
DNA-Binding Anticancer Drugs: One Target, Two Actions

{kind=link}
{kind=link}
Abstract
1. Introduction
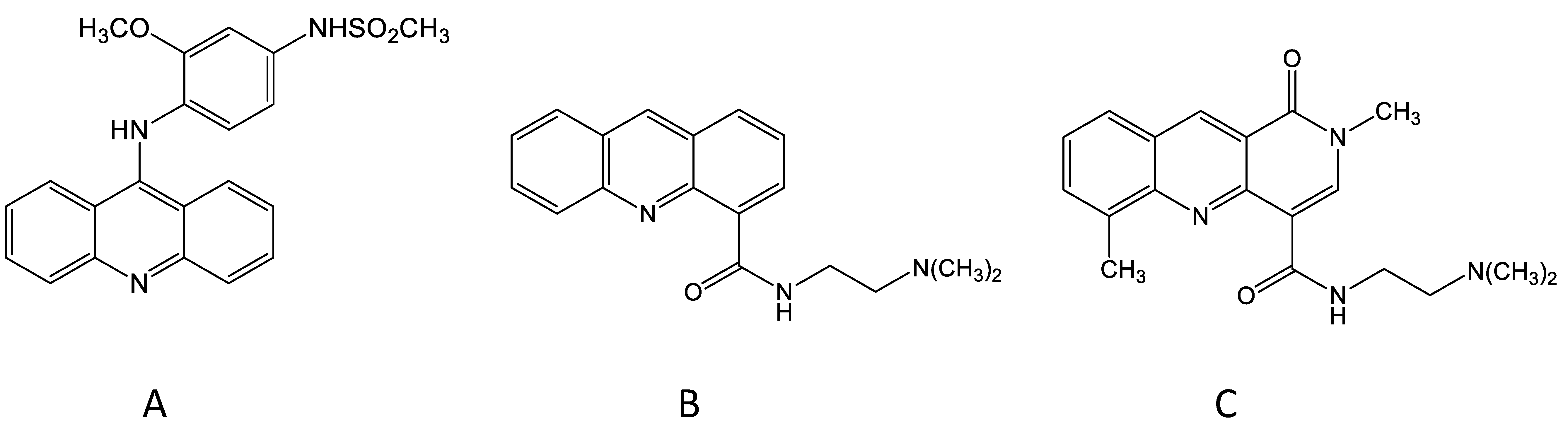
2. DNA Topoisomerases IIα and IIβ, the Molecular Targets of Amsacrine
3. The Discovery of N-[2-(Dimethylamino)-Ethyl]-Acridine-4-Carboxamide (DACA)
4. Toxicological Studies with DACA
5. The Development of the DACA Analogue SN 28049
6. Insights from In Vitro Studies Using Colon Carcinoma Lines
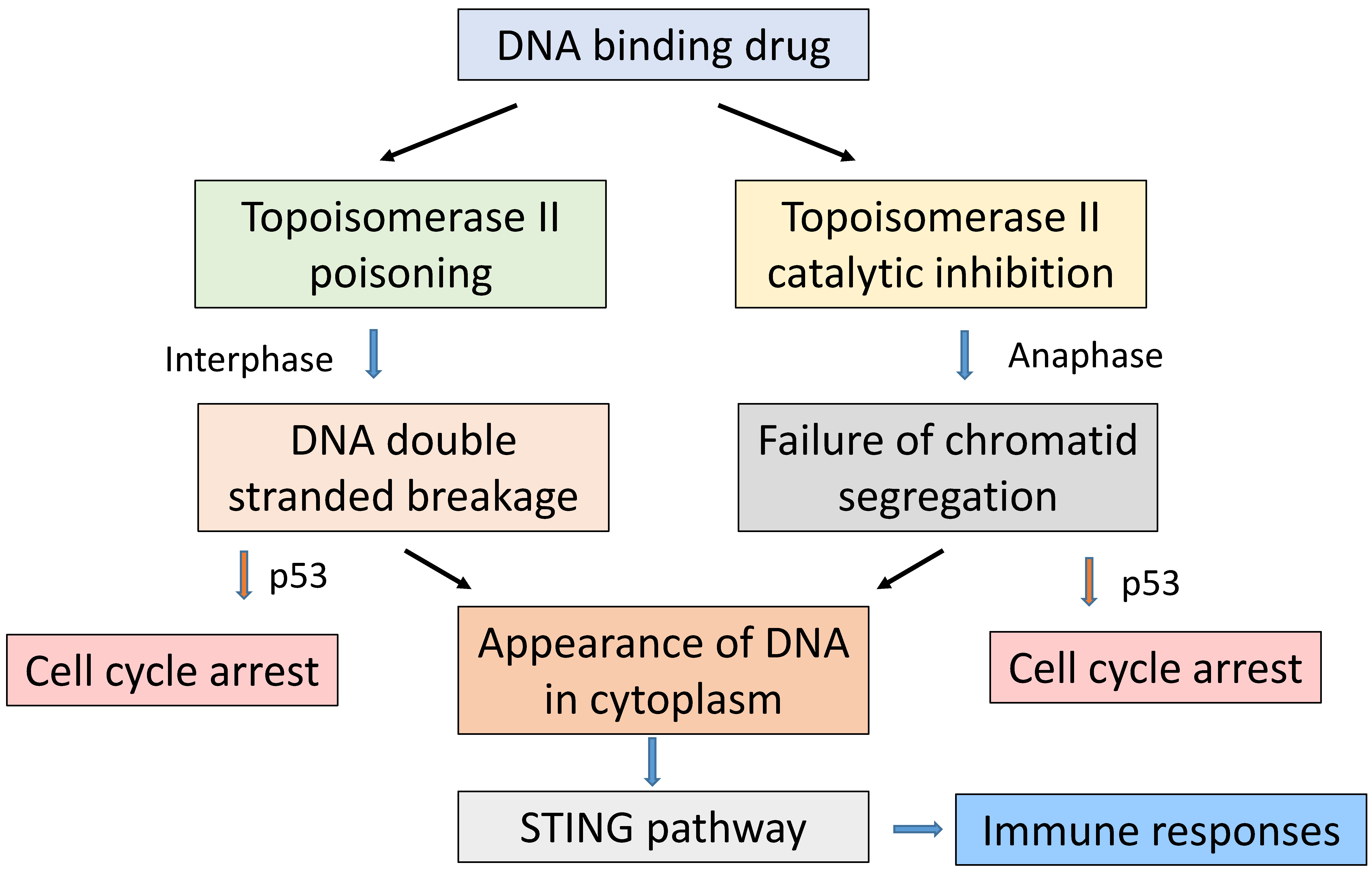
7. Role of Topoisomerase II
8. Pharmacokinetic Considerations in the Action of SN 28049
9. Immunological Considerations in the Action of SN 28049
10. Perspective
Funding
Acknowledgments
Conflicts of Interest
References
- Gatenby, R.A.; Brown, J.S. Integrating evolutionary dynamics into cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 675–686. [Google Scholar] [PubMed]
- Zitvogel, L.; Apetoh, L.; Ghiringhelli, F.; Kroemer, G. Immunological aspects of cancer chemotherapy. Nat. Rev. Immunol. 2008, 8, 59–73. [Google Scholar]
- Watson, J.D.; Crick, F.H. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature 1953, 171, 737–738. [Google Scholar]
- Cain, B.F.; Atwell, G.J. The experimental antitumour properties of three congeners of the acridylmethanesulphonanilide (AMSA) series. Eur. J. Cancer Clin. Oncol. 1974, 10, 539–549. [Google Scholar]
- Arlin, Z.A. Current status of amsacrine (AMSA) combination chemotherapy programs in acute leukemia. Cancer Treat. Rep. 1983, 67, 967–970. [Google Scholar] [PubMed]
- Arcamone, F. Properties of antitumor anthracyclines and new developments in their application: Cain Memorial Award Lecture. Cancer Res. 1985, 45, 5995–5999. [Google Scholar] [PubMed]
- Kohn, K.W.; Friedman, C.A.; Ewig, A.G.; Iqbal, Z.M. DNA chain growth during replication of asynchronous L1210 cells. Alkaline elution of large DNA segments from cells lysed on filters. Biochemistry 1974, 13, 4134–4139. [Google Scholar]
- Ralph, R.K. On the mechanism of action of 4’-[(9-acridinyl)-amino] methanesulphon-m-anisidide. Eur. J. Cancer Clin. Oncol. 1980, 16, 595–600. [Google Scholar]
