
Guanidine–Amide-Catalyzed Aza-Henry Reaction of Isatin-Derived Ketimines: Origin of Selectivity and New Catalyst Design


Abstract
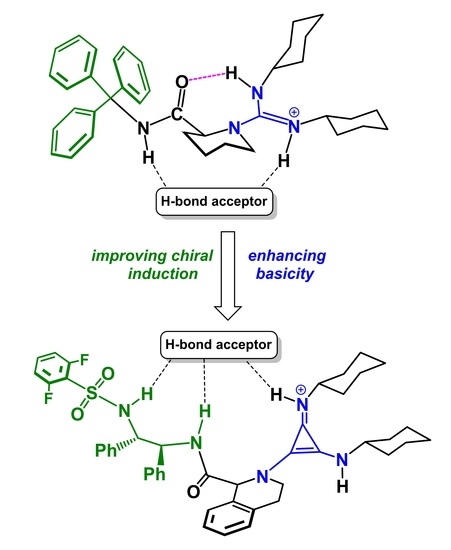
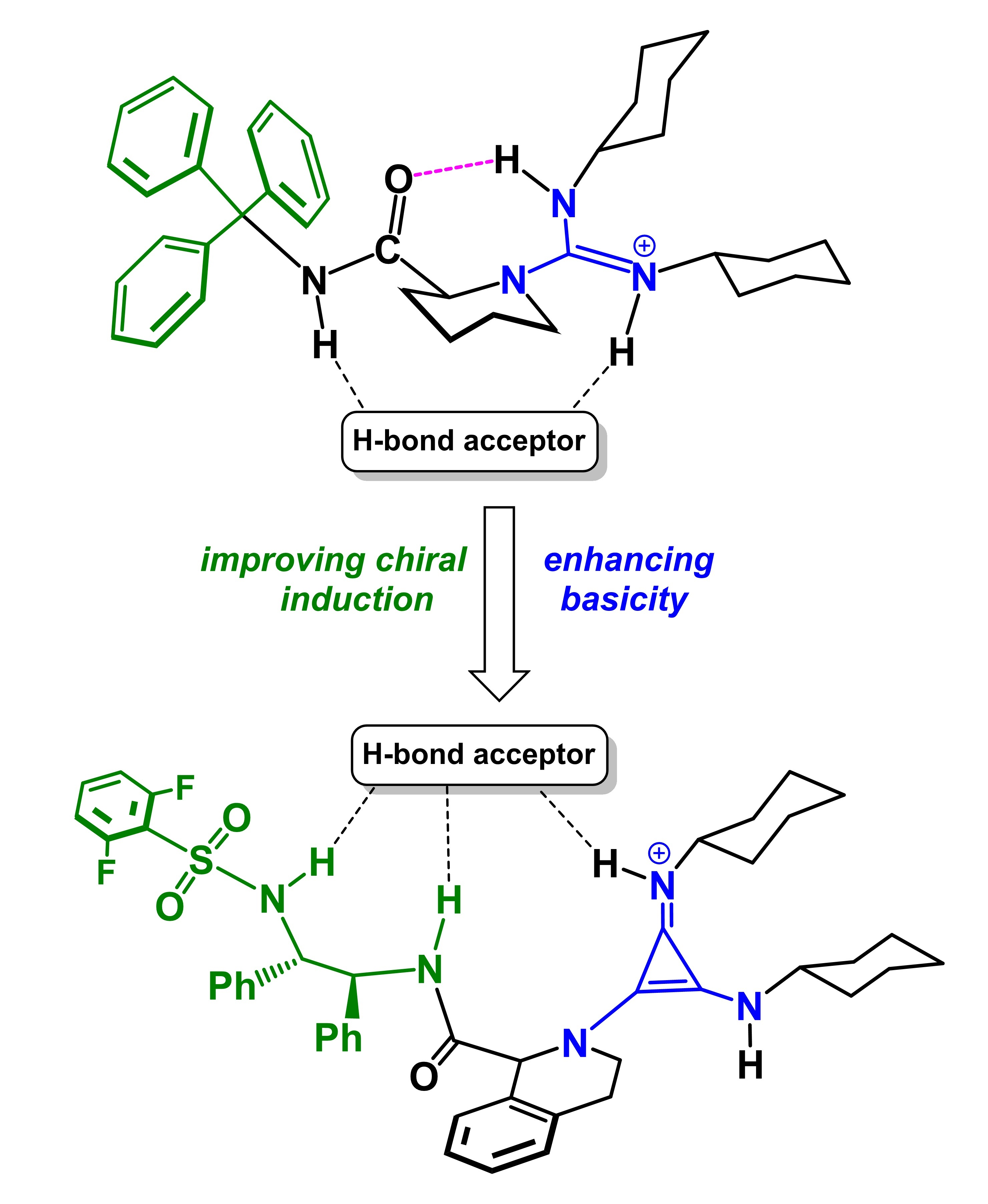
1. Introduction
2. Results and Discussion
2.1. Analysis of Reactivity at N Atoms in G1
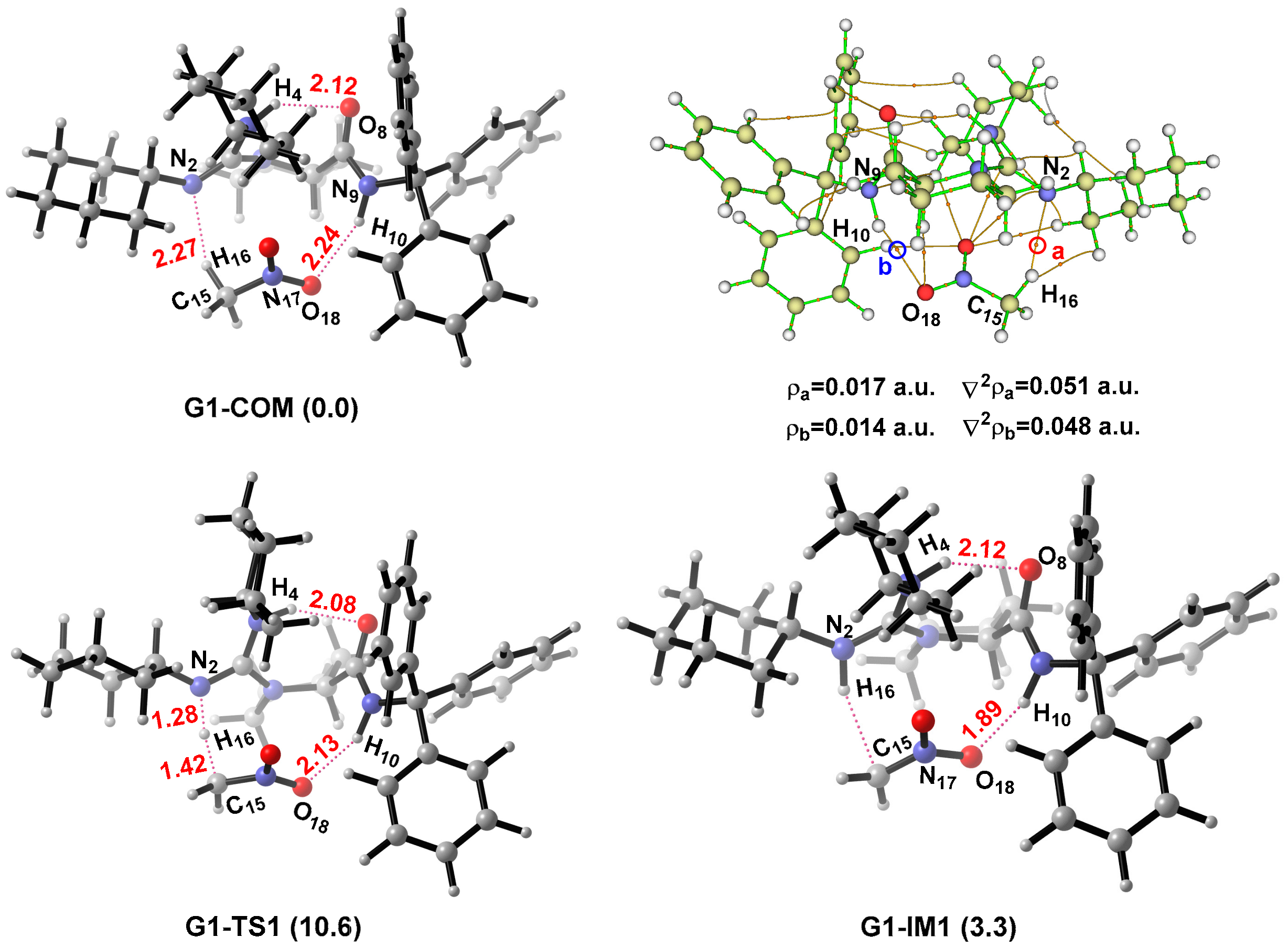
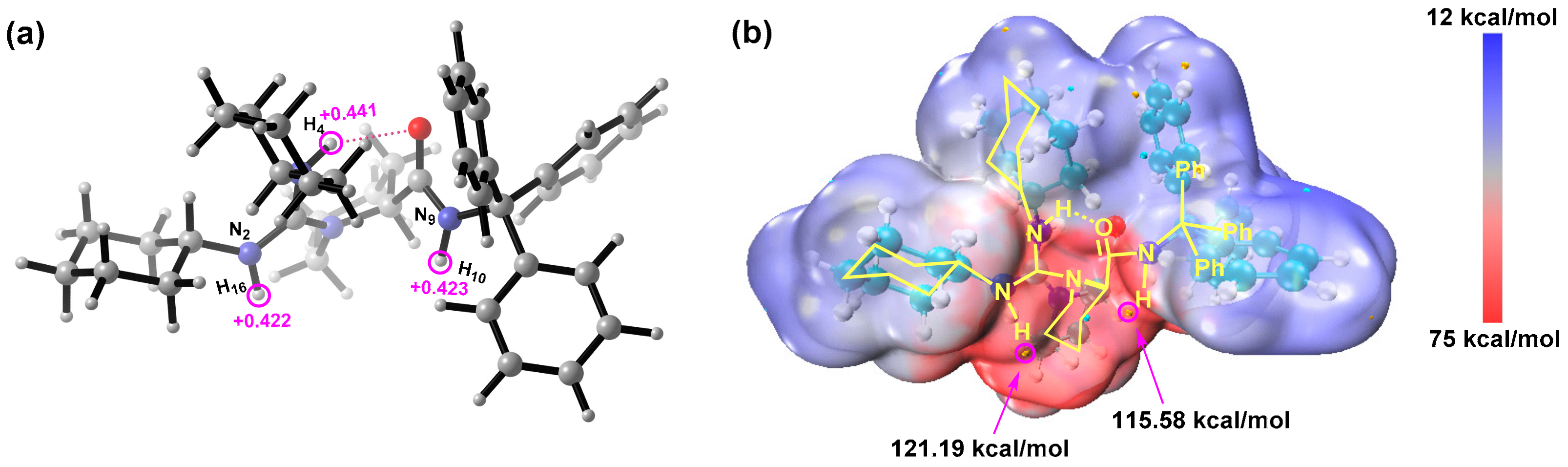
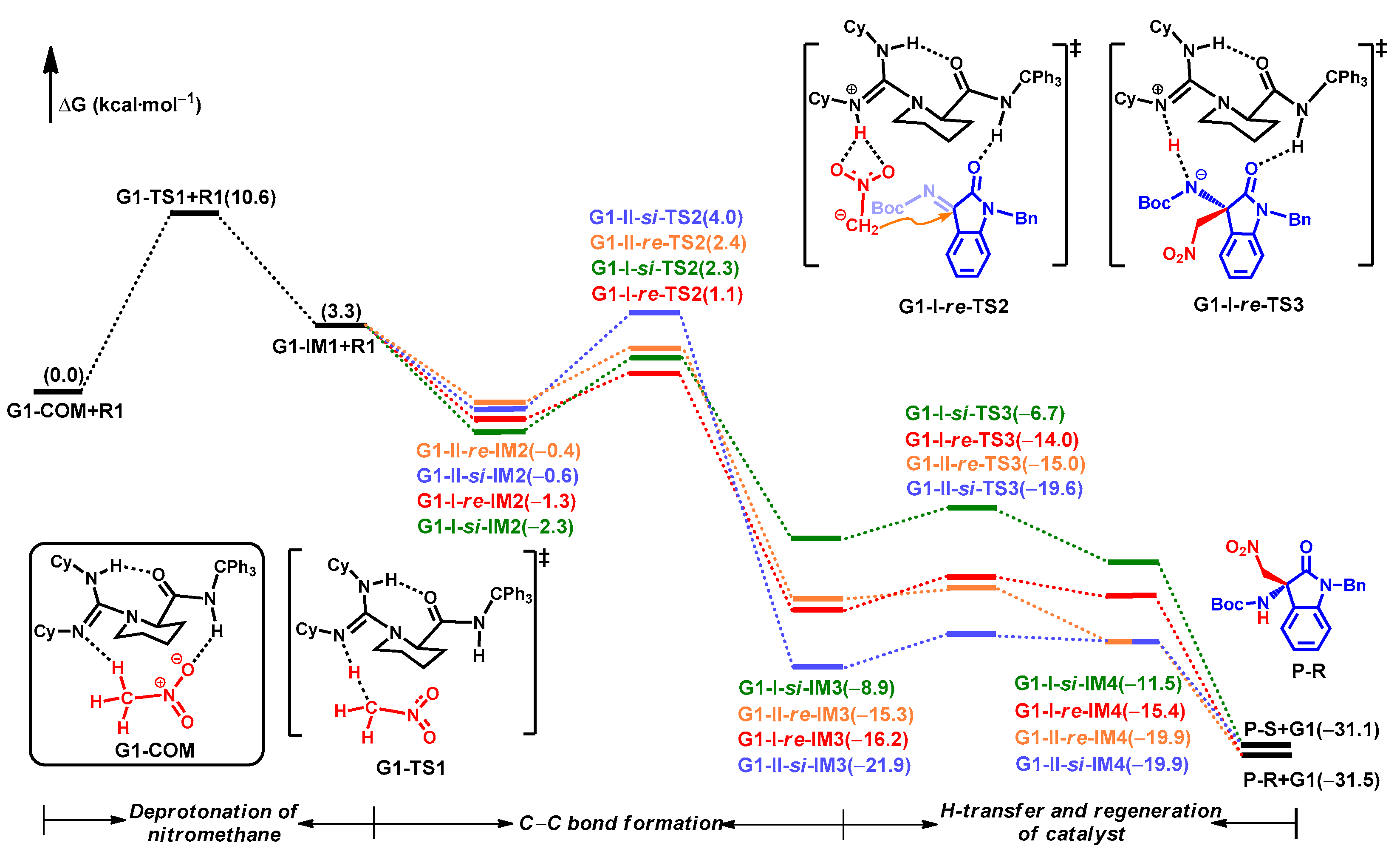
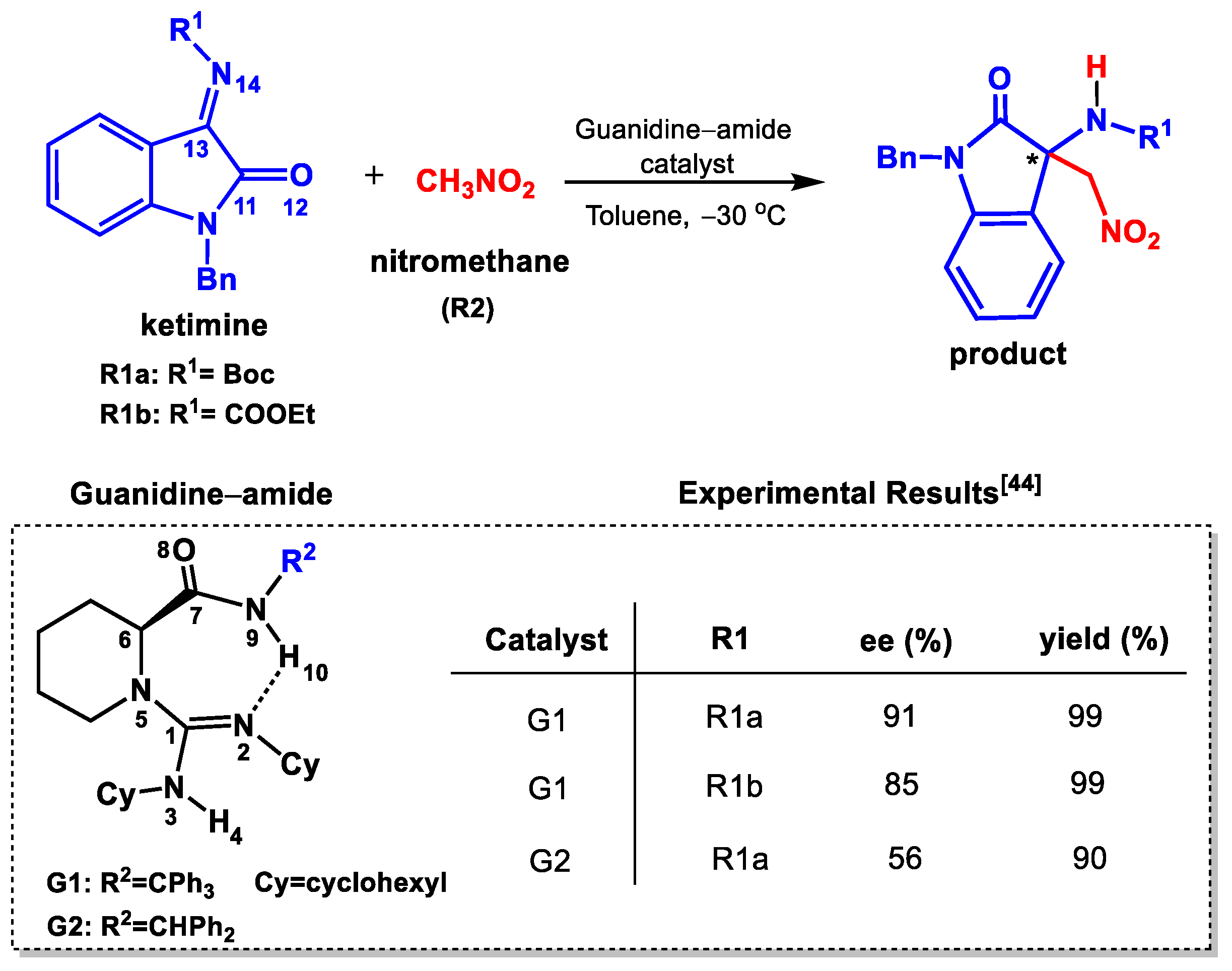
2.2. Catalytic Mechanism for R1a
2.2.1. Activation of CH3NO2
2.2.2. Formation of C–C Bond
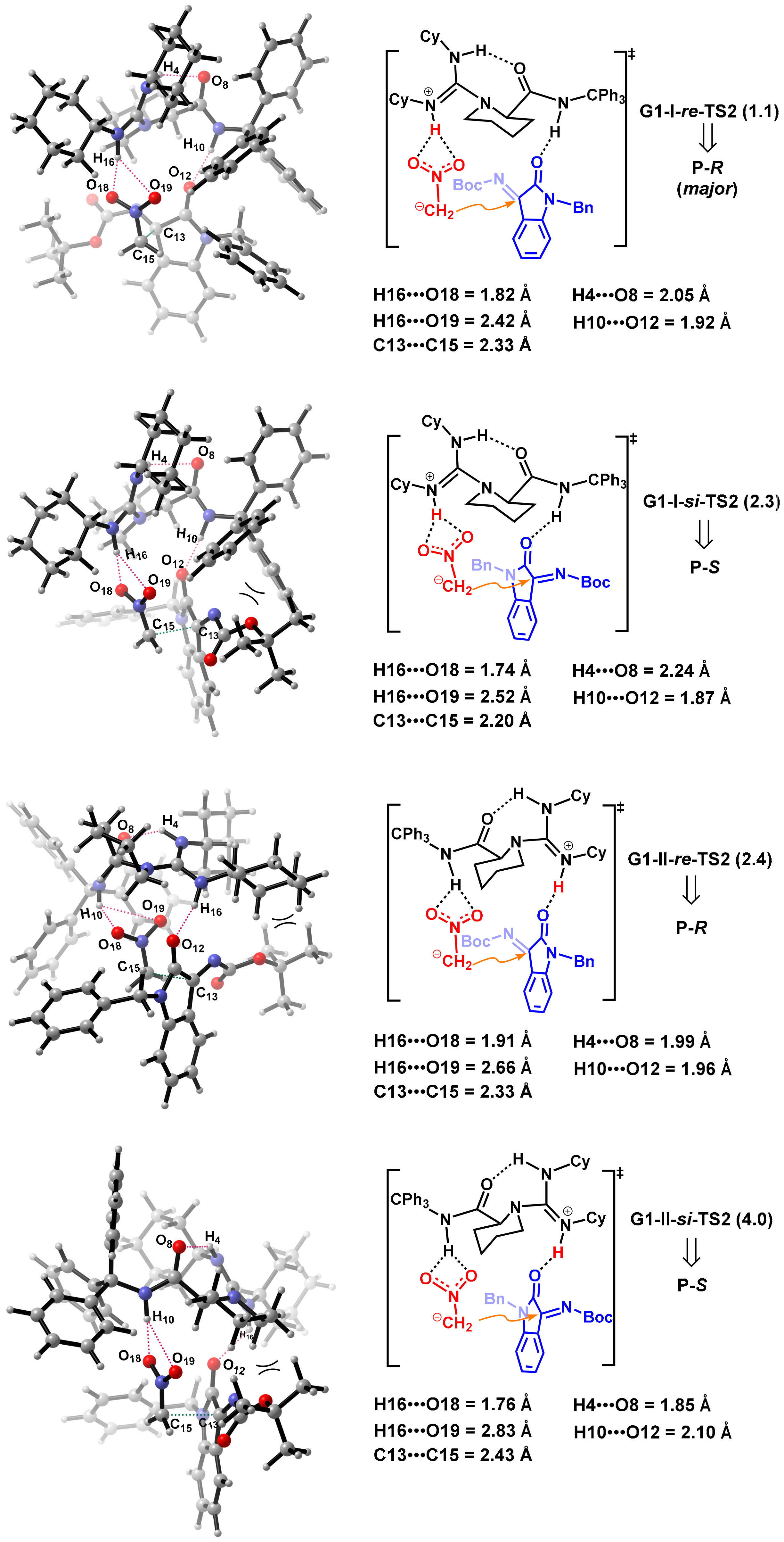
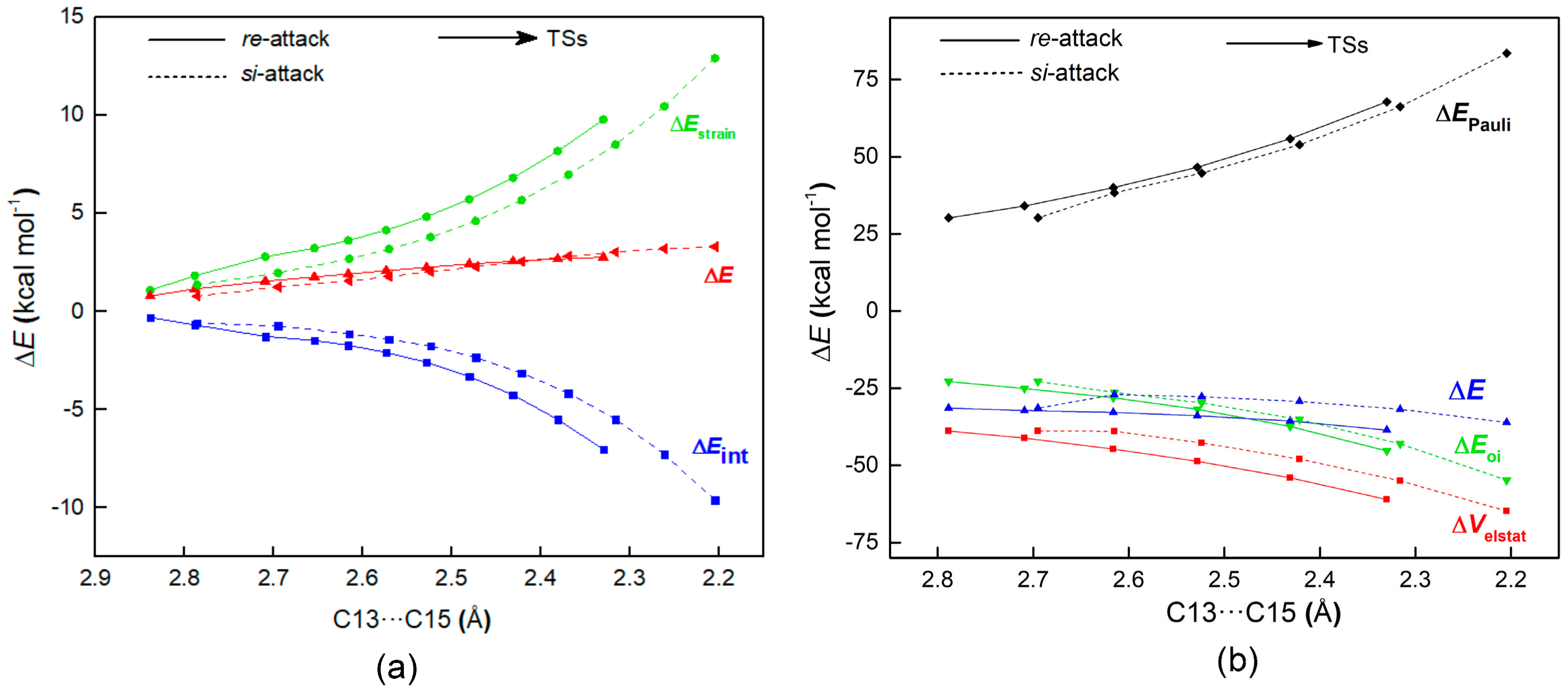
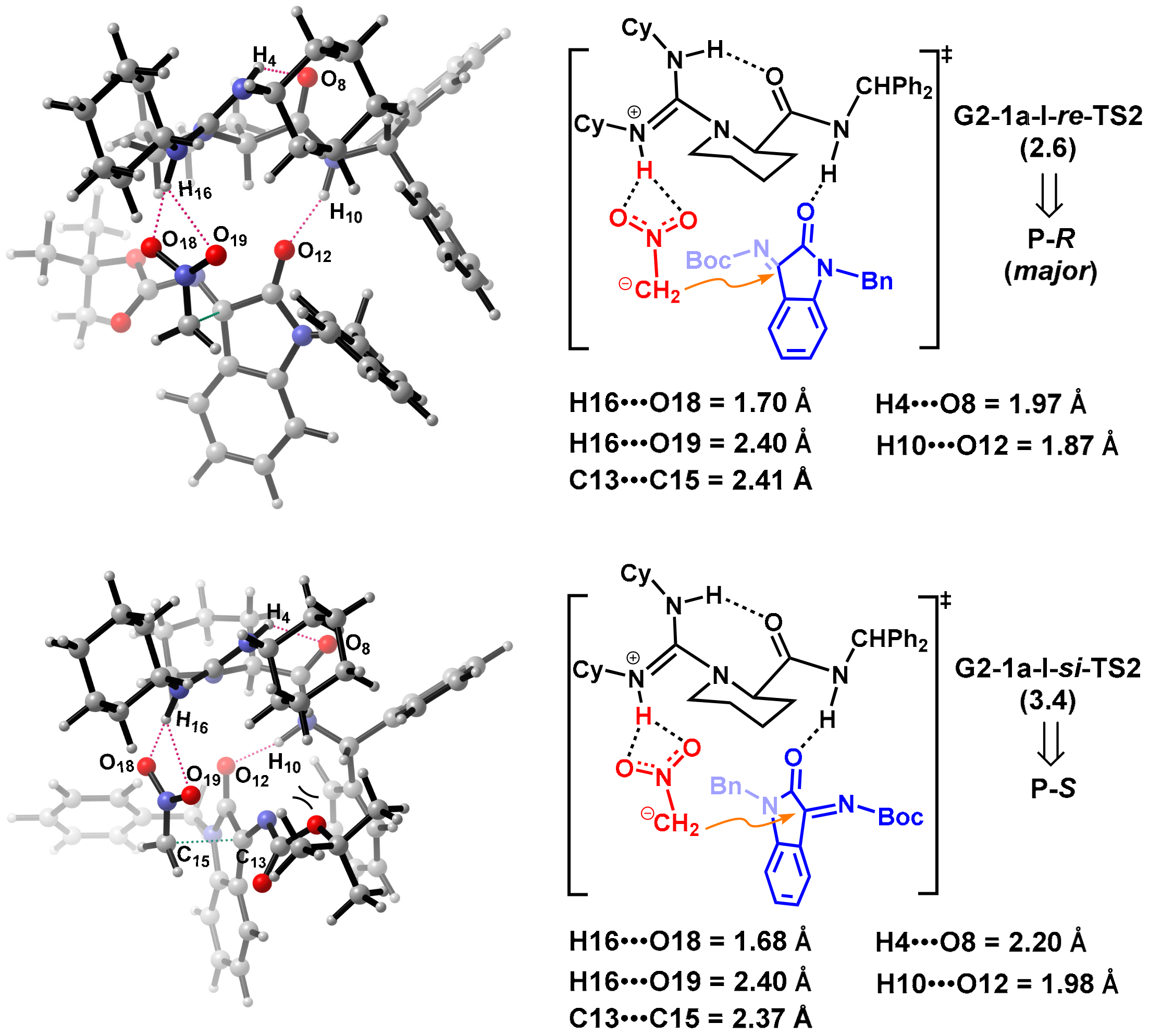
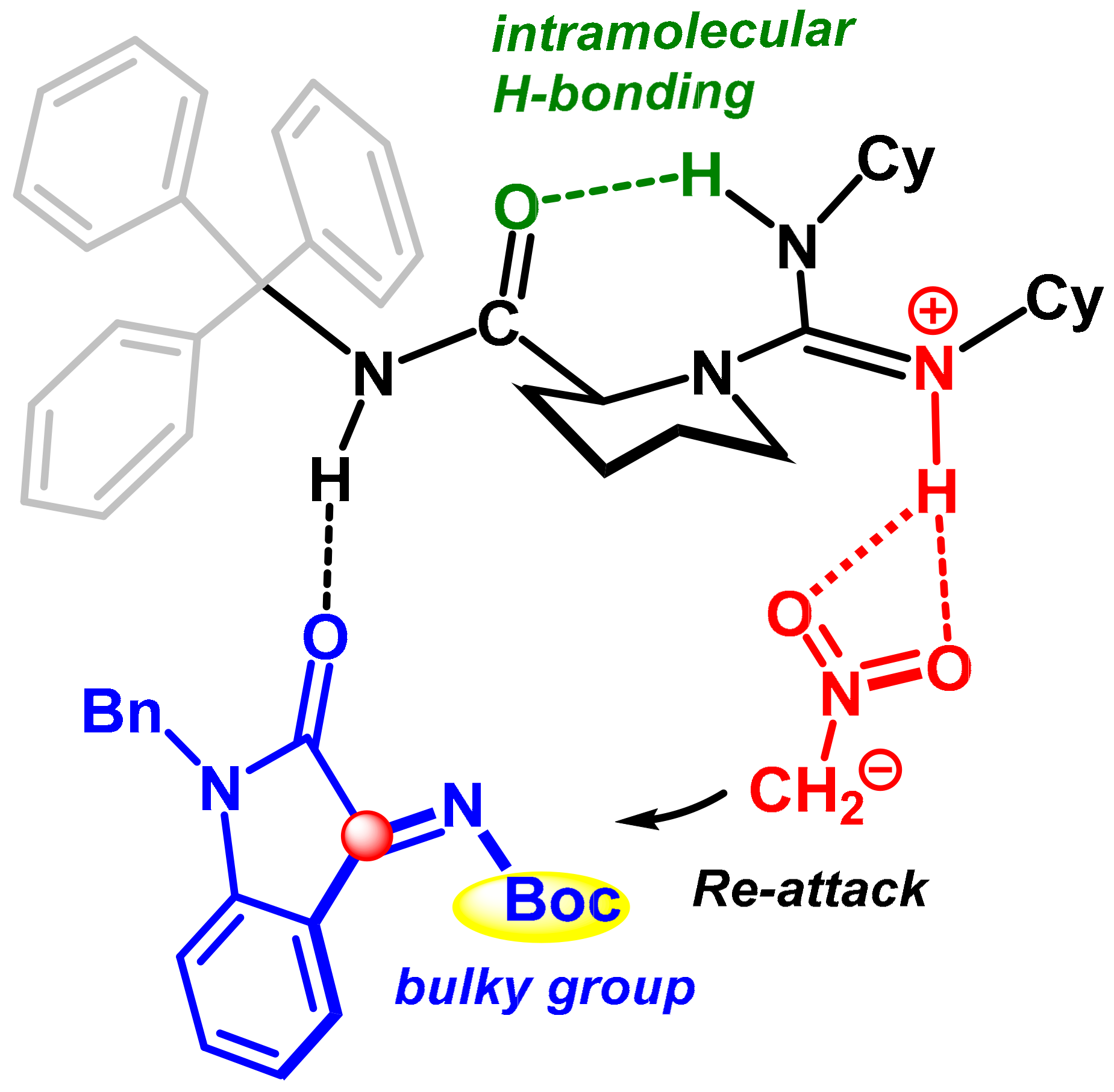
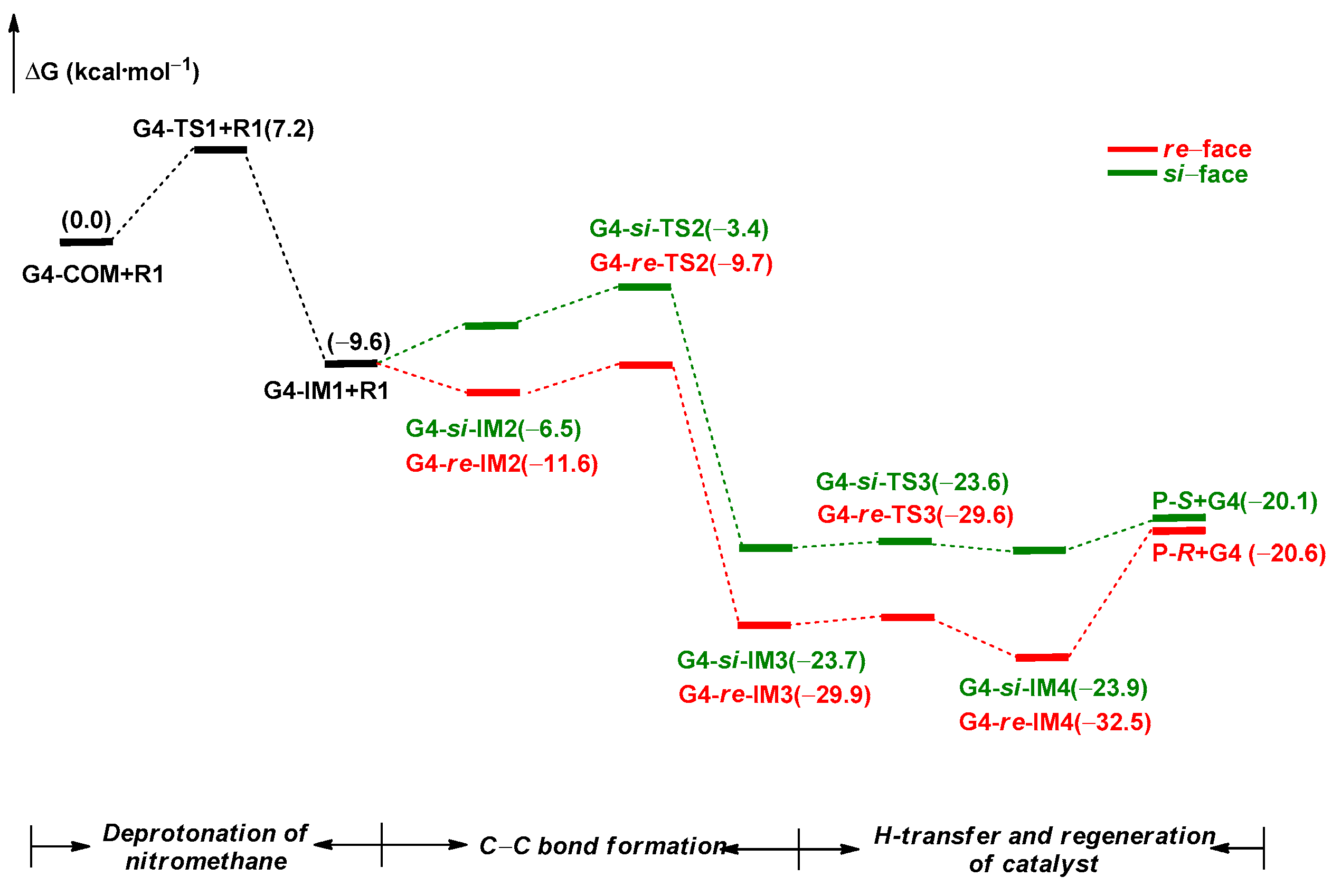
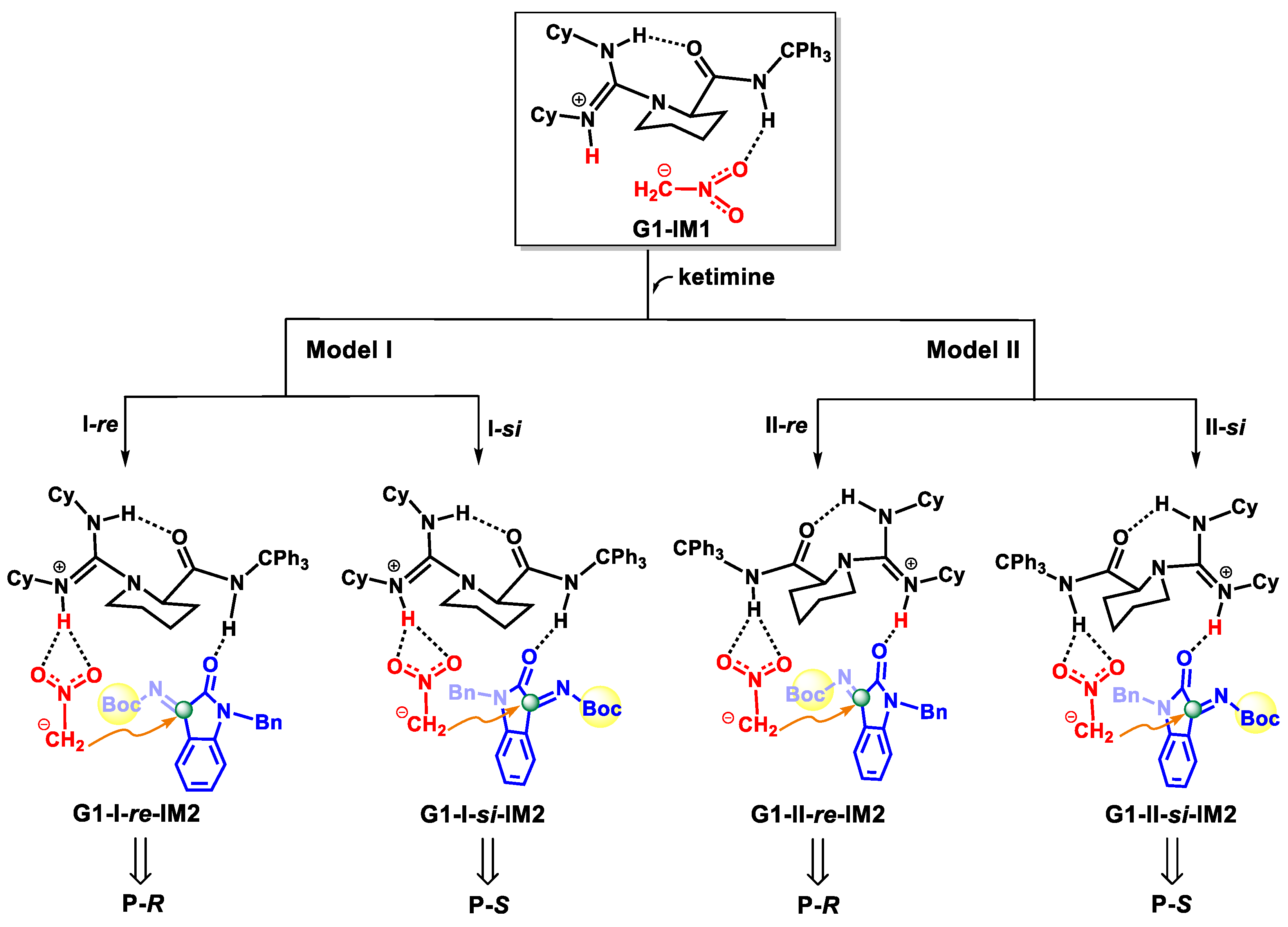
2.2.3. Origin of Stereoselectivity
2.2.4. Turnover Frequency (TOF) Analysis
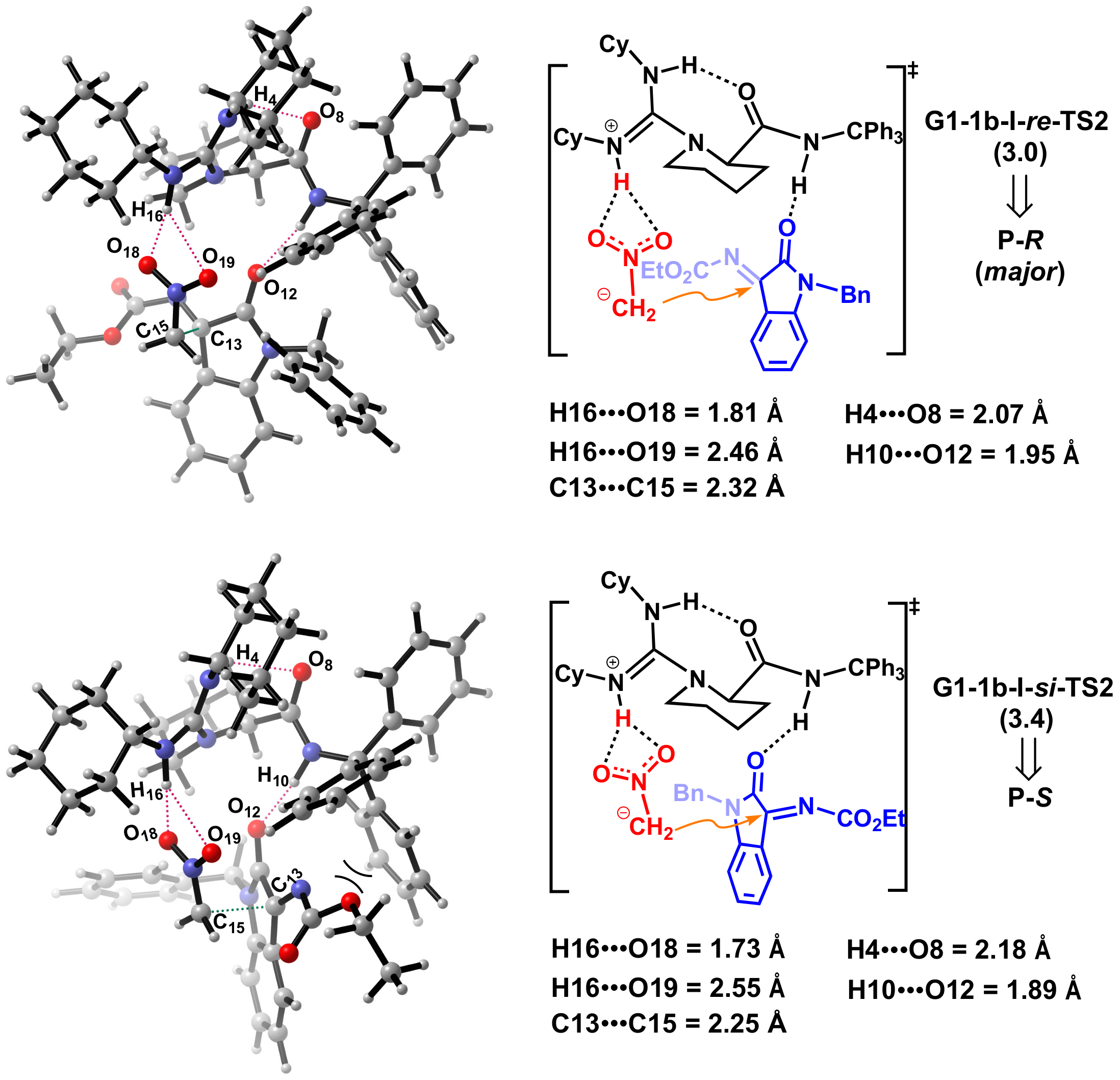
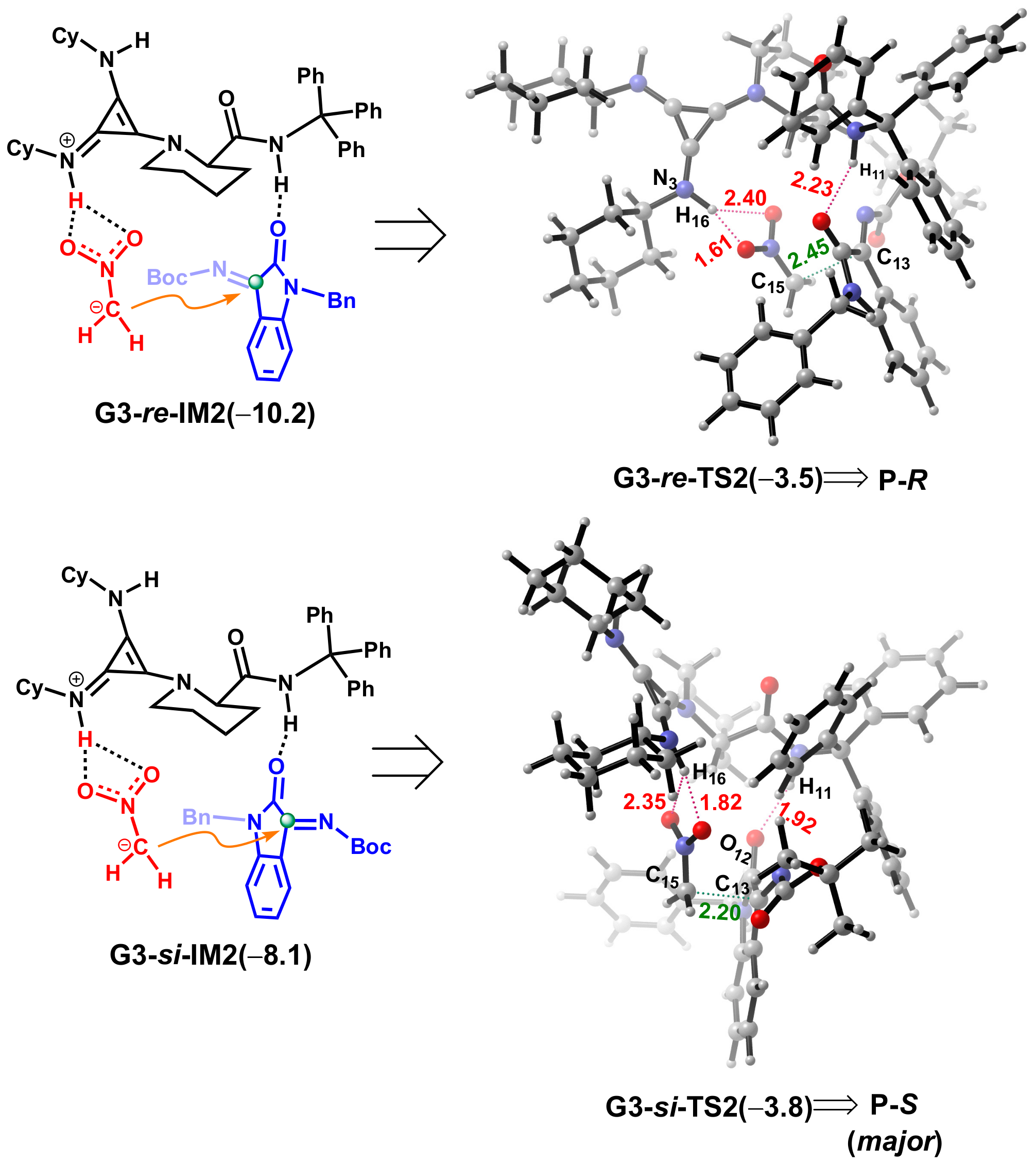
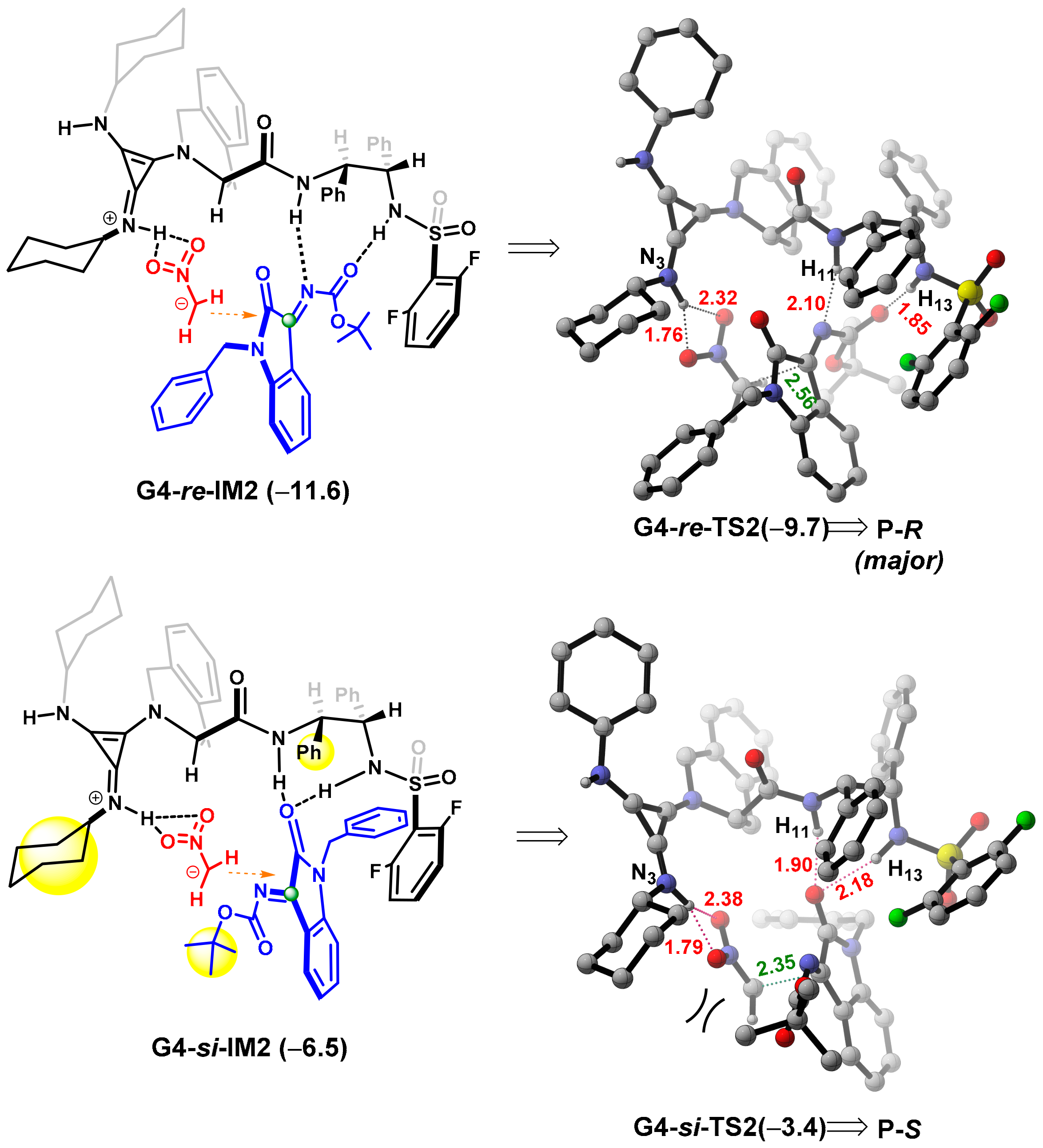
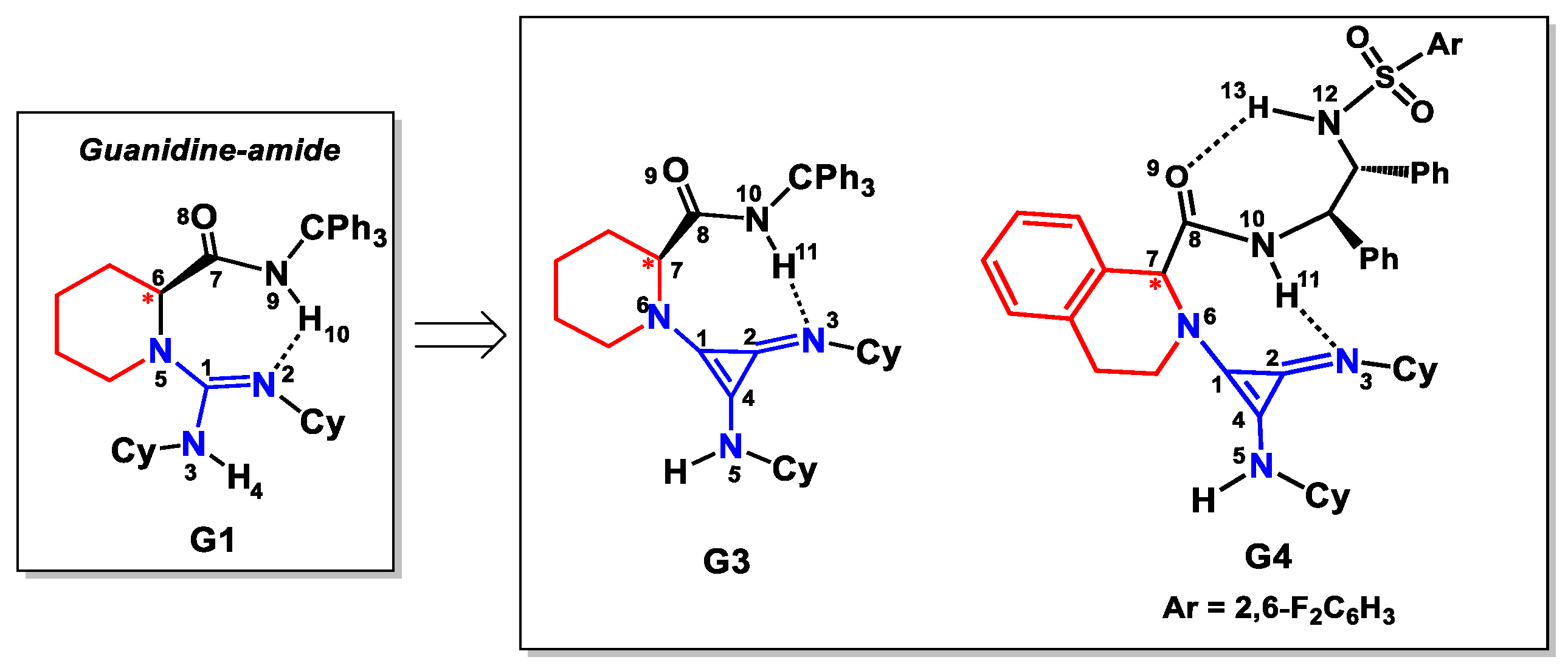
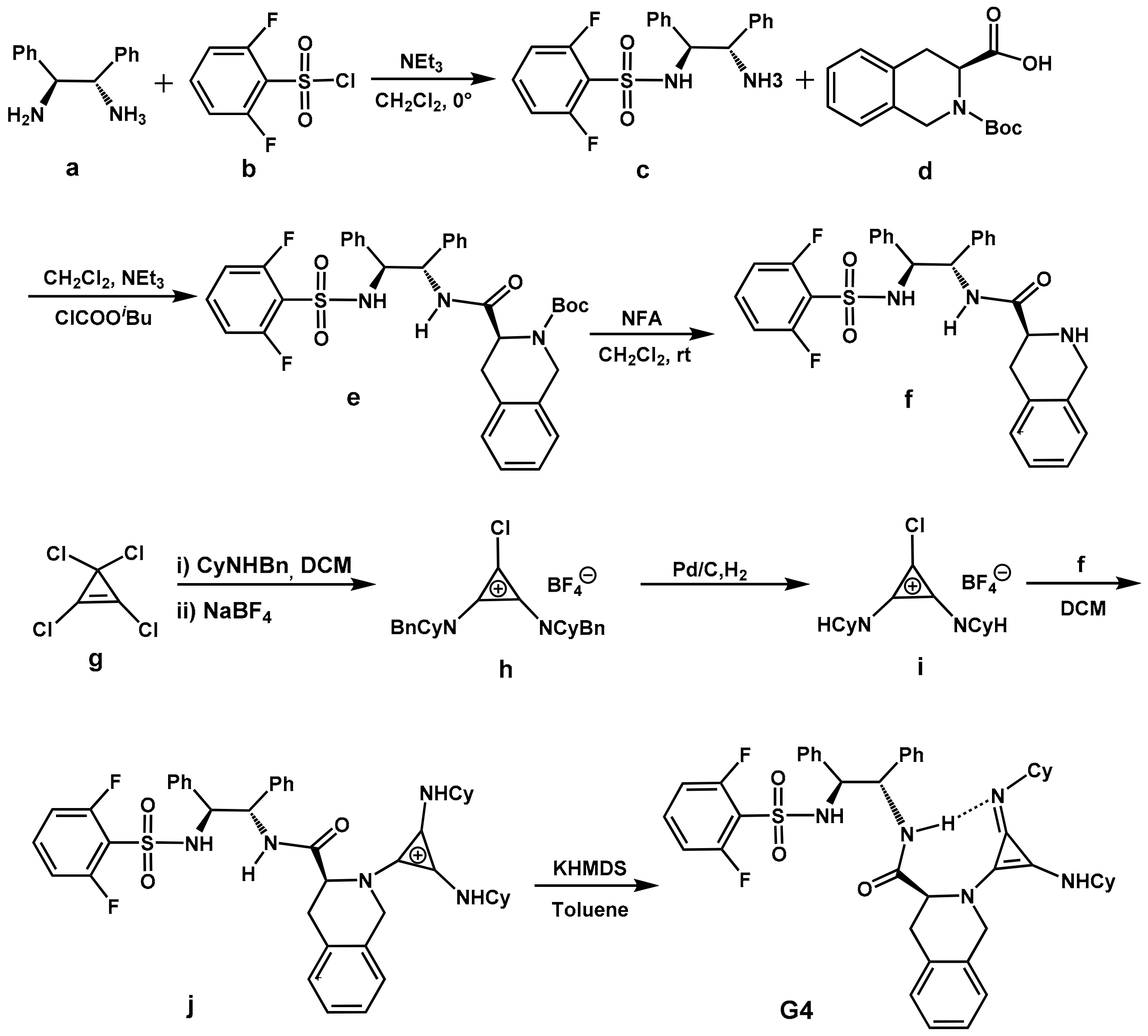
2.3. Design of New Catalysts
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Marques-Lopez, E.; Merino, P.; Tejero, T.; Herrera, R.P. Catalytic Enantioselective Aza-Henry Reactions. Eur. J. Org. Chem. 2009, 15, 2401–2420. [Google Scholar] [CrossRef]
- Nef, J.U. Ueber die Constitution der Salze der Nitroparaffine. Justus Liebigs Ann. Chem. 1894, 280, 263–291. [Google Scholar] [CrossRef]
- Ballini, R.; Petrini, M. Recent Synthetic Developments in the Nitro to Carbonyl Conversion (Nef Reaction). Tetrahedron 2004, 60, 1017–1047. [Google Scholar] [CrossRef]
- Krawczyk, H.; Albrecht, L.; Wojciechowski, J.; Wolf, W.M. Spontaneous Nef Reaction of 3-Aryl-2-(Diethoxyphosphoryl)-4-Nitroalkanoic Acids. Tetrahedron 2006, 62, 9135–9145. [Google Scholar] [CrossRef]
- Bhat, C.; Tilve, S.G. Henry-Nef Reaction: A Practical and Versatile Chiral Pool Route to 2-Substituted Pyrrolidine and Piperidine Alkaloids. Tetrahedron 2013, 69, 6129–6143. [Google Scholar] [CrossRef]
