
Replacing the Z-phenyl Ring in Tamoxifen® with a para-Connected NCN Pincer-Pt-Cl Grouping by Post-Modification †

 , ,
, ,  ,
,  and
and
Abstract
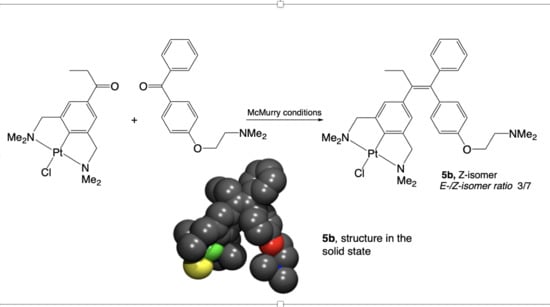
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
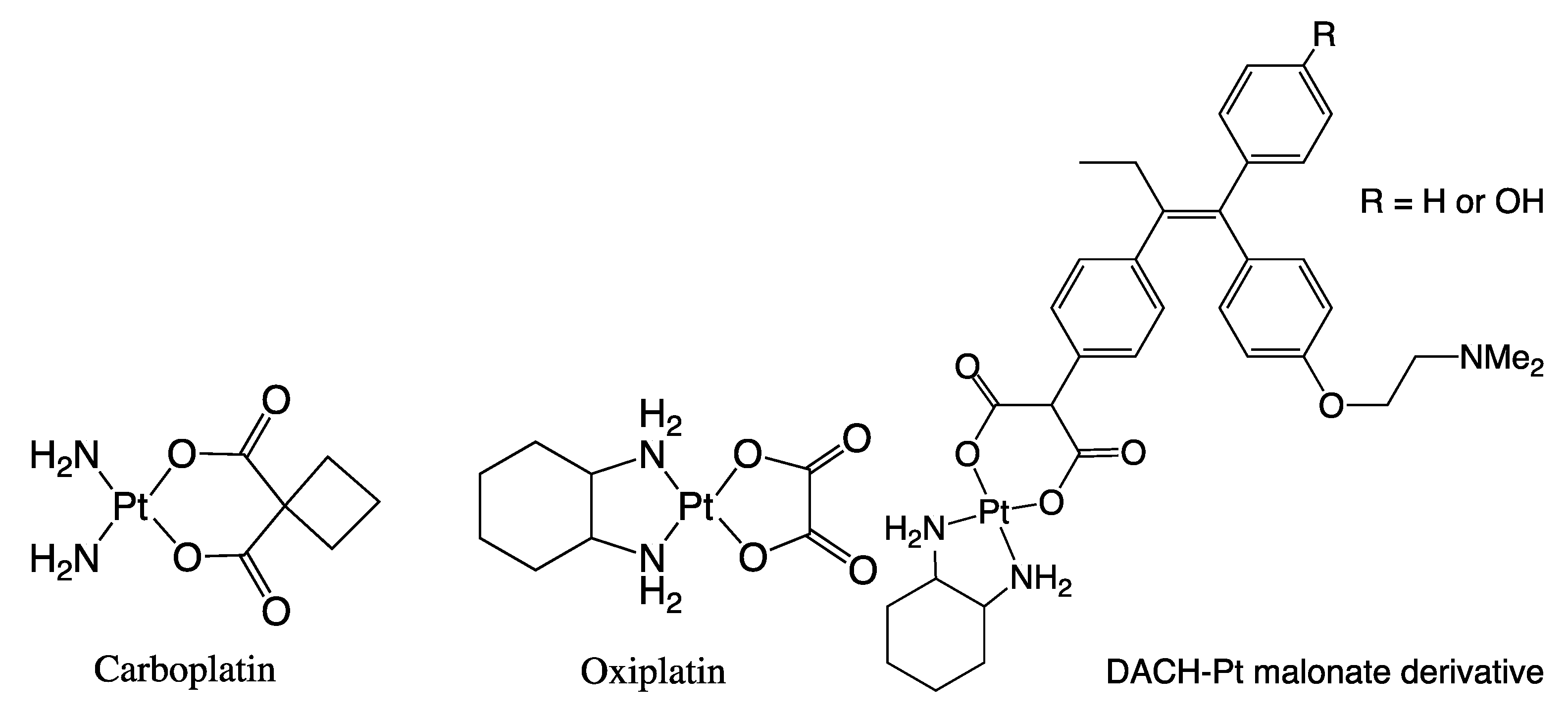
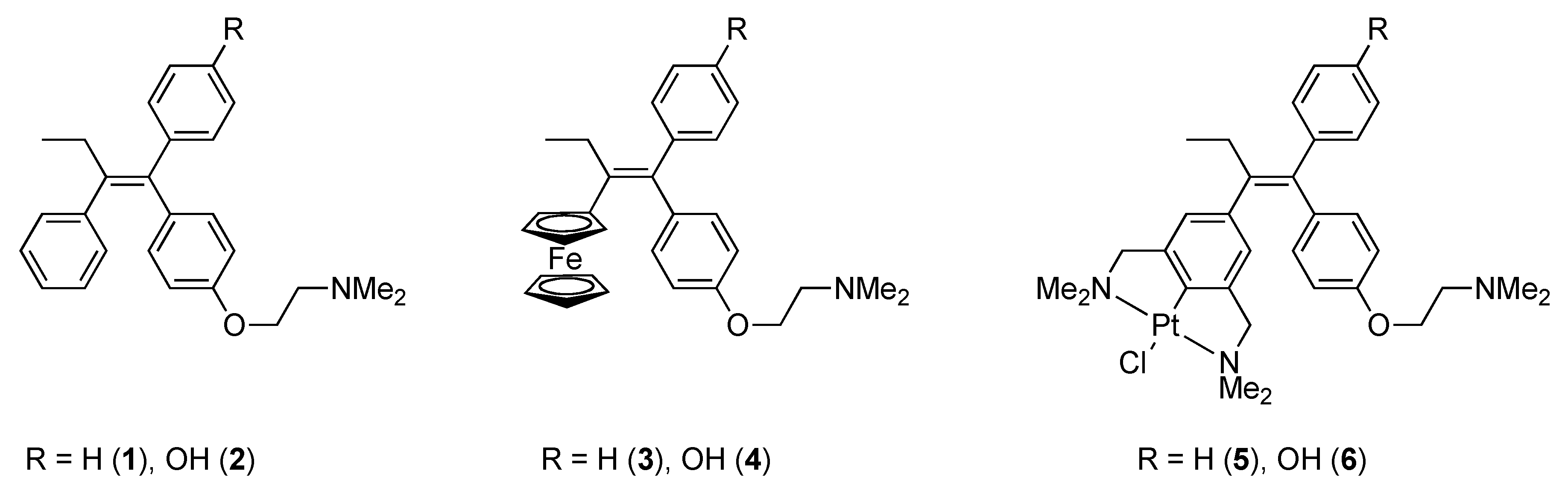
1. Introduction
2. Results
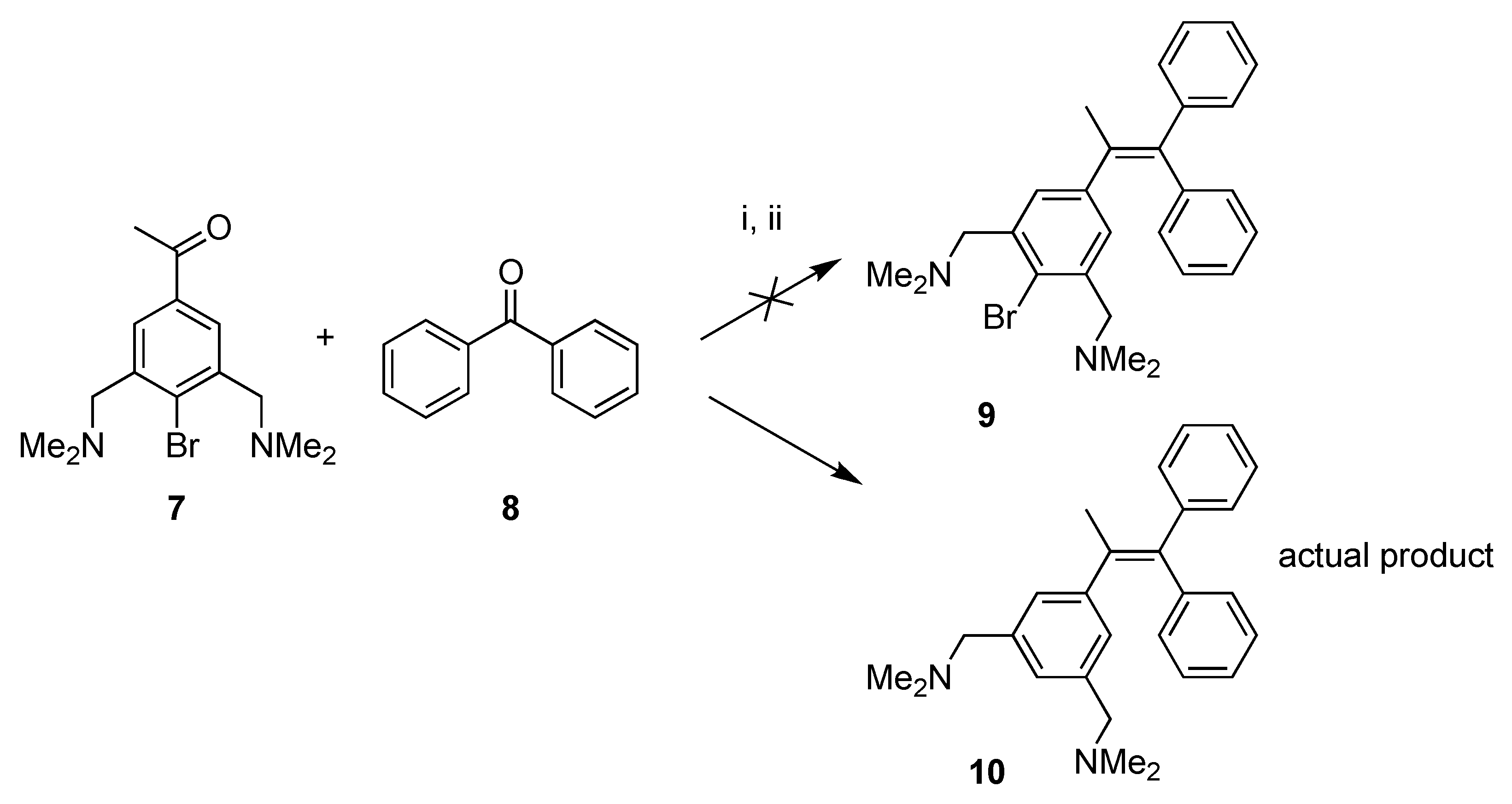
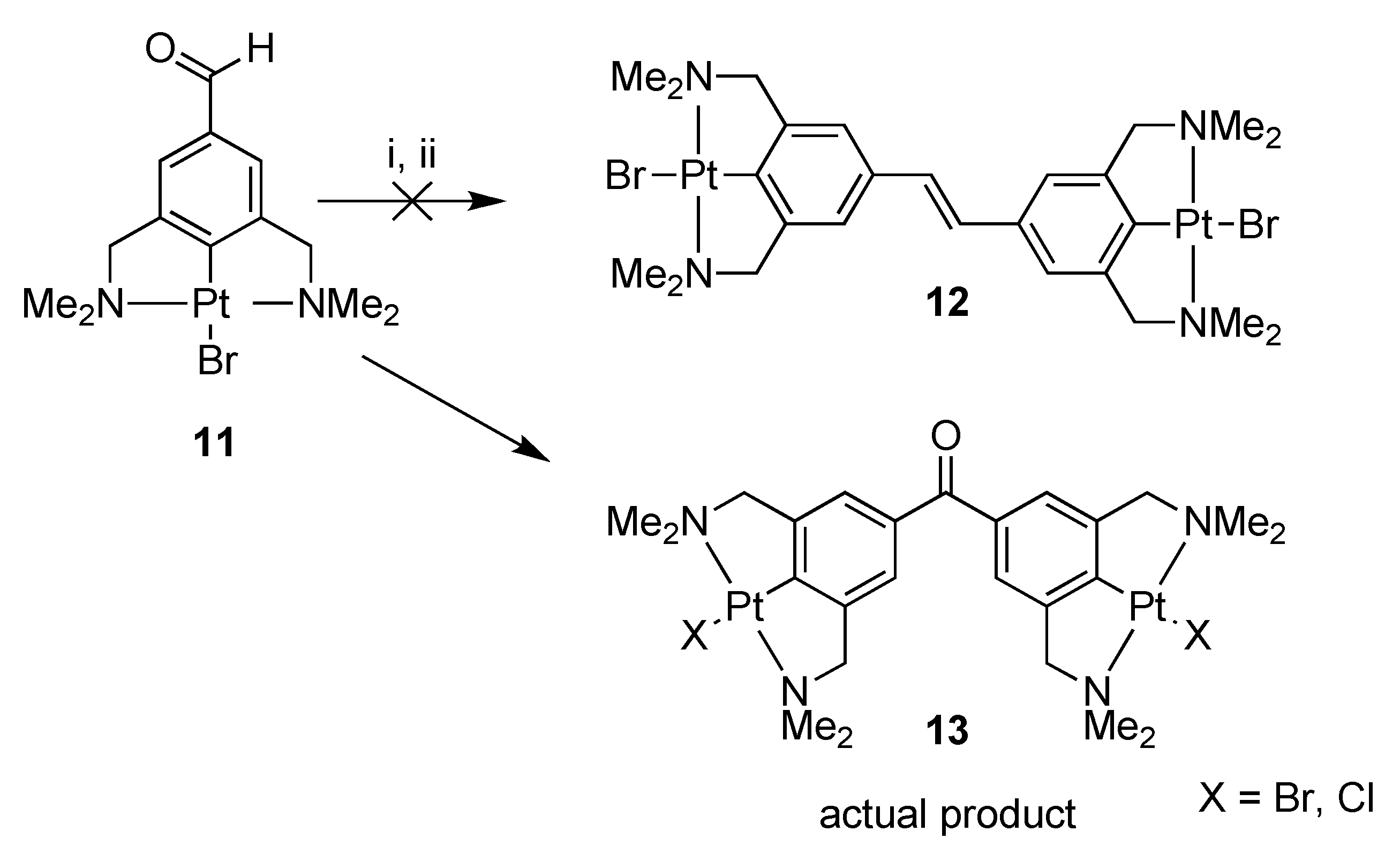
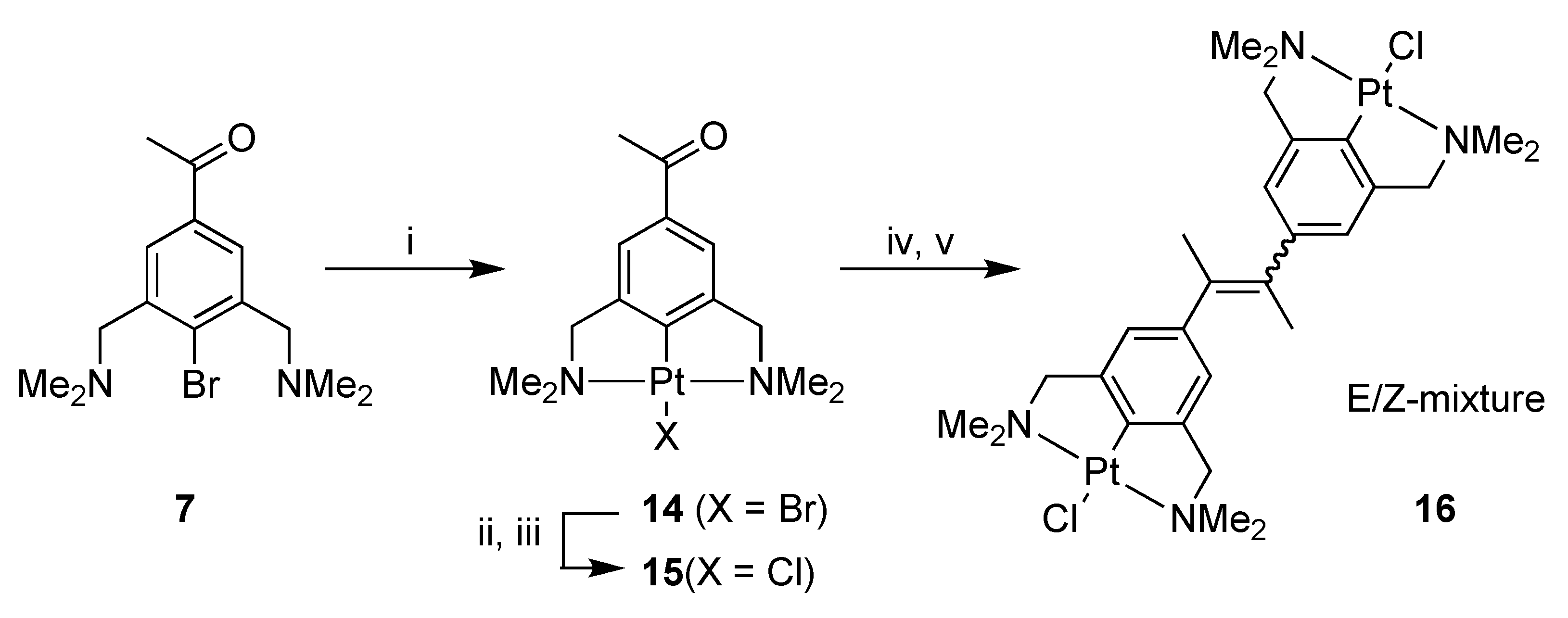
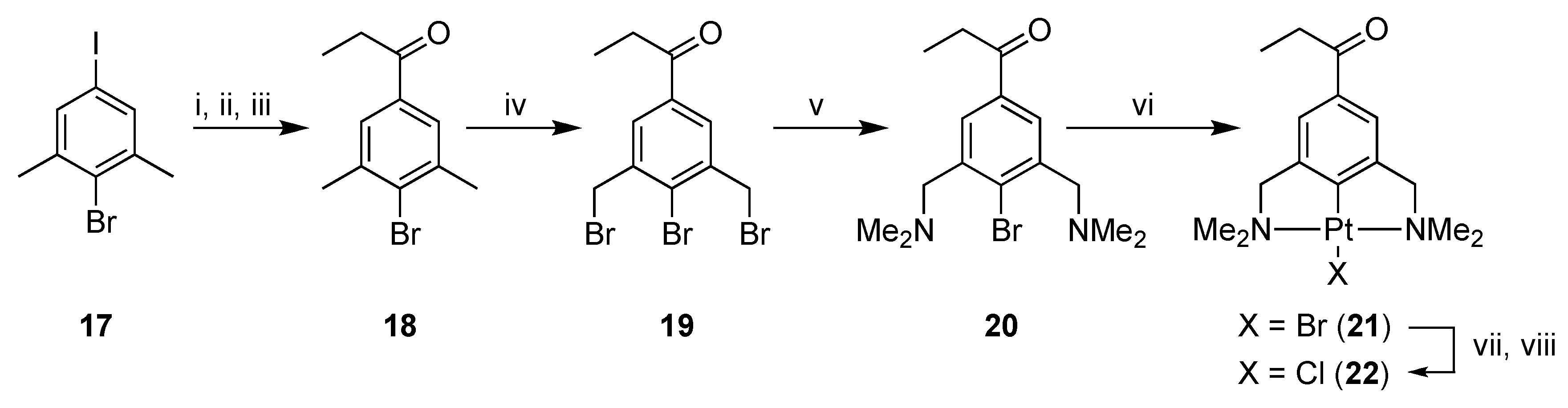
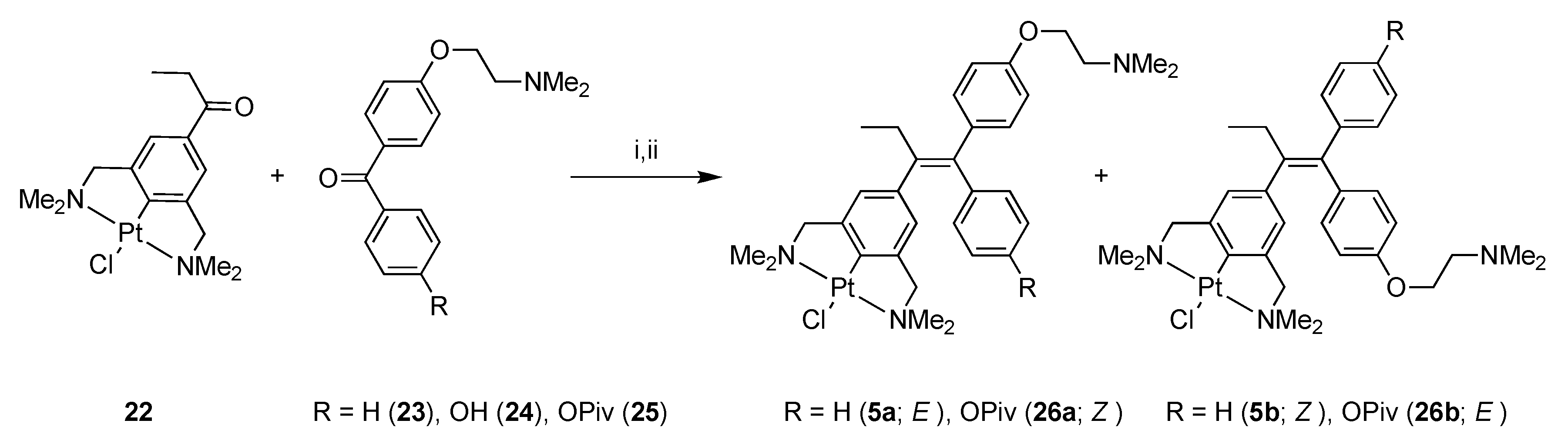
2.1. Synthesis
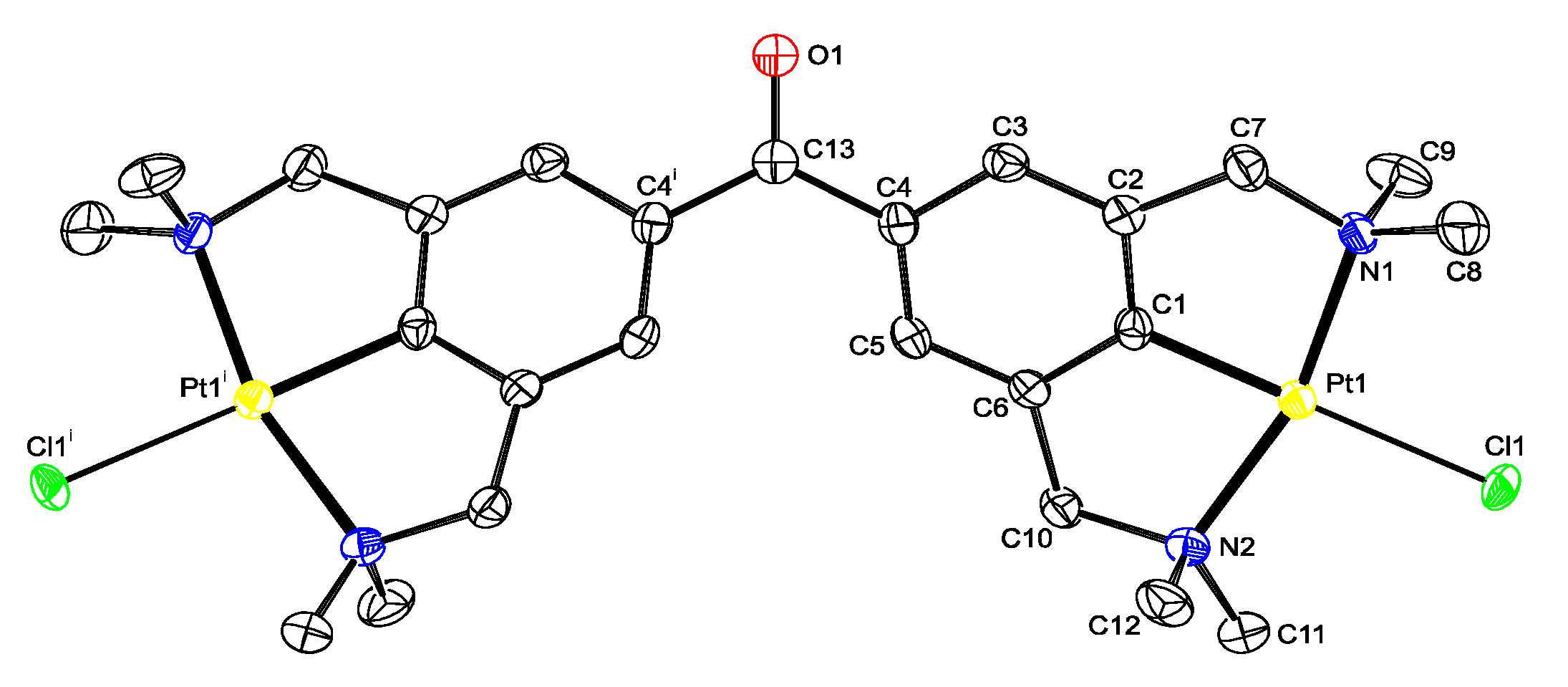
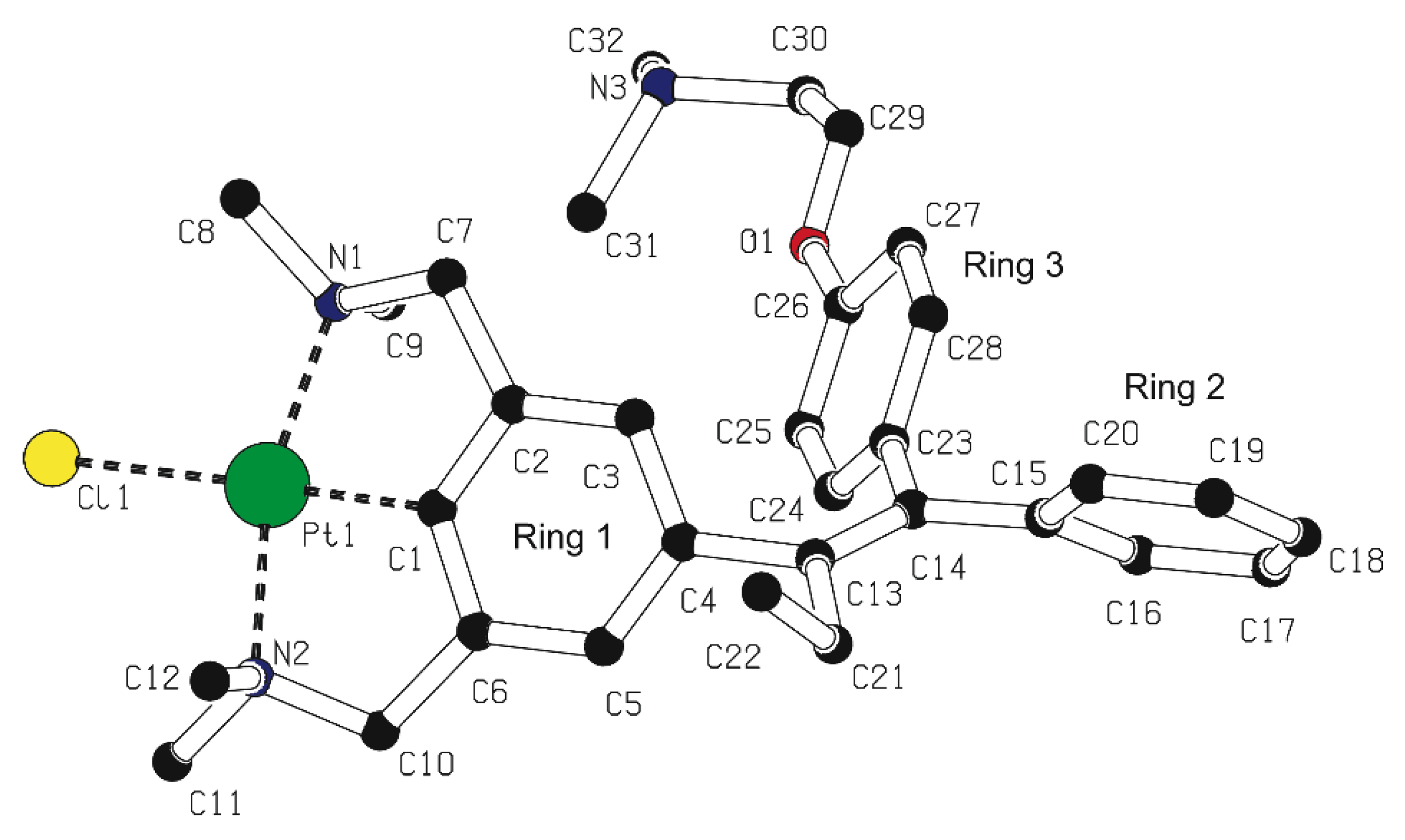
2.2. Structural Features of 13, and 5b in the Solid State
3. Discussion
4. Materials and Methods
4.1. Synthesis
4.1.1. General Information
4-Bromo-3,5-bis[(dimethylamino)methyl]acetophenone (7)
[PtBr(NCN-C(O)Me-4)] (14)
[PtCl(NCN-C(O)Me-4)] (15)
3,5-Dimethyl-4-bromopropiophenone (18)
3,5-Bis(bromomethyl)-4-bromopropiophenone (19)
3,5-Bis[(dimethylamino)methyl]-4-bromopropiophenone (20)
[PtBr(NCN-C(O)Et-4)] (21)
[PtCl(NCN-C(O)Et-4)] (22)
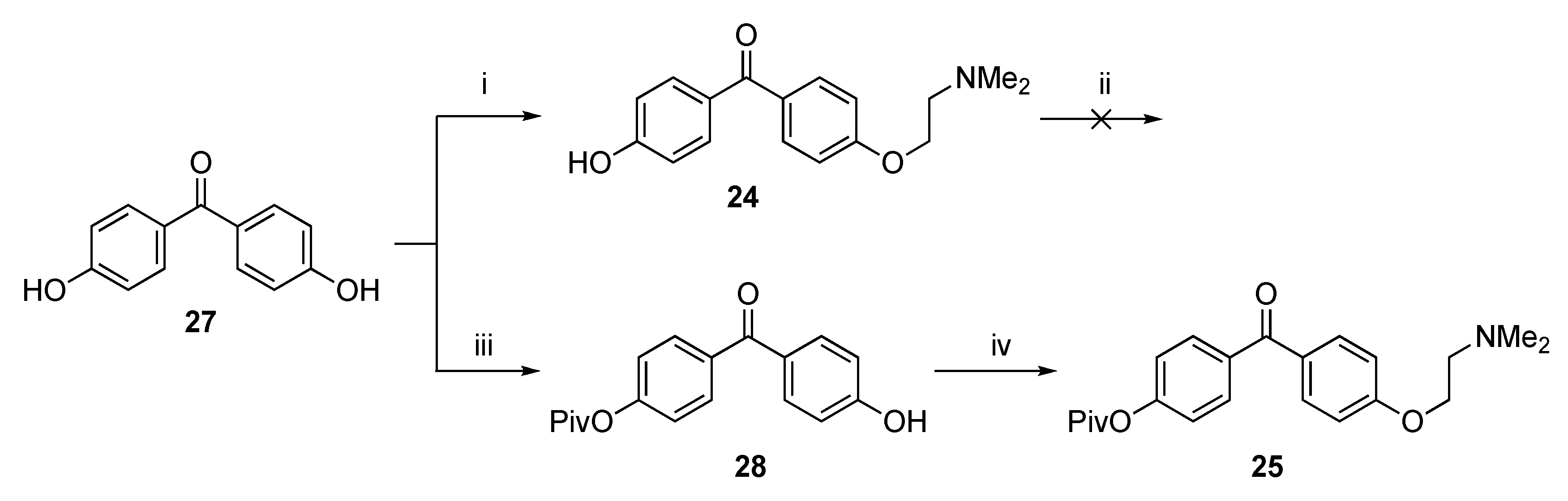
4-[2-(Dimethylamino)ethoxy]-4′-hydroxybenzophenone (24)
Attempted Protection of 24 with TBDMS-Cl
4-Trimethylacetoxy-4′-[2-(dimethylamino)ethoxy]benzophenone (25)
4.1.2. McMurry Reaction
Attempted Synthesis of 1-(1,1-Diphenyl-1-propenyl)-4-bromo-3,5-bis[(dimethylamino)methyl]Benzene (9); Formation of 1-(1,1-Diphenyl-1-propenyl)-3,5-bis[(dimethylamino)methyl]Benzene (10)
Attempted Synthesis of 3,3′,5,5′-Tetra(dimethylamino)methyl-4,4′-bisplatinumbromide-stilbene (12); Formation of 3,3′,5,5′-Tetra(dimethylaminomethyl)-4,4′-bisplatinum Halide-Benzophenone (13), (Halide = Br and Cl)
1-(2-(1-[(4-Dimethylaminoethoxy)phenyl]-1-phenyl-1-butenyl))-4-PtCl-3,5-bis[(dimethylamino)methyl]) Benzene (5)
1-(4-[2-(Dimethylamino)ethoxy]phenyl)-1-(4-trimethylacetoxyphenyl)-2-(NCN-PtCl)but-1-ene (26)
4.2. X-ray Crystal Structure Determination of Compound 13
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Collot, J.; Gradinaru, J.; Humbert, N.; Skander, M.; Zocchi, A.; Ward, T.R. Artificial Metalloenzymes for Enantioselective Catalysis Based on Biotin-Avidin. J. Am. Chem. Soc. 2003, 125, 9030–9031. [Google Scholar] [CrossRef]