- Nelson, E.M.; Tewey, K.; Liu, L.F. Mechanism of antitumor drug action: Poisoning of mammalian DNA topoisomerase II on DNA by 4’-(9-acridinylamino)-methanesulfon-m-anisidide. Proc. Natl. Acad. Sci. USA 1984, 81, 1361–1365. [Google Scholar] [PubMed]
- Liu, L.F.; Rowe, T.C.; Yang, L.; Tewey, K.M.; Chen, G.L. Cleavage of DNA by mammalian DNA topoisomerase II. J. Biol. Chem. 1983, 258, 15365–15370. [Google Scholar] [PubMed]
- Baguley, B.C.; Cain, B.F. Comparison of the in vivo and in vitro antileukemic activity of monosubstituted derivatives of 4’-(9-acridinylamino)methanesulfon-m- anisidide. Mol. Pharmacol. 1982, 22, 486–492. [Google Scholar]
- Baguley, B.C.; Finlay, G.J. Derivatives of amsacrine: Determinants required for high activity against Lewis lung carcinoma. J. Natl. Cancer Inst. 1988, 80, 195–199. [Google Scholar]
- Baguley, B.C.; Kernohan, A.R.; Wilson, W.R. Divergent activity of derivatives of amsacrine (m-AMSA) towards Lewis lung carcinoma and P388 leukaemia in mice. Eur. J. Cancer Clin. Oncol 1983, 19, 1607–1613. [Google Scholar]
- Denny, W.A.; Atwell, G.J.; Baguley, B.C. Potential antitumor agents. 40. Orally active 4,5-disubstituted derivatives of amsacrine. J. Med. Chem 1984, 27, 363–367. [Google Scholar] [PubMed]
- Harvey, V.J.; Hardy, J.R.; Smith, S.; Grove, W.; Baguley, B.C. Phase II study of the amsacrine analogue CI-921 (NSC 343499) in non- small cell lung cancer. Eur. J. Cancer 1991, 27, 1617–1620. [Google Scholar] [PubMed]
- Atwell, G.J.; Cain, B.F.; Baguley, B.C.; Finlay, G.J.; Denny, W.A. Potential antitumor agents. 43. Synthesis and biological activity of dibasic 9-aminoacridine-4-carboxamides, a new class of antitumor agent. J. Med. Chem. 1984, 27, 1481–1485. [Google Scholar] [PubMed]
- Atwell, G.J.; Rewcastle, G.W.; Baguley, B.C.; Denny, W.A. Potential antitumor agents. 50. In vivo solid-tumor activity of derivatives of N-[2-(dimethylamino)ethyl]acridine-4-carboxamide. J. Med. Chem. 1987, 30, 664–669. [Google Scholar] [PubMed]
