
Determination of Cd, Pb, and Cu in the Atmospheric Aerosol of Central East Antarctica at Dome C (Concordia Station)


 ,
,  ,
,  ,
,  , ,
, , 
Abstract

1. Introduction
2. Results and Discussion
2.1. Back Trajectories and Air Masses Arriving at Dome C
2.2. Metal Contents and Temporal Profiles
2.2.1. Cadmium
2.2.2. Lead
2.2.3. Copper
2.2.4. Relationship between Contaminated-on-Background Excesses of the Three Metals
- excess mass ratio from content in mass fraction Cd:Pb:Cu = 1:28:1600 (with Cu/Pb = ~57);
- excess mass ratio from content in atmospheric concentration Cd:Pb:Cu = 1:21:800 (with Cu/Pb = ~38).
2.3. Comparison with Antarctic Literature Data
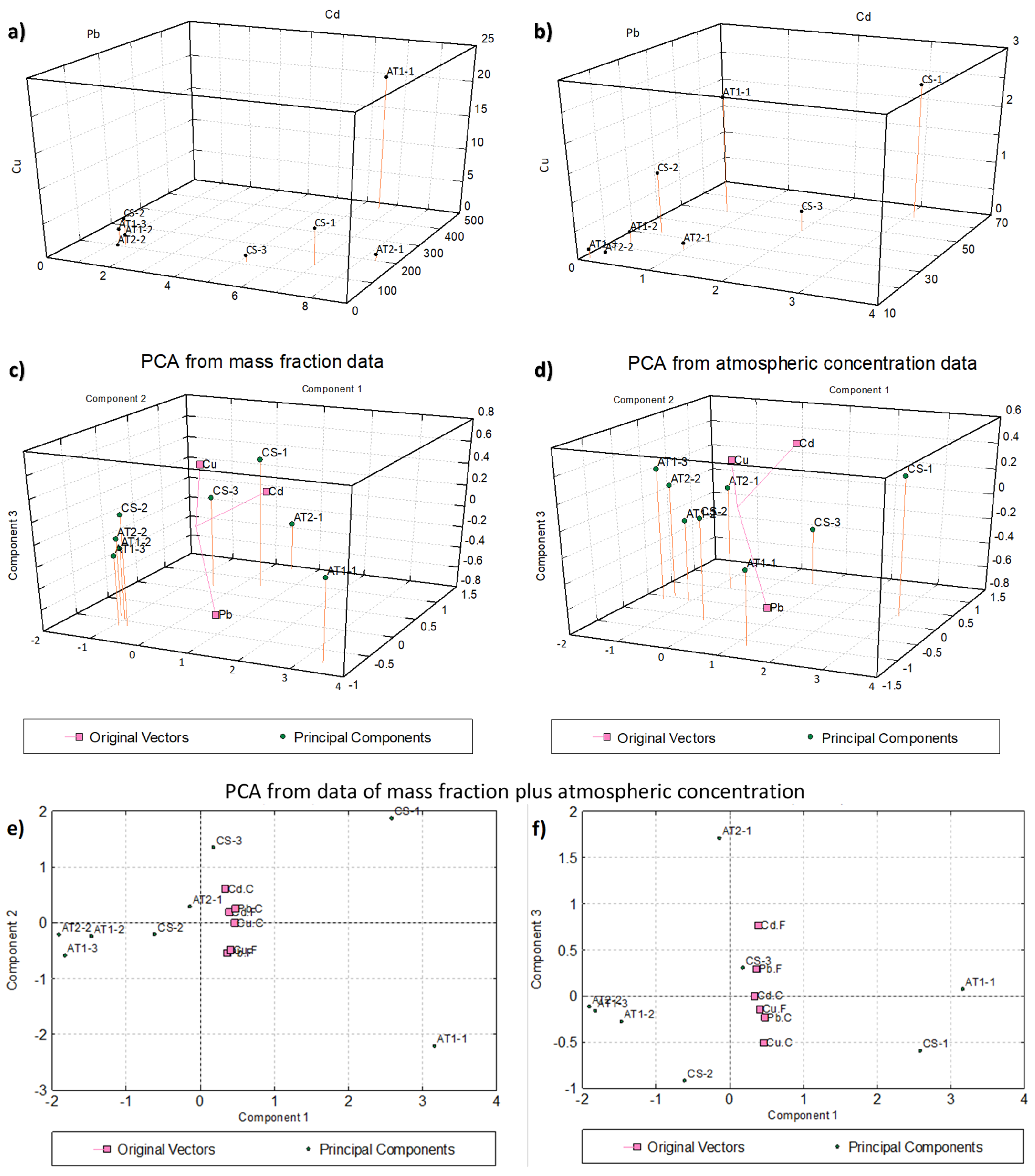
2.4. Statistical Analysis (Correlation and Principal Component Analysis)
2.5. Metal Contents in Station Fuels and Interpretation of Data
3. Materials and Methods
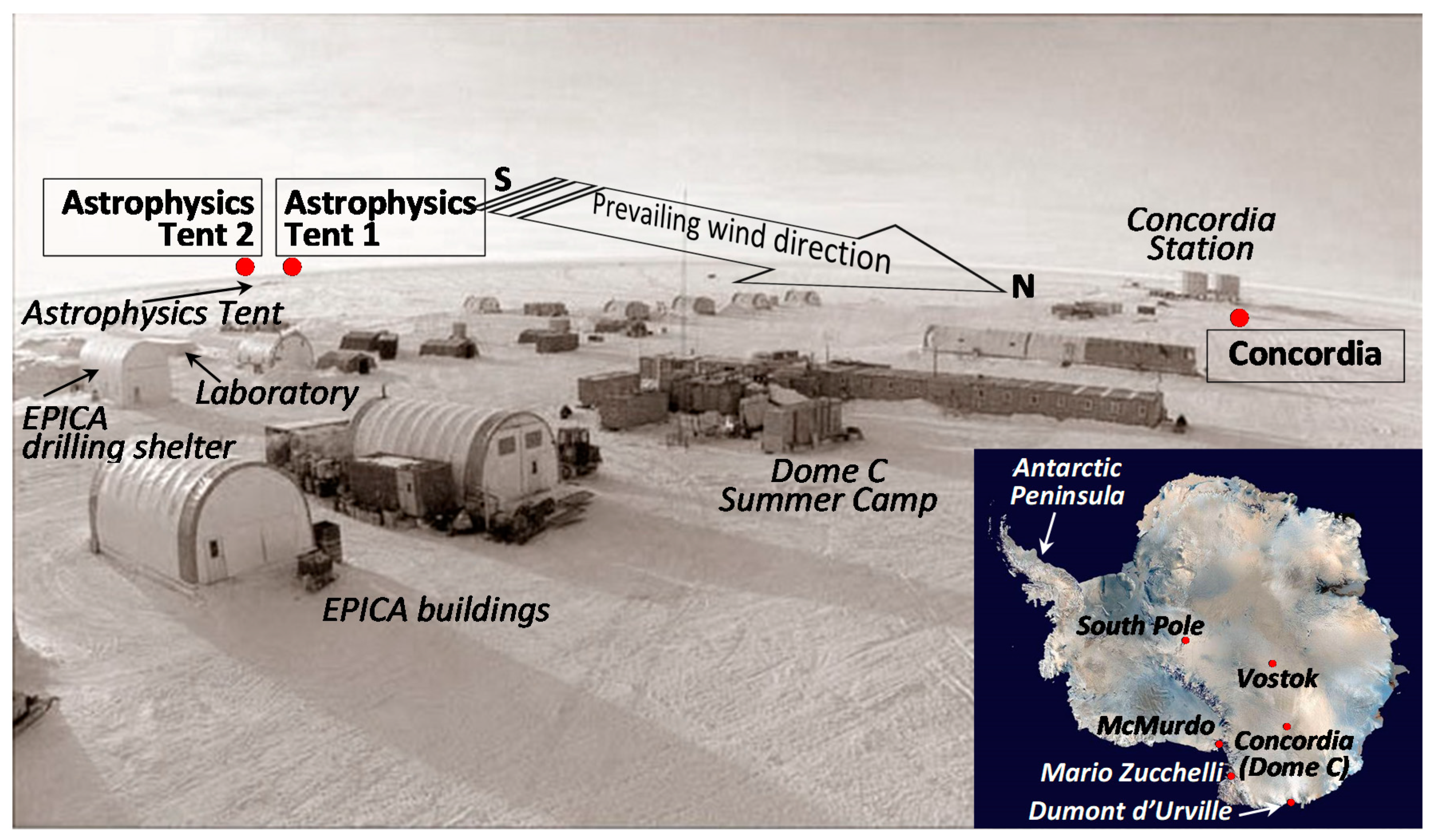
3.1. The Site and the Sampling Strategy
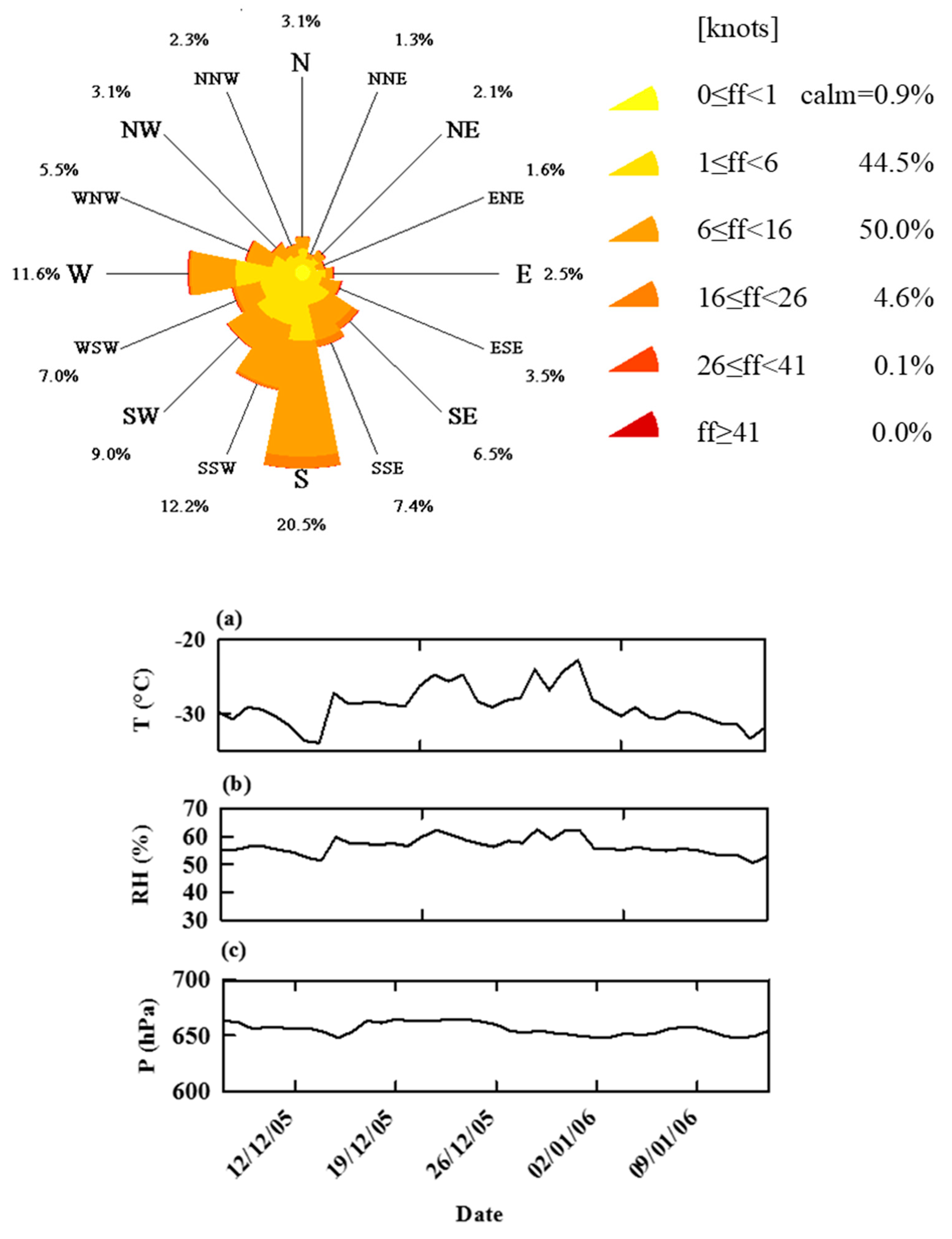
3.2. Meteorology and Air Backward Trajectories
3.3. Laboratories, Apparatus, Reagents, and General Procedures
3.3.1. Laboratories and Apparatus
3.3.2. Reagents and Standards
3.3.3. Decontamination Procedures
3.4. Sample Treatments
3.5. Voltammetric Analysis
3.6. Quality Control and Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Shaw, J.D.; Terauds, A.; Riddle, M.J.; Possingham, H.P.; Chown, S.L. Antarctica’s Protected Areas Are Inadequate, Unrepresentative, and at Risk. PLoS Biol. 2014, 12, e1001888. [Google Scholar] [CrossRef]
- Barbaro, E.; Zangrando, R.; Kirchgeorg, T.; Bazzano, A.; Illuminati, S.; Annibaldi, A.; Rella, S.; Truzzi, C.; Grotti, M.; Ceccarini, A.; et al. An integrated study of the chemical composition of Antarctic aerosol to investigate natural and anthropogenic sources. Environ. Chem. 2016, 13, 867–876. [Google Scholar] [CrossRef]
- Shaw, G.E. Aerosol transport from sources to ice sheets. In The Environmental Record in Glaciers and Ice Sheets; Oeschger, H., Langway, C.C., Eds.; John Wiley & Sons: New York, NY, USA, 1989; pp. 2251–2257. [Google Scholar]
- Boutron, C.F. Historical reconstruction of the earth’s past atmospheric environment from Greenland and Antarctic snow and ice cores. Environ. Rev. 1995, 3, 1–28. [Google Scholar] [CrossRef]
- Chemical Exchange between the Atmosphere and Polar Snow; Wolff, E.W., Bales, R.C., Eds.; Springer: Berlin/Heidelberg, Germany, 1996. [Google Scholar]
- Legrand, M.; Mayewski, P. Glaciochemistry of polar ice cores: A review. Rev. Geophys. 1997, 35, 219–243. [Google Scholar] [CrossRef]
- Planchon, F.A.M.; van de Velde, K.; Rosman, K.J.R.; Wolff, E.W.; Ferrari, C.P.; Boutron, C.F. One hundred fifty-year record of lead isotopes in Antarctic snow from Coats Land. Geochim. Cosmochim. Acta 2003, 67, 693–708. [Google Scholar] [CrossRef]
- Augustin, L.; Barbante, C.; Barnes, P.R.F.; Barnola, J.M.; Bigler, M.; Castellano, E.; Cattani, O.; Chappellaz, J.; Dahl-Jensen, D.; Delmonte, B.; et al. Eight glacial cycles from an Antarctic ice core. Nature 2004, 429, 623–628. [Google Scholar]
- Wolff, E.W.; Barbante, C.; Becagli, S.; Bigler, M.; Boutron, C.F.; Castellano, E.; de Angelis, M.; Federer, U.; Fischer, H.; Fundel, F.; et al. Changes in environment over the last 800,000 years from chemical analysis of the EPICA Dome C ice core. Quat. Sci. Rev. 2010, 29, 285–295. [Google Scholar] [CrossRef]
- Han, C.; Hur, S.; Burn, L.J.; Gabrielli, P.; Vallelonga, P.T.; Barbante, C.; Boutron, C.F.; Hong, S. Climate-related changes in lead isotopes over the past 800,000 years in the EPICA Dome C Antarctic ice core. In AGU Fall Meeting Abstracts; American Geophysical Union: Washington, DC, USA, 2013. [Google Scholar]
- Hong, S.; Han, C.H.; Hwang, H.J.; Soyol-Erdene, T.O.; Kang, J.H.; Hur, S.D.; Burn-Nunes, L.J.; Gabrielli, P.; Barbante, C.; Boutron, C.F. Trace elements and Pb isotope records in Dome C (East Antarctica) ice over the past 800,000 years. E3S Web Conf. 2013, 1, 23001. [Google Scholar] [CrossRef]
- Han, C.; Burn, L.; Vallelonga, P.; Hur, S.D.; Boutron, C.F.; Lee, S.; Hong, S. Lead Isotopic Constraints on the Provenance of Antarctic Dust and Relevant Atmospheric Circulation Patterns Prior to the Mid-Brunhes Event (~430 Kyr Ago). Sci. Rep. 2021. under review. [Google Scholar] [CrossRef]
- Shaw, G.E. Antarctic aerosols: A review. Rev. Geophys. 1988, 26, 89–112. [Google Scholar] [CrossRef]
- Artaxo, P.; Rabello, M.L.C.; Maenhaut, W.; Van Grieken, R. Trace elements and individual particle analysis of atmospheric aerosols from the Antarctic peninsula. Tellus B 1992, 44, 318–334. [Google Scholar] [CrossRef]
