
Analyzing Kinase Similarity in Small Molecule and Protein Structural Space to Explore the Limits of Multi-Target Screening

 , , , , and
, , , , and
Abstract
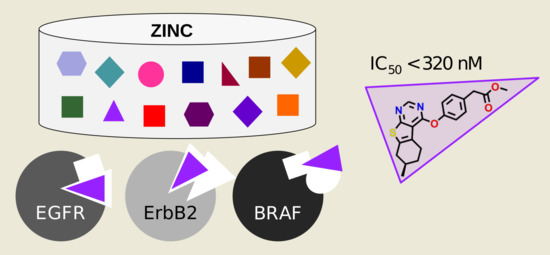
1. Introduction
2. Results and Discussion
2.1. Kinase Profiles
2.2. Virtual Screening against Kinase Profiles
2.3. Experimental Validation
2.4. Kinase Similarities
2.4.1. Ligand Profile Similarity (LigProfSim)
2.4.2. Pocket Sequence Similarity (PocSeqSim)
2.4.3. Interaction Fingerprint Similarity (IFPSim)
2.4.4. Pocket Structure Similarity (PocStrucSim)
2.4.5. Docking Rank Similarity (DockRankSim)
2.4.6. Comparison of Similarity Analyses
3. Data and Methods
3.1. Docking-Based Virtual Screening
3.2. Experimental Testing
3.2.1. DiscoverX KINOMEscan
3.2.2. Eurofins In Vitro Assay
3.2.3. IC Determination
3.3. Kinase Similarity Measures
3.3.1. Ligand Profile Similarity (LigProfSim)
3.3.2. Pocket Sequence Similarity (PocSeqSim)
3.3.3. Interaction Fingerprint Similarity (IFPSim)
3.3.4. Pocket Structure Similarity (PocStrucSim)
3.3.5. Docking Rank Similarity (DockRankSim)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2016, 16, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Roth, B.L.; Sheffler, D.J.; Kroeze, W.K. Magic shotguns versus magic bullets: Selectively non-selective drugs for mood disorders and schizophrenia. Nat. Rev. Drug Discov. 2004, 3, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Garuti, L.; Roberti, M.; Bottegoni, G. Multi-Kinase Inhibitors. Curr. Med. Chem. 2015, 22, 695–712. [Google Scholar] [CrossRef] [PubMed]
- Gentile, C.; Martorana, A.; Lauria, A.; Bonsignore, R. Kinase Inhibitors in Multitargeted Cancer Therapy. Curr. Med. Chem. 2017, 24, 1671–1686. [Google Scholar] [CrossRef] [PubMed]
- Moser, D.; Wisniewska, J.M.; Hahn, S.; Achenbach, J.; Buscató, E.; Klingler, F.M.; Hofmann, B.; Steinhilber, D.; Proschak, E. Dual-Target Virtual Screening by Pharmacophore Elucidation and Molecular Shape Filtering. ACS Med. Chem. Lett. 2012, 3, 155–158. [Google Scholar] [CrossRef]
- Schmidt, D.; Bernat, V.; Brox, R.; Tschammer, N.; Kolb, P. Identifying Modulators of CXC Receptors 3 and 4 with Tailored Selectivity Using Multi-Target Docking. ACS Chem. Biol. 2015, 10, 715–724. [Google Scholar] [CrossRef]
- Jaiteh, M.; Zeifman, A.; Saarinen, M.; Svenningsson, P.; Bréa, J.; Loza, M.I.; Carlsson, J. Docking Screens for Dual Inhibitors of Disparate Drug Targets for Parkinson’s Disease. J. Med. Chem. 2018, 61, 5269–5278. [Google Scholar] [CrossRef]
- Klebl, B.; Müller, G.; Hamacher, M.; Mannhold, R.; Kubinyi, H.; Folkers, G. Protein Kinases as Drug Targets, 49th ed.; Methods and Principles in Medicinal Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011. [Google Scholar] [CrossRef]
- Lin, J.; Gan, C.M.; Zhang, X.; Jones, S.; Sjöblom, T.; Wood, L.D.; Parsons, D.W.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; et al. A multidimensional analysis of genes mutated in breast and colorectal cancers. Genome Res. 2007, 17, 1304–1318. [Google Scholar] [CrossRef]
- Wood, L.D.; Parsons, D.W.; Jones, S.; Lin, J.; Sjöblom, T.; Leary, R.J.; Shen, D.; Boca, S.M.; Barber, T.; Ptak, J.; et al. The genomic landscapes of human breast and colorectal cancers. Science 2007, 318, 1108–1113. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef]