- Bridewell, D.J.; Finlay, G.J.; Baguley, B.C. Mechanism of cytotoxicity of N-[2-(dimethylamino)ethyl] acridine-4- carboxamide and of its 7-chloro derivative: The roles of topoisomerases I and II. Cancer Chemother. Pharmacol. 1999, 43, 302–308. [Google Scholar] [PubMed]
- Wakelin, L.P.; Atwell, G.J.; Rewcastle, G.W.; Denny, W.A. Relationships between DNA-binding kinetics and biological activity for the 9-aminoacridine-4-carboxamide class of antitumor agents. J. Med. Chem. 1987, 30, 855–861. [Google Scholar]
- Wakelin, L.P.; Adams, A.; Denny, W.A. Kinetic studies of the binding of acridinecarboxamide topoisomerase poisons to DNA: Implications for mode of binding of ligands with uncharged chromophores. J. Med. Chem. 2002, 45, 894–901. [Google Scholar]
- Baguley, B.C.; Zhuang, L.; Marshall, E. Experimental solid tumour activity of N-[2-(dimethylamino)ethyl]-acridine-4-carboxamide. Cancer Chemother. Pharmacol. 1995, 36, 244–248. [Google Scholar] [PubMed]
- McCrystal, M.R.; Evans, B.D.; Harvey, V.J.; Thompson, P.I.; Porter, D.J.; Baguley, B.C. Phase I study of the cytotoxic agent N-[2-(dimethylamino)ethyl]acridine-4-carboxamide. Cancer Chemother. Pharmacol. 1999, 44, 39–44. [Google Scholar] [PubMed]
- Leopold, W.R.; Nelson, J.M.; Plowman, J.; Jackson, R.C. Anthrapyrazoles, a new class of intercalating agents with high-level, broad spectrum activity against murine tumors. Cancer Res. 1985, 45, 5532–5539. [Google Scholar] [PubMed]
- Baguley, B.C. The development of new DNA intercalating anti-cancer drugs. In Horizons in Cancer Research; Watanabe, H.S., Ed.; Nova Publishers: New York, NY, USA, 2012; pp. 47–65. [Google Scholar]
- Chen, Y.Y.; Finlay, G.J.; Kirker, J.A.; Marshall, E.S.; Richardson, E.; Baguley, B.C. In vivo and in vitro assessment of the action of SN 28049, a benzonaphthyridine derivative targeting topoisomerase II, on the murine Colon 38 carcinoma. Invest. New Drugs 2011, 29, 1504–1510. [Google Scholar] [PubMed]
- Atwell, G.J.; Baguley, B.C.; Denny, W.A. Potential antitumor agents. 57. 2-Phenylquinoline-8-carboxamides as "minimal" DNA-intercalating antitumor agents with in vivo solid tumor activity. J. Med. Chem. 1989, 32, 396–401. [Google Scholar]
