
Anion–Anion Interactions in Aerogen-Bonded Complexes. Influence of Solvent Environment




Abstract
1. Introduction
2. Computational Methods
3. Results
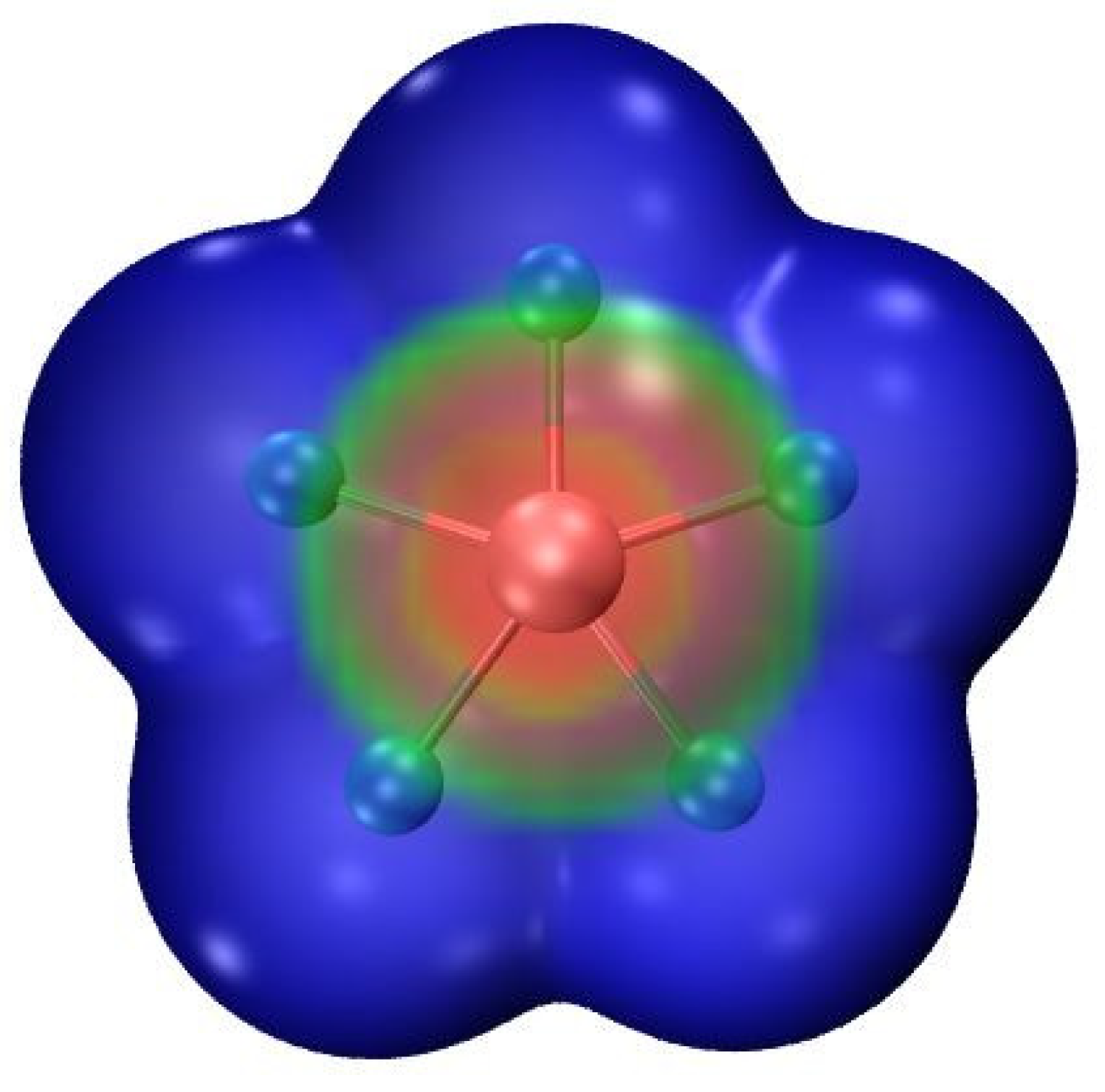
3.1. Monomers
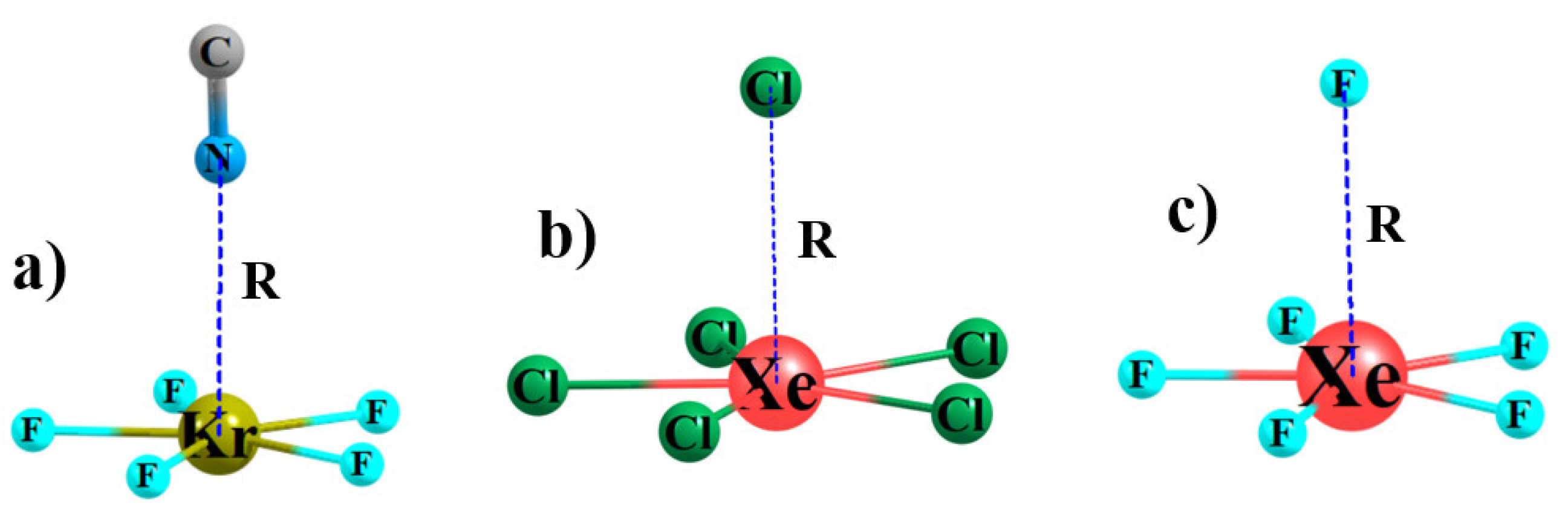
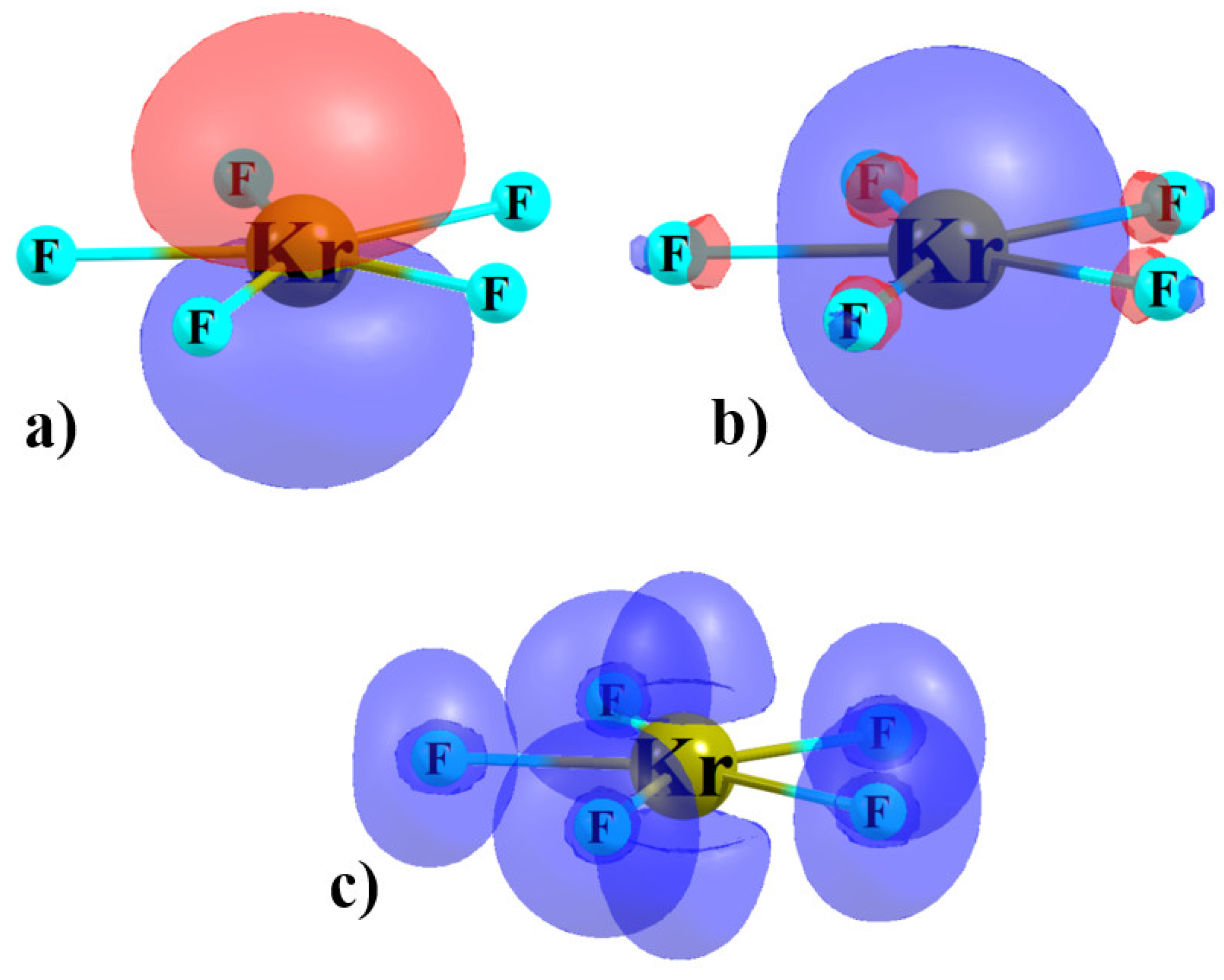
3.2. Complexes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen bonding: The sigma-hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef]
- Murray, J.S.; Lane, P.; Politzer, P. Expansion of the sigma-hole concept. J. Mol. Model. 2009, 15, 723–729. [Google Scholar] [CrossRef]
- Murray, J.S.; Lane, P.; Clark, T.; Riley, K.E.; Politzer, P. σ-Holes, π-holes and electrostatically-driven interactions. J. Mol. Model. 2012, 18, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.S.; Resnati, G.; Politzer, P. Close contacts and noncovalent interactions in crystals. Faraday Discuss. 2017, 203, 113–130. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S. Analysis of Halogen and Other sigma-Hole Bonds in Crystals. Crystals 2018, 8, 42. [Google Scholar] [CrossRef]
- Kolář, M.H.; Hobza, P. Computer Modeling of Halogen Bonds and Other σ-Hole Interactions. Chem. Rev. 2016, 116, 5155–5187. [Google Scholar] [CrossRef]
- Zhu, Z.D.; Xu, Z.J.; Zhu, W.L. Interaction Nature and Computational Methods for Halogen Bonding: A Perspective. J. Chem. Inf. Model. 2020, 60, 2683–2696. [Google Scholar] [CrossRef]
- Wang, R.J.; Li, Q.Z.; Scheiner, S. Complexes of HArF and AuX (X = F, Cl, Br, I). Comparison of H-bonds, halogen bonds, F-shared bonds and covalent bonds. Appl. Organomet. Chem. 2020, 34, e5891. [Google Scholar] [CrossRef]
- Mondal, S.; Manna, D.; Raja, K.; Mugesh, G. Halogen Bonding in Biomimetic Deiodination of Thyroid Hormones and their Metabolites and Dehalogenation of Halogenated Nucleosides. ChemBioChem 2020, 21, 911–923. [Google Scholar] [CrossRef]
- Rahman, F.U.; Tzeli, D.; Petsalakis, I.D.; Theodorakopoulos, G.; Ballester, P.; Rebek, J.; Yu, Y. Chalcogen Bonding and Hydrophobic Effects Force Molecules into Small Spaces. J. Am. Chem. Soc. 2020, 142, 5876–5883. [Google Scholar] [CrossRef]
- Kumar, V.; Xu, Y.J.; Bryce, D.L. Double Chalcogen Bonds: Crystal Engineering Stratagems via Diffraction and Multinuclear Solid-State Magnetic Resonance Spectroscopy. Chem. Eur. J. 2020, 26, 3275–3286. [Google Scholar] [CrossRef]
- Ho, P.C.; Wang, J.Z.; Meloni, F.; Vargas-Baca, I. Chalcogen bonding in materials chemistry. Coord. Chem. Rev. 2020, 422, 213464. [Google Scholar] [CrossRef]
- Scheiner, S. Coord.ation of a Central Atom by Multiple Intramolecular Pnicogen Bonds. Inorg. Chem. 2020, 59, 9315–9324. [Google Scholar] [CrossRef] [PubMed]
- Palanisamy, D. A quantum chemical perspective on the potency of electron donors and acceptors in pnicogen bonds (AS… N, P… N, N… N). J. Mol. Model. 2020, 26, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Elguero, J.; Del Bene, J.E.; Mo, O.; Montero-Campillo, M.M.; Yanez, M. Mutual Influence of Pnicogen Bonds and Beryllium Bonds: Energies and Structures in the Spotlight. J. Phys. Chem. A 2020, 124, 5871–5878. [Google Scholar] [CrossRef]
- Grabowski, S.J. Pnicogen and tetrel bonds-tetrahedral Lewis acid centres. Struct. Chem. 2019, 30, 1141–1152. [Google Scholar] [CrossRef]
- Scheiner, S. The ditetrel bond: Noncovalent bond between neutral tetrel atoms. Phys. Chem. Chem. Phys. 2020, 22, 16606–16614. [Google Scholar] [CrossRef] [PubMed]
- Roeleveld, J.J.; Lekanne Deprez, S.J.; Verhoofstad, A.; Frontera, A.; van der Vlugt, J.I.; Mooibroek, T.J. Engineering Crystals Using sp(3) -C Centred Tetrel Bonding Interactions. Chemistry 2020, 26, 10126–10132. [Google Scholar] [CrossRef]
