
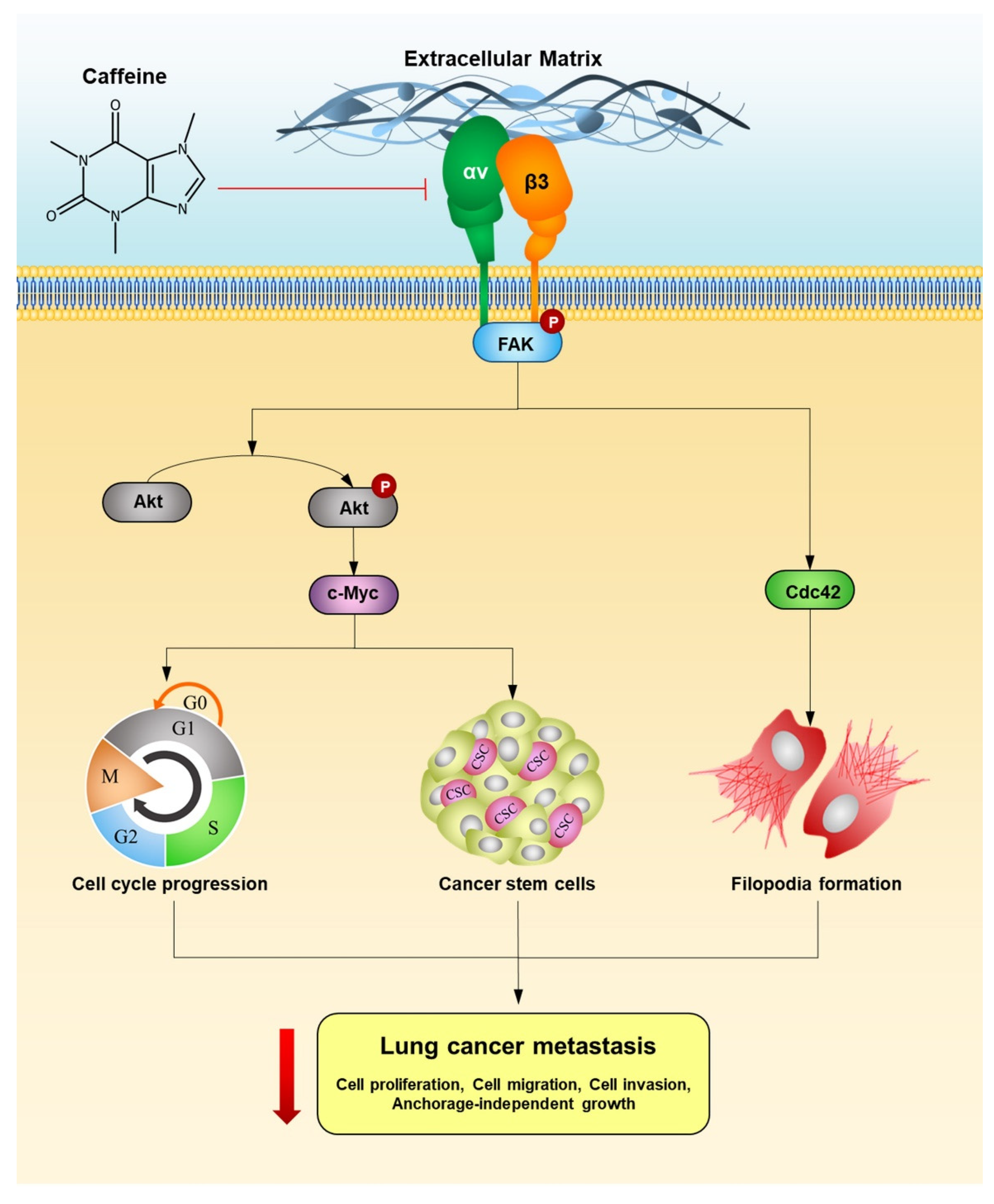
Caffeine Induces G0/G1 Cell Cycle Arrest and Inhibits Migration through Integrin αv, β3, and FAK/Akt/c-Myc Signaling Pathway

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
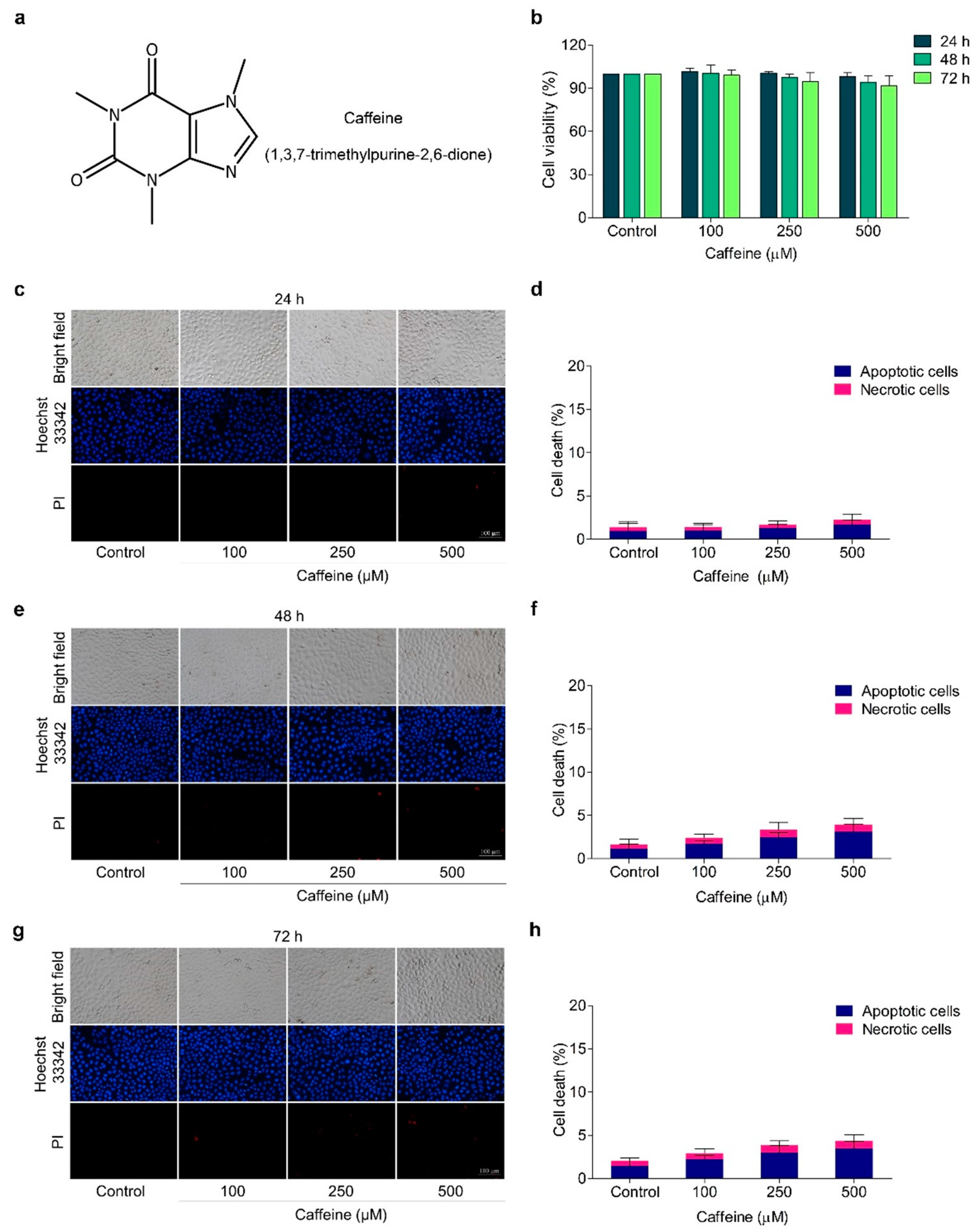
2.1. The Effect of Caffeine on Cell Viability in Human Lung Cancer NCI-H23 Cells
2.2. Caffeine Attenuates Cell Proliferation and Cell Cycle Progression
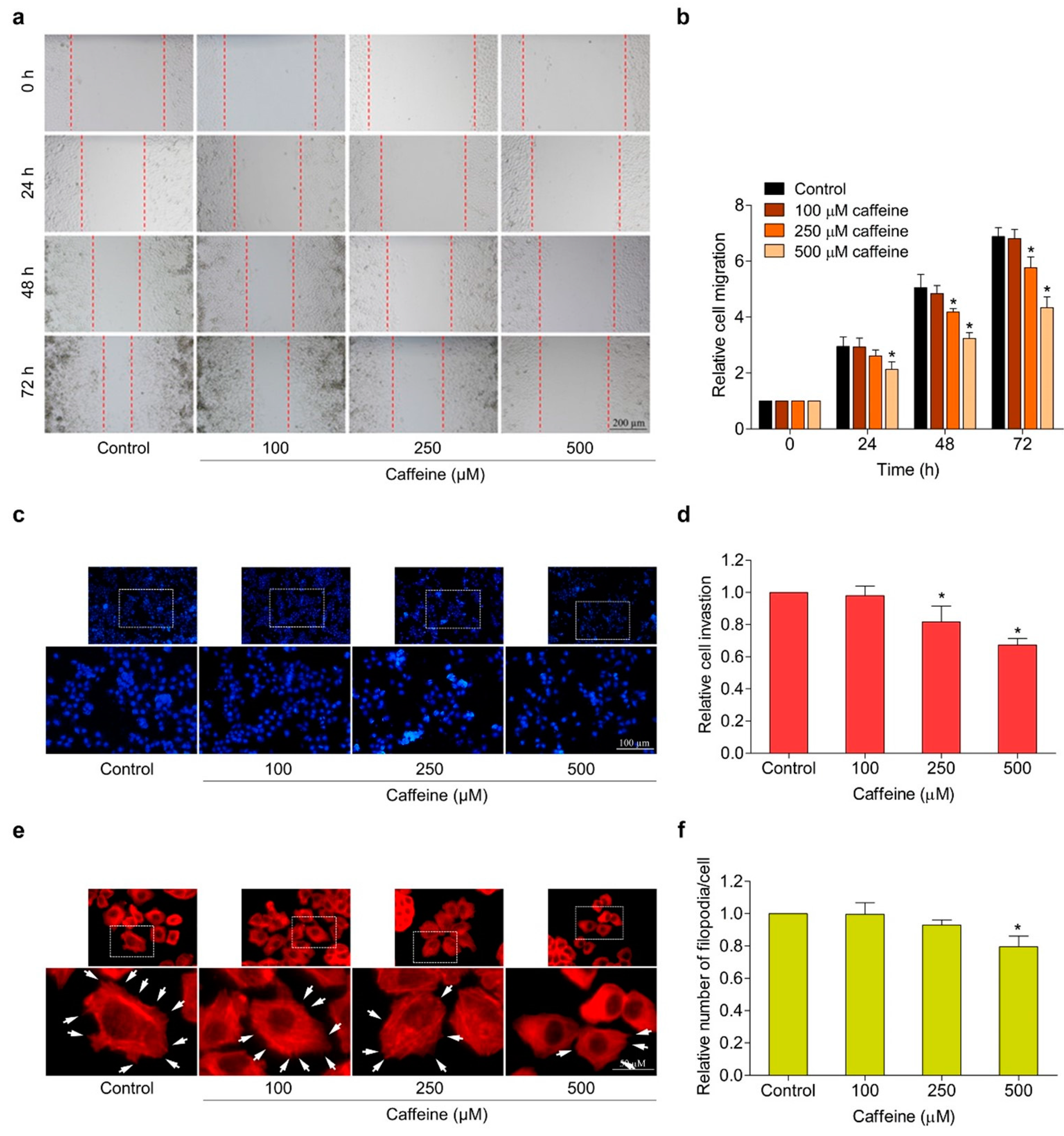
2.3. Caffeine Suppresses Migration, Invasion, and Filopodia Formation
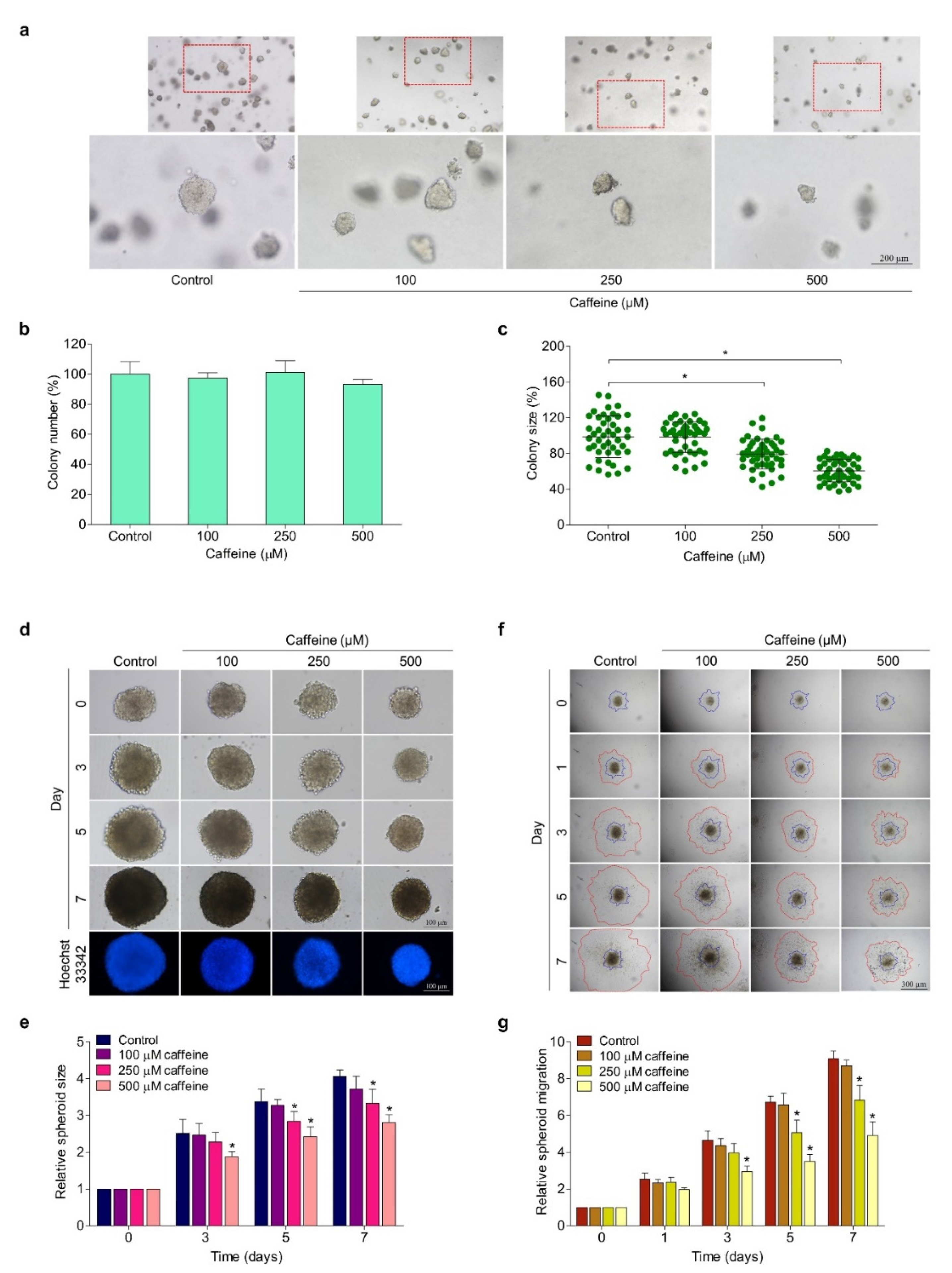
2.4. Caffeine Reduces Anchorage-Independent Growth and the CSC-like Phenotype of Hunam Lung Cancer NCI-H23 Cells
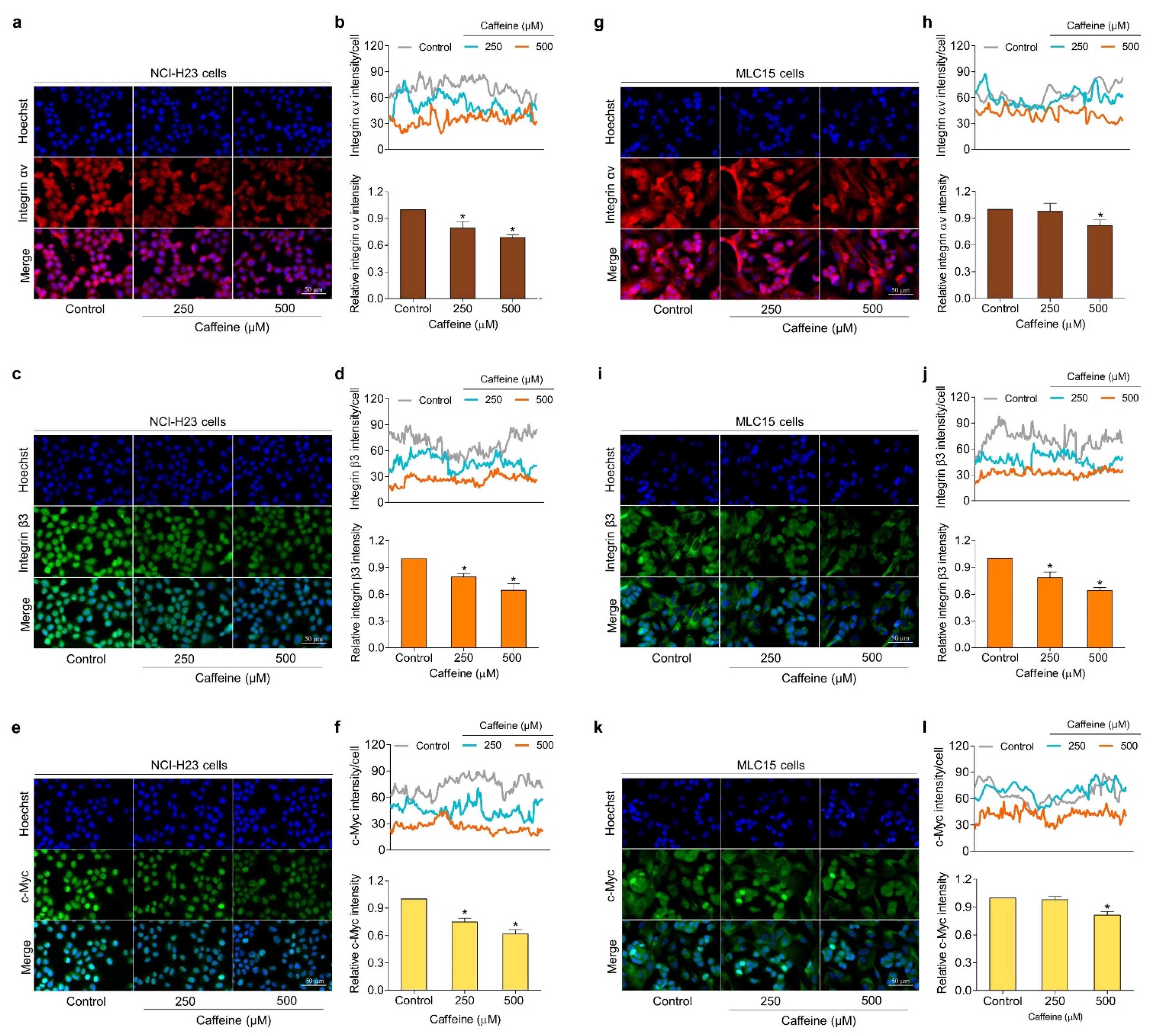
2.5. Caffeine Suppresses Metastasis-Related Signaling Pathways via Integrin Alteration
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Cultures
4.3. Cell Viability Assay
4.4. Differential Nuclear Staining Assay
4.5. Colony Formation Assay
4.6. Cell Cycle Analysis
4.7. Migration and Invasion Assays
4.8. Cell Morphology and Filopodia Characterization
4.9. Anchorage-Independent Growth Assay
4.10. Single Three-Dimensional (3D)Spheroid-Formation Assay
4.11. Tumor Spheroid-Based Migration Assay
4.12. Western Blot Analysis
4.13. Immunofluorescence Assay
4.14. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Barta, J.A.; Powell, C.A.; Wisnivesky, J.P. Global epidemiology of lung cancer. Ann. Glob. Health 2019, 85, 8. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Huysentruyt, L.C. On the origin of cancer metastasis. Crit. Rev. Oncog. 2013, 18, 43–73. [Google Scholar] [CrossRef] [PubMed]
- Guan, X. Cancer metastases: Challenges and opportunities. Acta Pharm. Sin. B 2015, 5, 402–418. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Condeelis, J. Regulation of the actin cytoskeleton in cancer cell migration and invasion. BBA Mol. Cell Res. 2007, 1773, 642–652. [Google Scholar] [CrossRef] [PubMed]
- Nagano, M.; Hoshino, D.; Koshikawa, N.; Akizawa, T.; Seiki, M. Turnover of focal adhesions and cancer cell migration. Int. J. Cell Biol. 2012, 2012, 310616. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, M.A.; Modzelewska, K.; Kwong, L.; Keely, P.J. Focal adhesion regulation of cell behavior. BBA Mol. Cell Res. 2004, 1692, 103–119. [Google Scholar] [CrossRef]
