
Diabetes and Cancer: Metabolic Association, Therapeutic Challenges, and the Role of Natural Products

 ,
, 
Abstract
1. Introduction
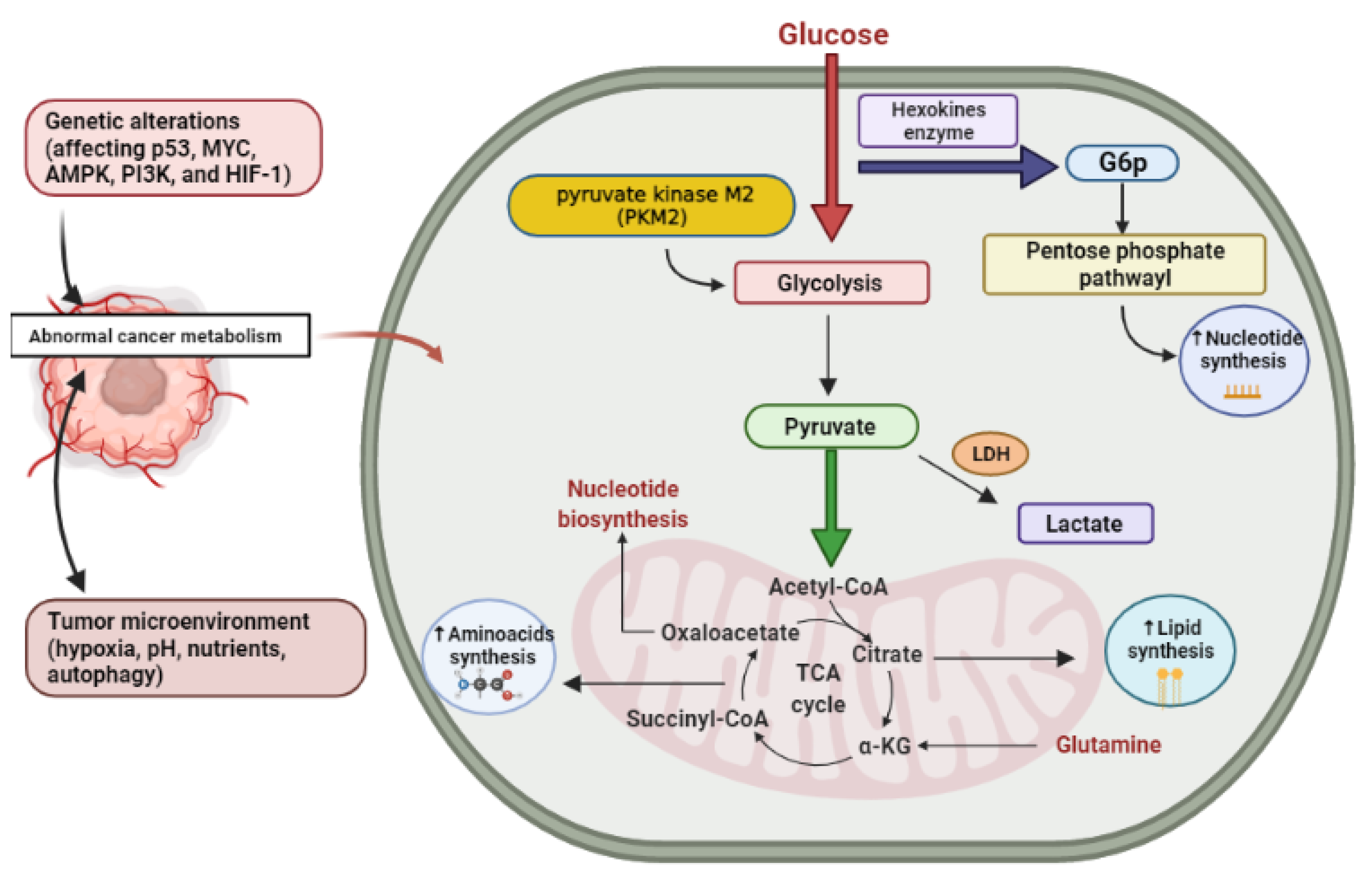
2. Cancer Altered Metabolism
2.1. Glucose
2.2. Glutamine
3. Diabetes Relationship with Metabolic Syndrome (MetS)
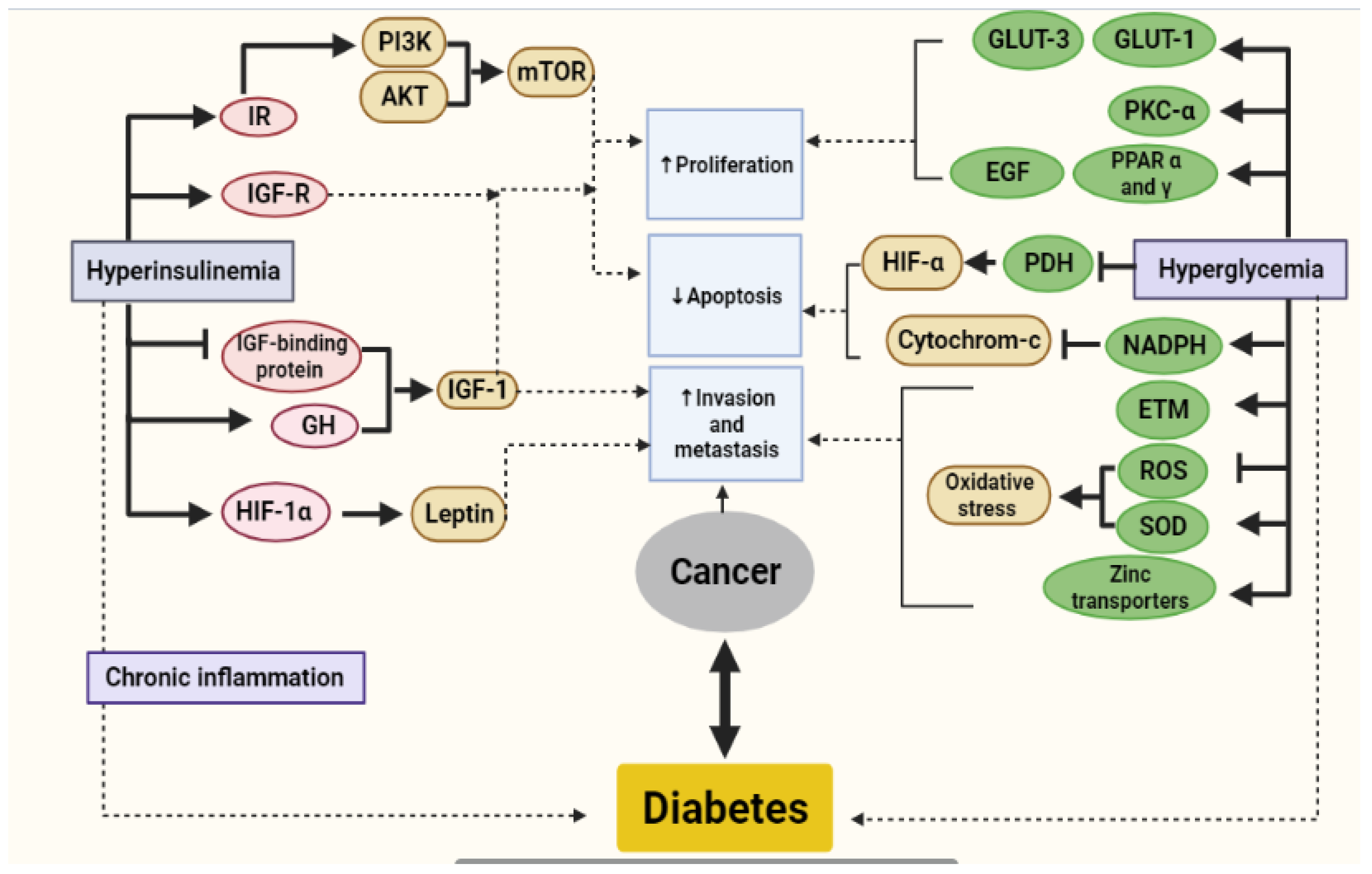
4. Metabolic Link between Diabetes and Cancer
4.1. Hyperinsulinemia
4.2. Hyperglycemia
4.3. Chronic Inflammation Due to Fat Imbalanced Metabolism
4.4. Correlation between DM and Specific Cancer Examples
4.4.1. Liver Cancer
4.4.2. Prostate Cancer
5. Impact of Diabetes and Obesity on Cancer
6. Therapeutic Challenges in Treating Patients with Diabetes and Cancer
6.1. Challenges Using Chemotherapeutic Agents
6.2. Challenges Using Glucocorticoids
6.3. Challenges of Using Cancer Treatments While on Glucose-Lowering Treatments
7. Suggested Therapies for Cancer and Diabetes Patients
8. Natural Products Targeting Diabetes and Cancer
8.1. Resveratrol
8.2. Curcumin
8.3. Thymoquinone
8.4. EGCG (Epigallocatechin Gallate)
8.5. Allicin
8.6. Emodin
8.7. Genistein
8.8. Parthenolide
8.9. Luteolin
8.10. Quercetin
8.11. Berberine
8.12. Phytosterols
9. The Controversy of Exogenous Antioxidants Administration in Cancer
10. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Zimmet, P.; Alberti, K.G.; Magliano, D.J.; Bennett, P.H. Diabetes mellitus statistics on prevalence and mortality: Facts and fallacies. Nat. Rev. Endocrinol. 2016, 12, 616–622. [Google Scholar] [CrossRef]
- Asmat, U.; Abad, K.; Ismail, K. Diabetes mellitus and oxidative stress—A concise review. Saudi Pharm. J. 2016, 24, 547–553. [Google Scholar] [CrossRef]
- Giri, B.; Dey, S.; Das, T.; Sarkar, M.; Banerjee, J.; Dash, S.K. Chronic hyperglycemia mediated physiological alteration and metabolic distortion leads to organ dysfunction, infection, cancer progression and other pathophysiological consequences: An update on glucose toxicity. Biomed. Pharmacother. 2018, 107, 306–328. [Google Scholar] [CrossRef]
- Wojciechowska, J.; Krajewski, W.; Bolanowski, M.; Kręcicki, T.; Zatoński, T. Diabetes and cancer: A review of current knowledge. Exp. Clin. Endocrinol. Diabetes 2016, 124, 263–275. [Google Scholar] [CrossRef]
- Suh, S.; Kim, K.-W. Diabetes and cancer: Cancer should be screened in routine diabetes assessment. Diabetes Metab. J. 2019, 43, 733. [Google Scholar] [CrossRef]
- De Martel, C.; Georges, D.; Bray, F.; Ferlay, J.; Clifford, G.M. Global burden of cancer attributable to infections in 2018: A worldwide incidence analysis. Lancet Glob. Health 2020, 8, e180–e190. [Google Scholar] [CrossRef]
- Cignarelli, A.; Genchi, V.A.; Caruso, I.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Diabetes and cancer: Pathophysiological fundamentals of a ‘dangerous affair’. Diabetes Res. Clin. Pract. 2018, 143, 378–388. [Google Scholar] [CrossRef]
- Lee, C.; An, D.; Park, J. Hyperglycemic memory in metabolism and cancer. Horm. Mol. Biol. Clin. Investig. 2016, 26, 77–85. [Google Scholar] [CrossRef]
- Ferroni, P.; Riondino, S.; Buonomo, O.; Palmirotta, R.; Guadagni, F.; Roselli, M. Type 2 diabetes and breast cancer: The interplay between impaired glucose metabolism and oxidant stress. Oxid. Med. Cell. Longev. 2015, 2015, 183928. [Google Scholar] [CrossRef]
- Srivastava, S.P.; Goodwin, J.E. Cancer biology and prevention in diabetes. Cells 2020, 9, 1380. [Google Scholar] [CrossRef]
- Sampayo, V.; Tofthagen, C. Hyperglycemia and Cancer: An algorithm to guide oncology nurses. Clin. J. Oncol. Nurs. 2017, 21, 345–352. [Google Scholar] [CrossRef]
- Ben, Q.; Xu, M.; Ning, X.; Liu, J.; Hong, S.; Huang, W.; Zhang, H.; Li, Z. Diabetes mellitus and risk of pancreatic cancer: A meta-analysis of cohort studies. Eur. J. Cancer 2011, 47, 1928–1937. [Google Scholar] [CrossRef]
- Elena, J.W.; Steplowski, E.; Yu, K.; Hartge, P.; Tobias, G.S.; Brotzman, M.J.; Chanock, S.J.; Stolzenberg-Solomon, R.Z.; Arslan, A.A.; Bueno-de-Mesquita, H.B. Diabetes and risk of pancreatic cancer: A pooled analysis from the pancreatic cancer cohort consortium. Cancer Causes Control 2013, 24, 13–25. [Google Scholar] [CrossRef]
- Villarreal-Garza, C.; Shaw-Dulin, R.; Lara-Medina, F.; Bacon, L.; Rivera, D.; Urzua, L.; Aguila, C.; Ramirez-Morales, R.; Santamaria, J.; Bargallo, E. Impact of diabetes and hyperglycemia on survival in advanced breast cancer patients. Exp. Diabetes Res. 2012, 2012, 732027. [Google Scholar] [CrossRef]
- Bjornsdottir, H.H.; Rawshani, A.; Rawshani, A.; Franzén, S.; Svensson, A.-M.; Sattar, N.; Gudbjörnsdottir, S. A national observation study of cancer incidence and mortality risks in type 2 diabetes compared to the background population over time. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Samuel, S.M.; Varghese, E.; Varghese, S.; Büsselberg, D. Challenges and perspectives in the treatment of diabetes associated breast cancer. Cancer Treat. Rev. 2018, 70, 98–111. [Google Scholar] [CrossRef]
- Zeng, L.; Zielinska, H.; Arshad, A.; Shield, J.; Bahl, A.; Holly, J.; Perks, C. Hyperglycaemia-induced chemoresistance in breast cancer cells: Role of the estrogen receptor. Endocr. Relat. Cancer 2016, 23, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Vigneri, P.; Frasca, F.; Sciacca, L.; Pandini, G.; Vigneri, R. Diabetes and cancer. Endocr. Relat. Cancer 2009, 16, 1103–1123. [Google Scholar] [CrossRef] [PubMed]
- Harding, J.L.; Andes, L.J.; Gregg, E.W.; Cheng, Y.J.; Weir, H.K.; Bullard, K.M.; Burrows, N.R.; Imperatore, G. Trends in cancer mortality among people with vs. without diabetes in the USA, 1988–2015. Diabetologia 2020, 63, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef]
- Villalba, M.; Rathore, M.G.; Lopez-Royuela, N.; Krzywinska, E.; Garaude, J.; Allende-Vega, N. From tumor cell metabolism to tumor immune escape. Int. J. Biochem. Cell Biol. 2013, 45, 106–113. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef]
- Danhier, P.; Bański, P.; Payen, V.L.; Grasso, D.; Ippolito, L.; Sonveaux, P.; Porporato, P.E. Cancer metabolism in space and time: Beyond the Warburg effect. Biochim. Biophys. Acta (BBA) Bioenerg. 2017, 1858, 556–572. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Matés, J.M.; Campos-Sandoval, J.A.; de Los Santos-Jiménez, J.; Márquez, J. Dysregulation of glutaminase and glutamine synthetase in cancer. Cancer Lett. 2019, 467, 29–39. [Google Scholar] [CrossRef]
- Bose, S.; Le, A. Glucose Metabolism in Cancer. Adv. Exp. Med. Biol. 2018, 1063, 3–12. [Google Scholar] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Patra, K.C.; Wang, Q.; Bhaskar, P.T.; Miller, L.; Wang, Z.; Wheaton, W.; Chandel, N.; Laakso, M.; Muller, W.J.; Allen, E.L. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 2013, 24, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, A.L.; Walton, Z.E.; Altman, B.J.; Stine, Z.E.; Dang, C.V. Seminars in Cell & Developmental Biology. In MYC and Metabolism on the Path to Cancer; Elsevier: Amsterdam, The Netherlands, 2015; pp. 11–21. [Google Scholar]
- Fadaka, A.; Ajiboye, B.; Ojo, O.; Adewale, O.; Olayide, I.; Emuowhochere, R. Biology of glucose metabolization in cancer cells. J. Oncol. Sci. 2017, 3, 45–51. [Google Scholar] [CrossRef]
- Feng, Y.; Xiong, Y.; Qiao, T.; Li, X.; Jia, L.; Han, Y. Lactate dehydrogenase A: A key player in carcinogenesis and potential target in cancer therapy. Cancer Med. 2018, 7, 6124–6136. [Google Scholar] [CrossRef]
- Palsson-McDermott, E.M.; Curtis, A.M.; Goel, G.; Lauterbach, M.A.; Sheedy, F.J.; Gleeson, L.E.; van den Bosch, M.W.; Quinn, S.R.; Domingo-Fernandez, R.; Johnston, D.G. Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. 2015, 21, 65–80. [Google Scholar] [CrossRef]
- Seton-Rogers, S. Feed it forward. Nat. Rev. Cancer 2011, 11, 461. [Google Scholar] [CrossRef]
- Daye, D.; Wellen, K.E. Seminars in Cell & Developmental Biology. In Metabolic Reprogramming in Cancer: Unraveling the Role of Glutamine in Tumorigenesis; Elsevier: Amsterdam, The Netherlands, 2012; pp. 362–369. [Google Scholar]
- Phan, L.M.; Yeung, S.-C.J.; Lee, M.-H. Cancer metabolic reprogramming: Importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol. Med. 2014, 11, 1. [Google Scholar] [PubMed]
- Hensley, C.T.; Wasti, A.T.; DeBerardinis, R.J. Glutamine and cancer: Cell biology, physiology, and clinical opportunities. J. Clin. Investig. 2013, 123, 3678–3684. [Google Scholar] [CrossRef]
- Deshmukh, A.; Deshpande, K.; Arfuso, F.; Newsholme, P.; Dharmarajan, A. Cancer stem cell metabolism: A potential target for cancer therapy. Mol. Cancer 2016, 15, 1–10. [Google Scholar] [CrossRef]
- Reaven, G.M. Role of insulin resistance in human disease. Diabetes 1988, 37, 1595–1607. [Google Scholar] [CrossRef] [PubMed]
- Alberti, K.; Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z.; Cleeman, J.I.; Donato, K.A.; Fruchart, J.-C.; James, W.P.T.; Loria, C.M.; Smith, S.C., Jr. Harmonizing the metabolic syndrome: A joint interim statement of the international diabetes federation task force on epidemiology and prevention; national heart, lung, and blood institute; American heart association; world heart federation; international atherosclerosis society; and international association for the study of obesity. Circulation 2009, 120, 1640–1645. [Google Scholar]
