
Petasis vs. Strecker Amino Acid Synthesis: Convergence, Divergence and Opportunities in Organic Synthesis

Abstract
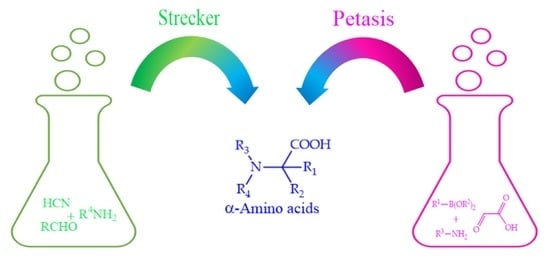
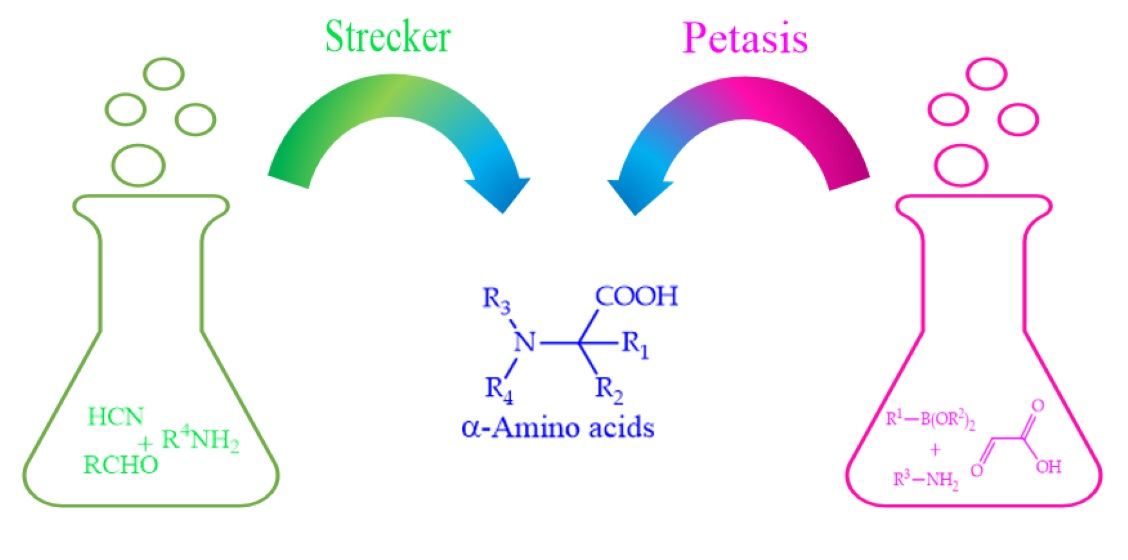{kind=link}
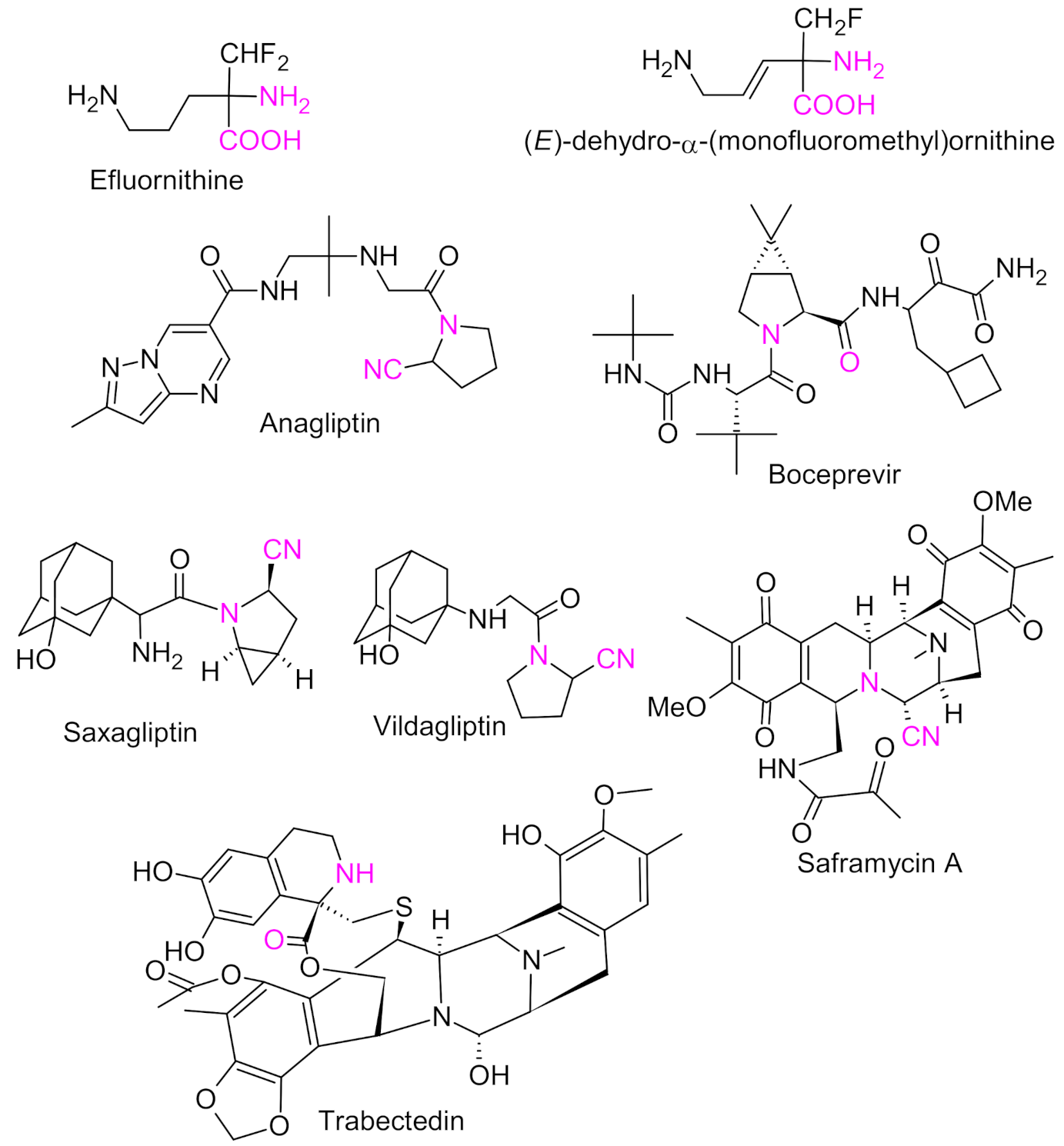{kind=link}
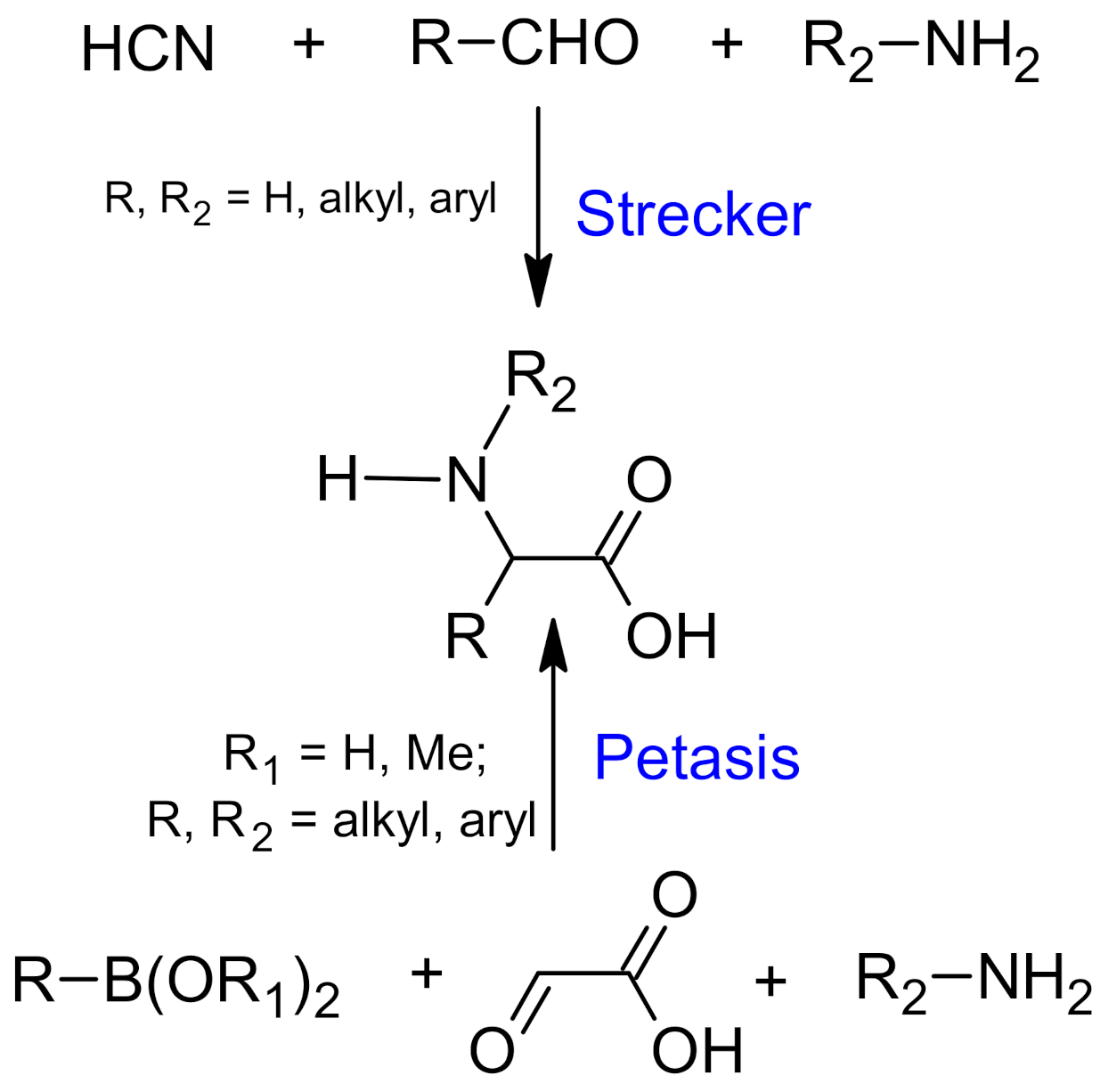{kind=link}
{kind=link}
{kind=link}
{kind=link}
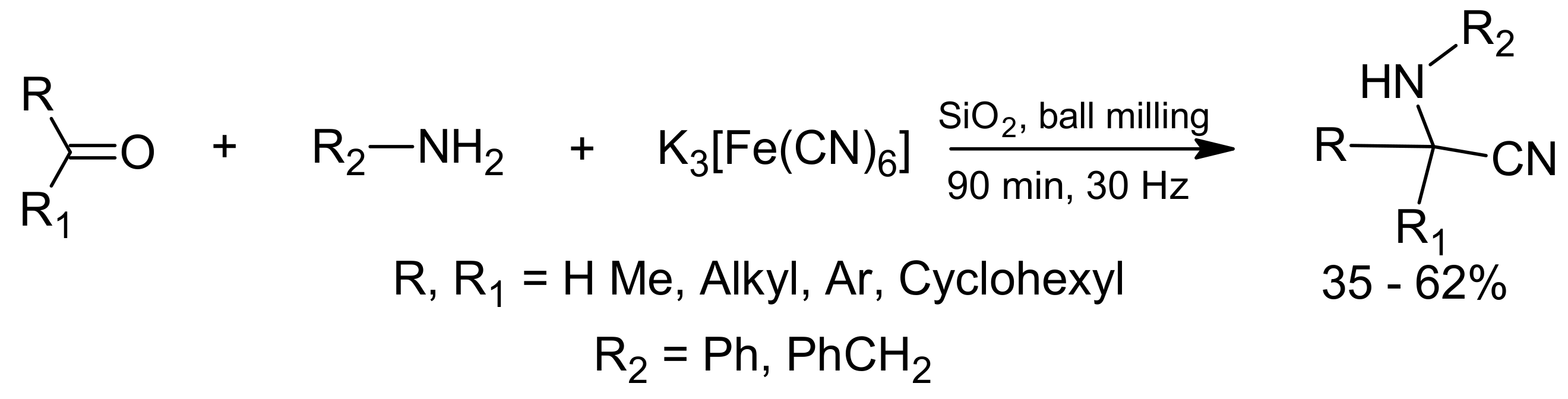{kind=link}
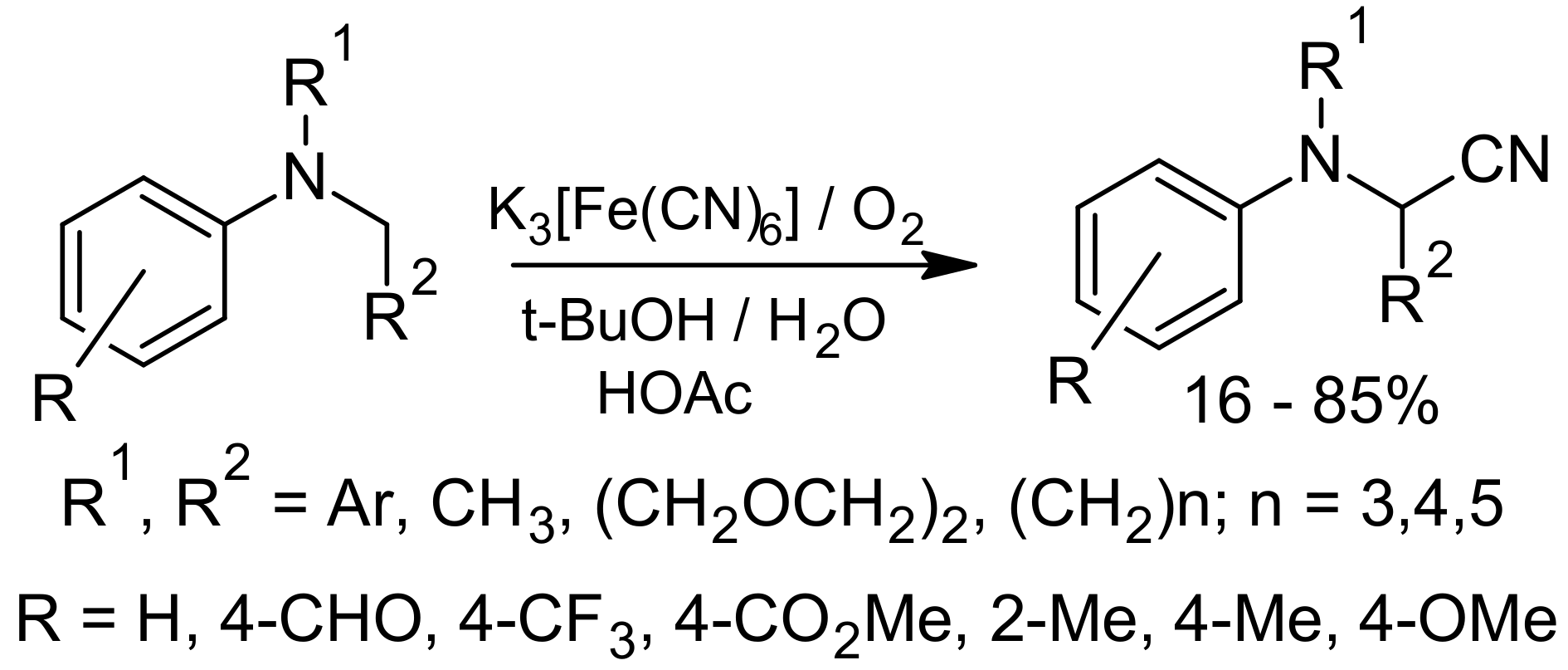{kind=link}
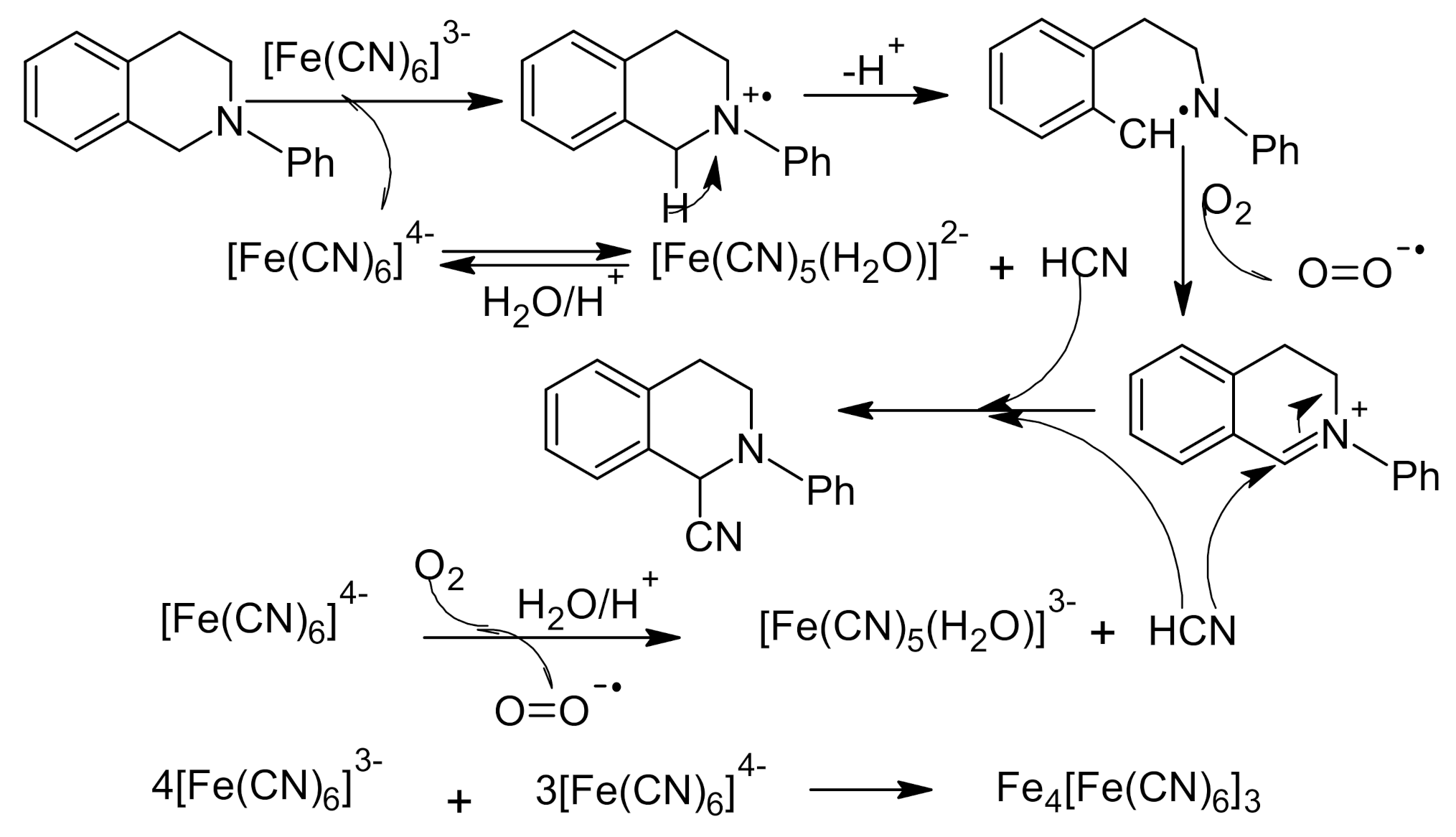{kind=link}
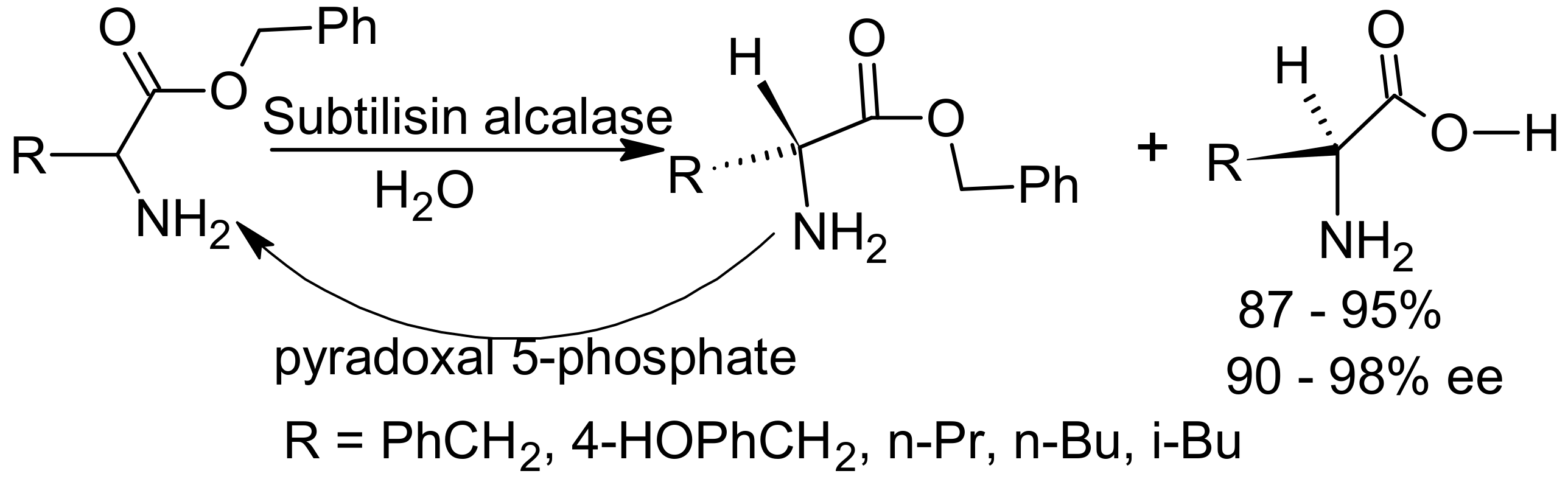{kind=link}
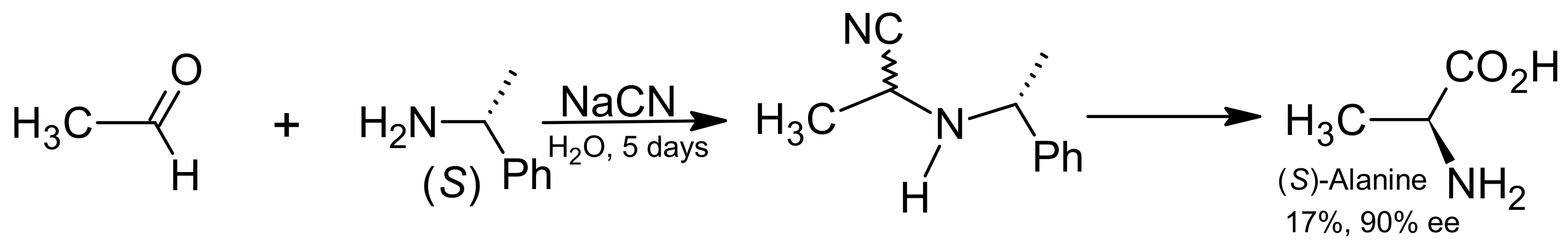{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
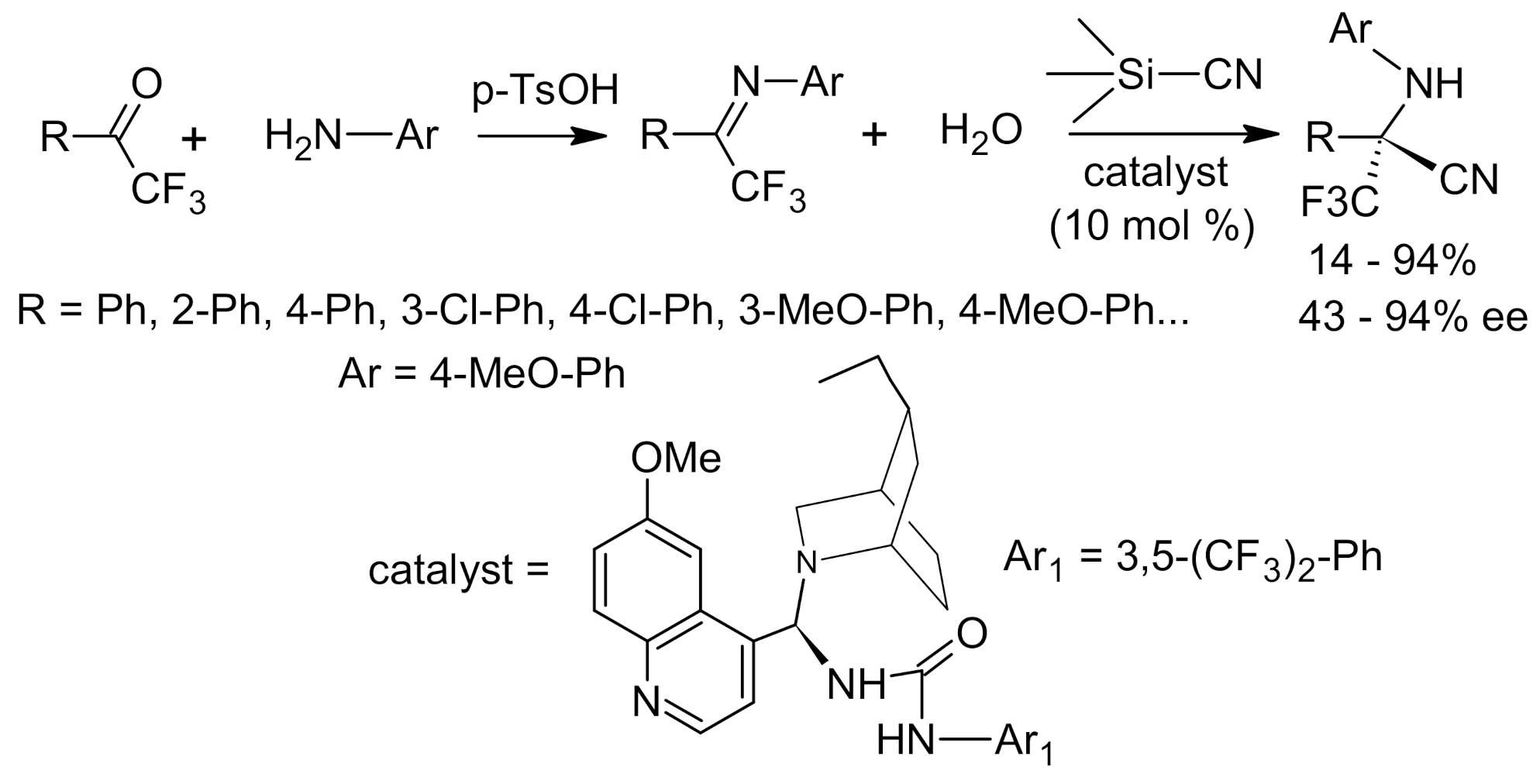{kind=link}
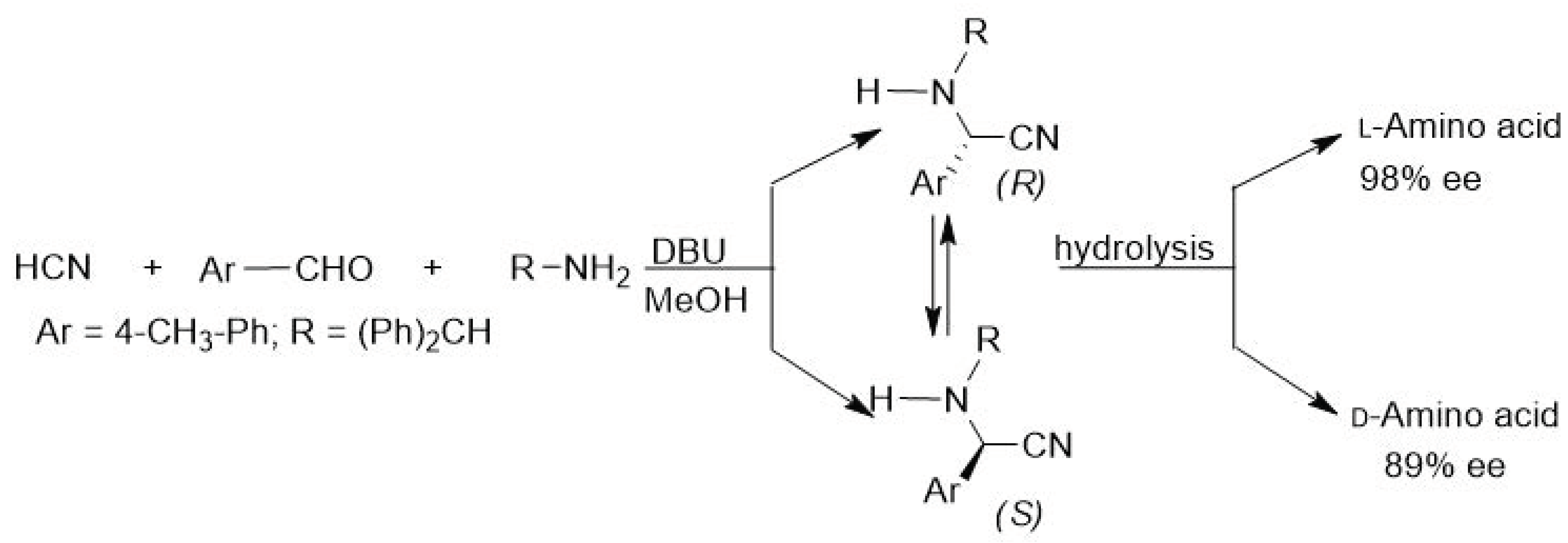{kind=link}
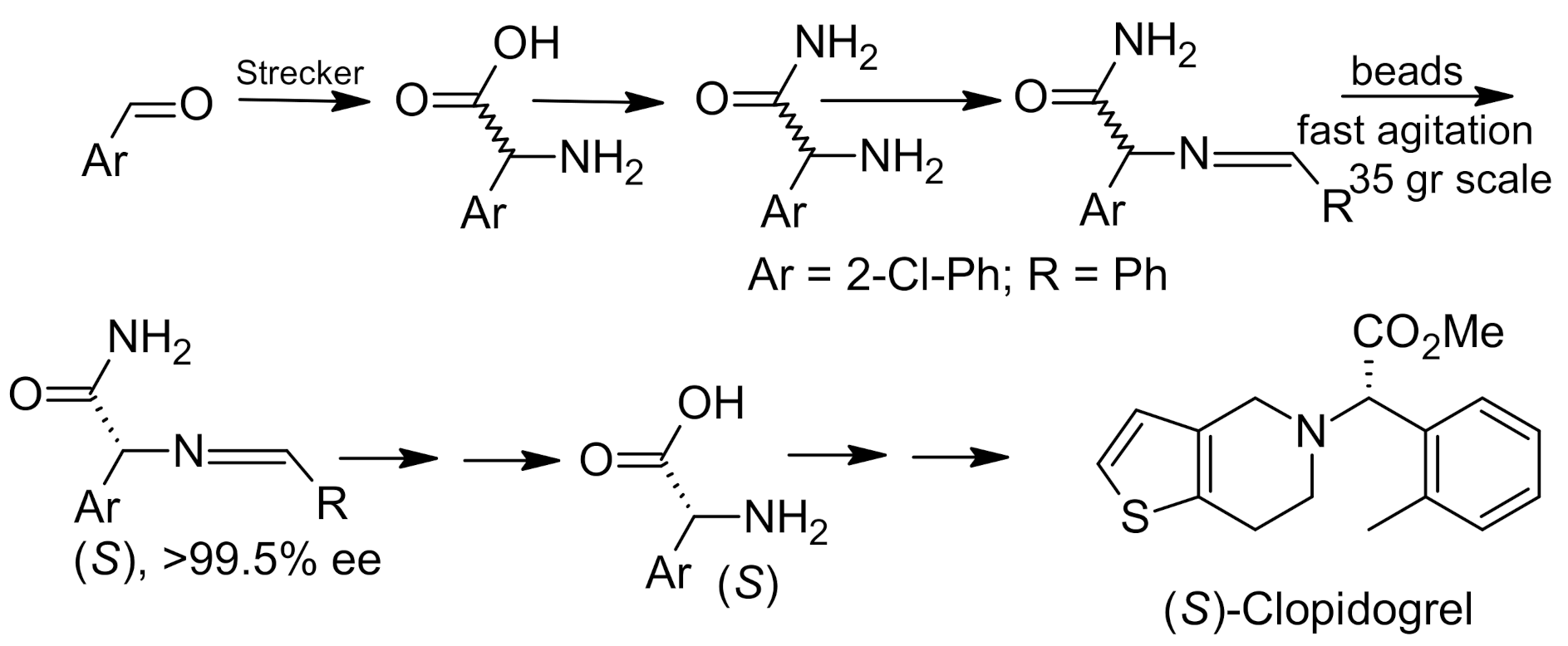{kind=link}
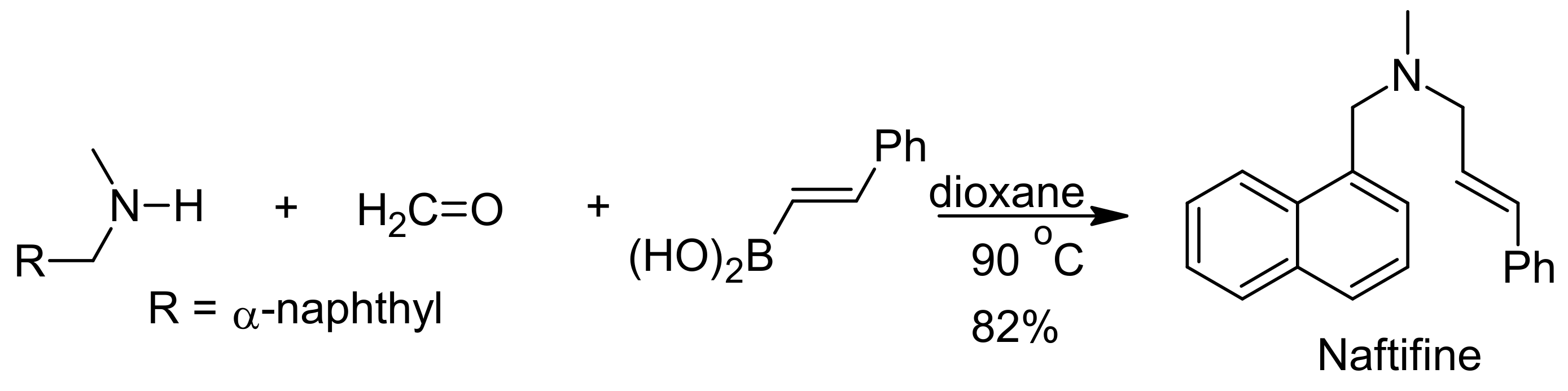{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
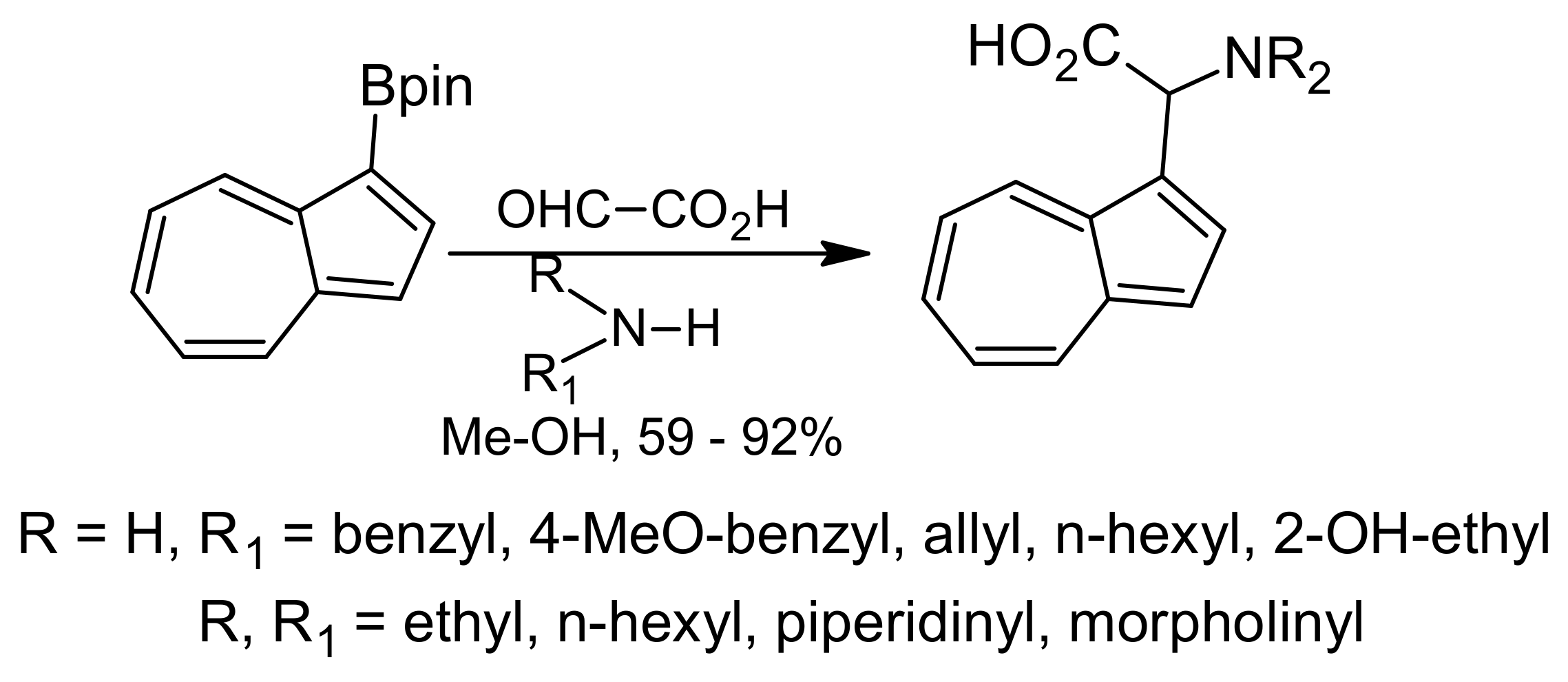{kind=link}
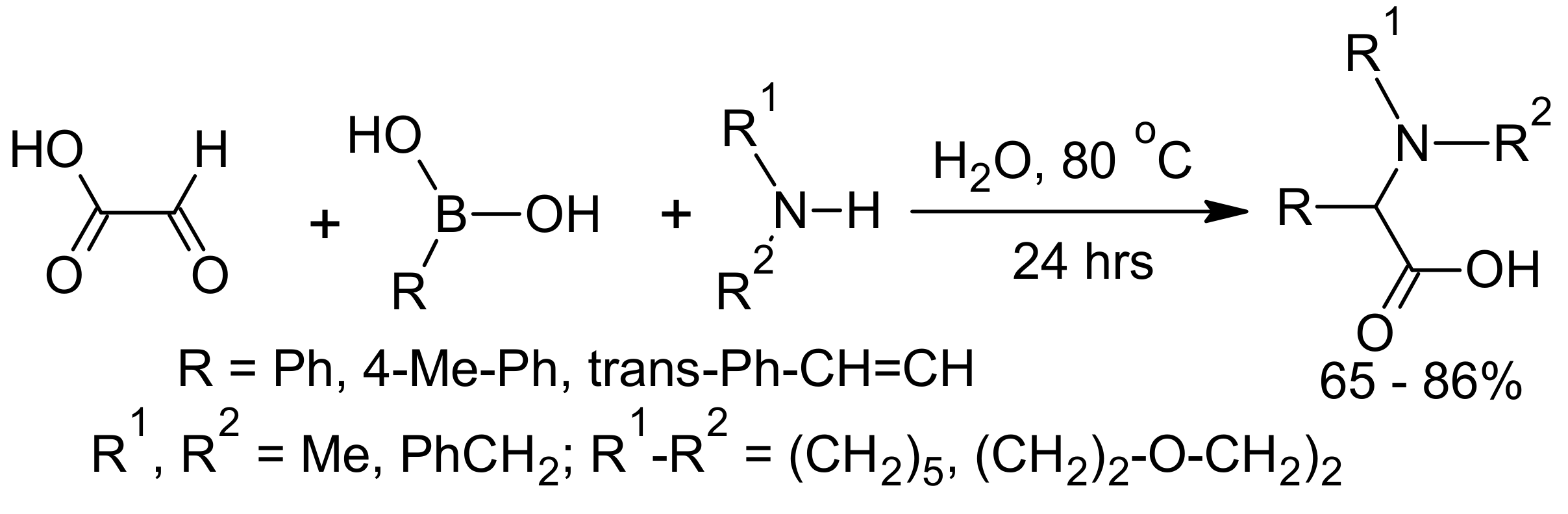{kind=link}
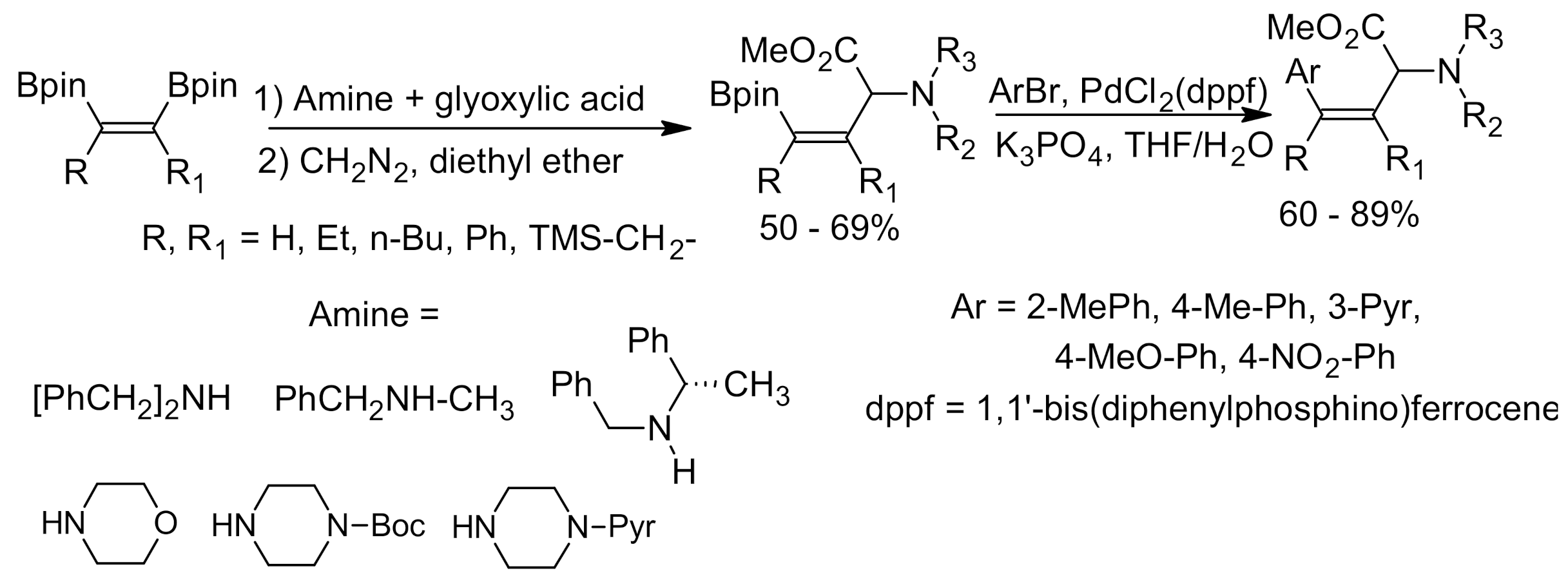{kind=link}
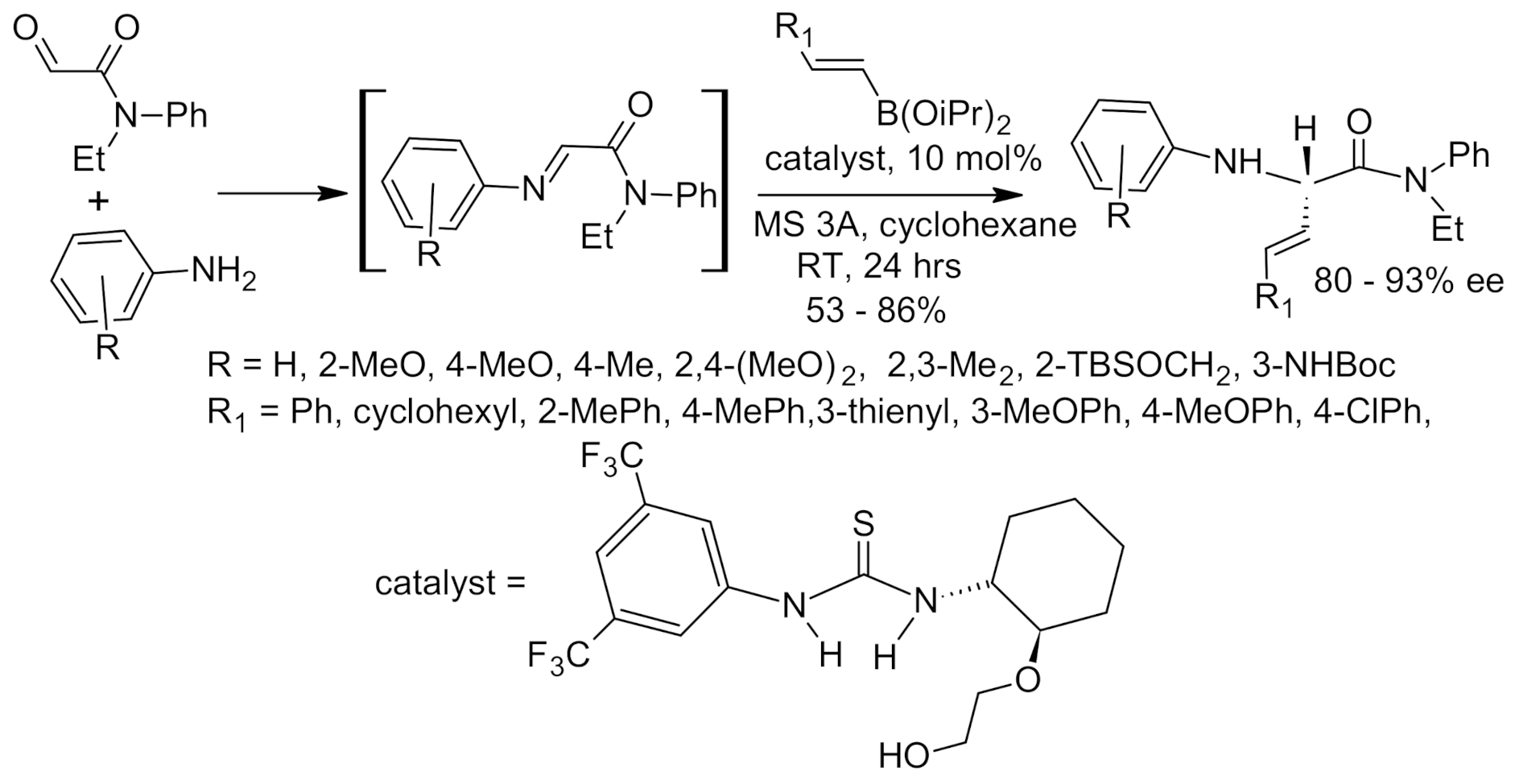{kind=link}
{kind=link}
{kind=link}
1. Introduction
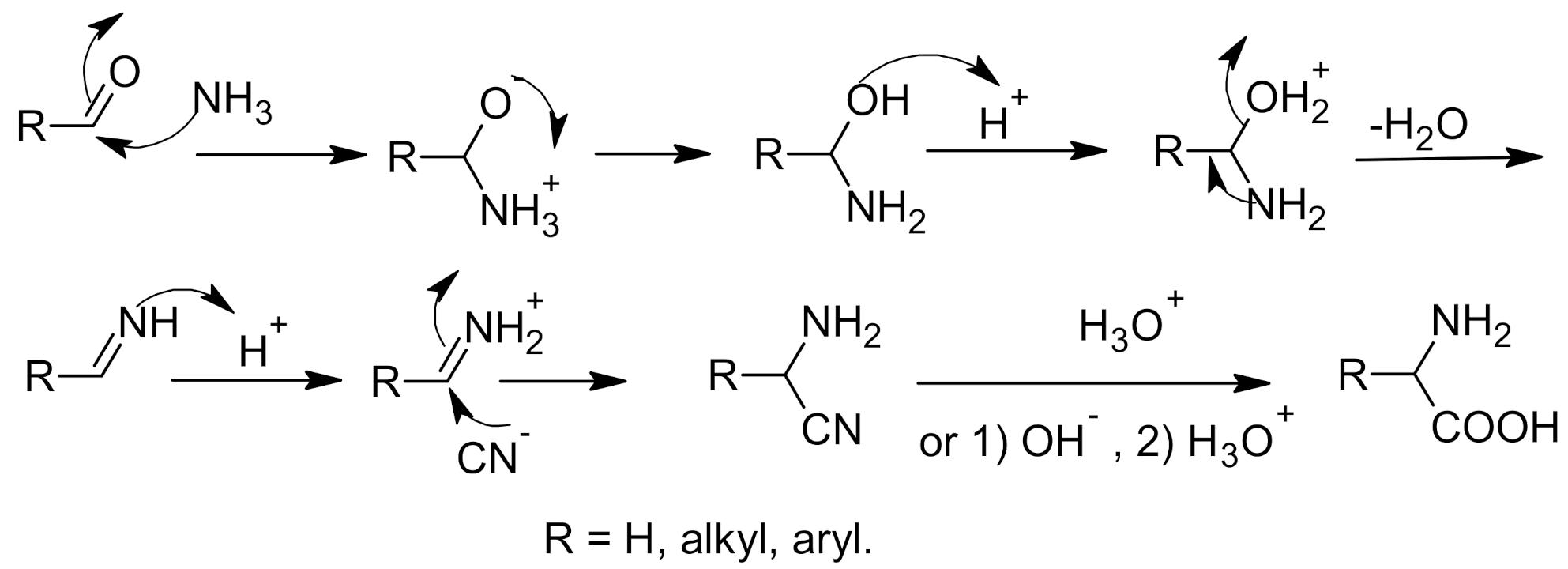
2. Strecker Reaction
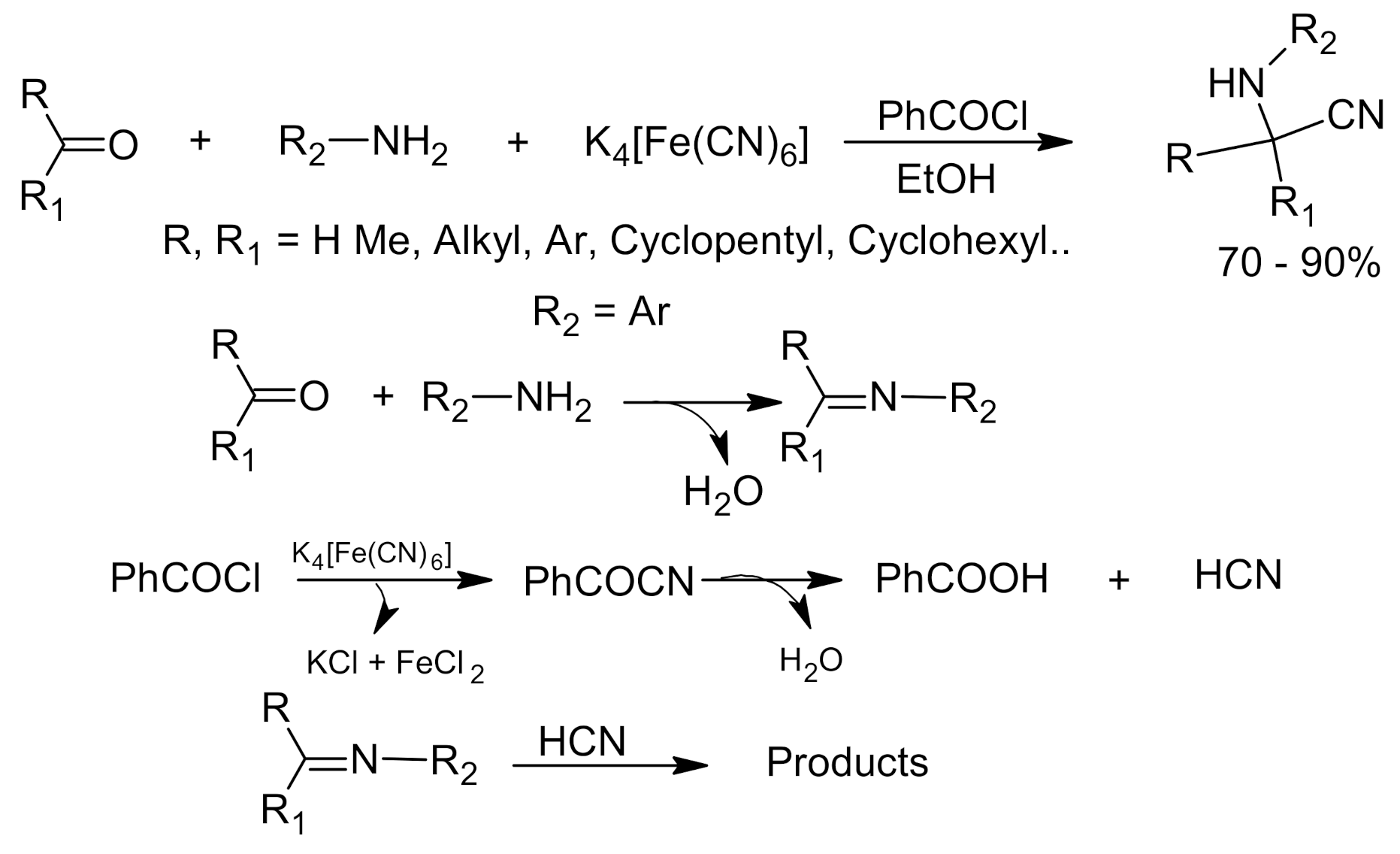
3. Green Strecker Reaction
4. Asymmetric Strecker Reaction
5. Petasis Reaction
6. Asymmetric Petasis Reaction
7. Discussion
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Bada, J. Strecker Synthesis. In Encyclopedia of Astrobiology; Gargaud, M., Amils, R., Quintanilla, J.C., Cleaves, H.J., II, Irvine, W.M., Pinti, D.L., Viso, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; p. 1603. [Google Scholar] [CrossRef]
- Strecker, A. Ueber die künstliche Bildung der Milchsäure und einen neuen, dem Glycocoll homologen Körper. Justus Liebigs Ann. Der Chem. 1850, 75, 27–45. [Google Scholar] [CrossRef]
- Strecker, A. Ueber einen neuen aus Aldehyd-Ammoniak und Blausäure entstehenden Körper. Justus Liebigs Ann. Der Chem. 1854, 91, 349–351. [Google Scholar] [CrossRef]
- Pascal, R. Bücherer–Bergs Synthesis. In Encyclopedia of Astrobiology; Gargaud, M., Amils, R., Quintanilla, J.C., Cleaves, H.J., II, Irvine, W.M., Pinti, D.L., Viso, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 221–222. [Google Scholar] [CrossRef]
- Monteiro, J.L.; Pieber, B.; Corrêa, A.G.; Kappe, C.O. Continuous synthesis of hydantoins: Intensifying the Bucherer–Bergs reaction. Synlett 2016, 27, 83–87. [Google Scholar] [CrossRef]
- Ashe, K.; Fernández-García, C.; Corpinot, M.K.; Coggins, A.J.; Bučar, D.-K.; Powner, M.W. Selective prebiotic synthesis of phosphoroaminonitriles and aminothioamides in neutral water. Commun. Chem. 2019, 2, 23. [Google Scholar] [CrossRef]
- Williams, R.M.; Hendrix, J.A. Asymmetric synthesis of arylglycines. Chem. Rev. 1992, 92, 889–917. [Google Scholar] [CrossRef]
- Ivanov, K.; Ivanova, S.; Georgieva, M.; Atanasov, P. Production and regulatory analytical control of amino acids include in food additives. Pharmacia 2014, 61, 48–54. [Google Scholar]