- Anderson, L.R.; Owens, T.W.; Naylor, M.J. Structural and mechanical functions of integrins. Biophys. Rev. 2014, 6, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Hood, J.D.; Cheresh, D.A. Role of integrins in cell invasion and migration. Nat. Rev. Cancer 2002, 2, 91–100. [Google Scholar] [CrossRef]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef]
- Dingemans, A.-M.C.; van den Boogaart, V.; Vosse, B.A.; van Suylen, R.-J.; Griffioen, A.W.; Thijssen, V.L. Integrin expression profiling identifies integrin alpha5 and beta1 as prognostic factors in early stage non-small cell lung cancer. Mol. Cancer 2010, 9, 152. [Google Scholar] [CrossRef] [PubMed]
- Caccavari, F.; Valdembri, D.; Sandri, C.; Bussolino, F.; Serini, G. Integrin signaling and lung cancer. Cell Adhes. Migr. 2010, 4, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.; Giancotti, F.G. Integrin signaling in cancer: Mechanotransduction, stemness, epithelial plasticity, and therapeutic resistance. Cancer Cell 2019, 35, 347–367. [Google Scholar] [CrossRef]
- van Nimwegen, M.J.; van de Water, B. Focal adhesion kinase: A potential target in cancer therapy. Biochem. Pharmacol. 2007, 73, 597–609. [Google Scholar] [CrossRef]
- Mitra, S.K.; Hanson, D.A.; Schlaepfer, D.D. Focal adhesion kinase: In command and control of cell motility. Nat. Rev. Mol. Cell Bio. 2005, 6, 56–68. [Google Scholar] [CrossRef]
- Ridley, A.J. Rho GTPase signalling in cell migration. Curr. Opin. Cell Biol. 2015, 36, 103–112. [Google Scholar] [CrossRef]
- Qiao, M.; Sheng, S.; Pardee, A.B. Metastasis and AKT activation. Cell Cycle 2008, 7, 2991–2996. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Weng, H.; Zhou, H.; Qu, L. Attacking c-Myc: Targeted and combined therapies for cancer. Curr. Pharm. Des. 2014, 20, 6543–6554. [Google Scholar] [CrossRef]
- Chanvorachote, P.; Sriratanasak, N.; Nonpanya, N. C-myc Contributes to Malignancy of Lung Cancer: A Potential Anticancer Drug Target. Anticancer Res. 2020, 40, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Bretones, G.; Delgado, M.D.; León, J. Myc and cell cycle control. Biochim. Biophys. Acta 2015, 1849, 506–516. [Google Scholar] [CrossRef]
