
5-Methoxybenzothiophene-2-Carboxamides as Inhibitors of Clk1/4: Optimization of Selectivity and Cellular Potency

 , ,
, ,  and
and
Abstract
1. Introduction
2. Results
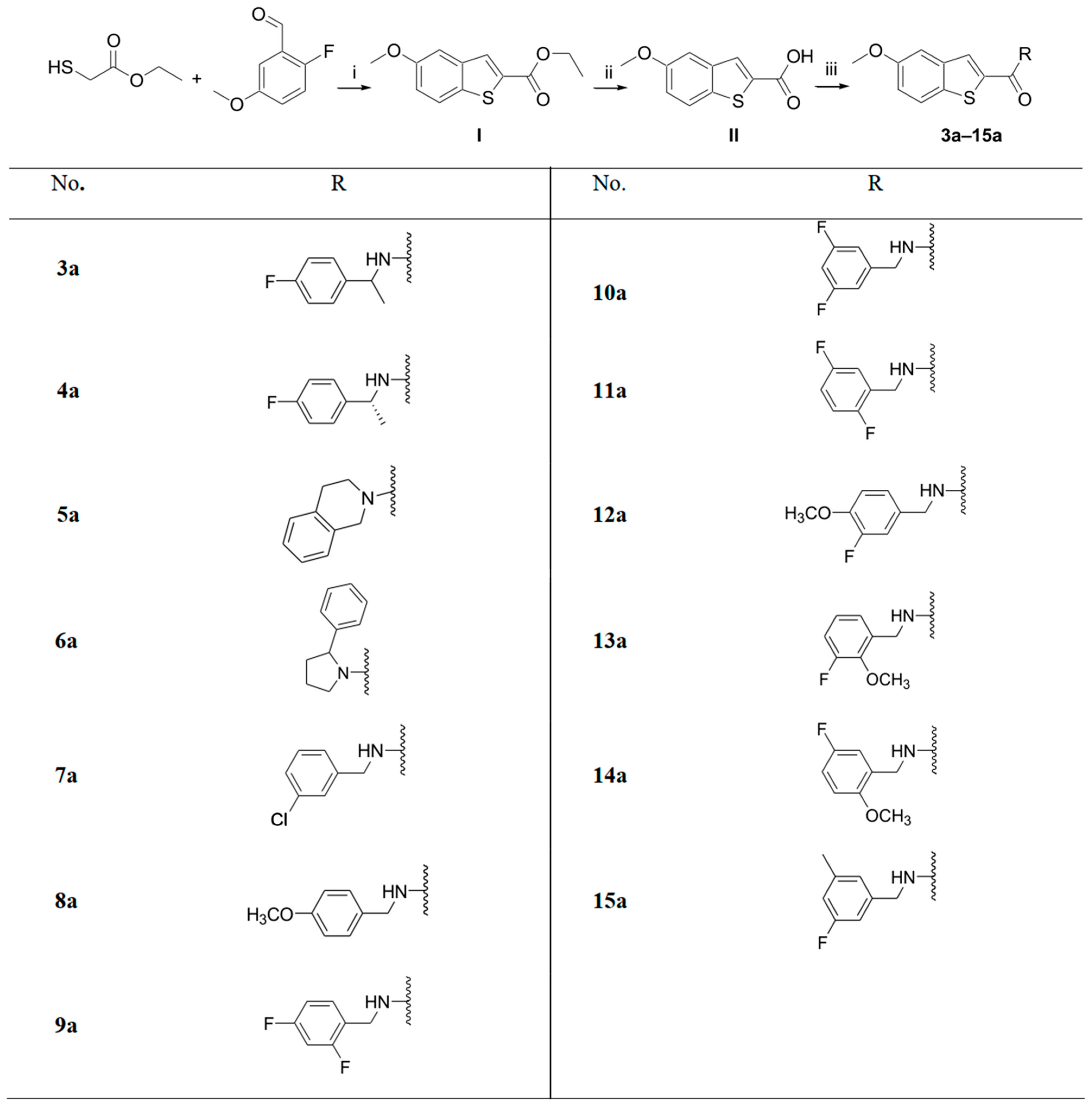
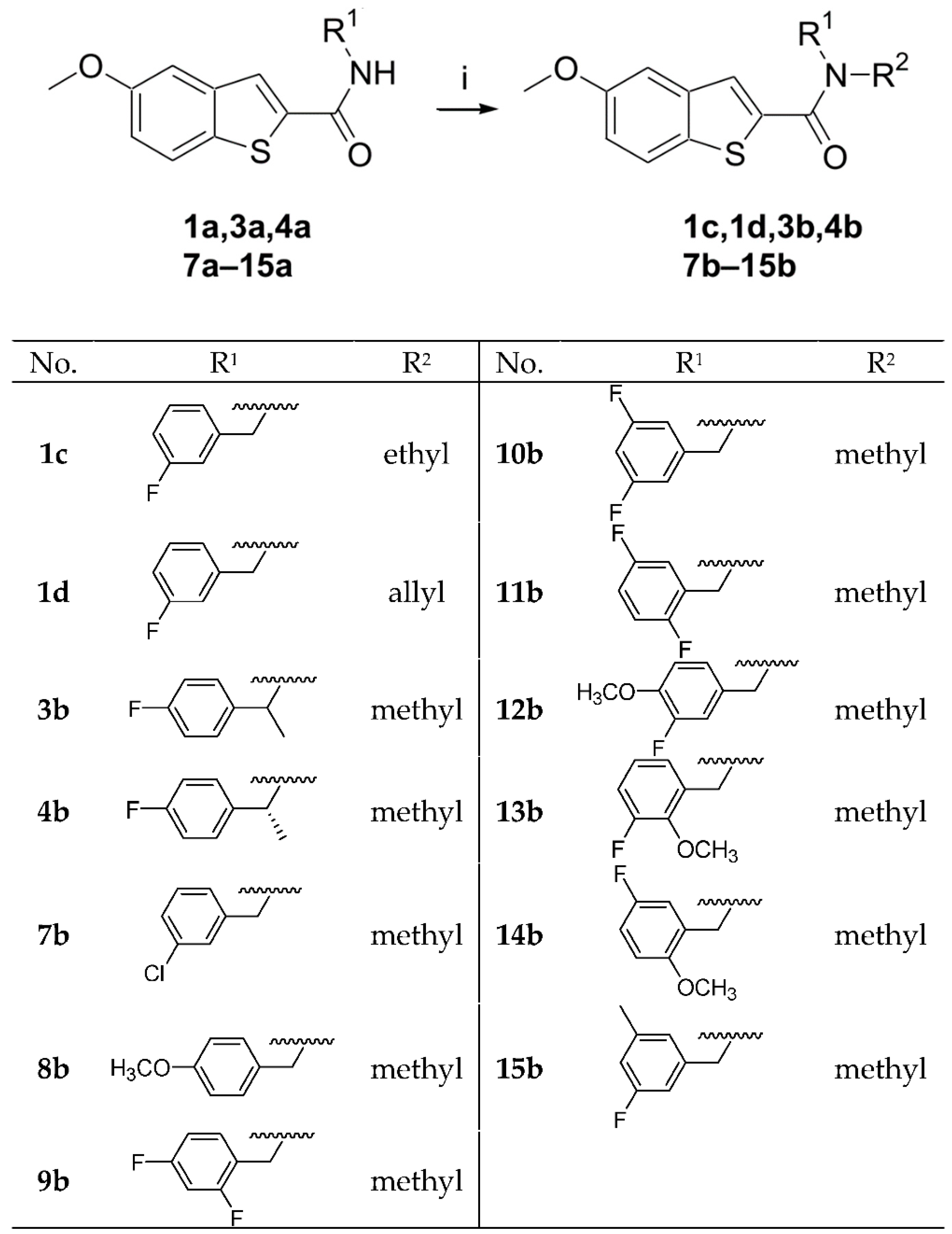
2.1. Chemistry
2.2. Biological Evaluation and Development of a Binding Model
2.2.1. In Vitro Clk1/Clk2 Inhibitory Activity
2.2.2. Development of a Binding Model Using Rigidified Analogues
2.2.3. Variation of the N-alkylation and Methylation of the Benzyl Position
2.2.4. Variation at the Benzyl Amide Function
2.2.5. Inhibition of Tumor Cell Growth In Vitro
2.2.6. Kinase Selectivity Profiling
3. Materials and Methods
3.1. Chemistry
3.1.1. General Synthetic Procedures
Procedure A, Synthesis of 5-Methoxybenzo[b]thiophene-2-carboxylic Acid Ethyl Ester (I)
Procedure B, Synthesis of 5-Methoxybenzo[b]thiophene-2-carboxylic Acid (II)
Procedure C, General Procedure for the Synthesis of 5-Methoxybenzo[b]thiophene-2-carboxylic Acid Amide Derivatives
Procedure D, General Procedure for the Synthesis of 5-Methoxybenzo[b]thiophene-2-carboxylic Acid Alkyl Amide Derivatives
3.1.2. Experimental Details and Synthesis of the Compounds
3.2. Biological Assays
3.2.1. Protein Kinases and Inhibition Assays
3.2.2. Antiproliferative Assay
3.3. Molecular Modeling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Iwai, K.; Yaguchi, M.; Nishimura, K.; Yamamoto, Y.; Tamura, T.; Nakata, D.; Dairiki, R.; Kawakita, Y.; Mizojiri, R.; Ito, Y. Anti-tumor efficacy of a novel CLK inhibitor via targeting RNA splicing and MYC-dependent vulnerability. EMBO Mol. Med. 2018, 10, e8289. [Google Scholar] [CrossRef] [PubMed]
- ElHady, A.K.; Abdel-Halim, M.; Abadi, A.H.; Engel, M. Development of Selective Clk1 and-4 Inhibitors for Cellular Depletion of Cancer-Relevant Proteins. J. Med. Chem. 2017, 60, 5377–5391. [Google Scholar] [CrossRef] [PubMed]
- Corkery, D.P.; Holly, A.C.; Lahsaee, S.; Dellaire, G. Connecting the speckles: Splicing kinases and their role in tumorigenesis and treatment response. Nucleus 2015, 6, 279–288. [Google Scholar] [CrossRef]
