toleman, M.A.; Eberl, M. A. A promising target for treatment of multidrug-resistant bacterial infections. Antimicrob. Agents Chemother. 2011, 55, 3635–3636. [Google Scholar] [CrossRef]
- Phu, N.H.; Day, N.P.J.; Tuan, P.Q.; Mai, N.T.H.; Chau, T.T.H.; Van Chuong, L.; Vinh, H.; Loc, P.P.; Sinh, D.X.; Hoa, N.T.T.; et al. Studies on new phosphonic acid antibiotics. III. Isolation and characterization of FR-31564, FR-32863 and FR-33289. J. Antibiot. 2020, 71. [Google Scholar] [CrossRef]
- Kuzuyama, T.; Shimizu, T.; Takahashi, S.; Seto, H. Fosmidomycin, a specific inhibitor of 1-deoxy-d-xylulose 5-phosphate reductoisomerase in the nonmevalonate pathway for terpenoid biosynthesis. Tetrahedron Lett. 1998, 39, 7913–7916. [Google Scholar] [CrossRef]
- Armstrong, C.M.; Meyers, D.J.; Imlay, L.S.; Meyers, C.F.; Odom, A.R. Resistance to the antimicrobial agent fosmidomycin and an FR900098 prodrug through mutations in the deoxyxylulose phosphate reductoisomerase gene (dxr). Antimicrob. Agents Chemother. 2015, 59, 5511–5519. [Google Scholar] [CrossRef] [PubMed]
- Jomaa, H.; Wiesner, J.; Sanderbrand, S.; Altincicek, B.; Weidemeyer, C.; Hintz, M. Inhibitors of the nonmevalonate pathway of isoprenoid biosynthesis as antimalarial drugs. Science 1999, 285, 1573–1577. [Google Scholar] [CrossRef] [PubMed]
- Murakawa, T.; Sakamoto, H.; Fukada, S.; Konishi, T. Pharmacokinetics of fosmidomycin, a new phosphonic acid antibiotic. Antimicrob. Agents Chemother. 1982, 21, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Lell, B.; Ruangweerayut, R.; Wiesner, J.; Missinou, M.A.; Schindler, A.; Baranek, T.; Hintz, M.; Hutchinson, D.; Jomaa, H.; Kremsner, P.G. Fosmidomycin, a novel chemotherapeutic agent for malaria. Antimicrob. Agents Chemother. 2003, 47, 735–738. [Google Scholar] [CrossRef] [PubMed]
- Missinou, M.A.; Borrmann, S.; Schindler, A.; Issifou, S.; Adegnika, A.A.; Matsiégui, P.-B.; Binder, R.; Lell, B.; Wiesner, J.; Baranek, T.; et al. Fosmidomycin for malaria. Lancet 2002, 360, 1941–1942. [Google Scholar] [CrossRef]
- Wiesner, J.; Henschker, D.; Hutchinson, D.B.; Beck, E.; Jomaa, H. In vitro and in vivo synergy of fosmidomycin, a novel antimalarial drug, with clindamycin. Antimicrob. Agents Chemother. 2002, 46, 2889–2894. [Google Scholar] [CrossRef]
- Ruengweerayut, R.; Looareesuwan, S.; Hutchinson, D.; Chauemung, A.; Banmairuroi, V.; Na-Bangchang, K. Assessment of the pharmacokinetics and dynamics of two combination regimens of fosmidomycin-clindamycin in patients with acute uncomplicated falciparum malaria. Malar. J. 2008, 7, 1–11. [Google Scholar] [CrossRef]
- Borrmann, S.; Adegnika, A.A.; Matsiegui, P.; Issifou, S.; Schindler, A.; Mawili-Mboumba, D.P.; Baranek, T.; Wiesner, J.; Jomaa, H.; Kremsner, P.G. Fosmidomycin-clindamycin for Plasmodium falciparum infections in African children. J. Infect. Dis. 2004, 189, 901–908. [Google Scholar] [CrossRef]
- Oyakhirome, S.; Issifou, S.; Pongratz, P.; Barondi, F.; Ramharter, M.; Kun, J.F.; Missinou, M.A.; Lell, B.; Kremsner, P.G. Randomized controlled trial of fosmidomycin-clindamycin versus sulfadoxine-pyrimethamine in the treatment of Plasmodium falciparum malaria. Antimicrob. Agents Chemother. 2007, 51, 1869–1871. [Google Scholar] [CrossRef]
- Borrmann, S.; Lundgren, I.; Oyakhirome, S.; Impouma, B.; Matsiegui, P.-B.; Adegnika, A.A.; Issifou, S.; Kun, J.F.J.; Hutchinson, D.; Wiesner, J.; et al. Fosmidomycin plus clindamycin for treatment of pediatric patients aged 1 to 14 years with Plasmodium falciparum malaria. Antimicrob. Agents Chemother. 2006, 50, 2713–2718. [Google Scholar] [CrossRef]