- Elbadawy, M.; Usui, T.; Yamawaki, H.; Sasaki, K. Emerging roles of C-Myc in cancer stem cell-related signaling and resistance to cancer chemotherapy: A potential therapeutic target against colorectal cancer. Int. J. Mol. Sci. 2019, 20, 2340. [Google Scholar] [CrossRef] [PubMed]
- Nehlig, A. Interindividual differences in caffeine metabolism and factors driving caffeine consumption. Pharmacol. Rev. 2018, 70, 384–411. [Google Scholar] [CrossRef] [PubMed]
- Glade, M.J. Caffeine—Not just a stimulant. Nutrition 2010, 26, 932–938. [Google Scholar] [CrossRef] [PubMed]
- Tazzeo, T.; Bates, G.; Roman, H.N.; Lauzon, A.-M.; Khasnis, M.D.; Eto, M.; Janssen, L.J. Caffeine relaxes smooth muscle through actin depolymerization. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2012, 303, L334–L342. [Google Scholar] [CrossRef] [PubMed]
- Saiki, S.; Sasazawa, Y.; Imamichi, Y.; Kawajiri, S.; Fujimaki, T.; Tanida, I.; Kobayashi, H.; Sato, F.; Sato, S.; Ishikawa, K.-I. Caffeine induces apoptosis by enhancement of autophagy via PI3K/Akt/mTOR/p70S6K inhibition. Autophagy 2011, 7, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Ku, B.M.; Lee, Y.K.; Jeong, J.Y.; Ryu, J.; Choi, J.; Kim, J.S.; Cho, Y.W.; Roh, G.S.; Kim, H.J.; Cho, G.J. Caffeine inhibits cell proliferation and regulates PKA/GSK3β pathways in U87MG human glioma cells. Mol. Cells 2011, 31, 275–279. [Google Scholar] [CrossRef]
- Chen, Y.; Chou, W.C.; Ding, Y.M.; Wu, Y.C. Caffeine inhibits migration in glioma cells through the ROCK-FAK pathway. Cell Physiol. Biochem. 2014, 33, 1888–1898. [Google Scholar] [CrossRef]
- Wang, Z.; Gu, C.; Wang, X.; Lang, Y.; Wu, Y.; Wu, X.; Zhu, X.; Wang, K.; Yang, H. Caffeine enhances the anti-tumor effect of 5-fluorouracil via increasing the production of reactive oxygen species in hepatocellular carcinoma. Med. Oncol. 2019, 36, 97. [Google Scholar] [CrossRef]
- Arjonen, A.; Kaukonen, R.; Ivaska, J. Filopodia and adhesion in cancer cell motility. Cell Adhes. Migr. 2011, 5, 421–430. [Google Scholar] [CrossRef]