- Kooistra, A.J.; Volkamer, A. Kinase-Centric Computational Drug Development. In Annual Reports in Medicinal Chemistry; Academic Press: Cambridge, MA, USA, 2017; Volume 50, pp. 263–299. [Google Scholar] [CrossRef]
- Sorgenfrei, F.A.; Fulle, S.; Merget, B. Kinome-Wide Profiling Prediction of Small Molecules. ChemMedChem 2018, 13, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- van Linden, O.P.J.; Kooistra, A.J.; Leurs, R.; de Esch, I.J.P.; de Graaf, C. KLIFS: A knowledge-based structural database to navigate kinase-ligand interaction space. J. Med. Chem. 2014, 57, 249–277. [Google Scholar] [CrossRef] [PubMed]
- Kooistra, A.J.; Kanev, G.K.; van Linden, O.P.; Leurs, R.; de Esch, I.J.; de Graaf, C. KLIFS: A structural kinase-ligand interaction database. Nucleic Acids Res. 2016, 44, D365–D371. [Google Scholar] [CrossRef]
- Kanev, G.K.; de Graaf, C.; Westerman, B.A.; de Esch, I.J.P.; Kooistra, A.J. KLIFS: An overhaul after the first 5 years of supporting kinase research. Nucleic Acids Res. 2020, 49, D562–D569. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. FDA Approved Kinase Inhibitors (‘-nibs’). Available online: http://www.brimr.org/PKI/PKIs.htm (accessed on 3 July 2020).
- Ung, P.M.U.; Rahman, R.; Schlessinger, A. Redefining the Protein Kinase Conformational Space with Machine Learning. Cell Chem. Biol. 2018, 25, 916–924. [Google Scholar] [CrossRef]
- Modi, V.; Dunbrack, R.L. Defining a new nomenclature for the structures of active and inactive kinases. Proc. Natl. Acad. Sci. USA 2019, 116, 6818–6827. [Google Scholar] [CrossRef]
- Karaman, M.W.; Herrgard, S.; Treiber, D.K.; Gallant, P.; Atteridge, C.E.; Campbell, B.T.; Chan, K.W.; Ciceri, P.; Davis, M.I.; Edeen, P.T.; et al. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008, 26, 127–132. [Google Scholar] [CrossRef]
- Holbro, T.; Hynes, N.E. ErbB receptors: Directing Key Signaling Networks Throughout Life. Annu. Rev. Pharmac. Toxic. 2004, 44, 195–217. [Google Scholar] [CrossRef]
- Seshadri, R.; Firgaira, F.A.; Horsfall, D.J.; McCaul, K.; Setlur, V.; Kitchen, P. Clinical significance of HER-2/neu oncogene amplification in primary breast cancer. The South Australian Breast Cancer Study Group. J. Clin. Oncol. 1993, 11, 1936–1942. [Google Scholar] [CrossRef]
- Klein, S.; Levitzki, A. Targeting the EGFR and the PKB pathway in cancer. Curr. Op. Cell Biol. 2009, 21, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.D.; Mansfield, P.F.; Akagi, M.; Takeda, A.; Liu, W.; Bucana, C.D.; Hicklin, D.J.; Ellis, L.M. Effects of combination anti-vascular endothelial growth factor receptor and anti-epidermal growth factor receptor therapies on the growth of gastric cancer in a nude mouse model. Eur. J. Cancer 2002, 38, 1133–1140. [Google Scholar] [CrossRef]
- McTigue, M.; Murray, B.W.; Chen, J.H.; Deng, Y.L.; Solowiej, J.; Kania, R.S. Molecular conformations, interactions, and properties associated with drug efficiency and clinical performance among VEGFR TK inhibitors. Proc. Natl. Acad. Sci. USA 2012, 109, 18281–18289. [Google Scholar] [CrossRef] [PubMed]
