
Multiplexed In Situ Protein Profiling with High-Performance Cleavable Fluorescent Tyramide


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
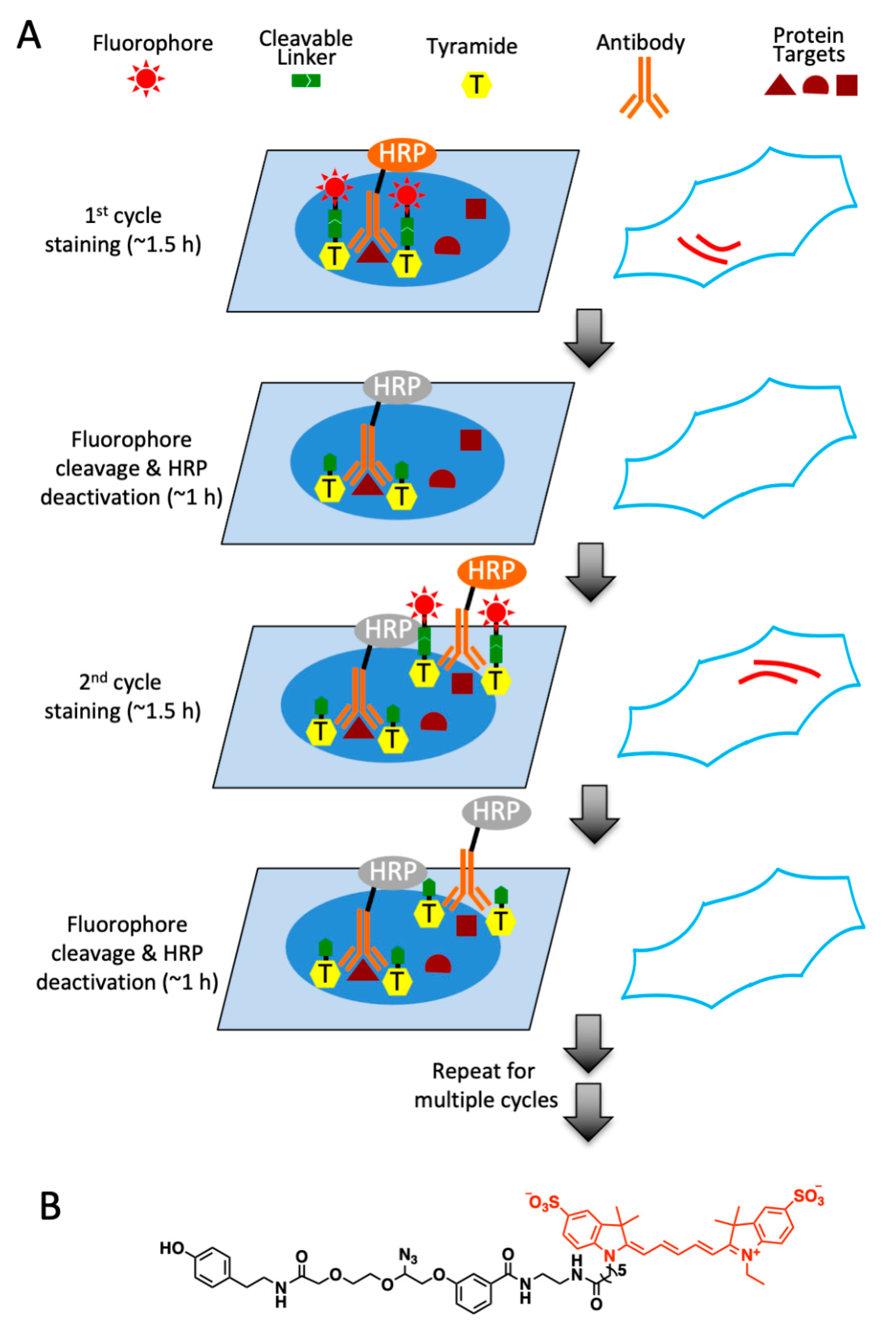
2.1. Platform Design
2.2. Design and Synthesis of High Performance CFT
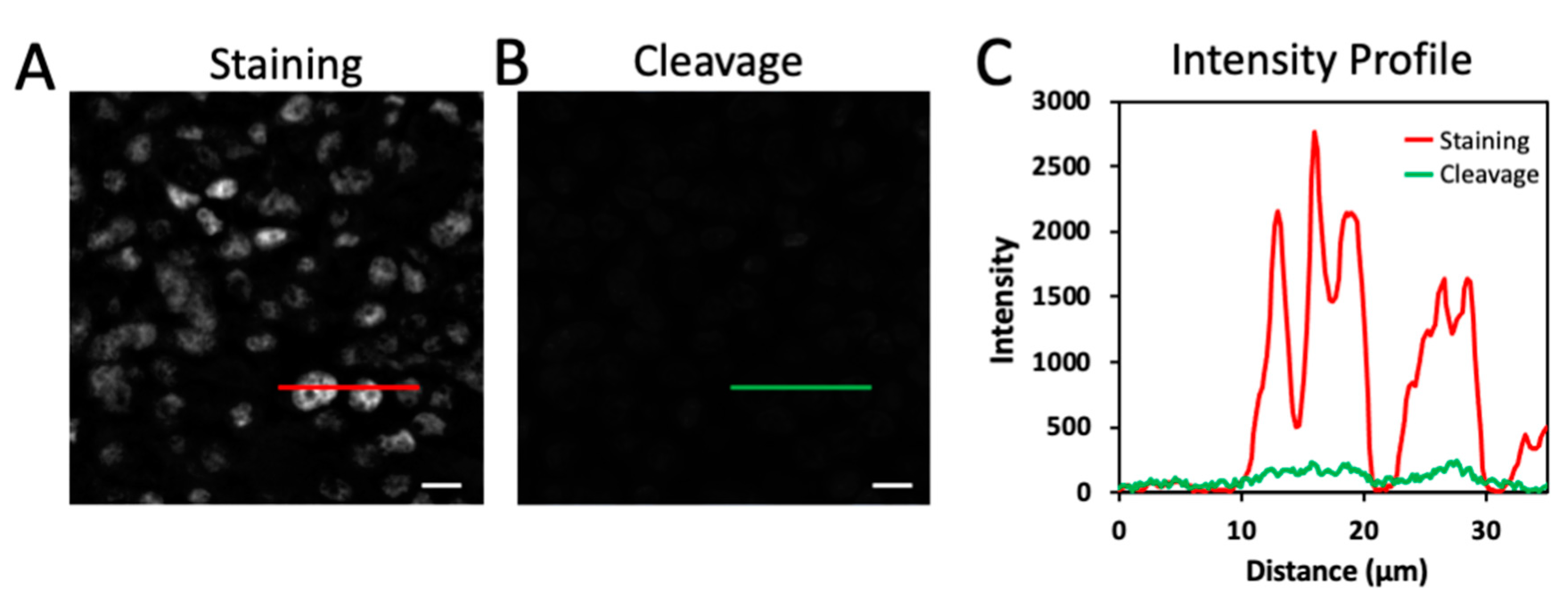
2.3. Efficient Fluorophore Cleavage While Preserving Epitope Integrity
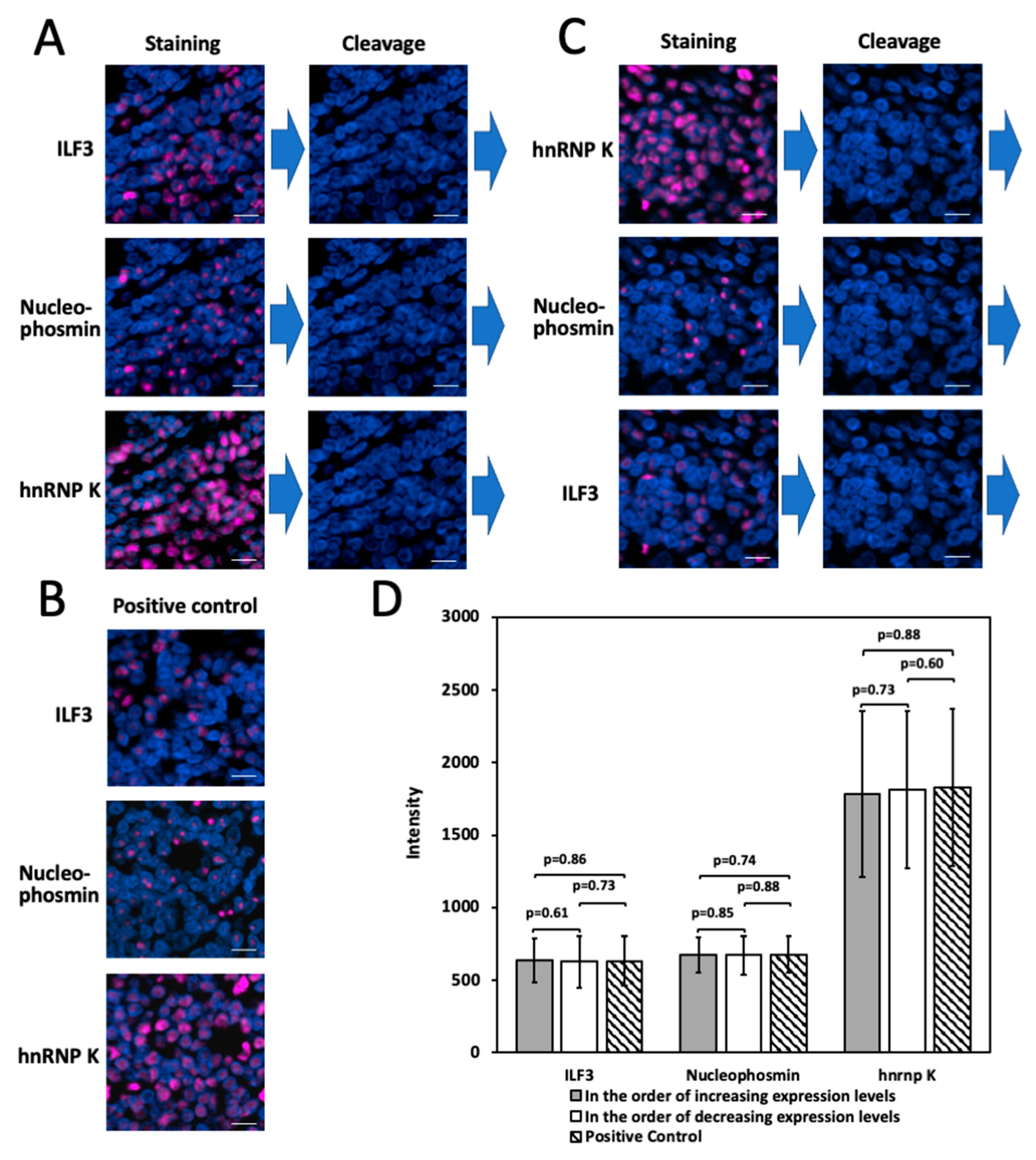
2.4. Simultaneous HRP Deactivation and Fluorophore Cleavage
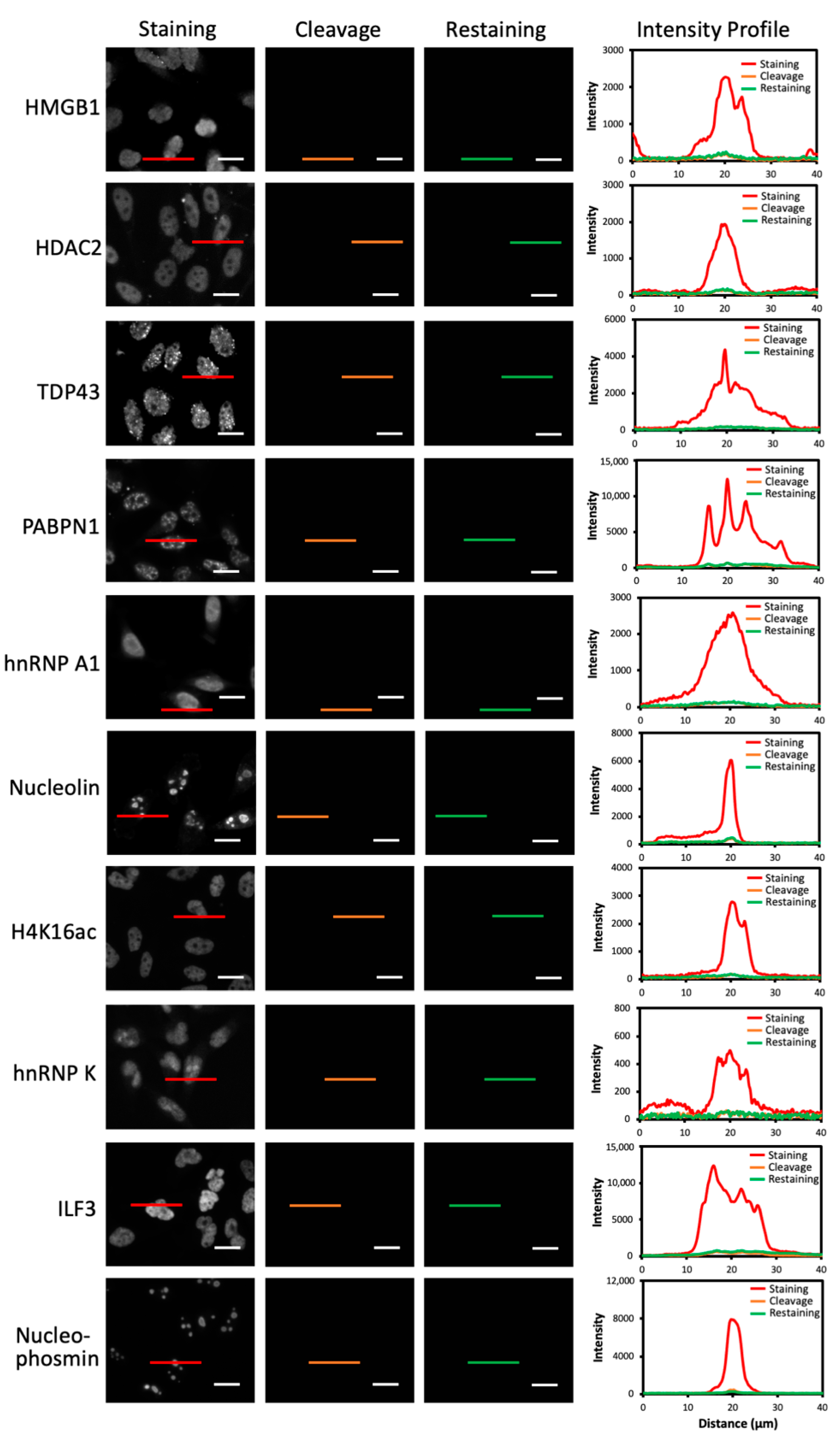
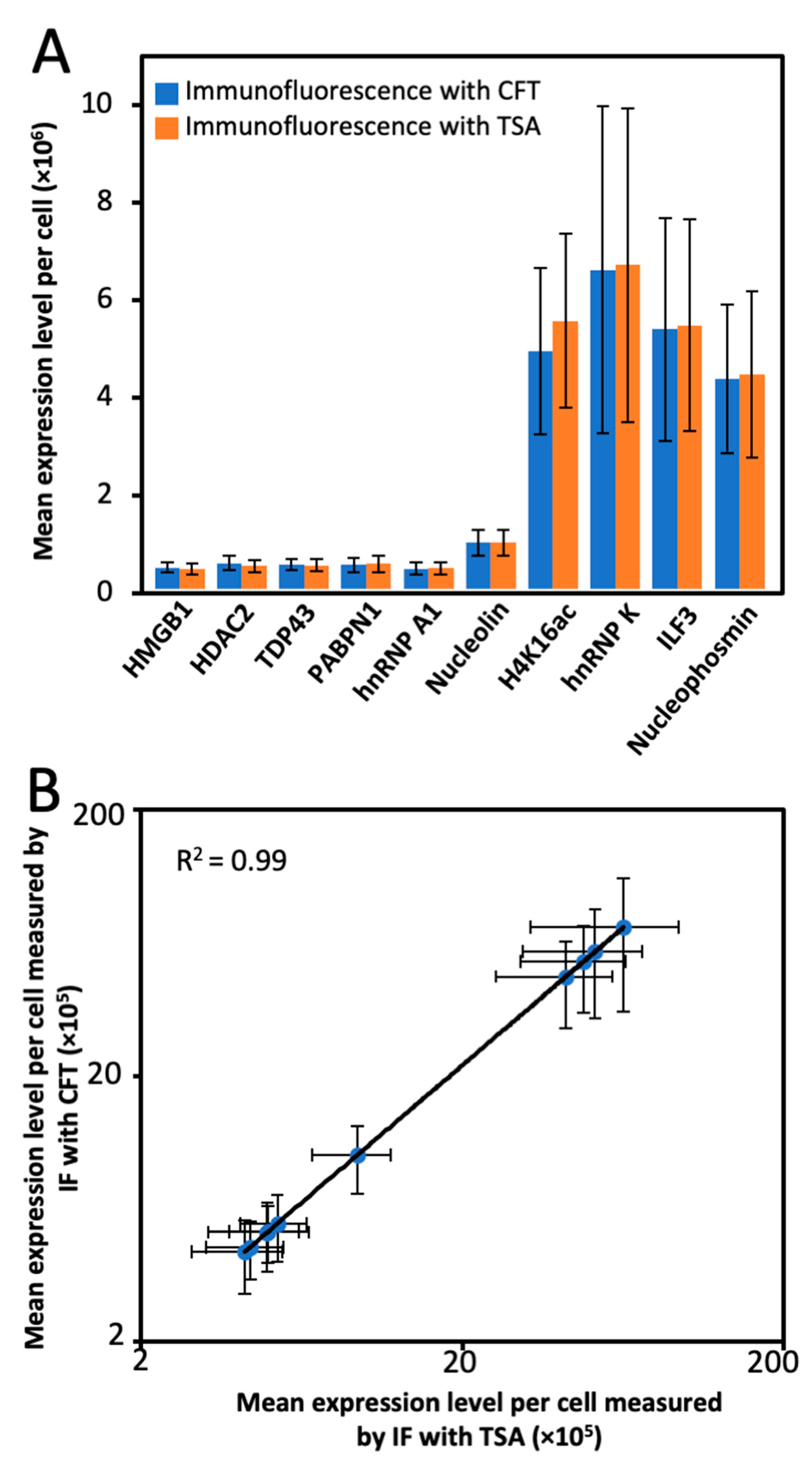
2.5. Multiplexed Protein Imaging in Cultured Cells
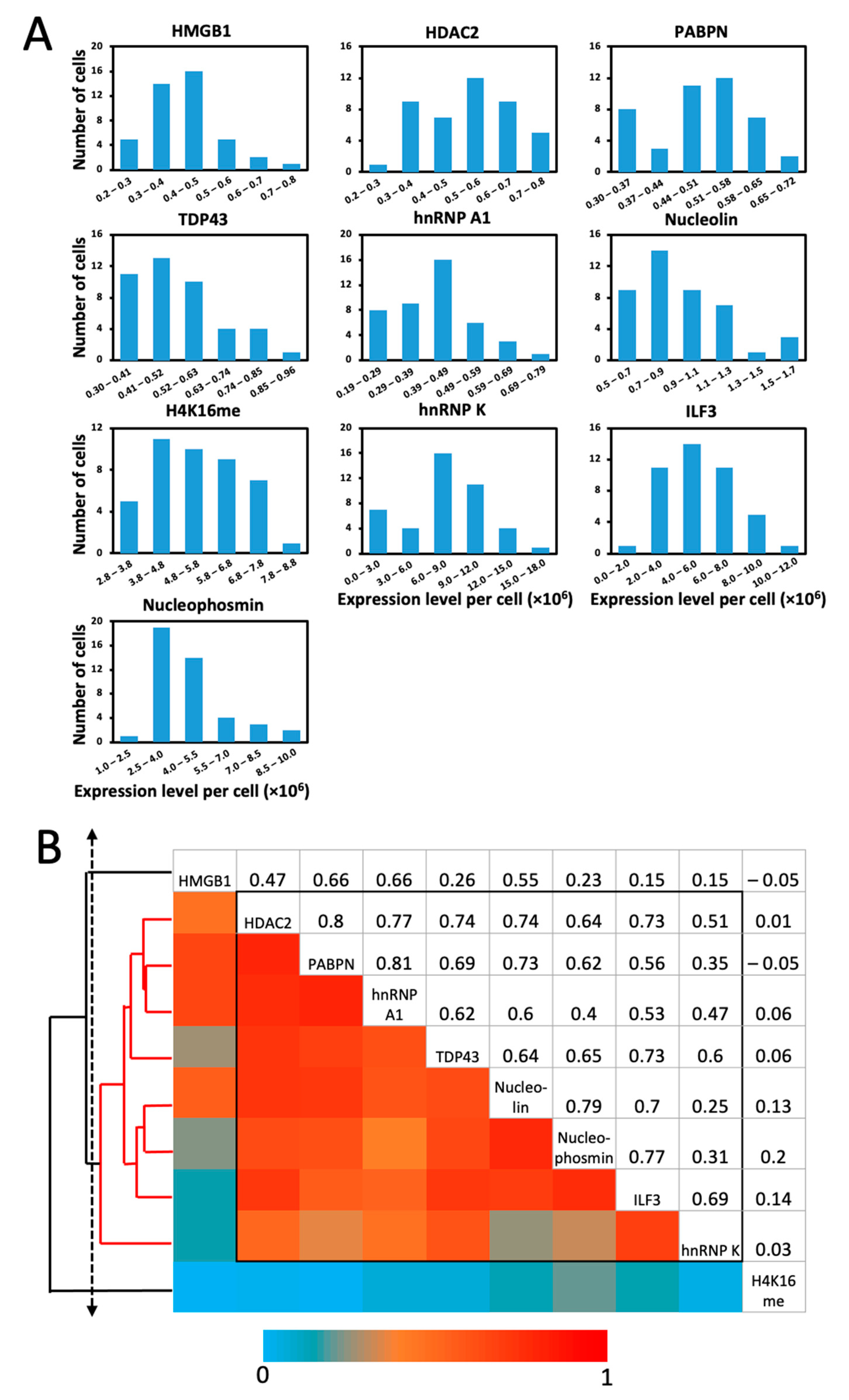
2.6. Single-Cell Protein Expression Heterogeneity and Correlation
2.7. Multiplexed In Situ Protien Profiling in FFPE Tissues
3. Materials and Methods
3.1. General Information
3.2. Synthesis of Tyramide-N3-Cy5
3.3. Deparaffinization and Antigen Retrieval of FFPE Tonsil Tissue
3.4. Protein Staining in FFPE Tonsil Tissue
3.5. Fluorophore Cleavage and HRP Deactivation
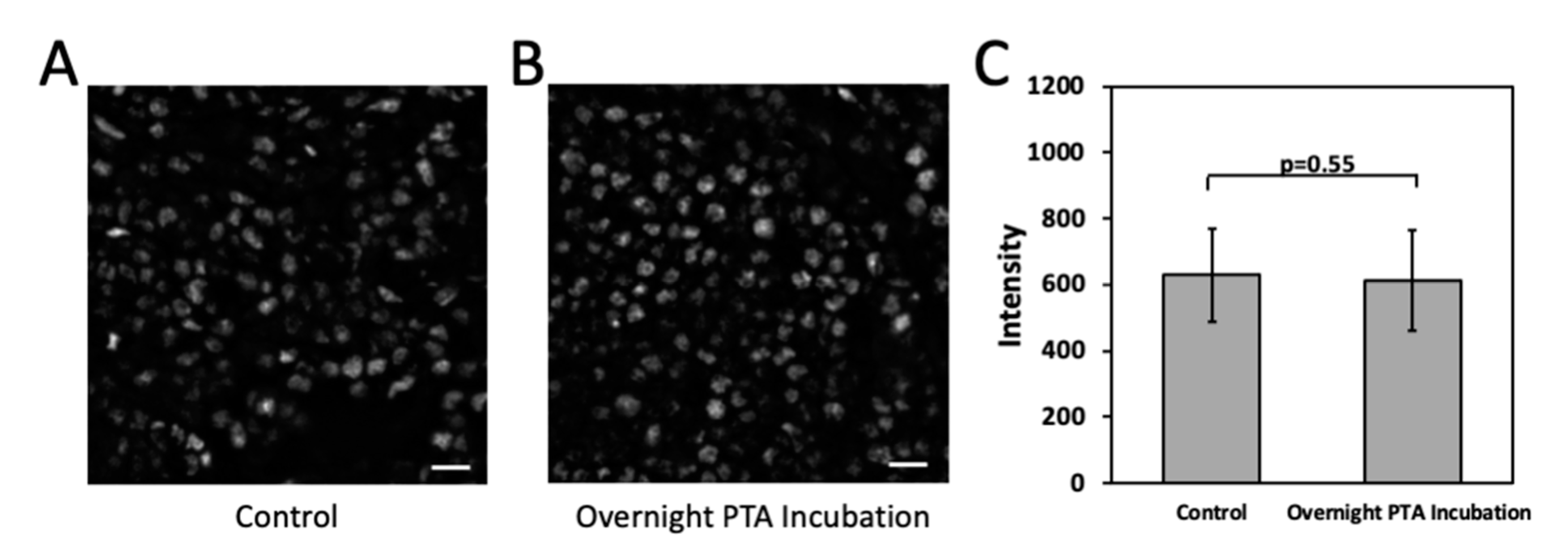
3.6. Effect of Cleavage by PTA
3.7. Cell Culture
3.8. Cell Fixation and Permeabilization
3.9. Immunofluorescence with CFT
3.10. Multiplexed Protein Imaging in Cells
3.11. Multiplexed Protein Imaging in FFPE Tonsil Tissue
3.12. Imaging and Data Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Steininger, R.J.; Rajaram, S.; Girard, L.; Minna, J.D.; Wu, L.F.; Altschuler, S.J. On comparing heterogeneity across biomarkers. Cytom. Part A 2015, 87, 558–567. [Google Scholar] [CrossRef]
- Junker, J.P.; Van Oudenaarden, A. Every cell is special: Genome-wide studies add a new dimension to single-cell biology. Cell 2014, 157, 8–11. [Google Scholar] [CrossRef] [PubMed]
