
Structure-Based Virtual Screening Identifies Multiple Stable Binding Sites at the RecA Domains of SARS-CoV-2 Helicase Enzyme


 ,
, 

Abstract
1. Introduction
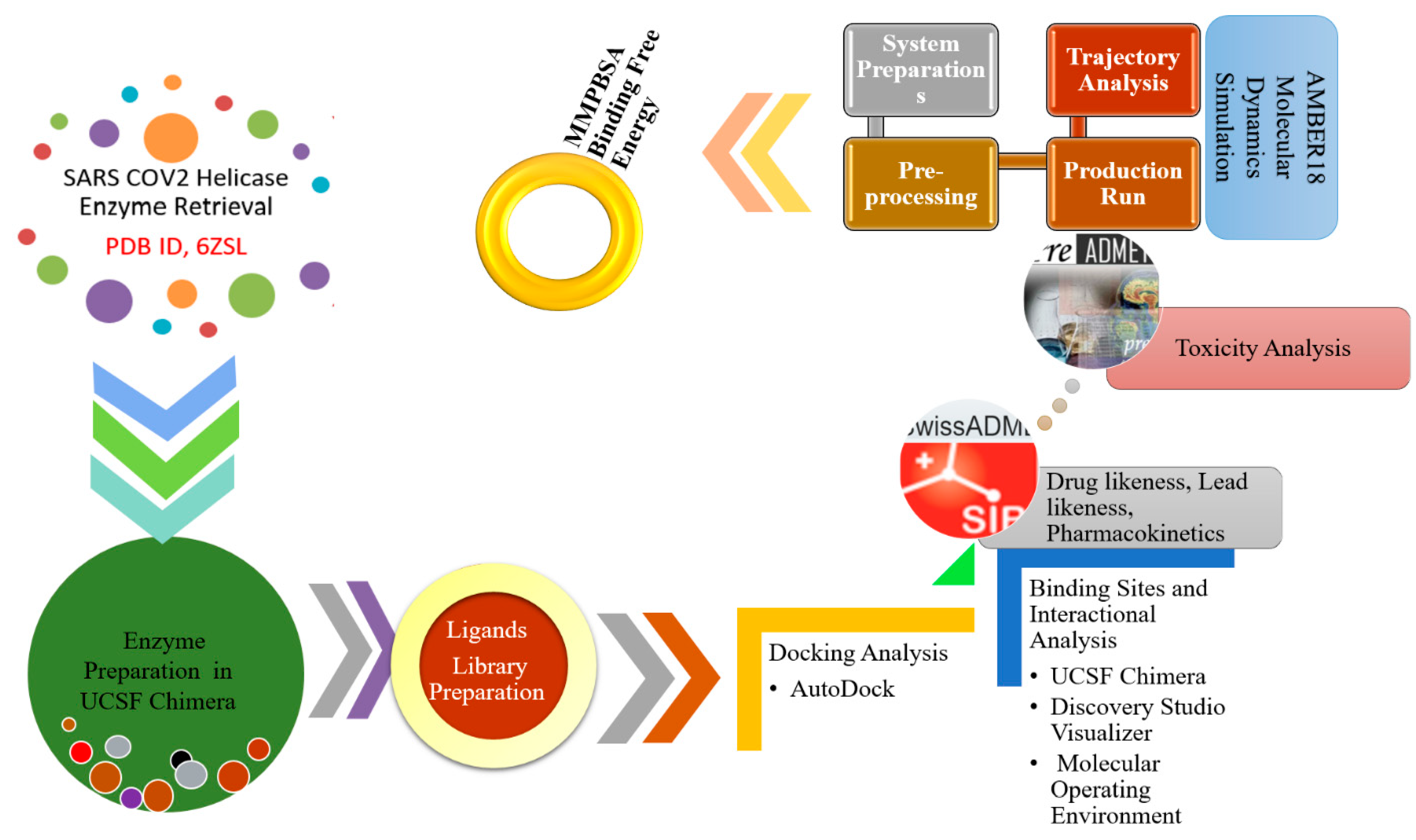
2. Materials and Methods
2.1. Preparation of the SARS-CoV-2 Helicase Structure
2.2. Phytochemicals Library Preparation
2.3. Binding Conformational Analysis
2.4. Molecular Dynamics (MD) Simulation
2.5. Free Binding Energy Calculations via MMPB/GBSA
3. Results and Discussions
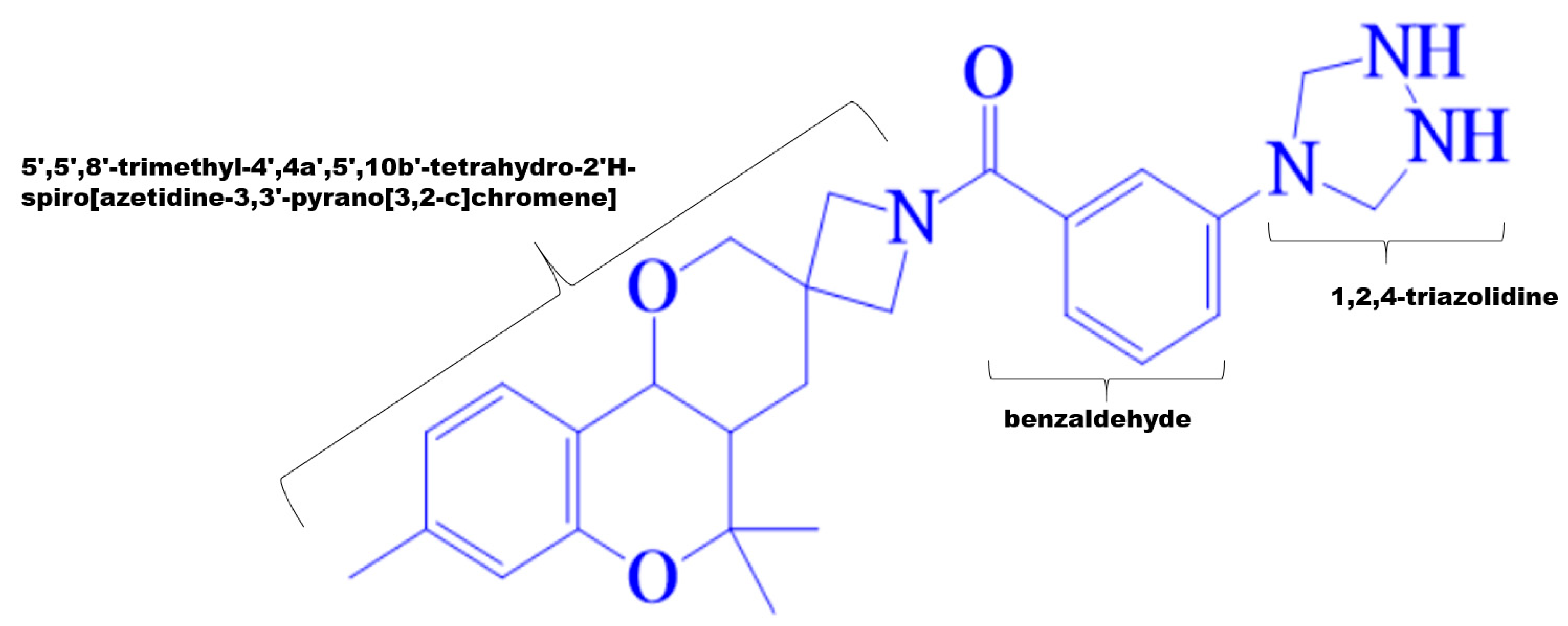
3.1. Virtual Screening of MPD3 Database
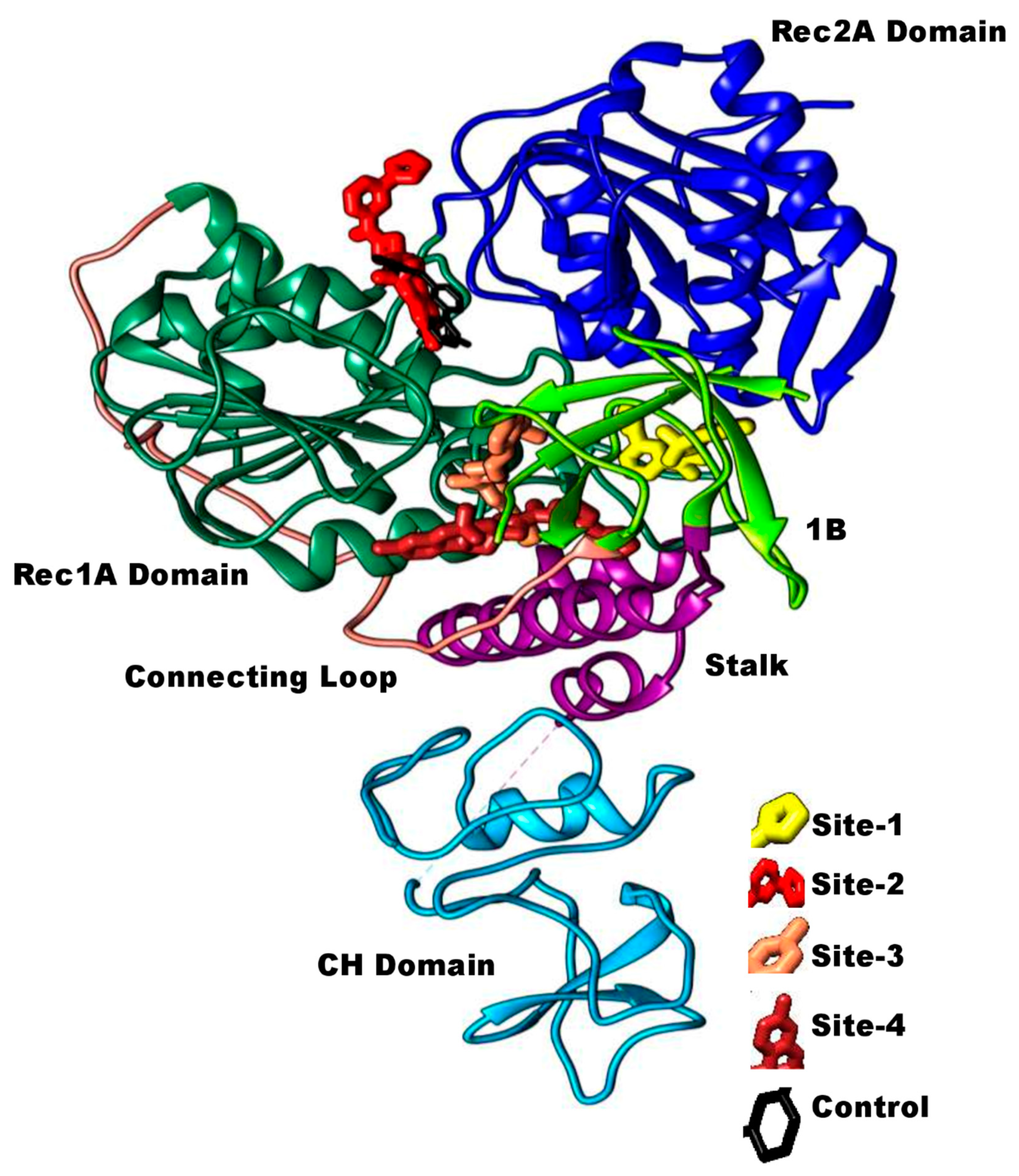
3.2. Comparative Binding Sites and Conformational Analysis
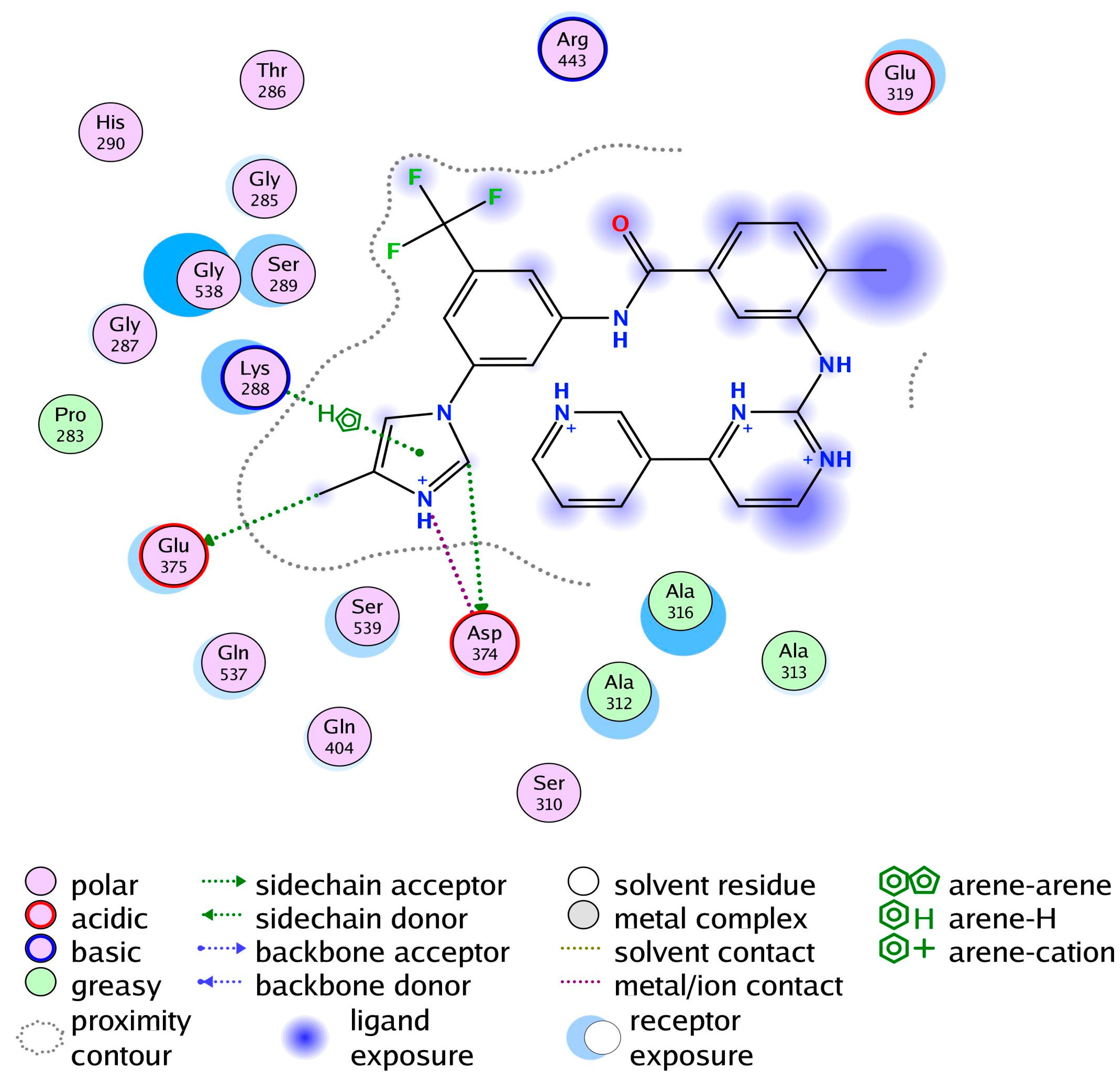
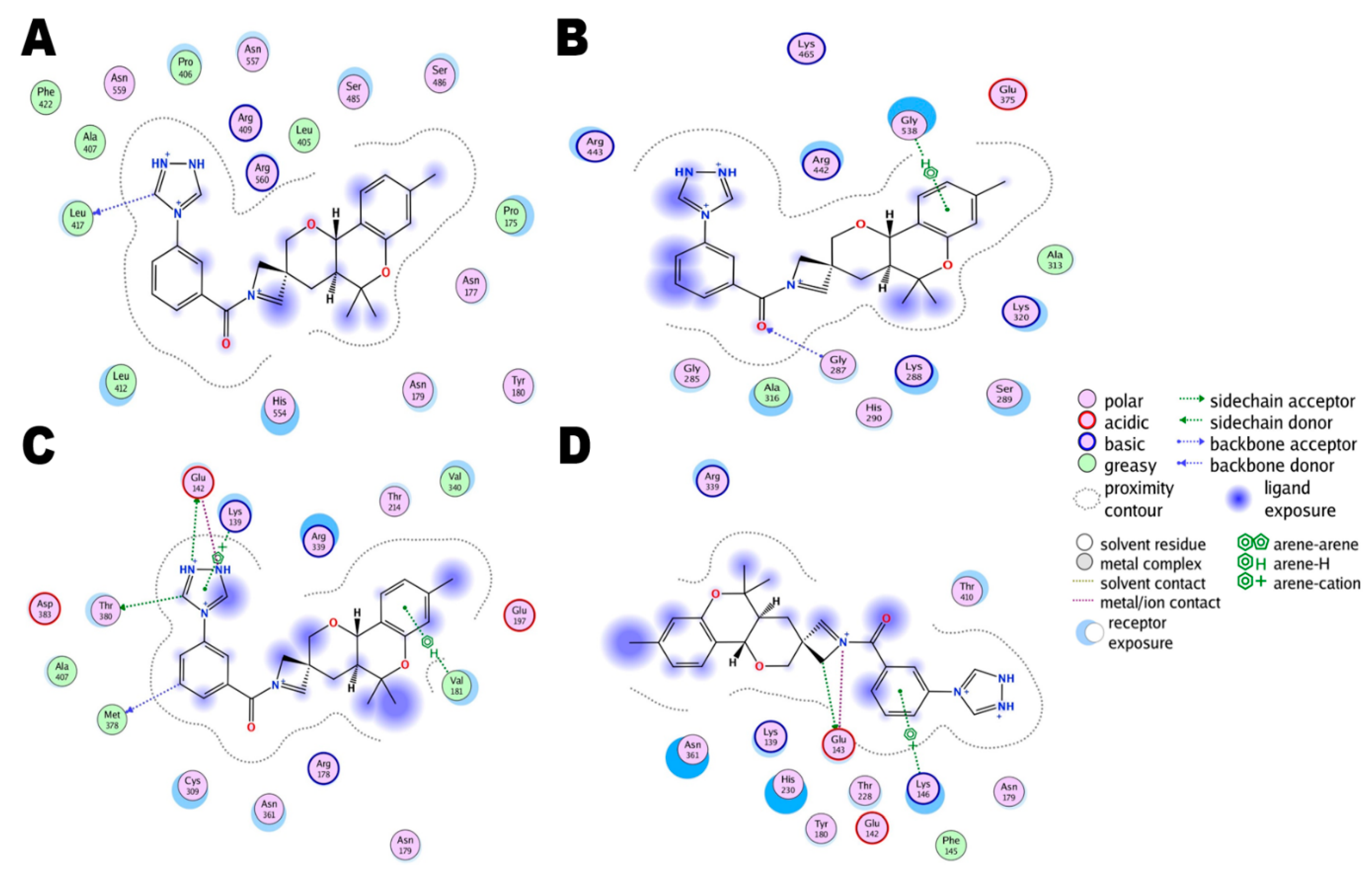
3.3. Comparative Chemical Interactions Analysis
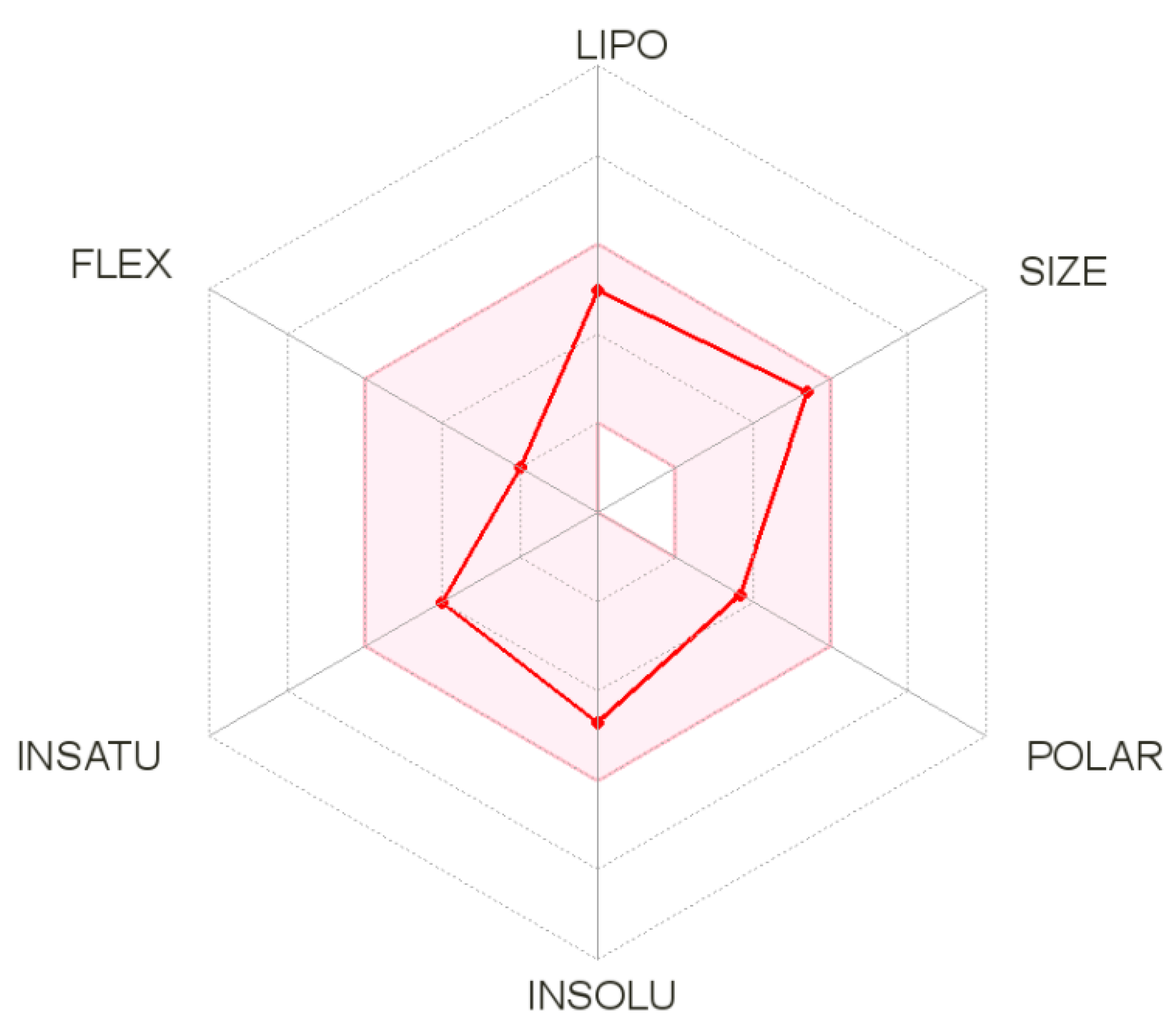
3.4. SwissADME Analysis
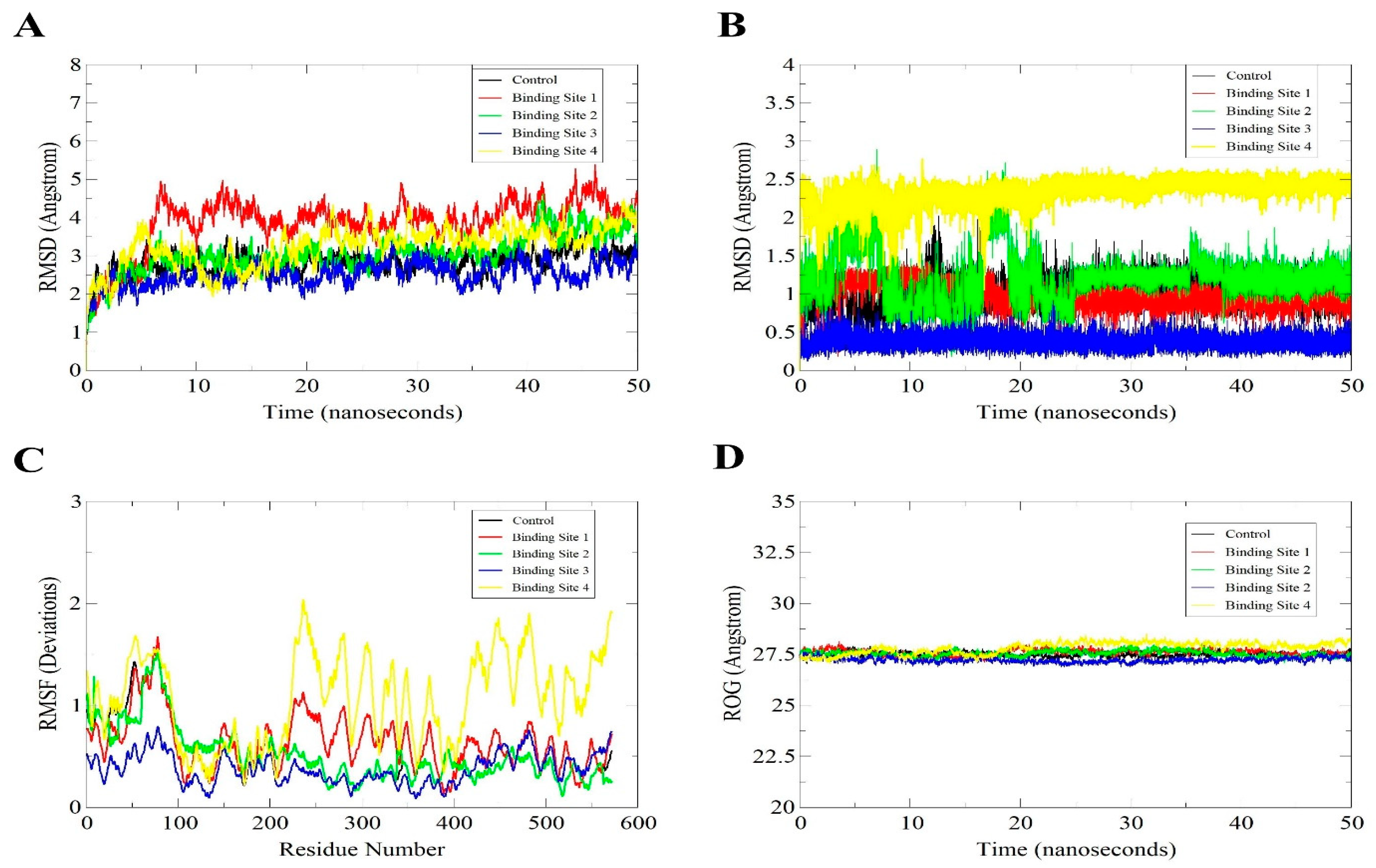
3.5. MD Simulation of the Docked Models for Structural Stability Analysis
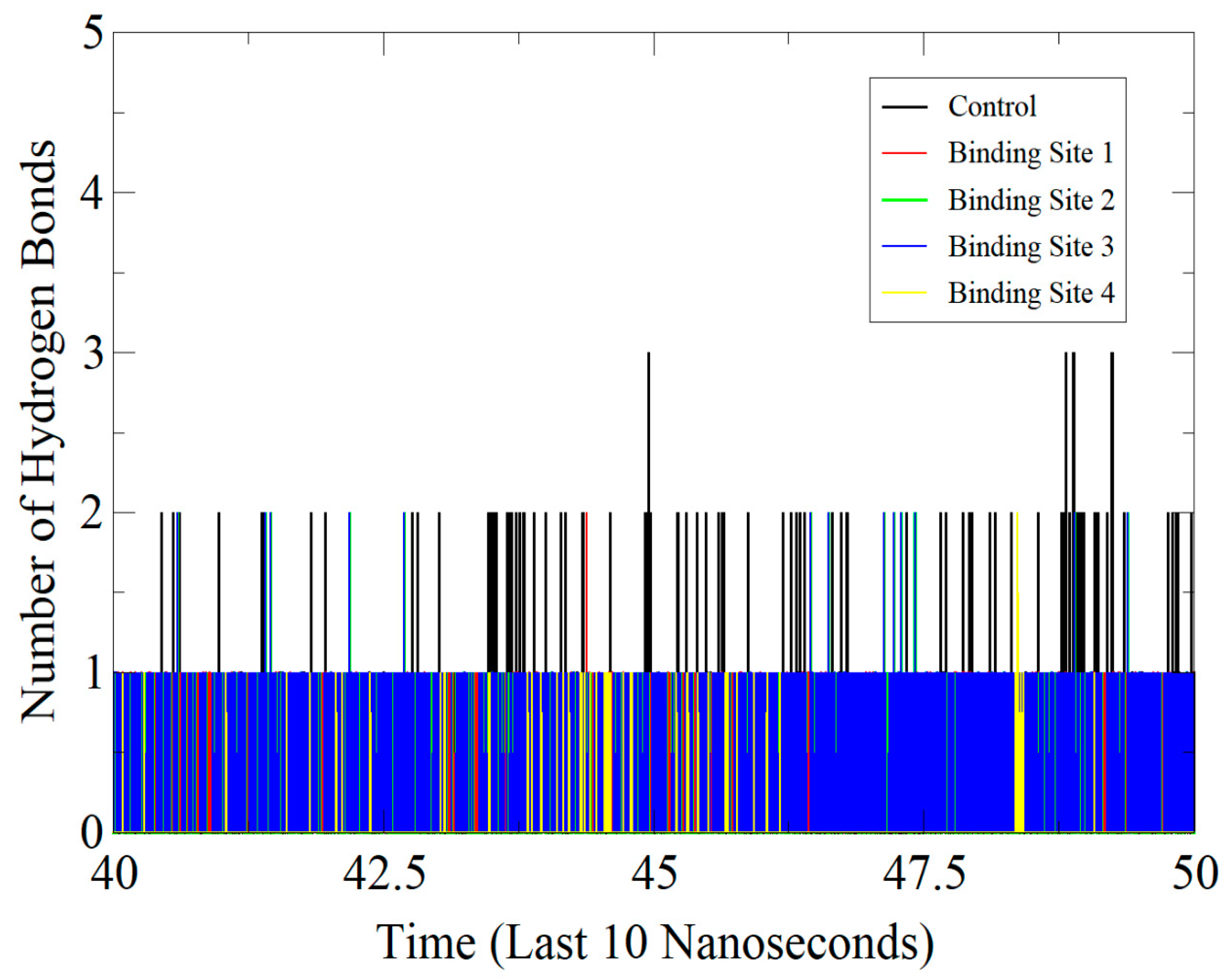
3.6. Protein-Inhibitor Stability Involving Hydrogen Bond Interactions
3.7. Determining Binding Free Energies
3.8. Residue Wise Energy Contribution
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A novel coronavirus from patients with pneumonia in china, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, Y.; Ye, D.; Liu, Q. Review of the 2019 novel coronavirus (sars-cov-2) based on current evidence. Int. J. Antimicrob. Agents 2020, 55, 105948. [Google Scholar] [CrossRef] [PubMed]
- Dar, H.A.; Waheed, Y.; Najmi, M.H.; Ismail, S.; Hetta, H.F.; Ali, A.; Muhammad, K. Multiepitope subunit vaccine design against covid-19 based on the spike protein of sars-cov-2: An in silico analysis. J. Immunol. Res. 2020, 2020, 8893483. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in wuhan, china. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical characteristics of coronavirus disease 2019 in china. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Zaim, S.; Chong, J.H.; Sankaranarayanan, V.; Harky, A. Covid-19 and multiorgan response. Curr. Probl. Cardiol. 2020, 45, 100618. [Google Scholar] [CrossRef]