- Lanaspa, M.; Moraleda, C.; Machevo, S.; González, R.; Serrano, B.; Macete, E.; Cisteró, P.; Mayor, A.; Hutchinson, D.; Kremsner, P.G.; et al. Inadequate efficacy of a new formulation of fosmidomycin-clindamycin combination in Mozambican children less than three years old with uncomplicated Plasmodium falciparum malaria. Antimicrob. Agents Chemother. 2012, 56, 2923–2928. [Google Scholar] [CrossRef] [PubMed]
- Borrmann, S.; Adegnika, A.A.; Moussavou, F.; Oyakhirome, S.; Esser, G.; Matsiegui, P.-B.; Ramharter, M.; Lundgren, I.; Kombila, M.; Issifou, S.; et al. Short-course regimens of artesunate-fosmidomycin in treatment of uncomplicated Plasmodium falciparum malaria. Antimicrob. Agents Chemother. 2005, 49, 3749–3754. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jelić, D.; Antolović, R. From erythromycin to azithromycin and new potential ribosome-binding antimicrobials. Antibiotics 2016, 5, 29. [Google Scholar] [CrossRef]
- Mutak, S. Azalides from azithromycin to new azalide derivatives. J. Antibiot. 2007, 60, 85–122. [Google Scholar] [CrossRef] [PubMed]
- Kannan, K.; Kanabar, P.; Schryer, D.; Florin, T.; Oh, E.; Bahroos, N.; Tenson, T.; Weissman, J.S.; Mankin, A.S. The general mode of translation inhibition by macrolide antibiotics. Proc. Natl. Acad. Sci. USA 2014, 111, 15958–15963. [Google Scholar] [CrossRef]
- Otoguro, K.; Ui, H.; Ishiyama, A.; Kobayashi, M.; Togashi, H.; Takahashi, Y.; Masuma, R.; Tanaka, H.; Tomoda, H.; Yamada, H.; et al. In vitro and in vivo antimalarial activities of a non-glycosidic 18-membered macrolide antibiotic, borrelidin, against drug-resistant strains of Plasmodia. J. Antibiot. 2003, 56, 727–728. [Google Scholar] [CrossRef]
- Ekland, E.H.; Schneider, J.; Fidock, D.A. Identifying apicoplast-targeting antimalarials using high-throughput compatible approaches. FASEB J. 2011, 25, 3583–3593. [Google Scholar] [CrossRef]
- Campbell, W.C. History of avermectin and ivermectin, with notes on the history of other macrocyclic lactone antiparasitic agents. Curr. Pharm. Biotechnol. 2012, 13, 853–865. [Google Scholar] [CrossRef]
- Crump, A.; ŌMura, S. Ivermectin, ‘Wonder drug’ from Japan: The human use perspective. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2011, 87, 13–28. [Google Scholar] [CrossRef]
- Nontasut, P.; Bussaratid, V.; Chullawichit, S.; Charoensook, N.; Visetsuk, K. Comparison of ivermectin and albendazole treatment for gnathostomiasis. Southeast Asian J. Trop. Med. Public Health 2000, 31, 374–377. [Google Scholar] [PubMed]
- Shinohara, E.H.; Martini, M.Z.; Neto, H.G.D.O.; Takahashi, A. Oral myiasis treated with ivermectin: Case report. Braz. Dent. J. 2004, 15, 79–81. [Google Scholar] [CrossRef] [PubMed]
- Currie, M.J.; Reynolds, G.J.; Glasgow, N.J.; Bowden, F.J. A pilot study of the use of oral ivermectin to treat head lice in primary school students in Australia. Pediatr. Dermatol. 2010, 27, 595–599. [Google Scholar] [CrossRef]
- Naquira, C.; Nalin, D.R.; Jimenez, G.; Guerra, J.G.; Neu, D.; Aziz, M.; Bernal, R. Ivermectin for human strongyloidiasis and other intestinal helminths. Am. J. Trop. Med. Hyg. 1989, 40, 304–309. [Google Scholar] [CrossRef]
- Lim, L.E.; Vilchèze, C.; Ng, C.; Jacobs, W.R.; Ramón-García, S.; Thompson, C.J. Anthelmintic avermectins kill Mycobacterium tuberculosis, including multidrug-resistant clinical strains. Antimicrob. Agents Chemother. 2013, 57, 1040–1046. [Google Scholar] [CrossRef]
- Campbell, W.C. Ivermemctin: An update. Parasitol. Today 1985, 1, 10–16. [Google Scholar] [CrossRef]
- Panchal, M.; Rawat, K.D.; Kumar, G.; Kibria, K.M.; Singh, S.S.; Kalamuddin, M.; Mohmmed, A.; Malhotra, P.; Tuteja, R. Plasmodium falciparum signal recognition particle components and anti-parasitic effect of ivermectin in blocking nucleo-cytoplasmic shuttling of SRP. Cell Death Dis. 2014, 5, e994-11. [Google Scholar] [CrossRef]
- Mectizan Donation Program. Available online: https://mectizan.org/news-resources/2015-annual-highlights/# (accessed on 26 January 2021).