- LaBaer, J.; Ramachandran, N. Protein microarrays as tools for functional proteomics. Curr. Opin. Chem. Biol. 2005, 9, 14–19. [Google Scholar] [CrossRef]
- Altelaar, F.M.; Munoz, J.; Heck, A.J.R. Next-generation proteomics: Towards an integrative view of proteome dynamics. Nat. Rev. Genet. 2012, 14, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Wang, S.; Dai, N.; Teo, Y.N.; Kool, E.T. Multispectral labeling of antibodies with polyfluorophores on a DNA backbone and application in cellular imaging. Proc. Natl. Acad. Sci. USA 2011, 108, 3493–3498. [Google Scholar] [CrossRef]
- Angelo, M.; Bendall, S.C.; Finck, R.; Hale, M.B.; Hitzman, C.; Borowsky, A.D.; Levenson, R.M.; Lowe, J.B.; Liu, S.D.; Zhao, S.; et al. Multiplexed ion beam imaging of human breast tumors. Nat. Med. 2014, 20, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Robertson, D.; Savage, K.; Reis-Filho, J.S.; Isacke, C.M. Multiple immunofluorescence labeling of formalin-fixed paraffin-embedded tissue. BMC Mol. Biol. 2008, 9, 1–10. [Google Scholar] [CrossRef]
- Mondal, M.; Liao, R.; Xiao, L.; Eno, T.; Guo, J. Highly Multiplexed Single-Cell In Situ Protein Analysis with Cleavable Fluorescent Antibodies. Angew. Chemie Int. Ed. 2017, 56, 2636–2639. [Google Scholar] [CrossRef]
- Schweller, R.M.; Zimak, J.; Duose, D.Y.; Qutub, A.A.; Hittelman, W.N.; Diehl, M.R. Multiplexed in situ immunofluorescence using dynamic DNA complexes. Angew. Chem. Int. Ed. Engl. 2012, 51, 9292–9296. [Google Scholar] [CrossRef]
- Duose, D.Y.; Schweller, R.M.; Zimak, J.; Rogers, A.R.; Hittelman, W.N.; Diehl, M.R. Configuring robust DNA strand displacement reactions for in situ molecular analyses. Nucleic Acids Res. 2012, 40, 3289–3298. [Google Scholar] [CrossRef] [PubMed]