- Davis, I.M.; Hunt, P.J.; Herrgard, S.; Ciceri, P.; Wodicka, M.L.; Pallares, G.; Hocker, M.; Treiber, K.D.; Zarrinkar, P.P.; Treiber, K.D. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, S.; Kuhn, D.; Klebe, G. A New Method to Detect Related Function among Proteins Independent of Sequence and Fold Homology. J. Mol. Biol. 2002, 323, 387–406. [Google Scholar] [CrossRef]
- Krotzky, T.; Fober, T.; Hüllermeier, E.; Klebe, G. Extended Graph-Based Models for Enhanced Similarity Search in Cavbase. IEEE/ACM Trans. Comput. Biol. Bioinform. 2014, 11, 878–890. [Google Scholar] [CrossRef]
- Wood, E.R.; Truesdale, A.T.; McDonald, O.B.; Yuan, D.; Hassell, A.; Dickerson, S.H.; Ellis, B.; Pennisi, C.; Horne, E.; Lackey, K.; et al. A Unique Structure for Epidermal Growth Factor Receptor Bound to GW572016 (Lapatinib). Cancer Res. 2004, 64, 6652–6659. [Google Scholar] [CrossRef]
- Aertgeerts, K.; Skene, R.; Yano, J.; Sang, B.C.; Zou, H.; Snell, G.; Jennings, A.; Iwamoto, K.; Habuka, N.; Hirokawa, A.; et al. Structural Analysis of the Mechanism of Inhibition and Allosteric Activation of the Kinase Domain of HER2 Protein. J. Biol. Chem. 2011, 286, 18756–18765. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Seto, M.; Banno, H.; Kawakita, Y.; Oorui, M.; Taniguchi, T.; Ohta, Y.; Tamura, T.; Nakayama, A.; Miki, H.; et al. Design and Synthesis of Novel Human Epidermal Growth Factor Receptor 2 (HER2)/Epidermal Growth Factor Receptor (EGFR) Dual Inhibitors Bearing a Pyrrolo[3,2-d]pyrimidine Scaffold. J. Med. Chem. 2011, 54, 8030–8050. [Google Scholar] [CrossRef]
- Wan, P.T.C.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Project, C.G.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef]
- Ren, L.; Wenglowsky, S.; Miknis, G.; Rast, B.; Buckmelter, A.J.; Ely, R.J.; Schlachter, S.; Laird, E.R.; Randolph, N.; Callejo, M.; et al. Non-oxime inhibitors of B-RafV600E kinase. Bioorg. Med. Chem. Lett. 2011, 21, 1243–1247. [Google Scholar] [CrossRef] [PubMed]
- Furet, P.; Guagnano, V.; Fairhurst, R.A.; Imbach-Weese, P.; Bruce, I.; Knapp, M.; Fritsch, C.; Blasco, F.; Blanz, J.; Aichholz, R.; et al. Discovery of NVP-BYL719 a potent and selective phosphatidylinositol-3 kinase alpha inhibitor selected for clinical evaluation. Bioorg. Med. Chem. Lett. 2013, 23, 3741–3748. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Ikemori-Kawada, M.; Jestel, A.; von König, K.; Funahashi, Y.; Matsushima, T.; Tsuruoka, A.; Inoue, A.; Matsui, J. Distinct Binding Mode of Multikinase Inhibitor Lenvatinib Revealed by Biochemical Characterization. ACS Med. Chem. Lett. 2015, 6, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Kolb, P.; Rosenbaum, D.M.; Irwin, J.J.; Fung, J.J.; Kobilka, B.K.; Shoichet, B.K. Structure-based discovery of β2-adrenergic receptor ligands. Proc. Natl. Acad. Sci. USA 2009, 106, 6843–6848. [Google Scholar] [CrossRef]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A Free Tool to Discover Chemistry for Biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef]
- Kuntz, I.D.; Meng, E.C.; Oatley, S.J.; Langridge, R.; Ferrin, T.E. A geometric approach to macromolecule-ligand interactions. J. Mol. Biol. 1982, 161, 269–288. [Google Scholar] [CrossRef]