- Ejere, H.O.D.; Schwartz, E.; Wormald, R. Ivermectin for onchocercal eye disease (river blindness). Cochrane Database Syst. Rev. 2012. [Google Scholar] [CrossRef]
- Kamgno, J.; Gardon, J.; Gardon-Wendel, N.; Ngangue, D.-; Duke, B.O.; Boussinesq, M. Adverse systemic reactions to treatment of onchocerciasis with ivermectin at normal and high doses given annually or three-monthly. Trans. R. Soc. Trop. Med. Hyg. 2004, 98, 496–504. [Google Scholar] [CrossRef]
- Omura, S.; Crump, A. Ivermectin: Panacea for resource-poor communities? Trends Parasitol. 2014, 30, 445–455. [Google Scholar] [CrossRef]
- Mackenzie, C.D.; Geary, T.G.; Gerlach, J.A. Possible pathogenic pathways in the adverse clinical events seen following ivermectin administration to onchocerciasis patients. Filaria J. 2003, 2. [Google Scholar] [CrossRef] [PubMed]
- Gardon, J.; Gardon-Wendel, N.; Ngangue, D.-; Kamgno, J.; Chippaux, J.-P.; Boussinesq, M. Serious reactions after mass treatment of onchocerciasis with ivermectin in an area endemic for Loa loa infection. Lancet 1997, 350, 18–22. [Google Scholar] [CrossRef]
- Alout, H.; Krajacich, B.J.; Meyers, J.I.; Grubaugh, N.D.; Brackney, D.E.; Kobylinski, K.C.; Ii, J.W.D.; Bolay, F.K.; Fakoli, L.S.; Diabaté, A.; et al. Evaluation of ivermectin mass drug administration for malaria transmission control across different West African environments. Malar. J. 2014, 417, 1–10. [Google Scholar] [CrossRef]
- Chaccour, C.; Barrio Ángel, I.; Gil Royo, A.G.; Urbistondo, D.M.; Slater, H.; Hammann, F.; Del Pozo, J.L. Screening for an ivermectin slow-release formulation suitable for malaria vector control. Malar. J. 2015, 102, 1–9. [Google Scholar] [CrossRef]
- Steketee, R.W.; Kuile, F.O.T. Ivermectin as a complementary strategy to kill mosquitoes and stop malaria transmission. Clin. Infect. Dis. 2015, 60, 366–368. [Google Scholar] [CrossRef][Green Version]
- Chaccour, C.; Lines, J.; Whitty, C. Effect of ivermectin on Anopheles gambiae mosquitoes fed on humans: The potential of oral insecticides in malaria control. J. Infect. Dis. 2010, 202, 113–116. [Google Scholar] [CrossRef]
- Foy, B.D.; Kobylinski, K.C.; da Silva, I.M.; Rasgon, J.L.; Sylla, M. Endectocides for malaria control. Trends Parasitol. 2011, 27, 423–428. [Google Scholar] [CrossRef]
- Pinilla, Y.T.; Lopes, S.C.P.; Sampaio, V.S.; Andrade, F.S.; Melo, G.C.; Orfanó, A.S.; Secundino, N.F.C.; Guerra, M.G.V.B.; Lacerda, M.V.G.; Kobylinski, K.C.; et al. Promising approach to reducing malaria transmission by ivermectin: Sporontocidal effect against Plasmodium vivax in the South American vectors Anopheles aquasalis and Anopheles darlingi. PLoS Negl. Trop. Dis. 2018, 12, 1–23. [Google Scholar] [CrossRef]
- Kobylinski, K.C.; Ubalee, R.; Ponlawat, A.; Nitatsukprasert, C.; Phasomkulsolsil, S.; Wattanakul, T.; Tarning, J.; Na-Bangchang, K.; McCardle, P.W.; Davidson, S.A.; et al. Ivermectin susceptibility and sporontocidal effect in Greater Mekong Subregion Anopheles. Malar. J. 2017, 16, 1–13. [Google Scholar] [CrossRef]
- Kobylinski, K.C.; Foy, B.D.; Richardson, J.H. Ivermectin inhibits and delays the development of Plasmodium falciparum in Anopheles gambiae. Am. J. Trop Med. Hyg. 2012, 381, 1–9. [Google Scholar]
