
Genome Mining of Pseudomonas Species: Diversity and Evolution of Metabolic and Biosynthetic Potential

 , , , and
, , , and
Abstract
1. Introduction
2. Results
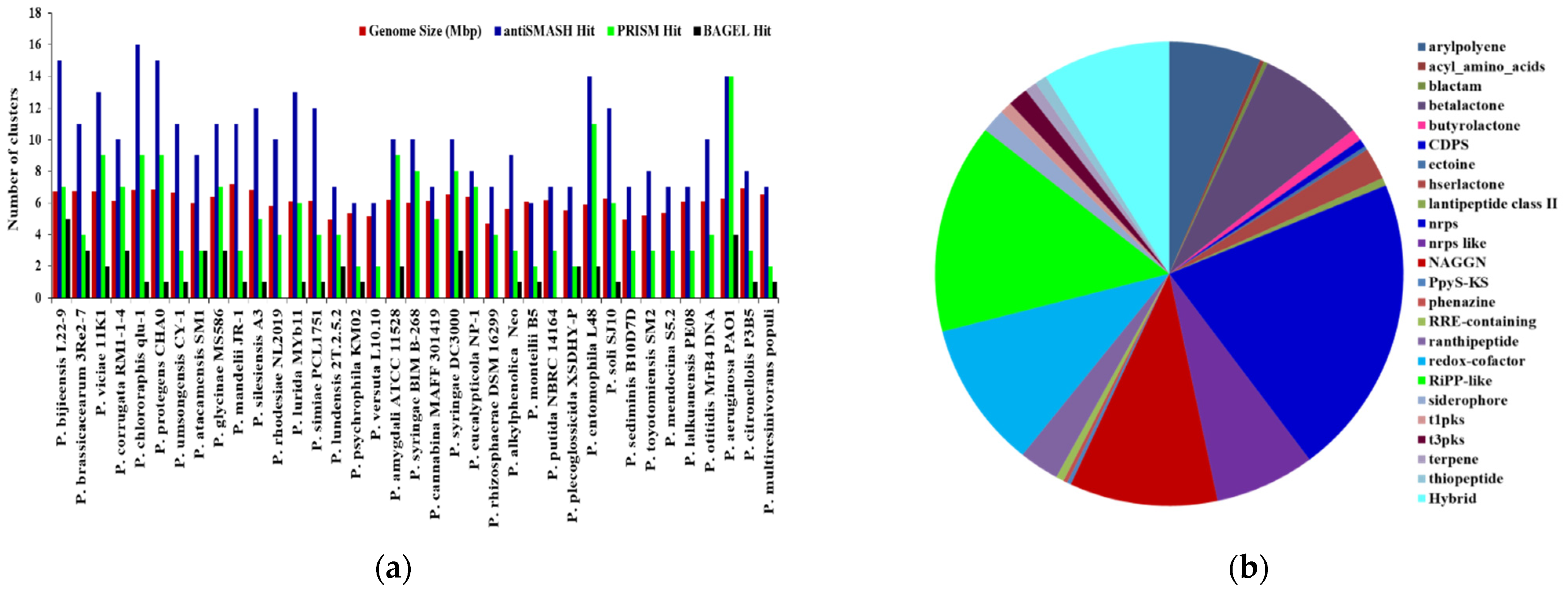
2.1. Distribution and Diversity of Biosynthetic Potential in Pseudomonas at Species Level
2.1.1. Putative BGC Prediction by antiSMASH in Pseudomonas Species Genomes
2.1.2. Putative BGC Prediction by PRISM in Pseudomonas Species Genomes
2.1.3. Putative BGC Prediction by BAGEL in Pseudomonas Species Genomes
2.1.4. KS and C Domain Determination in the Pseudomonas Genus Using NaPDoS
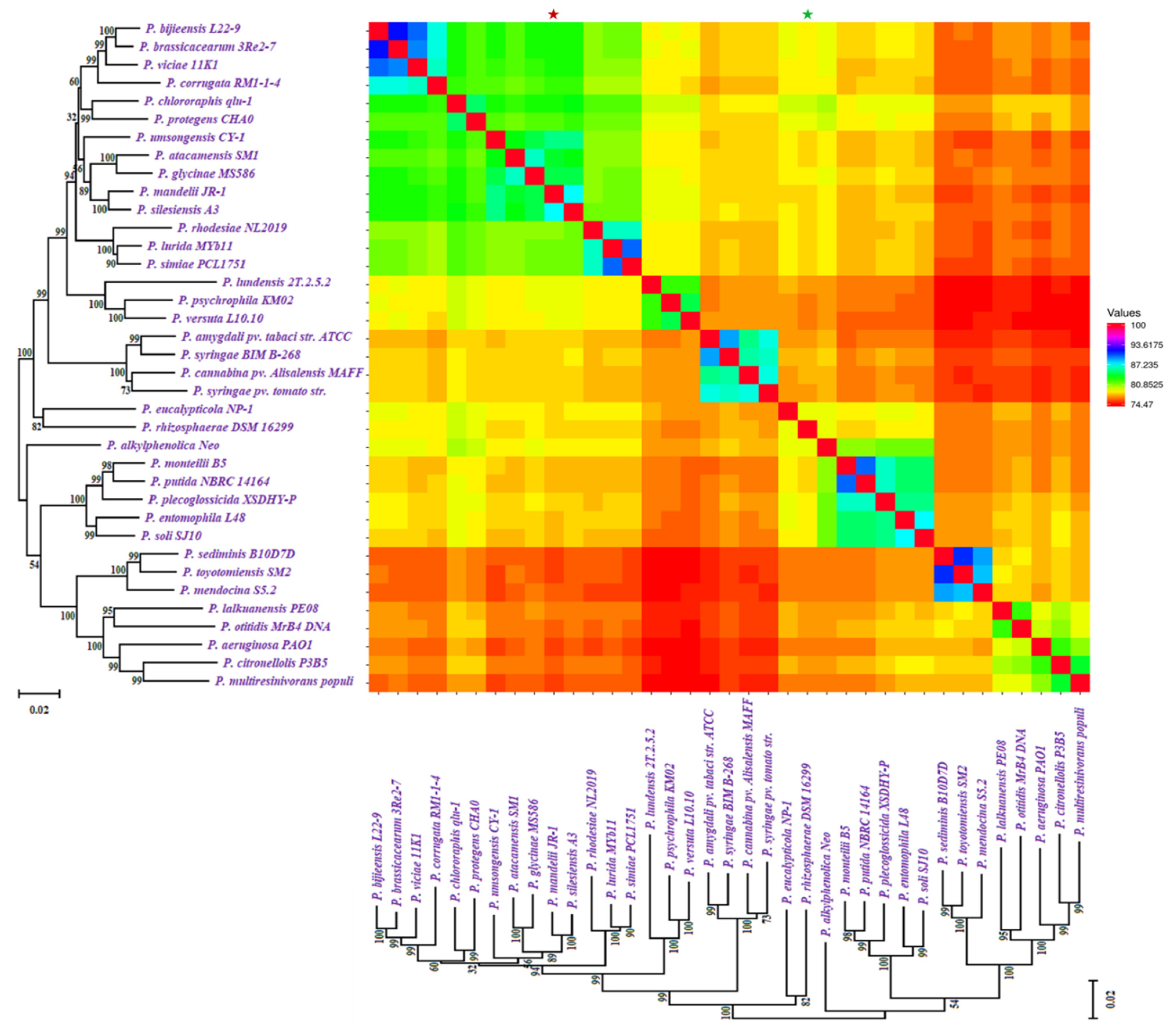
2.2. Whole-Genome Comparisons in Pseudomonas Species
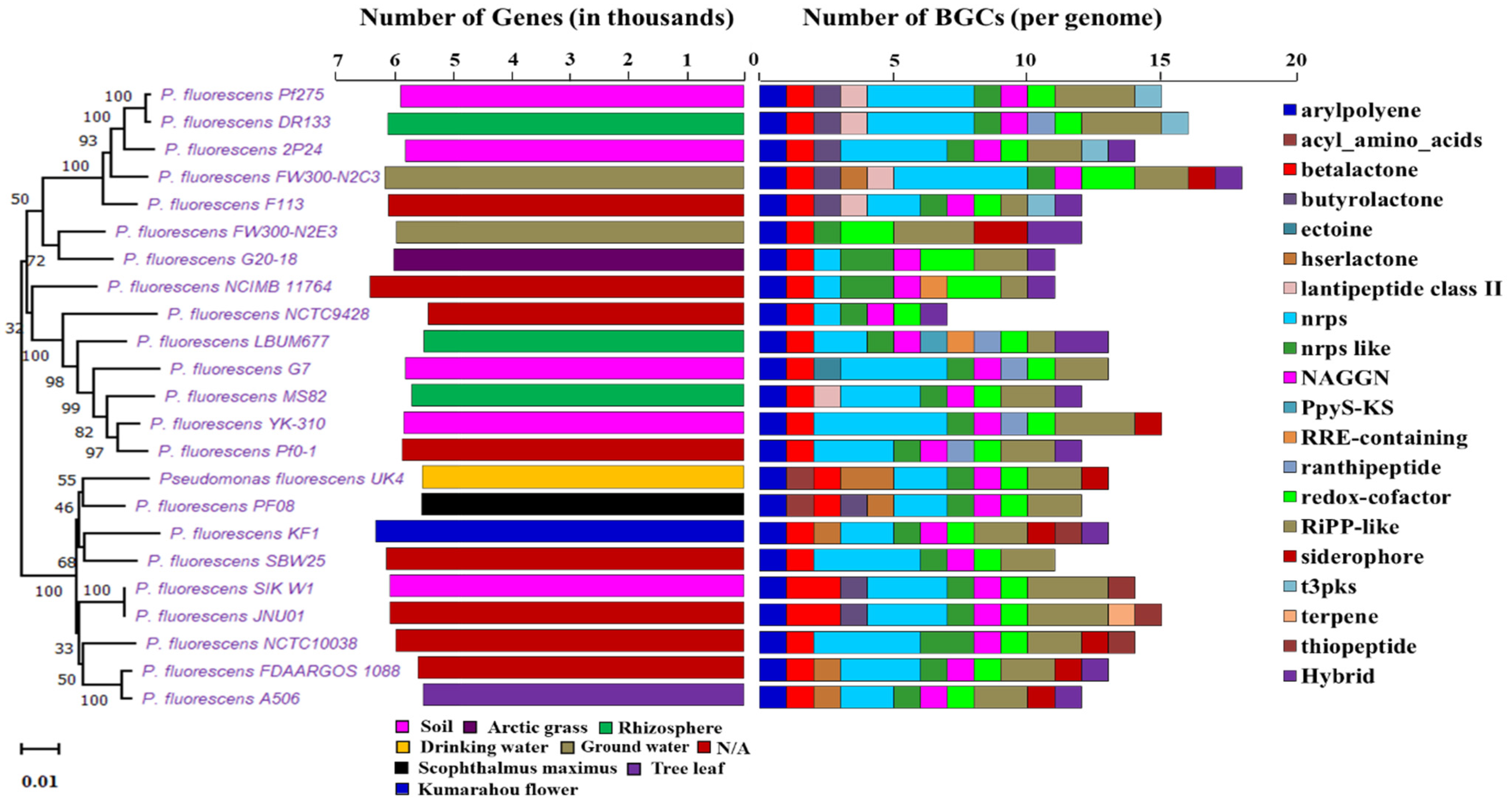
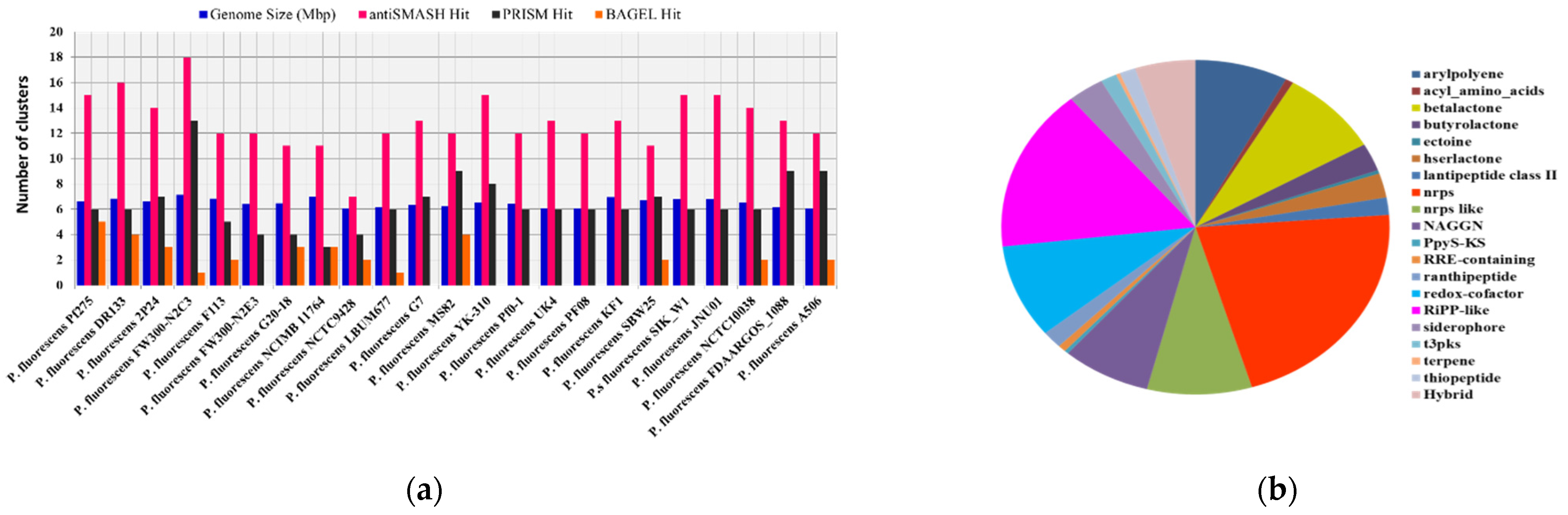
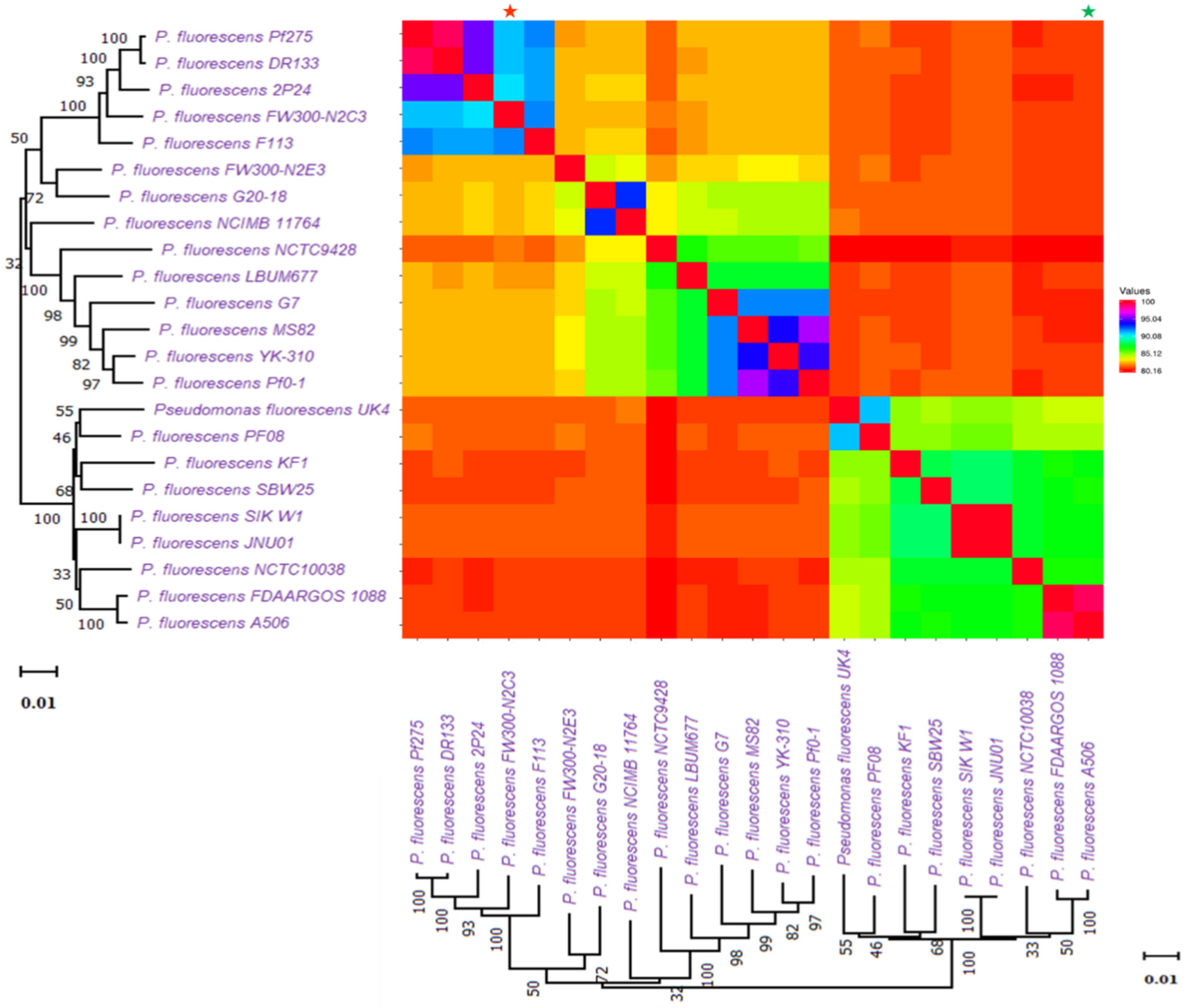
