
Bis-Cyclometalated Indazole and Benzimidazole Chiral-at-Iridium Complexes: Synthesis and Asymmetric Catalysis


Abstract
1. Introduction
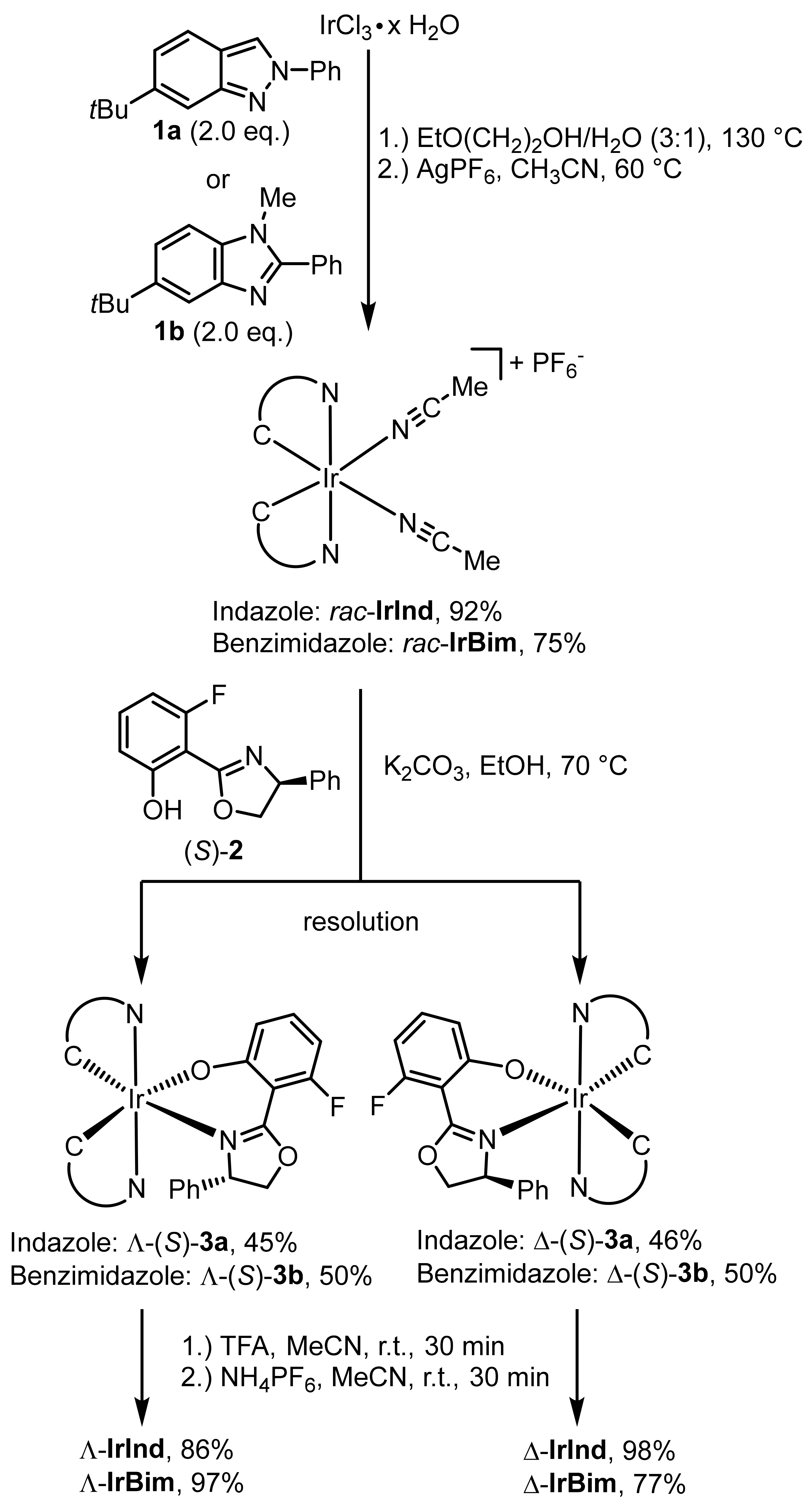
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Ritleng, V.; Sirlin, C.; Pfeffer, M. Ru-, Rh-, and Pd-Catalyzed C−C Bond Formation Involving C−H Activation and Addition on Unsaturated Substrates: Reactions and Mechanistic Aspects. Chem. Rev. 2002, 102, 1731–1770. [Google Scholar] [CrossRef]
- Albrecht, M. Cyclometalation Using d-Block Transition Metals: Fundamental Aspects and Recent Trends. Chem. Rev. 2010, 110, 576–623. [Google Scholar] [CrossRef] [PubMed]
- Djukic, J.-P.; Sortais, J.-B.; Barloy, L.; Pfeffer, M. Cycloruthenated Compounds—Synthesis and Applications. Eur. J. Inorg. Chem. 2009, 817–853. [Google Scholar] [CrossRef]
- Gaiddon, C.; Pfeffer, M. The Fate of Cycloruthenated Compounds: From C-H Activation to Innovative Anticancer Therapy. Eur. J. Inorg. Chem. 2017, 1639–1654. [Google Scholar] [CrossRef]
- Zhang, L.; Meggers, E. Steering Asymmetric Lewis Acid Catalysis Exclusively with Octahedral Metal-Centered Chirality. Acc. Chem. Res. 2017, 50, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Meggers, E. Asymmetric Photocatalysis with Bis-cyclometalated Rhodium Complexes. Acc. Chem. Res. 2019, 52, 833–847. [Google Scholar] [CrossRef] [PubMed]
- Huo, H.; Fu, C.; Harms, K.; Meggers, E. Asymmetric Catalysis with Substitutionally Labile yet Stereochemically Stable Chiral-at-Metal Iridium(III) Complex. J. Am. Chem. Soc. 2014, 136, 2990–2993. [Google Scholar] [CrossRef] [PubMed]
- Huo, H.; Shen, X.; Wang, C.; Zhang, L.; Röse, P.; Chen, L.-A.; Harms, K.; Marsch, M.; Hilt, G.; Meggers, E. Asymmetric photoredox transition-metal catalysis activated by visible light. Nature 2014, 515, 100–103. [Google Scholar] [CrossRef]
- Pierre, J.-L. Enantioselective creation of helical chirality in octahedral (OC-6) complexes. Recent advances. Coord. Chem. Rev. 1998, 178–180, 1183–1192. [Google Scholar] [CrossRef]
- Brunner, H. Optically Active Organometallic Compounds of Transition Elements with Chiral Metal Atoms. Angew. Chem. Int. Ed. 1999, 38, 1194–1208. [Google Scholar] [CrossRef]
- Knof, U.; Von Zelewsky, A. Predetermined Chirality at Metal Centers. Angew. Chem. Int. Ed. 1999, 38, 302–322. [Google Scholar] [CrossRef]
- Knight, P.D.; Scott, P. Predetermination of chirality at octahedral centres with tetradentate ligands: Prospects for enantioselective catalysis. Coord. Chem. Rev. 2003, 242, 125–143. [Google Scholar] [CrossRef]
- Ganter, C. Chiral organometallic half-sandwich complexes with defined metal configuration. Chem. Soc. Rev. 2003, 32, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Fontecave, M.; Hamelin, O.; Ménage, S. Chiral-at-Metal Complexes in Asymmetric Catalysis. Top. Organomet. Chem. 2005, 15, 271–288. [Google Scholar]