- Foy, B.D.; Alout, H.; Seaman, J.A.; Rao, S.; Magalhaes, T.; Wade, M.; Parikh, S.; Soma, D.D.; Sagna, A.B.; Fournet, F.; et al. Efficacy and risk of harms of repeat ivermectin mass drug administrations for control of malaria (RIMDAMAL): A cluster-randomised trial. Lancet 2019, 393, 1517–1526. [Google Scholar] [CrossRef]
- Dabira, E.D.; Soumare, H.M.; Lindsay, S.W.; Conteh, B.; Ceesay, F.; Bradley, J.; Kositz, C.; Broekhuizen, H.; Kandeh, B.; Fehr, A.E.; et al. Mass drug administration with high-dose ivermectin and dihydroartemisinin-piperaquine for malaria elimination in an area of low transmission with high coverage of malaria control interventions: Protocol for the massiv cluster randomized clinical trial. JMIR Res. Protoc. 2020, 9e20904. [Google Scholar] [CrossRef]
- De Carvalho, L.P.; Sandri, T.L.; De Melo, E.J.T.; Fendel, R.; Kremsner, P.G.; Mordmüller, B.; Held, J. Ivermectin impairs the development of sexual and asexual stages of Plasmodium falciparum in vitro. Antimicrob. Agents Chemother. 2019, 63, 1–9. [Google Scholar] [CrossRef]
- Mendes, A.M.; Albuquerque, I.S.; Machado, M.; Pissarra, J.; Meireles, P.; Prudêncio, M. Inhibition of Plasmodium liver infection by ivermectin. Antimicrob. Agents Chemother. 2017, 61, 1–8. [Google Scholar] [CrossRef]
- Metzger, W.G.; Theurer, A.; Pfleiderer, A.; Molnar, Z.; Maihöfer-Braatting, D.; Bissinger, A.L.; Sulyok, Z.; Köhler, C.; Egger-Adam, D.; Lalremruata, A.; et al. Ivermectin for causal malaria prophylaxis: A randomised controlled human infection trial. Trop. Med. Int. Heal. 2020, 25, 380–386. [Google Scholar] [CrossRef]
- Smit, M.R.; Ochomo, E.; Aljayyoussi, G.; Kwambai, T.; Abong’O, B.; Bayoh, N.; Gimnig, J.; Samuels, A.; Desai, M.; Phillips-Howard, P.A; et al. Efficacy and safety of high-dose ivermectin for reducing malaria transmission (IVERMAL): Protocol for a double-blind, randomized, placebo-controlled, dose-finding trial in Western Kenya. JMIR Res. Protoc. 2016, 5, e213. [Google Scholar] [CrossRef]
- Smit, M.R.; Ochomo, E.O.; Aljayyoussi, G.; Kwambai, T.K.; Abong’O, B.O.; Chen, T.; Bousema, T.; Slater, H.C.; Waterhouse, D.; Bayoh, N.M.; et al. Safety and mosquitocidal efficacy of high-dose ivermectin when co-administered with dihydroartemisinin-piperaquine in Kenyan adults with uncomplicated malaria (IVERMAL): A randomised, double-blind, placebo-controlled trial. Lancet Infect. Dis. 2018, 18, 615–626. [Google Scholar] [CrossRef]
- Muñoz, J.; Ballester, M.R.; Antonijoan, R.M.; Gich, I.; Rodríguez, M.; Colli, E.; Gold, S.; Krolewiecki, A.J. Safety and pharmacokinetic profile of fixed-dose ivermectin with an innovative 18mg tablet in healthy adult volunteers. PLoS Negl. Trop. Dis. 2018, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Guzzo, C.A.; Furtek, C.I.; Porras, A.G.; Chen, C.; Tipping, R.; Clineschmidt, C.M.; Sciberras, D.G.; Hsieh, J.Y.K.; Lasseter, K.C. Safety, tolerability, and pharmacokinetics of escalating high doses of ivermectin in healthy adults subjects. J. Clin. Pharmacol. 2002, 42, 1122–1133. [Google Scholar] [CrossRef]