- Mahmoudi, G.; Abedi, M.; Lawrence, S.E.; Zangrando, E.; Babashkina, M.G.; Klein, A.; Frontera, A.; Safin, D.A. Tetrel Bonding and Other Non-Covalent Interactions Assisted Supramolecular Aggregation in a New Pb(II) Complex of an Isonicotinohydrazide. Molecules 2020, 25, 4056. [Google Scholar] [CrossRef]
- Kumar, V.; Rodrigue, C.; Bryce, D.L. Short and Linear Intermolecular Tetrel Bonds to Tin. Cocrystal Engineering with Triphenyltin Chloride. Cryst. Growth Des. 2020, 20, 2027–2034. [Google Scholar] [CrossRef]
- Grabowski, S.J. Triel bond and coordination of triel centres—Comparison with hydrogen bond interaction. Coord. Chem. Rev. 2020, 407, 213171. [Google Scholar] [CrossRef]
- Grabowski, S.J. The Nature of Triel Bonds, a Case of B and Al Centres Bonded with Electron Rich Sites. Molecules 2020, 25, 2703. [Google Scholar] [CrossRef]
- Chi, Z.; Dong, W.; Li, Q.; Yang, X.; Scheiner, S.; Liu, S. Carbene triel bonds between TrR3 (Tr = B, Al) and N-heterocyclic carbenes. Int. J. Quantum Chem. 2019, 119, e25867. [Google Scholar] [CrossRef]
- Wang, R.J.; Wang, Z.; Yu, X.F.; Li, Q.Z. Synergistic and Diminutive Effects between Regium and Aerogen Bonds. ChemPhysChem 2020, 21, 2426–2431. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Mousavian, P.; Mohammadian-Sabet, F. Tuning of pnicogen and chalcogen bonds by an aerogen-bonding interaction: A comparative ab initio study. Mol. Phys. 2019, 117, 58–66. [Google Scholar] [CrossRef]
- Gao, M.; Cheng, J.; Li, W.; Xiao, B.; Li, Q. The aerogen–π bonds involving π systems. Chem. Phys. Lett. 2016, 651, 50–55. [Google Scholar] [CrossRef]
- Bauzá, A.; Frontera, A. Aerogen Bonding Interaction: A New Supramolecular Force? Angew. Chem. Int. Ed. 2015, 54, 7340–7343. [Google Scholar] [CrossRef]
- Ams, M.R.; Trapp, N.; Schwab, A.; Milic, J.V.; Diederich, F. Chalcogen Bonding “2S-2N Squares” versus Competing Interactions: Exploring the Recognition Properties of Sulfur. Chem. Eur. J. 2019, 25, 323–333. [Google Scholar] [CrossRef]
- Braga, D.; Grepioni, F.; Desiraju, G.R. Crystal engineering and organometallic architecture. Chem. Rev. 1998, 98, 1375–1405. [Google Scholar] [CrossRef] [PubMed]
- Braga, D.; Maini, L.; Grepioni, F. Crystal engineering of organometallic compounds through cooperative strong and weak hydrogen bonds: A simple route to mixed-metal systems. Angew. Chem. Int. Ed. 1998, 37, 2240–2242. [Google Scholar] [CrossRef]
- Dordevic, I.S.; Popadic, M.; Sarvan, M.; Petkovic-Benazzouz, M.; Janjic, G.V. Supramolecular insight into the substitution of sulfur by selenium, based on crystal structures, quantum-chemical calculations and biosystem recognition. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2020, 76, 122–136. [Google Scholar] [CrossRef]
- Fourmigue, M.; Dhaka, A. Chalcogen bonding in crystalline diselenides and selenocyanates: From molecules of pharmaceutical interest to conducting materials. Coord. Chem. Rev. 2020, 403, 213084. [Google Scholar] [CrossRef]
- Bauza, A.; Mooibroek, T.J.; Frontera, A. Towards design strategies for anion-pi interactions in crystal engineering. CrystEngComm 2016, 18, 10–23. [Google Scholar] [CrossRef]
- Bauza, A.; Seth, S.K.; Frontera, A. Tetrel bonding interactions at work: Impact on tin and lead coordination compounds. Coord. Chem. Rev. 2019, 384, 107–125. [Google Scholar] [CrossRef]
- Bonfant, G.; Melegari, M.; Balestri, D.; Mezzadri, F.; Marzaroli, V.; Bassanetti, I.; Marchio, L. Supramolecular Assemblies in Silver Complexes: Phase Transitions and the Role of the Halogen Bond. Inorg. Chem. 2020, 59, 4140–4149. [Google Scholar] [CrossRef]