- Ju, M.; Guan, W.Y.; Schomaker, J.M.; Harper, K.C. Sequential Reduction of Nitroalkanes Mediated by CS2 and Amidine/Guanidine Bases: A Controllable Nef Reaction. Org. Lett. 2019, 21, 8893–8898. [Google Scholar] [CrossRef]
- Lloyd, D.H.; Nichols, D.E. Nickel Boride/Hydrazine Hydrate: Reduction of Aromatic and Aliphatic Nitro Compounds. Synthesis of 4-(Benzyloxy)Indole and α-Alkyltryptamines. J. Org. Chem. 1986, 51, 4294–4295. [Google Scholar] [CrossRef]
- Lucet, D.; Gall, T.L.; Mioskowski, C. The Chemistry of Vicinal Diamines. Angew. Chem. Int. Ed. 1998, 37, 2581–2627. [Google Scholar] [CrossRef]
- Kotti, S.R.S.S.; Timmons, C.; Li, G.G. Vicinal Diamino Functionalities as Privileged Structural Elements in Biologically Active Compounds and Exploitation of Their Synthetic Chemistry. Chem. Biol. Drug. Des. 2006, 67, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Kizirian, J.C. Chiral Tertiary Diamines in Asymmetric Synthesis. Chem. Rev. 2008, 108, 140–205. [Google Scholar] [CrossRef]
- Yamada, K.; Moll, G.; Shibasaki, M. The First Enantioselective and Diastereoselective Catalytic Nitro-Mannich Reaction: A New Entry to Chiral Vicinal Diamines. Synlett 2001, SI, 980–982. [Google Scholar] [CrossRef]
- Palomo, C.; Oiarbide, M.; Halder, R.; Laso, A.; Lópze, R. Enantioselective Aza-Henry Reactions Assisted by Zn(II) and N-Methylephedrine. Angew. Chem. Int. Ed. 2006, 45, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Dudek, A.; Mlynarski, J. Iron-Catalyzed Asymmetric Nitro-Mannich Reaction. J. Org. Chem. 2017, 82, 11218–11224. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Harwood, S.J.; Gröger, H.; Shibasaki, M. The First Catalytic Asymmetric Nitro Mannich-Type Reaction Promoted by a New Heterobimetallic Complex. Angew. Chem. Int. Ed. 1999, 38, 3504–3506. [Google Scholar] [CrossRef]
- Zhang, S.; Li, Y.N.; Xu, Y.G.; Wang, Z.Y. Recent Progress in Copper Catalyzed Asymmetric Henry Reaction. Chin. Chem. Lett. 2018, 29, 873–883. [Google Scholar] [CrossRef]
- Okino, T.; Nakamura, S.; Furukawa, T.; Takemoto, Y. Enantioselective Aza-Henry Reaction Catalyzed by a Bifunctional Organocatalyst. Org. Lett. 2004, 6, 625–627. [Google Scholar] [CrossRef] [PubMed]