- Zrazhevskiy, P.; Gao, X. Quantum dot imaging platform for single-cell molecular profiling. Nat. Commun. 2013, 4, 1619. [Google Scholar] [CrossRef]
- Lin, J.R.; Fallahi-Sichani, M.; Sorger, P.K. Highly multiplexed imaging of single cells using a high-throughput cyclic immunofluorescence method. Nat. Commun. 2015, 6, 1–7. [Google Scholar] [CrossRef]
- Goltsev, Y.; Samusik, N.; Kennedy-Darling, J.; Bhate, S.; Hale, M.; Vazquez, G.; Black, S.; Nolan, G.P. Deep Profiling of Mouse Splenic Architecture with CODEX Multiplexed Imaging. Cell 2018, 174, 968–981.e15. [Google Scholar] [CrossRef]
- Giesen, C.; Wang, H.A.O.; Schapiro, D.; Zivanovic, N.; Jacobs, A.; Hattendorf, B.; Schüffler, P.J.; Grolimund, D.; Buhmann, J.M.; Brandt, S.; et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 2014, 11, 417–422. [Google Scholar] [CrossRef]
- Liao, R.; Pham, T.; Mastroeni, D.; Coleman, P.D.; Labaer, J.; Guo, J. Highly Sensitive and Multiplexed In-Situ Protein Profiling with Cleavable Fluorescent Streptavidin. Cells 2020, 9, 852. [Google Scholar] [CrossRef]
- Saka, S.K.; Wang, Y.; Kishi, J.Y.; Zhu, A.; Zeng, Y.; Xie, W.; Kirli, K.; Yapp, C.; Cicconet, M.; Beliveau, B.J.; et al. Immuno-SABER enables highly multiplexed and amplified protein imaging in tissues. Nat. Biotechnol. 2019, 37, 1080–1090. [Google Scholar] [CrossRef] [PubMed]
- Liao, R.; Mondal, M.; Nazaroff, C.D.; Mastroeni, D.; Coleman, P.D.; Labaer, J.; Guo, J. Highly Sensitive and Multiplexed Protein Imaging With Cleavable Fluorescent Tyramide Reveals Human Neuronal Heterogeneity. Front. Cell Dev. Biol. 2021, 8, 1–15. [Google Scholar] [CrossRef]
