
Synthesis of Tyrosol and Hydroxytyrosol Glycofuranosides and Their Biochemical and Biological Activities in Cell-Free and Cellular Assays

 ,
,
Abstract
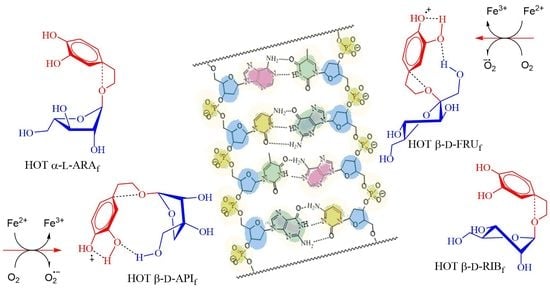
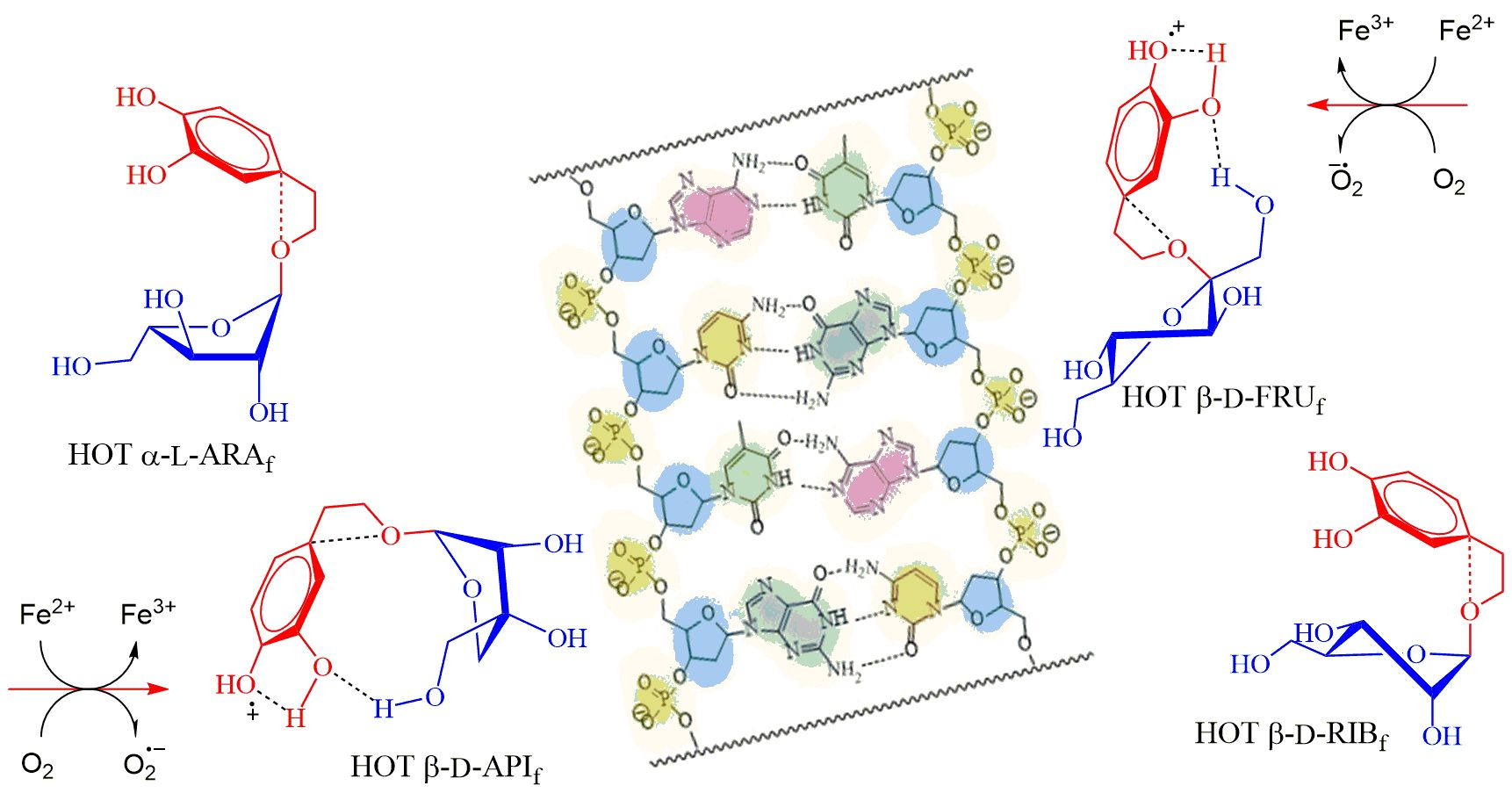
:
1. Introduction
2. Results and Discussion
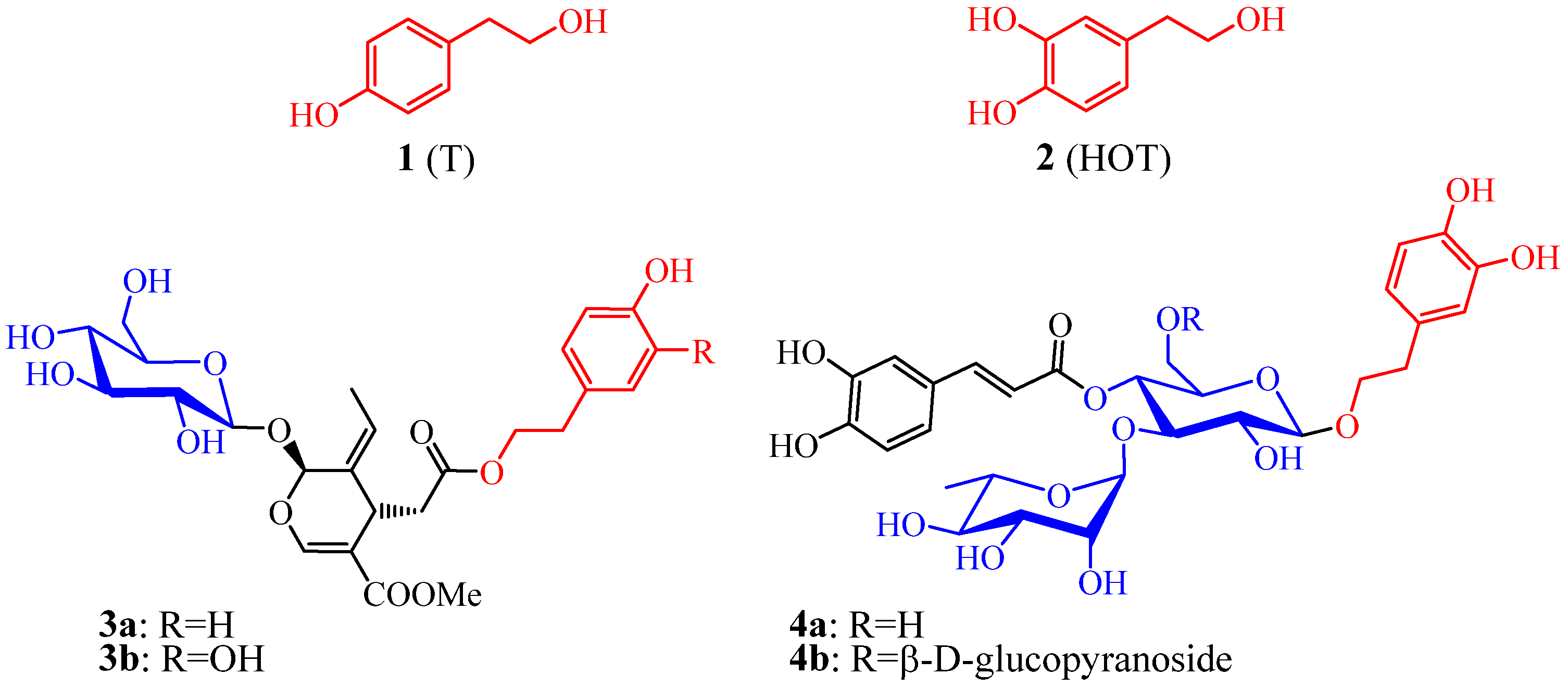
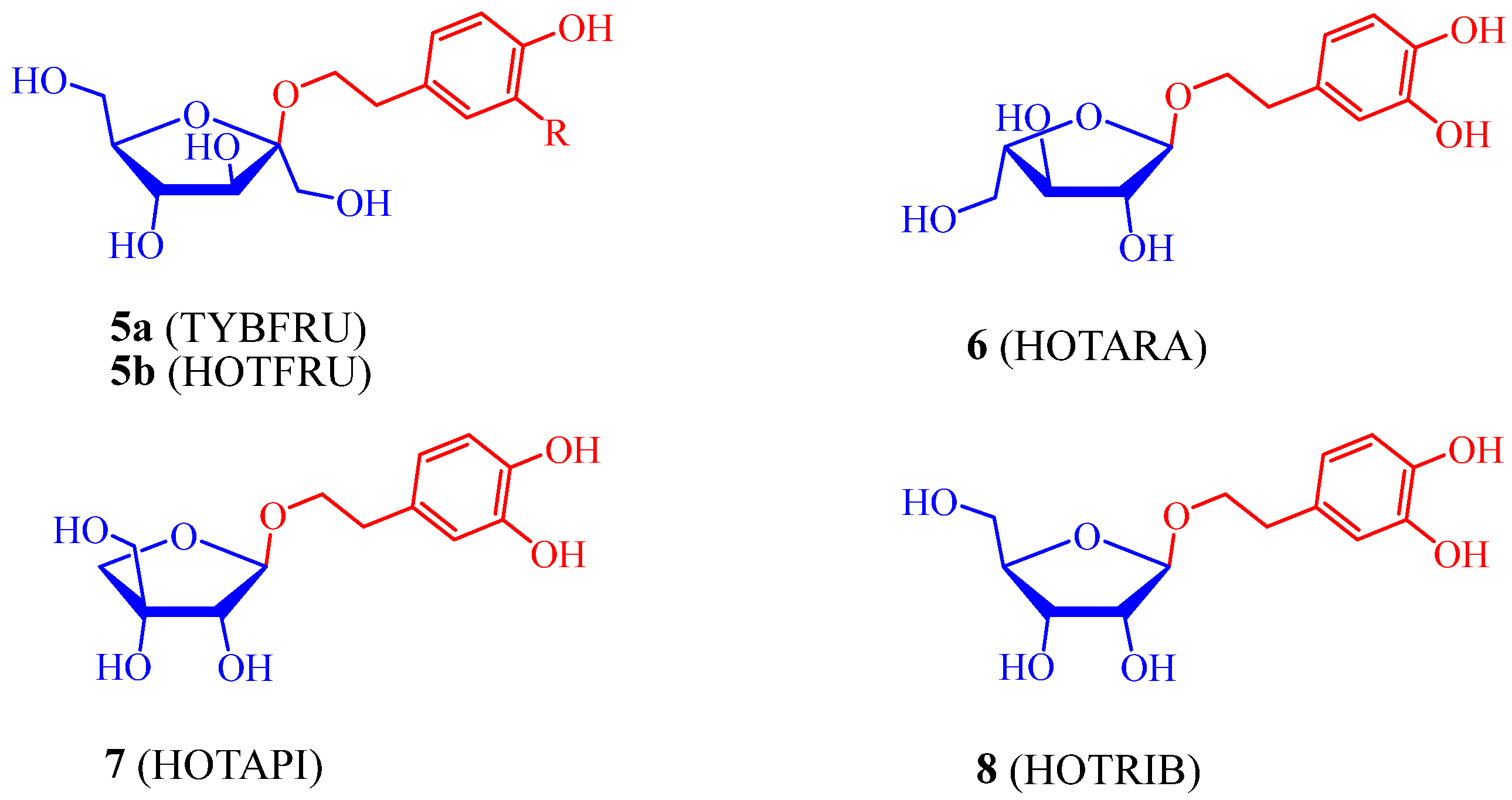
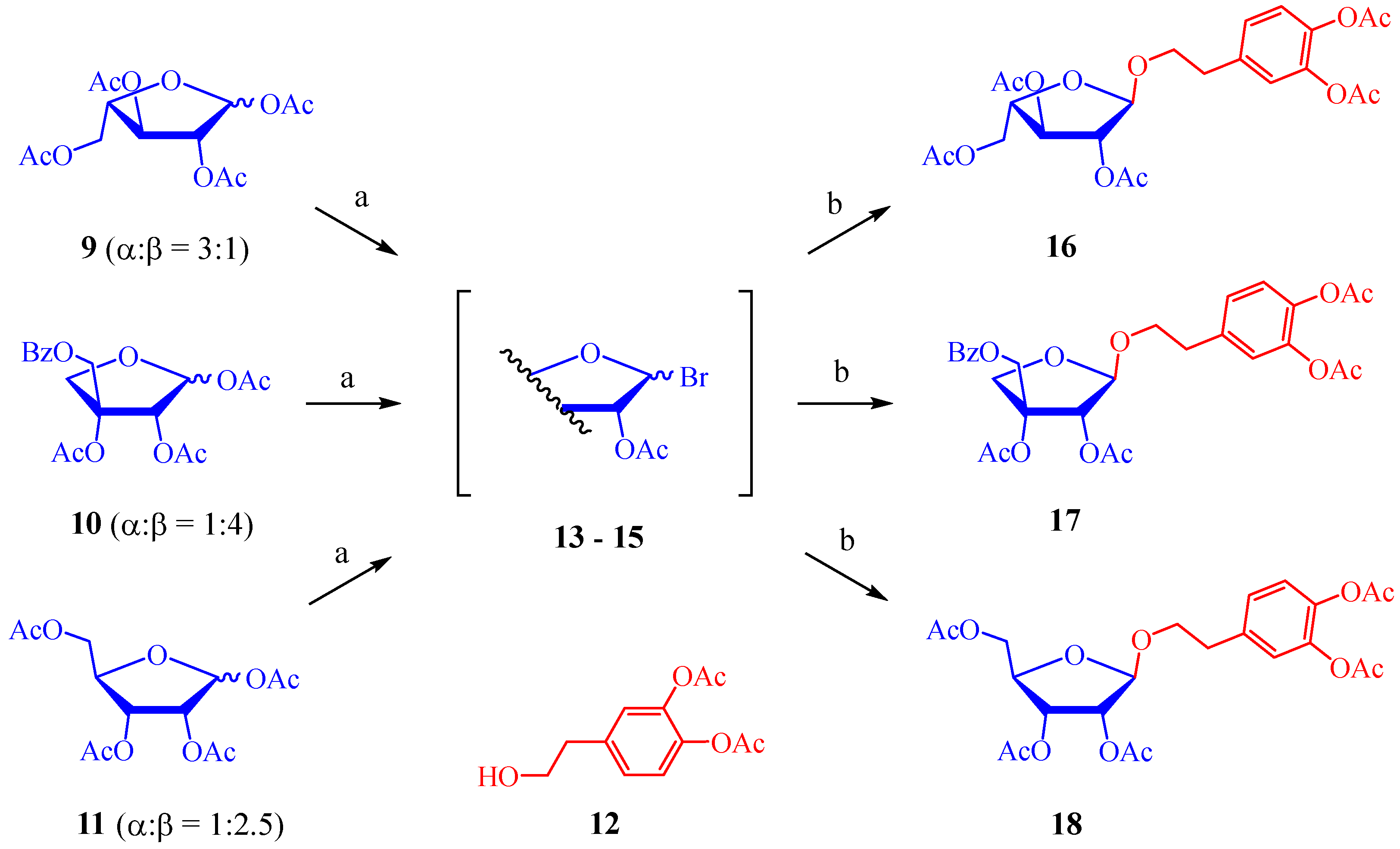
2.1. Synthesis of Hydroxytyrosol Glycofuranosides
2.2. Cell-Free Assays
2.2.1. Reducing Power Assay and DPPH Radical Scavenging Activity
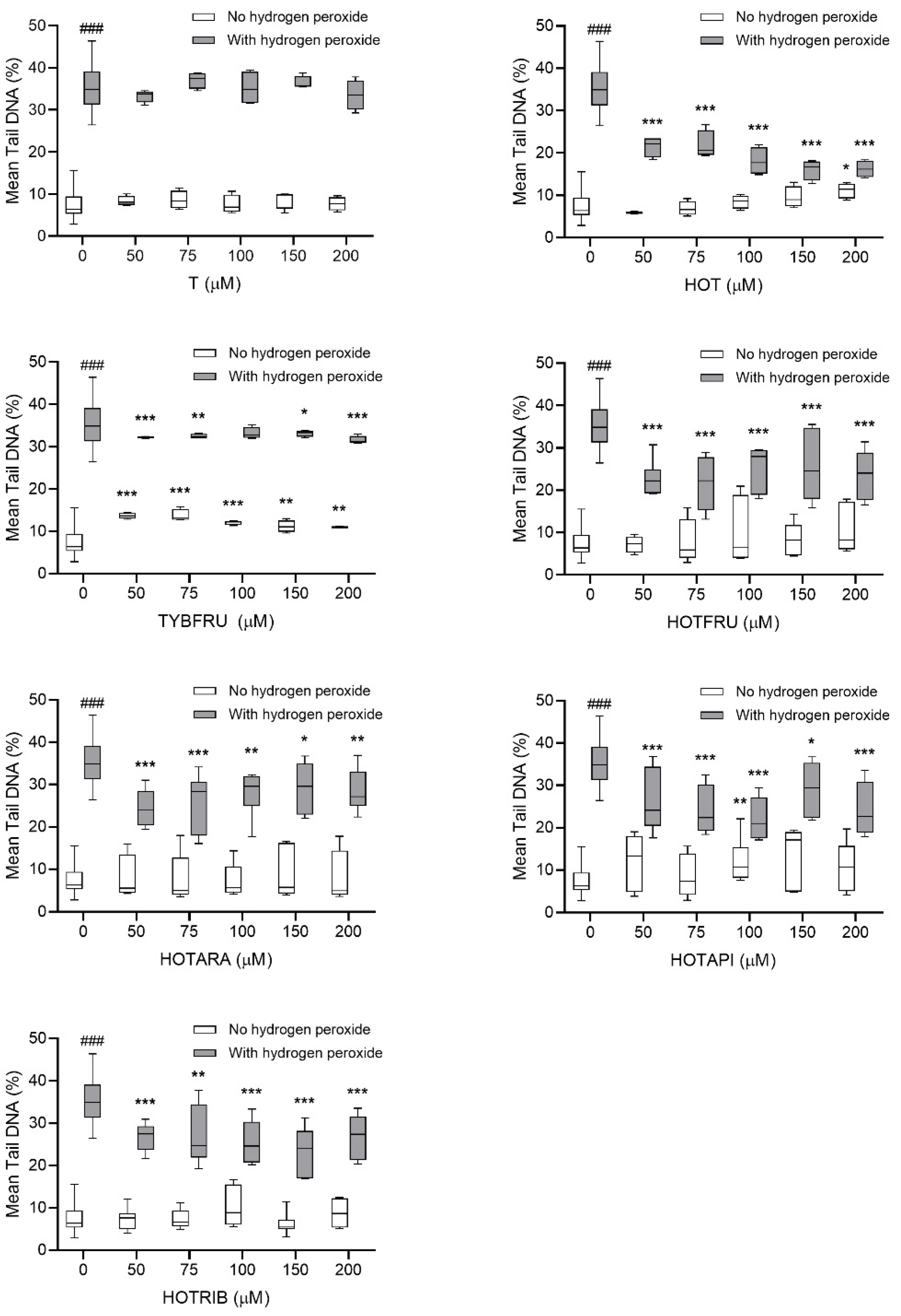
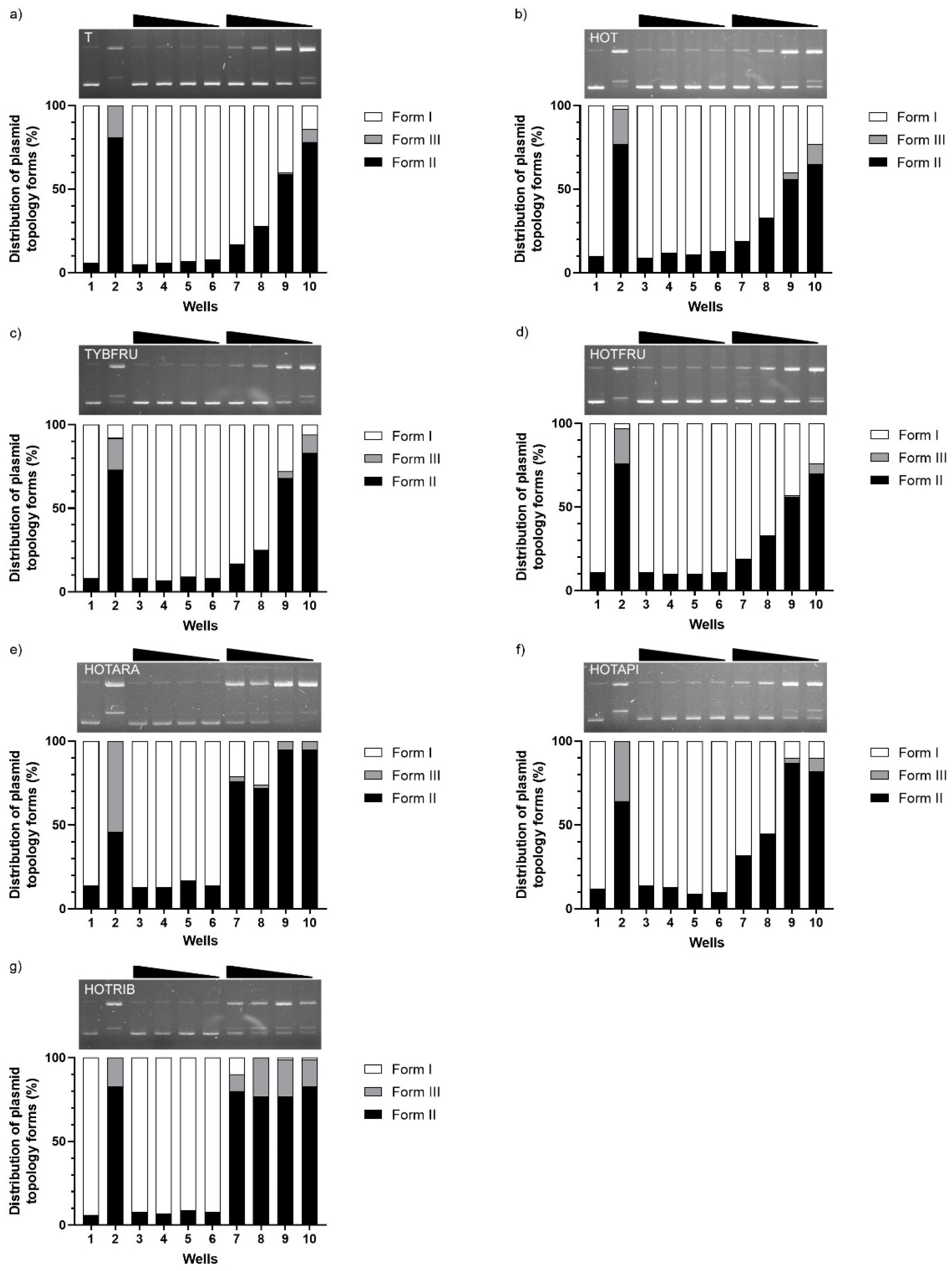
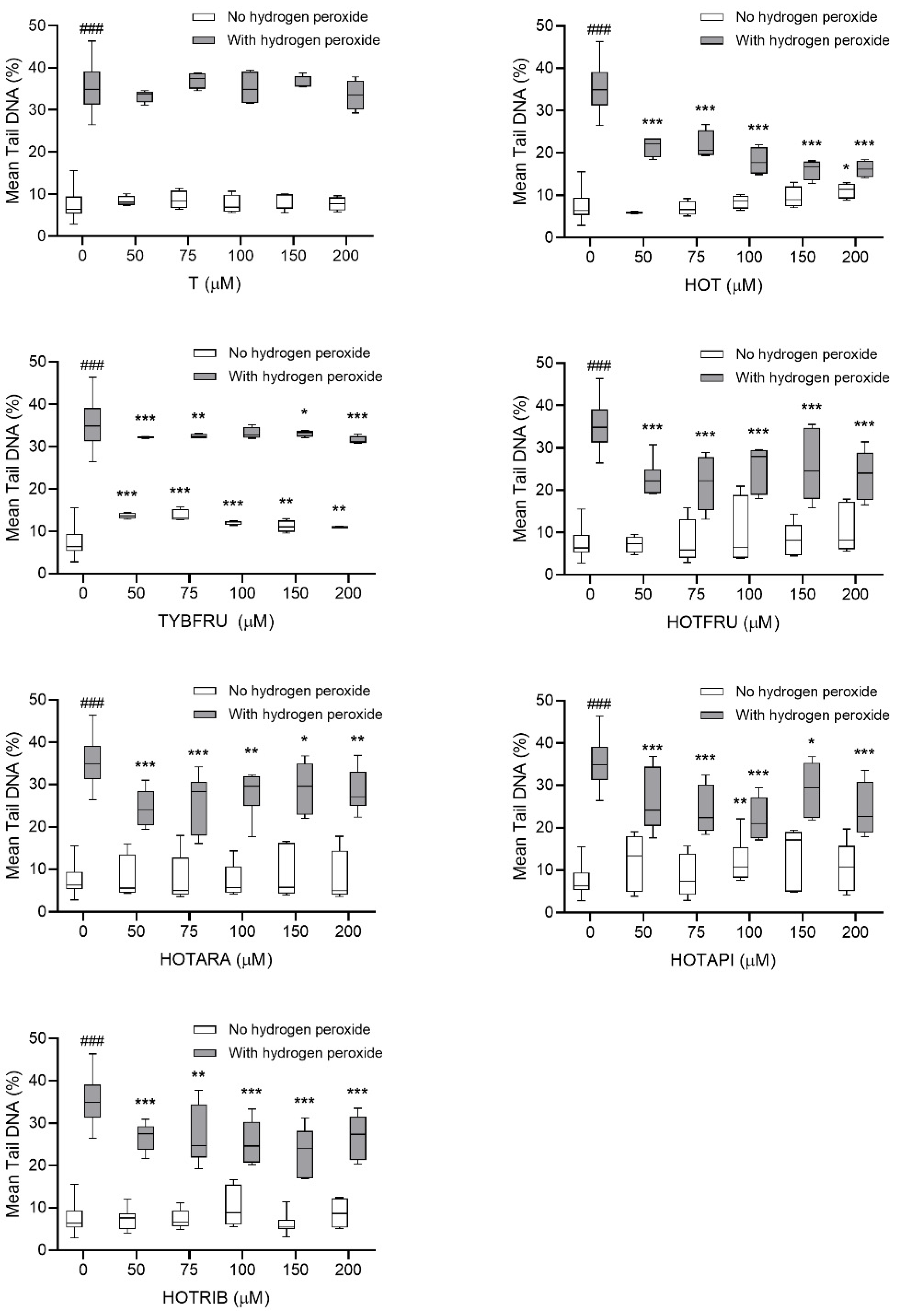
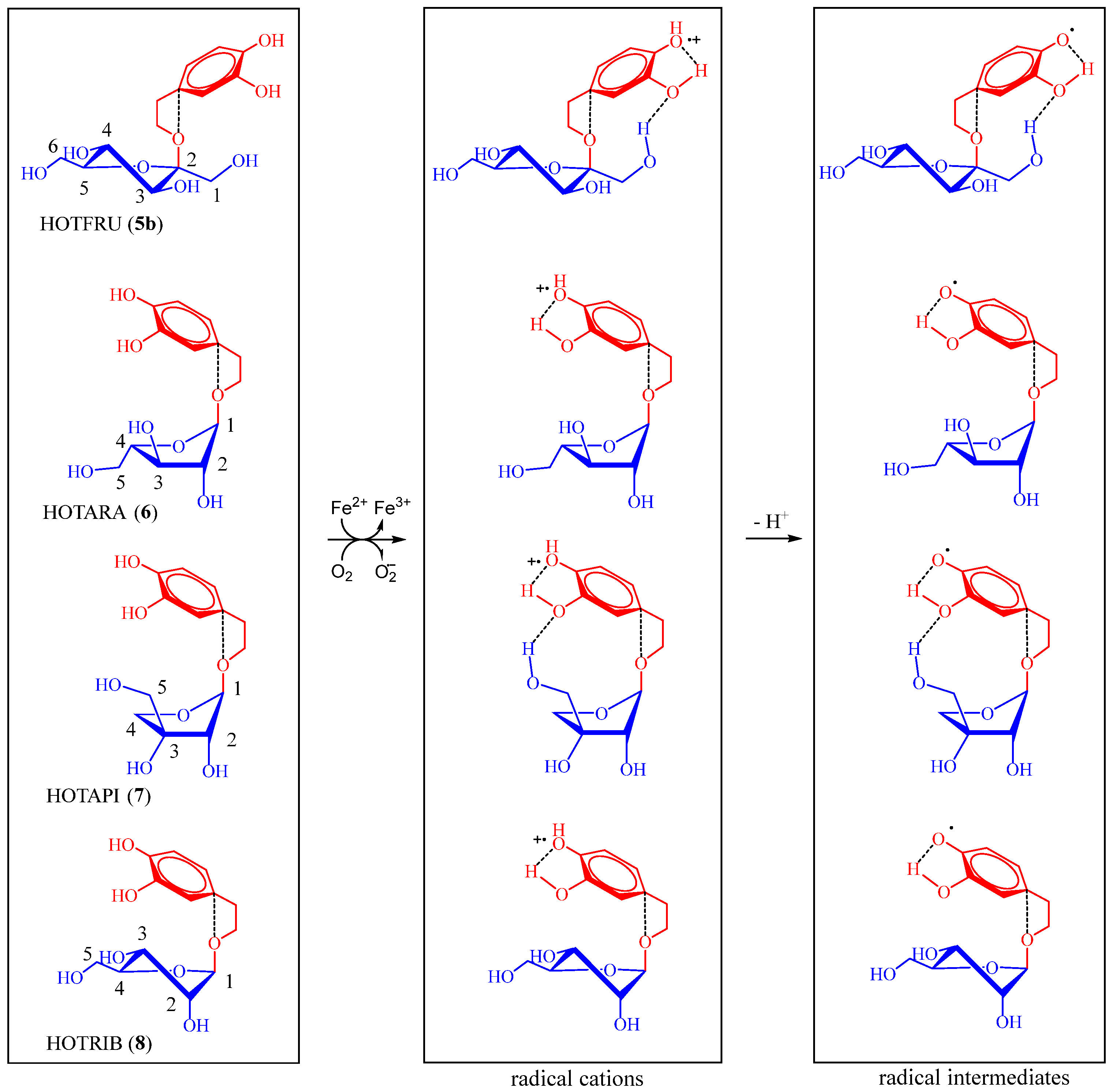
2.2.2. Assessment of DNA-Damaging/-Protective Potential
2.3. Cellular Assays
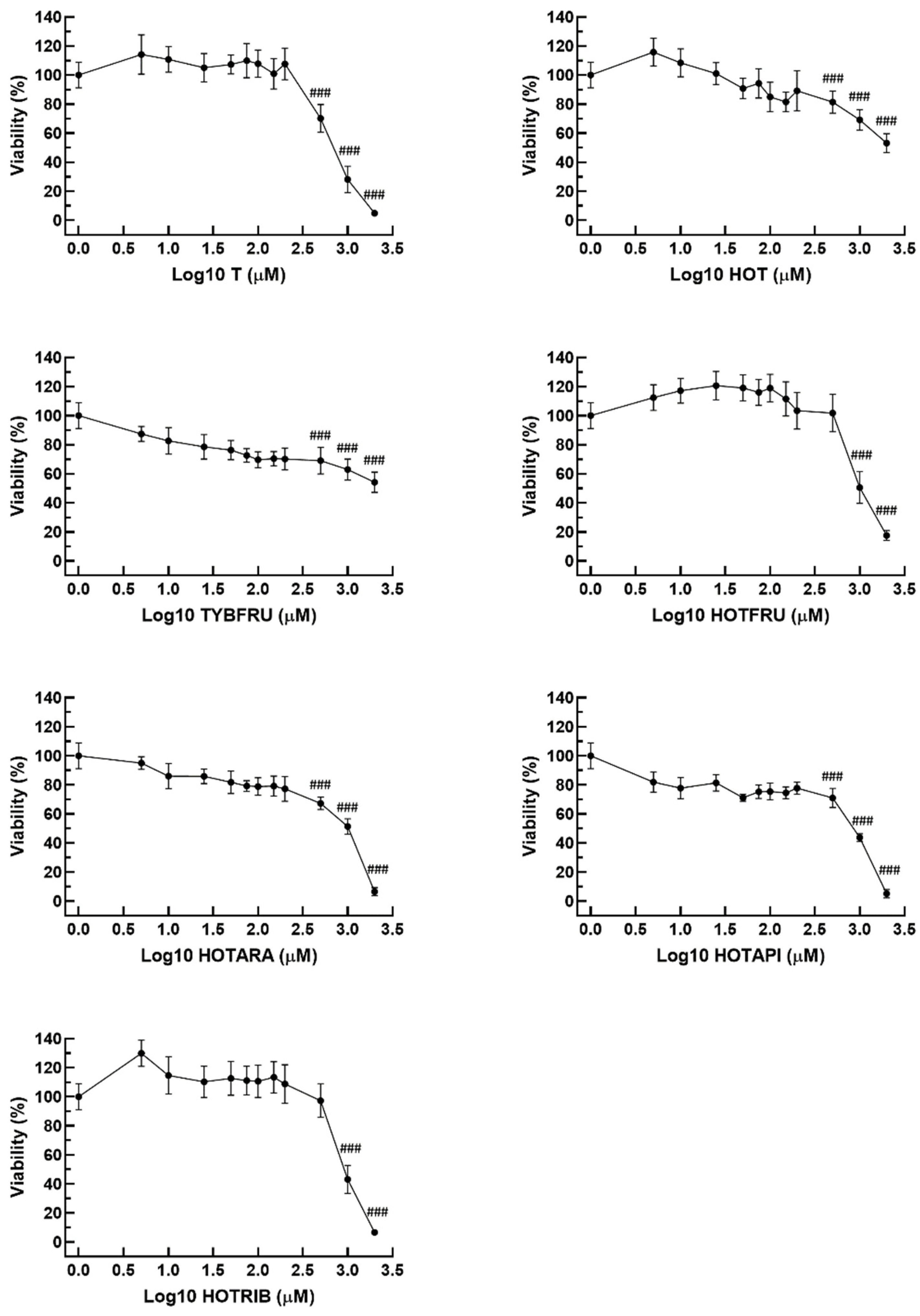
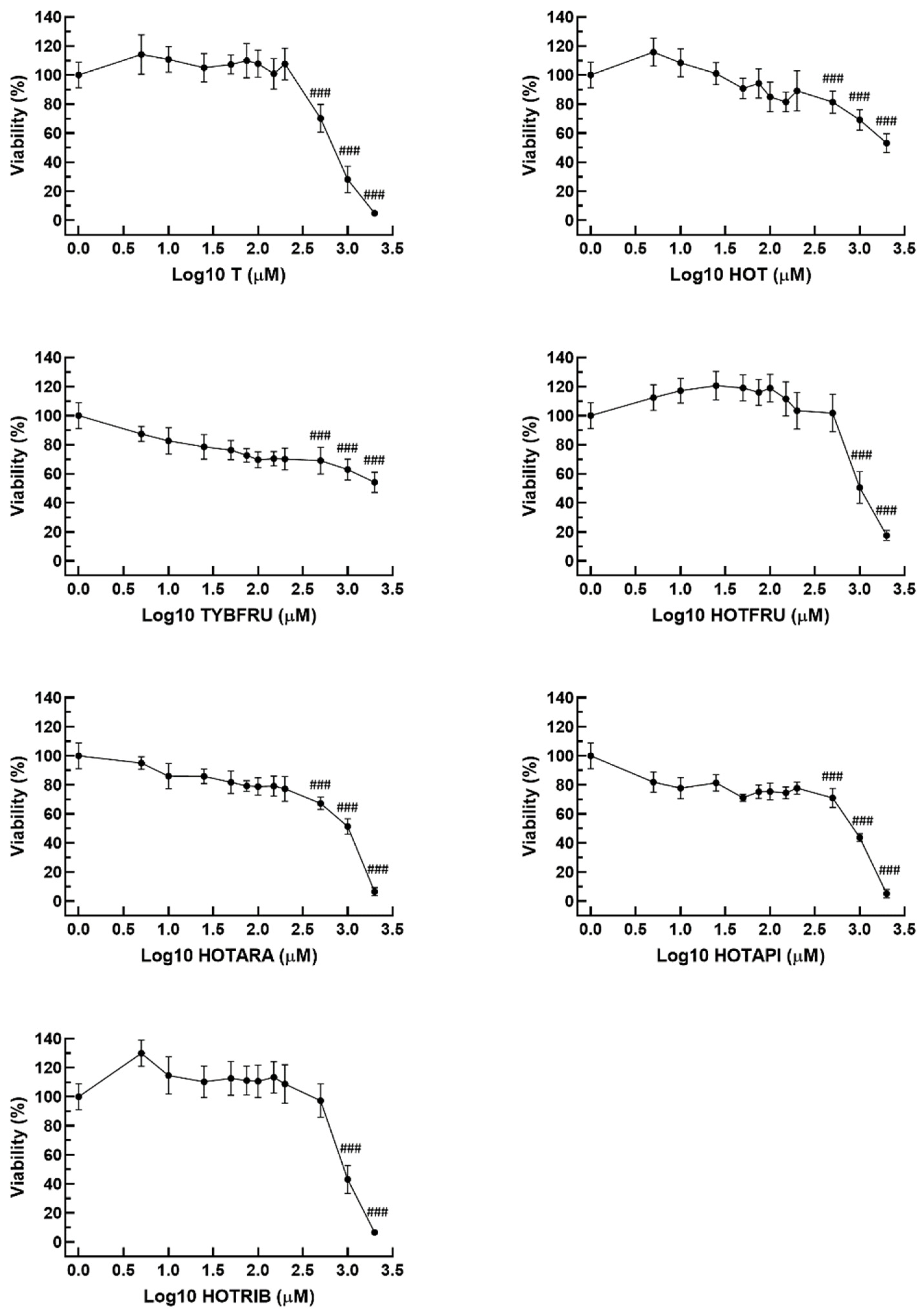
2.3.1. Cell Viability
2.3.2. Evaluation of Potential Protective Effects on HepG2 Cells
3. Materials and Methods
3.1. General
3.2. Glycosylation Methods
3.2.1. Preparation of Glycofuranosyl Bromides 13–15
3.2.2. General Procedure for 1,2-trans-Glycosylation under Conventional Heating Using Basic ZnCO3 as a Promoter. Method Δ
3.2.3. General Procedure for Microwave-Assisted Glycofuranosylation Using Basic ZnCO3 as a Promoter (Method MW)
3.2.4. Typical Preparative Procedure for Glycofuranosylation under Conventional Heating Using Basic ZnCO3
3.2.5. Characterization Data of Per-O-acylated Glycofuranosides 16–18
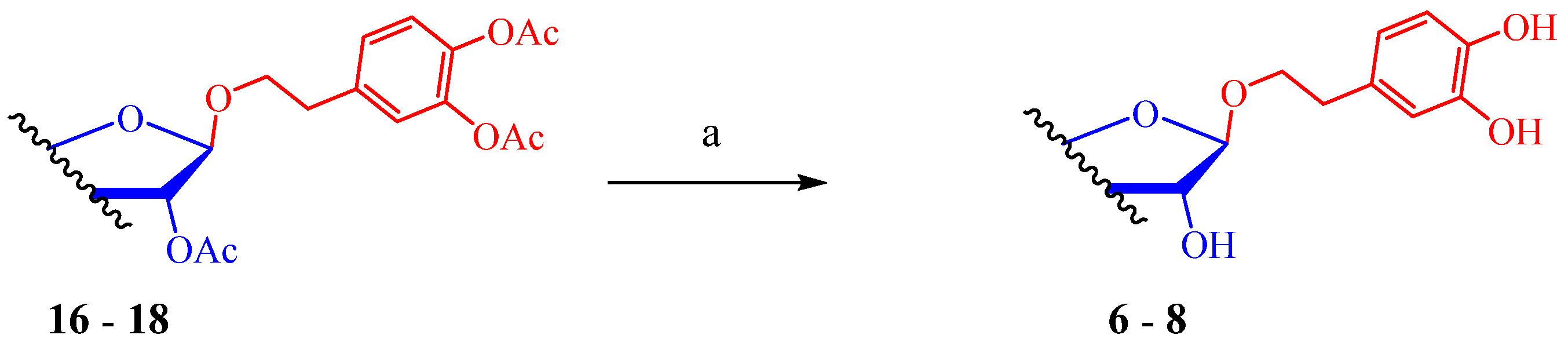
3.3. Deacylation of Per-O-Acylated Glycofuranosides 16–18
3.4. Biochemical Assays
3.4.1. Antioxidant Assays
3.4.2. DNA Topology Assay
3.5. Biological Assays In Vitro
3.5.1. HepG2 Cell Line
3.5.2. Cell Viability Assessment
3.5.3. Alkaline Comet Assay (Single-Cell Gel Electrophoresis; SCGE)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Medawar, E.; Huhn, S.; Villringer, A.; Witte, V.A. The effects of plant-based diets on the body and the brain: A systematic review. Transl. Psychiatry 2019, 9, 226. [Google Scholar] [CrossRef]
- Proshkina, E.; Shaposhnikov, M.; Moskalev, A. Genome-Protecting Compounds as Potential Geroprotectors. Int. J. Mol. Sci. 2020, 21, 4484. [Google Scholar] [CrossRef] [PubMed]
- Marković Karković, A.; Torić, J.; Barbarić, M.; Jakobušić Brala, C. Hydroxytyrosol, tyrosol and derivatives and their potential effects on human health. Molecules 2019, 24, 2001. [Google Scholar] [CrossRef] [Green Version]
- Tripoli, E.; Giammanco, M.; Tabacchi, G.; Di Majo, D.; Giammanco, S.; La Guardia, M. The phenolic compounds of olive oil: Structure, biological activity and beneficial effects on human health. Nutr. Res. Rev. 2005, 18, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, C.; Riguera, R. Phenylethanoid glycosides in plants: Structure and biological activity. Nat. Prod. Rep. 1994, 11, 591–606. [Google Scholar] [CrossRef]