- Yasufumi, O.; Tetsuro, S. Asymmetric Strecker Route toward the Synthesis of Biologically Active α,α-Disubstituted α-Amino Acids. Bull. Chem. Soc. Jpn. 2003, 76, 1115–1129. [Google Scholar] [CrossRef]
- Arasappan, A.; Venkatraman, S.; Padilla, A.I.; Wu, W.; Meng, T.; Jin, Y.; Wong, J.; Prongay, A.; Girijavallabhan, V.; George, N.F. Practical and efficient method for amino acid derivatives containing β-quaternary center: Application toward synthesis of hepatitis C virus NS3 serine protease inhibitors. Tetrahedron Lett. 2007, 48, 6343–6347. [Google Scholar] [CrossRef]
- Petasis, N.A.; Akritopoulou, I. The boronic acid mannich reaction: A new method for the synthesis of geometrically pure allylamines. Tetrahedron Lett. 1993, 34, 583–586. [Google Scholar] [CrossRef]
- Hu, X.; Ma, Y.; Li, Z. Eco-friendly synthesis of α-aminonitriles from ketones in PEG-400 medium using potassium Hexacyanoferrate(II) as cyanide source. J. Organomet. Chem. 2012, 705, 70–74. [Google Scholar] [CrossRef]
- Ivon, Y.M.; Tymtsunik, A.V.; Komarov, I.V.; Shishkin, O.V.; Grygorenko, O.O. Synthesis of a 2, 5-Diazabicyclo [2.2. 1] heptane-Derived α, β-Diamino Acid. Synth. Stuttg. 2015, 47, 1123–1130. [Google Scholar] [CrossRef]
- Van Hijfte, L.; Heydt, V.; Kolb, M. A versatile entry into the synthesis of α-(monofluoromethyl) amino acids: Preparation of α-(monofluoromethyl) serine and (E)-dehydro-α-(monofluoromethyl) ornithine. Tetrahedron Lett. 1993, 34, 4793–4796. [Google Scholar] [CrossRef]
- Razafindrabe, C.R.; Aubry, S.; Bourdon, B.; Andriantsiferana, M.; Pellet-Rostaing, S.; Lemaire, M. Synthesis of (±)-phthalascidin 650 analogue: New synthetic route to (±)-phthalascidin 622. Tetrahedron 2010, 66, 9061–9066. [Google Scholar] [CrossRef]
- Myers, A.G.; Kung, D.W. One-Step Construction of the Pentacyclic Skeleton of Saframycin A from a “Trimer” of α-Amino Aldehydes. Org. Lett. 2000, 2, 3019–3022. [Google Scholar] [CrossRef]
- Aoki, K.; Ijima, T.; Kamiyama, H.; Kamiko, K.; Terauchi, Y. Anagliptin decreases serum lathosterol level in patients with type 2 diabetes: A pilot study. Expert Opin. Pharmacother. 2015, 16, 1749–1754. [Google Scholar] [CrossRef] [PubMed]
- Augeri, D.J.; Robl, J.A.; Betebenner, D.A.; Magnin, D.R.; Khanna, A.; Robertson, J.G.; Wang, A.; Simpkins, L.M.; Taunk, P.; Huang, Q.; et al. Discovery and Preclinical Profile of Saxagliptin (BMS-477118): A Highly Potent, Long-Acting, Orally Active Dipeptidyl Peptidase IV Inhibitor for the Treatment of Type 2 Diabetes. J. Med. Chem. 2005, 48, 5025–5037. [Google Scholar] [CrossRef]
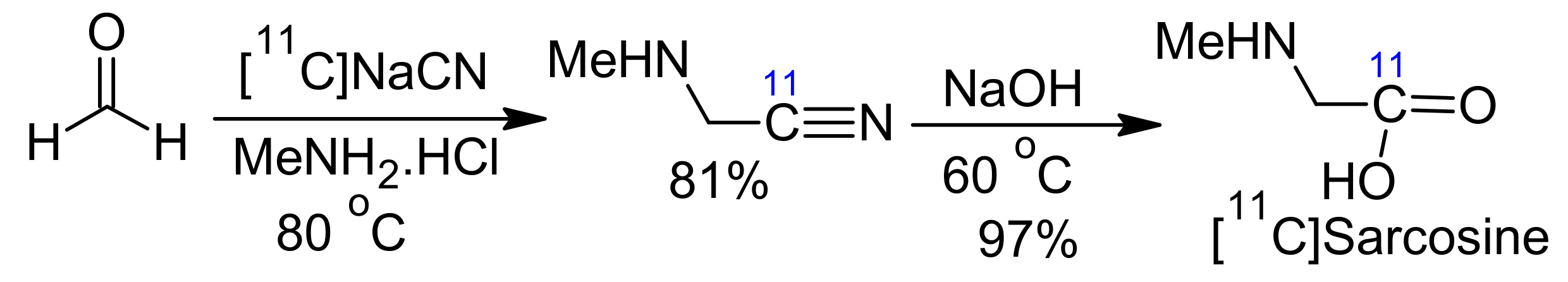
- Xing, J.; Brooks, A.F.; Fink, D.; Zhang, H.; Piert, M.R.; Scott, P.J.; Shao, X. High-yielding automated convergent synthesis of no-carrier-added [11C-carbonyl]-labeled amino acids using the Strecker Reaction. Synlett Acc. Rapid Commun. Synth. Org. Chem. 2017, 28, 371. [Google Scholar] [CrossRef][Green Version]
- Song, F.; Salter, R.; Weaner, L.E. A short synthesis of d-[1-14C]-serine of high enantiomeric purity. J. Label. Compd. Radiopharm. 2015, 58, 173–176. [Google Scholar] [CrossRef]