- Desguin, B.; Zhang, T.; Soumillion, P.; Hols, P.; Hu, J.; Hausinger, R.P. A tethered niacin-derived pincer complex with a nickel-carbon bond in lactate racemase. Science 2015, 349, 66–69. [Google Scholar] [CrossRef]
- Ho, C.-L.; Wong, W.-Y. Organometallic Versus Organic Molecules for Energy Conversion in Organic Light-Emitting Diodes and Solar Cells. In Organometallics and Related Molecules for Energy Conversion; Wong, W.-Y., Ed.; Springer: Berlin/Heidelberg, Germany, 2015; pp. 1–29. [Google Scholar] [CrossRef]
- Hussein, M.A.; Asiri, A.M. Organometallic Ferrocene- and Phosphorus-Containing Polymers: Synthesis and Characterization. Des. Monomers Polym. 2012, 15, 207–251. [Google Scholar] [CrossRef]
- Allardyce, C.S.; Dorcier, A.; Scolaro, C.; Dyson, P.J. Development of Organometallic (Organo-Transition Metal) Pharmaceuticals. Appl. Organometal. Chem. 2005, 19, 1–10. [Google Scholar] [CrossRef]
- Hirao, T.; Moriuchi, T. Advances in Bioorganometallic Chemistry; Elsevier: Amsterdam, The Netherlands, 2018; pp. 1–472. [Google Scholar]
- Bruijnincx, P.C.A.; Sadler, P.J. New trends for metal complexes with anticancer activity. Curr. Opin. Chem. Biol. 2008, 12, 197–206. [Google Scholar] [CrossRef]
- Jaouen, G.; Vessières, A.; Top, S. Ferrocifen type anticancer drugs. Chem. Soc. Rev. 2015, 44, 8802–8817. [Google Scholar] [CrossRef]
- Dehand, J.; Pfeffer, M. Cyclometallated Compounds. Coord. Chem. Rev. 1976, 18, 327–352. [Google Scholar] [CrossRef]
- Dehand, J.; Jordanov, J.; Pfeffer, M. Reactivity of primary amines co-ordinated to palladium and platinum. Part II. cyclometallation and rearrangement of the co-ordinated ligands. J. Chem. Soc. Dalton Trans. 1976, 1553–1556. [Google Scholar] [CrossRef]
- Abbenhuis, H.C.L.; Pfeffer, M.; Sutter, J.P.; De Cian, A.; Fischer, J.; Li, J.H.; Nelson, J.H. Carbon-Carbon and Carbon-Nitrogen Bond Formation Mediated by Ruthenium(II) Complexes: Synthesis of (1H)-Isoquinolinium Derivatives. Organometallics 1993, 12, 4464–4472. [Google Scholar] [CrossRef]
- Pfeffer, M.; Sutter, J.P.; Urriolabeitia, E.P. Ruthenium Mediated Carbon-Carbon and Carbon-Nitrogen bond formation: Parameters governing the reactivity of the metal centre. Bull. Soc. Chim. Fr. 1997, 134, 947–954. [Google Scholar]