- Meng, E.C.; Shoichet, B.K.; Kuntz, I.D. Automated docking with grid-based energy evaluation. J. Comp. Chem. 1992, 13, 505–524. [Google Scholar] [CrossRef]
- Shoichet, B.K.; Kuntz, I.D. Matching chemistry and shape in molecular docking. Protein Eng. Des. Sel. 1993, 6, 723–732. [Google Scholar] [CrossRef]
- Shoichet, B.K.; Leach, A.R.; Kuntz, I.D. Ligand solvation in molecular docking. Proteins 1999, 34, 4–16. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Shoichet, B.K. Rapid context-dependent ligand desolvation in molecular docking. J. Chem. Inf. Model. 2010, 50, 1561–1573. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Weber, W.; Bertics, P.J.; Gill, G.N. Immunoaffinity purification of the epidermal growth factor receptor. Stoichiometry of binding and kinetics of self-phosphorylation. J. Biol. Chem. 1984, 259, 14631–14636. [Google Scholar] [CrossRef]
- Quian, X.L.; Decker, S.J.; Greene, M.I. p185c-neu and epidermal growth factor receptor associate into a structure composed of activated kinases. Proc. Natl. Acad. Sci. USA 1992, 89, 1330–1334. [Google Scholar] [CrossRef] [PubMed]
- Sinnamon, R.H.; McDevitt, P.; Pietrak, B.L.; Leydon, V.R.; Xue, Y.; Lehr, R.; Qi, H.; Burns, M.; Elkins, P.; Ward, P.; et al. Baculovirus production of fully-active phosphoinositide 3-kinase alpha as a p85α–p110α fusion for X-ray crystallographic analysis with ATP competitive enzyme inhibitors. Protein Expr. Purif. 2010, 73, 167–176. [Google Scholar] [CrossRef]
- Kupcho, K.R.; Bruinsma, R.; Hallis, T.M.; Lasky, D.A.; Somberg, R.L.; Turek-Etienne, T.; Vogel, K.W.; Huwiler, K.G. Fluorescent Cascade and Direct Assays for Characterization of RAF Signaling Pathway Inhibitors. Curr. Chem. Genom. 2008, 1, 43–53. [Google Scholar] [CrossRef]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Felix, E.; Magarinos, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2018, 47, D930–D940. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Hendlich, M.; Rippmann, F.; Barnickel, G. LIGSITE: Automatic and efficient detection of potential small molecule-binding sites in proteins. J. Mol. Graph. Model. 1997, 15, 359–363. [Google Scholar] [CrossRef]
- Lyu, J.; Wang, S.; Balius, T.E.; Singh, I.; Levit, A.; Moroz, Y.S.; O’Meara, M.J.; Che, T.; Algaa, E.; Tolmachova, K.; et al. Ultra-large library docking for discovering new chemotypes. Nature 2019, 566, 224–229. [Google Scholar] [CrossRef]
- Chevillard, F.; Stotani, S.; Karawajczyk, A.; Hristeva, S.; Pardon, E.; Steyaert, J.; Tzalis, D.; Kolb, P. Interrogating dense ligand chemical space with a forward-synthetic library. Proc. Natl. Acad. Sci. USA 2019, 116, 11496–11501. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Kinase | Synonyms | UniProt ID | Group | Family |
|---|---|---|---|---|
| EGFR | ErbB1 | P00533 | TK | EGFR |
| ErbB2 | Her2 | P04626 | TK | EGFR |
| PI3K | PI3KCA, p110 | P42336 | Atypical | PIK |
| VEGFR2 | KDR | P35968 | TK | VEGFR |
| BRAF | - | P15056 | TKL | RAF |
| CDK2 | - | P24941 | CMGC | CDK |
| LCK | - | P06239 | TK | Src |
| MET | - | P08581 | TK | MET |
| p38 | MAPK14 | Q16539 | CMGC | MAPK |
| ID | Kinase Profile | No. of Tested Compounds |
|---|---|---|
| 1 | +EGFR+ErbB2—BRAF | 18 |
| 2 | +EGFR+PI3K—BRAF | 9 |
| 3 | +EGFR+VEGFR2—BRAF | 8 |