- Fu, G.; Pang, H.; Wong, Y. Naturally Occurring Phenylethanoid Glycosides: Potential Leads for New Therapeutics. Curr. Med. Chem. 2008, 15, 2592–2613. [Google Scholar] [CrossRef] [Green Version]
- Xue, Z.; Yang, B. Phenylethanoid glycosides: Research advances in their phytochemistry, pharmacological activity and pharmacokinetics. Molecules 2016, 21, 991. [Google Scholar] [CrossRef]
- Wu, L.; Georgiev, M.I.; Cao, H.; Nahar, L.; El-Seedi, H.R.; Sarker, S.D.; Xiao, J.; Lu, B. Therapeutic potential of phenylethanoid glycosides: A systematic review. Med. Res. Rev. 2020, 40, 2605–2649. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.Y.; Li, M.X.; Lin, T.; Qiu, Y.; Zhu, Y.T.; Li, X.L.; Tao, W.D.; Wang, P.; Ren, X.X.; Chen, L.P. A review on the structure and pharmacological activity of phenylethanoid glycosides. Eur. J. Med. Chem. 2021, 209, 112563. [Google Scholar] [CrossRef]
- Lo Giudice, V.; Faraone, I.; Bruno, M.R.; Ponticelli, M.; Labanca, F.; Bisaccia, D.; Massarelli, C.; Milella, L.; Todaro, L. Olive trees by-products as sources of bioactive and other industrially useful compounds: A systematic review. Molecules 2021, 26, 5081. [Google Scholar] [CrossRef] [PubMed]
- Carrara, M.; Kelly, M.T.; Roso, F.; Larroque, M.; Margout, D. Potential of Olive Oil Mill Wastewater as a Source of Polyphenols for the Treatment of Skin Disorders: A Review. J. Agric. Food Chem. 2021, 69, 7268–7284. [Google Scholar] [CrossRef]
- Vann, K.R.; Sedgeman, C.A.; Gopas, J.; Golan-Goldhirsh, A.; Osheroff, N. Effects of Olive Metabolites on DNA Cleavage Mediated by Human Type II Topoisomerases. Biochemistry 2015, 54, 4531–4541. [Google Scholar] [CrossRef] [PubMed]
- Rietjens, S.J.; Bast, A.; Haenen, G.R.M.M. New insights into controversies on the antioxidant potential of the olive oil antioxidant hydroxytyrosol. J. Agric. Food Chem. 2007, 55, 7609–7614. [Google Scholar] [CrossRef]
- Ciriminna, R.; Meneguzzo, F.; Fidalgo, A.; Ilharco, L.M.; Pagliaro, M. Extraction, benefits and valorization of olive polyphenols. Eur. J. Lipid Sci. Technol. 2016, 118, 503–511. [Google Scholar] [CrossRef]
- Robles-Almazan, M.; Pulido-Moran, M.; Moreno-Fernandez, J.; Ramirez-Tortosa, C.; Rodriguez-Garcia, C.; Quiles, J.L.; Ramirez-Tortosa, M. Hydroxytyrosol: Bioavailability, toxicity, and clinical applications. Food Res. Int. 2018, 105, 654–667. [Google Scholar] [CrossRef]
- De las Hazas, M.C.L.; Rubio, L.; Macia, A.; Motilva, M.J. Hydroxytyrosol: Emerging Trends in Potential Therapeutic Applications. Curr. Pharm. Des. 2018, 24, 2157–2179. [Google Scholar] [CrossRef]
- Bertelli, M.; Kiani, A.K.; Paolacci, S.; Manara, E.; Kurti, D.; Dhuli, K.; Bushati, V.; Miertus, J.; Pangallo, D.; Baglivo, M.; et al. Hydroxytyrosol: A natural compound with promising pharmacological activities. J. Biotechnol. 2020, 309, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Bernini, R.; Merendino, N.; Romani, A.; Velotti, F. Naturally Occurring Hydroxytyrosol: Synthesis and Anticancer Potential. Curr. Med. Chem. 2013, 20, 655–670. [Google Scholar] [CrossRef]