- Meggers, E. Asymmetric Synthesis of Octahedral Coordination Complexes. Eur. J. Inorg. Chem. 2011, 2911–2926. [Google Scholar] [CrossRef]
- Bauer, E.B. Chiral-at-metal complexes and their catalytic applications in organic synthesis. Chem. Soc. Rev. 2012, 41, 3153–3167. [Google Scholar] [CrossRef]
- Constable, E.C. Stereogenic metal centres—From Werner to supramolecular chemistry. Chem. Soc. Rev. 2013, 42, 1637–1651. [Google Scholar] [CrossRef] [PubMed]
- Von Zelewsky, A. Stereochemistry of Coordination Compounds: From Alfred Werner to the 21st Century. Chimia 2014, 68, 297–298. [Google Scholar] [CrossRef]
- Gong, L.; Chen, L.-A.; Meggers, E. Asymmetric Catalysis Mediated by the Ligand Sphere of Octahedral Chiral-at-Metal Complexes. Angew. Chem. Int. Ed. 2014, 53, 10868–10874. [Google Scholar] [CrossRef]
- Zhang, L.; Meggers, E. Stereogenic-Only-at-Metal Asymmetric Catalysts. Chem. Asian J. 2017, 12, 2335–2342. [Google Scholar] [CrossRef] [PubMed]
- Cruchter, T.; Larionov, V.A. Asymmetric catalysis with octahedral stereogenic-at-metal complexes featuring chiral ligands. Coord. Chem. Rev. 2018, 376, 95–113. [Google Scholar] [CrossRef]
- Coe, B.J.; Glenwright, S.J. Trans-effects in octahedral transition metal complexes. Coord. Chem. Rev. 2000, 203, 5–80. [Google Scholar] [CrossRef]
- See also: Li, Y.; Lei, M.; Yuan, W.; Meggers, E.; Gong, L. An N-heterocyclic carbene iridium catalyst with metal-centered chirality for enantioselective transfer hydrogenation of imines. Chem. Commun. 2017, 53, 8089–8092. [Google Scholar]
- Tamayo, A.B.; Garon, S.; Sajoto, T.; Djurovich, P.I.; Tsyba, I.M.; Bau, R.; Thompson, M.E. Cationic Bis-cyclometalated Iridium(III) Diimine Complexes and Their Use in Efficient Blue, Green, and Red Electroluminescent Devices. Inorg. Chem. 2005, 44, 8723–8732. [Google Scholar] [CrossRef] [PubMed]
- Shavaleev, N.M.; Scopelliti, R.; Grätzel, M.; Nazeeruddin, M.K. Phosphorescent cationic iridium(III) complexes with cyclometalating 1H-indazole and 2H-[1,2,3]-triazole ligands. Inorg. Chim. Acta 2012, 388, 84–87. [Google Scholar] [CrossRef]
- Zhao, K.-Y.; Shan, G.-G.; Fu, Q.; Su, Z.-M. Tuning Emission of AIE-Active Organometallic Ir(III) Complexes by Simple Modulation of Strength of Donor/Acceptor on Ancillary Ligands. Organometallics 2016, 35, 3996–4001. [Google Scholar] [CrossRef]
- Niu, Z.-G.; Han, H.-B.; Li, M.; Zhao, Z.; Chen, G.-Y.; Zheng, Y.-X.; Li, G.-N.; Zuo, J.-L. Tunable Emission Color of Iridium(III) Complexes with Phenylpyrazole Derivatives as the Main Ligands for Organic Light-Emitting Diodes. Organometallics 2018, 37, 3154–3164. [Google Scholar] [CrossRef]
- Huang, W.-S.; Lin, J.T.; Chien, C.-H.; Tao, Y.-T.; Sun, S.-S.; Wen, Y.-S. Highly Phosphorescent Bis-Cyclometalated Iridium Complexes Containing Benzoimidazole-Based Ligands. Chem. Mat. 2004, 16, 2480–2488. [Google Scholar] [CrossRef]