- Duthaler, U.; Suenderhauf, C.; Karlsson, M.O.; Hussner, J.; Zu Schwabedissen, H.M.; Krähenbühl, S.; Hammann, F. Population pharmacokinetics of oral ivermectin in venous plasma and dried blood spots in healthy volunteers. Br. J. Clin. Pharmacol. 2019, 85, 626–633. [Google Scholar] [CrossRef]
- Singh, L.; Fontinha, D.; Francisco, D.; Mendes, A.M.; Prudêncio, M.; Singh, K. Molecular design and synthesis of ivermectin hybrids targeting hepatic and erythrocytic stages of Plasmodium parasites. J. Med. Chem. 2020, 63, 1750–1762. [Google Scholar] [CrossRef]
- Spízek, J.; Rezanka, T. Lincosamides: Chemical structure, biosynthesis, mechanism of action, resistance and applications. Biochem. Pharmacol. 2017, 133, 20–28. [Google Scholar] [CrossRef]
- Kadlcik, S.; Kamenik, Z.; Vasek, D.; Nedved, M.; Janata, J. Elucidation of salicylate attachment in celesticetin biosynthesis opens the door to create a library of more efficient hybrid lincosamide antibiotics. Chem. Sci. 2017, 8, 3349–3355. [Google Scholar] [CrossRef] [PubMed]
- Tenson, T.; Lovmar, M.; Ehrenberg, M. The mechanism of action of macrolides, lincosamides and streptogramin B reveals the nascent peptide exit path in the ribosome. J. Mol. Biol. 2003, 330, 1005–1014. [Google Scholar] [CrossRef]
- Sasaki, E.; Lin, C.-I.; Lin, K.-Y.; Liu, H.-W. Construction of the octose 8-phosphate intermediate in Lincomycin A biosynthesis: Characterization of the reactions catalyzed by LmbR and LmbN. J. Am. Chem. Soc. 2012, 134, 17432–17435. [Google Scholar] [CrossRef]
- Lell, B.; Kremsner, P.G. Clindamycin as an antimalarial drug: Review of clinical trials. Antimicrob. Agents Chemother. 2007, 46, 2315–2320. [Google Scholar] [CrossRef]
- Held, J.; Westerman, R.; Kremsner, P.G. In vitro activity of mirincamycin (U24729A) against Plasmodium falciparum isolates from Gabon. Antimicrob. Agents Chemother. 2010, 54, 540–542. [Google Scholar] [CrossRef]
- Vento, T.J.; Cole, D.W.; Mende, K.; Calvano, T.P.; Rini, E.A.; Tully, C.C.; Zera, W.C.; Guymon, C.H.; Yu, X.; Cheatle, K.A.; et al. Multidrug-resistant gram-negative bacteria colonization of healthy US military personnel in the US and Afghanistan. BMC Infect Dis. 2013, 13, 1–12. [Google Scholar] [CrossRef]
- Vento, T.J.; Calvano, T.P.; Cole, D.W.; Mende, K.; Rini, E.A.; Tully, C.C.; Landrum, M.L.; Zera, W.; Guymon, C.H.; Yu, X.; et al. Staphylococcus aureus colonization of healthy military service members in the United States and Afghanistan. BMC Infect. Dis. 2013, 13, 1–9. [Google Scholar] [CrossRef]
- Phu, N.H.; Day, N.P.J.; Tuan, P.Q.; Mai, N.T.H.; Chau, T.T.H.; Van Chuong, L.; Vinh, H.; Loc, P.P.; Sinh, D.X.; Hoa, N.T.T.; et al. Concomitant bacteremia in adults with severe falciparum malaria. Clin. Infect. Dis. 2020, 71, 465–470. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
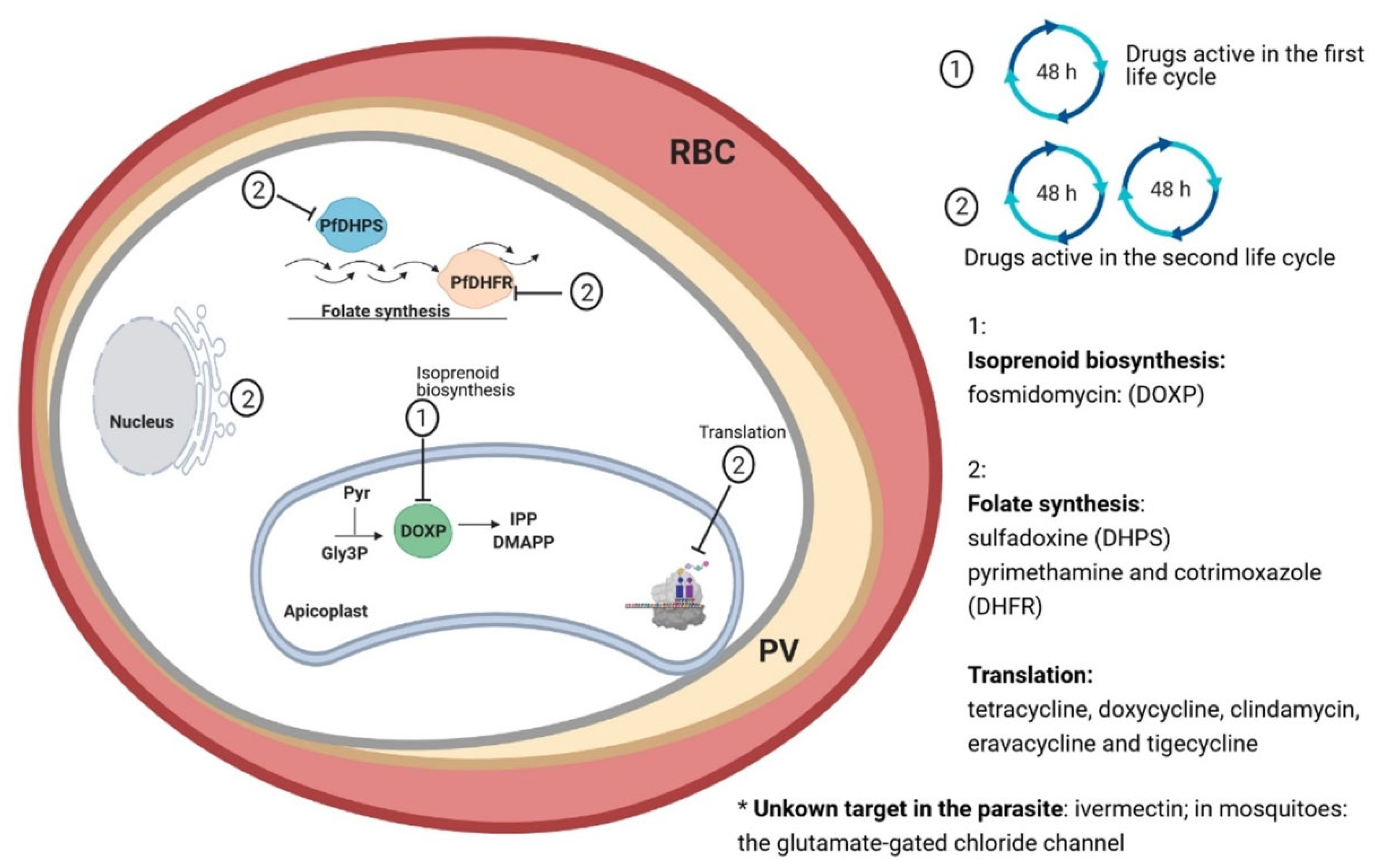{kind=link}
| Antibiotic | Outcomes – Pre-Clinical Data | |||
|---|---|---|---|---|
| In vitro (IC50) | Ref | In mice | Ref | |
| Tigecycline | One-cycle assay: 66 clinical isolates from Bangladesh with parasite density of 8311–13,735/µL: mean 699 nM (range: 496–986). | [68] | P. berghei infected mice (5 per group) were treated with 3.7, 11.1, 33.3 and 100 mg/kg for 4 days. Only the treatment with 100 mg/kg/day cured the mice on day 28. | [72] |