- Vilaplana-Pérez, C.; Auñón, D.; García-Flores, L.A.; Gil-Izquierdo, A. Hydroxytyrosol and Potential Uses in Cardiovascular Diseases, Cancer, and AIDS. Front. Nutr. 2014, 1, 1–11. [Google Scholar] [CrossRef] [Green Version]
- De Pablos, R.M.; Espinosa-Oliva, A.M.; Hornedo-Ortega, R.; Cano, M.; Arguelles, S. Hydroxytyrosol protects from aging process via AMPK and autophagy; a review of its effects on cancer, metabolic syndrome, osteoporosis, immune-mediated and neurodegenerative diseases. Pharmacol. Res. 2019, 143, 58–72. [Google Scholar] [CrossRef]
- Turck, D.; Bresson, J.; Burlingame, B.; Dean, T.; Fairweather-Tait, S.; Heinonen, M.; Hirsch-Ernst, K.I.; Mangelsdorf, I.; McArdle, H.J.; Naska, A.; et al. Safety of hydroxytyrosol as a novel food pursuant to Regulation (EC) No 258/97. EFSA J. 2017, 15, 4728. [Google Scholar] [CrossRef]
- Annunziata, F.; Contente, M.L.; Pinna, C.; Tamborini, L.; Pinto, A. Biocatalyzed flow oxidation of tyrosol to hydroxytyrosol and efficient production of their acetate esters. Antioxidants 2021, 10, 1142. [Google Scholar] [CrossRef]
- Kalampaliki, A.D.; Giannouli, V.; Skaltsounis, A.L.; Kostakis, I.K. A three-step, gram-scale synthesis of hydroxytyrosol, hydroxytyrosol acetate, and 3,4-dihydroxyphenylglycol. Molecules 2019, 24, 3239. [Google Scholar] [CrossRef] [Green Version]
- Bender, C.; Straßmann, S.; Heidrich, P. Cellular antioxidant effects and bioavailability of food supplements rich in hydroxytyrosol. Appl. Sci. 2021, 11, 4763. [Google Scholar] [CrossRef]
- Visioli, F.; Galli, C.; Grande, S.; Colonnelli, K.; Patelli, C.; Galli, G. Nutrient metabolism—Research communication hydroxytyrosol excretion differs between rats and humans and depends on the vehicle of administration. J. Nutr. 2003, 133, 2612–2615. [Google Scholar] [CrossRef]
- Granados-Principal, S.; Quiles, J.L.; Ramirez-Tortosa, C.L.; Sanchez-Rovira, P.; Ramirez-Tortosa, M.C. Hydroxytyrosol: From laboratory investigations to future clinical trials. Nutr. Rev. 2010, 68, 191–206. [Google Scholar] [CrossRef] [Green Version]
- Bernini, R.; Gilardini Montani, M.S.; Merendino, N.; Romani, A.; Velotti, F. Hydroxytyrosol-Derived Compounds: A Basis for the Creation of New Pharmacological Agents for Cancer Prevention and Therapy. J. Med. Chem. 2015, 58, 9089–9107. [Google Scholar] [CrossRef]
- Floris, B.; Galloni, P.; Conte, V.; Sabuzi, F. Tailored functionalization of natural phenols to improve biological activity. Biomolecules 2021, 11, 1325. [Google Scholar] [CrossRef]
- Monteiro, M.; Silva, A.F.R.; Resende, D.; Braga, S.S.; Coimbra, M.A.; Silva, A.M.S.; Cardoso, S.M. Strategies to broaden the applications of olive biophenols oleuropein and hydroxytyrosol in food products. Antioxidants 2021, 10, 444. [Google Scholar] [CrossRef]
- Tassano, E.; Alama, A.; Basso, A.; Dondo, G.; Galatini, A.; Riva, R.; Banfi, L. Conjugation of Hydroxytyrosol with Other Natural Phenolic Fragments: From Waste to Antioxidants and Antitumour Compounds. Eur. J. Org. Chem. 2015, 2015, 6710–6726. [Google Scholar] [CrossRef]