- Ryabov, A.D.; Sukharev, V.S.; Alexandrova, L.; Le Lagadec, R.; Pfeffer, M. New Synthesis and New Bio-Application of Cylometalated Ruthenium(II) Complexes for Fast Mediated Electron transfer with Peroxidase and Glucose Oxidase. Inorg. Chem. 2001, 40, 6529–6532. [Google Scholar] [CrossRef] [PubMed]
- Gaiddon, C.; Jeannequin, P.; Bishoff, P.; Pfeffer, M.; Sirlin, C.; Loeffler, J.P. Ruthenium(II)-derived organometallic compounds induce cytostatic and cytotoxic effects on mammalian cancer cell lines through p53-dependent and p53-independent mechanisms. J. Pharma. Exper. Ther. 2005, 315, 1403–1411. [Google Scholar] [CrossRef]
- Meng, X.; Leyva, L.M.; Jenny, M.; Gross, I.; Benosman, S.; Fricker, B.; Harlepp, S.; Hebraud, P.; Boos, A.; Wlosik, P.; et al. A ruthenium containing organometallic compound reduces tumor growth through induction of the endoplasmic reticulum stress gene CHOP. Cancer Res. 2009, 69, 5458–5466. [Google Scholar] [CrossRef] [PubMed]
- Licona, C.; Delhorme, J.-B.; Riegel, G.; Vidimar, V.; Cerón-Camacho, R.; Boff, B.; Venkatasamy, A.; Tomasetto, C.; da Silva Figueirdo Celestino Gomes, P.; Rognan, D.; et al. Anticancer activity of ruthenium and osmium cyclometalated compounds: Identification of ABCB1 and EGFR as resistance mechanisms. Inorg. Chem. Front. 2020, 7, 678–688. [Google Scholar] [CrossRef]
- Van Koten, G.; Leusink, A.J.; Noltes, J.G. Stable arylcopper compounds containing 2-(dimethylamino)methyl or 2-methoxymethyl groups at the aryl nucleus. J. Chem. Soc. Chem. Commun. 1970, 1107–1108. [Google Scholar] [CrossRef]
- Jastrzebski, J.T.B.H.; van Koten, G.; Goubitz, K.; Arlen, C.; Pfeffer, M. Synthesis of 8-(dimethylamino)-1-naphthyllithium Etherate: Its Structure in the Solid (X-ray) and in Solution (7Li and 1H NMR). J. Organomet. Chem. 1983, 246, C75–C79. [Google Scholar] [CrossRef]
- Wehman, E.; van Koten, G.; Knaap, C.T.; Ossor, H.; Pfeffer, M.; Spek, A.L. Synthesis of Cyclometalated (8-methoxynaphthyl)- and {8-(dimethylamino)naphthyl} Platinum Compounds and Their Reactivity Toward Electrophiles. X-ray Molecular Structures of Square- planar (8-methoxynaphthyl)platinum(II) Chloride bis (diethyl sulfide) and bis (8-methoxy naphthyl)platinum(II) Diethyl Sulfide and Octahedral fac-bis(8-methoxynaphthyl)Methyl platinum(IV) Iodide. Inorg. Chem. 1988, 27, 4409–4417. [Google Scholar] [CrossRef]
