
Structure Activity Relationship of Key Heterocyclic Anti-Angiogenic Leads of Promising Potential in the Fight against Cancer


Abstract
1. Introduction
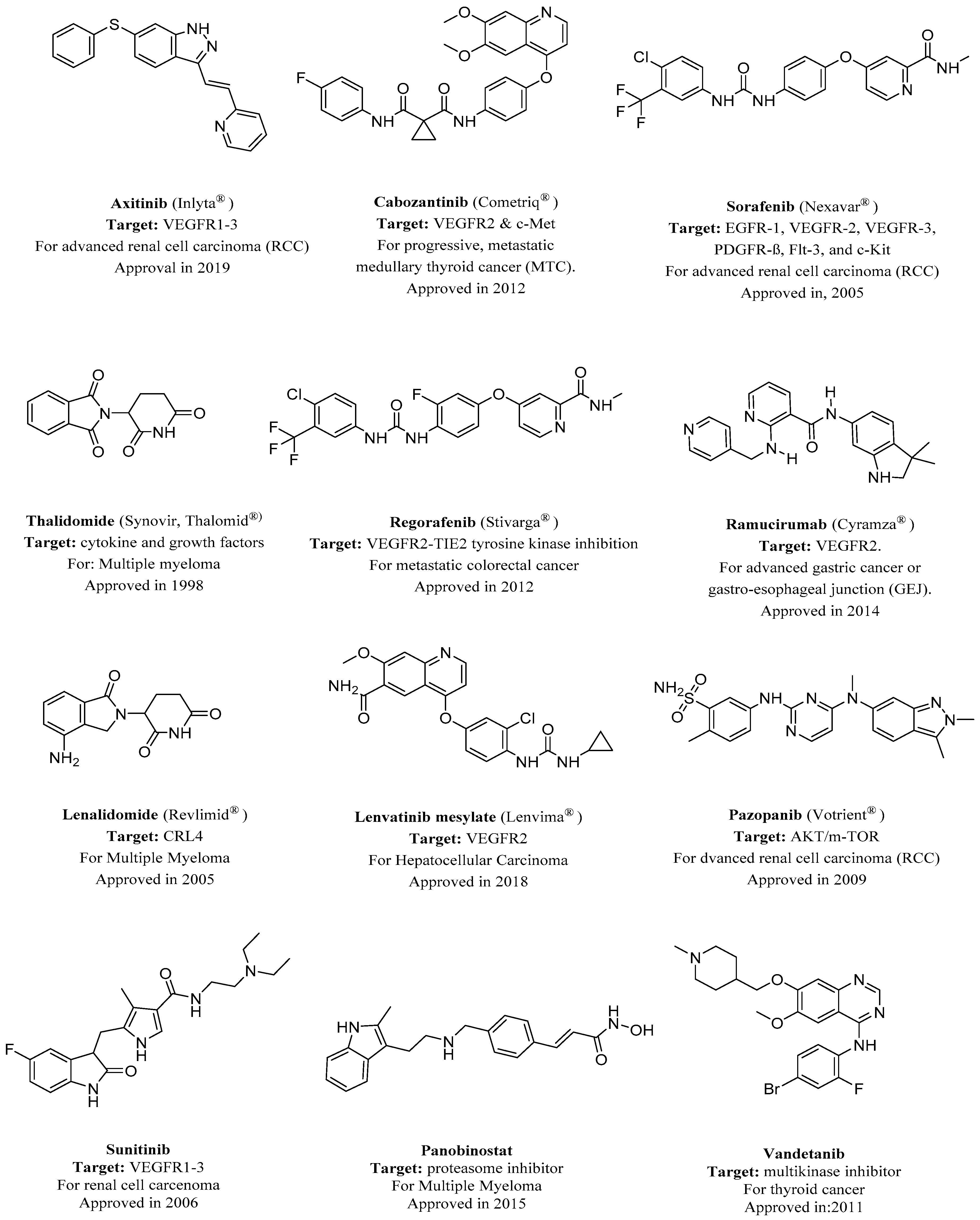
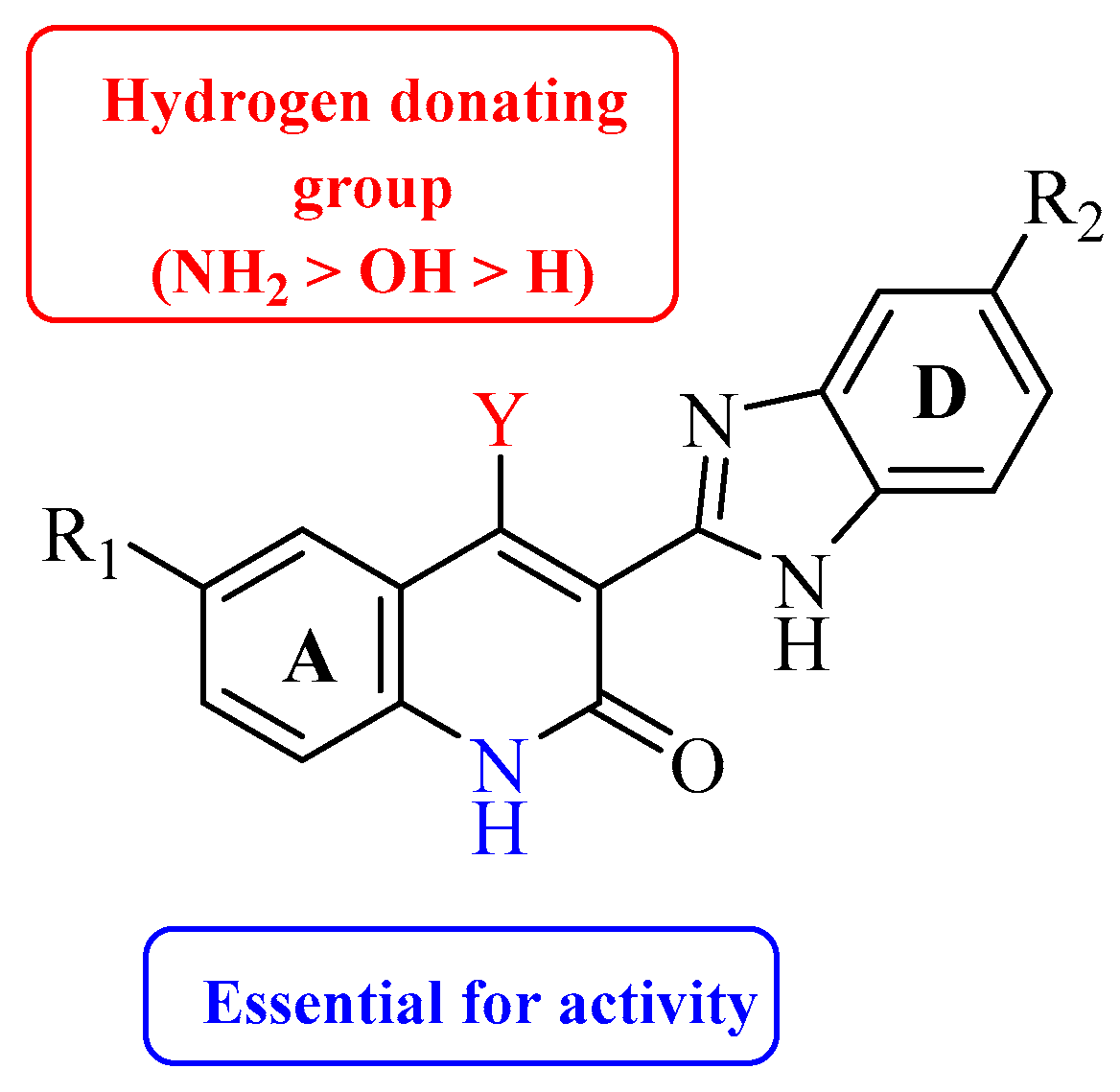
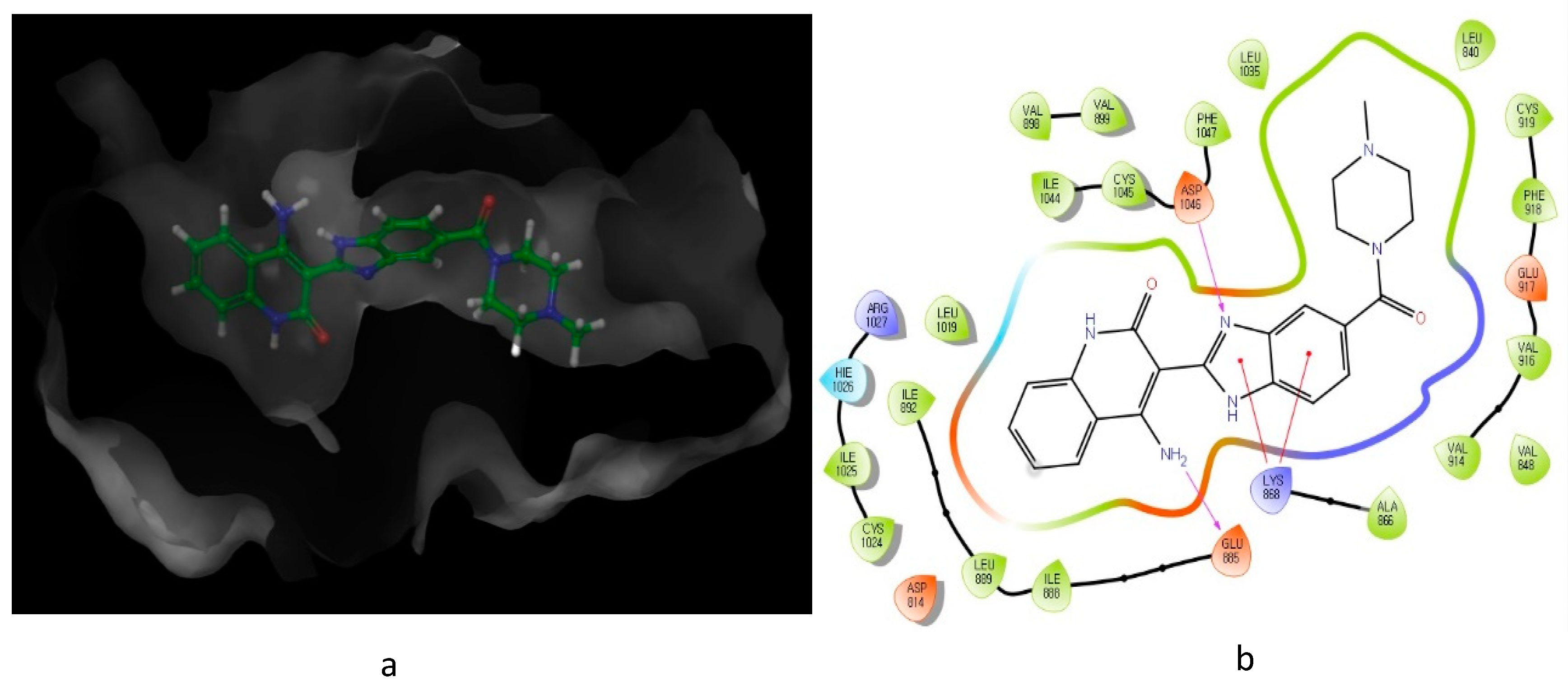
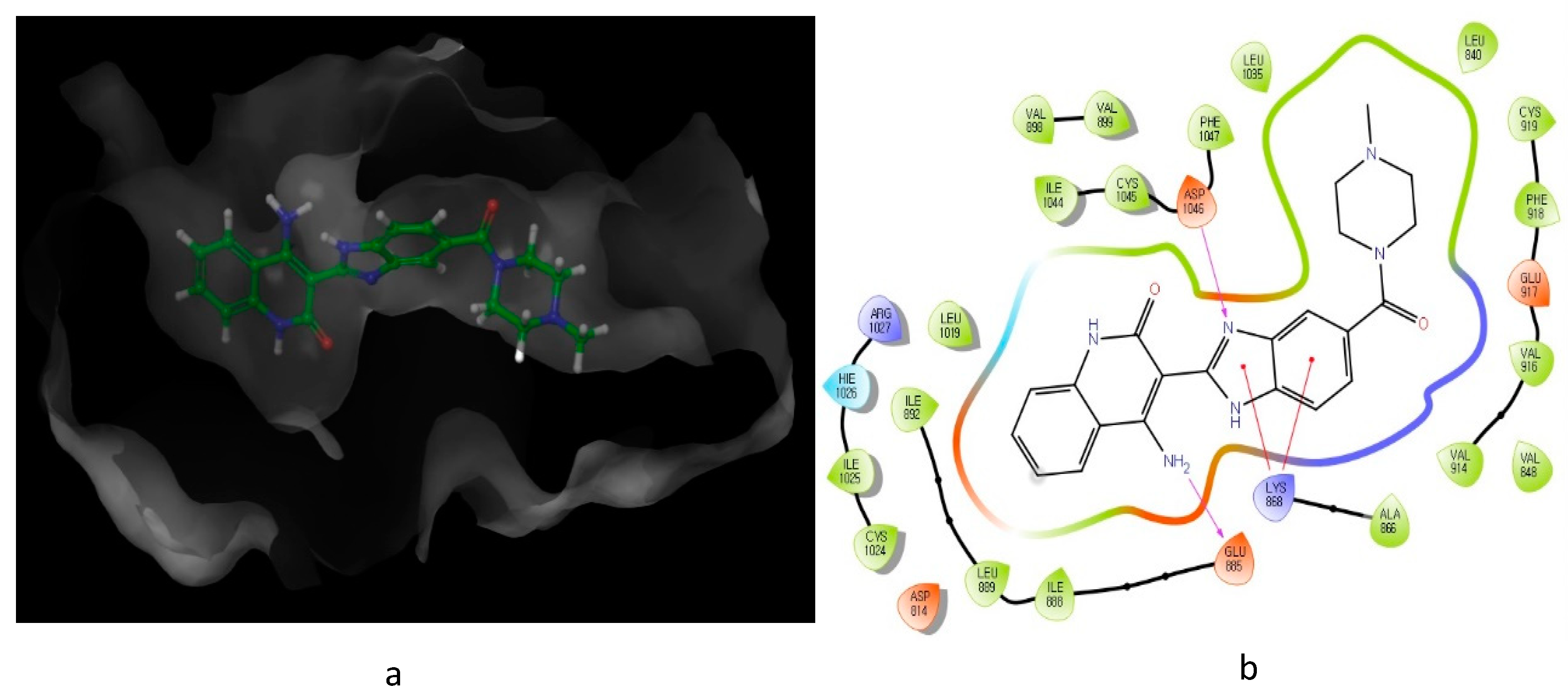
2. Nitrogen-Based Heterocycles
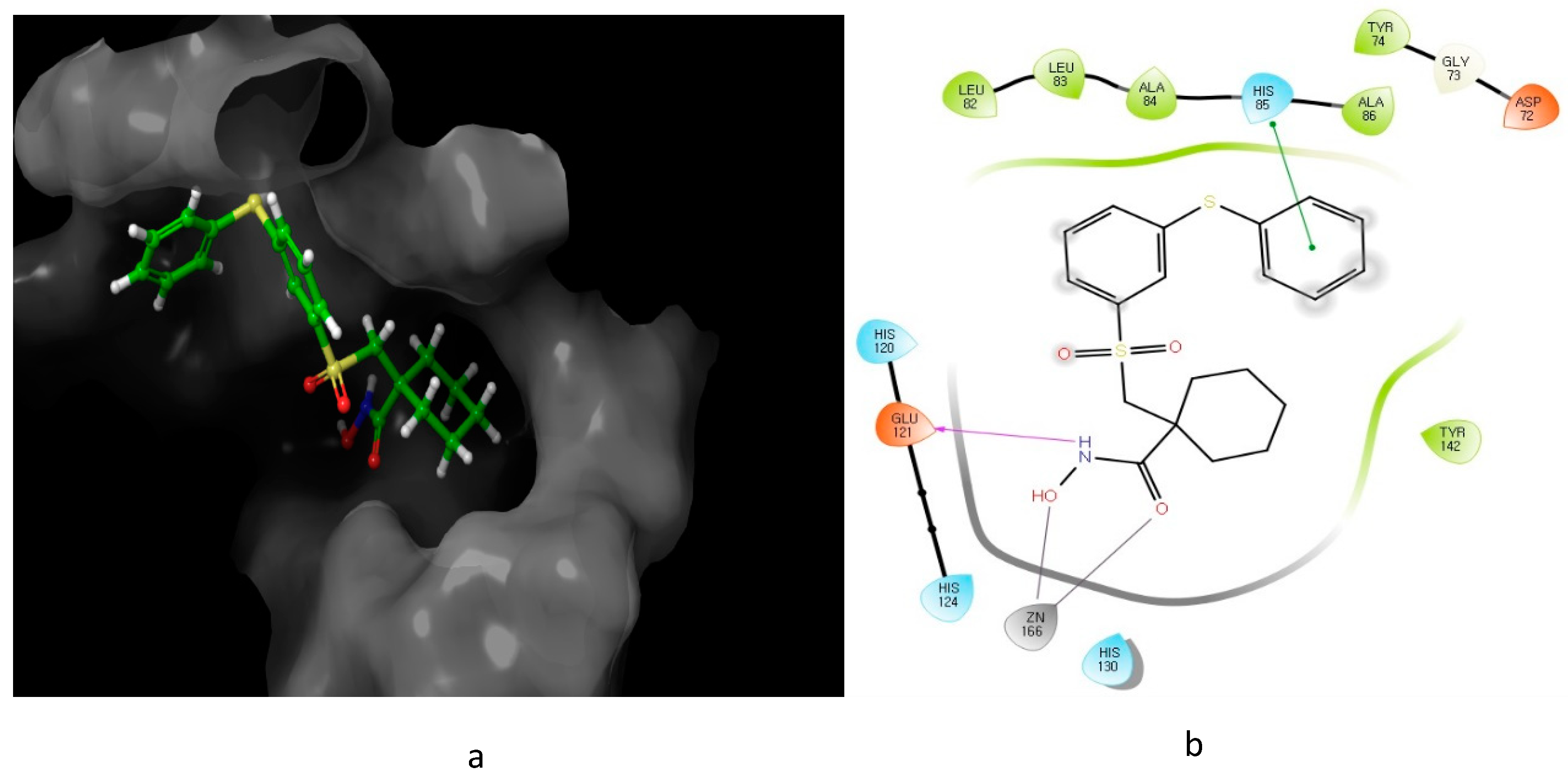
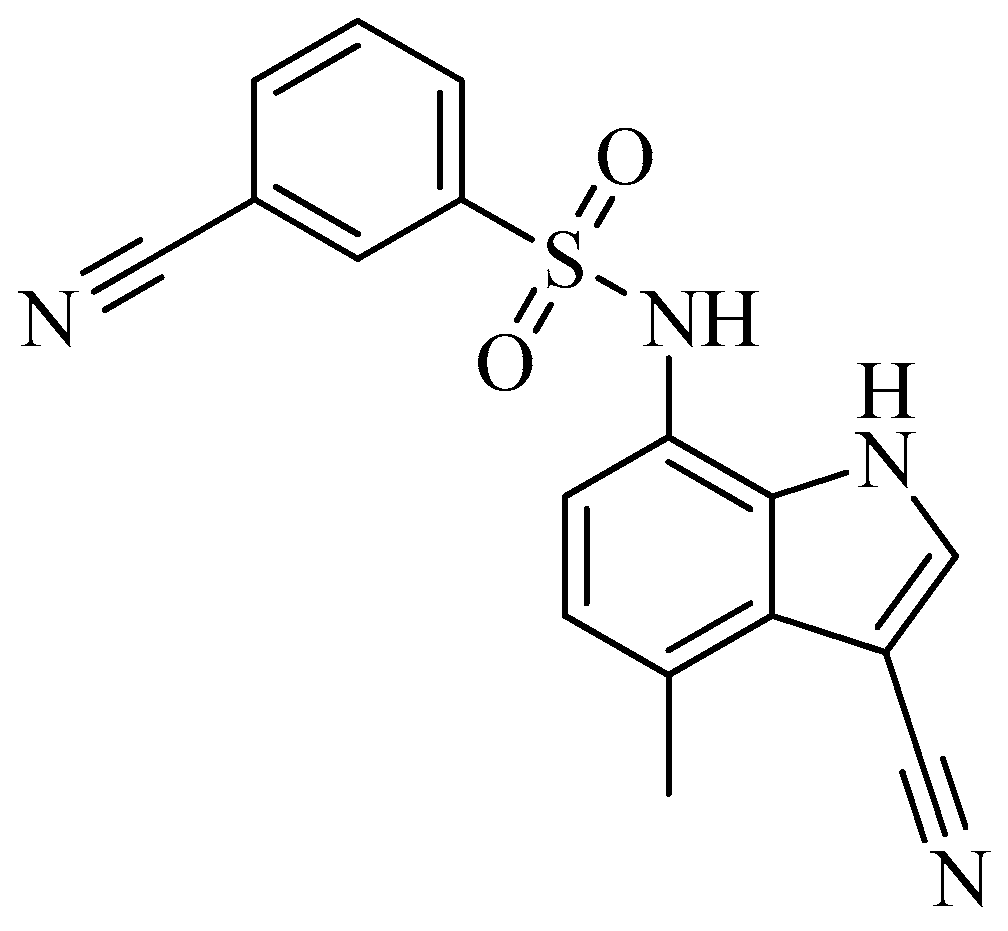
3. Heterocyclic Sulfonamides
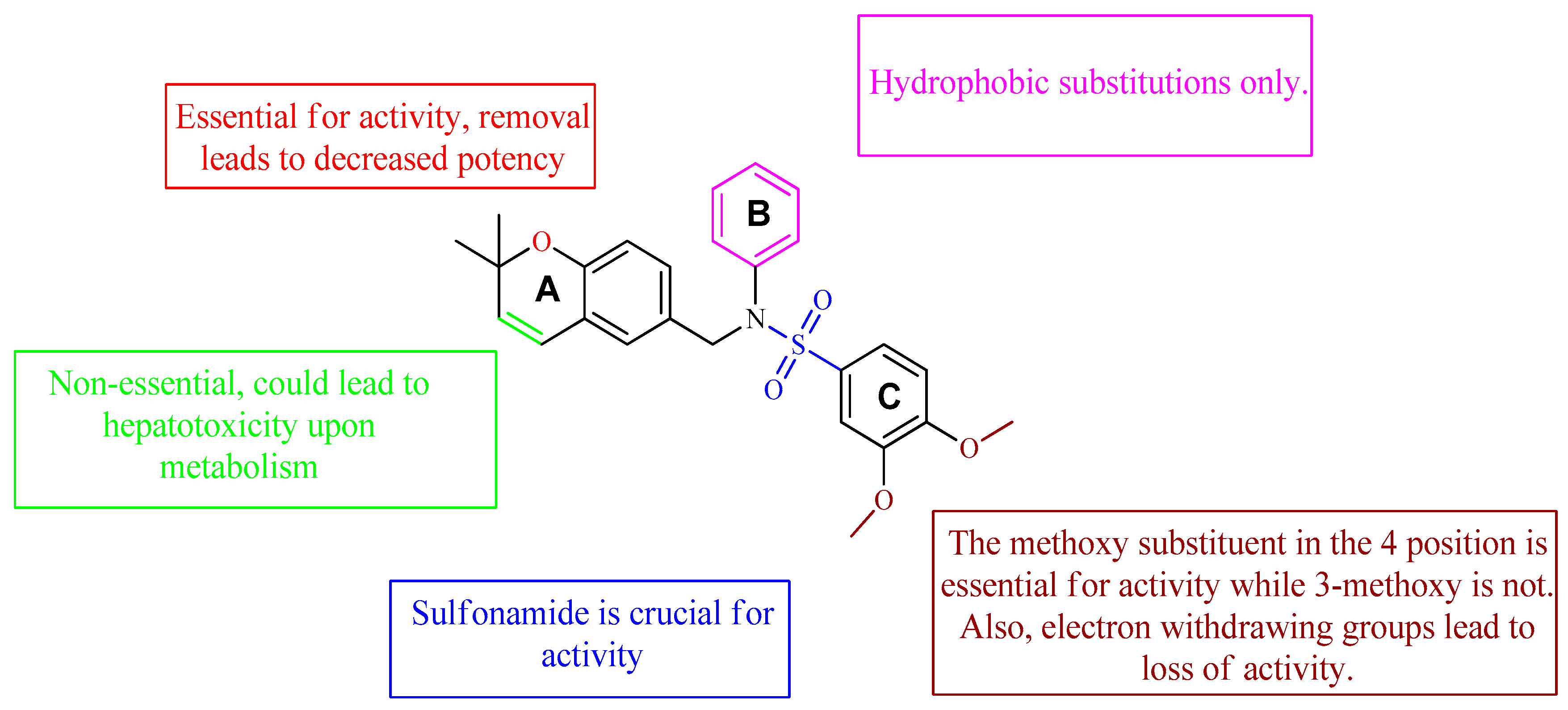
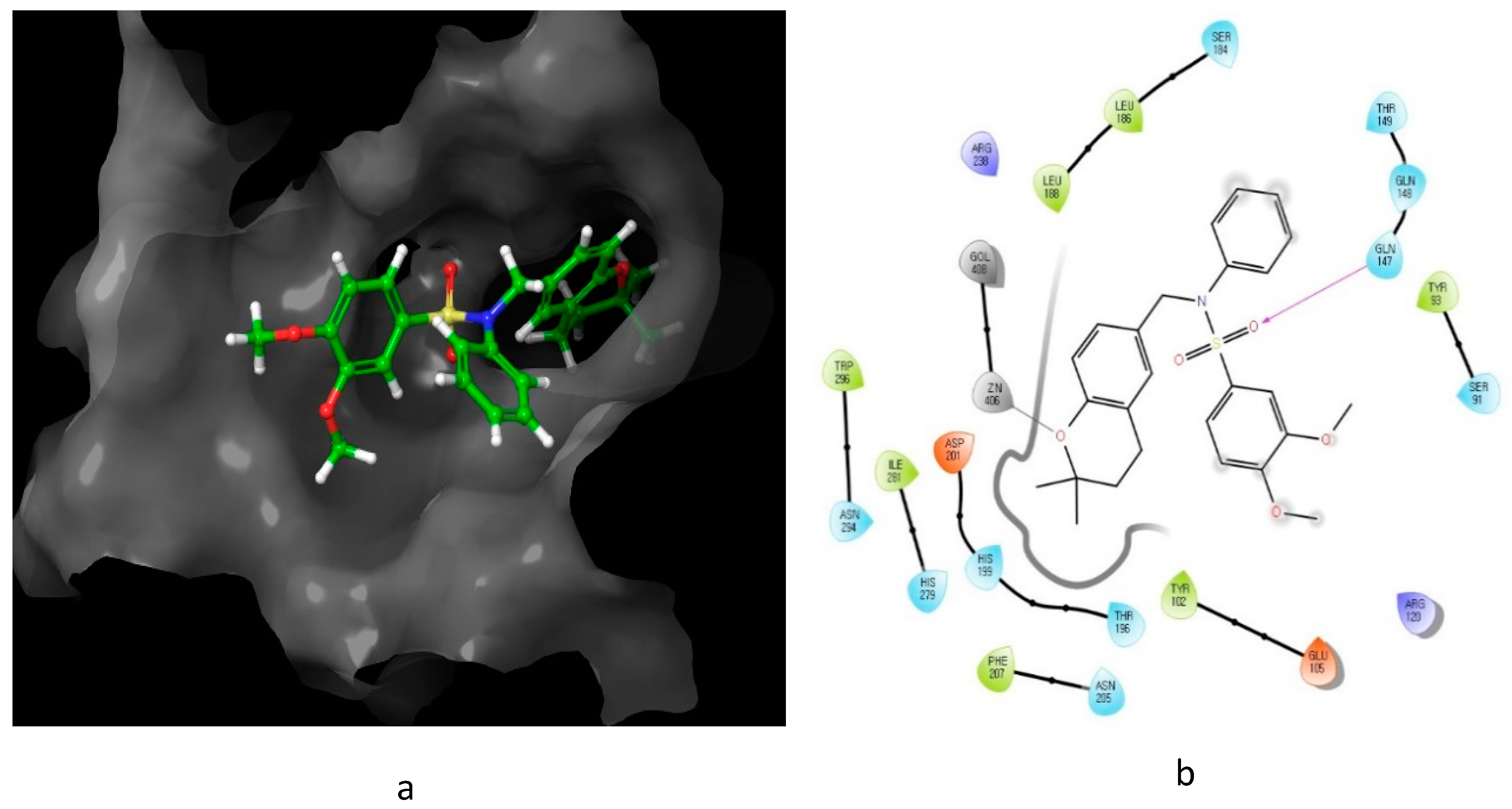
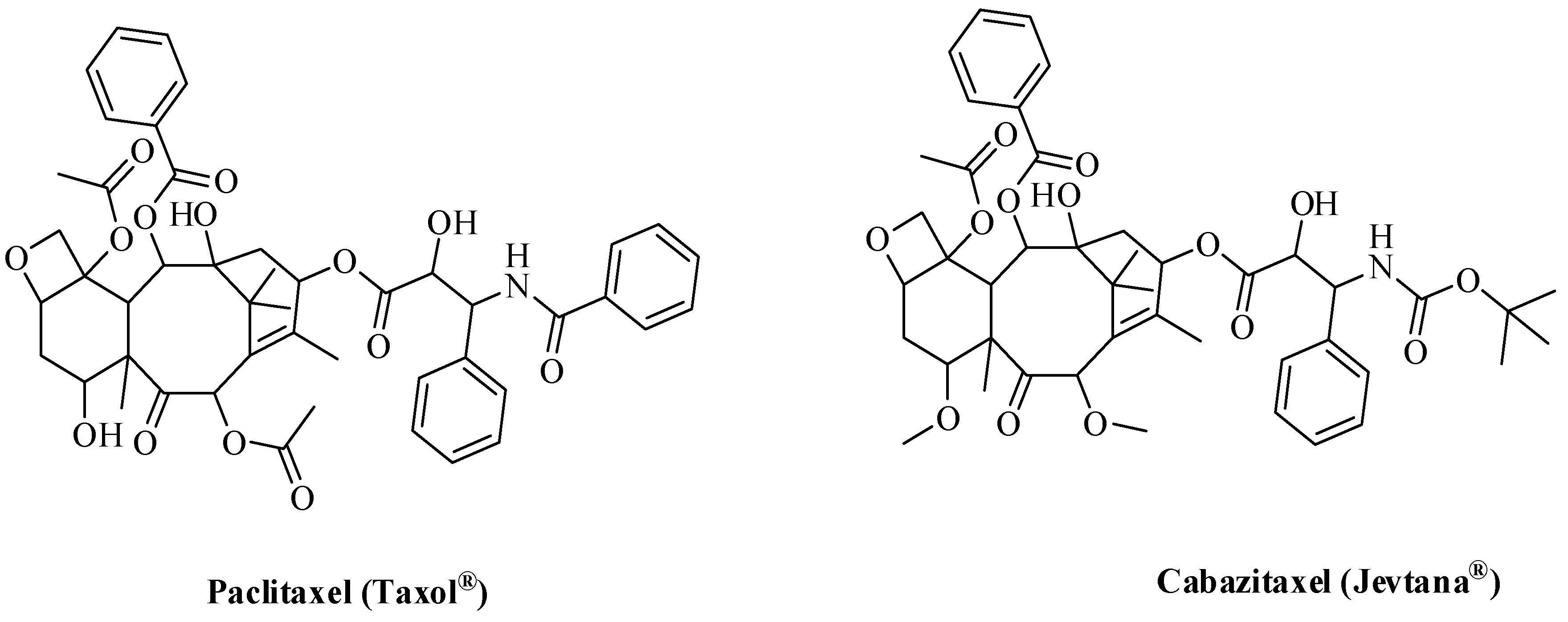
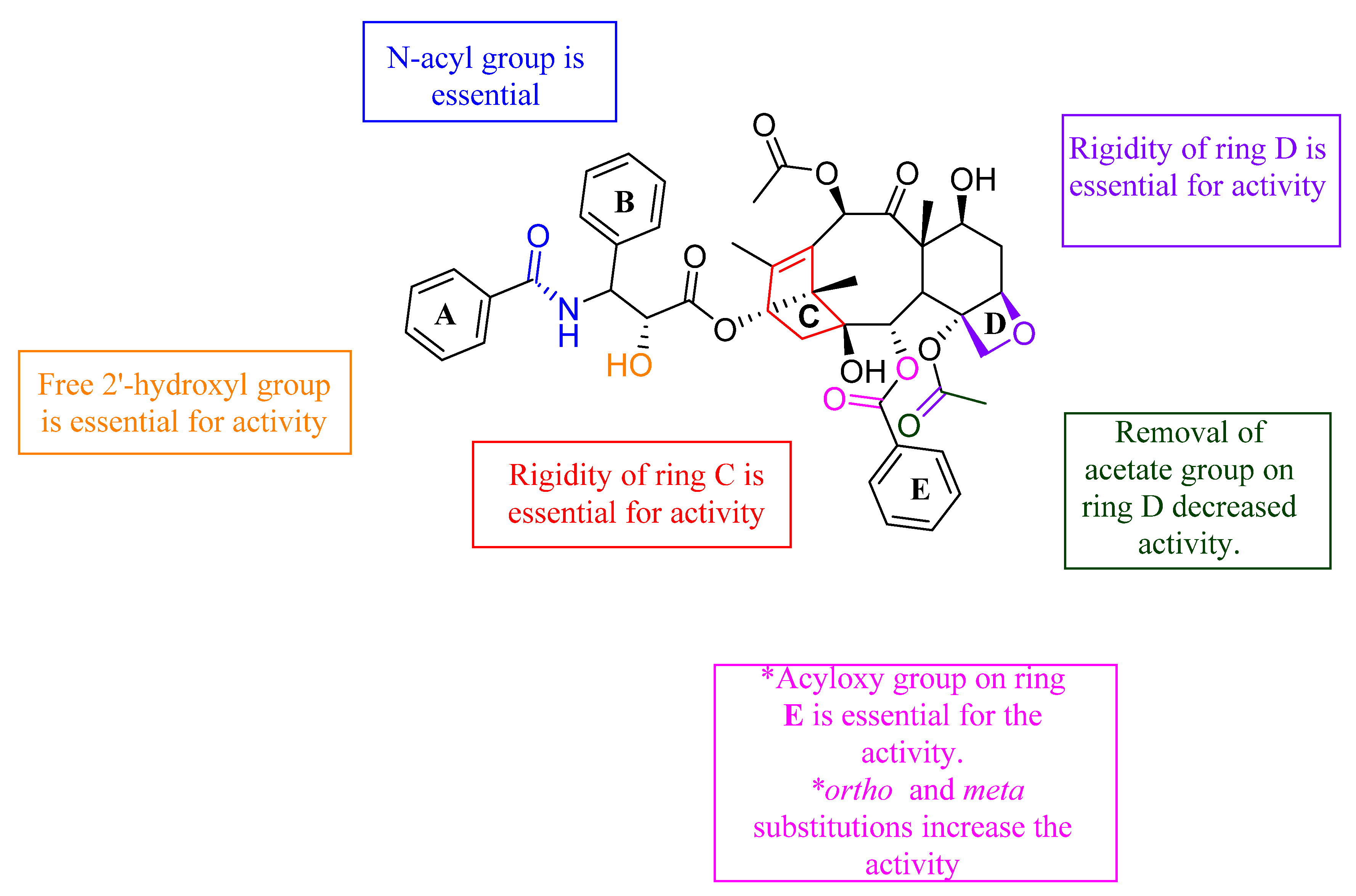
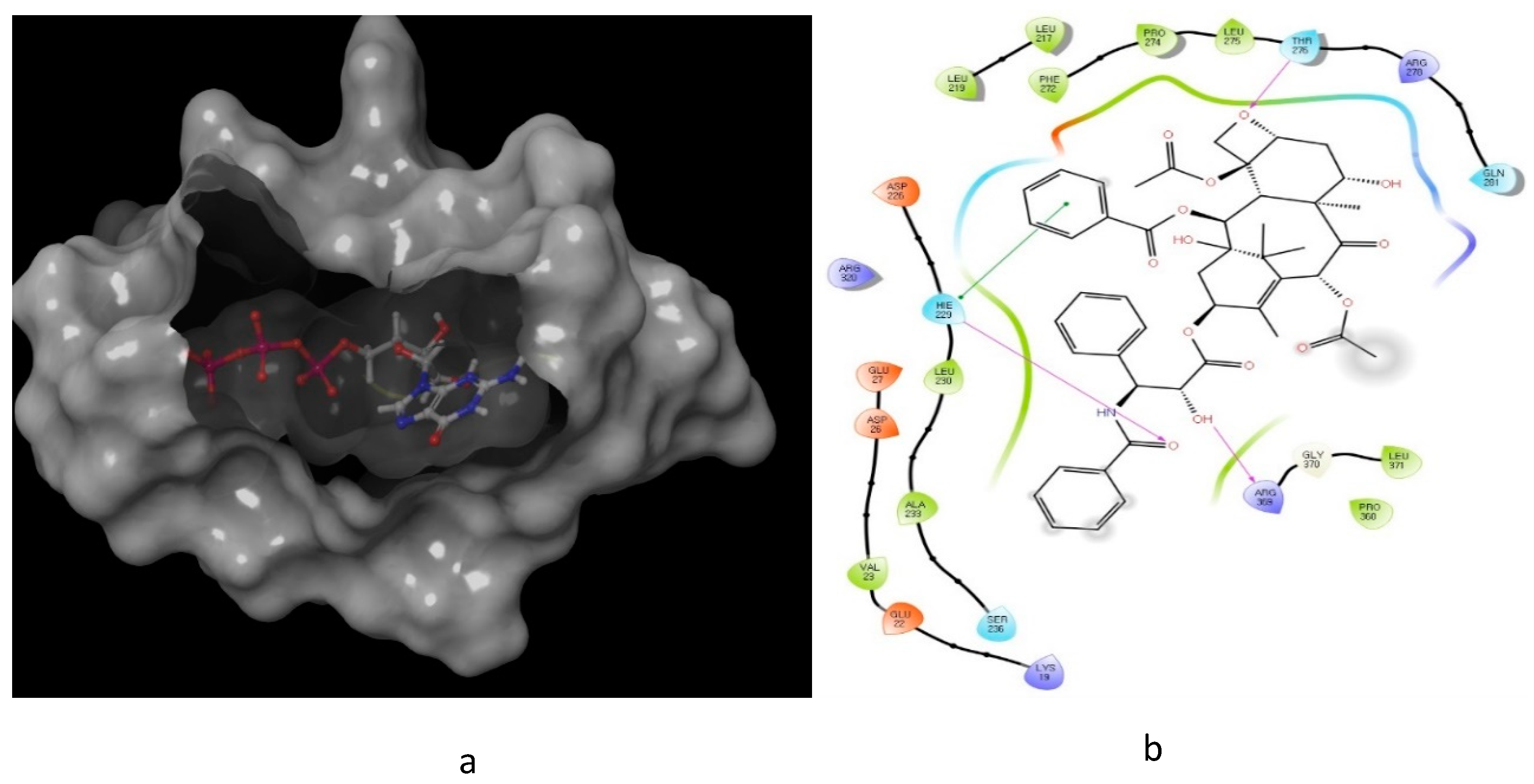
4. Oxygen-Based Heterocycles
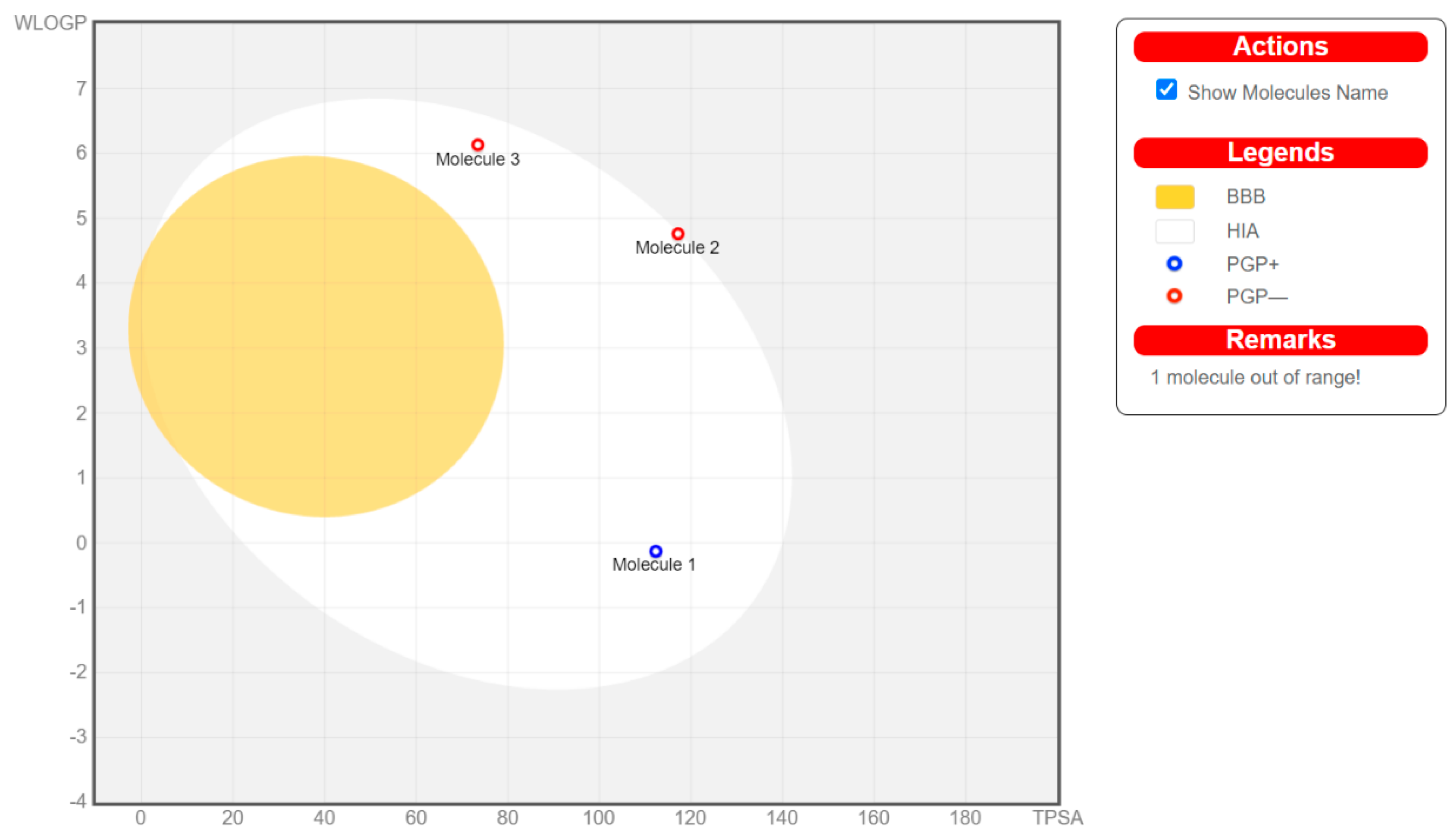
5. Drug Likeness and Absorption Distribution Metabolism Excretion (ADME) Prediction of the Chosen Inhibitors
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D.J. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A.J. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, H.; Ivaska, J. Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Drug Discov. 2018, 17, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Mashreghi, M.; Azarpara, H.; Bazaz, M.R.; Jaafari, M.R.; Masoudifar, A.; Mirzaei, H.; Jaafari, M.R. Angiogenesis biomarkers and their targeting ligands as potential targets for tumor angiogenesis. J. Cell. Physiol. 2018, 233, 2949–2965. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.; Fidler, I. Angiogenesis and metastasis. Eur. J. Cancer 1996, 32, 2451–2460. [Google Scholar] [CrossRef]
- Schättler, H.; Ledzewicz, U. Optimal Control for Mathematical Models of Cancer Therapies; Springer: Berlin/Heidelberg, Germany, 2015; Volume 42. [Google Scholar]
- McColl, B.K.; Stacker, S.A.; Achen, M.G. Molecular regulation of the VEGF family–inducers of angiogenesis and lymphangiogenesis. J. Pathol. Microbiol. Immunol. 2004, 112, 463–480. [Google Scholar] [CrossRef]