- Velusamy, M.; Thomas, K.R.J.; Chen, C.-H.; Lin, J.T.; Wen, Y.S.; Hsieh, W.-T.; Lai, C.-H.; Chou, P.-T. Synthesis, structure and electroluminescent properties of cyclometalated iridium complexes possessing sterically hindered ligands. Dalton Trans. 2007, 3025–3034. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhang, G.; Shih, H.-H.; Jiang, X.; Sun, P.; Pan, Y.; Cheng, C.-H. High-efficient phosphorescent iridium(III) complexes with benzimidazole ligand for organic light-emitting diodes: Synthesis, electrochemistry and electroluminescent properties. J. Organomet. Chem. 2009, 694, 2415–2420. [Google Scholar] [CrossRef]
- Chen, L.; Ding, J.; Cheng, Y.; Xie, Z.; Wang, L.; Jing, X.; Wang, F. Bipolar Heteroleptic Green Iridium Dendrimers Containing Oligocarbazole and Oxadiazole Dendrons for Bright and Efficient Nondoped Electrophosphorescent Devices. Chem. Asian J. 2011, 6, 1372–1380. [Google Scholar] [CrossRef] [PubMed]
- Shan, G.-G.; Li, H.-B.; Sun, H.-Z.; Cao, H.-T.; Zhu, D.-X.; Su, Z.-M. Enhancing the luminescence properties and stability of cationic iridium(III) complexes based on phenylbenzoimidazole ligand: A combined experimental and theoretical study. Dalton Trans. 2013, 42, 11056–11065. [Google Scholar] [CrossRef]
- Meggers, E. Chiral Auxiliaries as Emerging Tools for the Asymmetric Synthesis of Octahedral Metal Complexes. Chem. Eur. J. 2010, 16, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Wenzel, M.; Meggers, E. Chiral-Auxiliary-Mediated Asymmetric Synthesis of Ruthenium Polypyridyl Complexes. Acc. Chem. Res. 2013, 46, 2635–2644. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-A.; Xu, W.; Huang, B.; Ma, J.; Wang, L.; Xi, J.; Harms, K.; Gong, L.; Meggers, E. Asymmetric Catalysis with an Inert Chiral-at-Metal Iridium Complex. J. Am. Chem. Soc. 2013, 135, 10598–10601. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhang, X.; Huang, X.; Luo, S.; Meggers, E. Preparation of chiral-at-metal catalysts and their use in asymmetric photoredox chemistry. Nat. Protoc. 2018, 13, 605–632. [Google Scholar] [CrossRef] [PubMed]
- Grell, Y.; Hong, Y.; Huang, X.; Mochizuki, T.; Xie, X.; Harms, K.; Meggers, E. Chiral-at-Rhodium Catalyst Containing Two Different Cyclometalating Ligands. Organometallics 2019, 38, 3948–3954. [Google Scholar] [CrossRef]
- Steinlandt, P.S.; Zuo, W.; Harms, K.; Meggers, E. Bis-Cyclometalated Indazole Chiral-at-Rhodium Catalyst for Asymmetric Photoredox Cyanoalkylations. Chem. Eur. J. 2019, 25, 15333–15340. [Google Scholar] [CrossRef]
- Marchi, E.; Sinisi, R.; Bergamini, G.; Tragni, M.; Monari, M.; Bandini, M.; Ceroni, P. Easy Separation of Δ and Λ Isomers of Highly Luminescent [IrIII]-Cyclometalated Complexes Based on Chiral Phenol-Oxazoline Ancillary Ligands. Chem. Eur. J. 2012, 18, 8765–8773. [Google Scholar] [CrossRef]
- Ma, J.; Shen, X.; Harms, K.; Meggers, E. Expanding the family of bis-cyclometalated chiral-at-metal rhodium(III) catalysts with a benzothiazole derivative. Dalton Trans. 2016, 45, 8320–8323. [Google Scholar] [CrossRef]