- Cavallo, G.; Murray, J.S.; Politzer, P.; Pilati, T.; Ursini, M.; Resnati, G. Halogen bonding in hypervalent iodine and bromine derivatives: Halonium salts. IUCrJ 2017, 4, 411–419. [Google Scholar] [CrossRef]
- Chen, L.; Xiang, J.; Zhao, Y.; Yan, Q. Reversible Self-Assembly of Supramolecular Vesicles and Nanofibers Driven by Chalcogen-Bonding Interactions. J. Am. Chem. Soc. 2018, 140, 7079–7082. [Google Scholar] [CrossRef]
- Hung, M.K.; Tsai, K.W.; Sharma, S.; Lei, J.; Wu, J.Y.; Chen, S.A. Optoelectronic Properties of High Triplet sigma-pi-Conjugated Poly[(biphenyl group IV-A atom (C, Si, Ge, Sn)] Backbones. ACS Appl. Mater. Interfaces 2019, 11, 36895–36904. [Google Scholar] [CrossRef]
- Frontera, A.; Bauza, A. Regium-pi bonds: An Unexplored Link between Noble Metal Nanoparticles and Aromatic Surfaces. Chem. Eur. J. 2018, 24, 7228–7234. [Google Scholar] [CrossRef]
- Bhattacharyya, M.K.; Saha, U.; Dutta, D.; Frontera, A.; Verma, A.K.; Sharma, P.; Das, A. Unconventional DNA-relevant pi-stacked hydrogen bonded arrays involving supramolecular guest benzoate dimers and cooperative anion-pi/pi-pi/pi-anion contacts in coordination compounds of Co(ii) and Zn(ii) phenanthroline: Experimental and theoretical studies. New J. Chem. 2020, 44, 4504–4518. [Google Scholar]
- Bishop, A.; Brodbelt, J.S. Selective cleavage upon ETD of peptides containing disulfide or nitrogen-nitrogen bonds. Int. J. Mass Spectrom. 2015, 378, 127–133. [Google Scholar] [CrossRef]
- Frontera, A.; Bauzá, A. S⋅⋅⋅Sn Tetrel Bonds in the Activation of Peroxisome Proliferator-Activated Receptors (PPARs) by Organotin Molecules. Chem. Eur. J. 2018, 24, 16582–16587. [Google Scholar] [CrossRef] [PubMed]
- Kriz, K.; Fanfrlik, J.; Lepsik, M. Chalcogen Bonding in Protein-Ligand Complexes: PDB Survey and Quantum Mechanical Calculations. ChemPhysChem 2018, 19, 2540–2548. [Google Scholar] [CrossRef] [PubMed]
- Esrafili, M.D.; Mohammadian-Sabet, F.; Solimannejad, M. Single-electron aerogen bonds: Do they exist? Chem. Phys. Lett. 2016, 659, 196–202. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Vessally, E. A theoretical evidence for cooperative enhancement in aerogen-bonding interactions: Open-chain clusters of KrOF2 and XeOF. Chem. Phys. Lett. 2016, 662, 80–85. [Google Scholar] [CrossRef]
- Zierkiewicz, W.; Michalczyk, M.; Scheiner, S. Aerogen bonds formed between AeOF2 (Ae = Kr, Xe) and diazines: Comparisons between sigma-hole and pi-hole complexes. Phys. Chem. Chem. Phys. 2018, 20, 4676–4687. [Google Scholar] [CrossRef]
- Miao, J.J.; Xiong, Z.H.; Gao, Y. The effects of aerogen-bonding on the geometries and spectral properties of several small molecular clusters containing XeO3. J. Phys. Condens. Matter 2018, 30, 444001. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Song, B.; Gao, Y. Is Aerogen–π Interaction Capable of Initiating the Noncovalent Chemistry of Group 18? Chem. An Asian J. 2015, 10, 2615–2618. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Asadollahi, S. Strengthening of the halogen-bonding by an aerogen bond interaction: Substitution and cooperative effects in O3Z···NCX···NCY (Z = Ar, Kr, Xe; X = Cl, Br, I; Y = H, F, OH) complexes. Mol. Phys. 2016, 114, 2177–2186. [Google Scholar] [CrossRef]
- Bavafa, S.; Nowroozi, A.; Ebrahimi, A. Quantum chemical study of the nature of interactions between the boraphosphinine and alumaphosphinine with some of the mono- and divalent cations: Cation-pi or cation-lone pair? Struct. Chem. 2019, 30, 1887–1898. [Google Scholar] [CrossRef]
- Gomila, R.M.; Frontera, A. Covalent and Non-covalent Noble Gas Bonding Interactions in XeFn Derivatives (n = 2–6): A Combined Theoretical and ICSD Analysis. Front. Chem. 2020, 8, 395. [Google Scholar] [CrossRef]