- Albrecht, M.; van Koten, G. Platinum Group Organometallics Based on ‘Pincer’ Ligands: Sensors, Switchses and Catalysis. Angew. Chem. Int. Ed. Engl. 2001, 40, 3750–3781. [Google Scholar] [CrossRef]
- Wieczorek, B.; Dijkstra, H.P.; Egmond, M.R.; Klein Gebbink, R.J.M.; van Koten, G. Incorporating ECE-Pincer Metal Complexes as Functional Building Blocks in Semisynthetic Metalloenzymes, Supramolecular Polypeptide Hybrids, Tamoxifen Derivatives, Biomarkers and Sensors. J. Organomet. Chem. 2009, 694, 812–822. [Google Scholar] [CrossRef]
- van de Kuil, L.A.; Luitjes, J.; Grove, D.M.; Zwikker, J.W.; van der Linden, J.G.M.; Roelofsen, A.M.; Jenneskens, L.W.; Drenth, W.; van Koten, G. Electronic Tuning of Arylnickel(II) Complexes by Para Substitution of the Terdentate Monoanionic 2,6-Bis[(dimethylamino)methyl]phenyl Ligand. Organometallics 1994, 13, 468–477. [Google Scholar] [CrossRef]
- Valdés, H.; González-Sebastián, L.; Morales-Morales, D. Aromatic para-functionalized NCN pincer compounds. J. Organometal. Chem. 2017, 845, 229–257. [Google Scholar] [CrossRef]
- Rodríguez, G.; Albrecht, M.; Schoenmaker, J.; Ford, A.; Lutz, M.; Spek, A.L.; van Koten, G. Bifunctional Pincer-type Organometallics as Substrates for Organic Transformations and as Novel Building Blocks for Polymetallic Materials. J. Am. Chem. Soc. 2002, 124, 5127–5138. [Google Scholar] [CrossRef]
- Canty, A.J.; Ariafard, A.; van Koten, G. Computational Analysis of Mesomerism in para-Substituted mer- NCN Pincer Platinum(II) Complexes, Including its Relationships with Hammett σp Substituent Parameters. Chem. Eur. J. 2020, 67, 15629–15635. [Google Scholar] [CrossRef] [PubMed]
- Top, S.; Dauer, B.; Vaissermann, J.; Jaouen, G. Facile route to ferrocifen, 1-[4-(2-dimethylaminoethoxy)]-1-(phenyl-2-ferrocenyl-but- 1-ene), first organometallic analogue of tamoxifen, by the McMurry reaction. J. Organometal. Chem. 1997, 541, 355–361. [Google Scholar] [CrossRef]
- Top, S.; Kaloun, E.B.; Vessieres, A.; Leclercq, G.; Laios, I.; Ourevitch, I.; Deuschel, C.; McGlinchey, M.J.; Jaouen, G. Tamoxifen Derivatives for Delivery of the Antitumoral (DACH)Pt Group: Selective Synthesis by McMurry Coupling, and Biochemical Behaviour. ChemBioChem 2003, 4, 754–761. [Google Scholar] [CrossRef]
- McMurry, J.E. Carbonyl-coupling reactions using low-valent titanium. Chem. Rev. 1989, 89, 1513–1524. [Google Scholar] [CrossRef]