- Bandak, D.; Babii, O.; Vasiuta, R.; Komarov, I.V.; Mykhailiuk, P.K. Design and synthesis of novel 19F-amino acid: A promising 19F NMR label for peptide studies. Org. Lett. 2015, 17, 226–229. [Google Scholar] [CrossRef]
- Li, Z.; Ma, Y.; Xu, J.; Shi, J.; Cai, H. One-pot three-component synthesis of α-aminonitriles using potassium hexacyanoferrate(II) as an eco-friendly cyanide source. Tetrahedron Lett. 2010, 51, 3922–3926. [Google Scholar] [CrossRef]
- Poliakoff, M.; Licence, P. Green chemistry. Nature 2007, 450, 810–812. [Google Scholar] [CrossRef]
- Grundke, C.; Opatz, T. Strecker reactions with hexacyanoferrates as non-toxic cyanide sources. Green Chem. 2019, 21, 2362–2366. [Google Scholar] [CrossRef]
- Li, Z.; Li, R.; Zheng, H.; Wen, F.; Li, H.; Yin, J.; Yang, J. Hydrocyanation of sulfonylimines using potassium hexacyanoferrate(II) as an eco-friendly cyanide source. J. Braz. Chem. Soc. 2013, 24, 1739–1743. [Google Scholar] [CrossRef]
- Pechenyuk, S.I.; Domonov, D.P.; Shimkin, A.A.; Ivanov, Y.V. Thermal decomposition of iron cyano complexes in an inert atmosphere. Russ. Chem. Bull. 2015, 64, 322–328. [Google Scholar] [CrossRef]
- Kuhn, D.D.; Young, T.C. Photolytic degradation of hexacyanoferrate (II) in aqueous media: The determination of the degradation kinetics. Chemosphere 2005, 60, 1222–1230. [Google Scholar] [CrossRef]
- Bolm, C.; Mocci, R.; Schumacher, C.; Turberg, M.; Puccetti, F.; Hernández, J.G. Mechanochemical Activation of Iron Cyano Complexes: A Prebiotic Impact Scenario for the Synthesis of α-Amino Acid Derivatives. Angew. Chem. 2018, 130, 2447–2450. [Google Scholar] [CrossRef]
- Nauth, A.M.; Otto, N.; Opatz, T. α-Cyanation of Aromatic Tertiary Amines using Ferricyanide as a Non-Toxic Cyanide Source. Adv. Synth. Catal. 2015, 357, 3424–3428. [Google Scholar] [CrossRef]
- Li, Z.; Bohle, D.S.; Li, C.-J. Cu-catalyzed cross-dehydrogenative coupling: A versatile strategy for C–C bond formations via the oxidative activation of sp3 C–H bonds. Proc. Natl. Acad. Sci. USA 2006, 103, 8928–8933. [Google Scholar] [CrossRef]
- D’Este, M.; Alvarado-Morales, M.; Angelidaki, I. Amino acids production focusing on fermentation technologies–A review. Biotechnol. Adv. 2018, 36, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, S.; Yang, X.; Qiu, L.; Gao, B.; Li, R.; Chen, J. Reactive extraction of amino acids mixture in hydrolysate from cottonseed meal with di(2-ethylhexyl) phosphoric acid. J. Chem. Technol. Biotechnol. 2016, 91, 483–489. [Google Scholar] [CrossRef]
- Kim, Y.; Park, J.; Kim, M.-J. Dynamic Kinetic Resolution of Amines and Amino Acids by Enzyme–Metal Cocatalysis. ChemCatChem 2011, 3, 271–277. [Google Scholar] [CrossRef]
- Miyazawa, T. Enzymatic resolution of amino acids via ester hydrolysis. Amino Acids 1999, 16, 191–213. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-T.; Huang, W.-H.; Wang, K.-T. Resolution of Amino Acids in a Mixture of 2-Methyl-2-propanol/water (19:1) Catalyzed by Alcalase via in Situ Racemization of One Antipode Mediated by Pyridoxal 5-Phosphate. J. Org. Chem. 1994, 59, 7580–7581. [Google Scholar] [CrossRef]