- Lakka, H.-M.; Laaksonen, D.E.; Lakka, T.A.; Niskanen, L.K.; Kumpusalo, E.; Tuomilehto, J.; Salonen, J.T. The metabolic syndrome and total and cardiovascular disease mortality in middle-aged men. JAMA 2002, 288, 2709–2716. [Google Scholar] [CrossRef]
- Alberti, K.G.M.M.; Zimmet, P.; Shaw, J. Metabolic syndrome—A new world-wide definition. A consensus statement from the international diabetes federation. Diabet. Med. 2006, 23, 469–480. [Google Scholar] [CrossRef]
- Ford, E.S. Insulin resistance syndrome: The public health challenge. Endocr. Pract. 2003, 9, 23. [Google Scholar] [CrossRef]
- Ford, E.S.; Giles, W.H.; Dietz, W.H. Prevalence of the metabolic syndrome among US adults: Findings from the third National Health and Nutrition Examination Survey. JAMA 2002, 287, 356–359. [Google Scholar] [CrossRef] [PubMed]
- Nashar, K.; Egan, B.M. Relationship between chronic kidney disease and metabolic syndrome: Current perspectives. Diabetes Metab. Syndr. Obes. Targets Ther. 2014, 7, 421. [Google Scholar] [CrossRef]
- Cameron, A.J.; Shaw, J.E.; Zimmet, P.Z. The metabolic syndrome: Prevalence in worldwide populations. Endocrinol. Metab. Clin. 2004, 33, 351–375. [Google Scholar] [CrossRef]
- Ford, E.S.; Giles, W.H.; Mokdad, A.H. Increasing prevalence of the metabolic syndrome among US adults. Diabetes Care 2004, 27, 2444–2449. [Google Scholar] [CrossRef]
- Esposito, K.; Chiodini, P.; Colao, A.; Lenzi, A.; Giugliano, D. Metabolic syndrome and risk of cancer: A systematic review and meta-analysis. Diabetes Care 2012, 35, 2402–2411. [Google Scholar] [CrossRef]
- Alberti, K.G.M.; Zimmet, P.; Shaw, J. The metabolic syndrome—A new worldwide definition. Lancet 2005, 366, 1059–1062. [Google Scholar] [CrossRef]
- Duvnjak, L.; Duvnjak, M. The metabolic syndrome-an ongoing story. J. Physiol. Pharmacol. 2009, 60 (Suppl. S7), 19–24. [Google Scholar]
- Djiogue, S.; Nwabo Kamdje, A.H.; Vecchio, L.; Kipanyula, M.J.; Farahna, M.; Aldebasi, Y.; Seke Etet, P. Insulin resistance and cancer: The role of insulin and IGFs. Endocr. Relat. Cancer 2013, 20, R1–R17. [Google Scholar] [CrossRef]
- Cowey, S.; Hardy, R.W. The metabolic syndrome: A high-risk state for cancer? Am. J. Pathol. 2006, 169, 1505–1522. [Google Scholar] [CrossRef] [PubMed]
- Etet, P.F.S.; Vecchio, L.; Kamdje, A.H.N. Interactions between bone marrow stromal microenvironment and B-chronic lymphocytic leukemia cells: Any role for Notch, Wnt and Hh signaling pathways? Cell. Signal. 2012, 24, 1433–1443. [Google Scholar] [CrossRef]
- Pollak, M. The insulin and insulin-like growth factor receptor family in neoplasia: An update. Nat. Rev. Cancer 2012, 12, 159–169. [Google Scholar] [CrossRef]
- Sakurai, T.; Kudo, M. Signaling pathways governing tumor angiogenesis. Oncology 2011, 81 (Suppl. S1), 24–29. [Google Scholar] [CrossRef]
- Jee, S.H.; Kim, H.J.; Lee, J. Obesity, insulin resistance and cancer risk. Yonsei Med. J. 2005, 46, 449. [Google Scholar] [CrossRef]
- Calle, E.E.; Kaaks, R. Overweight, obesity and cancer: Epidemiological evidence and proposed mechanisms. Nat. Rev. Cancer 2004, 4, 579–591. [Google Scholar] [CrossRef]
- Saklayen, M.G. The Global Epidemic of the Metabolic Syndrome. Curr. Hypertens. Rep. 2018, 20, 12. [Google Scholar] [CrossRef] [PubMed]
- Flood, A.; Mai, V.; Pfeiffer, R.; Kahle, L.; Remaley, A.T.; Lanza, E.; Schatzkin, A. Elevated serum concentrations of insulin and glucose increase risk of recurrent colorectal adenomas. Gastroenterology 2007, 133, 1423–1429. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.M.; Stampfer, M.J.; Giovannucci, E.; Gann, P.H.; Ma, J.; Wilkinson, P.; Hennekens, C.H.; Pollak, M. Plasma insulin-like growth factor-I and prostate cancer risk: A prospective study. Science 1998, 279, 563–566. [Google Scholar] [CrossRef]
- Giovannucci, E.; Rimm, E.B.; Liu, Y.; Willett, W.C. Height, predictors of C-peptide and cancer risk in men. Int. J. Epidemiol. 2004, 33, 217–225. [Google Scholar] [CrossRef]
- Harman, S.M.; Metter, E.J.; Blackman, M.R.; Landis, P.K.; Carter, H.B. Serum levels of insulin-like growth factor I (IGF-I), IGF-II, IGF-binding protein-3, and prostate-specific antigen as predictors of clinical prostate cancer. J. Clin. Endocrinol. Metab. 2000, 85, 4258–4265. [Google Scholar] [CrossRef]
- Kaaks, R.; Lukanova, A.; Rinaldi, S.; Biessy, C.; Söderberg, S.; Olsson, T.; Stenman, U.-H.; Riboli, E.; Hallmans, G.; Stattin, P. Interrelationships between plasma testosterone, SHBG, IGF-I, insulin and leptin in prostate cancer cases and controls. Eur. J. Cancer Prev. 2003, 12, 309–315. [Google Scholar] [CrossRef]
- Verheus, M.; Peeters, P.H.; Rinaldi, S.; Dossus, L.; Biessy, C.; Olsen, A.; Tjønneland, A.; Overvad, K.; Jeppesen, M.; Clavel-Chapelon, F. Serum C-peptide levels and breast cancer risk: Results from the European prospective investigation into cancer and nutrition (EPIC). Int. J. Cancer 2006, 119, 659–667. [Google Scholar] [CrossRef]
- Gunter, M.J.; Hoover, D.R.; Yu, H.; Wassertheil-Smoller, S.; Manson, J.E.; Li, J.; Harris, T.G.; Rohan, T.E.; Xue, X.; Ho, G.Y. A prospective evaluation of insulin and insulin-like growth factor-I as risk factors for endometrial cancer. Cancer Epidemiol. Prev. Biomark. 2008, 17, 921–929. [Google Scholar] [CrossRef]
- Kourelis, T.V.; Siegel, R.D. Metformin and cancer: New applications for an old drug. Med. Oncol. 2012, 29, 1314–1327. [Google Scholar] [CrossRef]
- Uzunlulu, M.; Caklili, O.T.; Oguz, A. Association between metabolic syndrome and cancer. Ann. Nutr. Metab. 2016, 68, 173–179. [Google Scholar] [CrossRef]
- Belfiore, A. The role of insulin receptor isoforms and hybrid insulin/IGF-I receptors in human cancer. Curr. Pharm. Des. 2007, 13, 671–686. [Google Scholar] [CrossRef] [PubMed]
- Kiselyov, V.V.; Versteyhe, S.; Gauguin, L.; De Meyts, P. Harmonic oscillator model of the insulin and IGF1 receptors’ allosteric binding and activation. Mol. Syst. Biol. 2009, 5, 243. [Google Scholar] [CrossRef] [PubMed]
- Scapin, G.; Dandey, V.P.; Zhang, Z.; Prosise, W.; Hruza, A.; Kelly, T.; Mayhood, T.; Strickland, C.; Potter, C.S.; Carragher, B. Structure of the insulin receptor–insulin complex by single-particle cryo-EM analysis. Nature 2018, 556, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Andersen, D.K. Diabetes and pancreatic cancer. Endocr. Relat. Cancer 2012, 19, F9–F26. [Google Scholar] [CrossRef] [PubMed]
- Del Barco, S.; Vazquez-Martin, A.; Cufí, S.; Oliveras-Ferraros, C.; Bosch-Barrera, J.; Joven, J.; Martin-Castillo, B.; Menendez, J.A. Metformin: Multi-faceted protection against cancer. Oncotarget 2011, 2, 896. [Google Scholar] [CrossRef] [PubMed]
- Noto, H.; Goto, A.; Tsujimoto, T.; Osame, K.; Noda, M. Latest insights into the risk of cancer in diabetes. J. Diabetes Investig. 2013, 4, 225–232. [Google Scholar] [CrossRef]
- Anisimov, V.N. Metformin for aging and cancer prevention. Aging 2010, 2, 760. [Google Scholar] [CrossRef]
- Memmott, R.M.; Dennis, P.A. LKB1 and mammalian target of rapamycin as predictive factors for the anticancer efficacy of metformin. J. Clin. Oncol. 2009, 27, e226. [Google Scholar] [CrossRef]
- Baxter, R.; Brown, A.; Turtle, J. Association between serum insulin, serum somatomedin and liver receptors for human growth hormone in streptozotocin diabetes. Horm. Metab. Res. 1980, 12, 377–381. [Google Scholar] [CrossRef]
- Gallagher, E.J.; LeRoith, D. Diabetes, cancer, and metformin: Connections of metabolism and cell proliferation. Ann. N. Y. Acad. Sci. 2011, 1243, 54–68. [Google Scholar] [CrossRef]
- Friberg, E.; Mantzoros, C.S.; Wolk, A. Diabetes and risk of endometrial cancer: A population-based prospective cohort study. Cancer Epidemiol. Prev. Biomark. 2007, 16, 276–280. [Google Scholar] [CrossRef]
- Van Kruijsdijk, R.C.; Van Der Wall, E.; Visseren, F.L. Obesity and cancer: The role of dysfunctional adipose tissue. Cancer Epidemiol. Prev. Biomark. 2009, 18, 2569–2578. [Google Scholar] [CrossRef]
- Somasundar, P.; Alice, K.Y.; Vona-Davis, L.; McFadden, D.W. Differential effects of leptin on cancer in vitro. J. Surg. Res. 2003, 113, 50–55. [Google Scholar] [CrossRef]
- Adekola, K.; Rosen, S.T.; Shanmugam, M. Glucose transporters in cancer metabolism. Curr. Opin. Oncol. 2012, 24, 650. [Google Scholar] [CrossRef]
- Ryu, T.Y.; Park, J.; Scherer, P.E. Hyperglycemia as a risk factor for cancer progression. Diabetes Metab. J. 2014, 38, 330. [Google Scholar] [CrossRef]
- Fukada, T.; Yamasaki, S.; Nishida, K.; Murakami, M.; Hirano, T. Zinc homeostasis and signaling in health and diseases. JBIC J. Biol. Inorg. Chem. 2011, 16, 1123–1134. [Google Scholar] [CrossRef] [PubMed]
- Siebel, A.L.; Fernandez, A.Z.; El-Osta, A. Glycemic memory associated epigenetic changes. Biochem. Pharmacol. 2010, 80, 1853–1859. [Google Scholar] [CrossRef] [PubMed]
- Jee, S.H.; Ohrr, H.; Sull, J.W.; Yun, J.E.; Ji, M.; Samet, J.M. Fasting serum glucose level and cancer risk in Korean men and women. JAMA 2005, 293, 194–202. [Google Scholar] [CrossRef]
- Johnson, J.; Bowker, S. Intensive Glycaemic Control and Cancer Risk in Type 2 Diabetes: A Meta-Analysis of Major Trials; Springer: Berlin/Heidelberg, Germany, 2011. [Google Scholar]