- Deady, L.W.; Rodemann, T.; Zhuang, L.; Baguley, B.C.; Denny, W.A. Synthesis and cytotoxic activity of carboxamide derivatives of benzo[b][1,6]naphthyridines. J. Med. Chem. 2003, 46, 1049–1054. [Google Scholar]
- Drummond, C.J.; Finlay, G.J.; Broome, L.; Marshall, E.S.; Richardson, E.; Baguley, B.C. Action of SN 28049, a new DNA binding topoisomerase II-directed antitumour drug: Comparison with doxorubicin and etoposide. Invest. New Drugs 2011, 29, 1102–1110. [Google Scholar]
- Drummond, C.J. The Mechanism of Anti-Tumour Activity of the DNA Binding Agent SN 28049. Ph.D. Thesis, The University of Auckland, Auckland, New Zealand, 2007. [Google Scholar]
- Watanabe, Y.; Haugen-Strano, A.; Umar, A.; Yamada, K.; Hemmi, H.; Kikuchi, Y.; Takano, S.; Shibata, Y.; Barrett, J.C.; Kunkel, T.A.; et al. Complementation of an hMSH2 defect in human colorectal carcinoma cells by human chromosome 2 transfer. Mol. Carcinog. 2000, 29, 37–49. [Google Scholar]
- Clarke, D.J.; Johnson, R.T.; Downes, C.S. Topoisomerase II inhibition prevents anaphase chromatid segregation in mammalian cells independently of the generation of DNA strand breaks. J. Cell Sci. 1993, 105, 563–569. [Google Scholar]
- Downes, C.S.; Johnson, R.T. DNA topoisomerases and DNA repair. Bioessays 1988, 8, 179–184. [Google Scholar]
- Gemble, S.; Buhagiar-Labarchede, G.; Onclercq-Delic, R.; Fontaine, G.; Lambert, S.; Amor-Gueret, M. Topoisomerase IIalpha prevents ultrafine anaphase bridges by two mechanisms. Open. Biol. 2020, 10, 190259. [Google Scholar] [PubMed]
- Deming, P.B.; Cistulli, C.A.; Zhao, H.; Graves, P.R.; Piwnica-Worms, H.; Paules, R.S.; Downes, C.S.; Kaufmann, W.K. The human decatenation checkpoint. Proc. Natl. Acad. Sci. USA 2001, 98, 12044–12049. [Google Scholar] [CrossRef] [PubMed]
- Al-Ejeh, F.; Kumar, R.; Wiegmans, A.; Lakhani, S.R.; Brown, M.P.; Khanna, K.K. Harnessing the complexity of DNA-damage response pathways to improve cancer treatment outcomes. Oncogene 2010, 29, 6085–6098. [Google Scholar] [CrossRef] [PubMed]
