
New Quinoxaline Derivatives as Dual Pim-1/2 Kinase Inhibitors: Design, Synthesis and Biological Evaluation

 ,
,  , , , , ,
, , , , ,  ,
, 
Abstract
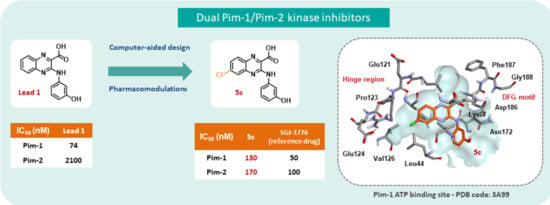
1. Introduction
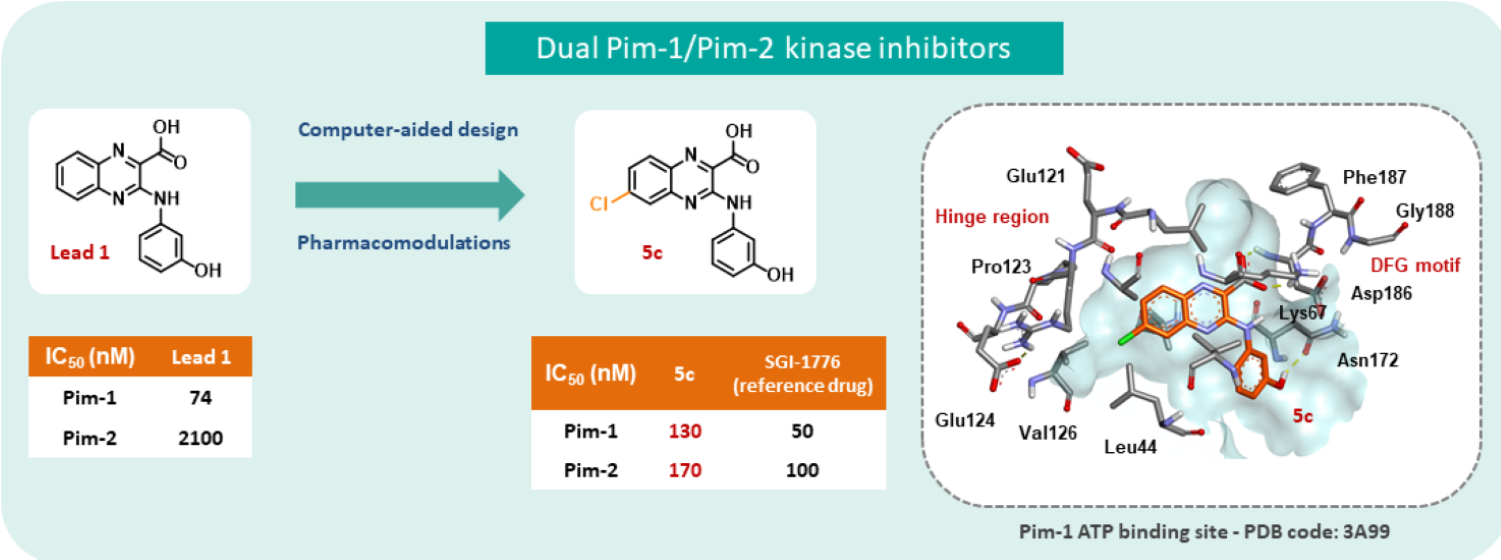
2. Results and Discussion
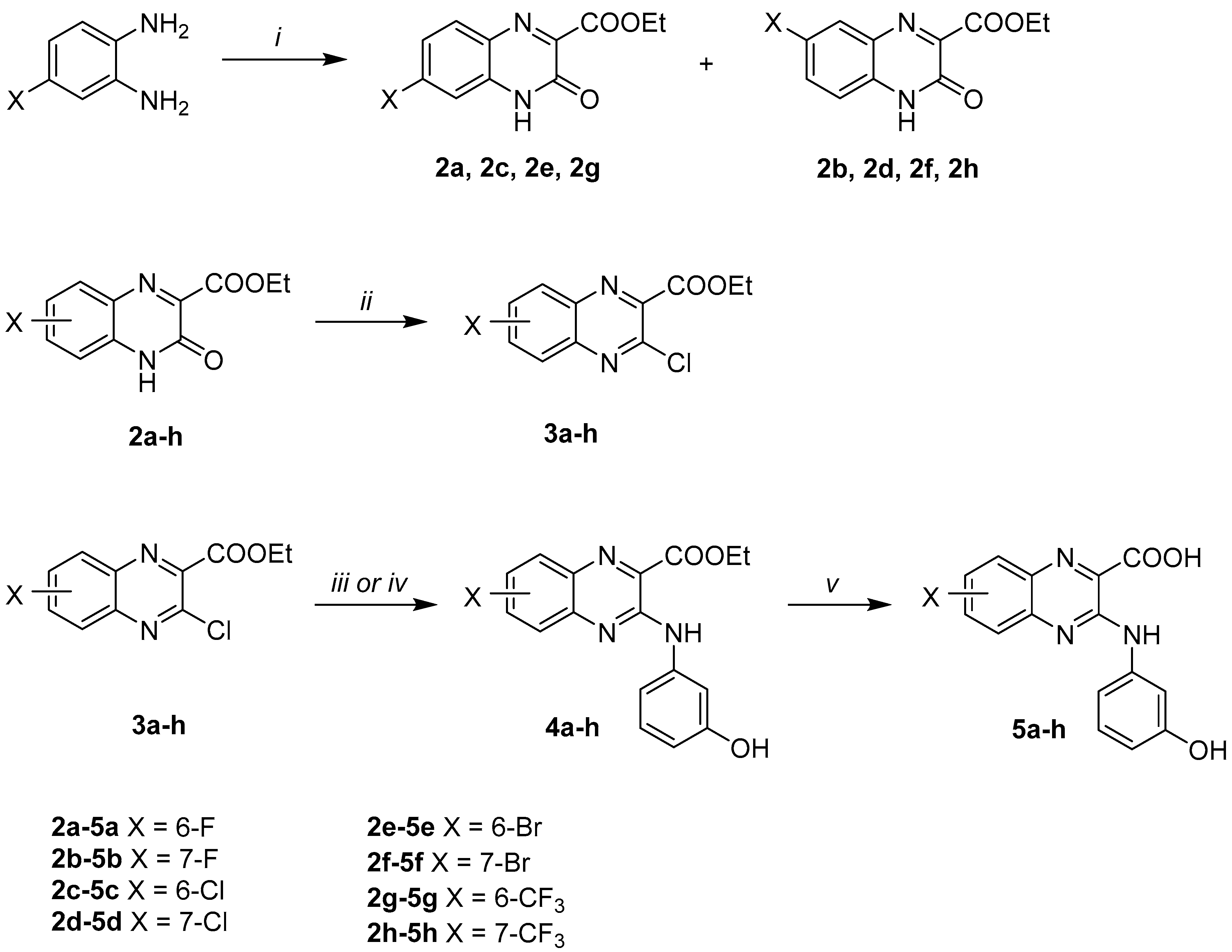
2.1. Synthesis
2.2. Enzymatic Assays
2.2.1. Pim-1 and Pim-2 Enzymatic Activity Inhibition
2.2.2. Selectivity over a Panel of Mammalian Protein Kinases
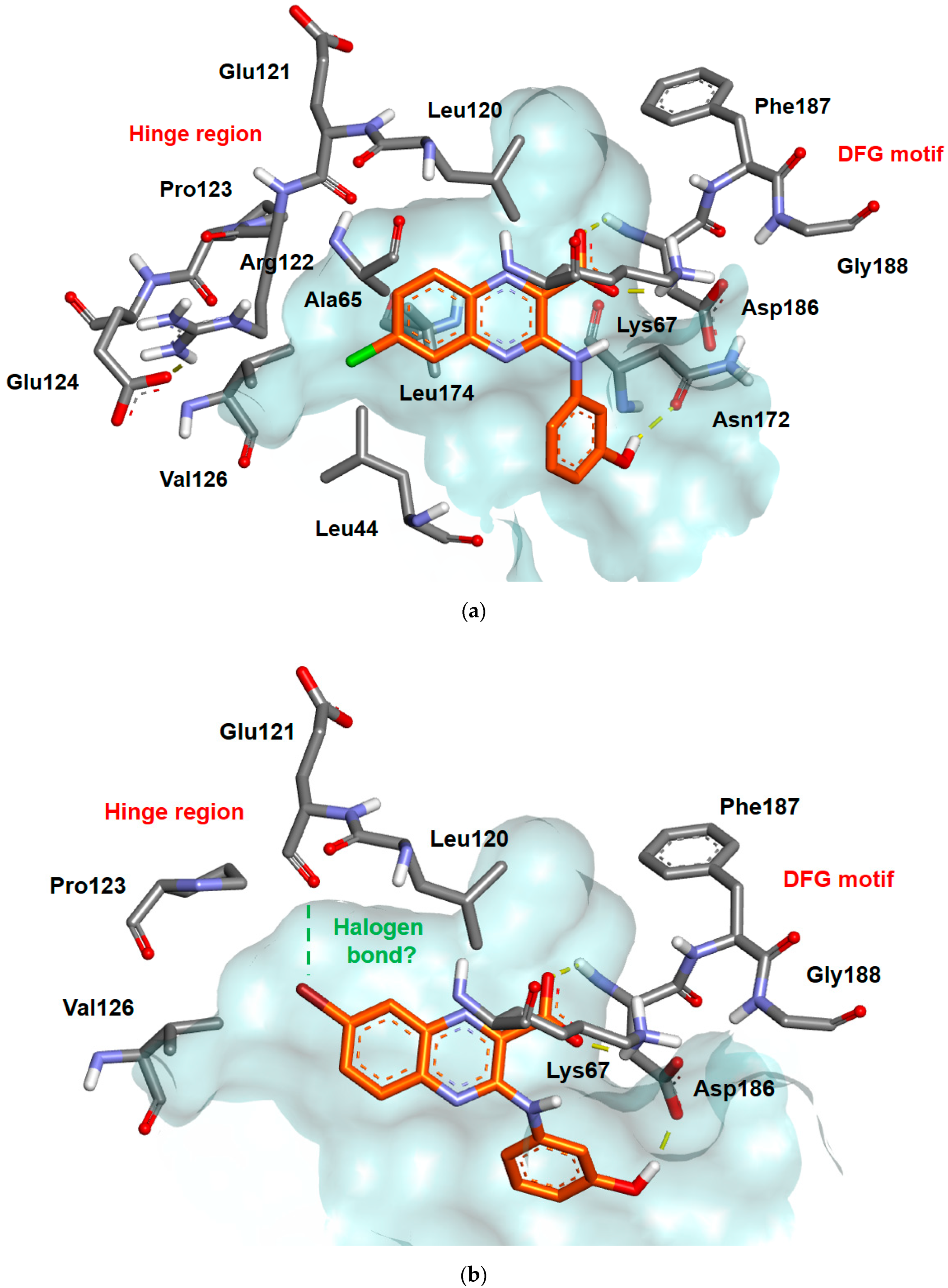
2.3. Docking Studies
2.4. In Vitro Cell-Based Assays
3. Materials and Methods
3.1. General Remarks
3.2. Chemistry
3.3. Docking Studies
3.4. Biology
3.4.1. Mammalian Protein Kinase Assays
3.4.2. Cell Cultures and Reagents
3.4.3. In Vitro Cell-Based Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Theo Cuypers, H.; Selten, G.; Quint, W.; Zijlstra, M.; Maandag, E.R.; Boelens, W.; van Wezenbeek, P.; Melief, C.; Berns, A. Murine Leukemia Virus-Induced T-Cell Lymphomagenesis: Integration of Proviruses in a Distinct Chromosomal Region. Cell 1984, 37, 141–150. [Google Scholar] [CrossRef]
- Bachmann, M.; Kosan, C.; Xing, P.X.; Montenarh, M.; Hoffmann, I.; Möröy, T. The Oncogenic Serine/Threonine Kinase Pim-1 Directly Phosphorylates and Activates the G2/M Specific Phosphatase Cdc25C. Int. J. Biochem. Cell Biol. 2006, 38, 430–443. [Google Scholar] [CrossRef] [PubMed]
- Narlik-Grassow, M.; Blanco-Aparicio, C.; Carnero, A. The PIM Family of Serine/Threonine Kinases in Cancer. Med. Res. Rev. 2014, 34, 136–159. [Google Scholar] [CrossRef] [PubMed]
- Nawijn, M.C.; Alendar, A.; Berns, A. For Better or for Worse: The Role of Pim Oncogenes in Tumorigenesis. Nat. Rev. Cancer 2010, 11, 23. [Google Scholar] [CrossRef] [PubMed]