- Correia, A.L.; Artaxo, P.; Maenhaut, W. Monitoring of atmospheric aerosol particles on the Antarctic Peninsula. Ann. Glaciol. 1998, 27, 560–564. [Google Scholar] [CrossRef]
- Hillamo, R.; Allegrini, I.; Sparapani, R.; Kerminen, V.M. Mass size distributions and precursor gas concentrations of major inorganic ions in Antarctic aerosol. Int. J. Environ. Anal. Chem. 1998, 71, 353–372. [Google Scholar] [CrossRef]
- Wolff, E.W.; Legrand, M.R.; Wagenbach, D. Coastal Antarctic aerosol and snowfall chemistry. J. Geophys. Res. Atmos. 1998, 103, 10927–10934. [Google Scholar] [CrossRef]
- Kerminen, V.M.; Teinila, K.; Hillamo, R. Chemistry of sea-salt particles in the summer Antarctic atmosphere. Atmos. Environ. 2000, 34, 2817–2825. [Google Scholar] [CrossRef]
- Teinila, K.; Kerminen, V.M.; Hillamo, R. A study of size-segregated aerosol chemistry in the Antarctic atmosphere. J. Geophys. Res. Atmos. 2000, 105, 3893–3904. [Google Scholar] [CrossRef]
- Jourdain, B.; Legrand, M. Year-round records of bulk and size-segregated aerosol composition and HCl and HNO3 levels in the Dumont d’Urville (coastal Antarctica) atmosphere: Implications for sea-salt aerosol fractionation in the winter and summer. J. Geophys. Res. Atmos. 2002, 107, ACH20-1–ACH20-13. [Google Scholar] [CrossRef]
- Rankin, A.M.; Wolff, E.W. A year-long record of size-segregated aerosol composition at Halley, Antarctica. J. Geophys. Res. 2003, 108, 108. [Google Scholar] [CrossRef]
- Gadhavi, H.; Jayaraman, A. Aerosol characteristics and aerosol radiative forcing over Maitri, Antarctica. Curr. Sci. 2004, 86, 296–304. [Google Scholar]
- Tomasi, C.; Vitale, V.; Lupi, A.; Di Carmine, C.; Campanelli, M.; Herber, A.; Treffeisen, R.; Stone, R.S.; Andrews, E.; Sharma, S.; et al. Aerosols in polar regions: A historical overview based on optical depth and in situ observations. J. Geophys. Res. Atmos. 2007, 112, D16205. [Google Scholar] [CrossRef]
- Asmi, E.; Frey, A.; Virkkula, A.; Ehn, M.; Manninen, H.E.; Timonen, H.; Tolonen-Kivimaki, O.; Aurela, M.; Hillamo, R.; Kulmala, M. Hygroscopicity and chemical composition of Antarctic sub-micrometre aerosol particles and observations of new particle formation. Atmos. Chem. Phys. 2010, 10, 4253–4271. [Google Scholar] [CrossRef]
- Chaubey, J.P.; Moorthy, K.K.; Babu, S.S.; Nair, S.V. The optical and physical properties of atmospheric aerosols over the Indian Antarctic stations during southern hemispheric summer of the International Polar Year 2007–2008. Ann. Geophys. 2011, 29, 109–121. [Google Scholar] [CrossRef]
- Teinilä, K.; Frey, A.; Hillamo, R.; Tülp, H.C.; Weller, R. A study of the sea-salt chemistry using size-segregated aerosol measurements at coastal Antarctic station Neumayer. Atmos. Environ. 2014, 96, 11–19. [Google Scholar] [CrossRef]
- Artaxo, P.; Andrade, F.; Maenhaut, W. Trace elements and receptor modelling of aerosols in the antarctic peninsula. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms. 1990, 49, 383–387. [Google Scholar] [CrossRef]
- Mazzera, D.M.; Lowenthal, D.H.; Chow, J.C.; Watson, J.G.; Grubisic, V. PM10 measurements at McMurdo Station, Antarctica. Atmos. Environ. 2001, 35, 1891–1902. [Google Scholar] [CrossRef]
- Truzzi, C.; Lambertucci, L.; Illuminati, S.; Annibaldi, A.; Scarponi, G. Direct gravimetric measurements of the mass of the antarctic aerosol collected by high volume sampler: PM10 summer seasonal variation at Terra Nova Bay. Ann. Chim. 2005, 95, 867–876. [Google Scholar] [CrossRef]
- Annibaldi, A.; Truzzi, C.; Illuminati, S.; Bassotti, E.; Scarponi, G. Determination of water-soluble and insoluble (dilute-HCl-extractable) fractions of Cd, Pb and Cu in Antarctic aerosol by square wave anodic stripping voltammetry: Distribution and summer seasonal evolution at Terra Nova Bay (Victoria Land). Anal. Bioanal. Chem. 2007, 387, 977–998. [Google Scholar] [CrossRef]
- Zoller, W.H.; Gladney, E.S.; Duce, R.A. Atmospheric concentrations and sources of trace metals at the South Pole. Science 1974, 183, 198–200. [Google Scholar] [CrossRef]
- Maenhaut, W.; Zoller, W.H. Determination of the chemical composition of the South Pole aerosol by instrumental neutron activation analysis. J. Radioanal. Chem. 1977, 37, 637–650. [Google Scholar] [CrossRef]
- Maenhaut, W.; Zoller, H.; Duce, R.A.; Hoffman, G.L. Concentration and Size Distribution Trace Elements in the South Polar Atmosphere. J. Geophys. Res. 1979, 84, 2421–2431. [Google Scholar] [CrossRef]