- Wang, J.; Liu, X.; Feng, X. Asymmetric strecker reactions. Chem. Rev. 2011, 111, 6947–6983. [Google Scholar] [CrossRef]
- Harada, K. Asymmetric Synthesis of α-Amino-acids by the Strecker Synthesis. Nature 1963, 200, 1201. [Google Scholar] [CrossRef]
- Kouznetsov, V.V.; Galvis, C.E.P. Strecker reaction and α-amino nitriles: Recent advances in their chemistry, synthesis, and biological properties. Tetrahedron 2018, 74, 773–810. [Google Scholar] [CrossRef]
- Kunz, H.; Sager, W.; Pfrengle, W.; Schanzenbach, D. Reversal of asymmetric induction in stereoselective strecker synthesis on galactosyl amine as the chiral matrix. Tetrahedron Lett. 1988, 29, 4397–4400. [Google Scholar] [CrossRef]
- Ma, D.; Tian, H.; Zou, G. Asymmetric Strecker-Type Reaction of α-Aryl Ketones. Synthesis of (S)-αM4CPG, (S)-MPPG, (S)-AIDA, and (S)-APICA, the Antagonists of Metabotropic Glutamate Receptors. J. Org. Chem. 1999, 64, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Ding, K. Synthesis of Enantiopure α,α-Disubstituted Amino Acids from the Asymmetric Strecker Reaction Products of Aldehydes. Org. Lett. 2000, 2, 2515–2517. [Google Scholar] [CrossRef]
- Robak, M.T.; Herbage, M.A.; Ellman, J.A. Synthesis and Applications of tert-Butanesulfinamide. Chem. Rev. 2010, 110, 3600–3740. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Xu, M.-H. Lewis acid promoted diastereoselective addition of TMSCN and TMSCF3 to isatin-derived N-sulfinyl ketimines: Synthesis of optically active tetrasubstituted 3-aminooxindoles. J. Org. Chem. 2014, 79, 7746–7751. [Google Scholar] [CrossRef]
- Cai, X.-H.; Xie, B. Recent advances in asymmetric Strecker reactions. Arkivoc 2014, 1, 205–248. [Google Scholar] [CrossRef]
- de Bruin, G.; Mock, E.D.; Hoogendoorn, S.; van den Nieuwendijk, A.M.; Mazurek, J.; van der Marel, G.A.; Florea, B.I.; Overkleeft, H.S. Enantioselective synthesis of adamantylalanine and carboranylalanine and their incorporation into the proteasome inhibitor bortezomib. Chem. Commun. 2016, 52, 4064–4067. [Google Scholar] [CrossRef] [PubMed]
- Iyer, M.S.; Gigstad, K.M.; Namdev, N.D.; Lipton, M. Asymmetric catalysis of the Strecker amino acid synthesis by a cyclic dipeptide. Amino Acids 1996, 11, 259–268. [Google Scholar] [CrossRef]
- Zuend, S.J.; Coughlin, M.P.; Lalonde, M.P.; Jacobsen, E.N. Scaleable catalytic asymmetric Strecker syntheses of unnatural α-amino acids. Nature, 2009; 461, 968–970. [Google Scholar] [CrossRef]
- Sigman, M.S.; Jacobsen, E.N. Enantioselective Addition of Hydrogen Cyanide to Imines Catalyzed by a Chiral (Salen)Al(III) Complex. J. Am. Chem. Soc. 1998, 120, 5315–5316. [Google Scholar] [CrossRef]
- Kobayashi, S.; Ishitani, H. Novel binuclear chiral zirconium catalysts used in enantioselective strecker reactions. Chirality Pharmacol. Biol. Chem. Conseq. Mol. Asymmetry 2000, 12, 540–543. [Google Scholar] [CrossRef]