- Mondal, M.; Liao, R.; Nazaroff, C.D.; Samuel, A.D.; Guo, J. Highly multiplexed single-cell in situ RNA and DNA analysis with bioorthogonal cleavable fluorescent oligonucleotides. Chem. Sci. 2018, 9, 2909–2917. [Google Scholar] [CrossRef] [PubMed]
- Jun, Y.W.; Kim, H.R.; Reo, Y.J.; Dai, M.; Ahn, K.H. Addressing the autofluorescence issue in deep tissue imaging by two-photon microscopy: The significance of far-red emitting dyes. Chem. Sci. 2017, 8, 7696–7704. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.H.; Yao, M.C.; Wu, H.Y.; Dong, J.; Ni, H.Y.; Kou, X.L.; Chang, L.; Luo, C.X.; Zhu, D.Y. HDAC2 (Histone deacetylase 2): A critical factor in environmental enrichment-mediated stroke recovery. J. Neurochem. 2020, 155, 679–696. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Ke, H.; Xu, H.; Wang, G.D.; Zhang, H.; Zou, L.; Xiang, S.; Li, M.; Peng, L.; Zhou, M.; et al. TDP-43 facilitates milk lipid secretion by post-transcriptional regulation of Btn1a1 and Xdh. Nat. Commun. 2020, 11, 341. [Google Scholar] [CrossRef] [PubMed]
- Harish, P.; Forrest, L.; Herath, S.; Dickson, G.; Malerba, A.; Popplewell, L. Inhibition of Myostatin Reduces Collagen Deposition in a Mouse Model of Oculopharyngeal Muscular Dystrophy (OPMD) With Established Disease. Front. Physiol. 2020, 11, 184. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.; Nie, H.; Yu, P.; Xie, B.; Wang, C.; Yang, F.; Wei, G.; Ni, T. HNRNPA1-mediated 3′ UTR length changes of HN1 contributes to cancer- and senescence-associated phenotypes. Aging 2019, 11, 4407–4437. [Google Scholar] [CrossRef] [PubMed]