- Hinchcliffe, E.H.; Day, C.A.; Karanjeet, K.B.; Fadness, S.; Langfald, A.; Vaughan, K.T.; Dong, Z. Chromosome missegregation during anaphase triggers p53 cell cycle arrest through histone H3.3 Ser31 phosphorylation. Nat. Cell Biol. 2016, 18, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Bridewell, D.J.; Finlay, G.J.; Baguley, B.C. Differential actions of aclarubicin and doxorubicin: The role of topoisomerase I. Oncol. Res. 1997, 9, 535–542. [Google Scholar]
- Chen, Y.Y.; Lukka, P.B.; Joseph, W.R.; Finlay, G.J.; Paxton, J.W.; McKeage, M.J.; Baguley, B.C. Selective cellular uptake and retention of SN 28049, a new DNA-binding topoisomerase II-directed antitumor agent. Cancer Chemother. Pharmacol. 2014, 74, 25–35. [Google Scholar] [CrossRef]
- Hurwitz, S.J.; Terashima, M.; Mizunuma, N.; Slapak, C.A. Vesicular anthracycline accumulation in doxorubicin-selected U-937 cells: Participation of lysosomes. Blood 1997, 89, 3745–3754. [Google Scholar]
- Markovits, J.; Pommier, Y.; Kerrigan, D.; Covey, J.M.; Tilchen, E.J.; Kohn, K.W. Topoisomerase II-mediated DNA breaks and cytotoxicity in relation to cell proliferation and the cell cycle in NIH 3T3 fibroblasts and L1210 leukemia cells. Cancer Res. 1987, 47, 2050–2055. [Google Scholar]
- Ng, K.W.; Marshall, E.A.; Bell, J.C.; Lam, W.L. cGAS-STING and Cancer: Dichotomous Roles in Tumor Immunity and Development. Trends. Immunol. 2018, 39, 44–54. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baguley, B.C.; Drummond, C.J.; Chen, Y.Y.; Finlay, G.J. DNA-Binding Anticancer Drugs: One Target, Two Actions. Molecules 2021, 26, 552. https://doi.org/10.3390/molecules26030552
Baguley BC, Drummond CJ, Chen YY, Finlay GJ. DNA-Binding Anticancer Drugs: One Target, Two Actions. Molecules. 2021; 26(3):552. https://doi.org/10.3390/molecules26030552
Chicago/Turabian StyleBaguley, Bruce C., Catherine J. Drummond, Ying Yi Chen, and Graeme J. Finlay. 2021. "DNA-Binding Anticancer Drugs: One Target, Two Actions" Molecules 26, no. 3: 552. https://doi.org/10.3390/molecules26030552
APA StyleBaguley, B. C., Drummond, C. J., Chen, Y. Y., & Finlay, G. J. (2021). DNA-Binding Anticancer Drugs: One Target, Two Actions. Molecules, 26(3), 552. https://doi.org/10.3390/molecules26030552