- Xu, G.-Q.; Liang, H.; Fang, J.; Jia, Z.-L.; Chen, J.-Q.; Xu, P.-F. Catalytic Enantioselective α-Fluorination of 2-Acyl Imidazoles via Iridium Complexes. Chem. Asian J. 2016, 11, 3355–3358. [Google Scholar] [CrossRef] [PubMed]
- Deng, T.; Thota, G.K.; Li, Y.; Kang, Q. Enantioselective conjugate addition of hydroxylamines to α,β-unsaturated 2-acyl imidazoles catalyzed by a chiral-at-metal Rh(III) complex. Chem. Front. 2017, 4, 573–577. [Google Scholar] [CrossRef]
- Liang, H.; Xu, G.-Q.; Feng, Z.-T.; Wang, Z.-Y.; Xu, P.-F. Dual Catalytic Switchable Divergent Synthesis: An Asymmetric Visible-Light Photocatalytic Approach to Fluorine-Containing γ-Keto Acid Frameworks. J. Org. Chem. 2019, 84, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Huo, H.; Wang, C.; Zhang, B.; Harms, K.; Meggers, E. Octahedral Chiral-at-Metal Iridium Catalysts: Versatile Chiral Lewis Acids for Asymmetric Conjugate Additions. Chem. Eur. J. 2015, 21, 9720–9726. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.A.; Fandrick, K.R.; Song, H.-J. Enantioselective Friedel−Crafts Alkylations of α,β-Unsaturated 2-Acyl Imidazoles Catalyzed by Bis(oxazolinyl)pyridine−Scandium(III) Triflate Complexes. J. Am. Chem. Soc. 2005, 127, 8942–8943. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.A.; Fandrick, K.R. Catalytic Enantioselective Pyrrole Alkylations of α,β-Unsaturated 2-Acyl Imidazoles. Org. Lett. 2006, 8, 2249–2252. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.A.; Fandrick, K.R.; Song, H.-J.; Scheidt, K.A.; Xu, R. Enantioselective Friedel−Crafts Alkylations Catalyzed by Bis(oxazolinyl)pyridine−Scandium(III) Triflate Complexes. J. Am. Chem. Soc. 2007, 129, 10029–10041. [Google Scholar] [CrossRef] [PubMed]
- Boersma, A.J.; Feringa, B.L.; Roelfes, G. Enantioselective Friedel–Crafts Reactions in Water Using a DNA-Based Catalyst. Angew. Chem. Int. Ed. 2009, 48, 3346–3348. [Google Scholar] [CrossRef] [PubMed]
- Shimada, N.; Stewart, C.; Tius, M.A. Asymmetric Nazarov Cyclizations. Tetrahedron 2011, 67, 5851–5870. [Google Scholar] [CrossRef] [PubMed]
- Raja, S.; Nakajima, M.; Rueping, M. Experimental and Computational Study of the Catalytic Asymmetric 4π-Electrocyclization of N-Heterocycles. Angew. Chem. Int. Ed. 2015, 54, 2762–2765. [Google Scholar] [CrossRef]
- Mietke, T.; Cruchter, T.; Larionov, V.A.; Faber, T.; Harms, K.; Meggers, E. Asymmetric Nazarov Cyclizations Catalyzed by Chiral-at-Metal Complexes. Adv. Synth. Catal. 2018, 360, 2093–2100. [Google Scholar] [CrossRef]
- In previous work we found that hexafluoroisopropanol (HFIP) is a very suitable solvent for this reaction. Since it is a weak acid, we speculate that it facilitates the release of the catalyst-bound product and thereby avoids product inhibition. See Ref. [51] for more details.