- Dubois, M.L.; Meller, A.; Samandi, S.; Brunelle, M.; Frion, J.; Brunet, M.A.; Toupin, A.; Beaudoin, M.C.; Jacques, J.F.; Lévesque, D.; et al. UBB pseudogene 4 encodes functional ubiquitin variants. Nat. Commun. 2020, 11, 1306. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ghorbani, M.; Weisz-Hubshman, M.; Rousseau, J.; Thiffault, I.; Schnur, R.E.; Breen, C.; Oegema, R.; Weiss, M.M.M.; Waisfisz, Q.; et al. Lysine acetyltransferase 8 is involved in cerebral development and syndromic intellectual disability. J. Clin. Invest. 2020, 130, 1431–1445. [Google Scholar] [CrossRef]
- Pellegrini, L.; Hauser, D.N.; Li, Y.; Mamais, A.; Beilina, A.; Kumaran, R.; Wetzel, A.; Nixon-Abell, J.; Heaton, G.; Rudenko, I.; et al. Proteomic analysis reveals co-ordinated alterations in protein synthesis and degradation pathways in LRRK2 knockout mice. Hum. Mol. Genet. 2018, 27, 3257–3271. [Google Scholar] [CrossRef]
- Watson, S.F.; Bellora, N.; MacIas, S. ILF3 contributes to the establishment of the antiviral type i interferon program. Nucleic Acids Res. 2020, 48, 116–129. [Google Scholar] [CrossRef]
- Chen, Q.; Xi, X.; Zeng, Y.; He, Z.; Zhao, J.; Li, Y. Acteoside inhibits autophagic apoptosis of retinal ganglion cells to rescue glaucoma-induced optic atrophy. J. Cell. Biochem. 2019, 120, 13133–13140. [Google Scholar] [CrossRef]
- Maugeri, N.; Campana, L.; Gavina, M.; Covino, C.; De Metrio, M.; Panciroli, C.; Maiuri, L.; Maseri, A.; D’Angelo, A.; Bianchi, M.E.; et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J. Thromb. Haemost. 2014, 12, 2074–2088. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Thul, P.J.; Akesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Ait Blal, H.; Alm, T.; Asplund, A.; Björk, L.; Breckels, L.M.; et al. A subcellular map of the human proteome. Science 2017, 356. [Google Scholar] [CrossRef]