| One-cycle assay: DW2: 568; 3D7: 332; 3 clinical isolates from Brazil: mean ~ 600 nM (range 344–726). | [69] | |||
| One-cycle assay: IC50 3D7: 2300 nM; Dd2: 2800 nM; Two-cycle assay: IC50: 3D7: 220 nM; Dd2: 173 nM; | [70] | |||
| Two-cycle assay: 23 clinical isolates from Gabon with a mean parasite density of 45,174/µL (range: 750–93,827): IC50: 160 nM (range: 114–223). | ||||
| Eravacycline | One-cycle assay: 3D7: IC50: 1996 nM; two-cycle assay: 14 nM. Thirty-three clinical isolates from Gabon with parasitemia at 0.05%. | [73] | ||
| One-day assay: IC50: 69 nM (range: 35–142); two-day assay: IC50: 29 nM (range: 13–157). | ||||
| Fosmidomycin | One-cycle assay: IC50 3D7:150 (range:100–240); HB3:71 (range: 46–140); Dd2:170 (range: 120–260); A2:150 (70–260); | [83] | Five P. vinckei infected mice were treated with 30 mg/kg daily for 8 days. All mice were cured on day 28. | [79] |
| One-cycle assay with 3D7: FMD showed synergism with CLD (FIC: 0.43); additive effect with doxycycline (FIC: 0.93), quinine (FIC: 0.93) and azithromycin (FIC: 0.84). | P. vinckei infected mice were treated for 2 days with 75 mg/kg FMD and 5 mg/kg CLD separately or in combination. The parasitemia of mice treated with FMD or CLD was 7.8 and 20% on day 3, respectively, while the controls had 42%. The combination reduced the parasitemia to 0.1% on day 3 and 0.2% on day 5. | [83] | ||
| Ivermectin | One-cycle assay: IC50 (mean) 3D7: 100 nM; Dd2: 110 nM; K1:365 nM; clinical isolates from Gabon (0.05% parasitemia): IC50 (mean) ~ 100 nM mature gametocytes: 500 nM. | [120] | Three × 10 mg/kg reduced ~ 80% of P. berghei load in mice 46 h after infection. | [121] |
| In vitro addition of 2 µM IVM impaired human hepatoma cells infection by P. berghei | [121] | |||
| Clinical Data | Ref |
|---|---|
| Cotrimoxazole | |
| Three-hundred HIV-infected Ugandan children received CTM prophylaxis, while 561 healthy children | [47] |
| were followed as control. After 11 months, only nine cases of malaria were diagnosed | |
| among children taking CTM prophylaxis, in comparison with 440 children in the control group. | |
| HIV-uninfected Ugandan children aged 6 weeks to 9 months breastfed on HIV-infected | [48] |
| mothers received CTM syrup (40 mg TM and 200 mg SFM) at the following doses: | |
| 2.5 mL/day for children ≤ 4 kg, 5 mL/day for children > 4–8 kg, and 10 mL/day for children | |
| > 8–15 kg. Children weighing 10–15 kg received CTM tablets (80 mg TM and 400 mg SFM) | |
| and were prescribed one tablet daily thereafter. After cessation of breastfeeding, | |
| HIV uninfected children were randomized to CTM prophylaxis (n = 87) | |
| or to continue daily CTM prophylaxis until 2 years of age (n = 98); 699 episodes of malaria | |
| in total: 299 episodes in the prophylaxis group and 400 episodes in the discontinued group. | |
| HIV-infected patients aged 18 years or older received CTM daily prophylaxis in Uganda. | [49] |
| Baseline incidence of malaria was 50 episodes per 100 person-years during a 154-day follow up | |
| (466 participants). CTM prophylaxis was associated with 9 episodes of malaria per 100 person-years | |