- Ting, A.; Schaus, S.E. Organocatalytic Asymmetric Mannich Reactions: New Methodology, Catalyst Design, and Synthetic Applications. Eur. J. Org. Chem. 2007, 2007, 5797–5815. [Google Scholar] [CrossRef]
- Belding, L.; Taimoory, S.M.; Dudding, T. Mirroring Enzymes: The Role of Hydrogen Bonding in an Asymmetric Organocatalyzed Aza-Henry Reaction-a DFT Study. ACS Catal. 2015, 5, 343–349. [Google Scholar] [CrossRef]
- Notz, W.; Tanaka, F.; Watanabe, S.-i.; Chowdari, N.S.; Turner, J.M.; Thayumanavan, R.; Barbas, C.F. The Direct Organocatalytic Asymmetric Mannich Reaction: Unmodified Aldehydes as Nucleophiles. J. Org. Chem. 2003, 68, 9624–9634. [Google Scholar] [CrossRef]
- Ochi, M.; Kawasaki, K.; Kataoka, H.; Uchio, Y.; Nishi, H. AG-041R, a Gastrin/CCK-B Antagonist, Stimulates Chondrocyte Proliferation and Metabolism in Vitro. Biochem. Biophys. Res. Commun. 2001, 283, 1118–1123. [Google Scholar] [CrossRef]
- Lin, H.; Danishefsky, S.J. Gelsemine: A Thought-Provoking Target for Total Synthesis. Angew. Chem. Int. Ed. 2003, 42, 36–51. [Google Scholar] [CrossRef]
- Oost, T.; Backfisch, G.; Bhowmik, S.; van Gaalen, M.M.; Geneste, H.; Hornberger, W.; Lubisch, W.; Netz, A.; Unger, L.; Wernet, W. Potent and Selective Oxindole-based Vasopressin 1b Receptor Antagonists with Improved Pharmacokinetic Properties. Bioorg. Med. Chem. Lett. 2011, 21, 3828–3831. [Google Scholar] [CrossRef] [PubMed]
- Berlinck, R.G.S.; Romminger, S. The Chemistry and Biology of Guanidine Natural Products. Nat. Prod. Rep. 2016, 33, 456–490. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Matsumura, E.; Masu, H. Bis(imidazolidine)pyridine-NiCl2 Catalyst for Nitro-Mannich Reaction of Isatin-Derived N-Boc Ketimines: Asymmetric Synthesis of Chiral 3-Substituted 3-Amino-2-oxindoles. Org. Lett. 2014, 16, 2768–2771. [Google Scholar] [CrossRef]