- Becskei, A.; Kaufmann, B.B.; van Oudenaarden, A. Contributions of low molecule number and chromosomal positioning to stochastic gene expression. Nat. Genet. 2005, 37, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Blake, W.J.; KAErn, M.; Cantor, C.R.; Collins, J.J. Noise in eukaryotic gene expression. Nature 2003, 422, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Elowitz, M.B.; Levine, A.J.; Siggia, E.D.; Swain, P.S. Stochastic gene expression in a single cell. Science 2002, 297, 1183–1186. [Google Scholar] [CrossRef] [PubMed]
- Golding, I.; Paulsson, J.; Zawilski, S.M.; Cox, E.C. Real-time kinetics of gene activity in individual bacteria. Cell 2005, 123, 1025–1036. [Google Scholar] [CrossRef] [PubMed]
- Ozbudak, E.M.; Thattai, M.; Kurtser, I.; Grossman, A.D.; van Oudenaarden, A. Regulation of noise in the expression of a single gene. Nat. Genet. 2002, 31, 69–73. [Google Scholar] [CrossRef]
- Raser, J.M.; O’Shea, E.K. Control of stochasticity in eukaryotic gene expression. Science 2004, 304, 1811–1814. [Google Scholar] [CrossRef]
- Rosenfeld, N.; Young, J.W.; Alon, U.; Swain, P.S.; Elowitz, M.B. Gene Regulation at the Single-Cell Level. Science 2005, 307, 1962–1965. [Google Scholar] [CrossRef]
- Raj, A.; Peskin, C.S.; Tranchina, D.; Vargas, D.Y.; Tyagi, S. Stochastic mRNA synthesis in mammalian cells. PLoS Biol. 2006, 4, 1707–1719. [Google Scholar] [CrossRef]
- Munsky, B.; Neuert, G.; van Oudenaarden, A. Using Gene Expression Noise to Understand Gene Regulation. Science 2012, 336, 183–187. [Google Scholar] [CrossRef]
- Eisen Michael, B.; Spellman Paul, T.; Brown Patrick, O.; Botstein, D. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA 1998, 95, 14863–14868. [Google Scholar] [CrossRef]