- Ahmed, N.; Escalona, R.; Leung, D.; Chan, E.; Kannourakis, G. Tumour microenvironment and metabolic plasticity in cancer and cancer stem cells: Perspectives on metabolic and immune regulatory signatures in chemoresistant ovarian cancer stem cells. Semin. Cancer Biol. 2018, 53, 265–281. [Google Scholar] [CrossRef]
- Fantozzi, A.; Gruber, D.C.; Pisarsky, L.; Heck, C.; Kunita, A.; Yilmaz, M.; Meyer-Schaller, N.; Cornille, K.; Hopfer, U.; Bentires-Alj, M.; et al. VEGF-Mediated Angiogenesis Links EMT-Induced Cancer Stemness to Tumor Initiation. Cancer Res. 2014, 74, 1566–1575. [Google Scholar] [CrossRef]
- Chung, A.S.; Lee, J.; Ferrara, N. Targeting the tumour vasculature: Insights from physiological angiogenesis. Nat. Rev. Cancer 2010, 10, 505–514. [Google Scholar] [CrossRef]
- Otrock, Z.K.; Mahfouz, R.A.; Makarem, J.A.; Shamseddine, A.I. Understanding the biology of angiogenesis: Review of the most important molecular mechanisms. Blood Cells Mol. Dis. 2007, 39, 212–220. [Google Scholar] [CrossRef]
- Fidler, I.J. Angiogenesis and cancer metastasis. Cancer J. 2000, 6, S134–S141. [Google Scholar] [PubMed]
- Nussenbaum, F.; Herman, I.M. Tumor Angiogenesis: Insights and Innovations. J. Oncol. 2010, 2010, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Dewhirst, M.W.; Secomb, T.W. Transport of drugs from blood vessels to tumour tissue. Nat. Rev. Cancer 2017, 17, 738–750. [Google Scholar] [CrossRef]
- Ronca, R.; Benkheil, M.; Mitola, S.; Struyf, S.; Liekens, S. Tumor angiogenesis revisited: Regulators and clinical implications. Med. Res. Rev. 2017, 37, 1231–1274. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Sun, H.; Wei, J.; Cen, B.; Dubois, R.N. CXCL1 Is Critical for Premetastatic Niche Formation and Metastasis in Colorectal Cancer. Cancer Res. 2017, 77, 3655–3665. [Google Scholar] [CrossRef]
- Miyake, M.; Goodison, S.; Lawton, A.; Gomes-Giacoia, E.; Rosser, C. Angiogenin promotes tumoral growth and angiogenesis by regulating matrix metallopeptidase-2 expression via the ERK1/2 pathway. Oncogene 2015, 34, 890–901. [Google Scholar] [CrossRef]