- Dhama, K.; Sharun, K.; Tiwari, R.; Dadar, M.; Malik, Y.S.; Singh, K.P.; Chaicumpa, W. Covid-19, an emerging coronavirus infection: Advances and prospects in designing and developing vaccines, immunotherapeutics, and therapeutics. Hum. Vaccin. Immunother. 2020, 16, 1232–1238. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, Z.; Alrefai, H.; Hetta, H.F.; H, A.K.; Munawar, N.; Abdul Rahman, S.; Elshaer, S.; Batiha, G.E.; Muhammad, K. Investigating virological, immunological, and pathological avenues to identify potential targets for developing covid-19 treatment and prevention strategies. Vaccines (Basel) 2020, 8, 443. [Google Scholar] [CrossRef] [PubMed]
- Elshabrawy, H.A. Sars-cov-2: An update on potential antivirals in light of sars-cov antiviral drug discoveries. Vaccines (Basel) 2020, 8, 335. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for sars-cov-2 and discovery of potential drugs by computational methods. Acta Pharm Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef]
- Amanat, F.; Krammer, F. Sars-cov-2 vaccines: Status report. Immunity 2020, 52, 583–589. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 2249. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.W.; Yuan, S.; Yuen, K.S.; Fung, S.Y.; Chan, C.P.; Jin, D.Y. Zoonotic origins of human coronaviruses. Int. J. Biol. Sci. 2020, 16, 1686–1697. [Google Scholar] [CrossRef] [PubMed]
- Hui, D.S.; E, I.A.; Madani, T.A.; Ntoumi, F.; Kock, R.; Dar, O.; Ippolito, G.; McHugh, T.D.; Memish, Z.A.; Drosten, C.; et al. The continuing 2019-ncov epidemic threat of novel coronaviruses to global health - the latest 2019 novel coronavirus outbreak in wuhan, china. Int. J. Infect. Dis. 2020, 91, 264–266. [Google Scholar] [CrossRef]
- Phan, T. Genetic diversity and evolution of sars-cov-2. Infect. Genet. Evol. 2020, 81, 104260. [Google Scholar] [CrossRef] [PubMed]
- Paules, C.I.; Marston, H.D.; Fauci, A.S. Coronavirus infections-more than just the common cold. JAMA 2020, 323, 707–708. [Google Scholar] [CrossRef] [PubMed]