| during 532-day follow-up (399 participants) (76% lower malaria rate), and CTM + ART | |
| was associated with 3.5 episodes per 100 person-years during a 126-day follow-up | |
| (1035 participants) (92% lower malaria rate). | |
| In Nigeria, a single dose of 8 mg/kg of TM + 40 mg/kg SFM cured all 42 children aged 5–12 years | [50] |
| with UFM on day 3. On day 14, all patients were still negative, but on day 67, 24 out of 36 patients were positive again. | |
| A total of 102 Nigerian children aged 0.5–12 years with UFM were treated with 20 mg/kg | [51] |
| CTM, twice daily for 5 days: on day 7, they had lower propensity to develop gametocytes | |
| than SP (34 versus 63%), checked by light microscopy. | |
| In Malawi, 205 children aged 0.5–5 years with UFM received CTM or SP for 5 days plus | [52] |
| ERY 125 mg 4 × day < 10 kg; 250 mg > 10 kg. Eighty-seven percent of children receiving CTM and 80% | |
| receiving SP reached adequate clinical responses on day 14. On day 7, gametocyte | |
| prevalence was 55% and 64% among children receiving CTM and SP, respectively. | |
| In Kenya, 500 participants ≥ 18 years old, HIV-positive, and taking first-line AS and | [57] |
| CTM were randomized to discontinue with CTM prophylaxis (STOP-CTM; 250 individuals) | |
| or continue (CTX; 250 individuals). Blood samples were collected at months 0, 3, 6, | |
| 9 and 12. The prevalence of mutant haplotypes associated with SP-resistant parasites | |
| in pfdhfr (51I/59R/108N) was 52% in the STOP-CTM arm versus 6.3% in the CTM arm. | |
| The pfdhps (437G/540E) was found in 57% in the STOP-CTM and 25% in the CTM arm. | |
| Fosmidomycin | |
| A total of 11 Gabonese and 15 Thai adults with UFM were treated with 1200 mg every 8 h for 7 days. | [81] |
| Seventy-eight percent of Gabonese and 22% of Thai patients were cured on day 28. | |
| A total of 27 Gabonese adults with UFM: 1–2 g every 8 h for 3, 4, or 5 days, | [82] |
| cure rates on day 14: 60, 88 and 89%, respectively. | |
| In Thailand, 70 patients with 15–61 years old with UFM were treated with two regimens of FMD in combination | [84] |
| with CLD. Group I: FMD (900 mg) and CLD (300 mg) every 6 h for 3 days | |
| (n = 25); Group II: FMD (1800 mg) and CLD (600 mg) every 12 h for 3 days (n = 54). | |
| The cure rates for Group I and Group II were 91.3 and 89.7% on day 28, respectively. | |
| A total of 36 Gabonese children 7–14 years with UFM were subjected to: FMD (30 mg/kg) + | [85] |
| CLD (5 mg/kg); FMD 30 mg/kg or CLD 5 mg/kg, every 12 h for 5 days. | |
| FMD + CLD or only CLD cured on day 28. | |
| A total of 105 Gabonese children aged 3–14 years with UFM received FMD (30 mg/kg) + | [86] |
| CLD (10 mg/kg) every 12 h for 3 days. 94% efficacy on day 28. | |
| A total of 51 Gabonese children 1–14 years old with UFM were treated with 3-day combination | [87] |
| of FMD (30 mg/kg) and CLD (10 mg/kg), respectively every 12 h. | |
| The cure rate on day 28 was only 62%. | |
| A total of 37 Mozambican children 6–36 months with UFM received 2 × day FMD (30 mg/kg) | [88] |
| and CLD (10 mg/kg): 45.9% cure on day 28. | |