- Renehan, A.G.; Zwahlen, M.; Egger, M. Adiposity and cancer risk: New mechanistic insights from epidemiology. Nat. Rev. Cancer 2015, 15, 484–498. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Hildebrandt, M.; Gu, J.; Tannir, N.; Matin, S.; Karam, J.; Wood, C.; Wu, X. MicroRNA profiling in clear cell renal cell carcinoma tissues potentially links tumorigenesis and recurrence with obesity. Br. J. Cancer 2017, 116, 77–84. [Google Scholar] [CrossRef]
- Giovannucci, E.; Harlan, D.M.; Archer, M.C.; Bergenstal, R.M.; Gapstur, S.M.; Habel, L.A.; Pollak, M.; Regensteiner, J.G.; Yee, D. Diabetes and Cancer: A Consensus Report. CA Cancer J. Clin. 2010, 60, 207–221. [Google Scholar] [CrossRef]
- Davila, J.; Morgan, R.; Shaib, Y.; McGlynn, K.; El-Serag, H. Diabetes increases the risk of hepatocellular carcinoma in the United States: A population based case control study. Gut 2005, 54, 533–539. [Google Scholar] [CrossRef]
- Lawson, D.; Gray, J.; McKillop, C.; Clarke, J.; Lee, F.; Patrick, R. Diabetes mellitus and primary hepatocellular carcinoma. Qjm Int. J. Med. 1986, 61, 945–955. [Google Scholar]
- Dyson, J.; Jaques, B.; Chattopadyhay, D.; Lochan, R.; Graham, J.; Das, D.; Aslam, T.; Patanwala, I.; Gaggar, S.; Cole, M. Hepatocellular cancer: The impact of obesity, type 2 diabetes and a multidisciplinary team. J. Hepatol. 2014, 60, 110–117. [Google Scholar] [CrossRef]
- Wang, C.; Wang, X.; Gong, G.; Ben, Q.; Qiu, W.; Chen, Y.; Li, G.; Wang, L. Increased risk of hepatocellular carcinoma in patients with diabetes mellitus: A systematic review and meta-analysis of cohort studies. Int. J. Cancer 2012, 130, 1639–1648. [Google Scholar] [CrossRef]
- Kampf, G. Efficacy of ethanol against viruses in hand disinfection. J. Hosp. Infect. 2018, 98, 331–338. [Google Scholar] [CrossRef]
- Noureddin, M.; Rinella, M.E. Nonalcoholic Fatty Liver Disease, Diabetes, Obesity, and Hepatocellular Carcinoma. Clin. Liver Dis. 2015, 19, 361–379. [Google Scholar] [CrossRef]
- Wiencke, J.K. Impact of race/ethnicity on molecular pathways in human cancer. Nat. Rev. Cancer 2004, 4, 79–84. [Google Scholar] [CrossRef]
- Wang, P.; Kang, D.; Cao, W.; Wang, Y.; Liu, Z. Diabetes mellitus and risk of hepatocellular carcinoma: A systematic review and meta-analysis. Diabetes/Metab. Res. Rev. 2012, 28, 109–122. [Google Scholar] [CrossRef]
- Qiao, G.; Le, Y.; Li, J.; Wang, L.; Shen, F. Glycogen Synthase Kinase-3β Is Associated with the Prognosis of Hepatocellular Carcinoma and May Mediate the Influence of Type 2 Diabetes Mellitus on Hepatocellular Carcinoma. PLoS ONE 2014, 9, e105624. [Google Scholar] [CrossRef]
- Tan, Y.; Wei, S.; Zhang, W.; Yang, J.; Yang, J.; Yan, L. Type 2 diabetes mellitus increases the risk of hepatocellular carcinoma in subjects with chronic hepatitis B virus infection: A meta-analysis and systematic review. Cancer Manag. Res. 2019, 11, 705–713. [Google Scholar] [CrossRef]
- Kasper, J.S.; Giovannucci, E. A Meta-analysis of Diabetes Mellitus and the Risk of Prostate Cancer. Cancer Epidemiol. Biomark. Prev. 2006, 15, 2056. [Google Scholar] [CrossRef] [PubMed]
- Bansal, D.; Bhansali, A.; Kapil, G.; Undela, K.; Tiwari, P. Type 2 diabetes and risk of prostate cancer: A meta-analysis of observational studies. Prostate Cancer Prostatic Dis. 2013, 16, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Waters, K.M.; Henderson, B.E.; Stram, D.O.; Wan, P.; Kolonel, L.N.; Haiman, C.A. Association of Diabetes With Prostate Cancer Risk in the Multiethnic Cohort. Am. J. Epidemiol. 2009, 169, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Kuriyama, S.; Kakizaki, M.; Yan, H.; Sone, T.; Nagai, M.; Sugawara, Y.; Ohmori-Matsuda, K.; Hozawa, A.; Nishino, Y.; et al. History of diabetes mellitus and the risk of prostate cancer: The Ohsaki Cohort Study. Cancer Causes Control 2010, 21, 1025–1032. [Google Scholar] [CrossRef] [PubMed]
- Ataey, A.; Jafarvand, E.; Adham, D.; Moradi-Asl, E. The relationship between obesity, overweight, and the human development index in world health organization eastern mediterranean region countries. J. Prev. Med. Public Health 2020, 53, 98. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.H.; LeRoith, D. Obesity, type 2 diabetes, and cancer: The insulin and IGF connection. Endocr. Relat. Cancer 2012, 19, F27–F45. [Google Scholar] [CrossRef]
- Gallagher, E.J.; LeRoith, D. Obesity and diabetes: The increased risk of cancer and cancer-related mortality. Physiol. Rev. 2015, 95, 727–748. [Google Scholar] [CrossRef]
- Kang, C.; LeRoith, D.; Gallagher, E.J. Diabetes, obesity, and breast cancer. Endocrinology 2018, 159, 3801–3812. [Google Scholar] [CrossRef]
- Pothuraju, R.; Rachagani, S.; Junker, W.M.; Chaudhary, S.; Saraswathi, V.; Kaur, S.; Batra, S.K. Pancreatic cancer associated with obesity and diabetes: An alternative approach for its targeting. J. Exp. Clin. Cancer Res. 2018, 37, 319. [Google Scholar] [CrossRef]
- Scully, T.; Ettela, A.; Gallagher, E.J.; LeRoith, D. Obesity, Type 2 Diabetes and Cancer Risk. Front. Oncol. 2020, 10, 3196. [Google Scholar]
- Perry, R.J.; Shulman, G.I. Mechanistic links between obesity, insulin, and cancer. Trends Cancer 2020, 6, 75–78. [Google Scholar] [CrossRef]
- Marques, R.G.; Fontaine, M.J.; Rogers, J. C-peptide: Much more than a byproduct of insulin biosynthesis. Pancreas 2004, 29, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Jenab, M.; Riboli, E.; Cleveland, R.J.; Norat, T.; Rinaldi, S.; Nieters, A.; Biessy, C.; Tjønneland, A.; Olsen, A.; Overvad, K. Serum C-peptide, IGFBP-1 and IGFBP-2 and risk of colon and rectal cancers in the European Prospective Investigation into Cancer and Nutrition. Int. J. Cancer 2007, 121, 368–376. [Google Scholar] [CrossRef]
- Ma, J.; Li, H.; Giovannucci, E.; Mucci, L.; Qiu, W.; Nguyen, P.L.; Gaziano, J.M.; Pollak, M.; Stampfer, M.J. Prediagnostic body-mass index, plasma C-peptide concentration, and prostate cancer-specific mortality in men with prostate cancer: A long-term survival analysis. Lancet Oncol. 2008, 9, 1039–1047. [Google Scholar] [CrossRef]
- Bloomgarden, Z. Diabetes and cancer. Diabetes Care 2001, 24, 780–781. [Google Scholar]
- Bloomgarden, Z.T. Second world congress on the insulin resistance syndrome. Diabetes Care 2005, 28, 1518–1523. [Google Scholar] [CrossRef]
- Richardson, L.C.; Pollack, L.A. Therapy insight: Influence of type 2 diabetes on the development, treatment and outcomes of cancer. Nat. Clin. Pract. Oncol. 2005, 2, 48–53. [Google Scholar] [CrossRef]
- Bertoni, A.G.; Saydah, S.; Brancati, F.L. Diabetes and the risk of infection-related mortality in the US. Diabetes Care 2001, 24, 1044–1049. [Google Scholar] [CrossRef] [PubMed]
- Theodoulou, M.; Seidman, A.D. Seminars in Oncology. In Cardiac Effects of Adjuvant Therapy for Early Breastcancer; Elsevier: Amsterdam, The Netherlands, 2003; pp. 730–739. [Google Scholar]
- Wildiers, H.; Highley, M.S.; de Bruijn, E.A.; van Oosterom, A.T. Pharmacology of anticancer drugs in the elderly population. Clin. Pharmacokinet. 2003, 42, 1213–1242. [Google Scholar] [CrossRef] [PubMed]
- Nichols, C.R. Testicular cancer. Curr. Probl. Cancer 1998, 22, 187–274. [Google Scholar] [CrossRef]
- Donnelly, R.; Emslie-Smith, A.M.; Gardner, I.D.; Morris, A.D. Vascular complications of diabetes. BMJ 2000, 320, 1062–1066. [Google Scholar] [CrossRef] [PubMed]
- Flatters, S.J.; Bennett, G.J. Ethosuximide reverses paclitaxel-and vincristine-induced painful peripheral neuropathy. Pain 2004, 109, 150–161. [Google Scholar] [CrossRef]
- Bonadonna, G.; Valagussa, P. Dose-response effect of adjuvant chemotherapy in breast cancer. N. Engl. J. Med. 1981, 304, 10–15. [Google Scholar] [CrossRef] [PubMed]
- DeVita, V.T.; Hubbard, S.M.; Longo, D.L. The chemotherapy of lymphomas: Looking back, moving forward—the Richard and Hinda Rosenthal Foundation award lecture. Cancer Res. 1987, 47, 5810–5824. [Google Scholar]
- Childs, B.B.; Cypress, M.; Spollett, G. Complete Nurse’s Guide to Diabetes Care; American Diabetes Association: Arlington County, VI, USA, 2017. [Google Scholar]
- Clement, S.; Braithwaite, S.; Magee, M.; Ahmann, A.; Smith, E.; Schafer, R.; Hirsch, I. Erratum: Management of diabetes and hyperglycemia in hospitals (Technical Review). Diabetes Care 2004, 27, 553–591. [Google Scholar] [CrossRef] [PubMed]
- Jiralerspong, S.; Palla, S.L.; Giordano, S.H.; Meric-Bernstam, F.; Liedtke, C.; Barnett, C.M.; Hsu, L.; Hung, M.-C.; Hortobagyi, G.N.; Gonzalez-Angulo, A.M. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J. Clin. Oncol. 2009, 27, 3297. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2015. CA Cancer J. Clin. 2015, 65, 5–29. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.X.; Yao, W.X.; Ge, J.; Peng, X.C.; Du, X.B.; Zhang, R.; Yao, B.; Xie, K.; Li, L.H.; Dong, H. Prognostic influence of metformin as first-line chemotherapy for advanced nonsmall cell lung cancer in patients with type 2 diabetes. Cancer 2011, 117, 5103–5111. [Google Scholar] [CrossRef] [PubMed]
- Volgi, J.R.; Baldwin, D., Jr. Glucocorticoid therapy and diabetes management. Nurs. Clin. N. Am. 2001, 36, 333–339. [Google Scholar]
- Braithwaite, S.S.; Barr, W.G.; Rahman, A.; Quddusi, S. Managing diabetes during glucocorticoid therapy: How to avoid metabolic emergencies. Postgrad. Med. 1998, 104, 163–176. [Google Scholar] [CrossRef]
- Hirsch, I.; Braithwaite, S.; Verderese, C. Practical Management of Inpatient Hyperglycemia; LakevilleConn Hilliard Publishing: Lakeville, CT, USA, 2005; pp. 1–41. [Google Scholar]