- Carvalho, F.M.; Kiametis, A.S.; de Araújo Oliveira, A.L.; Pirani, F.; Gargano, R. Spectroscopy, lifetime, and charge-displacement of the methanol-noble gas complexes: An integrated experimental-theoretical investigation. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2021, 246, 119049. [Google Scholar] [CrossRef] [PubMed]
- De Araujo Oliveira, A.L.; de Abreu Silva, M.; Pirani, F.; de Macedo, L.G.M.; Gargano, R. Hydrogen sulphide H2S and noble gases (Ng = He, Ne, Ar, Kr, Xe, Rn) complexes: A theoretical study of their dynamics, spectroscopy, and interactions. Int. J. Quantum Chem. 2020, 120, e26266. [Google Scholar] [CrossRef]
- Lu, Y.; Li, H.; Zhu, X.; Zhu, W.; Liu, H. How Does Halogen Bonding Behave in Solution? A Theoretical Study Using Implicit Solvation Model. J. Phys. Chem. A 2011, 115, 4467–4475. [Google Scholar] [CrossRef]
- Bania, K.K.; Guha, A.K.; Bhattacharyya, P.K.; Sinha, S. Effect of substituent and solvent on cation–π interactions in benzene and borazine: A computational study. Dalton Trans. 2014, 43, 1769–1784. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.S.; Zipse, H.; Sastry, G.N. Explicit Solvent Effect on Cation−π Interactions: A First Principle Investigation. J. Phys. Chem. B 2009, 113, 7225–7236. [Google Scholar] [CrossRef] [PubMed]
- Esrafili, M.D.; Sadr-Mousavi, A. Chalcogen bonds tuned by an N-H center dot center dot center dot pi or C-H center dot center dot center dot pi interaction: Investigation of substituent, cooperativity and solvent effects. Mol. Phys. 2017, 115, 1713–1723. [Google Scholar] [CrossRef]
- Mata, I.; Molins, E.; Alkorta, I.; Espinosa, E. The Paradox of Hydrogen-Bonded Anion–Anion Aggregates in Oxoanions: A Fundamental Electrostatic Problem Explained in Terms of Electrophilic···Nucleophilic Interactions. J. Phys. Chem. A 2014, 119, 183–194. [Google Scholar] [CrossRef]
- Shokri, A.; Ramezani, M.; Fattahi, A.; Kass, S.R. Electrostatically defying cation-cation clusters: Can likes attract in a low-polarity environment? J. Phys. Chem. A 2013, 117, 9252–9258. [Google Scholar] [CrossRef]
- Weinhold, F.; Klein, R.A. Anti-Electrostatic Hydrogen Bonds. Angew. Chem. Int. Ed. 2014, 53, 11214–11217. [Google Scholar] [CrossRef]
- Weinhold, F. Theoretical Prediction of Robust Second-Row Oxyanion Clusters in the Metastable Domain of Antielectrostatic Hydrogen Bonding. Inorg. Chem. 2018, 57, 2035–2044. [Google Scholar] [CrossRef]
- Frenking, G.; Caramori, G.F. No Need for a Re-examination of the Electrostatic Notation of the Hydrogen Bonding: A Comment. Angew. Chem. Int. Ed. 2015, 54, 2596–2599. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Mata, I.; Molins, E.; Espinosa, E. Charged versus Neutral Hydrogen-Bonded Complexes: Is There a Difference in the Nature of the Hydrogen Bonds? Chem. Eur. J. 2016, 22, 9226–9234. [Google Scholar] [CrossRef] [PubMed]
- Fatila, E.M.; Twum, E.B.; Sengupta, A.; Pink, M.; Karty, J.A.; Raghavachari, K.; Flood, A.H. Anions Stabilize Each Other inside Macrocyclic Hosts. Angew. Chem. Int. Ed. 2016, 55, 14057–14062. [Google Scholar] [CrossRef]
- Barbas, R.; Prohens, R.; Bauzá, A.; Franconetti, A.; Frontera, A. H-Bonded anion–anion complexes in fentanyl citrate polymorphs and solvates. Chem. Commun. 2019, 55, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Niemann, T.; Stange, P.; Strate, A.; Ludwig, R. When hydrogen bonding overcomes Coulomb repulsion: From kinetic to thermodynamic stability of cationic dimers. Phys. Chem. Chem. Phys. 2019, 21, 8215–8220. [Google Scholar] [CrossRef]
- White, N.G. Antielectrostatically hydrogen bonded anion dimers: Counter-intuitive, common and consistent. CrystEngComm 2019, 21, 4855–4858. [Google Scholar] [CrossRef]