- Richardson, W.H. Haloaromatic Substituted Olefins by the MC Murry Olefin Synthesis. Synth. Commun. 1981, 11, 895–899. [Google Scholar] [CrossRef]
- Donkervoort, J.G.; Jastrzebski, J.T.B.H.; Deelman, B.J.; Kooijman, H.; Veldman, N.; Spek, A.L.; van Koten, G. Novel Aryltitanium(IV) Complexes Containing η3-NCN-Pseudofacial- and η3-NCN-Meridional-Bonded [C6H3(CH2NMe2)2-2,6]- (Abbreviated as NCN) Ligands. The Crystal Structures of [TiCl2(η3-fac-NCN)(O-i-Pr)], [TiCl(η3-mer-NCN)(O-i-Pr)(OTf)] Containing an η1-O-Bonded Triflate Anion, and the Seven-Coordinated [Ti(η3-mer-NCN)(O-i-Pr)(OTf)2] Containing η1-O- and η2-O,O‘-Bonded Triflate Anions. Organometallics 1997, 16, 4174–4184. [Google Scholar] [CrossRef]
- Casado Lacabra, M.A.; Canty, A.M.; Lutz, M.; Patel, J.; Spek, A.L.; Sun, H.; van Koten, G. Mono(p-tolyl)platinum(II) and bis(p-tolyl)platinum(II) complexes of diethylsulfide as reagents for organoplatinum synthesis. Structures of [Pt(p-Tol)2(μ-SEt2)]2 and PtCl(p-Tol)(bpy) (bpy = 2,2′-bipyridine). Inorg. Chim. Acta 2016, 442, 167–171. [Google Scholar]
- Amijs, C.H.M.; van Klink, G.P.M.; van Koten, G. Carbon tetrachloride free benzylic brominations of methyl aryl halides. Green Chem. 2003, 5, 470–474. [Google Scholar] [CrossRef]
- Meegan, M.J.; Hughes, R.B.; Lloyd, D.G.; Williams, D.C.; Zisterer, D.M. Flexible Estrogen Receptor Modulators: Design, Synthesis, and Antagonistic Effects in Human MCF-7 Breast Cancer Cells. J. Med. Chem. 2001, 44, 1072–1084. [Google Scholar] [CrossRef]
- Kooijman, H.; Spek, A.L.; Batema, G.D.; van Koten, G. CSD Communications Is a Collection of Small Molecule Crystallographic Data Which Has Been Shared by Depositors. 2020. Available online: https://www.ccdc.cam.ac.uk/Community/csd-communications/CSDCommunicationsInformation/ (accessed on 24 March 2021).
- Gauthier, S.; Mailhot, J.; Labrie, J. New Highly Stereoselective Synthesis of (Z)-4-Hydroxytamoxifen and (Z)-4-Hydroxytoremifen via McMurry Reaction. J. Org. Chem. 1996, 61, 3890–3893. [Google Scholar] [CrossRef]
- Nikitin, K.; Ortin, Y.; Müller-Bunz, H.; Plamont, M.-A.; Jaouen, G.; Vessières, A.; McGlinchey, M.J. Organometallic SERMs (selective estrogen receptor modulators): Cobaltifens, the (cyclobutadiene)cobalt analogues of hydroxytamoxifen. J. Organometal. Chem. 2010, 695, 595–608. [Google Scholar] [CrossRef]