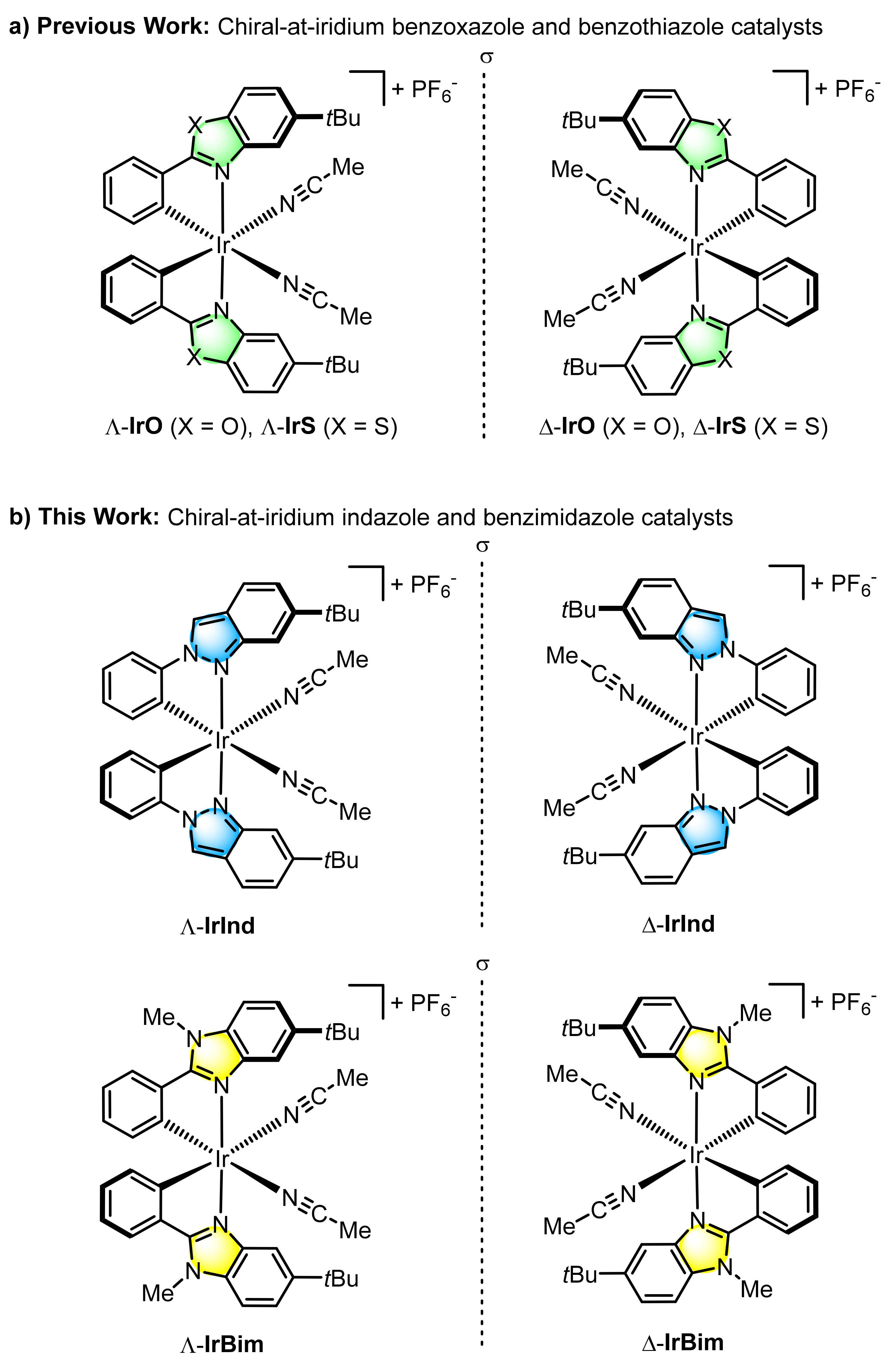{kind=link}
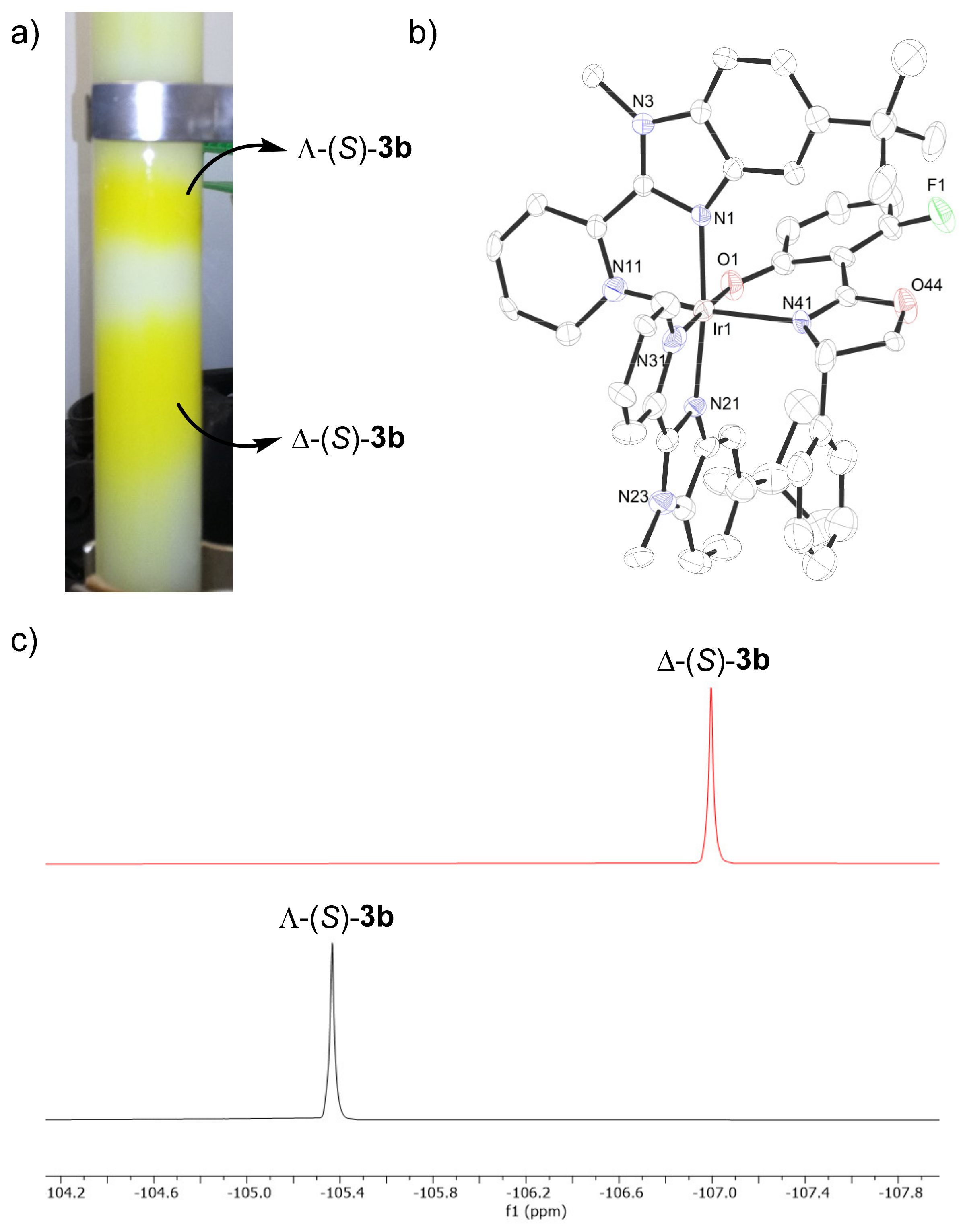{kind=link}
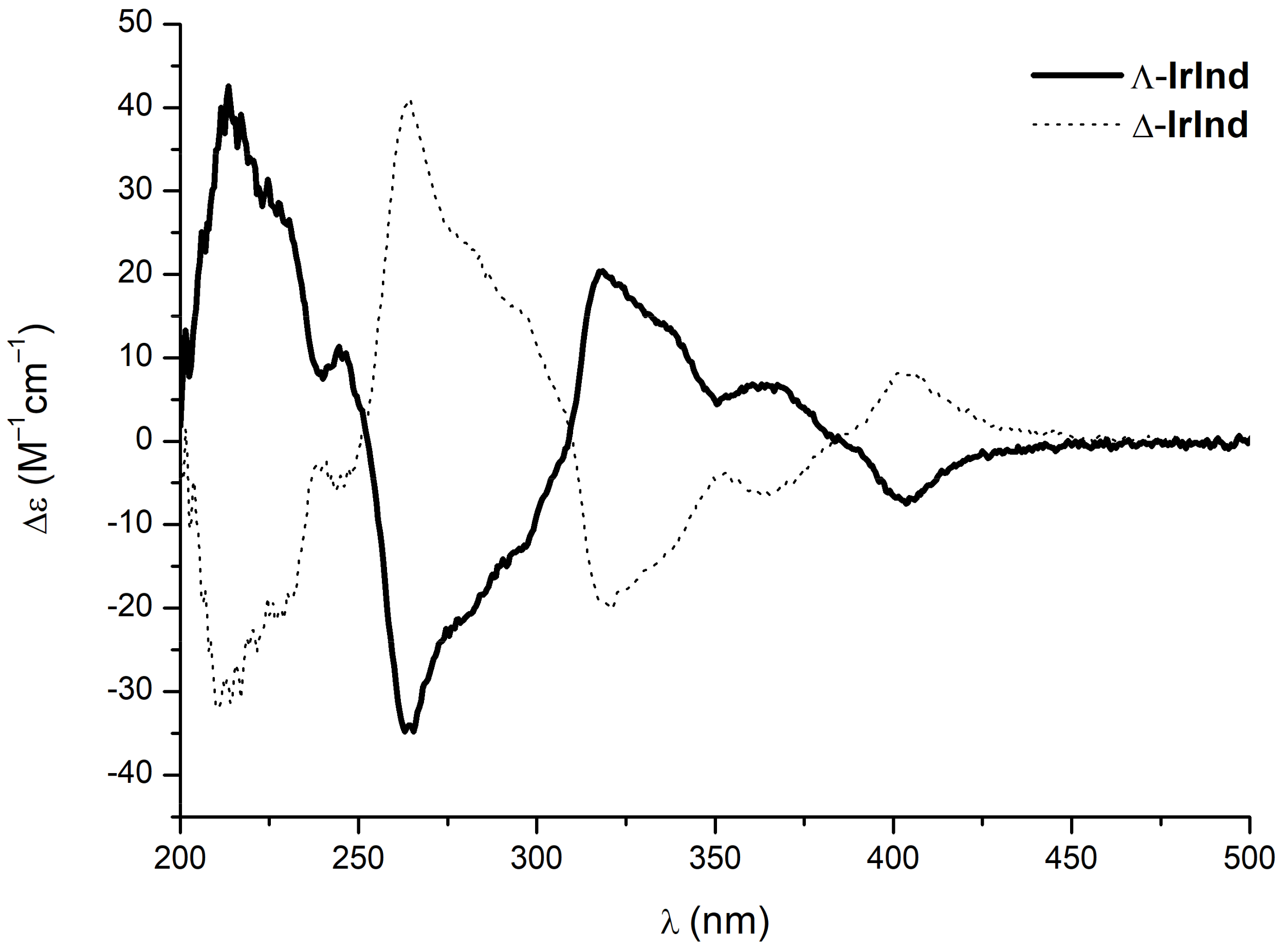{kind=link}
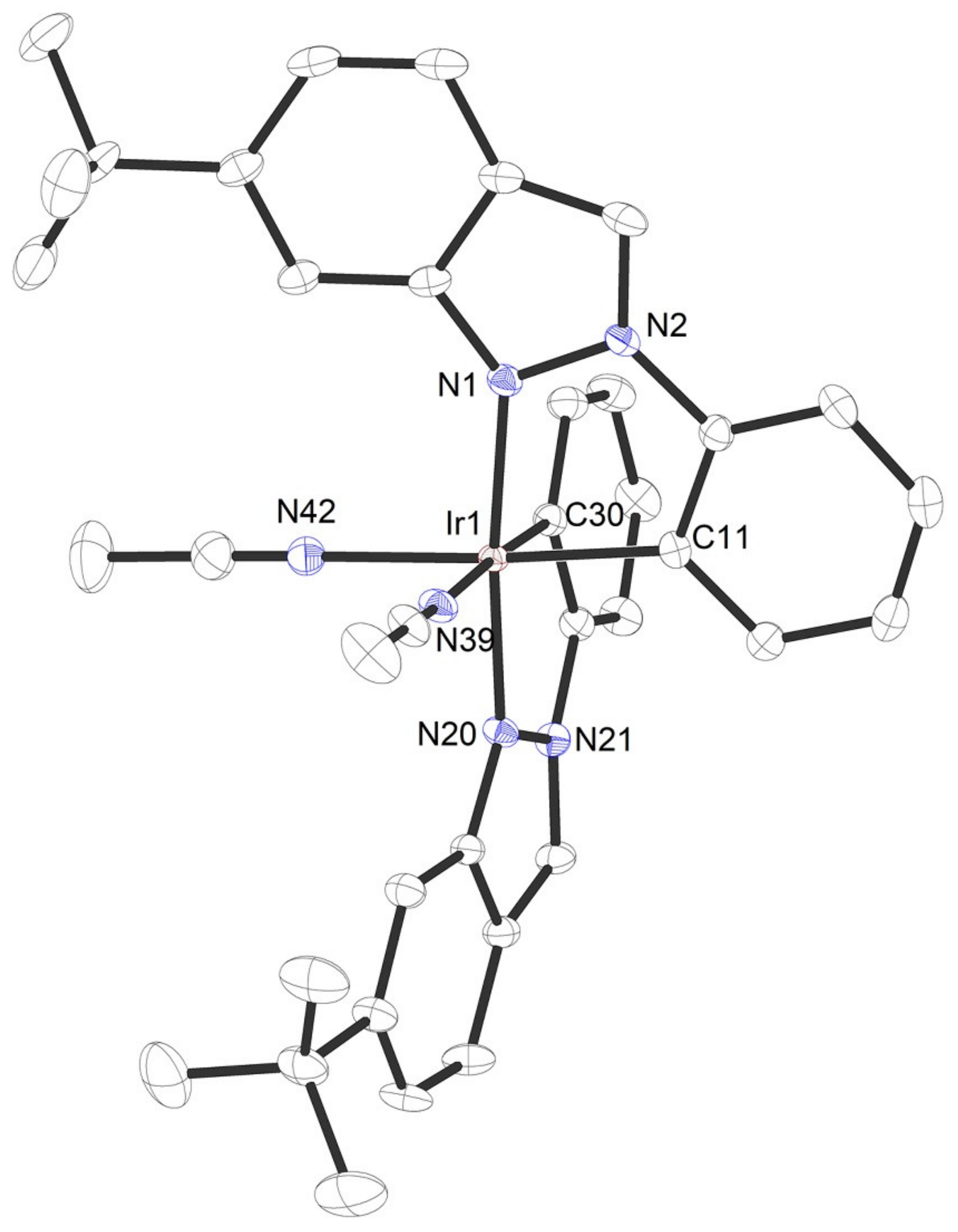{kind=link}
{kind=link}
 | |||||
| Entry | Catalyst | Loading (mol%) | Time (h) | Yieldb | ee c |
|---|---|---|---|---|---|
| 1 | Λ-IrInd | 2.0 | 29 | 84 (S) | 98.5 |
| 2 | Δ-IrBim | 2.0 | 49 | 81 (R) | 98 |
| 3 d | Λ-IrO | 1.0 | 20 | 97 (S) | 96 |