- Guadamillas, M.C.; Cerezo, A.; del Pozo, M.A. Overcoming anoikis–pathways to anchorage-independent growth in cancer. J. Cell Sci. 2011, 124, 3189–3197. [Google Scholar] [CrossRef] [PubMed]
- Srinual, S.; Chanvorachote, P.; Pongrakhananon, V. Suppression of cancer stem-like phenotypes in NCI-H460 lung cancer cells by vanillin through an Akt-dependent pathway. Int. J. Oncol. 2017, 50, 1341–1351. [Google Scholar] [CrossRef] [PubMed]
- Vinci, M.; Box, C.; Zimmermann, M.; Eccles, S.A. Tumor spheroid-based migration assays for evaluation of therapeutic agents. Methods Mol. Biol. 2013, 986, 253–266. [Google Scholar] [PubMed]
- Suhail, Y.; Cain, M.P.; Vanaja, K.; Kurywchak, P.A.; Levchenko, A.; Kalluri, R.; Kshitiz. Systems Biology of Cancer Metastasis. Cell Syst. 2019, 9, 109–127. [Google Scholar] [CrossRef]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal. Transduct. Target. Ther. 2020, 5, 28. [Google Scholar] [CrossRef]
- Shanmugam, M.K.; Lee, J.H.; Chai, E.Z.P.; Kanchi, M.M.; Kar, S.; Arfuso, F.; Dharmarajan, A.; Kumar, A.P.; Ramar, P.S.; Looi, C.Y.; et al. Cancer prevention and therapy through the modulation of transcription factors by bioactive natural compounds. Semin. Cancer Biol. 2016, 40–41, 35–47. [Google Scholar] [CrossRef]
- Jasiewicz, B.; Sierakowska, A. Chapter 15—Caffeine and its analogs, antioxidants and applications. In Aging, 2nd ed.; Preedy, V.R., Patel, V.B., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 155–164. [Google Scholar]
- Cheng, Y.-C.; Ding, Y.-M.; Hueng, D.-Y.; Chen, J.-Y.; Chen, Y. Caffeine suppresses the progression of human glioblastoma via cathepsin B and MAPK signaling pathway. J. Nutr. Biochem. 2016, 33, 63–72. [Google Scholar] [CrossRef]
- Venkata Charan Tej, G.N.; Neogi, K.; Verma, S.S.; Chandra Gupta, S.; Nayak, P.K. Caffeine-enhanced anti-tumor immune response through decreased expression of PD1 on infiltrated cytotoxic T lymphocytes. Eur. J. Pharmacol. 2019, 859, 172538. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, L.; Wan, Z.; He, Y.; Huang, H.; Xiang, H.; Wu, X.; Zhang, K.; Liu, Y.; Goodin, S.; et al. Atorvastatin and Caffeine in Combination Regulates Apoptosis, Migration, Invasion and Tumorspheres of Prostate Cancer Cells. Pathol. Oncol. Res. 2020, 26, 209–216. [Google Scholar] [CrossRef]