- Trence, D.L. Management of patients on chronic glucocorticoid therapy: An endocrine perspective. Prim. Care Clin. Off. Pract. 2003, 30, 593–605. [Google Scholar] [CrossRef]
- McCoubrie, R.; Jeffrey, D.; Paton, C.; Dawes, L. Managing diabetes mellitus in patients with advanced cancer: A case note audit and guidelines. Eur. J. Cancer Care 2005, 14, 244–248. [Google Scholar] [CrossRef]
- Medina-Bolivar, F.; Condori, J.; Rimando, A.M.; Hubstenberger, J.; Shelton, K.; O’Keefe, S.F.; Bennett, S.; Dolan, M.C. Production and secretion of resveratrol in hairy root cultures of peanut. Phytochemistry 2007, 68, 1992–2003. [Google Scholar] [CrossRef]
- Tian, B.; Liu, J. Resveratrol: A review of plant sources, synthesis, stability, modification and food application. J. Sci. Food Agric. 2020, 100, 1392–1404. [Google Scholar] [CrossRef]
- Lançon, A.; Hanet, N.; Jannin, B.; Delmas, D.; Heydel, J.-M.; Lizard, G.; Chagnon, M.-C.; Artur, Y.; Latruffe, N. Resveratrol in human hepatoma HepG2 cells: Metabolism and inducibility of detoxifying enzymes. Drug Metab. Dispos. 2007, 35, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Jeandet, P.; Douillet-Breuil, A.-C.; Bessis, R.; Debord, S.; Sbaghi, M.; Adrian, M. Phytoalexins from the Vitaceae: Biosynthesis, phytoalexin gene expression in transgenic plants, antifungal activity, and metabolism. J. Agric. Food Chem. 2002, 50, 2731–2741. [Google Scholar] [CrossRef]
- Salehi, B.; Mishra, A.P.; Nigam, M.; Sener, B.; Kilic, M.; Sharifi-Rad, M.; Fokou, P.V.T.; Martins, N.; Sharifi-Rad, J. Resveratrol: A double-edged sword in health benefits. Biomedicines 2018, 6, 91. [Google Scholar] [CrossRef]
- Li, L.; Qiu, R.L.; Lin, Y.; Cai, Y.; Bian, Y.; Fan, Y.; Gao, X.J. Resveratrol suppresses human cervical carcinoma cell proliferation and elevates apoptosis via the mitochondrial and p53 signaling pathways. Oncol. Lett. 2018, 15, 9845–9851. [Google Scholar] [CrossRef]
- Li, D.; Wang, G.; Jin, G.; Yao, K.; Zhao, Z.; Bie, L.; Guo, Y.; Li, N.; Deng, W.; Chen, X. Resveratrol suppresses colon cancer growth by targeting the AKT/STAT3 signaling pathway. Int. J. Mol. Med. 2019, 43, 630–640. [Google Scholar] [CrossRef]
- Öztürk, Y.; Günaydın, C.; Yalçın, F.; Nazıroğlu, M.; Braidy, N. Resveratrol enhances apoptotic and oxidant effects of paclitaxel through TRPM2 channel activation in DBTRG glioblastoma cells. Oxid. Med. Cell. Longev. 2019, 2019, 4619865. [Google Scholar] [CrossRef]
- Hoca, M.; Becer, E.; Kabadayı, H.; Yücecan, S.; Vatansever, H.S. The effect of resveratrol and quercetin on epithelial-mesenchymal transition in pancreatic cancer stem cell. Nutr. Cancer 2020, 72, 1231–1242. [Google Scholar] [CrossRef]
- Zhao, Y.; Cao, Y.; Sun, J.; Liang, Z.; Wu, Q.; Cui, S.; Zhi, D.; Guo, S.; Zhen, Y.; Zhang, S. Anti-breast cancer activity of resveratrol encapsulated in liposomes. J. Mater. Chem. B 2020, 8, 27–37. [Google Scholar] [CrossRef]
- Alobaedi, O.H.; Talib, W.H.; Basheti, I.A. Antitumor effect of thymoquinone combined with resveratrol on mice transplanted with breast cancer. Asian Pac. J. Trop. Med. 2017, 10, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Ismail, N.; Abdel–Mottaleb, Y.; Ahmed, A.A.E.; El-Maraghy, N.N. Novel combination of thymoquinone and resveratrol enhances anticancer effect on hepatocellular carcinoma cell line. Future J. Pharm. Sci. 2018, 4, 41–46. [Google Scholar] [CrossRef]
- Rai, G.; Mishra, S.; Suman, S.; Shukla, Y. Resveratrol improves the anticancer effects of doxorubicin in vitro and in vivo models: A mechanistic insight. Phytomedicine 2016, 23, 233–242. [Google Scholar] [CrossRef]
- Zhang, A.J.; Rimando, A.M.; Mizuno, C.S.; Mathews, S.T. α-Glucosidase inhibitory effect of resveratrol and piceatannol. J. Nutr. Biochem. 2017, 47, 86–93. [Google Scholar] [CrossRef]
- Rimando, A.M.; Nagmani, R.; Feller, D.R.; Yokoyama, W. Pterostilbene, a new agonist for the peroxisome proliferator-activated receptor α-isoform, lowers plasma lipoproteins and cholesterol in hypercholesterolemic hamsters. J. Agric. Food Chem. 2005, 53, 3403–3407. [Google Scholar] [CrossRef]
- Yang, D.K.; Kang, H.-S. Anti-diabetic effect of cotreatment with quercetin and resveratrol in streptozotocin-induced diabetic rats. Biomol. Ther. 2018, 26, 130. [Google Scholar] [CrossRef]
- Agarwal, A.; Kasinathan, A.; Ganesan, R.; Balasubramanian, A.; Bhaskaran, J.; Suresh, S.; Srinivasan, R.; Aravind, K.; Sivalingam, N. Curcumin induces apoptosis and cell cycle arrest via the activation of reactive oxygen species–independent mitochondrial apoptotic pathway in Smad4 and p53 mutated colon adenocarcinoma HT29 cells. Nutr. Res. 2018, 51, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Muangnoi, C.; Jithavech, P.; Ratnatilaka Na Bhuket, P.; Supasena, W.; Wichitnithad, W.; Towiwat, P.; Niwattisaiwong, N.; Haworth, I.S.; Rojsitthisak, P. A curcumin-diglutaric acid conjugated prodrug with improved water solubility and antinociceptive properties compared to curcumin. Biosci. Biotechnol. Biochem. 2018, 82, 1301–1308. [Google Scholar] [CrossRef] [PubMed]
- Talib, W.H.; Al-Hadid, S.A.; Ali, M.B.W.; Al-Yasari, I.H.; Abd Ali, M.R. Role of curcumin in regulating p53 in breast cancer: An overview of the mechanism of action. Breast Cancer Targets Ther. 2018, 10, 207. [Google Scholar] [CrossRef]
- Ismail, N.I.; Othman, I.; Abas, F.; H Lajis, N.; Naidu, R. Mechanism of apoptosis induced by curcumin in colorectal cancer. Int. J. Mol. Sci. 2019, 20, 2454. [Google Scholar] [CrossRef]
- Kunnumakkara, A.B.; Bordoloi, D.; Padmavathi, G.; Monisha, J.; Roy, N.K.; Prasad, S.; Aggarwal, B.B. Curcumin, the golden nutraceutical: Multitargeting for multiple chronic diseases. Br. J. Pharmacol. 2017, 174, 1325–1348. [Google Scholar] [CrossRef]
- Xu, X.-Y.; Meng, X.; Li, S.; Gan, R.-Y.; Li, Y.; Li, H.-B. Bioactivity, health benefits, and related molecular mechanisms of curcumin: Current progress, challenges, and perspectives. Nutrients 2018, 10, 1553. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.; Zaidi, S.F.; Cui, Z.G.; Zhou, D.; Saeed, S.A.; Inadera, H. Potential proapoptotic phytochemical agents for the treatment and prevention of colorectal cancer. Oncol. Lett. 2019, 18, 487–498. [Google Scholar] [CrossRef]
- Su, P.; Yang, Y.; Wang, G.; Chen, X.; Ju, Y. Curcumin attenuates resistance to irinotecan via induction of apoptosis of cancer stem cells in chemoresistant colon cancer cells. Int. J. Oncol. 2018, 53, 1343–1353. [Google Scholar] [CrossRef] [PubMed]
- Villegas, I.; Sánchez-Fidalgo, S.; de la Lastra, C.A. Chemopreventive effect of dietary curcumin on inflammation-induced colorectal carcinogenesis in mice. Mol. Nutr. Food Res. 2011, 55, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.M.; Lim, J.H.; Lee, T.J.; Park, J.-W.; Choi, K.S.; Kwon, T.K. Curcumin sensitizes tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis through reactive oxygen species-mediated upregulation of death receptor 5 (DR5). Carcinogenesis 2005, 26, 1905–1913. [Google Scholar] [CrossRef] [PubMed]
- Babushkina, E.A.; Belokopytova, L.V.; Grachev, A.M.; Meko, D.M.; Vaganov, E.A. Variation of the hydrological regime of Bele-Shira closed basin in Southern Siberia and its reflection in the radial growth of Larix sibirica. Reg. Environ. Chang. 2017, 17, 1725–1737. [Google Scholar] [CrossRef]
- Song, G.; Mao, Y.; Cai, Q.; Yao, L.; Ouyang, G.; Bao, S. Curcumin induces human HT-29 colon adenocarcinoma cell apoptosis by activating p53 and regulating apoptosis-related protein expression. Braz. J. Med. Biol. Res. 2005, 38, 1791–1798. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, L.; Wang, Y.; He, A.; Hu, H.; Zhang, J.; Han, M.; Huang, Y. Curcumin inhibits the proliferation and invasion of MG-63 cells through inactivation of the p-JAK2/p-STAT3 pathway. Oncotargets Ther. 2019, 12, 2011. [Google Scholar] [CrossRef]
- Tomeh, M.A.; Hadianamrei, R.; Zhao, X. A review of curcumin and its derivatives as anticancer agents. Int. J. Mol. Sci. 2019, 20, 1033. [Google Scholar] [CrossRef] [PubMed]
- Den Hartogh, D.J.; Gabriel, A.; Tsiani, E. Antidiabetic properties of curcumin I: Evidence from in vitro studies. Nutrients 2020, 12, 118. [Google Scholar] [CrossRef]
- Den Hartogh, D.J.; Gabriel, A.; Tsiani, E. Antidiabetic properties of curcumin II: Evidence from in vivo studies. Nutrients 2020, 12, 58. [Google Scholar] [CrossRef]
- Abdelsamia, E.M.; Khaleel, S.A.; Balah, A.; Baky, N.A.A. Curcumin augments the cardioprotective effect of metformin in an experimental model of type I diabetes mellitus; Impact of Nrf2/HO-1 and JAK/STAT pathways. Biomed. Pharmacother. 2019, 109, 2136–2144. [Google Scholar] [CrossRef]
- Dajani, E.; Shahwan, T.; Dajani, N. Overview of the preclinical pharmacological properties of Nigella sativa (black seeds): A complementary drug with historical and clinical significance. J. Physiol. Pharm. 2016, 67, 801–817. [Google Scholar]
- Younus, H. Molecular and Therapeutic: Actions of Thymoquinone; Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar]
- Ahmad, A.; Husain, A.; Mujeeb, M.; Khan, S.A.; Najmi, A.K.; Siddique, N.A.; Damanhouri, Z.A.; Anwar, F. A review on therapeutic potential of Nigella sativa: A miracle herb. Asian Pac. J. Trop. Biomed. 2013, 3, 337–352. [Google Scholar] [CrossRef]