- Guo, Y.; Zheng, C.; Xu, W.; Si, Y.; Dou, S.; Yang, Y. Free radical scavenging and hepatoprotective effects of salidroside analogs on CCl4-induced cytotoxicity in LO2 cells. Med. Chem. Res. 2013, 22, 2524–2530. [Google Scholar] [CrossRef]
- Horvathova, E.; Mastihubova, M.; Karnisova Potocka, E.; Kis, P.; Galova, E.; Sevcovicova, A.; Klapakova, M.; Hunakova, L.; Mastihuba, V. Comparative study of relationship between structure of phenylethanoid glycopyranosides and their activities using cell-free assays and human cells cultured in vitro. Toxicol. Vitr. 2019, 61, 104646. [Google Scholar] [CrossRef]
- Kis, P.; Mastihubová, M. A sustainable approach to phenylethanoid glycopyranosides: Study of glycosylations promoted by zinc salts. Sustain. Chem. Pharm. 2021, 24, 100537. [Google Scholar] [CrossRef]
- Mulani, S.K.; Guh, J.H.; Mong, K.K.T. A general synthetic strategy and the anti-proliferation properties on prostate cancer cell lines for natural phenylethanoid glycosides. Org. Biomol. Chem. 2014, 12, 2926–2937. [Google Scholar] [CrossRef]
- Shu, P.; Zhang, L.; Liu, A.; Li, J.; Liu, Q.; Sun, N.; Zhang, Y.; Wei, X.; Cui, M.; Ju, Z.; et al. Six Natural Phenylethanoid Glycosides: Total Synthesis, Antioxidant and Tyrosinase Inhibitory Activities. ChemistrySelect 2020, 5, 10817–10820. [Google Scholar] [CrossRef]
- Trincone, A.; Pagnotta, E.; Tramice, A. Enzymatic routes for the production of mono- and di-glucosylated derivatives of hydroxytyrosol. Bioresour. Technol. 2012, 115, 79–83. [Google Scholar] [CrossRef]
- Nieto-Domínguez, M.; De Eugenio, L.I.; Peñalver, P.; Belmonte-Reche, E.; Morales, J.C.; Poveda, A.; Jiménez-Barbero, J.; Prieto, A.; Plou, F.J.; Martínez, M.J. Enzymatic Synthesis of a Novel Neuroprotective Hydroxytyrosyl Glycoside. J. Agric. Food Chem. 2017, 65, 10526–10533. [Google Scholar] [CrossRef]
- Potocká, E.; Mastihubová, M.; Mastihuba, V. Enzymatic synthesis of tyrosol glycosides. J. Mol. Catal. B Enzym. 2015, 113, 23–28. [Google Scholar] [CrossRef]
- Karnišová Potocká, E.; Mastihubová, M.; Mastihuba, V. Enzymatic synthesis of tyrosol and hydroxytyrosol β-D-fructofuranosides. Biocatal. Biotransform. 2019, 37, 18–24. [Google Scholar] [CrossRef]
- Karnišová Potocká, E.; Mastihubová, M.; Mastihuba, V. Transrutinosylation of tyrosol by flower buds of Sophora japonica. Food Chem. 2021, 336, 127674. [Google Scholar] [CrossRef]
- Hollá, V.; Hill, R.; Antošová, M.; Polakovič, M. Design of immobilized biocatalyst and optimal conditions for tyrosol β-galactoside production. Bioprocess Biosyst. Eng. 2021, 44, 93–101. [Google Scholar] [CrossRef]
- Hollá, V.; Karkeszová, K.; Antošová, M.; Polakovič, M. Transglycosylation properties of a Kluyveromyces lactis enzyme preparation: Production of tyrosol β-fructoside using free and immobilized enzyme. Process Biochem. 2021, 110, 168–175. [Google Scholar] [CrossRef]