- Abd Ellah, N.H.; Gad, S.F.; Muhammad, K.; G, E.B.; Hetta, H.F. Nanomedicine as a promising approach for diagnosis, treatment and prophylaxis against covid-19. Nanomedicine (Lond) 2020, 15, 2085–2102. [Google Scholar] [CrossRef]
- Liu, L.; Wang, P.; Nair, M.S.; Yu, J.; Rapp, M.; Wang, Q.; Luo, Y.; Chan, J.F.; Sahi, V.; Figueroa, A.; et al. Potent neutralizing monoclonal antibodies directed to multiple epitopes on the sars-cov-2 spike. bioRxiv 2020. [Google Scholar]
- White, M.A.; Lin, W.; Cheng, X. Discovery of covid-19 inhibitors targeting the sars-cov-2 nsp13 helicase. J. Phys. Chem. Lett. 2020, 11, 9144–9151. [Google Scholar] [CrossRef]
- Habtemariam, S.; Nabavi, S.F.; Banach, M.; Berindan-Neagoe, I.; Sarkar, K.; Sil, P.C.; Nabavi, S.M. Should we try sars-cov-2 helicase inhibitors for covid-19 therapy? Arch. Med. Res. 2020, 51, 733–735. [Google Scholar] [CrossRef] [PubMed]
- Byrd, A.K.; Raney, K.D. Protein displacement by an assembly of helicase molecules aligned along single-stranded DNA. Nat. Struct Mol. Biol 2004, 11, 531–538. [Google Scholar] [CrossRef]
- Delagoutte, E.; von Hippel, P.H. Helicase mechanisms and the coupling of helicases within macromolecular machines. Part i: Structures and properties of isolated helicases. Q. Rev. Biophys 2002, 35, 431–478. [Google Scholar] [CrossRef]
- Kleymann, G.; Fischer, R.; Betz, U.A.; Hendrix, M.; Bender, W.; Schneider, U.; Handke, G.; Eckenberg, P.; Hewlett, G.; Pevzner, V.; et al. New helicase-primase inhibitors as drug candidates for the treatment of herpes simplex disease. Nat. Med. 2002, 8, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.U.; Froeyen, M. Structural elucidation of sars-cov-2 vital proteins: Computational methods reveal potential drug candidates against main protease, nsp12 polymerase and nsp13 helicase. J. Pharm Anal. 2020, 10, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Yan, L.; Ren, Z.; Wu, L.; Wang, J.; Guo, J.; Zheng, L.; Ming, Z.; Zhang, L.; Lou, Z.; et al. Delicate structural coordination of the severe acute respiratory syndrome coronavirus nsp13 upon atp hydrolysis. Nucleic Acids Res. 2019, 47, 6538–6550. [Google Scholar] [CrossRef]