- Capra, M.; Nuciforo, P.G.; Confalonieri, S.; Quarto, M.; Bianchi, M.; Nebuloni, M.; Boldorini, R.; Pallotti, F.; Viale, G.; Gishizky, M.L. Frequent alterations in the expression of serine/threonine kinases in human cancers. Cancer Res. 2006, 66, 8147–8154. [Google Scholar] [CrossRef] [PubMed]
- Duchon, A.; Herault, Y. DYRK1A, a dosage-sensitive gene involved in neurodevelopmental disorders, is a target for drug development in Down syndrome. Front. Behav. Neurosci. 2016, 10, 104. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, C.; Frank, D.; Will, R.; Jaschinski, C.; Frauen, R.; Katus, H.A.; Frey, N. DYRK1A is a novel negative regulator of cardiomyocyte hypertrophy. J. Biol. Chem. 2009, 284, 17320–17327. [Google Scholar] [CrossRef]
- Soppa, U.; Schumacher, J.; Florencio Ortiz, V.; Pasqualon, T.; Tejedor, F.; Becker, W. The Down syndrome-related protein kinase DYRK1A phosphorylates p27Kip1 and Cyclin D1 and induces cell cycle exit and neuronal differentiation. Cell Cycle 2014, 13, 2084–2100. [Google Scholar] [CrossRef]
- Yoshida, T.; Kim, J.H.; Carver, K.; Su, Y.; Weremowicz, S.; Mulvey, L.; Yamamoto, S.; Brennan, C.; Mei, S.; Long, H. CLK2 is an oncogenic kinase and splicing regulator in breast cancer. Cancer Res. 2015, 75, 1516–1526. [Google Scholar] [CrossRef] [PubMed]
- Loidreau, Y.; Marchand, P.; Dubouilh-Benard, C.; Nourrisson, M.-R.; Duflos, M.; Loaëc, N.; Meijer, L.; Besson, T. Synthesis and biological evaluation of N-aryl-7-methoxybenzo [b] furo [3,2-d] pyrimidin-4-amines and their N-arylbenzo [b] thieno [3,2-d] pyrimidin-4-amine analogues as dual inhibitors of CLK1 and DYRK1A kinases. Eur. J. Med. Chem. 2013, 59, 283–295. [Google Scholar] [CrossRef]
- Esvan, Y.J.; Zeinyeh, W.; Boibessot, T.; Nauton, L.; Théry, V.; Knapp, S.; Chaikuad, A.; Loaëc, N.; Meijer, L.; Anizon, F. Discovery of pyrido [3,4-g] quinazoline derivatives as CMGC family protein kinase inhibitors: Design, synthesis, inhibitory potency and X-ray co–crystal structure. Eur. J. Med. Chem. 2016, 118, 170–177. [Google Scholar] [CrossRef]
- Zeinyeh, W.; Esvan, Y.J.; Nauton, L.; Loaëc, N.; Meijer, L.; Théry, V.; Anizon, F.; Giraud, F.; Moreau, P. Synthesis and preliminary in vitro kinase inhibition evaluation of new diversely substituted pyrido [3,4-g] quinazoline derivatives. Bioorg. Med. Chem. Lett. 2016, 26, 4327–4329. [Google Scholar] [CrossRef]