- Al-Abd, A.M.; AlAmoudi, A.J.; Abdel-Naim, A.B.; Neamatallah, T.A.; Ashour, O.M. Anti-angiogenic agents for the treatment of solid tumors: Potential pathways, therapy and current strategies—A review. J. Adv. Res. 2017, 8, 591–605. [Google Scholar] [CrossRef]
- Zirlik, K.; Duyster, J. Anti-Angiogenics: Current Situation and Future Perspectives. Oncol. Res. Treat. 2018, 41, 166–171. [Google Scholar] [CrossRef]
- Akhtar, J.; Khan, A.A.; Ali, Z.; Haider, R.; Yar, M.S. Structure-activity relationship (SAR) study and design strategies of nitrogen-containing heterocyclic moieties for their anticancer activities. Eur. J. Med. Chem. 2017, 125, 143–189. [Google Scholar] [CrossRef]
- Kang, S.; Lee, J.M.; Jeon, B.; Elkamhawy, A.; Paik, S.; Hong, J.; Oh, S.-J.; Paek, S.H.; Lee, C.J.; Hassan, A.H.; et al. Repositioning of the antipsychotic trifluoperazine: Synthesis, biological evaluation and in silico study of trifluoperazine analogs as anti-glioblastoma agents. Eur. J. Med. Chem. 2018, 151, 186–198. [Google Scholar] [CrossRef]
- Elkamhawy, A.; Park, J.-E.; Cho, N.-C.; Sim, T.; Pae, A.N.; Roh, E.J. Discovery of a broad spectrum antiproliferative agent with selectivity for DDR1 kinase: Cell line-based assay, kinase panel, molecular docking, and toxicity studies. J. Enzym. Inhib. Med. Chem. 2016, 31, 158. [Google Scholar] [CrossRef] [PubMed]
- Kerru, N.; Gummidi, L.; Maddila, S.; Gangu, K.K.; Jonnalagadda, S.B. A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications. Molecules 2020, 25, 1909. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.-W.; Liu, D.-K.; Zhang, Q.-W.; Xu, Y.-G.; Shi, L. VEGFR-2 inhibitors and the therapeutic applications thereof: A patent review (2012–2016). Expert Opin. Ther. Patents 2017, 27, 987–1004. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Liu, C.-F.; Rao, G.-W. Anti-angiogenic Agents: A Review on Vascular Endothelial Growth Factor Receptor-2 (VEGFR-2) Inhibitors. Curr. Med. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.K.; Singhi, E.K.; Arroyo, J.P.; Ikizler, T.A.; Gould, E.R.; Brown, J.; Beckman, J.A.; Harrison, D.G.; Moslehi, J. Mechanisms of VEGF (Vascular Endothelial Growth Factor) Inhibitor–Associated Hypertension and Vascular Disease. Hypertension 2018, 71, e1–e8. [Google Scholar] [CrossRef]
- Farina, A.R.; Cappabianca, L.; Sebastiano, M.; Zelli, V.; Guadagni, S.; Mackay, A.R. Hypoxia-induced alternative splicing: The 11th Hallmark of Cancer. J. Exp. Clin. Cancer Res. 2020, 39, 1–30. [Google Scholar] [CrossRef]
- Yan, X.; Hui, Y.; Hua, Y.; Huang, L.; Wang, L.; Peng, F.; Tang, C.; Liu, D.; Song, J.; Wang, F. EG-VEGF silencing inhibits cell proliferation and promotes cell apoptosis in pancreatic carcinoma via PI3K/AKT/mTOR signaling pathway. Biomed. Pharmacother. 2019, 109, 762–769. [Google Scholar] [CrossRef]
- Schafer, C.M.; Gurley, J.M.; Kurylowicz, K.; Lin, P.K.; Chen, W.; Elliott, M.H.; Davis, G.E.; Bhatti, F.; Griffin, C.T. An inhibitor of endothelial ETS transcription factors promotes physiologic and therapeutic vessel regression. Proc. Natl. Acad. Sci. USA 2020, 117, 26494–26502. [Google Scholar] [CrossRef]
- Neufeld, G.; Cohen, T.; Gengrinovitch, S.; Poltorak, Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999, 13, 9–22. [Google Scholar] [CrossRef]
- Viallard, C.; Larrivée, B. Tumor angiogenesis and vascular normalization: Alternative therapeutic targets. Angiogenesis 2017, 20, 409–426. [Google Scholar] [CrossRef]
- Rosen, L.S. Clinical Experience with Angiogenesis Signaling Inhibitors: Focus on Vascular Endothelial Growth Factor (VEGF) Blockers. Cancer Control. 2002, 9, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2000, 103, 211–225. [Google Scholar] [CrossRef]
- Park, S.A.; Jeong, M.S.; Ha, K.-T.; Jang, S.B. Structure and function of vascular endothelial growth factor and its receptor system. BMB Rep. 2018, 51, 73–78. [Google Scholar] [CrossRef]