- Tursynbay, Y.; Zhang, J.; Li, Z.; Tokay, T.; Zhumadilov, Z.; Wu, D.; Xie, Y. Pim-1 Kinase as Cancer Drug Target: An Update. Biomed. Rep. 2016, 4, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, T.; Kitanaka, C.; Noguchi, K.; Muramatsu, T.; Asai, A.; Kuchino, Y. Physical and Functional Interactions between Pim-1 Kinase and Cdc25A Phosphatase: Implications for the pim-1-mediated activation of the c-myc signaling pathway. J. Biol. Chem. 1999, 274, 18659–18666. [Google Scholar] [CrossRef]
- Leung, C.O.; Wong, C.C.; Fan, D.N.; Kai, A.K.; Tung, E.K.; Xu, I.M.; Ng, I.O.; Lo, R.C. PIM1 Regulates Glycolysis and Promotes Tumor Progression in Hepatocellular Carcinoma. Oncotarget 2015, 6, 10880–10892. [Google Scholar] [CrossRef]
- Keane, N.A.; Reidy, M.; Natoni, A.; Raab, M.S.; O’Dwyer, M. Targeting the Pim Kinases in Multiple Myeloma. Blood Cancer J. 2015, 5, e325. [Google Scholar] [CrossRef]
- Amson, R.; Sigaux, F.; Przedborski, S.; Flandrin, G.; Givol, D.; Telerman, A. The Human Protooncogene Product P33pim Is Expressed during Fetal Hematopoiesis and in Diverse Leukemias. Proc. Natl. Acad. Sci. USA 1989, 86, 8857–8861. [Google Scholar] [CrossRef]
- Dhanasekaran, S.M.; Barrette, T.R.; Ghosh, D.; Shah, R.; Varambally, S.; Kurachi, K.; Pienta, K.J.; Rubin, M.A.; Chinnaiyan, A.M. Delineation of Prognostic Biomarkers in Prostate Cancer. Nature 2001, 412, 822–826. [Google Scholar] [CrossRef]
- Brasó-Maristany, F.; Filosto, S.; Catchpole, S.; Marlow, R.; Quist, J.; Francesch-Domenech, E.; Plumb, D.A.; Zakka, L.; Gazinska, P.; Liccardi, G.; et al. PIM1 Kinase Regulates Cell Death, Tumor Growth and Chemotherapy Response in Triple-Negative Breast Cancer. Nat. Med. 2016, 22, 1303–1313. [Google Scholar] [CrossRef] [PubMed]