- Aslan, M.; Afşar, E.; Kırımlıoglu, E.; Çeker, T.; Yılmaz, Ç. Antiproliferative Effects of Thymoquinone in MCF-7 Breast and HepG2 Liver Cancer Cells: Possible Role of Ceramide and ER Stress. Nutr. Cancer 2020, 73, 460–472. [Google Scholar] [CrossRef] [PubMed]
- Darakhshan, S.; Pour, A.B.; Colagar, A.H.; Sisakhtnezhad, S. Thymoquinone and its therapeutic potentials. Pharmacol. Res. 2015, 95, 138–158. [Google Scholar] [CrossRef] [PubMed]
- Laskar, A.A.; Khan, M.A.; Askari, F.; Younus, H. Thymoquinone binds and activates human salivary aldehyde dehydrogenase: Potential therapy for the mitigation of aldehyde toxicity and maintenance of oral health. Int. J. Biol. Macromol. 2017, 103, 99–110. [Google Scholar] [CrossRef]
- Motaghed, M.; Al-Hassan, F.M.; Hamid, S.S. Cellular responses with thymoquinone treatment in human breast cancer cell line MCF-7. Pharmacogn. Res. 2013, 5, 200. [Google Scholar]
- El-Sheikh, A.A.; Morsy, M.A.; Abdalla, A.M.; Hamouda, A.H.; Alhaider, I.A. Mechanisms of thymoquinone hepatorenal protection in methotrexate-induced toxicity in rats. Mediat. Inflamm. 2015, 2015, 859383. [Google Scholar] [CrossRef]
- Kundu, J.; Choi, B.Y.; Jeong, C.-H.; Kundu, J.K.; Chun, K.-S. Thymoquinone induces apoptosis in human colon cancer HCT116 cells through inactivation of STAT3 by blocking JAK2-and Src-mediated phosphorylation of EGF receptor tyrosine kinase. Oncol. Rep. 2014, 32, 821–828. [Google Scholar] [CrossRef] [PubMed]
- Woo, C.C.; Loo, S.Y.; Gee, V.; Yap, C.W.; Sethi, G.; Kumar, A.P.; Tan, K.H.B. Anticancer activity of thymoquinone in breast cancer cells: Possible involvement of PPAR-γ pathway. Biochem. Pharmacol. 2011, 82, 464–475. [Google Scholar] [CrossRef] [PubMed]
- Talib, W.H. Regressions of breast carcinoma syngraft following treatment with piperine in combination with thymoquinone. Sci. Pharm. 2017, 85, 27. [Google Scholar] [CrossRef]
- Pelegrin, S.; Galtier, F.; Chalançon, A.; Gagnol, J.P.; Barbanel, A.M.; Pélissier, Y.; Larroque, M.; Lepape, S.; Faucanié, M.; Gabillaud, I. Effects of Nigella sativa seeds (black cumin) on insulin secretion and lipid profile: A pilot study in healthy volunteers. Br. J. Clin. Pharmacol. 2019, 85, 1607–1611. [Google Scholar] [CrossRef]
- Khalil, P.; Masood, S.; Rehman, A.U.; Khalil, F.; Nawaf, J. Preventive Role of Thymoquinone against Certain Chronic Health Issues: A Review. Int. J. Nutr. Sci. 2020. [Google Scholar] [CrossRef]
- Durvasula, R.V.; Petermann, A.T.; Hiromura, K.; Blonski, M.; Pippin, J.; Mundel, P.; Pichler, R.; Griffin, S.; Couser, W.G.; Shankland, S.J. Activation of a local tissue angiotensin system in podocytes by mechanical strain. Kidney Int. 2004, 65, 30–39. [Google Scholar] [CrossRef]
- Montazeri, R.S.; Fatahi, S.; Sohouli, M.H.; Abu-Zaid, A.; Santos, H.O.; Găman, M.A.; Shidfar, F. The effect of nigella sativa on biomarkers of inflammation and oxidative stress: A systematic review and meta-analysis of randomized controlled trials. J. Food Biochem. 2021, e13625. [Google Scholar] [CrossRef]
- Lambert, J.D.; Lee, M.J.; Lu, H.; Meng, X.; Hong, J.J.; Seril, D.N.; Sturgill, M.G.; Yang, C.S. Epigallocatechin-3-gallate is absorbed but extensively glucuronidated following oral administration to mice. J. Nutr. 2003, 133, 4172–4177. [Google Scholar] [CrossRef]
- Nagle, D.G.; Ferreira, D.; Zhou, Y.-D. Epigallocatechin-3-gallate (EGCG): Chemical and biomedical perspectives. Phytochemistry 2006, 67, 1849–1855. [Google Scholar] [CrossRef]
- Rodríguez-Carrasco, Y.; Gaspari, A.; Graziani, G.; Santini, A.; Ritieni, A. Fast analysis of polyphenols and alkaloids in cocoa-based products by ultra-high performance liquid chromatography and Orbitrap high resolution mass spectrometry (UHPLC-Q-Orbitrap-MS/MS). Food Res. Int. 2018, 111, 229–236. [Google Scholar] [CrossRef]
- Chopade, V.; Phatak, A.; Upaganlawar, A.; Tankar, A. Green tea (Camellia sinensis): Chemistry, traditional, medicinal uses and its pharmacological activities-a review. Pharmacogn. Rev. 2008, 2, 157. [Google Scholar]
- Katiyar, S.K.; Elmets, C.A. Green tea polyphenolic antioxidants and skin photoprotection. Int. J. Oncol. 2001, 18, 1307–1313. [Google Scholar] [CrossRef]
- Musial, C.; Kuban-Jankowska, A.; Gorska-Ponikowska, M. Beneficial Properties of Green Tea Catechins. Int. J. Mol. Sci. 2020, 21, 1744. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-Q.; Lu, J.-L.; Liang, Y.-R.; Li, Q.-S. Suppressive effects of EGCG on cervical cancer. Molecules 2018, 23, 2334. [Google Scholar] [CrossRef]
- Ho, H.C.; Huang, C.C.; Lu, Y.T.; Yeh, C.M.; Ho, Y.T.; Yang, S.F.; Hsin, C.H.; Lin, C.W. Epigallocatechin-3-gallate inhibits migration of human nasopharyngeal carcinoma cells by repressing MMP-2 expression. J. Cell. Physiol. 2019, 234, 20915–20924. [Google Scholar] [CrossRef] [PubMed]
- Pal, D.; Sur, S.; Roy, R.; Mandal, S.; Kumar Panda, C. Epigallocatechin gallate in combination with eugenol or amarogentin shows synergistic chemotherapeutic potential in cervical cancer cell line. J. Cell. Physiol. 2019, 234, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.H.; Horng, C.T.; Lee, C.F.; Chiang, N.N.; Tsai, F.J.; Lu, C.C.; Chiang, J.H.; Hsu, Y.M.; Yang, J.S.; Chen, F.A. Epigallocatechin gallate sensitizes cisplatin-resistant oral cancer CAR cell apoptosis and autophagy through stimulating AKT/STAT3 pathway and suppressing multidrug resistance 1 signaling. Environ. Toxicol. 2017, 32, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Chuu, C.-P.; Chen, R.-Y.; Kokontis, J.M.; Hiipakka, R.A.; Liao, S. Suppression of androgen receptor signaling and prostate specific antigen expression by (−)-epigallocatechin-3-gallate in different progression stages of LNCaP prostate cancer cells. Cancer Lett. 2009, 275, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Li, W.; Chen, Z.; Guo, Q.; Wang, C.; Santhanam, R.K.; Chen, H. Inhibitory effect of epigallocatechin-3-O-gallate on α-glucosidase and its hypoglycemic effect via targeting PI3K/AKT signaling pathway in L6 skeletal muscle cells. Int. J. Biol. Macromol. 2019, 125, 605–611. [Google Scholar] [CrossRef]
- Lin, Y.; Shi, D.; Su, B.; Wei, J.; Găman, M.A.; Sedanur Macit, M.; Borges do Nascimento, I.J.; Guimaraes, N.S. The effect of green tea supplementation on obesity: A systematic review and dose–response meta-analysis of randomized controlled trials. Phytother. Res. 2020, 34, 2459–2470. [Google Scholar] [CrossRef]
- Lanzotti, V. The analysis of onion and garlic. J. Chromatogr. A 2006, 1112, 3–22. [Google Scholar] [CrossRef]
- Chen, H.; Zhu, B.; Zhao, L.; Liu, Y.; Zhao, F.; Feng, J.; Jin, Y.; Sun, J.; Geng, R.; Wei, Y. Allicin inhibits proliferation and invasion in vitro and in vivo via SHP-1-mediated STAT3 signaling in cholangiocarcinoma. Cell. Physiol. Biochem. 2018, 47, 641–653. [Google Scholar] [CrossRef]
- Huang, W.L.; Wu, S.F.; Xu, S.T.; Ma, Y.C.; Wang, R.; Jin, S.; Zhou, S. Allicin enhances the radiosensitivity of colorectal cancer cells via inhibition of NF-κB signaling pathway. J. Food Sci. 2020, 85, 1924–1931. [Google Scholar] [CrossRef]
- Yang, Z.; Du, J.; Zhu, J.; Rong, Y.; Chen, S.; Yu, L.; Deng, X.; Zhang, X.; Sheng, H.; Yang, L. Allicin Inhibits Proliferation by Decreasing IL-6 and IFN-β in HCMV-Infected Glioma Cells. Cancer Manag. Res. 2020, 12, 7305. [Google Scholar] [CrossRef]
- Schultz, C.R.; Gruhlke, M.C.; Slusarenko, A.J.; Bachmann, A.S. Allicin, a Potent New Ornithine Decarboxylase Inhibitor in Neuroblastoma Cells. J. Nat. Prod. 2020, 83, 2518–2527. [Google Scholar] [CrossRef] [PubMed]
- Jobani, B.M.; Najafzadeh, N.; Mazani, M.; Arzanlou, M.; Vardin, M.M. Molecular mechanism and cytotoxicity of allicin and all-trans retinoic acid against CD44+ versus CD117+ melanoma cells. Phytomedicine 2018, 48, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Țigu, A.B.; Toma, V.-A.; Moț, A.C.; Jurj, A.; Moldovan, C.S.; Fischer-Fodor, E.; Berindan-Neagoe, I.; Pârvu, M. The Synergistic Antitumor Effect of 5-Fluorouracil Combined with Allicin against Lung and Colorectal Carcinoma Cells. Molecules 2020, 25, 1947. [Google Scholar] [CrossRef] [PubMed]
- Arellano-Buendía, A.S.; Castañeda-Lara, L.G.; Loredo-Mendoza, M.L.; García-Arroyo, F.E.; Rojas-Morales, P.; Argüello-García, R.; Juárez-Rojas, J.G.; Tapia, E.; Pedraza-Chaverri, J.; Sánchez-Lozada, L.G. Effects of Allicin on Pathophysiological Mechanisms during the Progression of Nephropathy Associated to Diabetes. Antioxidants 2020, 9, 1134. [Google Scholar] [CrossRef]
- Mandal, S.K.; Das, A.; Dey, S.; Sahoo, U.; Bose, S.; Bose, A.; Dhiman, N.; Madan, S.; Ramadan, M.A. Bioactivities of Allicin and related organosulfur compounds from garlic: Overview of the literature since 2010. Egypt. J. Chem. 2019, 62, 1–11. [Google Scholar] [CrossRef]
- Li, L.; Sheng, X.; Zhao, S.; Zou, L.; Han, X.; Gong, Y.; Yuan, H.; Shi, L.; Guo, L.; Jia, T. Nanoparticle-encapsulated emodin decreases diabetic neuropathic pain probably via a mechanism involving P2X3 receptor in the dorsal root ganglia. Purinergic Signal. 2017, 13, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Zhao, X.; Cui, T. Full Length Research Paper Production of emodin from Aspergillus ochraceus at preparative scale. Afr. J. Biotechnol. 2010, 9. Available online: https://www.ajol.info/index.php/ajb/article/view/77969 (accessed on 9 April 2021).