- Motzer, R.J.; Hutson, T.E.; Cella, D.; Reeves, J.; Hawkins, R.; Guo, J.; Nathan, P.; Staehler, M.; De Souza, P.; Merchan, J.R.; et al. Pazopanib versus Sunitinib in Metastatic Renal-Cell Carcinoma. N. Engl. J. Med. 2013, 369, 722–731. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Maki, R.G.; Corless, C.L.; Antonescu, C.R.; Harlow, A.; Griffith, D.; Town, A.; McKinley, A.; Ou, W.-B.; Fletcher, J.A.; et al. Primary and Secondary Kinase Genotypes Correlate With the Biological and Clinical Activity of Sunitinib in Imatinib-Resistant Gastrointestinal Stromal Tumor. J. Clin. Oncol. 2008, 26, 5352–5359. [Google Scholar] [CrossRef] [PubMed]
- Scagliotti, G.V.; Govindan, R. Targeting Angiogenesis with Multitargeted Tyrosine Kinase Inhibitors in the Treatment of Non-Small Cell Lung Cancer. Oncology 2010, 15, 436–446. [Google Scholar] [CrossRef]
- Lin, Z.; Zhang, Q.; Luo, W. Angiogenesis inhibitors as therapeutic agents in cancer: Challenges and future directions. Eur. J. Pharmacol. 2016, 793, 76–81. [Google Scholar] [CrossRef]
- Liu, J.-C.; Narva, S.; Zhou, K.; Zhang, W. A Review on the Antitumor Activity of Various Nitrogenous-based Heterocyclic Compounds as NSCLC Inhibitors. Mini Rev. Med. Chem. 2019, 19, 1517–1530. [Google Scholar] [CrossRef]
- Garnock-Jones, K.P. Panobinostat: First global approval. Drugs 2015, 75, 695–704. [Google Scholar] [CrossRef]
- Renhowe, P.A.; Pecchi, S.; Shafer, C.M.; Machajewski, T.D.; Jazan, E.M.; Taylor, C.; Antonios-McCrea, W.; McBride, C.M.; Frazier, K.; Wiesmann, M.; et al. Design, Structure−Activity Relationships and in Vivo Characterization of 4-Amino-3-benzimidazol-2-ylhydroquinolin-2-ones: A Novel Class of Receptor Tyrosine Kinase Inhibitors. J. Med. Chem. 2009, 52, 278–292. [Google Scholar] [CrossRef]
- Kubo, H.; Fujiwara, T.; Jussila, L.; Hashi, H.; Ogawa, M.; Shimizu, K.; Awane, M.; Sakai, Y.; Takabayashi, A.; Alitalo, K.J.B. Involvement of vascular endothelial growth factor receptor-3 in maintenance of integrity of endothelial cell lining during tumor angiogenesis. J. Am. Soc. Hematol. 2000, 96, 546–553. [Google Scholar]