- Weirauch, U.; Beckmann, N.; Thomas, M.; Grünweller, A.; Huber, K.; Bracher, F.; Hartmann, R.K.; Aigner, A. Functional Role and Therapeutic Potential of the Pim-1 Kinase in Colon Carcinoma. Neoplasia 2013, 15, 783-IN28. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Li, Y.-Y. Use of Regulators and Inhibitors of Pim-1, a Serine/Threonine Kinase, for Tumour Therapy (Review). Mol. Med. Rep. 2014, 9, 2051–2060. [Google Scholar] [CrossRef] [PubMed]
- Isaac, M.; Siu, A.; Jongstra, J. The Oncogenic PIM Kinase Family Regulates Drug Resistance through Multiple Mechanisms. Drug Resist. Updat. 2011, 14, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Darby, R.A.J.; Unsworth, A.; Knapp, S.; Kerr, I.D.; Callaghan, R. Overcoming ABCG2-Mediated Drug Resistance with Imidazo-[1,2-b]-Pyridazine-Based Pim1 Kinase Inhibitors. Cancer Chemother. Pharmacol. 2015, 76, 853–864. [Google Scholar] [CrossRef]
- Mikkers, H.; Nawijn, M.; Allen, J.; Brouwers, C.; Verhoeven, E.; Jonkers, J.; Berns, A. Mice Deficient for All PIM Kinases Display Reduced Body Size and Impaired Responses to Hematopoietic Growth Factors. Mol. Cell. Biol. 2004, 24, 6104–6115. [Google Scholar] [CrossRef]
- Asati, V.; Mahapatra, D.K.; Bharti, S.K. PIM Kinase Inhibitors: Structural and Pharmacological Perspectives. Eur. J. Med. Chem. 2019, 172, 95–108. [Google Scholar] [CrossRef]
- Blanco-Aparicio, C.; Carnero, A. Pim Kinases in Cancer: Diagnostic, Prognostic and Treatment Opportunities. Biochem. Pharmacol. 2013, 85, 629–643. [Google Scholar] [CrossRef]
- Foulks, J.M.; Carpenter, K.J.; Luo, B.; Xu, Y.; Senina, A.; Nix, R.; Chan, A.; Clifford, A.; Wilkes, M.; Vollmer, D.; et al. A Small-Molecule Inhibitor of PIM Kinases as a Potential Treatment for Urothelial Carcinomas. Neoplasia 2014, 16, 403–412. [Google Scholar] [CrossRef]