- Shu, T.; Huang, M.; Wu, D.; Ren, Y.; Zhang, X.; Han, Y.; Mu, J.; Wang, R.; Qiu, Y.; Zhang, D.Y.; et al. Sars-coronavirus-2 nsp13 possesses ntpase and rna helicase activities that can be inhibited by bismuth salts. Virol. Sin. 2020, 35, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Briguglio, I.; Piras, S.; Corona, P.; Carta, A. Inhibition of rna helicases of ssrna(+) virus belonging to flaviviridae, coronaviridae and picornaviridae families. Int. J. Med. Chem. 2011, 2011, 213135. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.S.; Lee, J.; Lee, J.M.; Kim, Y.; Chin, Y.W.; Jee, J.G.; Keum, Y.S.; Jeong, Y.J. Identification of myricetin and scutellarein as novel chemical inhibitors of the sars coronavirus helicase, nsp13. Bioorg. Med. Chem. Lett. 2012, 22, 4049–4054. [Google Scholar] [CrossRef] [PubMed]
- Tanner, J.A.; Zheng, B.J.; Zhou, J.; Watt, R.M.; Jiang, J.Q.; Wong, K.L.; Lin, Y.P.; Lu, L.Y.; He, M.L.; Kung, H.F.; et al. The adamantane-derived bananins are potent inhibitors of the helicase activities and replication of sars coronavirus. Chem. Biol. 2005, 12, 303–311. [Google Scholar] [CrossRef]
- Lee, J.M.; Cho, J.B.; Ahn, H.C.; Jung, W.; Jeong, Y.J. A novel chemical compound for inhibition of sars coronavirus helicase. J. Microbiol. Biotechnol. 2017, 27, 2070–2073. [Google Scholar] [CrossRef]
- Cho, J.B.; Lee, J.M.; Ahn, H.C.; Jeong, Y.J. Identification of a novel small molecule inhibitor against sars coronavirus helicase. J. Microbiol. Biotechnol. 2015, 25, 2007–2010. [Google Scholar] [CrossRef]
- Adedeji, A.O.; Singh, K.; Kassim, A.; Coleman, C.M.; Elliott, R.; Weiss, S.R.; Frieman, M.B.; Sarafianos, S.G. Evaluation of ssya10-001 as a replication inhibitor of severe acute respiratory syndrome, mouse hepatitis, and middle east respiratory syndrome coronaviruses. Antimicrob. Agents Chemother. 2014, 58, 4894–4898. [Google Scholar] [CrossRef] [PubMed]
- Omolo, C.A.; Soni, N.; Fasiku, V.O.; Mackraj, I.; Govender, T. Update on therapeutic approaches and emerging therapies for sars-cov-2 virus. Eur. J. Pharmacol. 2020, 883, 173348. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; MacKerell, A.D., Jr. Computer-aided drug design methods. Methods Mol. Biol. 2017, 1520, 85–106. [Google Scholar]