- Dang, C.V.; Resar, L.M.; Emison, E.; Kim, S.; Li, Q.; Prescott, J.E.; Wonsey, D.; Zeller, K. Function of the c-Myc oncogenic transcription factor. Exp. Cell Res. 1999, 253, 63–77. [Google Scholar] [CrossRef]
- Gabay, M.; Li, Y.; Felsher, D.W. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb. Perspect. Med. 2014, 4, a014241. [Google Scholar] [CrossRef]
- García-Gutiérrez, L.; Delgado, M.D.; León, J. MYC Oncogene Contributions to Release of Cell Cycle Brakes. Genes 2019, 10, 244. [Google Scholar] [CrossRef]
- Zhou, L.; Wu, F.; Jin, W.; Yan, B.; Chen, X.; He, Y.; Yang, W.; Du, W.; Zhang, Q.; Guo, Y.; et al. Theabrownin Inhibits Cell Cycle Progression and Tumor Growth of Lung Carcinoma through c-myc-Related Mechanism. Front. Pharmacol. 2017, 8, 75. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhang, D.; Mao, J.; Ke, X.-X.; Zhang, R.; Yin, C.; Gao, N.; Cui, H. Morusin inhibits cell proliferation and tumor growth by down-regulating c-Myc in human gastric cancer. Oncotarget 2017, 8, 57187–57200. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; Yao, G.; Nevins, J.; You, L. Sensing and integration of Erk and PI3K signals by Myc. PLoS Comput. Biol. 2008, 4, e1000013. [Google Scholar] [CrossRef]
- Tsai, W.-B.; Aiba, I.; Long, Y.; Lin, H.-K.; Feun, L.; Savaraj, N.; Kuo, M.T. Activation of Ras/PI3K/ERK pathway induces c-Myc stabilization to upregulate argininosuccinate synthetase, leading to arginine deiminase resistance in melanoma cells. Cancer Res. 2012, 72, 2622–2633. [Google Scholar] [CrossRef]
- Domínguez-Cáceres, M.A.; García-Martínez, J.M.; Calcabrini, A.; González, L.; Porque, P.G.; León, J.; Martín-Pérez, J. Prolactin induces c-Myc expression and cell survival through activation of Src/Akt pathway in lymphoid cells. Oncogene 2004, 23, 7378–7390. [Google Scholar] [CrossRef]
- Li, Z.-H.; Zhou, Y.; Ding, Y.-X.; Guo, Q.-L.; Zhao, L. Roles of integrin in tumor development and the target inhibitors. Chin. J. Nat. Med. 2019, 17, 241–251. [Google Scholar] [CrossRef]