| A total of 50 Gabonese children with UFM were treated with AS-FMD (1 to 2 mg/kg and 30 mg/kg, | [89] |
| respectively), every 12 h on 2 or 4-day regimens. A 3-day regimen or longer achieved 100% cure on day 28. | |
| Ivermectin | |
| In London, 25 healthy volunteers received IVM (200µg/kg) or placebo. One day later, mosquitoes were fed | [113] |
| on volunteers and their mean survival was 2.3 days (IVM group) and 5.5 days (control group): | |
| mosquito mortality was 73, 84, and 89% on days 2, 3, and 4, respectively in the IVM group. | |
| No differences were found between the groups when mosquitos were fed 14 days after treatment. | |
| In Burkina Faso, healthy patients with at least 90 cm in height received a single dose | [118] |
| of IVM (150–200 µg/kg) and albendazole (400 mg) (control group n = 233). The intervention group (n = 330) received | |
| 5 more doses of IVM at 3-week intervals over the 18-week treatment phase. Incidence of | |
| malaria in the intervention group was 2 episodes per child and in the control group 2.39 episodes, | |
| showing that mass drug administration of IVM reduced malaria episodes during the transmission season. | |
| Controlled human malaria infection trial, in malaria naïve volunteers in Germany: 8 out 12 participants | [122] |
| received IVM 0.4 mg/kg once 2 h before being infected intravenously with 3200 P. falciparum sporozoites. | |
| No significant effect on parasitemia, showing that this dose of IVM has no major effect on the liver stage of P. falciparum. | |
| In Kenya, adults with UFM received 3 days of IVM at 300 (n = 48), 600 µg/kg (n = 47) or placebos (n = 46) + 3 days of DHA-PPQ. A. gambiae were fed with blood taken of patients on days 0.2 + 4 h, 7, 10, 14, 21, and 28 days post-treatment. Mosquito survival was checked daily until day 28 after feeding. Mosquito fed on blood taken 7 days after treatment showed the higher mortality rate of 96, 92, and 41%, to 600 µg/kg, 300 µg/kg, and placebo, respectively. | [124] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pessanha de Carvalho, L.; Kreidenweiss, A.; Held, J. Drug Repurposing: A Review of Old and New Antibiotics for the Treatment of Malaria: Identifying Antibiotics with a Fast Onset of Antiplasmodial Action. Molecules 2021, 26, 2304. https://doi.org/10.3390/molecules26082304
Pessanha de Carvalho L, Kreidenweiss A, Held J. Drug Repurposing: A Review of Old and New Antibiotics for the Treatment of Malaria: Identifying Antibiotics with a Fast Onset of Antiplasmodial Action. Molecules. 2021; 26(8):2304. https://doi.org/10.3390/molecules26082304
Chicago/Turabian StylePessanha de Carvalho, Lais, Andrea Kreidenweiss, and Jana Held. 2021. "Drug Repurposing: A Review of Old and New Antibiotics for the Treatment of Malaria: Identifying Antibiotics with a Fast Onset of Antiplasmodial Action" Molecules 26, no. 8: 2304. https://doi.org/10.3390/molecules26082304
APA StylePessanha de Carvalho, L., Kreidenweiss, A., & Held, J. (2021). Drug Repurposing: A Review of Old and New Antibiotics for the Treatment of Malaria: Identifying Antibiotics with a Fast Onset of Antiplasmodial Action. Molecules, 26(8), 2304. https://doi.org/10.3390/molecules26082304