- Slagt, M.Q.; Rodríguez, G.; Grutters, M.M.P.; Klein Gebbink, R.J.M.; Klopper, W.; Jenneskens, L.W.; Lutz, M.; Spek, A.L.; van Koten, G. Synthesis and Properties of para-Substituted NCN-Pincer Palladium and Platinum Complexes. Chem. Eur. J. 2004, 10, 1331–1344. [Google Scholar] [CrossRef]
- Top, S.; Tang, J.; Vessieres, A.; Carrez, D.; Provot, C.; Jaouen, G. Ferrocenyl hydroxytamoxifen: A prototype for a new range of oestradiol receptor site-directed cytotoxics. Chem. Commun. 1996, 955–956. [Google Scholar] [CrossRef]
- Vessières, A.; Top, S.; Beck, W.; Hillard, E.; Jaouen, G. Metal complex SERMs (selective oestrogen receptor modulators). The influence of different metal units on breast cancer cell antiproliferative effects. J. Chem. Soc. Dalton Trans. 2006, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Top, S.; Vessières, A.; Leclercq, G.; Quivy, J.; Tang, J.; Vaissermann, J.; Huche, M.; Jaouen, C. Synthesis, Biochemical Properties and Molecular Modelling Studies of Organometallic Specific Estrogen Receptor Modulators (SERMs), the Ferrocifens and Hydroxyferrocifens: Evidence for an Antiproliferative Effect of Hydroxyferrocifens on both Hormone-Dependent and Hormone-Independent Breast Cancer Cell Lines. Chem. Eur. J. 2003, 9, 5223–5236. [Google Scholar] [CrossRef]
- Jaouen, G.; Top, S.; Vessières, A.; Alberto, R. New paradigms for synthetic pathways inspired by bioorganometallic chemistry. J. Organomet. Chem. 2000, 600, 23–36. [Google Scholar] [CrossRef]
- Fus, F.; Yang, Y.; Lee, H.Z.; Top, S.; Carriere, M.; Bouron, A.; Pacureanu, A.; da Silva, J.C.; Salmain, M.; Vessières, A.; et al. Intracellular Localization of an Osmocenyl-Tamoxifen Derivative in Breast Cancer Cells Revealed by Synchrotron Radiation X-ray Fluorescence Nanoimaging. Angew. Chem. Int. Ed. Engl. 2019, 58, 3461–3465. [Google Scholar] [CrossRef]
- Dhingra, K. Antiestrogens–Tamoxifen, SERMs and Beyond. Investig. New Drugs 1999, 17, 285–311. [Google Scholar] [CrossRef]
- Top, S.; Vessières, A.; Cabestaing, C.; Laios, I.; Leclercq, G.; Provot, C.; Jaouen, G. Studies on organometallic selective estrogen receptor modulators. (SERMs) Dual activity in the hydroxy-ferrocifen series. J. Organomet. Chem. 2001, 637, 500–506. [Google Scholar] [CrossRef]