- Nascimento Junior, J.A.C.; Santos, A.M.; Quintans-Junior, L.J.; Walker, C.I.B.; Borges, L.P.; Serafini, M.R. Sars, mers and sars-cov-2 (covid-19) treatment: A patent review. Expert Opin. Ther. Pat. 2020, 30, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Mumtaz, A.; Ashfaq, U.A.; Ul Qamar, M.T.; Anwar, F.; Gulzar, F.; Ali, M.A.; Saari, N.; Pervez, M.T. Mpd3: A useful medicinal plants database for drug designing. Nat. Prod. Res. 2017, 31, 1228–1236. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. Ucsf chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with pyrx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar]
- Cagno, V.; Magliocco, G.; Tapparel, C.; Daali, Y. The tyrosine kinase inhibitor nilotinib inhibits sars-cov-2 in vitro. Basic Clin. Pharmacol. Toxicol. 2020. [Google Scholar] [CrossRef]
- Goodsell, D.S.; Morris, G.M.; Olson, A.J. Automated docking of flexible ligands: Applications of autodock. J. Mol. Recognit. 1996, 9, 1–5. [Google Scholar] [CrossRef]
- Maiorov, V.N.; Crippen, G.M. Significance of root-mean-square deviation in comparing three-dimensional structures of globular proteins. J. Mol. Biol. 1994, 235, 625–634. [Google Scholar] [CrossRef]
- Qasaymeh, R.M.; Rotondo, D.; Oosthuizen, C.B.; Lall, N.; Seidel, V. Predictive binding affinity of plant-derived natural products towards the protein kinase g enzyme of mycobacterium tuberculosis (mtpkng). Plants (Basel) 2019, 8, 477. [Google Scholar] [CrossRef] [PubMed]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal chemistry and the molecular operating environment (moe): Application of qsar and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. Swissadme: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Z.; Zhang, S.N.; Yang, X.Y. Combination of cheminformatics and bioinformatics to explore the chemical basis of the rhizomes and aerial parts of dioscorea nipponica makino. J. Pharm. Pharmacol. 2017, 69, 1846–1857. [Google Scholar] [CrossRef] [PubMed]