- Cunningham, W.C.; Zoller, W.H. The chemical composition of remote area aerosols. J. Aerosol Sci. 1981, 12, 367–384. [Google Scholar] [CrossRef]
- Tuncel, G.; Aras, N.K.; Zoller, W.H. Temporal variations and sources of elements in the South Pole atmosphere: 1. Nonenriched and moderately enriched elements. J. Geophys. Res. 1989, 94, 13025–13038. [Google Scholar] [CrossRef]
- Arimoto, R.; Hogan, A.; Grube, P.; Davis, D.; Webb, J.; Schloesslin, C.; Sage, S.; Raccah, F. Major ions and radionuclides in aerosol particles from the South Pole during ISCAT-2000. Atmos. Environ. 2004, 38, 5473–5484. [Google Scholar] [CrossRef]
- Arimoto, R.; Zeng, T.; Davis, D.; Wang, Y.; Khaing, H.; Nesbit, C.; Huey, G. Concentrations and sources of aerosol ions and trace elements during ANTCI-2003. Atmos. Environ. 2008, 42, 2864–2876. [Google Scholar] [CrossRef]
- De Mora, S.J.; Wylie, D.J.; Dick, A.L. Methanesulphonate and non-sea salt sulphate in aerosol, snow, and ice on the East Antarctic plateau. Antarct. Sci. 1997, 9, 46–55. [Google Scholar] [CrossRef]
- Fattori, I.; Becagli, S.; Bellandi, S.; Castellano, E.; Innocenti, M.; Mannini, A.; Severi, M.; Vitale, V.; Udisti, R. Chemical composition and physical features of summer aerosol at Terra Nova Bay and Dome C, Antarctica. J. Environ. Monit. 2005, 7, 1265–1274. [Google Scholar] [CrossRef] [PubMed]
- Becagli, S.; Castellano, E.; Cerri, O.; Chiari, M.; Lucarelli, F.; Marino, F.; Morganti, A.; Nava, S.; Rugi, F.; Severi, M.; et al. All year-round background aerosol at Dome C (Antarctica). Chemical composition of size-segregated samples collected during the 2004–2005 campaign. Conf. Proc. Ital. Phys. Soc. 2009, 97, 17–41. [Google Scholar]
- Udisti, R.; Dayan, U.; Becagli, S.; Busetto, M.; Frosini, D.; Legrand, M.; Lucarelli, F.; Preunkert, S.; Severi, M.; Traversi, R.; et al. Sea spray aerosol in central Antarctica. Present atmospheric behaviour and implications for paleoclimatic reconstructions. Atmos. Environ. 2012, 52, 109–120. [Google Scholar] [CrossRef]
- Annibaldi, A.; Truzzi, C.; Illuminati, S.; Scarponi, G. Direct gravimetric determination of aerosol mass concentration in central antarctica. Anal. Chem. 2011, 83, 143–151. [Google Scholar] [CrossRef]
- Budhavant, K.; Safai, P.D.; Rao, P.S.P. Sources and elemental composition of summer aerosols in the Larsemann Hills (Antarctica). Environ. Sci. Pollut. Res. 2015, 22, 2041–2050. [Google Scholar] [CrossRef]
- Rosman, K.J.R.; Chisholm, W.; Boutron, C.F.; Candelone, J.-P.; Patterson, C.C. Anthropogenic lead isotopes in Antarctica. Geophys. Res. Lett. 1994, 21, 2669–2672. [Google Scholar] [CrossRef]
- Toscano, G.; Gambaro, A.; Moret, I.; Capodaglio, G.; Turetta, C.; Cescon, P. Trace metals in aerosol at Terra Nova Bay, Antarctica. J. Environ. Monit. 2005, 7, 1275–1280. [Google Scholar] [CrossRef]
- Cullen, J.T.; Maldonado, M.T. Biogeochemistry of Cadmium and Its Release to the Environment. In Cadmium: From Toxicity to Essentiality; Sigel, A., Sigel, H., Sigel, R.K.O., Eds.; Springer: Dordrecht, The Netherlands, 2013; Chapter 2; pp. 31–62. ISBN 978-94-007-5179-8. [Google Scholar]
- Bazzano, A.; Soggia, F.; Grotti, M. Source identification of atmospheric particle-bound metals at Terra Nova Bay, Antarctica. Environ. Chem. 2015, 12, 245–252. [Google Scholar] [CrossRef]
- Planchon, F.A.M.; Boutron, C.F.; Barbante, C.; Cozzi, G.; Gaspari, V.; Wolff, E.W.; Ferrari, C.P.; Cescon, P. Changes in heavy metals in Antarctic snow from Coats Land since the mid-19th to the late-20th century. Earth Planet. Sci. Lett. 2002, 200, 207–222. [Google Scholar] [CrossRef]
- Mishra, V.K.; Kim, K.H.; Hong, S.; Lee, K. Aerosol composition and its sources at the King Sejong Station, Antarctic peninsula. Atmos. Environ. 2004, 38, 4069–4084. [Google Scholar] [CrossRef]
- Hur, S.D.; Cunde, X.; Hong, S.; Barbante, C.; Gabrielli, P.; Lee, K.; Boutron, C.F.; Ming, Y. Seasonal patterns of heavy metal deposition to the snow on Lambert Glacier basin, East Antarctica. Atmos. Environ. 2007, 41, 8567–8578. [Google Scholar] [CrossRef]
- Pacyna, J.M.; Pacyna, E.G. An assessment of global and regional emissions of trace metals to the atmosphere from anthropogenic sources worldwide. Environ. Rev. 2001, 9, 269–298. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Ten Chemicals of Major Health Concern. Preventing Disease Through Healthy Environments. Action Is Needed on Chemicals of Major Public Health Concern. 2010. Available online: https://www.who.int/ipcs/assessment/public_health/chemicals_phc/en/ (accessed on 16 February 2021).