- Hillard, E.; Vessières, A.; Thouin, G.; Jaouen, G.; Amotore, C. Ferrocene-Mediated Proton-Coupled Electron Transfer in a Series of Ferrocifen-Type Breast-Cancer Drug Candidates. Angew. Chem. Int. Ed. Eng. 2006, 45, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Fish, R.H.; Jaouen, G. Bioorganometallic Chemistry: Structural Diversity of Organometallic Complexes with Bioligands and Molecular Recognition Studies of Several Supramolecular Hosts with Biomolecules, Alkali-Metal Ions, and Organometallic Pharmaceuticals. Organometallics 2003, 22, 2166–2177. [Google Scholar] [CrossRef]
- Precigoux, G.; Courseille, C.; Geoffre, S. Hospital, [p-(Diméthylamino-2 éthoxy)phényl]-1 trans-Diphenyl-1,2 Butène-1 (Tamoxifène) (ICI-46474). Acta Crystallogr. Sect. B Struct. Sci. 1979, 35, 3070–3072. [Google Scholar] [CrossRef]
- Dyson, P.J.; Sava, G. Metal-based antitumour drugs in the post genomic era. J. Chem. Soc. Dalton Trans. 2006, 1929–1933. [Google Scholar] [CrossRef]
- Wong, E.; Giandomenico, C.M. Current Status of Platinum-Based Antitumor Drugs. Chem. Rev. 1999, 99, 2451–2466. [Google Scholar] [CrossRef]
- Rosenberg, B.; van Camp, L.; Krigas, T. Inhibition of Cell Division in Escherichia coli by Electrolysis Products from a Platinum Electrode. Nature 1965, 205, 698–699. [Google Scholar] [CrossRef] [PubMed]
- Alderden, R.A.; Hall, M.D.; Hambley, T.W. The Discovery and Development of Cisplatin. J. Chem. Educ. 2006, 83, 728–734. [Google Scholar] [CrossRef]
- Van Zutphen, S.; Reedijk, J. Targeting platinum anti-tumour drugs: Overview of strategies employed to reduce systemic toxicity. Coord. Chem. Rev. 2005, 249, 2845–2853. [Google Scholar] [CrossRef]
- Davies, P.J.; Veldman, N.; Grove, D.M.; Spek, A.L.; Lutz, B.T.G.; van Koten, G. Organoplatinum Building Blocks for one-Dimensional Hydrogen-Bonded Polymeric structures. Angew. Chem. Int. Ed. 1996, 35, 1959–1961. [Google Scholar] [CrossRef]
- Chuchuryukin, A.V.; Chase, P.A.; Mills, A.M.; Lutz, M.; Spek, A.L.; van Klink, G.P.M.; van Koten, G. Hydroxy- and Mercaptopyridine Pincer Platinum and Palladium Complexes Generated by Silver-Free Halide Abstraction. Inorg. Chem. 2006, 45, 2045–2054. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, B.; Snelders, D.J.M.; Dijkstra, H.P.; Versluis, K.; Lutz, M.; Spek, A.L.; Egmond, M.R.; Klein Gebbink, R.J.M.; van Koten, G. Coordination Chemistry in Water of a Free and a Lipase-Embedded Cationic NCN-Pincer Platinum Center with Neutral and Ionic Triarylphosphines. Organometallics 2012, 31, 2810–2820. [Google Scholar] [CrossRef]