- Jahan, S.; Sun, J.-M.; He, S.; Davie, J.R. Transcription-dependent association of HDAC2 with active chromatin. J. Cell. Physiol. 2018, 233, 1650–1657. [Google Scholar] [CrossRef]
- Lalmansingh, A.S.; Urekar, C.J.; Reddi, P.P. TDP-43 is a transcriptional repressor: The testis-specific mouse acrv1 gene is a TDP-43 target in vivo. J. Biol. Chem. 2011, 286, 10970–10982. [Google Scholar] [CrossRef]
- Jean-Philippe, J.; Paz, S.; Caputi, M. hnRNP A1: The Swiss army knife of gene expression. Int. J. Mol. Sci. 2013, 14, 18999–19024. [Google Scholar] [CrossRef] [PubMed]
- Abdelmohsen, K.; Gorospe, M. RNA-binding protein nucleolin in disease. RNA Biol. 2012, 9, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Box, J.K.; Paquet, N.; Adams, M.N.; Boucher, D.; Bolderson, E.; O’Byrne, K.J.; Richard, D.J. Nucleophosmin: From structure and function to disease development. BMC Mol. Biol. 2016, 17, 19. [Google Scholar] [CrossRef]
- Banerjee, A.; Apponi, L.H.; Pavlath, G.K.; Corbett, A.H. PABPN1: Molecular function and muscle disease. FEBS J. 2013, 280, 4230–4250. [Google Scholar] [CrossRef] [PubMed]
- Castella, S.; Bernard, R.; Corno, M.; Fradin, A.; Larcher, J.-C. Ilf3 and NF90 functions in RNA biology. Wiley Interdiscip. Rev. RNA 2015, 6, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Gao, F.-H. Role and molecular mechanism of heterogeneous nuclear ribonucleoprotein K in tumor development and progression. Biomed. Rep. 2016, 4, 657–663. [Google Scholar] [CrossRef]
- Liu, G.; Amin, S.; Okuhama, N.N.; Liao, G.; Mingle, L.A. A quantitative evaluation of peroxidase inhibitors for tyramide signal amplification mediated cytochemistry and histochemistry. Histochem. Cell Biol. 2006, 126, 283–291. [Google Scholar] [CrossRef]