- Zhao, W.; Qiao, B.; Tropp, J.; Pink, M.; Azoulay, J.D.; Flood, A.H. Linear Supramolecular Polymers Driven by Anion–Anion Dimerization of Difunctional Phosphonate Monomers Inside Cyanostar Macrocycles. J. Am. Chem. Soc. 2019, 141, 4980–4989. [Google Scholar] [CrossRef]
- Azofra, L.M.; Elguero, J.; Alkorta, I. Stabilisation of dianion dimers trapped inside cyanostar macrocycles. Phys. Chem. Chem. Phys. 2020, 22, 11348–11353. [Google Scholar] [CrossRef]
- Quinonero, D.; Alkorta, I.; Elguero, J. Cation-cation and anion-anion complexes stabilized by halogen bonds. Phys. Chem. Chem. Phys. 2016, 18, 27939–27950. [Google Scholar] [CrossRef]
- Wang, G.; Chen, Z.; Xu, Z.; Wang, J.; Yang, Y.; Cai, T.; Shi, J.; Zhu, W. Stability and Characteristics of the Halogen Bonding Interaction in an Anion–Anion Complex: A Computational Chemistry Study. J. Phys. Chem. B 2016, 120, 610–620. [Google Scholar] [CrossRef]
- Chalanchi, S.M.; Alkorta, I.; Elguero, J.; Quiñonero, D. Hydrogen Bond versus Halogen Bond in Cation–Cation Complexes: Effect of the Solvent. ChemPhysChem 2017, 18, 3462–3468. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Fu, Y.; Zhang, L.; Danovich, D.; Shaik, S.; Mo, Y. Hydrogen- and Halogen-Bonds between Ions of like Charges: Are They Anti-Electrostatic in Nature? J. Comput. Chem. 2018, 39, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Wang, G.; Xu, Z.; Chen, Z.; Wang, J.; Shi, J.; Zhu, W. Halogen bonding in differently charged complexes: Basic profile, essential interaction terms and intrinsic σ-hole. Phys. Chem. Chem. Phys. 2019, 21, 15106–15119. [Google Scholar] [CrossRef] [PubMed]
- Holthoff, J.M.; Engelage, E.; Weiss, R.; Huber, S.M. “Anti-Electrostatic” Halogen Bonding. Angew. Chem. Int. Ed. 2020, 59, 11150–11157. [Google Scholar] [CrossRef] [PubMed]
- Maxson, T.; Jalilov, A.S.; Zeller, M.; Rosokha, S.V. Halogen Bonding Between Anions: Association of Anion Radicals of Tetraiodo-p-benzoquinone with Iodide Anions. Angew. Chem. Int. Ed. 2020, 59, 17197–17201. [Google Scholar] [CrossRef]
- Ghosh, K.; Frontera, A.; Chattopadhyay, S. A theoretical insight on the anion⋯anion interactions observed in the solid state structure of a hetero-trinuclear complex. CrystEngComm 2021, 23, 1429–1438. [Google Scholar] [CrossRef]
- Wysokiński, R.; Michalczyk, M.; Zierkiewicz, W.; Scheiner, S. Anion–anion and anion–neutral triel bonds. Phys. Chem. Chem. Phys. 2021, 23, 4818–4828. [Google Scholar] [CrossRef]
- Scheiner, S.; Wysokiński, R.; Michalczyk, M.; Zierkiewicz, W. Pnicogen Bonds Pairing Anionic Lewis Acid with Neutral and Anionic Bases. J. Phys. Chem. A 2020, 124, 4998–5006. [Google Scholar] [CrossRef]
- Weinhold, F. Polyion Covalency: Exotic Species from the Unexplored World of Electrostatically Shielded Molecular Ion Chemistry. Angew. Chem. Int. Ed. 2017, 56, 14577–14581. [Google Scholar] [CrossRef]
- Quiñonero, D.; Alkorta, I.; Elguero, J. Metastable Dianions and Dications. ChemPhysChem 2020, 21, 1597–1607. [Google Scholar] [CrossRef] [PubMed]
- Zierkiewicz, W.; Wysokiński, R.; Michalczyk, M.; Scheiner, S. On the Stability of Interactions between Pairs of Anions—Complexes of MCl3− (M=Be, Mg, Ca, Sr, Ba) with Pyridine and CN−. ChemPhysChem 2020, 21, 870–877. [Google Scholar] [CrossRef] [PubMed]