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., 3rd; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Yang, L.; Skjevik, A.A.; Han Du, W.G.; Noodleman, L.; Walker, R.C.; Gotz, A.W. Data for molecular dynamics simulations of b-type cytochrome c oxidase with the amber force field. Data Brief. 2016, 8, 1209–1214. [Google Scholar] [CrossRef] [PubMed]
- Izaguirre, J.A.; Sweet, C.R.; Pande, V.S. Multiscale dynamics of macromolecules using normal mode langevin. Pac. Symp. Biocomput. 2010, 240–251. [Google Scholar]
- Harvey, M.J.; De Fabritiis, G. An implementation of the smooth particle mesh ewald method on gpu hardware. J. Chem. Theory Comput. 2009, 5, 2371–2377. [Google Scholar] [CrossRef]
- Van Bergen, L.A.; Alonso, M.; Pallo, A.; Nilsson, L.; De Proft, F.; Messens, J. Revisiting sulfur h-bonds in proteins: The example of peroxiredoxin ahpe. Sci. Rep. 2016, 6, 30369. [Google Scholar] [CrossRef] [PubMed]
- Roe, D.R.; Cheatham, T.E., 3rd. Ptraj and cpptraj: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. Vmd: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Miller, B.R., 3rd; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. Mmpbsa.Py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The mm/pbsa and mm/gbsa methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The vsgb 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins 2011, 79, 2794–2812. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Arnott, J.A.; Planey, S.L. The influence of lipophilicity in drug discovery and design. Expert Opin. Drug Discov. 2012, 7, 863–875. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Egan, W.J.; Merz, K.M., Jr.; Baldwin, J.J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef] [PubMed]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple selection criteria for drug-like chemical matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef]
- Whitty, A. Growing pains in academic drug discovery. Future Med. Chem. 2011, 3, 797–801. [Google Scholar] [CrossRef]
- Pace, C.N.; Fu, H.; Lee Fryar, K.; Landua, J.; Trevino, S.R.; Schell, D.; Thurlkill, R.L.; Imura, S.; Scholtz, J.M.; Gajiwala, K.; et al. Contribution of hydrogen bonds to protein stability. Protein Sci. 2014, 23, 652–661. [Google Scholar] [CrossRef] [PubMed]