- Duisenberg, A.J.M.; Kroon-Batenburg, L.M.J.; Schreurs, A.M.M. An intensity evaluation method: EVAL-14. J. Appl. Cryst. 2003, 36, 220–229. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Cryst. 2009, D65, 148–155. [Google Scholar] [CrossRef]
- Blessing, R.H. Data Reduction and Error Analysis for Accurate Single Crystal Diffraction Intensities. Crystallogr. Rev. 1987, 1, 3–58. [Google Scholar] [CrossRef]
- Beurskens, P.T.; Admiraal, G.; Beurskens, G.; Bosman, W.P.; Garcia-Granda, S.; Gould, R.O.; Smits, J.M.M.; Smykalla, C. The DIRDIF99 Program System; Technical Report of the Crystallography; Laboratory at University of Nijmegen, University of Nijmegen: Nijmegen, The Netherlands, 1999. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Spek, A.L. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Cryst. 2015, C71, 9–18. [Google Scholar]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Batema, G.D.; Korstanje, T.J.; Guillena, G.; Rodríguez, G.; Lutz, M.; van Klink, G.P.M.; Gossage, R.A.; van Koten, G. Replacing the Z-phenyl Ring in Tamoxifen® with a para-Connected NCN Pincer-Pt-Cl Grouping by Post-Modification . Molecules 2021, 26, 1888. https://doi.org/10.3390/molecules26071888
Batema GD, Korstanje TJ, Guillena G, Rodríguez G, Lutz M, van Klink GPM, Gossage RA, van Koten G. Replacing the Z-phenyl Ring in Tamoxifen® with a para-Connected NCN Pincer-Pt-Cl Grouping by Post-Modification . Molecules. 2021; 26(7):1888. https://doi.org/10.3390/molecules26071888
Chicago/Turabian StyleBatema, Guido D., Ties J. Korstanje, Gabriela Guillena, Gema Rodríguez, Martin Lutz, Gerard P. M. van Klink, Robert A. Gossage, and Gerard van Koten. 2021. "Replacing the Z-phenyl Ring in Tamoxifen® with a para-Connected NCN Pincer-Pt-Cl Grouping by Post-Modification " Molecules 26, no. 7: 1888. https://doi.org/10.3390/molecules26071888
APA StyleBatema, G. D., Korstanje, T. J., Guillena, G., Rodríguez, G., Lutz, M., van Klink, G. P. M., Gossage, R. A., & van Koten, G. (2021). Replacing the Z-phenyl Ring in Tamoxifen® with a para-Connected NCN Pincer-Pt-Cl Grouping by Post-Modification . Molecules, 26(7), 1888. https://doi.org/10.3390/molecules26071888