- Holmquist, M.; Blay, G.; Pedro, J.R. Highly Enantioselective Aza-Henry Reaction with Isatin N-Boc Ketimines. Chem. Commun. 2014, 50, 9309–9312. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Liu, X.H.; Wang, L.W.; Wang, J.; Feng, X.M. Highly Enantioselective Aza-Henry Reaction of Ketoimines Catalyzed by Chiral N, N′-Dioxide-Copper(I) Complexes. Org. Lett. 2008, 10, 5305–5308. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, L.; Fini, F.; Herrera, R.P.; Ricci, A.; Sgarzani, V. Enantioselective Aza-Henry Reaction Using Cinchona Organocatalysts. Tetrahedron 2006, 62, 375–380. [Google Scholar] [CrossRef]
- Bode, C.M.; Ting, A.; Schaus, S.E. A General Organic Catalyst for Asymmetric Addition of Stabilized Nucleophiles to Acyl Imines. Tetrahedron 2006, 62, 11499–11505. [Google Scholar] [CrossRef]
- Kumar, A.; Kuar, J.; Chimni, S.S.; Jassal, A.K. Organocatalytic Enantioselective Aza-Henry Reaction of Ketimines Derived from Isatins: Access to Optically Active 3-Amino-2-Oxindoles. RSC Adv. 2014, 4, 24816–24819. [Google Scholar] [CrossRef]
- Li, P.; Sun, D.W.; Jiang, M.; Liu, J.T. Asymmetric Aza-Henry Reaction of Fluoromethylated Imines Catalyzed by Cinchona-Derived Bifunctional Thiourea. Tetrahedron 2019, 75, 603–607. [Google Scholar] [CrossRef]
- Rampalakos, C.; Wulff, W.D. A Novel Bis-Thiourea Organocatalyst for the Asymmetric Aza-Henry Reaction. Adv. Synth. Catal. 2008, 350, 1785–1790. [Google Scholar] [CrossRef] [PubMed]
- Hajra, S.; Jana, B. Quinine-Based Trifunctional Organocatalyst for Tandem Aza-Henry Reaction-Cyclization: Asymmetric Synthesis of Spiroxindole-Pyrrolidine/Piperidines. Org. Lett. 2017, 19, 4778–4781. [Google Scholar] [CrossRef] [PubMed]
- Nunez, M.G.; Farley, A.J.; Dixon, D.J. Bifunctional Iminophosphorane Organocatalysts for Enantioselective Synthesis: Application to the Ketimine Nitro-Mannich Reaction. J. Am. Chem. Soc. 2013, 135, 16348–16351. [Google Scholar] [CrossRef]
- Xue, Y.S.; Wang, Y.H.; Cao, Z.Y.; Zhou, J.; Chen, Z.X. Computational Insight into the Cooperative Role of Non-covalent Interactions in the Aza-Henry Reaction Catalyzed by Quinine Derivatives: Mechanism and Enantioselectivity. Org. Biomol. Chem. 2016, 14, 9588–9597. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.D.; Liu, Y.; Liu, Y.X.; Wei, Z.L.; Cao, J.G.; Liang, D.P.; Lin, Y.J.; Duan, H.F. L-tert-Leucine Derived Urea-Ammonium Salts: Efficient Bifunctional Phase Transfer Catalysts for Highly Diastereo- and Enantioselective Aza-Henry Reaction of Isatin-derived N-Boc Ketimines with α-Aryl Nitromethanes. Tetrahedron 2019, 75, 2883–2892. [Google Scholar] [CrossRef]