- Shun-Hua, L.; Lin, L.; Ru-Nan, Y.; Liang, S.-D. Compounds of traditional Chinese medicine and neuropathic pain. Chin. J. Nat. Med. 2020, 18, 28–35. [Google Scholar]
- Talib, W.H.; Alsalahat, I.; Daoud, S.; Abutayeh, R.F.; Mahmod, A.I. Plant-Derived Natural Products in Cancer Research: Extraction, Mechanism of Action, and Drug Formulation. Molecules 2020, 25, 5319. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Yin, L.; Yang, J.; Shan, G. Emodin, an anthraquinone derivative from Rheum officinale Baill, enhances cutaneous wound healing in rats. Eur. J. Pharmacol. 2007, 567, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-C.; Chang, J.-H.; Tung, S.-F.; Wu, R.-T.; Foegh, M.L.; Chu, S.-H. Immunosuppressive effect of emodin, a free radical generator. Eur. J. Pharmacol. 1992, 211, 359–364. [Google Scholar] [CrossRef]
- Huei-Chen, H.; Shu-Hsun, C.; Chao, P.-D.L. Vasorelaxants from Chinese herbs, emodin and scoparone, possess immunosuppressive properties. Eur. J. Pharmacol. 1991, 198, 211–213. [Google Scholar] [CrossRef]
- Kaneshiro, T.; Morioka, T.; Inamine, M.; Kinjo, T.; Arakaki, J.; Chiba, I.; Sunagawa, N.; Suzui, M.; Yoshimi, N. Anthraquinone derivative emodin inhibits tumor-associated angiogenesis through inhibition of extracellular signal-regulated kinase 1/2 phosphorylation. Eur. J. Pharmacol. 2006, 553, 46–53. [Google Scholar] [CrossRef]
- Zhu, S.; Wang, Y.; Wang, X.; Li, J.; Hu, F. Emodin inhibits ATP-induced IL-1β secretion, ROS production and phagocytosis attenuation in rat peritoneal macrophages via antagonizing P2X7 receptor. Pharm. Biol. 2014, 52, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.-C.; Chung, J.-G. Anticancer potential of emodin. BioMedicine 2012, 2, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Fu, J.; Yin, X.; Cao, S.; Li, X.; Lin, L.; Huyiligeqi; Ni, J. Emodin: A review of its pharmacology, toxicity and pharmacokinetics. Phytother. Res. 2016, 30, 1207–1218. [Google Scholar] [CrossRef]
- Mukund, V.; Mukund, D.; Sharma, V.; Mannarapu, M.; Alam, A. Genistein: Its role in metabolic diseases and cancer. Crit. Rev. Oncol. Hematol. 2017, 119, 13–22. [Google Scholar] [CrossRef]
- Spagnuolo, C.; Russo, G.L.; Orhan, I.E.; Habtemariam, S.; Daglia, M.; Sureda, A.; Nabavi, S.F.; Devi, K.P.; Loizzo, M.R.; Tundis, R. Genistein and cancer: Current status, challenges, and future directions. Adv. Nutr. 2015, 6, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Chodon, D.; Banu, S.M.; Padmavathi, R.; Sakthisekaran, D. Inhibition of cell proliferation and induction of apoptosis by genistein in experimental hepatocellular carcinoma. Mol. Cell. Biochem. 2007, 297, 73. [Google Scholar] [CrossRef] [PubMed]
- Tatsuta, M.; Iishi, H.; Baba, M.; Yano, H.; Uehara, H.; Nakaizumi, A. Attenuation by genistein of sodium-chloride-enhanced gastric carcinogenesis induced by N-methyl-N′-nitro-N-nitrosoguanidine in Wistar rats. Int. J. Cancer 1999, 80, 396–399. [Google Scholar] [CrossRef]
- Estrela, J.M.; Mena, S.; Obrador, E.; Benlloch, M.; Castellano, G.; Salvador, R.; Dellinger, R.W. Polyphenolic phytochemicals in cancer prevention and therapy: Bioavailability versus bioefficacy. J. Med. Chem. 2017, 60, 9413–9436. [Google Scholar] [CrossRef]
- Yousefi, H.; Alihemmati, A.; Karimi, P.; Alipour, M.R.; Habibi, P.; Ahmadiasl, N. Effect of genistein on expression of pancreatic SIRT1, inflammatory cytokines and histological changes in ovariectomized diabetic rat. Iran. J. Basic Med. Sci. 2017, 20, 423. [Google Scholar]
- Luo, J.; Wang, A.; Zhen, W.; Wang, Y.; Si, H.; Jia, Z.; Alkhalidy, H.; Cheng, Z.; Gilbert, E.; Xu, B. Phytonutrient genistein is a survival factor for pancreatic β-cells via GPR30-mediated mechanism. J. Nutr. Biochem. 2018, 58, 59–70. [Google Scholar] [CrossRef]
- Talib, W.H.; Al Kury, L.T. Parthenolide inhibits tumor-promoting effects of nicotine in lung cancer by inducing P53-dependent apoptosis and inhibiting VEGF expression. Biomed. Pharmacother. 2018, 107, 1488–1495. [Google Scholar] [CrossRef]
- Williams, B.; Lees, F.; Tsangari, H.; Hutchinson, M.; Perilli, E.; Crotti, T. Assessing the Effects of Parthenolide on Inflammation, Bone Loss, and Glial Cells within a Collagen Antibody-Induced Arthritis Mouse Model. Mediat. Inflamm. 2020, 2020, 6245798. [Google Scholar] [CrossRef] [PubMed]
- Aljancic, I.; Vajs, V.; Bulatovic, V.; Menkovic, N.; Milosavljevic, S. Parthenolide from the aerial parts of Tanacetum larvatum. Biochem. Syst. Ecol. 2001, 29, 655–658. [Google Scholar] [CrossRef]
- Che, S.-T.; Bie, L.; Li, X.; Qi, H.; Yu, P.; Zuo, L. Parthenolide inhibits the proliferation and induces the apoptosis of human uveal melanoma cells. Int. J. Ophthalmol. 2019, 12, 1531. [Google Scholar] [CrossRef] [PubMed]
- Tadić, V.; Živković, J.; Bigović, D.; Žugić, A. Variation of parthenolide and phenolic compounds content in different parts of Tanacetum parthenium (L.) Schulz Bip. Asteraceae during 18 months storage. Lek. Sirovine 2019, 35–39. [Google Scholar] [CrossRef]
- Pajak, B.; Gajkowska, B.; Orzechowski, A. Molecular basis of parthenolide-dependent proapoptotic activity in cancer cells. Folia Histochem. Cytobiol. 2008, 46, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Liu, L.; Lee, S.-O.; Kim, Y.-T.; You, K.-R.; Kim, D.-G. Susceptibility of cholangiocarcinoma cells to parthenolide-induced apoptosis. Cancer Res. 2005, 65, 6312–6320. [Google Scholar] [CrossRef] [PubMed]
- Carlisi, D.; Buttitta, G.; Di Fiore, R.; Scerri, C.; Drago-Ferrante, R.; Vento, R.; Tesoriere, G. Parthenolide and DMAPT exert cytotoxic effects on breast cancer stem-like cells by inducing oxidative stress, mitochondrial dysfunction and necrosis. Cell Death Dis. 2016, 7, e2194. [Google Scholar] [CrossRef] [PubMed]
- Nakabayashi, H.; Shimizu, K. Involvement of Akt/NF-κB pathway in antitumor effects of parthenolide on glioblastoma cells in vitro and in vivo. BMC Cancer 2012, 12, 1–11. [Google Scholar] [CrossRef]
- Yip-Schneider, M.T.; Wu, H.; Stantz, K.; Agaram, N.; Crooks, P.A.; Schmidt, C.M. Dimethylaminoparthenolide and gemcitabine: A survival study using a genetically engineered mouse model of pancreatic cancer. BMC Cancer 2013, 13, 194. [Google Scholar] [CrossRef]
- Hao, Q.; Wang, B.; Zhang, W.; Qiu, W.; Liu, Q.; Li, X. NF-κB inhibitor parthenolide promotes renal tubules albumin uptake in type 2 diabetic nephropathy. Chin. Med. Sci. J. 2020, 35, 31–42. [Google Scholar]
- Kim, C.Y.; Kang, B.; Hong, J.; Choi, H.-S. Parthenolide inhibits lipid accumulation via activation of Nrf2/Keap1 signaling during adipocyte differentiation. Food Sci. Biotechnol. 2020, 29, 431–440. [Google Scholar] [CrossRef]
- Imran, M.; Rauf, A.; Abu-Izneid, T.; Nadeem, M.; Shariati, M.A.; Khan, I.A.; Imran, A.; Orhan, I.E.; Rizwan, M.; Atif, M. Luteolin, a flavonoid, as an anticancer agent: A review. Biomed. Pharmacother. 2019, 112, 108612. [Google Scholar] [CrossRef]
- Lim, S.H.; Jung, S.K.; Byun, S.; Lee, E.J.; Hwang, J.A.; Seo, S.G.; Kim, Y.A.; Yu, J.G.; Lee, K.W.; Lee, H.J. Luteolin suppresses UVB-induced photoageing by targeting JNK1 and p90RSK2. J. Cell. Mol. Med. 2013, 17, 672–680. [Google Scholar] [CrossRef]
- Wang, H.; Yang, L.; Zu, Y.; Zhao, X. Microwave-assisted simultaneous extraction of luteolin and apigenin from tree peony pod and evaluation of its antioxidant activity. Sci. World J. 2014, 2014, 506971. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Kuang, G.; Wan, J.; Zhang, X.; Li, H.; Gong, X.; Li, H. Luteolin suppresses the metastasis of triple-negative breast cancer by reversing epithelial-to-mesenchymal transition via downregulation of β-catenin expression. Oncol. Rep. 2017, 37, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.-Q.; Li, M.-H.; Qin, Y.-M.; Jiang, H.-Y.; Zhang, X.; Wu, M.-H. Luteolin inhibits tumorigenesis and induces apoptosis of non-small cell lung cancer cells via regulation of MicroRNA-34a-5p. Int. J. Mol. Sci. 2018, 19, 447. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Lim, T.; Han, M.S.; Lee, S.-H.; Baek, S.H.; Nan, H.-Y.; Lee, C. Anticancer effect of luteolin is mediated by downregulation of TAM receptor tyrosine kinases, but not interleukin-8, in non-small cell lung cancer cells. Oncol. Rep. 2017, 37, 1219–1226. [Google Scholar] [CrossRef]
- Yu, Q.; Zhang, M.; Ying, Q.; Xie, X.; Yue, S.; Tong, B.; Wei, Q.; Bai, Z.; Ma, L. Decrease of AIM2 mediated by luteolin contributes to non-small cell lung cancer treatment. Cell Death Dis. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Shi, R.; Wang, X.; Shen, H.-M. Luteolin, a flavonoid with potential for cancer prevention and therapy. Curr. Cancer Drug Targets 2008, 8, 634–646. [Google Scholar] [CrossRef]
- Horinaka, M.; Yoshida, T.; Shiraishi, T.; Nakata, S.; Wakada, M.; Nakanishi, R.; Nishino, H.; Matsui, H.; Sakai, T. Luteolin induces apoptosis via death receptor 5 upregulation in human malignant tumor cells. Oncogene 2005, 24, 7180–7189. [Google Scholar] [CrossRef]
- Cai, X.; Ye, T.; Liu, C.; Lu, W.; Lu, M.; Zhang, J.; Wang, M.; Cao, P. Luteolin induced G2 phase cell cycle arrest and apoptosis on non-small cell lung cancer cells. Toxicol. Vitr. 2011, 25, 1385–1391. [Google Scholar] [CrossRef]
- Ambasta, R.K.; Jha, S.K.; Kumar, D.; Sharma, R.; Jha, N.K.; Kumar, P. Comparative study of anti-angiogenic activities of luteolin, lectin and lupeol biomolecules. J. Transl. Med. 2015, 13, 307. [Google Scholar] [CrossRef]