- Míguez, N.; Ramírez-Escudero, M.; Gimeno-Pérez, M.; Poveda, A.; Jiménez-Barbero, J.; Ballesteros, A.O.; Fernández-Lobato, M.; Sanz-Aparicio, J.; Plou, F.J. Fructosylation of Hydroxytyrosol by the β-Fructofuranosidase from Xanthophyllomyces dendrorhous: Insights into the Molecular Basis of the Enzyme Specificity. ChemCatChem 2018, 10, 4892–4901. [Google Scholar] [CrossRef]
- Khymenets, O.; Joglar, J.; Clapés, P.; Parella, T.; Covas, M.I.; De La Torre, R. Biocatalyzed synthesis and structural characterization of monoglucuronides of hydroxytyrosol, tyrosol, homovanillic alcohol, and 3-(4′-hydroxyphenyl) propanol. Adv. Synth. Catal. 2006, 348, 2155–2162. [Google Scholar] [CrossRef]
- Bassanini, I.; Krejzová, J.; Panzeri, W.; Monti, D.; Křen, V.; Riva, S. A Sustainable One-Pot, Two-Enzyme Synthesis of Naturally Occurring Arylalkyl Glucosides. ChemSusChem 2017, 10, 2040–2045. [Google Scholar] [CrossRef]
- Green, J.W. The Glycofuranosides. Adv. Carbohydr. Chem. Biochem. 1967, 21, 95–142. [Google Scholar] [CrossRef]
- Taha, H.A.; Richards, M.R.; Lowary, T.L. Conformational analysis of furanoside-containing mono- and oligosaccharides. Chem. Rev. 2013, 113, 1851–1876. [Google Scholar] [CrossRef]
- Rahim, M.A.; Saeed, F.; Khalid, W.; Hussain, M.; Anjum, F.M. Functional and nutraceutical properties of fructo-oligosaccharides derivatives: A review. Int. J. Food Prop. 2021, 24, 1588–1602. [Google Scholar] [CrossRef]
- Konishi, T.; Ishii, T. The Origin and Functions of Arabinofuranosyl Residues in Plant Cell Walls. Trends Glycosci. Glycotechnol. 2012, 24, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Pičmanová, M.; Møller, B.L. Apiose: One of nature’s witty games. Glycobiology 2016, 26, 430–442. [Google Scholar] [CrossRef] [Green Version]
- Peltier, P.; Euzen, R.; Daniellou, R.; Nugier-Chauvin, C.; Ferrières, V. Recent knowledge and innovations related to hexofuranosides: Structure, synthesis and applications. Carbohydr. Res. 2008, 343, 1897–1923. [Google Scholar] [CrossRef] [Green Version]
- Chlubnova, I.; Legentil, L.; Dureau, R.; Pennec, A.; Almendros, M.; Daniellou, R.; Nugier-Chauvin, C.; Ferrières, V. Specific and non-specific enzymes for furanosyl-containing conjugates: Biosynthesis, metabolism, and chemo-enzymatic synthesis. Carbohydr. Res. 2012, 356, 44–61. [Google Scholar] [CrossRef]
- Dumon, C.; Song, L.; Bozonnet, S.; Fauré, R.; O’Donohue, M.J. Progress and future prospects for pentose-specific biocatalysts in biorefining. Process Biochem. 2012, 47, 346–357. [Google Scholar] [CrossRef]
- Numan, M.T.; Bhosle, N.B. α-L-arabinofuranosidases: The potential applications in biotechnology. J. Ind. Microbiol. Biotechnol. 2006, 33, 247–260. [Google Scholar] [CrossRef]
- Manoochehri, H.; Hosseini, N.F.; Saidijam, M.; Taheri, M.; Rezaee, H.; Nouri, F. A review on invertase: Its potentials and applications. Biocatal. Agric. Biotechnol. 2020, 25, 101599. [Google Scholar] [CrossRef]