- Chiarugi, P.; Giannoni, E. Anoikis: A necessary death program for anchorage-dependent cells. Biochem. Pharmacol. 2008, 76, 1352–1364. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-N.; Koo, K.H.; Sung, J.Y.; Yun, U.-J.; Kim, H. Anoikis Resistance: An Essential Prerequisite for Tumor Metastasis. Int. J. Cell Biol. 2012, 2012, 306879. [Google Scholar] [CrossRef] [PubMed]
- Baum, O.; Hlushchuk, R.; Forster, A.; Greiner, R.; Clézardin, P.; Zhao, Y.; Djonov, V.; Gruber, G. Increased invasive potential and up-regulation of MMP-2 in MDA-MB-231 breast cancer cells expressing the β3 integrin subunit. Int. J. Oncol. 2007, 30, 325–332. [Google Scholar] [PubMed]
- Desgrosellier, J.S.; Barnes, L.A.; Shields, D.J.; Huang, M.; Lau, S.K.; Prévost, N.; Tarin, D.; Shattil, S.J.; Cheresh, D.A. An integrin αvβ3–c-Src oncogenic unit promotes anchorage-independence and tumor progression. Nat. Med. 2009, 15, 1163–1169. [Google Scholar] [CrossRef]
- Fong, Y.-C.; Liu, S.-C.; Huang, C.-Y.; Li, T.-M.; Hsu, S.-F.; Kao, S.-T.; Tsai, F.-J.; Chen, W.-C.; Chen, C.-Y.; Tang, C.-H. Osteopontin increases lung cancer cells migration via activation of the αvβ3 integrin/FAK/Akt and NF-κB-dependent pathway. Lung Cancer 2009, 64, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Dolinschek, R.; Hingerl, J.; Benge, A.; Zafiu, C.; Schüren, E.; Ehmoser, E.K.; Lössner, D.; Reuning, U. Constitutive activation of integrin αvβ3 contributes to anoikis resistance of ovarian cancer cells. Mol. Oncol. 2021, 15, 503–522. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Tan, H.; Dong, X.; Xu, Z.; Shi, C.; Han, X.; Jiang, H.; Krissansen, G.W.; Sun, X. Antisense integrin αV and β3 gene therapy suppresses subcutaneously implanted hepatocellular carcinomas. Digestive Liver Dis. 2007, 39, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: Mechanistic findings and clinical applications. Nat. Rev. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Bode, A.M.; Dong, Z.; Lee, M.-H. AKT as a therapeutic target for cancer. Cancer Res. 2019, 79, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Machesky, L.M.; Li, A. Fascin: Invasive filopodia promoting metastasis. Commun. Integr. Biol. 2010, 3, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Mattila, P.K.; Lappalainen, P. Filopodia: Molecular architecture and cellular functions. Nat. Rev. Mol. Cell Biol. 2008, 9, 446–454. [Google Scholar] [CrossRef]