- Stack, E.C.; Wang, C.; Roman, K.A.; Hoyt, C.C. Multiplexed immunohistochemistry, imaging, and quantitation: A review, with an assessment of Tyramide signal amplification, multispectral imaging and multiplex analysis. Methods 2014, 70, 46–58. [Google Scholar] [CrossRef] [PubMed]
- van de Corput, M.P.; Dirks, R.W.; van Gijlswijk, R.P.; van de Rijke, F.M.; Raap, A.K. Fluorescence in situ hybridization using horseradish peroxidase-labeled oligodeoxynucleotides and tyramide signal amplification for sensitive DNA and mRNA detection. Histochem. Cell Biol. 1998, 110, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Wei, W.; Su, Y.; Kim, J.; Shin, Y.S.; Mai, W.X.; Nathanson, D.A.; Heath, J.R. Chemical methods for the simultaneous quantitation of metabolites and proteins from single cells. J. Am. Chem. Soc. 2015, 137, 4066–4069. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pham, T.; Liao, R.; Labaer, J.; Guo, J. Multiplexed In Situ Protein Profiling with High-Performance Cleavable Fluorescent Tyramide. Molecules 2021, 26, 2206. https://doi.org/10.3390/molecules26082206
Pham T, Liao R, Labaer J, Guo J. Multiplexed In Situ Protein Profiling with High-Performance Cleavable Fluorescent Tyramide. Molecules. 2021; 26(8):2206. https://doi.org/10.3390/molecules26082206
Chicago/Turabian StylePham, Thai, Renjie Liao, Joshua Labaer, and Jia Guo. 2021. "Multiplexed In Situ Protein Profiling with High-Performance Cleavable Fluorescent Tyramide" Molecules 26, no. 8: 2206. https://doi.org/10.3390/molecules26082206
APA StylePham, T., Liao, R., Labaer, J., & Guo, J. (2021). Multiplexed In Situ Protein Profiling with High-Performance Cleavable Fluorescent Tyramide. Molecules, 26(8), 2206. https://doi.org/10.3390/molecules26082206