- Cortes, J.; Tamura, K.; DeAngelo, D.J.; de Bono, J.; Lorente, D.; Minden, M.; Uy, G.L.; Kantarjian, H.; Chen, L.S.; Gandhi, V.; et al. Phase I Studies of AZD1208, a Proviral Integration Moloney Virus Kinase Inhibitor in Solid and Haematological Cancers. Br. J. Cancer 2018, 118, 1425–1433. [Google Scholar] [CrossRef]
- Raab, M.S.; Thomas, S.K.; Ocio, E.M.; Guenther, A.; Goh, Y.-T.; Talpaz, M.; Hohmann, N.; Zhao, S.; Xiang, F.; Simon, C.; et al. The First-in-Human Study of the Pan-PIM Kinase Inhibitor PIM447 in Patients with Relapsed and/or Refractory Multiple Myeloma. Leukemia 2019, 33, 2924–2933. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Mandiyan, V.; Suzuki, Y.; Zhang, C.; Rice, J.; Tsai, J.; Artis, D.R.; Ibrahim, P.; Bremer, R. Crystal Structures of Proto-Oncogene Kinase Pim1: A Target of Aberrant Somatic Hypermutations in Diffuse Large Cell Lymphoma. J. Mol. Biol. 2005, 348, 183–193. [Google Scholar] [CrossRef]
- Bullock, A.N.; Russo, S.; Amos, A.; Pagano, N.; Bregman, H.; Debreczeni, J.É.; Lee, W.H.; von Delft, F.; Meggers, E.; Knapp, S. Crystal Structure of the PIM2 Kinase in Complex with an Organoruthenium Inhibitor. PLoS ONE 2009, 4, e7112. [Google Scholar] [CrossRef] [PubMed]
- Koblish, H.; Li, Y.; Shin, N.; Hall, L.; Wang, Q.; Wang, K.; Covington, M.; Marando, C.; Bowman, K.; Boer, J.; et al. Preclinical Characterization of INCB053914, a Novel Pan-PIM Kinase Inhibitor, Alone and in Combination with Anticancer Agents, in Models of Hematologic Malignancies. PLoS ONE 2018, 13, e0199108. [Google Scholar] [CrossRef] [PubMed]
- Czardybon, W.; Windak, R.; Gołas, A.; Gałęzowski, M.; Sabiniarz, A.; Dolata, I.; Salwińska, M.; Guzik, P.; Zawadzka, M.; Gabor-Worwa, E.; et al. A Novel, Dual Pan-PIM/FLT3 Inhibitor SEL24 Exhibits Broad Therapeutic Potential in Acute Myeloid Leukemia. Oncotarget 2018, 9. [Google Scholar] [CrossRef]