- Ambasta, R.K.; Gupta, R.; Kumar, D.; Bhattacharya, S.; Sarkar, A.; Kumar, P. Can luteolin be a therapeutic molecule for both colon cancer and diabetes? Brief. Funct. Genom. 2019, 18, 230–239. [Google Scholar] [CrossRef]
- Rauf, A.; Imran, M.; Khan, I.A.; ur-Rehman, M.; Gilani, S.A.; Mehmood, Z.; Mubarak, M.S. Anticancer potential of quercetin: A comprehensive review. Phytother. Res. 2018, 32, 2109–2130. [Google Scholar] [CrossRef] [PubMed]
- Grande, F.; Parisi, O.I.; Mordocco, R.A.; Rocca, C.; Puoci, F.; Scrivano, L.; Quintieri, A.M.; Cantafio, P.; Ferla, S.; Brancale, A. Quercetin derivatives as novel antihypertensive agents: Synthesis and physiological characterization. Eur. J. Pharm. Sci. 2016, 82, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Iacopetta, D.; Grande, F.; Caruso, A.; Mordocco, R.A.; Plutino, M.R.; Scrivano, L.; Ceramella, J.; Muià, N.; Saturnino, C.; Puoci, F. New insights for the use of quercetin analogs in cancer treatment. Future Med. Chem. 2017, 9, 2011–2028. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.K.; Bang, M.H.; Kim, E.S.; Kang, N.E.; Jung, K.C.; Cho, H.J.; Park, J.H. Quercetin decreases the expression of ErbB2 and ErbB3 proteins in HT-29 human colon cancer cells. J. Nutr. Biochem. 2005, 16, 155–162. [Google Scholar] [CrossRef]
- Granado-Serrano, A.B.; Martín, M.A.; Bravo, L.; Goya, L.; Ramos, S. Quercetin induces apoptosis via caspase activation, regulation of Bcl-2, and inhibition of PI-3-kinase/Akt and ERK pathways in a human hepatoma cell line (HepG2). J. Nutr. 2006, 136, 2715–2721. [Google Scholar] [CrossRef]
- Liao, H.; Bao, X.; Zhu, J.; Qu, J.; Sun, Y.; Ma, X.; Wang, E.; Guo, X.; Kang, Q.; Zhen, Y. O-Alkylated derivatives of quercetin induce apoptosis of MCF-7 cells via a caspase-independent mitochondrial pathway. Chem. Biol. Interact. 2015, 242, 91–98. [Google Scholar] [CrossRef]
- Ranganathan, S.; Halagowder, D.; Sivasithambaram, N.D. Quercetin suppresses twist to induce apoptosis in MCF-7 breast cancer cells. PLoS ONE 2015, 10, e0141370. [Google Scholar] [CrossRef]
- Minaei, A.; Sabzichi, M.; Ramezani, F.; Hamishehkar, H.; Samadi, N. Co-delivery with nano-quercetin enhances doxorubicin-mediated cytotoxicity against MCF-7 cells. Mol. Biol. Rep. 2016, 43, 99–105. [Google Scholar] [CrossRef]
- Chang, J.-H.; Lai, S.-L.; Chen, W.-S.; Hung, W.-Y.; Chow, J.-M.; Hsiao, M.; Lee, W.-J.; Chien, M.-H. Quercetin suppresses the metastatic ability of lung cancer through inhibiting Snail-dependent Akt activation and Snail-independent ADAM9 expression pathways. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2017, 1864, 1746–1758. [Google Scholar] [CrossRef]
- Ali, H.; Dixit, S. Quercetin attenuates the development of 7, 12-dimethyl benz (a) anthracene (DMBA) and croton oil-induced skin cancer in mice. J. Biomed. Res. 2015, 29, 139. [Google Scholar]
- Bule, M.; Abdurahman, A.; Nikfar, S.; Abdollahi, M.; Amini, M. Antidiabetic effect of quercetin: A systematic review and meta-analysis of animal studies. Food Chem. Toxicol. 2019, 125, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, P.; Vijayakumar, S.; Kothandaraman, S.; Palani, M. Anti-diabetic activity of quercetin extracted from Phyllanthus emblica L. fruit: In silico and in vivo approaches. J. Pharm. Anal. 2018, 8, 109–118. [Google Scholar] [CrossRef]
- An, Y.-W.; Jin, H.-T.; Yuan, B.; Wang, J.-C.; Wang, C.; Liu, H.-Q. Research progress of berberine mediated photodynamic therapy. Oncol. Lett. 2021, 21, 1–10. [Google Scholar] [CrossRef]
- Yun, D.; Yoon, S.Y.; Park, S.J.; Park, Y.J. The Anticancer Effect of Natural Plant Alkaloid Isoquinolines. Int. J. Mol. Sci. 2021, 22, 1653. [Google Scholar] [CrossRef]
- Xiong, P.; Niu, L.; Talaei, S.; Kord-Varkaneh, H.; Clark, C.C.; Găman, M.-A.; Rahmani, J.; Dorosti, M.; Mousavi, S.M.; Zarezadeh, M. The effect of berberine supplementation on obesity indices: A dose–response meta-analysis and systematic review of randomized controlled trials. Complement. Ther. Clin. Pract. 2020, 39, 101113. [Google Scholar] [CrossRef]
- Costea, T.; Hudiță, A.; Ciolac, O.-A.; Gălățeanu, B.; Ginghină, O.; Costache, M.; Ganea, C.; Mocanu, M.-M. Chemoprevention of colorectal cancer by dietary compounds. Int. J. Mol. Sci. 2018, 19, 3787. [Google Scholar] [CrossRef] [PubMed]
- Ramprasath, V.R.; Awad, A.B. Role of phytosterols in cancer prevention and treatment. J. Aoac Int. 2015, 98, 735–738. [Google Scholar] [CrossRef]
- Xiong, M.; Huang, Y.; Liu, Y.; Huang, M.; Song, G.; Ming, Q.; Ma, X.; Yang, J.; Deng, S.; Wen, Y. Antidiabetic activity of ergosterol from Pleurotus ostreatus in KK-Ay mice with spontaneous type 2 diabetes mellitus. Mol. Nutr. Food Res. 2018, 62, 1700444. [Google Scholar] [CrossRef]
- Oei, A.L.; Sweep, F.C.; Thomas, C.M.; Boerman, O.C.; Massuger, L.F. The use of monoclonal antibodies for the treatment of epithelial ovarian cancer. Int. J. Oncol. 2008, 32, 1145–1157. [Google Scholar] [CrossRef]
- Pourianezhad, F.; Tahmasebi, S.; Nikfar, S.; Mirhoseini, M.; Abdusi, V. Review on feverfew, a valuable medicinal plant. J. Herbmed Pharmacol. 2016, 5. [Google Scholar]
- Carlisi, D.; D’Anneo, A.; Angileri, L.; Lauricella, M.; Emanuele, S.; Santulli, A.; Vento, R.; Tesoriere, G. Parthenolide sensitizes hepatocellular carcinoma cells to TRAIL by inducing the expression of death receptors through inhibition of STAT3 activation. J. Cell. Physiol. 2011, 226, 1632–1641. [Google Scholar] [CrossRef] [PubMed]
- Godic, A.; Poljšak, B.; Adamic, M.; Dahmane, R. The role of antioxidants in skin cancer prevention and treatment. Oxid. Med. Cell. Longev. 2014, 2014, 860479. [Google Scholar] [CrossRef] [PubMed]
- Westerlund, A.; Steineck, G.; Bälter, K.; Stattin, P.; Grönberg, H.; Hedelin, M. Dietary supplement use patterns in men with prostate cancer: The Cancer Prostate Sweden study. Ann. Oncol. 2011, 22, 967–972. [Google Scholar] [CrossRef] [PubMed]
- Willcox, J.K.; Ash, S.L.; Catignani, G.L. Antioxidants and prevention of chronic disease. Crit. Rev. Food Sci. Nutr. 2004, 44, 275–295. [Google Scholar] [CrossRef]
- Alpha-Tocopherol Beta Carotene Cancer Prevention Study Group. The effect of vitamin E and beta carotene on the incidence of lung cancer and other cancers in male smokers. N. Engl. J. Med. 1994, 330, 1029–1035. [Google Scholar] [CrossRef]
- Goodman, M.; Bostick, R.M.; Kucuk, O.; Jones, D.P. Clinical trials of antioxidants as cancer prevention agents: Past, present, and future. Free Radic. Biol. Med. 2011, 51, 1068–1084. [Google Scholar] [CrossRef]
- Middha, P.; Weinstein, S.J.; Männistö, S.; Albanes, D.; Mondul, A.M. β-carotene supplementation and lung cancer incidence in the alpha-tocopherol, Beta-carotene cancer prevention study: The role of tar and nicotine. Nicotine Tob. Res. 2019, 21, 1045–1050. [Google Scholar] [CrossRef]
- Narita, S.; Saito, E.; Sawada, N.; Shimazu, T.; Yamaji, T.; Iwasaki, M.; Ishihara, J.; Takachi, R.; Shibuya, K.; Inoue, M. Dietary consumption of antioxidant vitamins and subsequent lung cancer risk: The J apan P ublic H ealth C enter-based prospective study. Int. J. Cancer 2018, 142, 2441–2460. [Google Scholar] [CrossRef]
- Klein, E.A.; Thompson, I.M.; Tangen, C.M.; Crowley, J.J.; Lucia, M.S.; Goodman, P.J.; Minasian, L.M.; Ford, L.G.; Parnes, H.L.; Gaziano, J.M. Vitamin E and the risk of prostate cancer: The Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2011, 306, 1549–1556. [Google Scholar] [CrossRef]
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Bergo, M.O. Antioxidants accelerate lung cancer progression in mice. Sci. Transl. Med. 2014, 6, 221ra15. [Google Scholar] [CrossRef]
- Wiel, C.; Le Gal, K.; Ibrahim, M.X.; Jahangir, C.A.; Kashif, M.; Yao, H.; Ziegler, D.V.; Xu, X.; Ghosh, T.; Mondal, T. BACH1 stabilization by antioxidants stimulates lung cancer metastasis. Cell 2019, 178, 330–345. [Google Scholar] [CrossRef] [PubMed]
- Le Gal, K.; Ibrahim, M.X.; Wiel, C.; Sayin, V.I.; Akula, M.K.; Karlsson, C.; Dalin, M.G.; Akyürek, L.M.; Lindahl, P.; Nilsson, J. Antioxidants can increase melanoma metastasis in mice. Sci. Transl. Med. 2015, 7, 308re8. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Heaney, M.L.; Gardner, J.R.; Karasavvas, N.; Golde, D.W.; Scheinberg, D.A.; Smith, E.A.; O’Connor, O.A. Vitamin C antagonizes the cytotoxic effects of antineoplastic drugs. Cancer Res. 2008, 68, 8031–8038. [Google Scholar] [CrossRef] [PubMed]
- Fukui, M.; Yamabe, N.; Zhu, B.T. Resveratrol attenuates the anticancer efficacy of paclitaxel in human breast cancer cells in vitro and in vivo. Eur. J. Cancer 2010, 46, 1882–1891. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Sun, C.; Zhou, B.; Xing, H.; Ma, D.; Chen, G.; Weng, D. Low concentration of quercetin antagonizes the cytotoxic effects of anti-neoplastic drugs in ovarian cancer. PLoS ONE 2014, 9, e100314. [Google Scholar] [CrossRef]
- Wu, Y.J.; Muldoon, L.L.; Neuwelt, E.A. The chemoprotective agent N-acetylcysteine blocks cisplatin-induced apoptosis through caspase signaling pathway. J. Pharmacol. Exp. Ther. 2005, 312, 424–431. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Risk Factors | NCEP/ATP III | WHO 1999 | IDF |
| (3 of 5 criteria necessary) [41,42] | (impaired glucose regulation or hyperinsulinemia and 2 or more criteria necessary) A | (increased waist circumference plus any 2 of other 4 criteria) [50] | |