- Sohtome, Y.; Nagasawa, K. Dynamic Asymmetric Organocatalysis: Cooperative Effects of Weak Interactions and Conformational Flexibility in Asymmetric Organocatalysts. Chem. Commun. 2012, 48, 7777–7789. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Loh, W.T.; Zhang, Y.; Chen, T.; Ma, T.; Liu, H.; Wang, J.; Tan, C.H. Chiral Guanidinium Salt Catalyzed Enantioselective Phospha-Mannich Reactions. Angew. Chem. Int. Ed. 2009, 48, 7387–7390. [Google Scholar] [CrossRef]
- Corey, E.J.; Grogan, M.J. Enantioselective Synthesis of α-Amino Nitriles from N-Benzhydryl Imines and HCN with a Chiral Bicyclic Guanidine as Catalyst. Org. Lett. 1999, 1, 157–160. [Google Scholar] [CrossRef]
- Cho, B.; Wong, M.W. Unconventional Bifunctional Lewis-Brønsted Acid Activation Mode in Bicyclic Guanidine-Catalyzed Conjugate Addition Reactions. Molecules 2015, 20, 15108–15121. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.S.; Jiang, D.F.; Jiang, H.; Kee, C.W.; Hirao, H.; Nishimura, T.; Wong, M.W.; Tan, C.H. Mechanistic Insights into Bicyclic Guanidine-Catalyzed Reactions from Microscopic and Macroscopic Perspectives. J. Org. Chem. 2015, 80, 5745–5752. [Google Scholar] [CrossRef]
- Li, J.; Jiang, W.Y.; Han, K.L.; He, G.Z.; Li, C. Density Functional Study on the Mechanism of Bicyclic Guanidine-Catalyzed Strecker Reaction. J. Org. Chem. 2003, 68, 8786–8789. [Google Scholar] [CrossRef]
- Kee, C.W.; Peh, K.Q.E.; Wong, M.W. Coupling Reactions of Alkynyl Indoles and CO2 by Bicyclic Guanidine: Origin of Catalytic Activity? Chem. Asian J. 2017, 12, 1780–1789. [Google Scholar] [CrossRef] [PubMed]
- Cho, B.; Tan, C.H.; Wong, M.W. Sequential Catalytic Role of Bifunctional Bicyclic Guanidine in Asymmetric Phospha-Michael Reaction. Org. Biomol. Chem. 2011, 9, 4550–4557. [Google Scholar] [CrossRef]