- Illuminati, S.; Annibaldi, A.; Truzzi, C.; Libani, G.; Mantini, C.; Scarponi, G. Determination of water-soluble, acid-extractable and inert fractions of Cd, Pb and Cu in Antarctic aerosol by square wave anodic stripping voltammetry after sequential extraction and microwave digestion. J. Electroanal. Chem. 2015, 755, 182–196. [Google Scholar] [CrossRef]
- Wedepohl, K.H. The composition of the continental crust. Geochim. Cosmochim. Acta 1995, 59, 1217–1232. [Google Scholar] [CrossRef]
- Rudnick, R.L.; Gao, S. Composition of the Continental Crust. In Treatise on Geochemistry, 2nd ed.; Holland, H.D., Turekian, K.K., Eds.; Elsevier Ltd.: Amsterdam, The Netherlands, 2014; Volume 4, Chapter 4.1; pp. 1–51. ISBN 9780080983004. [Google Scholar]
- Wolff, E.W.; Suttie, E.D.; Peel, D.A. Antarctic snow record of cadmium, copper, and zinc content during the twentieth century. Atmos. Environ. 1999, 33, 1535–1541. [Google Scholar] [CrossRef]
- Hong, S.; Boutron, C.F.; Gabrielli, P.; Barbante, C.; Ferrari, C.P.; Petit, J.R.; Lee, K.; Lipenkov, V.Y. Past natural changes in Cu, Zn and Cd in Vostok Antarctic ice dated back to the penultimate interglacial period. Geophys. Res. Lett. 2004, 31, L20111. [Google Scholar] [CrossRef]
- Lee, K.; Hur, S.D.; Hou, S.; Hong, S.; Qin, X.; Ren, J.; Liu, Y.; Rosman, K.J.R.; Barbante, C.; Boutron, C.F. Atmospheric pollution for trace elements in the remote high-altitude atmosphere in central Asia as recorded in snow from Mt. Qomolangma (Everest) of the Himalayas. Sci. Total Environ. 2008, 404, 171–181. [Google Scholar] [CrossRef]
- Gabrielli, P.; Barbante, C.; Boutron, C.; Cozzi, G.; Gaspari, V.; Planchon, F.; Ferrari, C.; Turetta, C.; Hong, S.; Cescon, P. Variations in atmospheric trace elements in Dome C (East Antarctica) ice over the last two climatic cycles. Atmos. Environ. 2005, 39, 6420–6429. [Google Scholar] [CrossRef]
- Zreda-Gostynska, G.; Kyle, P.R.; Finnegan, D.; Prestbo, K.M. Volcanic gas emissions from Mount Erebus and their impact on the Antarctic environment. J. Geophys. Res. Solid Earth 1997, 102, 15039–15055. [Google Scholar] [CrossRef]
- Nriagu, J.O. A global assessment of natural sources of atmospheric trace metals. Nature 1989, 338, 47–49. [Google Scholar] [CrossRef]
- Rosman, K.J.R.; Chisholm, W.; Boutron, C.F.; Candelone, J.-P.; Görlach, U. Isotopic evidence for the source of lead in Greenland snows since the late 1960s. Nature 1993, 362, 333–335. [Google Scholar] [CrossRef]
- Rosman, K.J.R.; Chisholm, W.; Hong, S.; Candelone, J.-P.; Boutron, C.F. Lead from Carthaginian and Roman Spanish mines isotopically identified in Greenland ice dated from 600 B.C. to 300 A.D. Environ. Sci. Technol. 1997, 31, 3413–3416. [Google Scholar] [CrossRef]
- Wolff, E.W.; Suttie, E.D. Antarctic snow record of southern hemisphere lead pollution. Geophys. Res. Lett. 1994, 21, 781–784. [Google Scholar] [CrossRef]
- Sangster, D.F.; Outridge, P.M.; Davis, W.J. Stable lead isotope characteristics of lead ore deposits of environmental significance. Environ. Rev. 2000, 8, 115–147. [Google Scholar] [CrossRef]
- Rosman, K.J.R. Natural isotopic variations in lead in polar snow and ice as indicators of source regions. In Environmental Contamination in Antarctica: A Challenge to Analytical Chemistry; Caroli, S., Cescon, P., Walton, D.W.H., Eds.; Elsevier: Oxford, UK, 2001; pp. 87–106. [Google Scholar]
- McConnell, J.R.; Lamorey, G.W.; Hutterli, M.A. A 250-year high-resolution record of Pb flux and crustal enrichment in central Greenland. Geophys. Res. Lett. 2002, 29, 45-1–45-4. [Google Scholar] [CrossRef]
- McConnell, J.R.; Edwards, R. Coal burning leaves toxic heavy metal legacy in the Arctic. Proc. Natl. Acad. Sci. USA 2008, 105, 12140–12144. [Google Scholar] [CrossRef]
- Görlach, U.; Boutron, C.F. Variations in heavy metals concentrations in Antarctic snows from 1940 to 1980. J. Atmos. Chem. 1992, 14, 205–222. [Google Scholar] [CrossRef]
- Boutron, C.F.; Patterson, C.C.; Lorius, C.; Petrov, V.N.; Barkov, N.T. Atmospheric Lead in Antarctic Ice. Ann. Glaciol. 1988, 10, 5–9. [Google Scholar] [CrossRef]
- Wolff, E.W.; Peel, D.A. Closer to a true value for heavy metal concentrations in recent Antarctic snow by improved contamination control. Ann. Glaciol. 1985, 7, 61–69. [Google Scholar] [CrossRef]
- Boutron, C.F.; Patterson, C.C. Relative levels of natural and anthropogenic lead in recent Antarctic snow. J. Geophys. Res. 1987, 92, 8454–8464. [Google Scholar] [CrossRef]
- Dick, A.L. Trace Elements in Antarctic Snow and Air. Ph.D. Thesis, British Antarctic Survey, Council for National Academic Awards, Cambridge, UK, 1987. [Google Scholar]
- Mcconnell, J.R.; Maselli, O.J.; Sigl, M.; Vallelonga, P.; Neumann, T.; Anschütz, H.; Bales, R.C.; Curran, M.A.J.; Das, S.B.; Edwards, R.; et al. Antarctic-wide array of high-resolution ice core records reveals pervasive lead pollution began in 1889 and persists today. Sci. Rep. 2014, 4, 4–7. [Google Scholar] [CrossRef]
- Candelone, J.-P.; Hong, S.; Pellone, C.; Boutron, C.F. Post-Industrial Revolution changes in large-scale atmospheric pollution of the northern hemisphere by heavy metals as documented in central Greenland snow and ice. J. Geophys. Res. Atmos. 1995, 100, 16605–16616. [Google Scholar] [CrossRef]
- Barbante, C.; Turetta, C.; Capodaglio, G.; Scarponi, G. Recent Decrease in the Lead Concentration of Antarctic Snow. Int. J. Environ. Anal. Chem. 1997, 68, 457–477. [Google Scholar] [CrossRef]
- Vallelonga, P.; van de Velde, K.; Candelone, J.-P.; Morgan, V.I.; Boutron, C.F.; Rosman, K.J.R. The lead pollution history of Law Dome, Antarctica, from isotopic measurements on ice cores: 1500 AD to 1989 AD. Earth Planet. Sci. Lett. 2002, 204, 291–306. [Google Scholar] [CrossRef]
- Nriagu, J.O. Global inventory of natural and anthropogenic emissions of trace metals to the atmosphere. Nature 1979, 279, 409–411. [Google Scholar] [CrossRef]
- Basile, I.; Grousset, F.E.; Revel, M.; Petit, J.R.; Biscaye, P.E.; Barkov, N.I. Patagonian origin of glacial dust deposited in East Antarctica (Vostok and Dome C) during glacial stages 2, 4 and 6. Earth Planet. Sci. Lett. 1997, 146, 573–589. [Google Scholar] [CrossRef]
- Lunt, D.J.; Valdes, P.J. Dust transport to Dome C, Antarctica, at the Last Glacial Maximum and present day. Geophys. Res. Lett. 2001, 28, 295–298. [Google Scholar] [CrossRef]
- Lunt, D.J.; Valdes, P.J. The modern dust cycle: Comparison of model results with observations and study of sensitivities. J. Geophys. Res. Atmos. 2002, 107, AAC 1-1–AAC 1-16. [Google Scholar] [CrossRef]
- Delmonte, B.; Petit, J.; Maggi, V. Glacial to Holocene implications of the new 27000-year dust record from the EPICA Dome C (East Antarctica) ice core. Clim. Dyn. 2002, 18, 647–660. [Google Scholar] [CrossRef]
- Röthlisberger, R.; Mulvaney, R.; Wolff, E.W.; Hutterli, M.A.; Bigler, M.; Sommer, S.; Jouzel, J. Dust and sea salt variability in central East Antarctica (Dome C) over the last 45 kyrs and its implications for southern high-latitude climate. Geophys. Res. Lett. 2002, 29, 24. [Google Scholar] [CrossRef]
- Gaiero, D.M. Dust provenance in Antarctic ice during glacial periods: From where in southern South America? Geophys. Res. Lett. 2007, 34, 1–6. [Google Scholar] [CrossRef]
- Delmonte, B.; Delmas, R.J.; Petit, J.R. Comment on “Dust provenance in Antarctic ice during glacial periods: From where in southern South America?” by D. M. Gaiero. Geophys. Res. Lett. 2008, 35, 22–24. [Google Scholar] [CrossRef]
- Lambert, F.; Delmonte, B.; Petit, J.R.; Bigler, M.; Kaufmann, P.R.; Hutterli, M.A.; Stocker, T.F.; Ruth, U.; Steffensen, J.P.; Maggi, V. Dust—Climate couplings over the past 800,000 years from the EPICA Dome C ice core. Nature 2008, 452, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Ginoux, P.; Ramaswamy, V. Distribution, transport, and deposition of mineral dust in the Southern Ocean and Antarctica: Contribution of major sources. J. Geophys. Res. Atmos. 2008, 113, 1–15. [Google Scholar] [CrossRef]
- Marino, F.; Castellano, E.; Ceccato, D.; De Deckker, P.; Delmonte, B.; Ghermandi, G.; Maggi, V.; Petit, J.R.; Revel-Rolland, M.; Udisti, R. Defining the geochemical composition of the EPICA Dome C ice core dust during the last glacial-interglacial cycle. Geochem. Geophys. Geosyst. 2008, 9, Q10018. [Google Scholar] [CrossRef]
- Delmonte, B.; Paleari, C.I.; Andò, S.; Garzanti, E.; Andersson, P.S.; Petit, J.R.; Crosta, X.; Narcisi, B.; Baroni, C.; Salvatore, M.C.; et al. Causes of dust size variability in central East Antarctica (Dome B): Atmospheric transport from expanded South American sources during Marine Isotope Stage 2. Quat. Sci. Rev. 2017, 168, 55–68. [Google Scholar] [CrossRef]
- Gassó, S.; Stein, A.; Marino, F.; Castellano, E.; Udisti, R.; Ceratto, J. A combined observational and modeling approach to study modern dust transport from the Patagonia desert to East Antarctica. Atmos. Chem. Phys. 2010, 10, 8287–8303. [Google Scholar] [CrossRef]
- Rowley, P.D.; Williams, P.L.; Schmidt, D.L.; Reynolds, R.L.; Ford, A.B.; Clark, A.H.; Farrar, E.; McBride, S.L. Copper mineralization along the Lassiter Coast of the Antarctic Peninsula. Econ. Geol. 1975, 70, 982–987. [Google Scholar] [CrossRef]
- Rowley, P.D.; Williams, P.L.; Douglas, E.P. Mineral Occurrences of Antarctica. In Petroleum and Mined Resources of Antarctica; U.S. Geological Survey Circular 909; Behrendt, J.C., Ed.; U.S. Geological Survey: Washington, DC, USA, 1983; pp. 25–49. [Google Scholar]
- U.S. Congress Office of Technology Assessment. Potential Mineral Resources in Antarctica. In Polar Prospects: A Minerals Treaty for Antarctica; US Government Printing Office: Washington, DC, USA, 1989; Chapter 4; pp. 91–122. [Google Scholar]
- Ericksen, G.E. Metallogenic provinces of southeastern Pacific region. In Circum-Pacific Energy and Mineral Resources; Memoir no. 25; Halbouty, M.T., Maher, J.C., Lian, H.M., Eds.; American Association of Petroleum Geologists: Tulsa, OH, USA, 1976; pp. 527–538. ISBN 9781629812113. [Google Scholar]
- Cunningham, C.G.; Zappettini, E.O.; Vivallo, S.W.; Celada, C.M.; Quispe, J.; Singer, D.A.; Briskey, J.A.; Sutphin, D.M.; Gajardo, M.M.; Diaz, A.; et al. Quantitative Mineral Resource Assessment of Copper, Molybdenum, Gold, and Silver in Undiscovered Porphyry Copper Deposits in the Andes Mountains of South America; USGS Open File Report Series 2008-1253; USGS: Washington, DC, USA, 2008; 28 p.