- Sadhukhan, A.; Saravanan, S.; Khan, N.-u.H.; Kureshy, R.I.; Abdi, S.H.; Bajaj, H.C. Modified Asymmetric Strecker Reaction of Aldehyde with Secondary Amine: A Protocol for the Synthesis of S-Clopidogrel (An Antiplatelet Agent). J. Org. Chem. 2012, 77, 7076–7080. [Google Scholar] [CrossRef]
- Qiu, X.-L.; Qing, F.-L. Recent Advances in the Synthesis of Fluorinated Amino Acids. Eur. J. Org. Chem. 2011, 2011, 3261–3278. [Google Scholar] [CrossRef]
- Liu, Y.-L.; Yin, X.-P.; Zhou, J. Internally Reuse Waste: Catalytic Asymmetric One-Pot Strecker Reaction of Fluoroalkyl Ketones, Anilines and TMSCN by Sequential Catalysis. Chin. J. Chem. 2018, 36, 321–328. [Google Scholar] [CrossRef]
- Kawasaki, T.; Takamatsu, N.; Aiba, S.; Tokunaga, Y. Spontaneous formation and amplification of an enantioenriched α-amino nitrile: A chiral precursor for Strecker amino acid synthesis. Chem. Commun. 2015, 51, 14377–14380. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, S.; Yoshimura, K.; Yamazaki, Y.; Takamatsu, N.; Kuraishi, T.; Aiba, S.; Tokunaga, Y.; Kawasaki, T. Asymmetric Strecker Reaction Arising from the Molecular Orientation of an Achiral Imine at the Single-Crystal Face: Enantioenriched l-and d-Amino Acids. Angew. Chem. Int. Ed. 2017, 56, 1055–1058. [Google Scholar] [CrossRef] [PubMed]
- Sögütoglu, L.-C.; Steendam, R.R.; Meekes, H.; Vlieg, E.; Rutjes, F.P. Viedma ripening: A reliable crystallisation method to reach single chirality. Chem. Soc. Rev. 2015, 44, 6723–6732. [Google Scholar] [CrossRef] [PubMed]
- Viedma, C. Chiral symmetry breaking during crystallization: Complete chiral purity induced by nonlinear autocatalysis and recycling. Phys. Rev. Lett. 2005, 94, 065504. [Google Scholar] [CrossRef] [PubMed]
- Baglai, I.; Leeman, M.; Wurst, K.; Kaptein, B.; Kellogg, R.M.; Noorduin, W.L. The Strecker reaction coupled to Viedma ripening: A simple route to highly hindered enantiomerically pure amino acids. Chem. Commun. 2018, 54, 10832–10834. [Google Scholar] [CrossRef]
- van der Meijden, M.W.; Leeman, M.; Gelens, E.; Noorduin, W.L.; Meekes, H.; van Enckevort, W.J.P.; Kaptein, B.; Vlieg, E. Kellogg, R.M. Attrition-Enhanced Deracemization in the Synthesis of Clopidogrel-A Practical Application of a New Discovery. Org. Process. Res. Dev. 2009, 13, 1195–1198. [Google Scholar] [CrossRef]
- Jumbam, N.D.; Masamba, W. Bio-Catalysis in Multicomponent Reactions. Molecules 2020, 25, 5935. [Google Scholar] [CrossRef]
- Vongvilai, P.; Ramström, O. Dynamic Asymmetric Multicomponent Resolution: Lipase-Mediated Amidation of a Double Dynamic Covalent System. J. Am. Chem. Soc. 2009, 131, 14419–14425. [Google Scholar] [CrossRef]
- Chrzanowska, M.; Grajewska, A.; Meissner, Z.; Rozwadowska, M.; Wiatrowska, I. A concise synthesis of tetrahydroisoquinoline-1-carboxylic acids using a Petasis reaction and Pomeranz–Fritsch–Bobbitt cyclization sequence. Tetrahedron 2012, 68, 3092–3097. [Google Scholar] [CrossRef]