- Fang, B.; Liu, X.H.; Zhao, J.N.; Tang, Y.; Lin, L.L.; Feng, X.M. Chiral Bifunctional Guanidine-catalyzed Enantioselective Aza-Henry Reaction of Isatin-derived Ketimines. J. Org. Chem. 2015, 80, 3332–3338. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.D.; Liu, X.H.; Feng, X.M. Chiral Organobases: Properties and Applications in Asymmetric Catalysis. Chin. Chem. Lett. 2018, 29, 1201–1208. [Google Scholar] [CrossRef]
- Dong, S.X.; Feng, X.M.; Liu, X.H. Chiral Guanidines and Their Derivatives in Asymmetric Synthesis. Chem. Soc. Rev. 2018, 47, 8525–8540. [Google Scholar] [CrossRef]
- Chou, H.; Leow, D.; Tan, C.H. Recent Advances in Chiral Guanidine-Catalyzed Enantioselective Reactions. Chem. Asian J. 2019, 14, 3803–3822. [Google Scholar] [CrossRef]
- Liu, X.H.; Lin, L.L.; Feng, X.M. Amide-based Bifunctional Organocatalysts in Asymmetric Reactions. Chem. Commun. 2009, 41, 6145–6158. [Google Scholar] [CrossRef]
- Li, J.; Su, Z.S.; Wang, J.M.; Hu, C.W. Mechanistic Investigations on Asymmetric N-H Insertion of Amines Catalyzed by Palladium-chiral Guanidine Complex. J. Catal. 2018, 364, 426–436. [Google Scholar] [CrossRef]
- Li, J.; Meng, X.X.; Hu, C.W.; Su, Z.S. Cooperative Catalysis of Chiral Guanidine and Rh2(OAc)4 in Asymmetric O-H Insertion of Carboxylic Acid: A Theoretical Investigation. J. Org. Chem. 2019, 84, 15020–15031. [Google Scholar] [CrossRef]
- Emamian, S.; Lv, T.; Kruse, H.; Emamian, H. Exploring Nature and Predicting Strength of Hydrogen Bonds: A Correlation Analysis between Atoms-in-Molecules Descriptors, Binding Energies, and Energy Components of Symmetry-Adapted Perturbation Theory. J. Comput. Chem. 2019, 40, 2868–2881. [Google Scholar] [CrossRef]
- Manzetti, S.; Lv, T. The Geometry and Electronic Structure of Aristolochic Acid: Possible Implications for a Frozen Resonance. J. Phys. Org. Chem. 2013, 26, 473–483. [Google Scholar] [CrossRef]
- Schneebeli, S.T.; Hall, M.L.; Breslow, R.; Friesner, R. Quantitative DFT Modeling of the Enantiomeric Excess for Dioxirane-catalyzed Epoxidations. J. Am. Chem. Soc. 2009, 131, 3965–3973. [Google Scholar] [CrossRef] [PubMed]