- Sillitoe, R.H. Copper Provinces. In Geology and Genesis of Major Copper Deposits and Districts of the World: A Tribute to Richard H. Sillitoe; Special Publications of The Society of Economic Geologists Series 16; Hedenquist, J.W., Harris, M., Camus, F., Eds.; Cenveo Publisher Services: Lancaster, PA, USA, 2012; Chapter 1; pp. 1–18. [Google Scholar]
- Barbaro, E.; Padoan, S.; Kirchgeorg, T.; Zangrando, R.; Toscano, G.; Barbante, C.; Gambaro, A. Particle size distribution of inorganic and organic ions in coastal and inland Antarctic aerosol. Environ. Sci. Pollut. Res. 2017, 24, 2724–2733. [Google Scholar] [CrossRef] [PubMed]
- Barbante, C.; Bellomi, T.; Mezzadri, G.; Cescon, P.; Scarponi, G.; Morel, C.; Jay, S.; Van De Velde, K.; Ferrari, C.; Boutron, C.F. Direct determination of heavy metals at picogram per gram levels in Greenland and Antarctic snow by double focusing inductively coupled plasma mass spectrometry. J. Anal. At. Spectrom. 1997, 12, 925–931. [Google Scholar] [CrossRef]
- Kennicutt, M.C., II; Chown, S.L.; Cassano, J.J.; Liggett, D.; Massom, R.; Peck, L.S.; Rintoul, S.R.; Storey, J.W.V.; Vaughan, D.G.; Wilson, T.J.; et al. Six priorities for Antarctic science. Nature 2014, 512, 23–25. [Google Scholar] [CrossRef] [PubMed]
- Kennicutt, M.C.; Chown, S.L.; Cassano, J.J.; Liggett, D.; Peck, L.S.; Massom, R.; Rintoul, S.R.; Storey, J.; Vaughan, D.G.; Wilson, T.J.; et al. A roadmap for Antarctic and Southern Ocean science for the next two decades and beyond. Antarct. Sci. 2015, 27, 3–18. [Google Scholar] [CrossRef]
- Borbély-Kiss, I.; Koltay, E.; Szabó, G.Y.; Bozó, L.; Tar, K. Composition and sources of urban and rural atmospheric aerosol in Eastern Hungary. J. Aerosol Sci. 1999, 30, 369–391. [Google Scholar] [CrossRef]
- Buchanan, C.M.; Beverland, I.J.; Heal, M.R. The influence of weather-type and long-range transport on airborne particle concentrations in Edinburgh, UK. Atmos. Environ. 2002, 36, 5343–5354. [Google Scholar] [CrossRef]
- Abdalmogith, S.S.; Harrison, R.M. The use of trajectory cluster analysis to examine the long-range transport of secondary inorganic aerosol in the UK. Atmos. Environ. 2005, 39, 6686–6695. [Google Scholar] [CrossRef]
- Borge, R.; Lumbreras, J.; Vardoulakis, S.; Kassomenos, P.; Rodríguez, E. Analysis of long-range transport influences on urban PM10 using two-stage atmospheric trajectory clusters. Atmos. Environ. 2007, 41, 4434–4450. [Google Scholar] [CrossRef]
- Nyanganyura, D.; Makarau, A.; Mathuthu, M.; Meixner, F.X. A five-day back trajectory climatology for Rukomechi research station (northern Zimbabwe) and the impact of large-scale atmospheric flows on concentrations of airborne coarse and fine particulate mass. S. Afr. J. Sci. 2008, 104, 43–52. [Google Scholar]
- Fleming, Z.L.; Monks, P.S.; Manning, A.J. Review: Untangling the influence of air-mass history in interpreting observed atmospheric composition. Atmos. Res. 2012, 104–105, 1–39. [Google Scholar] [CrossRef]
- Kottmeier, C.; Fay, B. Trajectories in the Antarctic lower troposphere. J. Geophys. Res. Atmos. 1998, 103, 10947–10959. [Google Scholar] [CrossRef]
- Scarchilli, C.; Frezzotti, M.; Ruti, P.M. Snow precipitation at four ice core sites in East Antarctica: Provenance, seasonality and blocking factors. Clim. Dyn. 2011, 37, 2107–2125. [Google Scholar] [CrossRef]
- Markle, B.R.; Bertler, N.A.N.; Sinclair, K.E.; Sneed, S.B. Synoptic variability in the Ross Sea region, Antarctica, as seen from back-trajectory modeling and ice core analysis. J. Geophys. Res. Atmos. 2012, 117, D02113. [Google Scholar] [CrossRef]
- Barbaro, E.; Zangrando, R.; Vecchiato, M.; Piazza, R.; Cairns, W.R.L.; Capodaglio, G.; Barbante, C.; Gambaro, A. Free amino acids in Antarctic aerosol: Potential markers for the evolution and fate of marine aerosol. Atmos. Chem. Phys. 2015, 15, 5457–5469. [Google Scholar] [CrossRef]
- Elvidge, A.D.; Renfrew, I.A.; King, J.C.; Orr, A.; Lachlan-Cope, T.A.; Weeks, M.; Gray, S.L. Foehn jets over the Larsen C Ice Shelf, Antarctica. Q. J. R. Meteorol. Soc. 2015, 141, 698–713. [Google Scholar] [CrossRef]
- Vecchiato, M.; Argiriadis, E.; Zambon, S.; Barbante, C.; Toscano, G.; Gambaro, A.; Piazza, R. Persistent Organic Pollutants (POPs) in Antarctica: Occurrence in continental and coastal surface snow. Microchem. J. 2015, 119, 75–82. [Google Scholar] [CrossRef]
- Illuminati, S.; Bau, S.; Annibaldi, A.; Mantini, C.; Libani, G.; Truzzi, C.; Scarponi, G. Evolution of size-segregated aerosol mass concentration during the Antarctic summer at Northern Foothills, Victoria Land. Atmos. Environ. 2016, 125, 212–221. [Google Scholar] [CrossRef]
- Zangrando, R.; Barbaro, E.; Vecchiato, M.; Kehrwald, N.M.; Barbante, C.; Gambaro, A. Levoglucosan and phenols in Antarctic marine, coastal and plateau aerosols. Sci. Total Environ. 2016, 544, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Truzzi, C.; Annibaldi, A.; Illuminati, S.; Mantini, C.; Scarponi, G. Chemical fractionation by sequential extraction of Cd, Pb, and Cu in Antarctic atmospheric particulate for the characterization of aerosol composition, sources, and summer evolution at Terra Nova Bay, Victoria Land. Air Qual. Atmos. Health 2017, 10, 783–798. [Google Scholar] [CrossRef]
- Hong, S.B.; Yoon, Y.J.; Becagli, S.; Gim, Y.; Chambers, S.D.; Park, K.T.; Park, S.J.; Traversi, R.; Severi, M.; Vitale, V.; et al. Seasonality of aerosol chemical composition at King Sejong Station (Antarctic Peninsula) in 2013. Atmos. Environ. 2020, 223, 117185. [Google Scholar] [CrossRef]
- Marina-Montes, C.; Pérez-Arribas, L.V.; Escudero, M.; Anzano, J.; Cáceres, J.O. Heavy metal transport and evolution of atmospheric aerosols in the Antarctic region. Sci. Total Environ. 2020, 721, 137702. [Google Scholar] [CrossRef]
- Illuminati, S.; Annibaldi, A.; Bau, S.; Scarchilli, C.; Ciardini, V.; Grigioni, P.; Girolametti, F.; Vagnoni, F.; Scarponi, G.; Truzzi, C. Seasonal Evolution of Size-Segregated Particulate Mercury in the Atmospheric Aerosol over Terra Nova Bay, Antarctica. Molecules 2020, 25, 3971. [Google Scholar] [CrossRef]
- Peel, D.A.; Wolff, E.W. Recent Variations in Heavy Metal Concentrations in Firn and Air from the Antarctic Peninsula. Ann. Glaciol. 1982, 3, 255–259. [Google Scholar] [CrossRef]
- Dick, A.L.; Peel, D.A. Trace Elements in Antarctic Air and Snowfall. Ann. Glaciol. 1985, 7, 12–19. [Google Scholar] [CrossRef]
- Dick, A.L. Concentrations and sources of metals in the Antarctic Peninsula aerosol. Geochim. Cosmochim. Acta 1991, 55, 1827–1836. [Google Scholar] [CrossRef]
- Völkening, J.; Baumann, H.; Heumann, K.G. Atmospheric distribution of particulate lead over the Atlantic Ocean from Europe to Antarctica. Atmos. Environ. 1988, 22, 1169–1174. [Google Scholar] [CrossRef]
- Wagenbach, D.; Gorlach, U.; Moser, K.; Munnich, K.O. Coastal Antarctic aerosol: The seasonal pattern of its chemical composition and radionuclide content. Tellus Ser. B 1988, 40, 426–436. [Google Scholar] [CrossRef][Green Version]
- Rädlein, N.; Heumann, K.G. Trace Analysis of Heavy Metals in Aerosols Over the Atlantic Ocean from Antarctica to Europe. Int. J. Environ. Anal. Chem. 1992, 48, 127–150. [Google Scholar] [CrossRef]
- Chen, L.; Yu, Q.; Yang, S. A study of aerosol chemistry in the atmosphere over oceans Part III: Forms and air-sea fluxes of metal. Atmos. Sin. 1994, 18, 215–223. [Google Scholar]
- Rädlein, N.; Heumann, K.G. Size fractionated impactor sampling of aerosol particles over the Atlantic Ocean from Europe to Antarctica as a methodology for source identification of Cd, Pb, Tl, Ni, Cr, and Fe. Fresenius J. Anal. Chem. 1995, 352, 748–755. [Google Scholar] [CrossRef]