2.3. Distribution and Evolution of Secondary Metabolites in Pseudomonas fluorescence at Subspecies Level
3. Discussion
4. Materials and Methods
4.1. Collection of Genome Sequences
4.2. Phylogeny and Whole Genome Comparisons
4.3. Computational Approaches for the Identification of Gene Clusters Potentially Encoding Secondary Metabolites
4.3.1. antiSMASH 6.0
4.3.2. PRISM 4
4.3.3. BAGEL4
4.3.4. NaPDoS-Analysis of C and KS Domains from NRPS and PKS Clusters
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Condurso, H.L.; Bruner, S.D. Structure and noncanonical chemistry of nonribosomal peptide biosynthetic machinery. Nat. Prod. Rep. 2012, 29, 1099–1110. [Google Scholar] [CrossRef]
- Hur, G.H.; Vickery, C.R.; Burkart, M.D. Explorations of catalytic domains in non-ribosomal peptide synthetase enzymology. Nat. Prod. Rep. 2012, 29, 1074–1098. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.T. Insights into the chemical logic and enzymatic machinery of NRPS assembly lines. Nat. Prod. Rep. 2016, 33, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Hertweck, C. The biosynthetic logic of polyketide diversity. Angew. Chem. Int. Ed. 2009, 48, 4688–4716. [Google Scholar] [CrossRef]
- Piel, J. Biosynthesis of polyketides by trans-AT polyketide synthases. Nat. Prod. Rep. 2010, 27, 996–1047. [Google Scholar] [CrossRef] [PubMed]
- Dunbar, K.L.; Mitchell, D.A. Revealing nature’s synthetic potential through the study of ribosomal natural product biosynthesis. ACS Chem. Biol. 2013, 8, 473–487. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Arnison, P.G.; Bibb, M.J.; Bierbaum, G.; Bowers, A.A.; Bugni, T.S.; Bulaj, G.; Camarero, J.A.; Campopiano, D.J.; Challis, G.L.; Clardy, J. Ribosomally synthesized and