| 4 | +VEGFR2 | 4 |
| Compound | P | Research Lab | Unit | EGFR | ErbB2 | ErbB2insYVMA | BRAF |
|---|---|---|---|---|---|---|---|
| DS39984 | 1 | DiscoverX | % ctrl. activity at 10 µM | 17 | 21 | n.d. | – |
| DS39984 | 1 | Eurofins | % inhib. at 20 µM ± s.d. | – | n.d. | – | |
| DS39984 | 1 | Rauh Lab | IC ± s.d. | nM | n.d. | nM | n.d. |
| K001MM011 | 4 | DiscoverX | % ctrl. activity at 10 µM | 1.4 | 53 | n.d. | – |
| Kinase | Promiscuity | Mean (±s.d.) | |||
|---|---|---|---|---|---|
| LigProfSim | PocSeqSim | IFPSim | PocStrucSim | ||
| EGFR | 0.59 | 0.37 () | 0.50 () | 0.81 () | 0.50 () |
| ErbB2 | 0.62 | 0.37 () | 0.50 () | 0.64 () | 0.38 () |
| PI3K | 0.65 | 0.18 () | 0.23 () | 0.61 () | 0.33 () |
| BRAF | 0.82 | 0.56 () | 0.42 () | 0.82 () | 0.52 () |
| CDK2 | 0.55 | 0.33 () | 0.40 () | 0.80 () | 0.49 () |
| LCK | 0.63 | 0.34 () | 0.45 () | 0.82 () | 0.50 () |
| MET | 0.79 | 0.31 () | 0.45 () | 0.79 () | 0.46 () |
| p38 | 0.77 | 0.43 () | 0.44 () | 0.82 () | 0.47 () |
| VEGFR2 | 0.70 | 0.51 () | 0.46 () | 0.83 () | 0.50 () |
| Kinase (Family/Group) | # Compounds | # Structures | ||
|---|---|---|---|---|
| Actives | Tested | IFPSim | PocStrucSim | |
| EGFR (TK/EGFR) | 3382 | 5702 | 150 | 15 |
| ErbB2 (TK/EGFR) | 1048 | 1690 | 2 | 2 |
| PI3K (Atypical/PIK) | 2706 | 4150 | 26 | 2 |
| VEGFR2 (TK/VEGFR) | 5197 | 7426 | 41 | 13 |
| BRAF (TKL/RAF) | 2968 | 3625 | 69 | 25 |
| CDK2 (CMGC/CDK) | 837 | 1520 | 377 | 43 |
| LCK (TK/Src) | 976 | 1552 | 34 | 29 |
| MET (TK(MET)) | 2248 | 2851 | 70 | 11 |
| p38(CMGC/MAPK) | 2753 | 3581 | 196 | 36 |
| Total | 22,115 | 32,097 | 965 | 176 |
| Kinase | PDB | DFG | αC |
|---|---|---|---|
| EGFR | 1XKK | in | out |
| EGFR | 3POZ | in | out |
| ErbB2 | 3PP0 | in | in |
| ErbB2 | 3RCD | in | out-like |
| BRAF | 1UWH | out | out |
| BRAF | 3PPK | in | in |
| PI3K | 4JPS | in | in |
| VEGFR2 | 2P2H | in | out |
| VEGFR2 | 3WZD | in | out |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmidt, D.; Scharf, M.M.; Sydow, D.; Aßmann, E.; Martí-Solano, M.; Keul, M.; Volkamer, A.; Kolb, P. Analyzing Kinase Similarity in Small Molecule and Protein Structural Space to Explore the Limits of Multi-Target Screening. Molecules 2021, 26, 629. https://doi.org/10.3390/molecules26030629
Schmidt D, Scharf MM, Sydow D, Aßmann E, Martí-Solano M, Keul M, Volkamer A, Kolb P. Analyzing Kinase Similarity in Small Molecule and Protein Structural Space to Explore the Limits of Multi-Target Screening. Molecules. 2021; 26(3):629. https://doi.org/10.3390/molecules26030629
Chicago/Turabian StyleSchmidt, Denis, Magdalena M. Scharf, Dominique Sydow, Eva Aßmann, Maria Martí-Solano, Marina Keul, Andrea Volkamer, and Peter Kolb. 2021. "Analyzing Kinase Similarity in Small Molecule and Protein Structural Space to Explore the Limits of Multi-Target Screening" Molecules 26, no. 3: 629. https://doi.org/10.3390/molecules26030629
APA StyleSchmidt, D., Scharf, M. M., Sydow, D., Aßmann, E., Martí-Solano, M., Keul, M., Volkamer, A., & Kolb, P. (2021). Analyzing Kinase Similarity in Small Molecule and Protein Structural Space to Explore the Limits of Multi-Target Screening. Molecules, 26(3), 629. https://doi.org/10.3390/molecules26030629