- Huang, Z.Q.; Ji, W.D.; Tang, R.K.; Huang, R.T.; Yang, X.L.; Yu, T.; Zhang, G.X. Chemical composition of marine aerosol in Western Pacific East Indian Ocean Southern Ocean and Prydz Bay and its source discrimination. Ocean. Taiwan Strait 2003, 22, 505–517. [Google Scholar]
- Xu, G.; Gao, Y. Atmospheric trace elements in aerosols observed over the Southern Ocean and coastal East Antarctica. Polar Res. 2014, 33, 23973. [Google Scholar] [CrossRef]
- Shuhui, Z.; Liqi, C.; Hongmei, L. Characteristics of trace metals in marine aerosols and their source identification over the Southern Ocean. Adv. Polar Sci. 2015, 26, 203–214. [Google Scholar] [CrossRef]
- Khandekar, R.N.; Tripathi, R.M.; Raghunath, R.; Sathe, A.P.; Nambi, K.S.V. Heavy metal concentrations in air, water and rock samples at Antarctica during 1989-1990. Curr. Sci. India 1992, 63, 201–204. [Google Scholar]
- Huang, Z.Q.; Ji, W.D.; Yang, X.L.; Huang, R.T.; Tang, R.K.; Yu, T.; Zhang, G.X. The chemical composition of marine aerosol over Zhongshan Station in Antarctica and its sources discrimination in 1998. Acta Ocean. Sin. 2005, 27, 59–66. [Google Scholar]
- Huang, Z.Q.; Ji, W.D.; Yang, X.L. The chemical composition of aerosol over Zhongshan Station in Antarctica and its sources discrimination. J. Oceanogr. Taiwan Strait 2003, 22, 334–346. [Google Scholar]
- Dalla Riva, S.; Abelmoschi, M.L.; Chiantore, M.; Grotti, M.; Magi, E.; Soggia, F. Biogeochemical cycling of Pb in the coastal marine environment at Terra Nova Bay, Ross Sea. Antarct. Sci. 2003, 15, 425–432. [Google Scholar] [CrossRef]
- Jolliffe, I.T. Principal Components Analysis, 2nd ed.; Springer: New York, NY, USA, 2002; ISBN 978-0-387-22440-4. [Google Scholar]
- Robbins, W.K.; Walker, H.H. Analysis of Petroleum for Trace Metals—Determination of Trace Quantities of Cadmium in Petroleum by Atomic Absorption Spectrometry. Anal. Chem. 1975, 47, 1269–1275. [Google Scholar] [CrossRef]
- Shumway, L.A. Trace Element and Polycyclic Aromatic Hydrocarbon Analyses of Jet Engine Fuels: Jet A, JP5, and JP8; SSC San Diego Technical Report 1845; Space and Naval Warfare (SPAWAR) Systems Center: San Diego, CA, USA, 2000; 33p. [Google Scholar]
- Saint’Pierre, T.D.; Felicidade Dias, L.; Pozebon, D.; Aucélio, R.Q.; Curtius, A.J.; Welz, B. Determination of Cu, Mn, Ni and Sn in gasoline by electrothermal vaporization inductively coupled plasma mass spectrometry, and emulsion sample introduction. Spectrochim. Acta Part B At. Spectrosc. 2002, 57, 1991–2001. [Google Scholar] [CrossRef]
- Chaves, E.S.; Lepri, F.G.; Silva, J.S.A.; De Quadros, D.P.C.; Saint’Pierre, T.D.; Curtius, A.J. Determination of Co, Cu, Fe, Mn, Ni and V in diesel and biodiesel samples by ETV-ICP-MS. J. Environ. Monit. 2008, 10, 1211–1216. [Google Scholar] [CrossRef]
- Blakey, S.; Rye, L.; Wilson, C.W. Aviation gas turbine alternative fuels: A review. Proc. Combust. Inst. 2011, 33, 2863–2885. [Google Scholar] [CrossRef]
- Cassella, R.J.; Brum, D.M.; Lima, C.F.; Fonseca, T.C.O. Stabilization of aviation gasoline as detergent emulsion for lead determination by electrothermal atomic absorption spectrometry. Fuel Process. Technol. 2011, 92, 933–938. [Google Scholar] [CrossRef][Green Version]
- Nomngongo, P.N.; Ngila, J.C.; Musyoka, S.M.; Msagati, T.A.M.; Moodley, B. A solid phase extraction procedure based on electrospun cellulose-g-oxolane-2,5-dione nanofibers for trace determination of Cd, Cu, Fe, Pb and Zn in gasoline samples by ICP-OES. Anal. Methods 2013, 5, 3000–3008. [Google Scholar] [CrossRef]
- Ebrahimzadeh, H.; Kasaeian, M.; Khalilzadeh, A.; Moazzen, E. New magnetic polymeric nanoparticles for extraction of trace cadmium ions and the determination of cadmium content in diesel oil samples. Anal. Methods 2014, 6, 4617–4624. [Google Scholar] [CrossRef]
- Kolling, L.; Zmozinski, A.V.; Rodrigues Vale, M.G.; Messias da Silva, M. The use of dried matrix spot for determination of Pb and Ni in automotive gasoline by solid sampling high-resolution continuum source graphite furnace atomic absorption spectrometry. Talanta 2019, 205, 120105. [Google Scholar] [CrossRef]
- Aguirre, M.Á.; Canals, A.; López-García, I.; Hernández-Córdoba, M. Determination of cadmium in used engine oil, gasoline and diesel by electrothermal atomic absorption spectrometry using magnetic ionic liquid-based dispersive liquid-liquid microextraction. Talanta 2020, 220, 121395. [Google Scholar] [CrossRef]
- Vicentino, P.O.; Cassella, R.J.; Leite, D.; Resano, M. Extraction induced by microemulsion breaking as a novel tool for the simultaneous determination of Cd, Mn, Pb and Sb in gasoline samples by ICP-MS and discrete sample introduction. Talanta 2020, 206, 120230. [Google Scholar] [CrossRef]
- Grotti, M.; Soggia, F.; Ardini, F.; Magi, E. Major and trace element partitioning between dissolved and particulate phases in Antarctic surface snow. J. Environ. Monit. 2011, 13, 2511–2520. [Google Scholar] [CrossRef] [PubMed]
- Grotti, M.; Soggia, F.; Ardini, F.; Magi, E.; Becagli, S.; Traversi, R.; Udisti, R. Year-round record of dissolved and particulate metals in surface snow at Dome Concordia (East Antarctica). Chemosphere 2015, 138, 916–923. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Kim, Y.; Boutron, C.F.; Ferrari, C.P.; Petit, J.R.; Barbante, C.; Rosman, K.; Lipenkov, V.Y. Climate-related variations in lead concentrations and sources in Vostok Antarctic ice from 65,000 to 240,000 years BP. Geophys. Res. Lett. 2003, 30, 2138. [Google Scholar] [CrossRef]
- Barbante, C.; Turetta, C.; Gambaro, A.; Capodaglio, G.; Scarponi, G. Sources and origins of aerosols reaching Antarctica as revealed by lead concentration profiles in shallow snow. Ann. Glaciol. 1998, 27, 674–678. [Google Scholar] [CrossRef]
- Fossat, E. The Concordia Station on the Antarctic Plateau: The Best Site on Earth for the 21st Century Astronomers. J. Astrophys. Astr. 2005, 26, 349–357. [Google Scholar] [CrossRef][Green Version]
- Stein, A.F.; Draxler, R.R.; Rolph, G.D.; Stunder, B.J.B.; Cohen, M.D.; Ngan, F. NOAA’s HYSPLIT atmospheric transport and dispersion modeling system. Bull. Am. Meteorol. Soc. 2015, 96, 2059–2077. [Google Scholar] [CrossRef]
- Rolph, G.; Stein, A.; Stunder, B. Real-time Environmental Applications and Display sYstem: READY. Environ. Model. Softw. 2017, 95, 210–228. [Google Scholar] [CrossRef]
- Prospero, J.M.; Charlson, R.J.; Mohnen, V.; Jaenicke, R.; Delany, A.C.; Moyers, J.; Zoller, W.; Rahn, K. The atmospheric aerosol system: An overview. Rev. Geophys. 1983, 21, 1607–1629. [Google Scholar] [CrossRef]
- Harris, J.M. An analysis of 5-day midtropospheric flow patterns for the South Pole: 1985-1989. Tellus B 1992, 44, 409–421. [Google Scholar] [CrossRef][Green Version]
- Chance, R.; Jickells, T.D.; Baker, A.R. Atmospheric trace metal concentrations, solubility and deposition fluxes in remote marine air over the south-east Atlantic. Mar. Chem. 2015, 177, 45–56. [Google Scholar] [CrossRef]
- Truzzi, C.; Lambertucci, L.; Gambini, G.; Scarponi, G. Optimization of square wave anodic stripping voltammetry (SWASV) for the simultaneous determination of Cd, Pb, and Cu in seawater and comparison with differential pulse anodic stripping voltammetry (DPASV). Ann. Chim. 2002, 92, 313–326. [Google Scholar] [PubMed]
- Truzzi, C.; Annibaldi, A.; Illuminati, S.; Bassotti, E.; Scarponi, G. Square-wave anodic-stripping voltammetric determination of Cd, Pb, and Cu in a hydrofluoric acid solution of siliceous spicules of marine sponges (from the Ligurian Sea, Italy, and the Ross Sea, Antarctica). Anal. Bioanal. Chem. 2008, 392, 247–262. [Google Scholar] [CrossRef]
- National Institute of Standards and Technologies (NIST). Standard Reference Material (SRM) 1648 for Urban Particulate Matter; Certificate Issue Date: 28 April 1998, Expiration of Certification 31 December 2008; NIST: Gaithersburg, MD, USA, 1998.