- Wu, P.; Givskov, M.; Nielsen, T.E. Reactivity and Synthetic Applications of Multicomponent Petasis Reactions. Chem. Rev. 2019, 119, 11245–11290. [Google Scholar] [CrossRef] [PubMed]
- Boguszewski, P.A.; Davies, J.W.; Marsh, P.A.; Williamson, M. MEDI 309-Polymer assisted, high throughput methods for Petasis and Ugi reactions. In Abstracts of Papers of the American Chemical Society; The American Chemical Society: Washington, DC, USA, 2008. [Google Scholar]
- Zhang, J.; Yun, F.; Xie, R.; Cheng, C.; Chen, G.; Li, J.; Tang, P.; Yuan, Q. Petasis three-component reaction accelerated by trifluoroacetic acid: Synthesis of indoline-derived glycines. Tetrahedron Lett. 2016, 57, 3916–3919. [Google Scholar] [CrossRef]
- Frauenlob, R.; García, C.; Bradshaw, G.A.; Burke, H.M.; Bergin, E. A Copper-Catalyzed Petasis Reaction for the Synthesis of Tertiary Amines and Amino Esters. J. Org. Chem. 2012, 77, 4445–4449. [Google Scholar] [CrossRef]
- Cornier, P.G.; Delpiccolo, C.M.L.; Boggián, D.B.; Mata, E.G. Solid-phase Petasis multicomponent reaction for the generation of β-lactams 3-substituted with non-proteinogenic α-amino acids. Tetrahedron Lett. 2013, 54, 4742–4745. [Google Scholar] [CrossRef]
- Potowski, M.; Esken, R.; Brunschweiger, A. Translation of the copper/bipyridine-promoted Petasis reaction to solid phase-coupled DNA for encoded library synthesis. Biorg. Med. Chem. 2020, 28, 115441. [Google Scholar] [CrossRef] [PubMed]
- Petasis, N.A.; Goodman, A.; Zavialov, I.A. A new synthesis of α-arylglycines from aryl boronic acids. Tetrahedron 1997, 53, 16463–16470. [Google Scholar] [CrossRef]
- Liepouri, F.; Bernasconi, G.; Petasis, N.A. Component-Selective and Stereocontrolled One-Step Three-Component Reaction among Aldehydes, Amines, and Allenyl Boronic Acids or Allenyl Pinacolboronates. Org. Lett. 2015, 17, 1628–1631. [Google Scholar] [CrossRef]
- Murafuji, T.; Tasaki, Y.; Fujinaga, M.; Tao, K.; Kamijo, S.; Ishiguro, K. Blue Amino Acids Derived from Azulen-1-ylboronic Acid Pinacol Ester via the Petasis Reaction. Synthesis 2017, 49, 1037–1042. [Google Scholar] [CrossRef]
- Petasis, N.A.; Zavialov, I.A. A New and Practical Synthesis of α-Amino Acids from Alkenyl Boronic Acids. J. Am. Chem. Soc. 1997, 119, 445–446. [Google Scholar] [CrossRef]
- Tao, C.-Z.; Zhang, Z.-T.; Wu, J.-W.; Li, R.-H.; Cao, Z.-L. Synthesis of unnatural N-glycosyl α-amino acids via Petasis reaction. Chin. Chem. Lett. 2014, 25, 532–534. [Google Scholar] [CrossRef]
- Naskar, D.; Roy, A.; Seibel, W.L.; Portlock, D.E. Hydroxylamines and sulfinamide as amine components in the Petasis boronic acid–Mannich reaction: Synthesis of N-hydroxy or alkoxy-α-aminocarboxylicacids and N-(tert-butyl sulfinyl)-α-amino carboxylicacids. Tetrahedron Lett. 2003, 44, 8865–8868. [Google Scholar] [CrossRef]
- Nielsen, S.D.; Smith, G.P.; Begtrup, M.; Kristensen, J.L. Synthesis of N-alkylated amino acids using fluorous-tagged hydroxylamines. Tetrahedro 2011, 67, 5261–5267. [Google Scholar] [CrossRef]
- Dhudshia, B.; Tiburcio, J.; Thadani, A.N. Diastereoselective allylation and crotylation of N-unsubstituted imines derived from ketones. Chem. Commun. 2005, 5551–5553. [Google Scholar] [CrossRef]