- Akbar Samadani, A.; Keymoradzdeh, A.; Shams, S.; Soleymanpour, A.; Elham Norollahi, S.; Vahidi, S.; Rashidy-Pour, A.; Ashraf, A.; Mirzajani, E.; Khanaki, K.; et al. Mechanisms of cancer stem cell therapy. Clinica Chimica Acta 2020, 510, 581–592. [Google Scholar] [CrossRef]
- Nassar, D.; Blanpain, C. Cancer Stem Cells: Basic Concepts and Therapeutic Implications. Annu. Rev. Pathol. 2016, 11, 47–76. [Google Scholar] [CrossRef] [PubMed]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.-G.; Lee, S.-H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018, 2018, 5416923. [Google Scholar] [CrossRef] [PubMed]
- Chopra, A.S.; Liu, X.; Liu, H. Chapter 13-Cancer Stem Cells: Metastasis and Evasion From the Host Immune Responses. In Cancer Stem Cells; Liu, H., Lathia, J.D., Eds.; Academic Press: Boston, MA, USA, 2016; pp. 341–366. [Google Scholar]
- Heng, W.S.; Gosens, R.; Kruyt, F.A.E. Lung cancer stem cells: Origin, features, maintenance mechanisms and therapeutic targeting. Biochem. Pharmacol. 2019, 160, 121–133. [Google Scholar] [CrossRef]
- Sumkhemthong, S.; Chamni, S.; Ecoy, G.U.; Taweecheep, P.; Suwanborirux, K.; Prompetchara, E.; Chanvorachote, P.; Chaotham, C. Jorunnamycin A Suppresses Stem-Like Phenotypes and Sensitizes Cisplatin-Induced Apoptosis in Cancer Stem-Like Cell-Enriched Spheroids of Human Lung Cancer Cells. Mar. Drugs 2021, 19, 261. [Google Scholar] [CrossRef] [PubMed]
- Phiboonchaiyanan, P.P.; Chanvorachote, P. Suppression of a cancer stem-like phenotype mediated by alpha-lipoic acid in human lung cancer cells through down-regulation of β-catenin and Oct-4. Cell Oncol. 