- McMahon, G. VEGF receptor signaling in tumor angiogenesis. Oncologist 2000, 5, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Stetler-Stevenson, W.G. Matrix metalloproteinases in angiogenesis: A moving target for therapeutic intervention. J. Clin. Investig. 1999, 103, 1237–1241. [Google Scholar] [CrossRef] [PubMed]
- Gialeli, C.; Theocharis, A.D.; Karamanos, N.K. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 2011, 278, 16–27. [Google Scholar] [CrossRef]
- Spuul, P.; Daubon, T.; Pitter, B.; Alonso, F.; Fremaux, I.; Kramer, I.; Montañez, E.; Génot, E. VEGF-A/Notch-Induced Podosomes Proteolyse Basement Membrane Collagen-IV during Retinal Sprouting Angiogenesis. Cell Rep. 2016, 17, 484–500. [Google Scholar] [CrossRef]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- González-Avila, G.; Sommer, B.; Mendoza-Posada, D.A.; Ramos, C.; Garcia-Hernandez, A.A.; Falfan-Valencia, R. Matrix metalloproteinases participation in the metastatic process and their diagnostic and therapeutic applications in cancer. Crit. Rev. Oncol. 2019, 137, 57–83. [Google Scholar] [CrossRef]
- Baeriswyl, V.; Christofori, G. The angiogenic switch in carcinogenesis. Semin. Cancer Biol. 2009, 19, 329–337. [Google Scholar] [CrossRef]
- Becker, D.P.; Villamil, C.I.; Barta, T.E.; Bedell, L.J.; Boehm, T.L.; DeCrescenzo, G.A.; Freskos, J.N.; Getman, D.P.; Hockerman, S.; Heintz, R.J.; et al. Synthesis and Structure−Activity Relationships of β- and α-Piperidine Sulfone Hydroxamic Acid Matrix Metalloproteinase Inhibitors with Oral Antitumor Efficacy. J. Med. Chem. 2005, 48, 6713–6730. [Google Scholar] [CrossRef]
- Solution Structure and Backbone Dynamics of the Catalytic Domain of Matrix Metalloproteinase-2 Complexed with a Hydroxamic Acid Inhibitor. Available online: http://www.rcsb.org/structure/1HOV (accessed on 1 November 2020).