| Fasting glucose | ≥110 mg/dL | ≥110 mg/dL | ≥110 mg/dL |
| Prandial glucose | >140 mg/dL | ||
| Hyperinsulinemia | Fasting serum insulin: third quartile for the control group | ||
| Hypertriglyceridemia B | ≥150 mg/dL | ≥150 mg/dL | ≥150 mg/dL |
| Low HDL-C | <40 mg/dL <50 mg/dL | <35 mg/dL <39 mg/dL | <40 mg/dL <50 mg/dL |
| Men Women | |||
| Abdominal obesity | >102 cm >88 cm | waist: hip ratio >0.9 in >0.85 in | ≥94 cm ≥80 cm |
| Men Women | |||
| Hypertension B | ≥130/≥85 mm Hg | ≥140/≥90 mm Hg | ≥130/≥85 mm |
| Micro-albuminuria | ≥20 μg/min |
| Natural Products | Mechanism of Action in Cancer | Mechanism of Action in Diabetes |
|---|---|---|
| Resveratrol | ↑ caspase-3 and caspase-9, p53, X-associated protein Bcl-2 in human cervical carcinoma [141] ↓ signaling pathway of AKT/STAT3 in colon cancer cells [142] ↑ TRPM2 channel in glioblastoma cells [143] control the expressions of vimentin, CTA-2, IL-1β, TNF- α, and N-cadherin in pancreatic cancer cells [144] ↑ Bcl-2 activity, p53, Bax, and caspase expression in breast cancer [145] ↓ tumor volume in Ehrlich ascites carcinoma cells [148] | ↓ α-glucosidase, carbohydrate absorption, and post-prandial blood glucose response in mice [149] ↑ muscle and liver glucose absorption ↓ adipose tissue and liver inflammation ↑ adaptive thermogenesis capability ↓ pancreatic insulin secretion, and mimic caloric restriction effects [149] ↑ serum blood glucose, insulin level, and dyslipidemia in diabetic rats [151] |
| Curcumin | ↑ apoptotic ligand-inducing tumor-necrosis-factor-related apoptosis (TRAIL) pathways ↑ death receptor factor 5in HT-29 and HCT-116 colon cancer cells [161] ↑ Fas-mediated apoptotic pathway, caspase 8 activation, Bax expression ↓ Bcl-2 in HT-29 colon cancer [162,163], and HCT-116 [155] ↓ JAK/STAT signal in osteosarcoma [164] interfere with nuclear factor κ (NFκB), epidermal growth factor receptor (EGFR), and mitogen-activated protein kinase (MAPK) in prostate cancer [165] | ↑ adipocyte differentiation substantially, glucose levels, and the activity of human peroxisome proliferator-activated receptor (PPAR)-gamma ligand-binding [166] ↓ glucose uptake, leptin levels, NF-kB p65 nuclear fraction, phospho-JNK1/2, phospho-IRS-1(S), MMP-2, MMP-9 and VEGF protein in 3T3-L1 adipocytes [166] ↓ levels of urinary enzymes (acid phosphatase, alkaline phosphatase (ALP), aspartate aminotransferase (AST), and alanine aminotransferase (ALT)) [167] ↓ JAK/STAT signalling pathway ↓ myocardial degeneration, lipid peroxidation levels, IL-6, creatine kinase-MB (CK-MB), troponin I, and tumor growth factor-β1 (TGF-β1) in co-treatment with metformin [168] |
| Thymoquinone | ↓ cell arrest at various stages in MCF-7 cell line [175] ↓ serum TNF- α, IL-6, and iNOS enzyme production and improve histopathological outputs in Wistar rats [176] ↓ phosphorylation of EGFR to tyrosine-1173 residues and JAK2 in HCT 116 [177] ↑ cytotoxicity in the xenograft mouse model of breast cancer [178] ↑ apoptosis by reducing antiapoptotic protein expression [179] | ↓ blood glucose level, triglycerides and cholesterol concentrations ↑ high-density lipoprotein, insulin sensitivity and pancreatic β-cell regeneration [180] maintain the integrity of pancreatic β-cells by enhancing oxidative stress [181] |
| EGCG | ↓ MMP-2 through modulation the Src signaling pathway in cervical cancer [190] ↓ cyclinD1 ↑ LIMD1, RBSP3, and p16 cell-cycle inhibitors at the G1/S cell cycle level in cervical cancer cell line [192] ↑ AKT/STAT3 pathway, apoptosis in cisplatin-resistant oral cancer cell [193] | ↓ α-glucosidase ↑ glucose uptake ↑ GLUT4 translocation to plasma membrane through PI3K/AKT signaling pathway in L6 skeletal muscle cells [195] |
| Allicin | ↓ cell migration, STAT3 ↑ SHP-1, apoptosis in cholangiocarcinoma cell [198] ↓ signaling pathway of NF-κ in colorectal cancer [199] ↓ cytokine release ↑ p53 and radiotherapy sensitivity enhancement in glioma cells [200] ↑ apoptosis ↓ ornithine decarboxylase in neuroblastoma Cells. [201] ↓ MMP-9 mRNA expression ↑ cyclin D1 in melanoma cells [202] | ↑ insulin levels ↓ hyperglycemia, (GLUT4) and IRSs expression [204] ↑ Nrf2 ↓ Keap1, HIF-1α, SBP, and VEGF [204] ↓ autoantibodies and anti-islet cell antibodies (ICA) for type 1 diabetes (IDDM) [205] |
| Emodin | ↑ apoptosis, cell cycle arrest, HIF-1α, glutathione phase I and II, and glutathione S-transferase P, N-acetyltransferase ↓ angiogenesis, invasion, migration, formation of chemical-induced carcinogen-DNA adducts, HER2/neu, CKII kinase, and p34cdc2 kinase in human cancer cells [215] ↓ ERK phosphorylation in epithelial ovarian cancer [266] | ↓ cellular FLICE-inhibitory protein (cFLIP) and p38MAPK pathway ↑ activate PPARγ pathway, and modulate AMPK signalling pathway [216] |
| Genistein | ↓ proliferation, angiogenesis and metastasis ↑ apoptosis leading to tumor growth reduction in hepatocellular and gastric cancer models of Wistar rats [219,220] ↓ COX-2-stimulating factors such as activated protein-1 (AP-1) and Nf-κB in pancreatic, colon, breast, and lung cancer [217] | modulate the proliferation of β-cells and the secretion of insulin [222] ↑ cAMP signaling, and the mass of islets in diabetic mice [223] |
| Parthenolide | interrupt DNA replication [267] ↓ STAT3 ↑ apoptotic pathway [268] ↑ p53 and reactive oxygen species (ROS) [228] ↓ breast cancer stem-like cells by stimulating oxidative stress and necrosis [231] ↓ cell growth of glioblastoma cells [232] | ↓ inflammation and remodel the impaired insulin signaling pathway ↑ cubilin and albumin uptake expression [234] modulate reactive oxygen species production, and control the Nrf2- (Keap1) pathway [235] ↓ adipogenic factors (PPARc and C/EBPa) and its target protein FABP4 production [235] |
| Luteolin | ↑ apoptosis pathways and death receptor 5 expression [244] ↑ JNK ↓ NF-κB, mediate the cellular growth inhibition and G2 arrest [245] ↓ proliferation, angiogenesis, VEGF receptor, PI3K/Akt and PI3K/p70S6 kinase pathways [246] ↓ cell proliferation and stimulate apoptosis in H460 and A549 cells [240,241,242] | regulate diabetes through mammalian target of rapamycin (mTOR), cytokine, AMPK, and p53 [247] |
| Quercetin | ↑ apoptosis ↓epidermal growth factor receptor (EGFR) expression [251] ↑ caspase-3 and-9 ↓ Bcl-2, Bcl-xL [252] ↑ G1 phase arrest in breast cancer cell lines [253,254] ↓ cell invasion and migration ↑ apoptosis in human lung carcinoma A549 cells [256] | ↓ serum glucose level modulate hepatic gene expressions ↓ α-glucosidase activity, and PPAR-γ ↑ insulin action [259] |
| Berberine | ↓ survivin and Bcl2 ↑ apoptosis, p53, and Bax expression in gastric cancer cells [260] ↑ JNK phosphorylation, cytochrome c and AIF, and caspase-3 ↓ mitochondrial membrane potential, and Bcl-2 expression in breast cancer [261] | ↓ blood glucose ↓ risk of metabolic syndrome, levels of hemoglobin A1C and triglyceride ↓ leptin and resistin secretion ↑ insulin sensitivity and weight loss ↑ lipid metabolism and mRNA expression of adiponectin [262] |
| Phytosterols | ↓ cell growth and metastasis ↓ tumor size in athymic mice injected with MCF-7 cells [264] | ↑ GLUT4 translocation ↑ PI3K/Akt and PKC pathways ↓ fasting blood glucose levels [265] |
| Combination Treatment | Results | Ref. |
|---|---|---|
| Vitamin C + doxorubicin | Increased resistance to treatment in cell lines of chronic myelogenous leukemia (K562) and lymphoma (RL) | [282] |
| Larger tumors in mice with RL cell xenografts | ||
| Resveratrol + paclitaxel | Decreased antitumor action of paclitaxel in human breast tumor cells | [283] |
| Quercetin at low doses + cisplatin, 5-FU, taxol or pirarubicin | Decreased efficiency of the treatment in athymic nude mice with ovarian tumor cells (C13*) xenografts | [284] |
| N-acetylcysteine before or up to 1 h after the drug + cisplatin | Blockade of proapoptotic effect of cisplatin in human ovarian carcinoma cells (SKOV3), human SCLC tumor cells (B.5 LX-1), human glioblastoma cells (U87), and rat fibroblasts | [285] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Talib, W.H.; Mahmod, A.I.; Abuarab, S.F.; Hasen, E.; Munaim, A.A.; Haif, S.K.; Ayyash, A.M.; Khater, S.; AL-Yasari, I.H.; Kury, L.T.A. Diabetes and Cancer: Metabolic Association, Therapeutic Challenges, and the Role of Natural Products. Molecules 2021, 26, 2179. https://doi.org/10.3390/molecules26082179
Talib WH, Mahmod AI, Abuarab SF, Hasen E, Munaim AA, Haif SK, Ayyash AM, Khater S, AL-Yasari IH, Kury LTA. Diabetes and Cancer: Metabolic Association, Therapeutic Challenges, and the Role of Natural Products. Molecules. 2021; 26(8):2179. https://doi.org/10.3390/molecules26082179
Chicago/Turabian StyleTalib, Wamidh H., Asma Ismail Mahmod, Sara Feras. Abuarab, Eliza Hasen, Amer A. Munaim, Shatha Khaled Haif, Amani Marwan Ayyash, Samar Khater, Intisar Hadi AL-Yasari, and Lina T. Al Kury. 2021. "Diabetes and Cancer: Metabolic Association, Therapeutic Challenges, and the Role of Natural Products" Molecules 26, no. 8: 2179. https://doi.org/10.3390/molecules26082179
APA StyleTalib, W. H., Mahmod, A. I., Abuarab, S. F., Hasen, E., Munaim, A. A., Haif, S. K., Ayyash, A. M., Khater, S., AL-Yasari, I. H., & Kury, L. T. A. (2021). Diabetes and Cancer: Metabolic Association, Therapeutic Challenges, and the Role of Natural Products. Molecules, 26(8), 2179. https://doi.org/10.3390/molecules26082179