2017, 40, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Seguin, L.; Kato, S.; Franovic, A.; Camargo, M.F.; Lesperance, J.; Elliott, K.C.; Yebra, M.; Mielgo, A.; Lowy, A.M.; Husain, H. An integrin β 3–KRAS–RalB complex drives tumour stemness and resistance to EGFR inhibition. Nat. Cell Biol. 2014, 16, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Desgrosellier, J.S.; Lesperance, J.; Seguin, L.; Gozo, M.; Kato, S.; Franovic, A.; Yebra, M.; Shattil, S.J.; Cheresh, D.A. Integrin αvβ3 Drives Slug Activation and Stemness in the Pregnant and Neoplastic Mammary Gland. Dev. Cell 2014, 30, 295–308. [Google Scholar] [CrossRef]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal. Transduc. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef]
- Li, Q.-Q.; Xie, Y.-K.; Wu, Y.; Li, L.-L.; Liu, Y.; Miao, X.-B.; Liu, Q.-Z.; Yao, K.-T.; Xiao, G.-H. Sulforaphane inhibits cancer stem-like cell properties and cisplatin resistance through miR-214-mediated downregulation of c-MYC in non-small cell lung cancer. Oncotarget 2017, 8, 12067–12080. [Google Scholar] [CrossRef] [PubMed]
- Geissmann, Q. OpenCFU, a New Free and Open-Source Software to Count Cell Colonies and Other Circular Objects. PLoS ONE 2013, 8, e54072. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meisaprow, P.; Aksorn, N.; Vinayanuwattikun, C.; Chanvorachote, P.; Sukprasansap, M. Caffeine Induces G0/G1 Cell Cycle Arrest and Inhibits Migration through Integrin αv, β3, and FAK/Akt/c-Myc Signaling Pathway. Molecules 2021, 26, 7659. https://doi.org/10.3390/molecules26247659
Meisaprow P, Aksorn N, Vinayanuwattikun C, Chanvorachote P, Sukprasansap M. Caffeine Induces G0/G1 Cell Cycle Arrest and Inhibits Migration through Integrin αv, β3, and FAK/Akt/c-Myc Signaling Pathway. Molecules. 2021; 26(24):7659. https://doi.org/10.3390/molecules26247659
Chicago/Turabian StyleMeisaprow, Pichitchai, Nithikoon Aksorn, Chanida Vinayanuwattikun, Pithi Chanvorachote, and Monruedee Sukprasansap. 2021. "Caffeine Induces G0/G1 Cell Cycle Arrest and Inhibits Migration through Integrin αv, β3, and FAK/Akt/c-Myc Signaling Pathway" Molecules 26, no. 24: 7659. https://doi.org/10.3390/molecules26247659
APA StyleMeisaprow, P., Aksorn, N., Vinayanuwattikun, C., Chanvorachote, P., & Sukprasansap, M. (2021). Caffeine Induces G0/G1 Cell Cycle Arrest and Inhibits Migration through Integrin αv, β3, and FAK/Akt/c-Myc Signaling Pathway. Molecules, 26(24), 7659. https://doi.org/10.3390/molecules26247659