- Tazarki, H.; Zeinyeh, W.; Esvan, Y.J.; Knapp, S.; Chatterjee, D.; Schröder, M.; Joerger, A.C.; Khiari, J.; Josselin, B.; Baratte, B. New pyrido [3,4-g] quinazoline derivatives as CLK1 and DYRK1A inhibitors: Synthesis, biological evaluation and binding mode analysis. Eur. J. Med. Chem. 2019, 166, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, C.; Miralinaghi, P.; Mariano, M.; Hartmann, R.W.; Engel, M. Hydroxybenzothiophene ketones are efficient pre-mRNA splicing modulators due to dual inhibition of Dyrk1A and Clk1/4. ACS Med. Chem. Lett. 2014, 5, 963–967. [Google Scholar] [CrossRef]
- Bendjeddou, L.Z.; Loaëc, N.; Villiers, B.; Prina, E.; Späth, G.F.; Galons, H.; Meijer, L.; Oumata, N. Exploration of the imidazo [1,2-b] pyridazine scaffold as a protein kinase inhibitor. Eur. J. Med. Chem. 2017, 125, 696–709. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.M.; Centanni, M.; Virili, C.; Miccoli, M.; Ferrari, P.; Ruffilli, I.; Ragusa, F.; Antonelli, A.; Fallahi, P. Sunitinib in the treatment of thyroid cancer. Curr. Med. Chem. 2019, 26, 963–972. [Google Scholar] [CrossRef]
- Murár, M.; Dobiaš, J.; Šramel, P.; Addová, G.; Hanquet, G.; Boháč, A. Novel CLK1 inhibitors based on N-aryloxazol-2-amine skeleton-a possible way to dual VEGFR2 TK/CLK ligands. Eur. J. Med. Chem. 2017, 126, 754–761. [Google Scholar] [CrossRef] [PubMed]
- Araki, S.; Dairiki, R.; Nakayama, Y.; Murai, A.; Miyashita, R.; Iwatani, M.; Nomura, T.; Nakanishi, O. Inhibitors of CLK protein kinases suppress cell growth and induce apoptosis by modulating pre-mRNA splicing. PLoS ONE 2015, 10, e0116929. [Google Scholar] [CrossRef] [PubMed]
- Bullock, A.N.; Das, S.; Debreczeni, J.É.; Rellos, P.; Fedorov, O.; Niesen, F.H.; Guo, K.; Papagrigoriou, E.; Amos, A.L.; Cho, S. Kinase domain insertions define distinct roles of CLK kinases in SR protein phosphorylation. Structure 2009, 17, 352–362. [Google Scholar] [CrossRef]
- Prak, K.; Kriston-Vizi, J.; Chan, A.E.; Luft, C.; Costa, J.R.; Pengo, N.; Ketteler, R. Benzobisthiazoles represent a novel scaffold for kinase inhibitors of CLK family members. Biochemistry 2016, 55, 608–617. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, C.; Kail, D.; Mariano, M.; Empting, M.; Weber, N.; Paul, T.; Hartmann, R.W.; Engel, M. Design and synthesis of a library of lead-like 2, 4-bisheterocyclic substituted thiophenes as selective Dyrk/Clk inhibitors. PLoS ONE 2014, 9, e87851. [Google Scholar] [CrossRef] [PubMed]