- Diehl, A.M.; Ouadoudi, O.; Andreadou, E.; Manolikakes, G. Sulfonamides as Amine Component in the Petasis-Borono Mannich Reaction: A Concise Synthesis of α-Aryl-and α-Alkenylglycine Derivatives. Synthesis 2018, 50, 3936–3946. [Google Scholar] [CrossRef]
- Candeias, N.R.; Cal, P.M.S.D.; André, V.; Duarte, M.T.; Veiros, L.F.; Gois, P.M.P. Water as the reaction medium for multicomponent reactions based on boronic acids. Tetrahedron 2010, 66, 2736–2745. [Google Scholar] [CrossRef]
- Ishiyama, T.; Matsuda, N.; Murata, M.; Ozawa, F.; Suzuki, A.; Miyaura, N. Platinum(0)-Catalyzed Diboration of Alkynes with Tetrakis(alkoxo)diborons: An Efficient and Convenient Approach to cis-Bis(boryl)alkenes. Organometallics 1996, 15, 713–720. [Google Scholar] [CrossRef]
- Sridhar, T.; Berrée, F.; Sharma, G.V.M.; Carboni, B. Regio- and Stereocontrolled Access to γ-Boronated Unsaturated Amino Esters and Derivatives from (Z)-Alkenyl 1,2-Bis(boronates). J. Org. Chem. 2014, 79, 783–789. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, Y.; Miyabe, H.; Takemoto, Y. Catalytic Enantioselective Petasis-Type Reaction of Quinolines Catalyzed by a Newly Designed Thiourea Catalyst. J. Am. Chem. Soc. 2007, 129, 6686–6687. [Google Scholar] [CrossRef] [PubMed]
- Lou, S.; Schaus, S.E. Asymmetric Petasis Reactions Catalyzed by Chiral Biphenols. J. Am. Chem. Soc. 2008, 130, 6922–6923. [Google Scholar] [CrossRef] [PubMed]
- Churches, Q.I.; Stewart, H.E.; Cohen, S.B.; Shröder, A.; Turner, P.; Hutton, C.A. Stereoselectivity of the Petasis reaction with various chiral amines and styrenylboronic acids. Pure Appl. Chem. 2008, 80, 687–694. [Google Scholar] [CrossRef]
- Churches, Q.I.; White, J.M.; Hutton, C.A. Synthesis of β,γ-Dihydroxyhomotyrosines by a Tandem Petasis–Asymmetric Dihydroxylation Approach. Org. Lett. 2011, 13, 2900–2903. [Google Scholar] [CrossRef]
- Li, Y.; Xu, M.-H. Lewis Acid Promoted Highly Diastereoselective Petasis Borono-Mannich Reaction: Efficient Synthesis of Optically Active β,γ-Unsaturated α-Amino Acids. Org. Lett. 2012, 14, 2062–2065. [Google Scholar] [CrossRef]
- Inokuma, T.; Suzuki, Y.; Sakaeda, T.; Takemoto, Y. Synthesis of Optically Active N-Aryl Amino Acid Derivatives through the Asymmetric Petasis Reaction Catalyzed by a Novel Hydroxy–Thiourea Catalyst. Chem. An Asian J. 2011, 6, 2902–2906. [Google Scholar] [CrossRef]
- Morozova, V.A.; Beletskaya, I.P.; Titanyuk, I.D. Synthesis of enantiopure cyclic amino acid derivatives via a sequential diastereoselective Petasis reaction/ring closing olefin metathesis process. Tetrahedron: Asymmetry 2017, 28, 349–354. [Google Scholar] [CrossRef]
- Bułyszko, I.; Chrzanowska, M.; Grajewska, A. Rozwadowska, M.D. Synthesis of (+)-6,7-Dimethoxy-1,2,3,4-tetrahydroisoquinoline-1-carboxylic Acid, a Diastereoselective Approach. Eur. J. Org. Chem. 2015, 2015, 383–388. [Google Scholar] [CrossRef]