- Gurung, A.B. In silico structure modelling of sars-cov-2 nsp13 helicase and nsp14 and repurposing of fda approved antiviral drugs as dual inhibitors. Gene Rep. 2020, 21, 100860. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Physicochemical Properties | Pharmacokinetics | ||
|---|---|---|---|
| Formula | C26H32N4O3 | GI absorption | High |
| Molecular weight | 448.56 g/mol | BBB permeant | Yes |
| Num. heavy atoms | 33 | P-gp substrate | Yes |
| Num. arom. heavy atoms | 12 | CYP1A2 inhibitor | No |
| Fraction Csp3 | 0.5 | CYP2C19 inhibitor | No |
| Num. rotatable bonds | 3 | CYP2C9 inhibitor | No |
| Num. H-bond acceptors | 5 | CYP2D6 inhibitor | Yes |
| Num. H-bond donors | 2 | CYP3A4 inhibitor | Yes |
| Molar Refractivity | 140.83 | Log Kp (skin permeation) | −6.77 cm/s |
| TPSA | 66.07 Ų | Druglikeness | |
| Lipophilicity | Lipinski | Yes; 0 violation | |
| Log Po/w (iLOGP) | 3.5 | Ghose | No; 1 violation: MR > 130 |
| Log Po/w (XLOGP3) | 3.19 | Veber | Yes |
| Log Po/w (WLOGP) | 1.37 | Egan | Yes |
| Log Po/w (MLOGP) | 2.92 | Muegge | Yes |
| Log Po/w (SILICOS-IT) | 2.68 | Bioavailability Score | 0.55 |
| Consensus Log Po/w | 2.73 | Medicinal Chemistry | |
| Water Solubility | PAINS | 0 alert | |
| Log S (ESOL) | −4.7 | Brenk | 0 alert |
| Solubility | 8.91 × 10−3 mg/mL; 1.99 × 10−5 mol/L | Leadlikeness | No; 1 violation: MW > 350 |
| Class | Moderately soluble | Synthetic accessibility | 5.04 |
| Log S (Ali) | −4.25 | Toxicity and Mutagenicity | |
| Solubility | 2.53 × 10−2 mg/mL; 5.64 × 10−5 mol/L | Carcino Mouse | Negative |
| Class | Moderately soluble | Carcino Rat | Negative |
| Log S (SILICOS-IT) | −6.44 | Daphnia | 0.08 |
| Solubility | 1.64 × 10−4 mg/mL; 3.66 × 10−7 mol/L | hERG Inhibition | Medium risk |
| Class | Poorly soluble | Ames test | Mutagen |
| Method | Energy Component | Control | Binding Site 1 | Binding Site 2 | Binding Site 3 | Binding Site 4 |
|---|---|---|---|---|---|---|
| MMGBSA | van der Waals Energy | −66.6851 | −65.4398 | −49.3253 | −58.7992 | −58.3329 |
| Electrostatic Energy | −53.9084 | −12.9283 | −21.8973 | −14.2816 | −8.3200 | |