| 4 e | Λ-IrS | 1.0 | 40 | 94 (S) | 99 |
 | |||||
| Entry | Catalyst (mol%) | Time (h) | Yieldb | trans/cis | ee c |
|---|---|---|---|---|---|
| 1 d | Λ-IrS (2.0) | 7 | 75% (1R,2S) | 15:1 | 93 |
| 2 | Λ-IrInd (2.0) | 24 | 46% (1R,2S) | 11:1 | 96 |
| 3 | Δ-IrBim (2.0) | 24 | 73% (1S,2R) | 12.5:1 | 94 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brunen, S.; Grell, Y.; Steinlandt, P.S.; Harms, K.; Meggers, E. Bis-Cyclometalated Indazole and Benzimidazole Chiral-at-Iridium Complexes: Synthesis and Asymmetric Catalysis. Molecules 2021, 26, 1822. https://doi.org/10.3390/molecules26071822
Brunen S, Grell Y, Steinlandt PS, Harms K, Meggers E. Bis-Cyclometalated Indazole and Benzimidazole Chiral-at-Iridium Complexes: Synthesis and Asymmetric Catalysis. Molecules. 2021; 26(7):1822. https://doi.org/10.3390/molecules26071822
Chicago/Turabian StyleBrunen, Sebastian, Yvonne Grell, Philipp S. Steinlandt, Klaus Harms, and Eric Meggers. 2021. "Bis-Cyclometalated Indazole and Benzimidazole Chiral-at-Iridium Complexes: Synthesis and Asymmetric Catalysis" Molecules 26, no. 7: 1822. https://doi.org/10.3390/molecules26071822
APA StyleBrunen, S., Grell, Y., Steinlandt, P. S., Harms, K., & Meggers, E. (2021). Bis-Cyclometalated Indazole and Benzimidazole Chiral-at-Iridium Complexes: Synthesis and Asymmetric Catalysis. Molecules, 26(7), 1822. https://doi.org/10.3390/molecules26071822