- Karnišová Potocká, E.; Mastihubová, M.; Mastihuba, V. Apiose-relevant glycosidases. Catalysts 2021, 11, 1251. [Google Scholar] [CrossRef]
- Kaur, A.P.; Bhardwaj, S.; Dhanjal, D.S.; Nepovimova, E.; Cruz-Martins, N.; Kuča, K.; Chopra, C.; Singh, R.; Kumar, H.; Șen, F.; et al. Plant prebiotics and their role in the amelioration of diseases. Biomolecules 2021, 11, 440. [Google Scholar] [CrossRef]
- Torres-Mendoza, D.; González, J.; Ortega-Barría, E.; Heller, M.V.; Capson, T.L.; McPhail, K.; Gerwick, W.H.; Cubilla-Rios, L. Weakly Antimalarial Flavonol Arabinofuranosides from Calycolpus warszewiczianus. J. Nat. Prod. 2006, 69, 826–828. [Google Scholar] [CrossRef]
- Park, Y.J.; Kim, D.M.; Jeong, M.H.; Yu, J.S.; So, H.M.; Bang, I.J.; Kim, H.R.; Kwon, S.H.; Kim, K.H.; Chung, K.H. (−)-Catechin-7-O-β-D-Apiofuranoside Inhibits Hepatic Stellate Cell Activation by Suppressing the STAT3 Signaling Pathway. Cells 2019, 9, 30. [Google Scholar] [CrossRef] [Green Version]
- Mastihubová, M.; Poláková, M. A selective and mild glycosylation method of natural phenolic alcohols. Beilstein J. Org. Chem. 2016, 12, 524–530. [Google Scholar] [CrossRef] [Green Version]
- Dudíková, J.; Mastihubová, M.; Mastihuba, V.; Kolarova, N. Exploration of transfructosylation activity in cell walls from Cryptococcus laurentii for production of functionalised β-D-fructofuranosides. J. Mol. Catal. B Enzym. 2007, 45, 27–33. [Google Scholar] [CrossRef]
- Kis, P.; Potocká, E.; Mastihuba, V.; Mastihubová, M. Efficient chemoenzymatic synthesis of 4-nitrophenyl β-D-apiofuranoside and its use in screening of β-D-apiofuranosidases. Carbohydr. Res. 2016, 430, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Mastihuba, V.; Karnišová Potocká, E.; Uhliariková, I.; Kis, P.; Kozmon, S.; Mastihubová, M. Reaction mechanism of β-apiosidase from Aspergillus aculeatus. Food Chem. 2019, 274, 543–546. [Google Scholar] [CrossRef]
- Imamura, A.; Lowary, T. Chemical Synthesis of Furanose Glycosides. Trends Glycosci. Glycotechnol. 2011, 23, 134–152. [Google Scholar] [CrossRef] [Green Version]
- Gallo-Rodriguez, C.; Kashiwagi, G.A. Selective Glycosylations with Furanosides. In Selective Glycosylation: Synthetic Methods and Catalysts; Bennett, C.S., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2017; pp. 297–326. ISBN 9783527696239. [Google Scholar]
- Backinowsky, L.V.; Nepogod’ev, S.A.; Shashkov, A.S.; Kochetkov, N.K. 1,2-O-cyanoalkylidene derivatives of furanoses as 1,2-trans-glycosylating agents. Carbohydr. Res. 1985, 138, 41–54. [Google Scholar] [CrossRef]
- Ho, P.-T. Branched-chain sugars. Reaction of furanoses with formaldehyde: A simple synthesis of D- and L-apiose. Can. J. Chem. 1979, 57, 381–383. [Google Scholar] [CrossRef] [Green Version]
- Hammerschmidt, F.; Öhler, E.; Polsterer, J.-P.; Zbiral, E.; Balzarini, J.; Declercq, E. Ein einfacher Weg zu D-Apio-β-D-furanosyl- und 2′-Desoxyapio-β-D-furanosylnucleosiden. Liebigs Ann. 1995, 1995, 551–558. [Google Scholar] [CrossRef]