- National Institute of Standards and Technologies (NIST). Standard Reference Material (SRM) 1648a for Urban Particulate Matter; Current Certificate Issue Date: 6 October 2020; Expiration of Certification 1 October 2027; NIST: Gaithersburg, MD, USA, 2020.
- National Research Council of Canada (NRCC). Seawater Reference Material for Trace Metals, NASS-6; NRCC: Ottawa, ON, Canada, 2010.
- Annibaldi, A.; Illuminati, S.; Truzzi, C.; Finale, C.; Scarponi, G. Soluble/insoluble (dilute-HCl-extractable) fractionation of Cd, Pb and Cu in Antarctic snow and its relationship with metal fractionations in the aerosol. E3S Web Conf. 2013, 1, 23006. [Google Scholar] [CrossRef]
- Illuminati, S.; Annibaldi, A.; Truzzi, C.; Finale, C.; Scarponi, G. Square-wave anodic-stripping voltammetric determination of Cd, Pb and Cu in wine: Set-up and optimization of sample pre-treatment and instrumental parameters. Electrochim. Acta 2013, 104, 148–161. [Google Scholar] [CrossRef]
- Wold, S. Cross-Validatory Estimation of the Number of Components in Factor and Principal Components Models. Technometrics 1978, 20, 397–405. [Google Scholar] [CrossRef]
- Annibaldi, A.; Truzzi, C.; Illuminati, S.; Scarponi, G. Recent sudden decrease of lead in Adriatic coastal seawater during the years 2000–2004 in parallel with the phasing out of leaded gasoline in Italy. Mar. Chem. 2009, 113, 238–249. [Google Scholar] [CrossRef]
- Annibaldi, A.; Illuminati, S.; Truzzi, C.; Scarponi, G. SWASV speciation of Cd, Pb and Cu for the determination of seawater contamination in the area of the Nicole shipwreck (Ancona coast, Central Adriatic Sea). Mar. Pollut. Bull. 2011, 62, 2813–2821. [Google Scholar] [CrossRef]
- Annibaldi, A.; Truzzi, C.; Illuminati, S.; Bassotti, E.; Finale, C.; Scarponi, G. First Systematic Voltammetric Measurements of Cd, Pb, and Cu in Hydrofluoric Acid-Dissolved Siliceous Spicules of Marine Sponges: Application to Antarctic Specimens. Anal. Lett. 2011, 44, 2792–2807. [Google Scholar] [CrossRef]
- Illuminati, S.; Truzzi, C.; Annibaldi, A.; Migliarini, B.; Carnevali, O.; Scarponi, G. Cadmium bioaccumulation and metallothionein induction in the liver of the Antarctic teleost Trematomus bernacchii during an on-site short-term exposure to the metal via seawater. Toxicol. Environ. Chem. 2010, 92, 617–640. [Google Scholar] [CrossRef]
- Illuminati, S.; Annibaldi, A.; Romagnoli, T.; Libani, G.; Antonucci, M.; Scarponi, G.; Totti, C.; Truzzi, C. Distribution of Cd, Pb and Cu between dissolved fraction, inorganic particulate and phytoplankton in seawater of Terra Nova Bay (Ross Sea, Antarctica) during austral summer 2011–12. Chemosphere 2017, 185, 1122–1135. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sampling Point | Sample No. | Mass Fraction (±SD) | Atmospheric Concentration (±SD) | |||||
|---|---|---|---|---|---|---|---|---|
| Cd (µg g−1) | Pb (mg g−1) | Cu (mg g−1) | Cd (pg m−3) | Pb (pg m−3) | Cu (ng m−3) | |||
| Concordia | 1 | 7.0 ± 1.2 | 0.14 ± 0.02 | 5.4 ± 0.9 | 3.1 ± 0.4 | 62 ± 7 | 2.4 ± 0.3 | |
| 2 | 1.4 ± 0.4 | 0.11 ± 0.01 | 3.9 ± 0.6 | 0.40 ± 0.13 | 31 ± 3 | 1.1 ± 0.2 | ||
| 3 | 5.2 ± 0.5 | 0.11 ± 0.01 | 0.92 ± 0.06 | 2.0 ± 0.2 | 44 ± 6 | 0.36 ± 0.04 | ||
| Astrophysics T. 1 | 1 | 6.6 ± 1.6 | 0.47 ± 0.06 | 20 ± 2 | 0.70 ± 0.18 | 50 ± 7 | 2.1 ± 0.3 | |
| 2 | 1.3 ± 0.4 | 0.12 ± 0.01 | 1.06 ± 0.14 | 0.25 ± 0.09 | 23 ± 2 | 0.20 ± 0.03 | ||
| 3 | 1.0 ± 0.5 | 0.13 ± 0.02 | 1.53 ± 0.12 | 0.09 ± 0.04 | 12 ± 1 | 0.14 ± 0.02 | ||
| Astrophysics T. 2 | 1 | 8.4 ± 1.0 | 0.20 ± 0.03 | 0.88 ± 0.28 | 0.98 ± 0.14 | 24 ± 4 | 0.10 ± 0.03 | |
| 2 | 1.3 ± 0.4 | 0.10 ± 0.01 | 0.17 ± 0.03 | 0.20 ± 0.06 | 15 ± 2 | 0.027 ± 0.004 | ||
| Area Station/site | Period | Metal Atmospheric Concentration, pg m−3 | Reference | ||
|---|---|---|---|---|---|
| Cd Average ± SD (min-max) | Pb Average ± SD (min-max) | Cu Average ± SD (min-max) | |||
| Antarctic Plateau | |||||
| Dome C | Summer 2005–2006 | 0.24 ± 0.13 a (0.09–3.1) | 21 ± 8 a (12–62) | 120 ± 70 a (27–2400) | This work |
| South Pole | Summer 1970–1971 | - | 630 ± 300 (<190–1200) | 36 ± 19 (25–64) | [31] |
| South Pole | Summer 1974–1975 | <15 | - | 29 ± 17 | [32] |
| South Pole | Summer 1974–1975 | ≤18 | 27 ± 10 (W) b 76 ± 40 (N) b | 29 ± 17 | [33] |
| South Pole | Summers 1971–1978 | 49 ± 38 | - | 59 ± 47 | [34] |
| Winters 1975–1976 | <200 | - | 79 ± 16 | ||
| South Pole | Summers 1979–1983 | 110 ± 60 | - | 190 ± 130 | [35] |
| Winters 1979–1983 | 50 ± 40 | - | 130 ± 80 | ||
| South Pole | November 2000–January 2001 | - | <32 c | - | [36] |
| South Pole | November 2000–January 2001 | - | 30 ± 10 | - | [37] |
| South Pole | November–December 2003 | - | 180 ± 40 | - | [37] |
| Antarctic Peninsula | |||||
| Spaatz Island Plateau site Rothera | Summer 1979–1980 | 3.5 ± 0.8 3.1 ± 2.1 5.4 ± 0.7 | 226 ± 35 79 ± 24 153 ± 60 | - - - | [119] |
| Beethoven Peninsula | Summer 1982–1983 | <1.1 | <8.5 | 11.3 ± 6.2 | [120] |
| Gipps Ice Rice | Summer 1984–1985 | 0.06 ± 0.1 | 4.7 ± 0.8 | 1.0 ± 1.0 | [121] |
| Comandante Ferraz | Summers1985–1988 | - | 800 d | 690 d | [14] |
| Winters 1985–1988 | - | 830 d | 1200 d | ||
| Overall 1985–1988 | - | 830 d | 990 d | ||
| Comandante Ferraz | Summers1985–1993 | 1200 d | 3450 d | [15] | |
| Winters 1985–1993 | - | 970 d | 2740 d | ||
| Overall 1985–1993 | - | 1060 d | 3010 d | ||
| King Sejong | January 2000–December 2001 | 1.3 ± 3 (0.07–16.7) | 41 ± 54.5 (0.71–232) | 143 ± 471 (2.0–2413) | [49] |
| Antarctic Coast or Southern Ocean | |||||
| Ekström Ice Shelf | April, May, December 1983 | - | 17 ± 9 | - | [122] |
| Georg von Neumayer | Febuary 1983–December 1984 | - | 11 ± 1.5 | - | [123] |