- Wysokiński, R.; Zierkiewicz, W.; Michalczyk, M.; Scheiner, S. Anion…Anion Attraction in Complexes of MCl3− (M = Zn, Cd, Hg) with CN−. ChemPhysChem 2020, 21, 1119–1125. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. B 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Christe, K.O.; Curtis, E.C.; Dixon, D.A.; Mercier, H.P.; Sanders, J.C.P.; Schrobilgen, G.J. The Pentafluoroxenate(Iv) Anion, Xef5—the 1st Example of a Pentagonal Planar Ax5 Species. J. Am. Chem. Soc. 1991, 113, 3351–3361. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian-Basis Sets for Use in Correlated Molecular Calculations. The Atoms Boron through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Moller, C.; Plesset, M.S. Note on an approximation treatment for many-electron systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef]
- Peterson, K.A. Systematically convergent basis sets with relativistic pseudopotentials. I. Correlation consistent basis sets for the post-d group 13-15 elements. J. Chem. Phys. 2003, 119, 11099–11112. [Google Scholar] [CrossRef]
- Costa, P.J. The halogen bond: Nature and applications. Phys. Sci. Rev. 2017, 2. [Google Scholar] [CrossRef]
- Devore, D.P.; Ellington, T.L.; Shuford, K.L. Interrogating the Interplay between Hydrogen and Halogen Bonding in Graphitic Carbon Nitride Building Blocks. J. Phys. Chem. A 2020, 124, 10817–10825. [Google Scholar] [CrossRef]
- Hong, Y.M.; Lu, Y.X.; Zhu, Z.D.; Xu, Z.J.; Liu, H.L. Metalloids as halogen bond acceptors: A combined crystallographic data and theoretical investigation. Chem. Phys. Lett. 2020, 745, 137270. [Google Scholar] [CrossRef]
- Saberinasab, M.; Salehzadeh, S.; Solimannejad, M. The effect of a strong cation center dot center dot center dot pi interaction on a weak selenium center dot center dot center dot pi interaction: A theoretical study. Comput. Theor. Chem. 2016, 1092, 41–46. [Google Scholar] [CrossRef]
- Scheiner, S. Comparison of Bifurcated Halogen with Hydrogen Bonds. Molecules 2021, 26, 350. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. Carbon as an electron donor atom. Polyhedron 2021, 193, 114905. [Google Scholar] [CrossRef]
- Scheiner, S. Origins and properties of the tetrel bond. Phys. Chem. Chem. Phys. 2021, 23, 5702–5717. [Google Scholar] [CrossRef] [PubMed]
- Spada, L.; Gou, Q.; Geboes, Y.; Herrebout, W.A.; Melandri, S.; Caminati, W. Rotational Study of Dimethyl Ether-Chlorotrifluoroethylene: Lone Pair center dot center dot center dot pi Interaction Links the Two Subunits. J. Phys. Chem. A 2016, 120, 4939–4943. [Google Scholar] [CrossRef] [PubMed]
- Tondro, T.; Roohi, H. Substituent effects on the halogen and pnictogen bonds characteristics in ternary complexes 4-YPhNH2 center dot center dot center dot PH2F center dot center dot center dot ClX (Y = H, F, CN, CHO, NH2, CH3, NO2 and OCH3, and X = F, OH, CN, NC, FCC and NO2): A theoretical study. J. Chem. Sci. 2020, 132, 1–21. [Google Scholar]
- Yang, J.M.; Yu, Q.W.; Yang, F.L.; Lu, K.; Yan, C.X.; Dou, W.; Yang, L.Z.; Zhou, P.P. Competition and cooperativity of hydrogen-bonding and tetrel-bonding interactions involving triethylene diamine (DABCO), H2O and CO2 in air. New J. Chem. 2020, 44, 2328–2338. [Google Scholar] [CrossRef]
- Zhao, Q. Mutual influence of tetrel and halogen bonds between XCN (X=Cl, Br) and 4-TF3-pyridine (T=C, Si, Ge). J. Mol. Model. 2020, 26, 1–8. [Google Scholar] [CrossRef]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. Calculation of Small Molecular Interactions by Differences of Separate Total Energies—Some Procedures with Reduced Errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Keith, A.T. AIMAll (Version 14.11.23); TK Gristmill Software: Overland Park, KS, USA, 2014. [Google Scholar]