- Kam, B.L.; Barascut, J.-L.; Imbach, J.-L. A general method of synthesis and isolation, and an n.m.r-spectroscopic study, of tetra-O-acetyl-D-aldopentofuranoses. Carbohydr. Res. 1979, 69, 135–142. [Google Scholar] [CrossRef]
- Gravier-Pelletier, C.; Ginisty, M.; Le Merrer, Y. A versatile scaffold for a library of liposidomycins analogues: A crucial and potent glycosylation step. Tetrahedron Asymmetry 2004, 15, 189–193. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Fotakis, G.; Timbrell, J.A. In vitro cytotoxicity assays: Comparison of LDH, neutral red, MTT and protein assay in hepatoma cell lines following exposure to cadmium chloride. Toxicol. Lett. 2006, 160, 171–177. [Google Scholar] [CrossRef]
- Tutino, V.; Caruso, M.G.; Messa, C.; Perri, E.; Notarnicola, M. Antiproliferative, antioxidant and anti-inflammatory effects of hydroxytyrosol on human hepatoma HepG2 and Hep3B cell lines. Anticancer Res. 2012, 32, 5371–5377. [Google Scholar]
- Kuwajima, H.; Takai, Y.; Takaishi, K.; Inoue, K. Synthesis of 13C-Labeled Possible Intermediates in the Biosynthesis of Phenylethanoid Derivatives, Cornoside and Rengyosides. Chem. Pharm. Bull. 1998, 46, 581–586. [Google Scholar] [CrossRef] [Green Version]
- Yen, G.C.; Chen, H.Y. Antioxidant Activity of Various Tea Extracts in Relation to Their Antimutagenicity. J. Agric. Food Chem. 1995, 43, 27–32. [Google Scholar] [CrossRef]
- Zhao, J.; Liu, T.; Ma, L.; Yan, M.; Gu, Z.; Huang, Y.; Xu, F.; Zhao, Y. Antioxidant and preventive effects of extract from Nymphaea candida flower on in vitro immunological liver injury of rat primary hepatocyte cultures. Evid. Based Complement. Altern. Med. 2011, 2011, 497673. [Google Scholar] [CrossRef] [Green Version]
- Sharma, O.P.; Bhat, T.K. DPPH antioxidant assay revisited. Food Chem. 2009, 113, 1202–1205. [Google Scholar] [CrossRef]
- Čipák, L.; Miadoková, E.; Dingová, H.; Kogan, G.; Novotný, L.; Rauko, P. Comparative DNA protectivity and antimutagenicity studies using DNA-topology and Ames assays. Toxicol. Vitr. 2001, 15, 677–681. [Google Scholar] [CrossRef]
- Majer, B.J.; Mersch-Sundermann, V.; Darroudi, F.; Laky, B.; de Wit, K.; Knasmüller, S. Genotoxic effects of dietary and lifestyle related carcinogens in human derived hepatoma (HepG2, Hep3B) cells. Mutat. Res. 2004, 551, 153–166. [Google Scholar] [CrossRef]
- Singh, N.P.; McCoy, M.T.; Tice, R.R.; Schneider, E.L. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp. Cell Res. 1988, 175, 184–191. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Donor | Heating 1 ZnCO3 (equiv) | Time (min) | Product | Yield 3 (%) | α:β 4 |
|---|---|---|---|---|---|---|
| 1 | 13 | Δ, 0.44 | 80 | 16 | 70 | α only |
| 2 | 13 | MW, 0.35 | 6 | 16 | 30 | 75:25 |
| 3 | 14 | Δ, 0.44 | 50 | 17 | 67 | β only |
| 4 | 14 | MW, 0.44 | 6 | 17 | 61 | 21:79 |
| 5 | 14 | MW 2, 0.44 | 2.3 | 17 | 37 | 12:88 |
| 6 | 15 | Δ, 0.44 | 50 | 18 | 67 | β only |