- Oyallon, B.; Brachet-Botineau, M.; Logé, C.; Bonnet, P.; Souab, M.; Robert, T.; Ruchaud, S.; Bach, S.; Berthelot, P.; Gouilleux, F.; et al. Structure-Based Design of Novel Quinoxaline-2-Carboxylic Acids and Analogues as Pim-1 Inhibitors. Eur. J. Med. Chem. 2018, 154, 101–109. [Google Scholar] [CrossRef]
- Le, B.T.; Kumarasiri, M.; Adams, J.R.; Yu, M.; Milne, R.; Sykes, M.J.; Wang, S. Targeting Pim Kinases for Cancer Treatment: Opportunities and Challenges. Future Med. Chem. 2015, 7, 35–53. [Google Scholar] [CrossRef]
- Mahesh, R.; Dhar, A.K.; Sasank, T.V.N.V.T.; Thirunavukkarasu, S.; Devadoss, T. Citric Acid: An Efficient and Green Catalyst for Rapid One Pot Synthesis of Quinoxaline Derivatives at Room Temperature. Chin. Chem. Lett. 2011, 22, 389–392. [Google Scholar] [CrossRef]
- Mahesh, R.; Devadoss, T.; Dhar, A.K.; Venkatesh, S.M.; Mundra, S.; Pandey, D.K.; Bhatt, S.; Jindal, A.K. Ligand-Based Design, Synthesis, and Pharmacological Evaluation of 3-Methoxyquinoxalin-2-Carboxamides as Structurally Novel Serotonin Type-3 Receptor Antagonists. Arch. Pharm. (Weinheim) 2012, 345, 687–694. [Google Scholar] [CrossRef]
- Takano, Y.; Shiga, F.; Asano, J.; Ando, N.; Uchiki, H.; Fukuchi, K.; Anraku, T. Design, Synthesis, and AMPA Receptor Antagonistic Activity of a Novel 6-Nitro-3-Oxoquinoxaline-2-Carboxylic Acid with a Substituted Phenyl Group at the 7 Position. Bioorg. Med. Chem. 2005, 13, 5841–5863. [Google Scholar] [CrossRef]
- Takano, Y.; Shiga, F.; Asano, J.; Hori, W.; Fukuchi, K.; Anraku, T.; Uno, T. Design and Synthesis of Novel 7-Heterocycle-6-Trifluoromethyl-3-Oxoquinoxaline-2-Carboxylic Acids Bearing a Substituted Phenyl Group as Superior AMPA Receptor Antagonists with Good Physicochemical Properties. Bioorg. Med. Chem. 2006, 14, 776–792. [Google Scholar] [CrossRef] [PubMed]
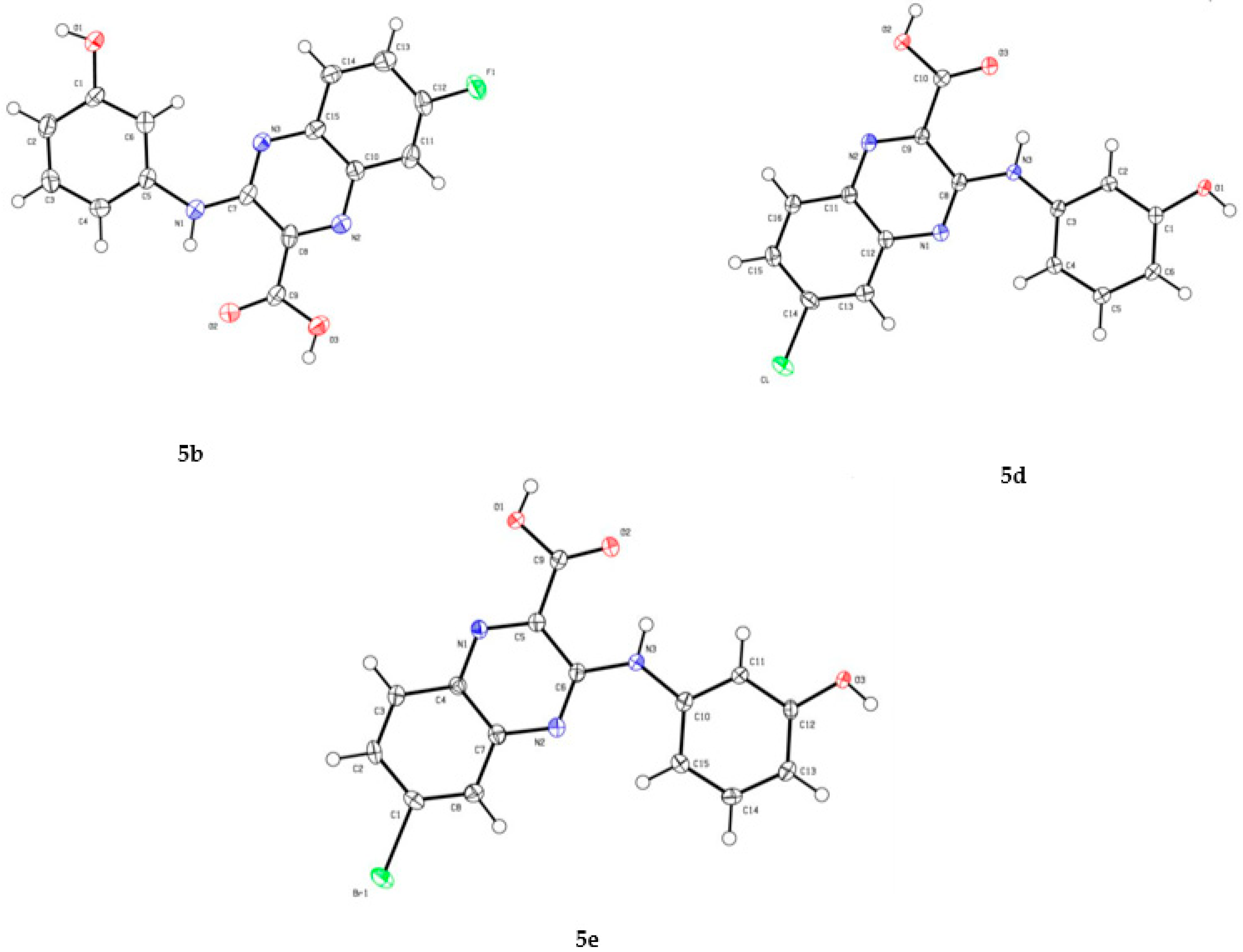