- Fernández, I.; Bickelhaupt, F.M. The Activation Strain Model and Molecular Orbital Theory: Understanding and Designing Chemical Reactions. Chem. Soc. Rev. 2014, 43, 4953–4967. [Google Scholar] [CrossRef]
- Bickelhaupt, F.M.; Houk, K.N. Analyzing Reaction Rates with the Distortion/Interaction-Activation Strain Model. Angew. Chem. Int. Ed. 2017, 56, 10070–10086. [Google Scholar] [CrossRef]
- Kozuch, S.; Shaik, S. A Combined Kinetic-Quantum Mechanical Model for Assessment of Catalytic Cycles: Application to Cross-Coupling and Heck Reactions. J. Am. Chem. Soc. 2006, 128, 3355–3365. [Google Scholar] [CrossRef] [PubMed]
- Bandar, J.S.; Lambert, T.H. Enantioselective Brønsted Base Catalysis with Chiral Cyclopropenimines. J. Am. Chem. Soc. 2012, 134, 5552–5555. [Google Scholar] [CrossRef]
- Guest, M.; Sueur, R.L.; Pilkington, M.; Dudding, T. Development of an Unsymmetrical Cyclopropenimine-Guanidine Platform for Accessing Strongly Basic Proton Sponges and Boron-Difluoride Diaminonaphthalene Fluorophores. Chem. Eur. J. 2020, 26, 8608–8620. [Google Scholar] [CrossRef]
- Ruan, S.; Lin, X.B.; Xie, L.H.; Lin, L.L.; Feng, X.M.; Liu, X.H. Asymmetric Synthesis of 3-Aminodihydrocoumarins via the Chiral Guanidine Catalyzed Cascade Reaction of Azlactones. Org. Chem. Front. 2018, 5, 32–35. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Eectron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C.; Schlegel, H.B. An Improved Algorithm for Reaction-path Following. J. Chem. Phys. 1989, 90, 2154–2161. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular Interactions from a Natural Bond Orbital, Donor-acceptor Viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Carlosa, L.R.; Loroa, H.; Lagob, A.F.; Dávalos, J.Z. Gas-phase Proton Afnity and Basicity of Hydroxybenzophenones. Chem. Phys. Lett. 2018, 713, 132–136. [Google Scholar] [CrossRef]
- Wolters, L.P.; Bickelhaupt, F.M. The Activation Strain Model and Molecular Orbital Theory. WIREs Comput. Mol. Sci. 2015, 5, 324–343. [Google Scholar] [CrossRef]
- Ess, D.H.; Houk, K.N. Distortion/Interaction Energy Control of 1,3-Dipolar Cycloaddition Reactivity. J. Am. Chem. Soc. 2007, 129, 10646–10647. [Google Scholar] [CrossRef]