| 7 | 15 | MW, 0.44 | 6 | 18 | 48 | 20:80 |
| Compound | Concentrations (mM) | Reducing Power 1 | DPPH Scavenging 1 |
|---|---|---|---|
| (Absorbance) | (%) | ||
| T (1) | 10 | 0.089 ± 0.042 | 6.710 ± 2.907 |
| 1 | 0.094 ± 0.012 | 2.120 ± 1.310 | |
| 0.1 | 0.048 ± 0.027 | 0.955 ± 0.090 | |
| 0.01 | 0.051 ± 0.043 | 0.000 ± 0.000 | |
| HOT (2) | 10 | nd | 82.710 ± 8.200 |
| 1 | 1.891 ± 0.702 | 81.137 ± 7.947 | |
| 0.1 | 0.259 ± 0.054 | 15.763 ± 1.488 | |
| 0.01 | 0.046 ± 0.040 | 0.000 ± 0.000 | |
| TYBFRU (5a) | 10 | 0.147 ± 0.038 | 3.920 ± 1.240 |
| 1 | 0.099 ± 0.037 | 1.410 ± 0.040 | |
| 0.1 | 0.051 ± 0.029 | 0.000 ± 0.000 | |
| 0.01 | 0.051 ± 0.035 | 0.000 ± 0.000 | |
| HOTFRU (5b) | 10 | nd | 84.170 ± 14.467 |
| 1 | 1.788 ± 0.227 | 78.980 ± 13.150 | |
| 0.1 | 0.285 ± 0.001 | 14.260 ± 20.167 | |
| 0.01 | 0.116 ± 0.051 | 5.870 ± 8.301 | |
| HOTARA (6) | 10 | nd | 81.788 ± 2.861 |
| 1 | 1.527 ± 0.589 | 73.895 ± 7.768 | |
| 0.1 | 0.276 ± 0.093 | 5.600 ± 3.652 | |
| 0.01 | 0.167 ± 0.122 | 2.143 ± 3.439 | |
| HOTAPI (7) | 10 | nd | 82.513 ± 3.338 |
| 1 | 1.883 ± 0.923 | 74.678 ± 11.425 | |
| 0.1 | 0.274 ± 0.081 | 5.230 ± 3.775 | |
| 0.01 | 0.124 ± 0.038 | 1.265 ± 1.189 | |
| HOTRIB (8) | 10 | nd | 83.210 ± 9.270 |
| 1 | 2.304 ± 0.045 | 75.440 ± 19.640 | |
| 0.1 | 0.364 ± 0.045 | 16.950 ± 10.530 | |
| 0.01 | 0.135 ± 0.057 | 9.615 ± 13.600 | |
| GA | 10 | nd | 91.609 ± 6.011 |
| 1 | 2.151 ± 0.652 | 88.454 ± 6.240 | |
| 0.1 | 0.309 ± 0.010 | 25.720 ± 11.839 | |
| 0.01 | 0.108 ± 0.064 | 3.088 ± 4.963 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kis, P.; Horváthová, E.; Gálová, E.; Ševčovičová, A.; Antalová, V.; Karnišová Potocká, E.; Mastihuba, V.; Mastihubová, M. Synthesis of Tyrosol and Hydroxytyrosol Glycofuranosides and Their Biochemical and Biological Activities in Cell-Free and Cellular Assays. Molecules 2021, 26, 7607. https://doi.org/10.3390/molecules26247607
Kis P, Horváthová E, Gálová E, Ševčovičová A, Antalová V, Karnišová Potocká E, Mastihuba V, Mastihubová M. Synthesis of Tyrosol and Hydroxytyrosol Glycofuranosides and Their Biochemical and Biological Activities in Cell-Free and Cellular Assays. Molecules. 2021; 26(24):7607. https://doi.org/10.3390/molecules26247607
Chicago/Turabian StyleKis, Peter, Eva Horváthová, Eliška Gálová, Andrea Ševčovičová, Veronika Antalová, Elena Karnišová Potocká, Vladimír Mastihuba, and Mária Mastihubová. 2021. "Synthesis of Tyrosol and Hydroxytyrosol Glycofuranosides and Their Biochemical and Biological Activities in Cell-Free and Cellular Assays" Molecules 26, no. 24: 7607. https://doi.org/10.3390/molecules26247607
APA StyleKis, P., Horváthová, E., Gálová, E., Ševčovičová, A., Antalová, V., Karnišová Potocká, E., Mastihuba, V., & Mastihubová, M. (2021). Synthesis of Tyrosol and Hydroxytyrosol Glycofuranosides and Their Biochemical and Biological Activities in Cell-Free and Cellular Assays. Molecules, 26(24), 7607. https://doi.org/10.3390/molecules26247607