| Polar Solvation Energy | 69.4213 | 32.2840 | 39.1614 | 33.5678 | 24.6442 | |
| Non-polar Solvation Energy | −6.5520 | −5.6700 | −4.1186 | −5.3352 | −5.5330 | |
| Gas Phase Energy | −120.5934 | −78.3681 | −71.2226 | −73.0808 | −66.6529 | |
| Solvation Energy | 62.8693 | 26.6139 | 35.0428 | 28.2325 | 19.1112 | |
| Total Binding Energy | −57.7241 | −51.7542 | −36.1798 | −44.8483 | −47.5417 | |
| MMPBSA | van der Waals Energy | −66.6851 | −65.4398 | −49.3253 | −58.7992 | −58.3329 |
| Electrostatic Energy | −53.9084 | −12.9283 | −21.8973 | −14.2816 | −8.3200 | |
| Polar Solvation Energy | 83.5288 | 36.4641 | 42.6764 | 42.5941 | 31.4819 | |
| Non-polar Solvation Energy | −4.4635 | −3.8817 | −3.6419 | −3.6784 | −3.9953 | |
| Gas Phase Energy | −120.5934 | −78.3681 | −71.2226 | −73.0808 | −66.6529 | |
| Solvation Energy | 79.0654 | 32.5824 | 39.0346 | 38.9157 | 27.4866 | |
| Total Binding Energy | −41.5280 | −45.7857 | −32.1881 | −34.1651 | −39.1663 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, S.; Waheed, Y.; Ismail, S.; Bhatti, S.; Abbasi, S.W.; Muhammad, K. Structure-Based Virtual Screening Identifies Multiple Stable Binding Sites at the RecA Domains of SARS-CoV-2 Helicase Enzyme. Molecules 2021, 26, 1446. https://doi.org/10.3390/molecules26051446
Ahmad S, Waheed Y, Ismail S, Bhatti S, Abbasi SW, Muhammad K. Structure-Based Virtual Screening Identifies Multiple Stable Binding Sites at the RecA Domains of SARS-CoV-2 Helicase Enzyme. Molecules. 2021; 26(5):1446. https://doi.org/10.3390/molecules26051446
Chicago/Turabian StyleAhmad, Sajjad, Yasir Waheed, Saba Ismail, Saadia Bhatti, Sumra Wajid Abbasi, and Khalid Muhammad. 2021. "Structure-Based Virtual Screening Identifies Multiple Stable Binding Sites at the RecA Domains of SARS-CoV-2 Helicase Enzyme" Molecules 26, no. 5: 1446. https://doi.org/10.3390/molecules26051446
APA StyleAhmad, S., Waheed, Y., Ismail, S., Bhatti, S., Abbasi, S. W., & Muhammad, K. (2021). Structure-Based Virtual Screening Identifies Multiple Stable Binding Sites at the RecA Domains of SARS-CoV-2 Helicase Enzyme. Molecules, 26(5), 1446. https://doi.org/10.3390/molecules26051446