- Cambridge Crystallographic Data Centre. Supplementary X-ray Crystallographic Data (CCDC-2051184, CCDC-2051185 and CCDC-2051186); Cambridge Crystallographic Data Centre: Cambridge, UK, 2020; Available online: https://www.ccdc.cam.ac.uk/ (accessed on 18 December 2020).
- Zegzouti, H.; Zdanovskaia, M.; Hsiao, K.; Goueli, S.A. ADP-Glo: A Bioluminescent and Homogeneous ADP Monitoring Assay for Kinases. ASSAY Drug Dev. Technol. 2009, 7, 560–572. [Google Scholar] [CrossRef] [PubMed]
- Bissantz, C.; Kuhn, B.; Stahl, M. A Medicinal Chemist’s Guide to Molecular Interactions. J. Med. Chem. 2010, 53, 5061–5084. [Google Scholar] [CrossRef]
- Mizuki, M.; Schwäble, J.; Steur, C.; Choudhary, C.; Agrawal, S.; Sargin, B.; Steffen, B.; Matsumura, I.; Kanakura, Y.; Böhmer, F.D.; et al. Suppression of Myeloid Transcription Factors and Induction of STAT Response Genes by AML-Specific Flt3 Mutations. Blood 2003, 101, 3164–3173. [Google Scholar] [CrossRef]
- Kim, K.-T.; Baird, K.; Ahn, J.-Y.; Meltzer, P.; Lilly, M.; Levis, M.; Small, D. Pim-1 Is up-Regulated by Constitutively Activated FLT3 and Plays a Role in FLT3-Mediated Cell Survival. Blood 2005, 105, 1759–1767. [Google Scholar] [CrossRef]
- Zhang, X.; Yu, H.; Wang, F.; Han, Y.; Yang, W. Pim-2 Modulates Aerobic Glycolysis and Energy Production during the Development of Colorectal Tumors. Int. J. Med. Sci. 2015, 12, 487–493. [Google Scholar] [CrossRef]
- Tripos Associates, Inc. SYBYL-X 1.3; Tripos Associates, Inc.: St. Louis, MO, USA, 2013. [Google Scholar]
- Clark, M.; Cramer, R.D.; Van Opdenbosch, N. Validation of the General Purpose Tripos 5.2 Force Field. J. Comput. Chem. 1989, 10, 982–1012. [Google Scholar] [CrossRef]
- Morishita, D.; Takami, M.; Yoshikawa, S.; Katayama, R.; Sato, S.; Kukimoto-Niino, M.; Umehara, T.; Shirouzu, M.; Sekimizu, K.; Yokoyama, S.; et al. Cell-Permeable Carboxyl-Terminal P27 Kip1 Peptide Exhibits Anti-Tumor Activity by Inhibiting Pim-1 Kinase. J. Biol. Chem. 2011, 286, 2681–2688. [Google Scholar] [CrossRef]