- Hopffgarten, M.V.; Frenking, G. Energy Decomposition Analysis. WIREs Comput. Mol. Sci. 2012, 2, 43–62. [Google Scholar] [CrossRef]
- Te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Guerra, C.F.; Gisbergen, S.J.A.V.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Beddall, P.M. Virial Field Relationship for Molecular Charge Distributions and the Spatial Partitioning of Molecular Properties. J. Chem. Phys. 1972, 56, 3320–3329. [Google Scholar] [CrossRef]
- Bianchi, R.; Gervasio, G.; Marabello, D. Experimental Electron Density Analysis of Mn2(CO)10: Metal-Metal and Metal-Ligand Bond Characterization. Inorg. Chem. 2000, 39, 2360–2366. [Google Scholar] [CrossRef] [PubMed]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Path | TDI | TDTS | TOF (s−1) | Product |
|---|---|---|---|---|
| I-re | G1-1a-I-re-IM2 | G1-1a-I-re-TS2 | 2.06 × 1010 | P-R (major) |
| I-si | G1-1a-I-si-IM2 | G1-1a-I-si-TS2 | 2.98 × 108 | P-S |
| II-re | G1-1a-II-re-IM2 | G1-1a-II-re-TS2 | 1.24 × 1010 | P-R |
| II-si | G1-1a-II-si-IM2 | G1-1a-II-si-TS2 | 1.03 × 1010 | P-S |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, J.; Tang, D.; Hu, C.; Su, Z. Guanidine–Amide-Catalyzed Aza-Henry Reaction of Isatin-Derived Ketimines: Origin of Selectivity and New Catalyst Design. Molecules 2021, 26, 1965. https://doi.org/10.3390/molecules26071965
He J, Tang D, Hu C, Su Z. Guanidine–Amide-Catalyzed Aza-Henry Reaction of Isatin-Derived Ketimines: Origin of Selectivity and New Catalyst Design. Molecules. 2021; 26(7):1965. https://doi.org/10.3390/molecules26071965
Chicago/Turabian StyleHe, Jiajia, Dianyong Tang, Changwei Hu, and Zhishan Su. 2021. "Guanidine–Amide-Catalyzed Aza-Henry Reaction of Isatin-Derived Ketimines: Origin of Selectivity and New Catalyst Design" Molecules 26, no. 7: 1965. https://doi.org/10.3390/molecules26071965
APA StyleHe, J., Tang, D., Hu, C., & Su, Z. (2021). Guanidine–Amide-Catalyzed Aza-Henry Reaction of Isatin-Derived Ketimines: Origin of Selectivity and New Catalyst Design. Molecules, 26(7), 1965. https://doi.org/10.3390/molecules26071965