- Feng, Y.; Likos, J.J.; Zhu, L.; Woodward, H.; Munie, G.; McDonald, J.J.; Stevens, A.M.; Howard, C.P.; De Crescenzo, G.A.; Welsch, D.; et al. Solution structure and backbone dynamics of the catalytic domain of matrix metalloproteinase-2 complexed with a hydroxamic acid inhibitor. Biochimica et Biophysica Acta 2002, 1598, 10–23. [Google Scholar] [CrossRef]
- Gulçin, I.; Taslimi, P. Sulfonamide inhibitors: A patent review 2013-present. Expert Opin. Ther. Patents 2018, 28, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Awadallah, F.M.; Bua, S.; Mahmoud, W.R.; Nada, H.H.; Nocentini, A.; Supuran, C.T. Inhibition studies on a panel of human carbonic anhydrases with N1-substituted secondary sulfonamides incorporating thiazolinone or imidazolone-indole tails. J. Enzym. Inhib. Med. Chem. 2018, 33, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Al-Sanea, M.M.; Elkamhawy, A.; Paik, S.; Bua, S.; Lee, S.H.; AbdelGawad, M.A.; Roh, E.J.; Eldehna, W.M.; Supuran, C.T. Synthesis and biological evaluation of novel 3-(quinolin-4-ylamino)benzenesulfonamides as carbonic anhydrase isoforms I and II inhibitors. J. Enzym. Inhib. Med. Chem. 2019, 34, 1457–1464. [Google Scholar] [CrossRef] [PubMed]
- Al-Sanea, M.M.; Elkamhawy, A.; Paik, S.; Lee, K.; El Kerdawy, A.M.; Abbas, B.S.N.; Roh, E.J.; Eldehna, W.M.; Elshemy, H.A.; Bakr, R.B.; et al. Sulfonamide-based 4-anilinoquinoline derivatives as novel dual Aurora kinase (AURKA/B) inhibitors: Synthesis, biological evaluation and in silico insights. Bioorg. Med. Chem. 2020, 28, 115525. [Google Scholar] [CrossRef] [PubMed]
- Owa, T.; Nagasu, T. Novel sulphonamide derivatives for the treatment of cancer. Expert Opin. Ther. Patents 2000, 10, 1725–1740. [Google Scholar] [CrossRef]
- Supuran, C.T. Exploring the multiple binding modes of inhibitors to carbonic anhydrases for novel drug discovery. Expert Opin. Drug Discov. 2020, 15, 671–686. [Google Scholar] [CrossRef]
- Funahashi, Y.; Sugi, N.H.; Semba, T.; Yamamoto, Y.; Hamaoka, S.; Tsukahara-Tamai, N.; Ozawa, Y.; Tsuruoka, A.; Nara, K.; Takahashi, K. Sulfonamide derivative, E7820, is a unique angiogenesis inhibitor suppressing an expression of integrin α2 subunit on endothelium. Cancer Res. 2002, 62, 6116–6123. [Google Scholar]
- Pan, L.; Zhao, Y.; Yuan, Z.; Qin, G. Research advances on structure and biological functions of integrins. SpringerPlus 2016, 5, 1–11. [Google Scholar] [CrossRef]
- Hytönen, V.P.; Wehrle-Haller, B. Mechanosensing in cell–matrix adhesions–Converting tension into chemical signals. Exp. Cell Res. 2016, 343, 35–41. [Google Scholar] [CrossRef]
- Kechagia, J.Z.; Ivaska, J.; Roca-Cusachs, P. Integrins as biomechanical sensors of the microenvironment. Nat. Rev. Mol. Cell Biol. 2019, 20, 457–473. [Google Scholar] [CrossRef]
- Mizejewski, G.J. Role of integrins in cancer: Survey of expression patterns. Proc. Soc. Exp. Boil. Med. 1999, 222, 124–138. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Lu, D.; Scully, M.; Kakkar, V. The Role of Integrins in Cancer and the Development of Anti-Integrin Therapeutic Agents for Cancer Therapy. Perspect. Med. Chem. 2008, 2, 57–73. [Google Scholar] [CrossRef]
- Yousefi, H.; Vatanmakanian, M.; Mahdiannasser, M.; Mashouri, L.; Alahari, N.V.; Monjezi, M.R.; Ilbeigi, S.; Alahari, S.K. Understanding the role of integrins in breast cancer invasion, metastasis, angiogenesis, and drug resistance. Oncogene 2021, 1–21. [Google Scholar] [CrossRef]
- Yoshimura, K.; Meckel, K.F.; Laird, L.S.; Chia, C.Y.; Park, J.-J.; Olino, K.L.; Tsunedomi, R.; Harada, T.; Iizuka, N.; Hazama, S.; et al. Integrin α2 Mediates Selective Metastasis to the Liver. Cancer Res. 2009, 69, 7320–7328. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; De Noronha, R.G.; Devi, N.S.; Jabbar, A.A.; Kaluz, S.; Liu, Y.; Mooring, S.R.; Nicolaou, K.; Wang, B.; Van Meir, E.G. Sulfonamides as a new scaffold for hypoxia inducible factor pathway inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 5528–5532. [Google Scholar] [CrossRef]
- Hatzikirou, H.; Basanta, D.; Simon, M.; Schaller, K.; Deutsch, A. ‘Go or Grow’: The key to the emergence of invasion in tumour progression? Math. Med. Biol. 2012, 29, 49–65. [Google Scholar] [CrossRef]
- Lu, J.; Tan, M.; Cai, Q. The Warburg effect in tumor progression: Mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett. 2015, 356, 156–164. [Google Scholar] [CrossRef]
- Bacon, A.; Harris, A.L. Hypoxia-inducible factors and hypoxic cell death in tumour physiology. Ann. Med. 2004, 36, 530–539. [Google Scholar] [CrossRef]
- Ferguson, J.; De Los Santos, Z.; Devi, N.; Van Meir, E.; Zingales, S.K.; Wang, B. Examining the structure-activity relationship of benzopyran-based inhibitors of the hypoxia inducible factor-1 pathway. Bioorg. Med. Chem. Lett. 2017, 27, 1731–1736. [Google Scholar] [CrossRef]
- Bhattarai, D.; Xu, X.; Lee, K. Hypoxia-inducible factor-1 (HIF-1) inhibitors from the last decade (2007 to 2016): A “structure–activity relationship” perspective. Med. Res. Rev. 2018, 38, 1404–1442. [Google Scholar] [CrossRef]
- Delost, M.D.; Smith, D.T.; Anderson, B.J.; Njardarson, J.T. From Oxiranes to Oligomers: Architectures of U.S. FDA Approved Pharmaceuticals Containing Oxygen Heterocycles. J. Med. Chem. 2018, 61, 10996–11020. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Chen, J.; Jiang, M.; Zhang, N.; Na, K.; Luo, C.; Zhang, R.; Sun, M.; Lin, G.; Zhang, R.; et al. Paclitaxel–Paclitaxel Prodrug Nanoassembly as a Versatile Nanoplatform for Combinational Cancer Therapy. ACS Appl. Mater. Interfaces 2016, 8, 33506–33513. [Google Scholar] [CrossRef] [PubMed]
- Nogales, E. Structural insights into microtubule function. Ann. Rev. Biophys. Biomol. Struct. 2001, 30, 397–420. [Google Scholar] [CrossRef] [PubMed]
- Petrášek, J.; Schwarzerová, K. Actin and microtubule cytoskeleton interactions. Curr. Opin. Plant. Biol. 2009, 12, 728–734. [Google Scholar] [CrossRef] [PubMed]
- Knossow, M.; Campanacci, V.; Khodja, L.A.; Gigant, B. The Mechanism of Tubulin Assembly into Microtubules: Insights from Structural Studies. iScience 2020, 23, 9. [Google Scholar] [CrossRef]
- Vemu, A.; Atherton, J.; Spector, J.O.; Moores, C.A.; Roll-Mecak, A. Tubulin isoform composition tunes microtubule dynamics. Mol. Biol. Cell 2017, 28, 3564–3572. [Google Scholar] [CrossRef]
- Martin, M.; Akhmanova, A. Coming into Focus: Mechanisms of Microtubule Minus-End Organization. Trends Cell Biol. 2018, 28, 574–588. [Google Scholar] [CrossRef]