- Tahtouh, T.; Elkins, J.M.; Filippakopoulos, P.; Soundararajan, M.; Burgy, G.; Durieu, E.; Cochet, C.; Schmid, R.S.; Lo, D.C.; Delhommel, F. Selectivity, cocrystal structures, and neuroprotective properties of leucettines, a family of protein kinase inhibitors derived from the marine sponge alkaloid leucettamine B. J. Med. Chem. 2012, 55, 9312–9330. [Google Scholar] [CrossRef] [PubMed]
- Anastassiadis, T.; Deacon, S.W.; Devarajan, K.; Ma, H.; Peterson, J.R. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- Cuny, G.D.; Robin, M.; Ulyanova, N.P.; Patnaik, D.; Pique, V.; Casano, G.; Liu, J.-F.; Lin, X.; Xian, J.; Glicksman, M.A. Structure–activity relationship study of acridine analogs as haspin and DYRK2 kinase inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 3491–3494. [Google Scholar] [CrossRef] [PubMed]
- McLauchilan, H.; ELlliott, M.; Cohen, P. The specificities of protein kinase inhibitors: An update. Biochem. J. 2003, 371, 199–204. [Google Scholar]
- Rosenthal, A.S.; Tanega, C.; Shen, M.; Mott, B.T.; Bougie, J.M.; Nguyen, D.-T.; Misteli, T.; Auld, D.S.; Maloney, D.J.; Thomas, C.J. Potent and selective small molecule inhibitors of specific isoforms of Cdc2-like kinases (Clk) and dual specificity tyrosine-phosphorylation-regulated kinases (Dyrk). Bioorg. Med. Chem. Lett. 2011, 21, 3152–3158. [Google Scholar] [CrossRef] [PubMed]
- Coombs, T.C.; Tanega, C.; Shen, M.; Wang, J.L.; Auld, D.S.; Gerritz, S.W.; Schoenen, F.J.; Thomas, C.J.; Aubé, J. Small-molecule pyrimidine inhibitors of the cdc2-like (Clk) and dual specificity tyrosine phosphorylation-regulated (Dyrk) kinases: Development of chemical probe ML315. Bioorg. Med. Chem. Lett. 2013, 23, 3654–3661. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Phoa, A.F.; Abbassi, R.H.; Hoque, M.; Reekie, T.A.; Font, J.S.; Ryan, R.M.; Stringer, B.W.; Day, B.W.; Johns, T.G. Structural optimization and pharmacological evaluation of inhibitors targeting dual-specificity tyrosine phosphorylation-regulated kinases (DYRK) and CDC-like kinases (CLK) in glioblastoma. J. Med. Chem. 2017, 60, 2052–2070. [Google Scholar] [CrossRef] [PubMed]