- Jones, G.; Willet, P.; Glen, R.C. Development and Validation of a Genetic Algorithm for Flexible Docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Dassault Systèmes. Dassault Systèmes BIOVIA, Discovery Studio Visualizer, v19.1.0.18287; Dassault Systèmes: San Diego, CA, USA, 2019. [Google Scholar]
- Ponte, A.L.; Ribeiro-Fleury, T.; Chabot, V.; Gouilleux, F.; Langonné, A.; Hérault, O.; Charbord, P.; Domenech, J. Granulocyte-Colony-Stimulating Factor Stimulation of Bone Marrow Mesenchymal Stromal Cells Promotes CD34+ Cell Migration Via a Matrix Metalloproteinase-2-Dependent Mechanism. Stem Cells Dev. 2012, 21, 3162–3172. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
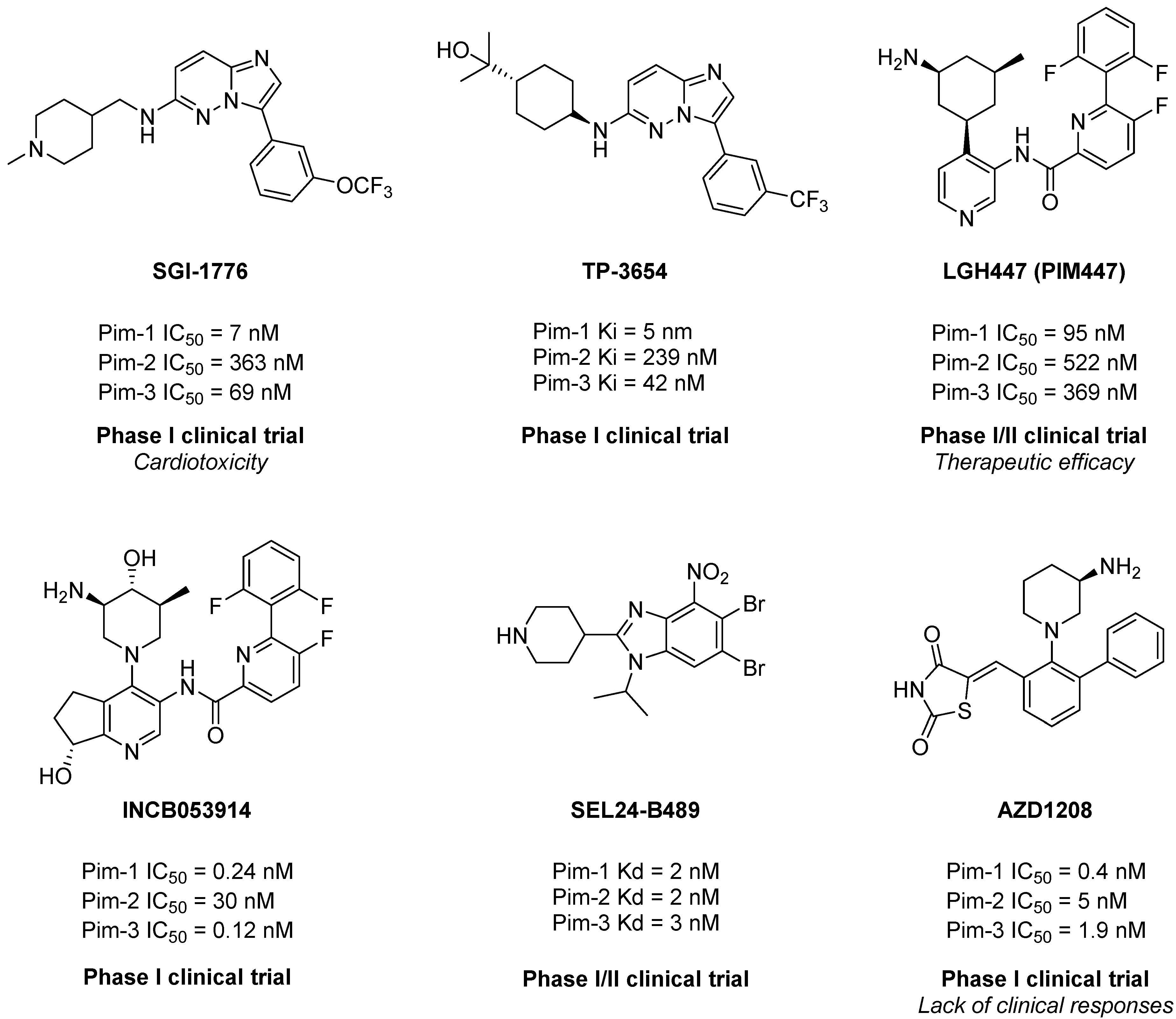
| Entry | Compound | X | HsPim-1 IC50 (µM) 1 | HsPim-2 IC50 (µM) 1 |
| 1 | 1 | H | 0.074 | 2.10 |
| 2 | 5a | 6-F | 0.17 | 0.76 |
| 3 | 5b | 7-F | 1.24 | 4.20 |
| 4 | 5c | 6-Cl | 0.13 | 0.17 |
| 5 | 5d | 7-Cl | 2.10 | 2.40 |
| 6 | 5e | 6-Br | 0.16 | 0.58 |
| 7 | 5f | 7-Br | 0.18 | 2.20 |
| 8 | 5g | 6-CF3 | 0.20 | 1.80 |
| 9 | 5h | 7-CF3 | 3.85 | 6.40 |
| 10 | SGI-1776 | 0.05 | 0.10 | |
 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Compound | X | Kinase Enzymatic IC50 (µM) 1 | ||||||||
| Pim-1 | Pim-2 | DYRK1A | CDK5/p25 | CDK9/CyclinT | Haspin | CLK1 | CK1ε | GSK3β | |||
| 1 | 1 | H | 0.07 | 2.10 | 0.27 | >10 | >10 | >10 | >10 | >10 | >10 |
| 2 | 5c | 6-Cl | 0.13 | 0.17 | 2.58 | >10 | >10 | >10 | >10 | >10 | 2.80 |
| 3 | 5e | 6-Br | 0.16 | 0.58 | 1.98 | >10 | >10 | >10 | >10 | >10 | 3.22 |
| 4 | SGI-1776 | 0.05 | 0.10 | 3.80 | 9.53 | 1.08 | 0.05 | 0.43 | 6.54 | >10 | |
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Compound | X | EC50 (µM) 1 | |||