| Antarctic Ocean (Atlantic) includes Georg von Neumayer and Filchner Ice Shelf | Summer 1989–1990 | 26 ± 18 | 328 ± 269 | 163 ± 85 | [124] |
| Antarctic Ocean | Summer 1986–1987 | 20 | 1100 | 2300 | [125] |
| Antarctic Ocean | 1989–1990 | 105 | 269 | - | [126] |
| Southern Ocean | Summer 1999–2000 | 40 | 230 | 700 | [127] |
| Southern Ocean coastal | Summer 2010–2011 | 4 (0–20) | - | - | [128] |
| Coastal East Antarctica | Summer 2010–2011 | 17(0–50) | - | - | [128] |
| Southern Ocean | Summer 2011–2012 | 17 ± 29 | 127 ± 98 | 957 ± 638 | [129] |
| McMurdo Hut Point (downwind) | Summer 1995–1996 Summer 1996–1997 Total summers 1995–1997 | - - - | 1244 ± 2513 389 ± 541 851 ± 1915 | 194 ± 260 183 ± 104 189 ± 202 | [28] |
| McMurdo Radar Sat (upwind) | Summer 1995–1996 Summer 1996–1997 Total summers 1995–1997 | - - - | 620 ± 1048 300 ± 438 470 ± 827 | 243 ± 304 152 ± 141 200 ± 244 | [28] |
| Maitri | Summer 1990 | - | 306 ± 68 | 704 ± 339 | [130] |
| Zhongshan Station | March 1998–Febuary 1999 | 70 | 559 | 1280 | [131] |
| Zhongshan Station | March 1998–November 1999 | 47 | 431 | 778 | [132] |
| Prydz Bay | Summer 1999–2000 | 30 | 80 | 460 | [127] |
| Mario Zucchelli (formerly Terra Nova Bay) Faraglione Camp | Summer 2000–2001 | - | 16.7 (9.7–38.7) | 422 (86–641) | [45] |
| Summer 2001–2002 | - | 15.0 (6.8–48.7) | 394 (121–1102) | [45] | |
| Summer 2010–2011 | - | 22.5 e | 32.5 e | [47] | |
| Summers 1988–2002 | - | 10 ± 3 | - | [133] | |
| Summer 2000–2001 | 3.4 ± 2.2 f (0.55–6.3) | 24 ± 17 f (8.7–48) | 266 ± 103 f (72–365) | [30] | |
| Summer 2000–2001 | 0.14–19 g | 17–36 g | 177–436 g | [53] | |
| Summer 2000–2001 | 9.5 ± 13.1 g (0.93–39) | 33 ± 16 g (17–60) | 340 ± 150 g (88–480) | [115] | |
| Larsemann Hills Antarctic Coast | Summer 2009–2010 | - | 260 ± 180 | 2190 ± 530 | [43] |
| Larsemann Hills Southern Ocean | Summer 2009–2010 | - | 580 ± 190 | 9020 ± 5800 | [43] |
| Metals | Mass Fraction | Atmospheric Concentration | |||
|---|---|---|---|---|---|
| r | p-Value (*) | r | p-Value (*) | ||
| Cd-Pb | 0.50 | 0.10 | 0.83 | 0.006 | |
| Cd-Cu | 0.38 | 0.17 | 0.57 | 0.071 | |
| Pb-Cu | 0.93 | 0.0004 | 0.87 | 0.003 | |
| Metals | Background Samples | Contamination Samples | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mass Fraction | Atmospheric Conc. | Mass Fraction | Atmospheric Conc. | ||||||||
| r | p-Value * | r | p-Value* | r | p-Value * | r | p-Value * | ||||
| Cd-Pb | −0.69 | 0.16 | 0.97 | 0.016 | 0.12 | 0.44 | 0.65 | 0.19 | |||
| Cd-Cu | 0.33 | 0.34 | 0.86 | 0.072 | −0.084 | 0.46 | 0.34 | 0.33 | |||
| Pb-Cu | 0.17 | 0.42 | 0.88 | 0.062 | 0.94 | 0.032 | 0.87 | 0.064 | |||
| Regression | Total Dataset | Separate Datasets | ||
|---|---|---|---|---|
| from Mass Fraction from atmospheric conc. | Background Samples from Mass Fraction from atmospheric conc. | Contamination Samples from Mass Fraction from atmospheric conc. | ||
| Pb vs. Cd | 14 | 64 | 9.4 | |
| 20 | −58 | 15 | ||
| Cu vs. Cd | 510 | 3300 | 370 | |
| 810 | 3000 | −570 | ||
| Cu vs. Pb | 47 | 51 | 65 | |
| 49 | 18 | 52 | ||
| Fuel | Metal Contents µg metal/kg fuel | Metal Ratios µg metal A/µg metal B | |||||
|---|---|---|---|---|---|---|---|
| Cd | Pb | Cu | Pb/Cd | Cu/Cd | Cu/Pb | ||
| SAB diesel fuel | 0.7 ± 0.2 | 10 ± 1 | 70 ± 7 | 14 ± 4 | 100 ± 30 | 7 ± 1 | |
| Unleaded gasoline | 1.0 ± 0.2 | 65 ± 3 | 101 ± 9 | 65 ± 13 | 101 ± 22 | 1.6 ± 0.2 | |
| Jet A-1 fuel | 7 ± 3 | 80 ± 2 | 192 ± 17 | 11 ± 5 | 27 ± 12 | 2.4 ± 0.2 | |
| Field Blank Filter | Metal Concentrations (Mean ± SD) | ||
|---|---|---|---|
| Cd (ng L−1) | Pb (ng L−1) | Cu (µg L−1) | |
| Concordia, blank 1 | 10.0 ± 0.3 (n = 3) | 84 ± 5 (n = 4) | 0.39 ± 0.02 (n = 5) |
| Concordia, blank 2 | 9.4 ± 0.8 (n = 4) | 82 ± 8 (n = 4) | 0.34 ± 0.03 (n = 4) |
| Astrophysics T. 1, blank 1 | 10.1 ± 1.0 (n = 3) | 75 ± 7 (n = 3) | 0.34 ± 0.02 (n = 3) |
| Astrophysics T. 1, blank 2 | 9.4 ± 1.8 (n = 3) | 80 ± 9 (n = 3) | 0.40 ± 0.03 (n = 3) |
| Astrophysics T. 2, blank 1 | 10.7 ± 0.3 (n = 3) | 82 ± 4 (n = 3) | 0.34 ± 0.02 (n = 4) |
| Astrophysics T. 2, blank 2 | 10.0 ± 1.8 (n = 3) | 76 ± 9 (n = 4) | 0.37 ± 0.03 (n = 3) |
| Weighted mean ± SD i.e., blank to be subtracted | 10 ± 1 (n = 19) | 80 ± 7 (n = 21) | 0.36 ± 0.02 (n = 22) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Illuminati, S.; Annibaldi, A.; Truzzi, C.; Mantini, C.; Conca, E.; Malandrino, M.; Giglione, G.; Fanelli, M.; Scarponi, G. Determination of Cd, Pb, and Cu in the Atmospheric Aerosol of Central East Antarctica at Dome C (Concordia Station). Molecules 2021, 26, 1997. https://doi.org/10.3390/molecules26071997
Illuminati S, Annibaldi A, Truzzi C, Mantini C, Conca E, Malandrino M, Giglione G, Fanelli M, Scarponi G. Determination of Cd, Pb, and Cu in the Atmospheric Aerosol of Central East Antarctica at Dome C (Concordia Station). Molecules. 2021; 26(7):1997. https://doi.org/10.3390/molecules26071997
Chicago/Turabian StyleIlluminati, Silvia, Anna Annibaldi, Cristina Truzzi, Caterina Mantini, Eleonora Conca, Mery Malandrino, Giada Giglione, Matteo Fanelli, and Giuseppe Scarponi. 2021. "Determination of Cd, Pb, and Cu in the Atmospheric Aerosol of Central East Antarctica at Dome C (Concordia Station)" Molecules 26, no. 7: 1997. https://doi.org/10.3390/molecules26071997
APA StyleIlluminati, S., Annibaldi, A., Truzzi, C., Mantini, C., Conca, E., Malandrino, M., Giglione, G., Fanelli, M., & Scarponi, G. (2021). Determination of Cd, Pb, and Cu in the Atmospheric Aerosol of Central East Antarctica at Dome C (Concordia Station). Molecules, 26(7), 1997. https://doi.org/10.3390/molecules26071997