- Mott, B.T.; Tanega, C.; Shen, M.; Maloney, D.J.; Shinn, P.; Leister, W.; Marugan, J.J.; Inglese, J.; Austin, C.P.; Misteli, T. Evaluation of substituted 6-arylquinazolin-4-amines as potent and selective inhibitors of cdc2-like kinases (Clk). Bioorg. Med. Chem. Lett. 2009, 19, 6700–6705. [Google Scholar] [CrossRef]
- Fedorov, O.; Huber, K.; Eisenreich, A.; Filippakopoulos, P.; King, O.; Bullock, A.N.; Szklarczyk, D.; Jensen, L.J.; Fabbro, D.; Trappe, J. Specific CLK inhibitors from a novel chemotype for regulation of alternative splicing. Chem. Biol. 2011, 18, 67–76. [Google Scholar] [CrossRef]
- Mariano, M.; Hartmann, R.W.; Engel, M. Systematic diversification of benzylidene heterocycles yields novel inhibitor scaffolds selective for Dyrk1A, Clk1 and CK2. Eur. J. Med. Chem. 2016, 112, 209–216. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Cpd. No. | R | Clk1% Inhibition at 100 nM a | IC50 (nM) b | Clk2% Inhibition at 250 nM a | IC50 (nM) b | Cpd. No. | R | Clk1% Inhibition at 100 nM a | IC50 (nM)b | Clk2 % Inhibition at 250 nM a | IC50 (nM) b |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1ac |  | 33.9 | n.d. | 29.8 | n.d. | 4a |  | 54 | 114 ± 5.4 | n.i. | n.d. |
| 1bc |  | 80 | 6.9 ± 0.12 | -- | 19.1 ± 0.5 | 4b |  | n.i. | n.d. | n.i. | n.d. |
| 1c |  | 50.2 | 100 | 42.4 | n.d. | 5a |  | n.i. | n.d. | n.i. | n.d. |
| 1d |  | n.i. | n.d. | n.i. | n.d. | 6a |  | n.i. | n.d. | n.i. | n.d. |
| 2ac |  | 23.5 | n.d. | n.d. | n.d. | 7a |  | 34.7 | n.d. | 55 | n.d. |
| 2bc |  | 28.7 | n.d. | n.d. | n.d. | 7b |  | 88.5 | 6.2 ± 0.1 | 73.4 | 104 ± 4.2 |
| 3a |  | 28.7 | n.d. | n.i. | n.d. | 8a |  | n.i. | n.d. | n.i. | n.d. |
| 3b |  | n.i. | n.d. | n.i. | n.d. | 8b |  | 45.5 | n.d. | 23.7 | n.d. |

| Cpd. No. | R | Clk1% Inhibition at 100 nM a | IC50 (nM) b | Clk2% Inhibition at 250 nM a | IC50 (nM) b | Cpd. No. | R | Clk1% Inhibition at 100 nM a | IC50 (nM) b | Clk2% Inhibition at 250 nM a | IC50 (nM) b |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 9a |  | n.i. | n.d. | 25.8 | n.d. | 12b |  | 82.8 | 16.8 ± 1.2 | 39.7 | n.d. |
| 9b |  | 8.5 | n.d. | 16.9 | n.d. | 13a |  | 12.5 | n.d. | 26.9 | n.d. |