- Koolmeister, T.; Södergren, M.; Scobie, M. The first example of chiral induction using homochiral boronic esters in the Petasis reaction. Tetrahedron Lett. 2002, 43, 5969–5970. [Google Scholar] [CrossRef]
- Wang, Y.; Guan, R.; Sivaguru, P.; Cong, X.; Bi, X. Silver-Catalyzed anti-Markovnikov Hydroboration of C–C Multiple Bonds. Org. Lett. 2019, 21, 4035–4038. [Google Scholar] [CrossRef]
- Agahi, R.; Challinor, A.J.; Carter, N.B.; Thomas, S.P. Earth-abundant metal catalysis enabled by counterion activation. Org. Lett. 2019, 21, 993–997. [Google Scholar] [CrossRef]
- Jang, W.J.; Song, S.M.; Moon, J.H.; Lee, J.Y.; Yun, J. Copper-catalyzed enantioselective hydroboration of unactivated 1, 1-disubstituted alkenes. J. Am. Chem. Soc. 2017, 139, 13660–13663. [Google Scholar] [CrossRef] [PubMed]
- Joshi-Pangu, A.; Ma, X.; Diane, M.; Iqbal, S.; Kribs, R.J.; Huang, R.; Wang, C.-Y.; Biscoe, M.R. Palladium-catalyzed borylation of primary alkyl bromides. J. Org. Chem. 2012, 77, 6629–6633. [Google Scholar] [CrossRef] [PubMed]
- Dudnik, A.S.; Fu, G.C. Nickel-catalyzed coupling reactions of alkyl electrophiles, including unactivated tertiary halides, to generate carbon–boron bonds. J. Am. Chem. Soc. 2012, 134, 10693–10697. [Google Scholar] [CrossRef]
- Muncipinto, G.; Moquist, P.N.; Schreiber, S.L.; Schaus, S.E. Catalytic Diastereoselective Petasis Reactions. Angew. Chem. Int. Ed. 2011, 50, 8172–8175. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.-C.; Wang, Y.; Li, E.-Q.; Cui, H.; Duan, Z. Enantio- and Diastereoselective Synthesis of β-Aryl-β-pyrazolyl α-Amino Acid Esters via Copper-Catalyzed Reaction of Azomethine Ylides with Benzylidenepyrazolones. Adv. Synth. Catal. 2019, 361, 1389–1393. [Google Scholar] [CrossRef]
- Menor-Salván, C. From the dawn of organic chemistry to astrobiology: Urea as a foundational component in the origin of nucleobases and nucleotides. In Prebiotic Chemistry and Chemical Evolution of Nucleic Acids; Springer: Berlin/Heidelberg, Germany, 2018; pp. 85–142. [Google Scholar] [CrossRef]
- Hall, D.G. Structure, properties, and preparation of boronic acid derivatives. Overview of their reactions and applications. Boronic Acids 2005, 1. [Google Scholar] [CrossRef]




























Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masamba, W. Petasis vs. Strecker Amino Acid Synthesis: Convergence, Divergence and Opportunities in Organic Synthesis. Molecules 2021, 26, 1707. https://doi.org/10.3390/molecules26061707
Masamba W. Petasis vs. Strecker Amino Acid Synthesis: Convergence, Divergence and Opportunities in Organic Synthesis. Molecules. 2021; 26(6):1707. https://doi.org/10.3390/molecules26061707
Chicago/Turabian StyleMasamba, Wayiza. 2021. "Petasis vs. Strecker Amino Acid Synthesis: Convergence, Divergence and Opportunities in Organic Synthesis" Molecules 26, no. 6: 1707. https://doi.org/10.3390/molecules26061707
APA StyleMasamba, W. (2021). Petasis vs. Strecker Amino Acid Synthesis: Convergence, Divergence and Opportunities in Organic Synthesis. Molecules, 26(6), 1707. https://doi.org/10.3390/molecules26061707