| MV4-11 2 | HCT-116 2 | HS-27a 2 | MSC 2 | |||
| 1 | 1 | H | 61.2 ± 3.9 | 32.9 ± 10.2 | 84.8 ± 4.0 | 43.6 ± 11.5 |
| 2 | 5c | 6-Cl | 35.5 ± 1.1 | 45.3 ± 1.0 | 86.3 ± 3.3 | 63.2 ± 13.1 |
| 3 | 5e | 6-Br | 32.9 ± 9.6 | 40.7 ± 0.1 | >100 | >100 |
| 4 | SGI-1776 | 0.03 ± 0.003 | 4.4 ± 1.7 | 11.3 ± 4.2 | 4.0 ± 1.1 | |
Sample Availability: Samples of the compounds are available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oyallon, B.; Brachet-Botineau, M.; Logé, C.; Robert, T.; Bach, S.; Ibrahim, S.; Raoul, W.; Croix, C.; Berthelot, P.; Guillon, J.; et al. New Quinoxaline Derivatives as Dual Pim-1/2 Kinase Inhibitors: Design, Synthesis and Biological Evaluation. Molecules 2021, 26, 867. https://doi.org/10.3390/molecules26040867
Oyallon B, Brachet-Botineau M, Logé C, Robert T, Bach S, Ibrahim S, Raoul W, Croix C, Berthelot P, Guillon J, et al. New Quinoxaline Derivatives as Dual Pim-1/2 Kinase Inhibitors: Design, Synthesis and Biological Evaluation. Molecules. 2021; 26(4):867. https://doi.org/10.3390/molecules26040867
Chicago/Turabian StyleOyallon, Bruno, Marie Brachet-Botineau, Cédric Logé, Thomas Robert, Stéphane Bach, Sajida Ibrahim, William Raoul, Cécile Croix, Pascal Berthelot, Jean Guillon, and et al. 2021. "New Quinoxaline Derivatives as Dual Pim-1/2 Kinase Inhibitors: Design, Synthesis and Biological Evaluation" Molecules 26, no. 4: 867. https://doi.org/10.3390/molecules26040867
APA StyleOyallon, B., Brachet-Botineau, M., Logé, C., Robert, T., Bach, S., Ibrahim, S., Raoul, W., Croix, C., Berthelot, P., Guillon, J., Pinaud, N., Gouilleux, F., Viaud-Massuard, M.-C., & Denevault-Sabourin, C. (2021). New Quinoxaline Derivatives as Dual Pim-1/2 Kinase Inhibitors: Design, Synthesis and Biological Evaluation. Molecules, 26(4), 867. https://doi.org/10.3390/molecules26040867