- Janke, C.; Magiera, M.M. The tubulin code and its role in controlling microtubule properties and functions. Nat. Rev. Mol. Cell Biol. 2020, 21, 307–326. [Google Scholar] [CrossRef]
- Checchi, P.M.; Nettles, J.H.; Zhou, J.; Snyder, J.P.; Joshi, H.C. Microtubule-interacting drugs for cancer treatment. Trends Pharmacol. Sci. 2003, 24, 361–365. [Google Scholar] [CrossRef]
- Banerjee, S.; Hwang, D.-J.; Li, W.; Miller, D.D. Current Advances of Tubulin Inhibitors in Nanoparticle Drug Delivery and Vascular Disruption/Angiogenesis. Molecules 2016, 21, 1468. [Google Scholar] [CrossRef]
- Bates, D.; Eastman, A. Microtubule destabilising agents: Far more than just antimitotic anticancer drugs. Br. J. Clin. Pharmacol. 2017, 83, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, E.; Andre, N.; Braguer, D. Targeting Microtubules to Inhibit Angiogenesis and Disrupt Tumour Vasculature:Implications for Cancer Treatment. Curr. Cancer Drug Targets 2007, 7, 566–581. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, T.; Yang, C.; Norris, A.; Glass, T.; Bane, S.; Ravindra, R.; Banerjee, A.; Metaferia, B.; Thomas, S.L.; Giannakakou, P.; et al. Evaluation of the Tubulin-Bound Paclitaxel Conformation: Synthesis, Biology, and SAR Studies of C-4 to C-3‘ Bridged Paclitaxel Analogues. J. Med. Chem. 2007, 50, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Żwawiak, J.; Zaprutko, L.J. A brief history of taxol. J. Med. Sci. 2014, 83, 47–52. [Google Scholar]
- Dynamic and Asymmetric Fluctuations in the Microtubule Wall Captured by High-Resolution Cryoelectron Microscopy. Available online: http://www.rcsb.org/structure/6WVM (accessed on 1 October 2020).
- Debs, G.E.; Cha, M.; Liu, X.; Huehn, A.R.; Sindelar, C.V. Dynamic and asymmetric fluctuations in the microtubule wall captured by high-resolution cryoelectron microscopy. Proc. Nat. Acad. Sci. USA 2020, 117, 16976–16984. [Google Scholar] [CrossRef]
- 6WVM—High Curvature Lateral Interaction within a 13-Protofilament, Taxol Stabilized Microtubule. Available online: http://www.rcsb.org/structure/6WVM (accessed on 18 January 2021).
- Jürgen, B.J.F. Computer-aided drug discovery. F1000Res 2015, 4, 630. [Google Scholar] [CrossRef]
- Elkamhawy, A.; Kim, N.Y.; Hassan, A.H.; Park, J.-E.; Paik, S.; Yang, J.-E.; Oh, K.-S.; Lee, B.H.; Lee, M.Y.; Shin, K.J.; et al. Thiazolidine-2,4-dione-based irreversible allosteric IKK-β kinase inhibitors: Optimization into in vivo active anti-inflammatory agents. Eur. J. Med. Chem. 2020, 188, 111955. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, V.B.; Resende, J.; Rodrigues, P.F.; Bononi, F.C.; Benevenuto, C.G.; Taft, C.A. Computer-aided drug design and ADMET predictions for identification and evaluation of novel potential farnesyltransferase inhibitors in cancer therapy. J. Mol. Graph. Model. 2010, 28, 513–523. [Google Scholar] [CrossRef]
- Elkamhawy, A.; Paik, S.; Kim, H.J.; Park, J.-H.; Londhe, A.M.; Lee, K.; Pae, A.N.; Park, K.D.; Roh, E.J. Discovery of N-(1-(3-fluorobenzoyl)-1H-indol-5-yl)pyrazine-2-carboxamide: A novel, selective, and competitive indole-based lead inhibitor for human monoamine oxidase B. J. Enzym. Inhib. Med. Chem. 2020, 35, 1568–1580. [Google Scholar] [CrossRef]
- Elkamhawy, A.; Kim, N.Y.; Hassan, A.H.; Park, J.-E.; Yang, J.-E.; Elsherbeny, M.H.; Paik, S.; Oh, K.-S.; Lee, B.H.; Lee, M.Y.; et al. Optimization study towards more potent thiazolidine-2,4-dione IKK-β modulator: Synthesis, biological evaluation and in silico docking simulation. Bioorg. Chem. 2019, 92, 103261. [Google Scholar] [CrossRef]
- Elkamhawy, A.; Paik, S.; Hassan, A.H.; Lee, Y.S.; Roh, E.J. Hit discovery of 4-amino- N -(4-(3-(trifluoromethyl)phenoxy)pyrimidin-5-yl)benzamide: A novel EGFR inhibitor from a designed small library. Bioorg. Chem. 2017, 75, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Xu, S.; Zhu, Z.; Xu, J. Multi-target design strategies for the improved treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2019, 176, 228–247. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Kerns, E.H. Drug-Like Properties: Concepts, Structure Design and Methods from ADME to Toxicity Optimization; Academic Press: Cambridge, MA, USA, 2015. [Google Scholar]
- Daina, A.; Zoete, V. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Alavijeh, M.S.; Chishty, M.; Qaiser, M.Z.; Palmer, A.M. Drug metabolism and pharmacokinetics, the blood-brain barrier, and central nervous system drug discovery. NeuroRX 2005, 2, 554–571. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Y | R1 | R2 | VEGFR-2 (IC50 μM) | PDGFRβ (IC50 μM) |
|---|---|---|---|---|---|
| 1 | OH | H | H | 0.24 | 0.020 |
| 2 | NH2 | H | H | 0.058 | 0.010 |
| 3 | NHMe | H | H | 0.22 | 0.030 |
| 4 |  | H | H | 0.17 | 0.0002 |
| 5 | H |  | H | 0.017 | 0.0003 |
| 6 | NH2 |  | H | 0.005 | 0.0001 |
| 7 | NH2 | H | Me | 0.22 | 0.030 |
| 8 | NH2 | H |  | 0.057 | 0.005 |
| 9 | NH2 | H |  | 0.027 | 0.002 |
| 10 | NH2 | H |  | 0.026 | 0.0009 |
 | ||||||
| Ki (nM) | ||||||
| Compound | MMP-1 | MMP-2 | MMP-3 | MMP-9 | MMP-13 | Cmax |
| 11 (β-sulfone) | 800 | 0.4 | 17.5 | 1 | 0.6 | 1372 |
| 12 (α-sulfone) | 435 | ˂0.1 | 18.1 | 0.3 | 0.15 | 3119 |
 | |||
| Compound | R1 | R2 | LN229-HRE-Lux (IC50 μM) |
| 13 |  |  | 0.6 μM |
| 14 |  |  | 0.6 μM |
| 15 |  |  | 1.3 μM |
| 16 |  |  | 3.3 μM |
| 17 |  |  | >25 μM |
| 18 |  |  | 0.5 μM |
 | |||||
| Compound | R1 | R2 | R3 | R4 | IC50 PC3 (nM) |
| 19 |  |  | H | H | 550 |
| 20 |  |  | H | H | 320 |
| 21 |  |  | H | H | 128 |
| 22 |  |  | H | H | 55 |
| 23 |  |  | H | H | 53 |
| 24 |  |  | H | H | 4600 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nada, H.; Elkamhawy, A.; Lee, K. Structure Activity Relationship of Key Heterocyclic Anti-Angiogenic Leads of Promising Potential in the Fight against Cancer. Molecules 2021, 26, 553. https://doi.org/10.3390/molecules26030553
Nada H, Elkamhawy A, Lee K. Structure Activity Relationship of Key Heterocyclic Anti-Angiogenic Leads of Promising Potential in the Fight against Cancer. Molecules. 2021; 26(3):553. https://doi.org/10.3390/molecules26030553
Chicago/Turabian StyleNada, Hossam, Ahmed Elkamhawy, and Kyeong Lee. 2021. "Structure Activity Relationship of Key Heterocyclic Anti-Angiogenic Leads of Promising Potential in the Fight against Cancer" Molecules 26, no. 3: 553. https://doi.org/10.3390/molecules26030553
APA StyleNada, H., Elkamhawy, A., & Lee, K. (2021). Structure Activity Relationship of Key Heterocyclic Anti-Angiogenic Leads of Promising Potential in the Fight against Cancer. Molecules, 26(3), 553. https://doi.org/10.3390/molecules26030553