| 10a |  | 62.2 | 53.5 ± 3.6 | 30 | n.d. | 13b |  | 17.6 | n.d. | 17 | n.d. |
| 10b |  | 88.8 | 12.7 ± 0.9 | 74.7 | 125 ± 7 | 14a |  | 43.2 | n.d. | 24.5 | n.d. |
| 11a |  | 31.8 | n.d. | 3 | n.d. | 14b |  | 49.2 | n.d. | 44 | n.d. |
| 11b |  | 71.2 | 33.6 ± 1.8 | 52.7 | 103 ± 5 | 15a |  | 42 | n.d. | 20 | n.d. |
| 12a |  | n.i. | n.d. | 27 | n.d. | 15b |  | 87.8 | 10.9 ± 0.6 | 72.5 | 167 ± 9 |
| Cpd. No. | GI50 (μM) a | Clk1 Inhibition IC50 (nM) | Selectivity Factor for Clk1 over Clk2 |
|---|---|---|---|
| 1b | 0.63 | 6.9 | 2.7 |
| 7b | 8.3 | 6.2 | 16 |
| 10b | 0.43 | 12.7 | 10 |
| 12b | 12.12 | 16.8 | >15 |
| 15b | 7.92 | 10.9 | 15 |
| 10b | |
|---|---|
| Kinase | % Inhibition at 1 μM a |
| CDK5/p25 | 3 |
| DYRK1A b | 75% @ 0.75 µM IC50 = 355 ± 20 nM |
| DYRK1B | 70 |
| DYRK2 | 19 |
| DYRK3 | 3 |
| Clk1 | 80 |
| Clk3 | 48 |
| Clk4 | 100 |
| PIM1 | 6 |
| CK1 γ2 | 2 |
| CK2 α1 | 5 |
| Haspin | 0 |
| MLCK2 | 3 |
| STK17A (DRAK1) | 25 |
| SRPK1 | 4 |
| SRPK2 | 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
ElHady, A.K.; El-Gamil, D.S.; Chen, P.-J.; Hwang, T.-L.; Abadi, A.H.; Abdel-Halim, M.; Engel, M. 5-Methoxybenzothiophene-2-Carboxamides as Inhibitors of Clk1/4: Optimization of Selectivity and Cellular Potency. Molecules 2021, 26, 1001. https://doi.org/10.3390/molecules26041001
ElHady AK, El-Gamil DS, Chen P-J, Hwang T-L, Abadi AH, Abdel-Halim M, Engel M. 5-Methoxybenzothiophene-2-Carboxamides as Inhibitors of Clk1/4: Optimization of Selectivity and Cellular Potency. Molecules. 2021; 26(4):1001. https://doi.org/10.3390/molecules26041001
Chicago/Turabian StyleElHady, Ahmed K., Dalia S. El-Gamil, Po-Jen Chen, Tsong-Long Hwang, Ashraf H. Abadi, Mohammad Abdel-Halim, and Matthias Engel. 2021. "5-Methoxybenzothiophene-2-Carboxamides as Inhibitors of Clk1/4: Optimization of Selectivity and Cellular Potency" Molecules 26, no. 4: 1001. https://doi.org/10.3390/molecules26041001
APA StyleElHady, A. K., El-Gamil, D. S., Chen, P.-J., Hwang, T.-L., Abadi, A. H., Abdel-Halim, M., & Engel, M. (2021). 5-Methoxybenzothiophene-2-Carboxamides as Inhibitors of Clk1/4: Optimization of Selectivity and Cellular Potency. Molecules, 26(4), 1001. https://doi.org/10.3390/molecules26041001