- Kitaura, K.; Morokuma, K. A new energy decomposition scheme for molecular interactions within the Hartree-Fock approximation. Int. J. Quantum Chem. 1976, 10, 325–340. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Su, P.; Li, H. Energy decomposition analysis of covalent bonds and intermolecular interactions. J. Chem. Phys. 2009, 131, 014102. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Quantitative analysis of molecular surface based on improved Marching Tetrahedra algorithm. J. Mol. Graph. Model. 2012, 38, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. Model. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Grant, D.J.; Wang, T.H.; Dixon, D.A.; Christe, K.O. Heats of Formation of XeF3+, XeF3-, XeF5+, XeF7+, XeF7-, and XeF8 from High Level Electronic Structure Calculations. Inorg. Chem. 2010, 49, 261–270. [Google Scholar] [CrossRef]
- Scheiner, S. Understanding noncovalent bonds and their controlling forces. J. Chem. Phys. 2020, 153, 140901. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| r(Ae-X) a, Å | VS,max, kcal/mol | |
|---|---|---|
| Water (ε = 78.4) | ||
| KrF5− | 2.034 | −67.1 |
| XeF5− | 2.066 | −61.3 |
| XeCl5− | 2.728 | −51.7 |
| DMF (ε = 37.2) | ||
| KrF5− | 2.034 | −67.2 |
| XeF5− | 2.066 | −61.4 |
| XeCl5− | 2.729 | −51.8 |
| THF (ε = 7.4) | ||
| KrF5− | 2.036 | −67.5 |
| XeF5− | 2.066 | −62.0 |
| XeCl5− | 2.730 | −52.4 |
| An=F− | An=Cl− | An=CN− | ||||
|---|---|---|---|---|---|---|
| R(Ae···An) | Δr(Ae-X) | R(Ae···An) | Δr(Ae-X) | R(Ae···An) | Δr(Ae-X) | |
| Water (ε = 78.4) | ||||||
| KrF5− | 2.848 | −0.001 | 3.406 | 0.000 | 3.083 | −0.001 |
| XeF5− | 2.981 | 0.006 | 3.564 | 0.003 | 3.296 | 0.003 |
| XeCl5− | 2.894 | −0.008 | 3.458 | −0.006 | 3.165 | −0.008 |
| DMF (ε = 37.2) | ||||||
| KrF5− | 2.839 | −0.001 | 3.409 | 0.000 | 3.086 | −0.001 |
| XeF5− | 2.979 | 0.006 | 3.563 | 0.003 | 3.296 | 0.003 |
| XeCl5− | 2.888 | −0.007 | 3.461 | −0.007 | 3.169 | −0.008 |
| THF (ε = 7.4) | ||||||
| KrF5− | 2.860 | −0.001 | a | a | 3.104 | −0.001 |
| XeF5− | 2.939 | 0.008 | 3.658 | 0.004 | 3.304 | 0.004 |
| XeCl5− | 2.837 | −0.008 | 3.438 | 0.003 | 3.170 | −0.007 |
| Eint | Eb | |||||
|---|---|---|---|---|---|---|
| F− | Cl− | NC− | F− | Cl− | NC− | |
| Water (ε = 78.4) | ||||||
| KrF5− | −0.56 | −0.62 | −0.96 | −0.50 | −0.63 | −0.98 |
| XeF5− | −0.74 | −0.43 | −0.69 | −0.64 | −0.41 | −0.68 |
| XeCl5− | −1.51 | −1.81 | −2.05 | −1.49 | −1.81 | −2.04 |
| DMF (ε = 37.2) | ||||||
| KrF5− | 0.50 | 0.33 | −0.09 | 0.58 | 0.33 | −0.09 |
| XeF5− | 0.22 | 0.52 | 0.20 | 0.35 | 0.56 | 0.24 |
| XeCl5− | −0.57 | −0.85 | −1.14 | −0.52 | −0.84 | −1.13 |
| THF (ε = 7.4) | ||||||
| KrF5− | 8.36 | a | 7.04 | 8.63 | a | 7.12 |
| XeF5− | 7.30 | 7.32 | 6.69 | 7.71 | 7.45 | 6.85 |
| XeCl5− | 6.02 | 5.80 | 5.19 | 6.15 | 5.87 | 5.26 |
| AeX5− | R | Eint | Eex | % | Eelec | Erep | Epol | % | Edisp | % |
|---|---|---|---|---|---|---|---|---|---|---|
| Water (ε = 78.4) | ||||||||||
| KrF5− | F− | −6.17 | −5.41 | 2.4 | 202.99 | 16.96 | −215.18 | 95.2 | −5.53 | 2.4 |
| Cl− | −3.05 | −3.89 | 1.9 | 189.54 | 12.44 | −195.79 | 95.5 | −5.35 | 2.6 | |
| NC− | −3.30 | −3.87 | 2.0 | 182.56 | 12.40 | −189.76 | 95.7 | −4.63 | 2.3 | |
| XeF5− | F− | −5.84 | −6.71 | 3.0 | 198.54 | 19.20 | −211.35 | 94.5 | −5.52 | 2.5 |
| Cl− | −2.61 | −4.41 | 2.2 | 186.30 | 12.87 | −192.43 | 95.4 | −4.94 | 2.4 | |
| NC− | −2.23 | −7.32 | 3.7 | 177.30 | 20.20 | −186.96 | 93.6 | −5.45 | 2.7 | |
| XeCl5− | F− | −8.26 | −8.51 | 4.0 | 180.92 | 24.98 | −197.67 | 92.3 | −7.98 | 3.7 |
| Cl− | −4.64 | −6.03 | 3.1 | 171.40 | 18.29 | −180.01 | 92.6 | −8.29 | 4.3 | |
| NC− | −4.43 | −5.67 | 3.0 | 165.20 | 17.24 | −174.31 | 93.3 | −6.89 | 3.7 | |
| DMF (ε = 37.2) | ||||||||||
| KrF5− | F− | −5.20 | −5.59 | 2.5 | 200.76 | 17.48 | −212.25 | 95.0 | −5.60 | 2.5 |
| Cl− | −2.05 | −3.88 | 1.9 | 187.67 | 12.38 | −192.92 | 95.5 | −5.30 | 2.6 | |
| NC− | −2.33 | −3.85 | 2.0 | 180.76 | 12.34 | −186.99 | 95.7 | −4.59 | 2.3 | |
| XeF5− | F− | −4.92 | −6.78 | 3.1 | 196.39 | 19.38 | −208.38 | 94.4 | −5.53 | 2.5 |
| Cl− | −1.68 | −4.44 | 2.2 | 184.37 | 12.94 | −189.62 | 95.3 | −4.93 | 2.5 | |
| NC− | −1.33 | −7.34 | 3.7 | 175.49 | 20.24 | −184.29 | 93.5 | −5.44 | 2.8 | |
| XeCl5− | F− | −7.45 | −8.71 | 4.1 | 178.74 | 25.49 | −194.98 | 92.1 | −7.99 | 3.8 |
| Cl− | −3.75 | −6.00 | 3.1 | 169.61 | 18.18 | −177.34 | 92.6 | −8.20 | 4.3 | |
| NC− | −3.58 | −5.63 | 3.1 | 163.54 | 17.11 | −171.78 | 93.2 | −6.82 | 3.7 | |
| THF (ε = 7.4) | ||||||||||
| KrF5− | F− | 2.51 | −5.38 | 2.7 | 185.63 | 16.71 | −189.21 | 94.7 | −5.24 | 2.6 |
| Cl− | not stable | |||||||||
| NC− | 4.73 | −3.74 | 2.1 | 167.44 | 11.92 | −166.57 | 95.4 | −4.32 | 2.5 | |
| XeF5− | F− | 1.75 | −7.86 | 3.9 | 180.17 | 22.30 | −186.94 | 93.1 | −5.93 | 3.0 |
| Cl− | 5.35 | −3.52 | 2.0 | 170.97 | 10.40 | −168.30 | 95.6 | −4.20 | 2.4 | |
| NC− | 5.26 | −7.37 | 4.2 | 162.15 | 20.21 | −164.45 | 92.9 | −5.28 | 3.0 | |
| XeCl5− | F− | −1.57 | −10.50 | 5.4 | 162.70 | 30.18 | −175.65 | 90.3 | −8.30 | 4.3 |
| Cl− | 2.65 | −6.19 | 3.6 | 156.42 | 18.47 | −158.19 | 91.8 | −7.86 | 4.6 | |
| NC− | 2.55 | −5.87 | 3.5 | 151.02 | 17.65 | −153.58 | 92.5 | −6.67 | 4.0 | |
| Ediss | Rdiss | Ediss | Rdiss | Ediss | Rdiss | |
|---|---|---|---|---|---|---|
| R=F− | R=Cl− | R=NC− | ||||
| DMF (ε = 37.2) | ||||||
| KrF5− | 1.89 | 5.24 | 1.75 | 6.01 | negative Eint | |
| XeF5− | 2.00 | 4.38 | 1.47 | 6.16 | 2.17 | 6.50 |
| THF (ε = 7.4) | ||||||
| KrF5− | 0.97 | 3.66 | a | 1.25 | 4.30 | |
| XeF5− | 1.43 | 3.94 | 0.30 | 4.66 | 1.01 | 4.50 |
| XeCl5− | 2.50 | 4.04 | 2.09 | 4.97 | 2.73 | 4.57 |
| Ta | ρ | ∇ 2ρ | H | ρ | ∇2ρ | H | ρ | ∇ 2ρ | H |
|---|---|---|---|---|---|---|---|---|---|
| Water (ε = 78.4) | |||||||||
| An=F− | An=Cl− | An=NC− | |||||||
| KrF5− | 0.017 | 0.074 | 0.002 | 0.010 | 0.034 | 0.002 | 0.012 | 0.047 | 0.002 |
| XeF5− | 0.018 | 0.070 | 0.001 | 0.010 | 0.033 | 0.001 | 0.011 | 0.039 | 0.002 |
| XeCl5− | 0.020 | 0.080 | 0.001 | 0.012 | 0.040 | 0.001 | 0.014 | 0.050 | 0.002 |
| DMF (ε = 37.2) | |||||||||
| An=F− | An=Cl− | An=NC− | |||||||
| KrF5− | 0.017 | 0.075 | 0.002 | 0.010 | 0.034 | 0.002 | 0.012 | 0.047 | 0.002 |
| XeF5− | 0.018 | 0.070 | 0.001 | 0.010 | 0.033 | 0.001 | 0.011 | 0.039 | 0.002 |
| XeCl5− | 0.020 | 0.081 | 0.001 | 0.012 | 0.039 | 0.002 | 0.014 | 0.049 | 0.002 |
| THF (ε = 7.4) | |||||||||
| An=F− | An=Cl− | An=NC− | |||||||
| KrF5− | 0.016 | 0.071 | 0.002 | a | 0.012 | 0.045 | 0.002 | ||
| XeF5− | 0.019 | 0.075 | 0.001 | 0.009 | 0.025 | 0.001 | 0.011 | 0.038 | 0.001 |
| XeCl5− | 0.023 | 0.090 | 0.001 | 0.013 | 0.037 | 0.001 | 0.014 | 0.049 | 0.002 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grabarz, A.; Michalczyk, M.; Zierkiewicz, W.; Scheiner, S. Anion–Anion Interactions in Aerogen-Bonded Complexes. Influence of Solvent Environment. Molecules 2021, 26, 2116. https://doi.org/10.3390/molecules26082116
Grabarz A, Michalczyk M, Zierkiewicz W, Scheiner S. Anion–Anion Interactions in Aerogen-Bonded Complexes. Influence of Solvent Environment. Molecules. 2021; 26(8):2116. https://doi.org/10.3390/molecules26082116
Chicago/Turabian StyleGrabarz, Anna, Mariusz Michalczyk, Wiktor Zierkiewicz, and Steve Scheiner. 2021. "Anion–Anion Interactions in Aerogen-Bonded Complexes. Influence of Solvent Environment" Molecules 26, no. 8: 2116. https://doi.org/10.3390/molecules26082116
APA StyleGrabarz, A., Michalczyk, M., Zierkiewicz, W., & Scheiner, S. (2021). Anion–Anion Interactions in Aerogen-Bonded Complexes. Influence of Solvent Environment. Molecules, 26(8), 2116. https://doi.org/10.3390/molecules26082116