post-translationally modified peptide natural products: Overview and recommendations for a universal nomenclature. Nat. Prod. Rep. 2013, 30, 108–160. [Google Scholar] [CrossRef] [PubMed]
- Letzel, A.-C.; Pidot, S.J.; Hertweck, C. Genome mining for ribosomally synthesized and post-translationally modified peptides (RiPPs) in anaerobic bacteria. BMC Genom. 2014, 15, 983. [Google Scholar] [CrossRef]
- Hudson, G.A.; Mitchell, D.A. RiPP antibiotics: Biosynthesis and engineering potential. Curr. Opin. Microbiol. 2018, 45, 61–69. [Google Scholar] [CrossRef] [PubMed]
- McCranie, E.K.; Bachmann, B.O. Bioactive oligosaccharide natural products. Nat. Prod. Rep. 2014, 31, 1026–1042. [Google Scholar] [CrossRef] [PubMed]
- Flatt, P.M.; Mahmud, T. Biosynthesis of aminocyclitol-aminoglycoside antibiotics and related compounds. Nat. Prod. Rep. 2007, 24, 358–392. [Google Scholar] [CrossRef]
- Massingill, J.L., Jr.; Hodgkins, J.E. Alkaloids of bacteria. Phytochemistry 1967, 6, 977–982. [Google Scholar] [CrossRef]
- Zotchev, S.B. Alkaloids from marine bacteria. Adv. Bot. Res. 2013, 68, 301–333. [Google Scholar]
- Mullowney, M.W.; McClure, R.A.; Robey, M.T.; Kelleher, N.L.; Thomson, R.J. Natural products from thioester reductase containing biosynthetic pathways. Nat. Prod. Rep. 2018, 35, 847–878. [Google Scholar] [CrossRef] [PubMed]
- Cane, D.E.; Ikeda, H. Exploration and mining of the bacterial terpenome. Acc. Chem. Res. 2012, 45, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Christianson, D.W. Structural biology and chemistry of the terpenoid cyclases. Chem. Rev. 2006, 106, 3412–3442. [Google Scholar] [CrossRef] [PubMed]
- Baunach, M.; Franke, J.; Hertweck, C. Terpenoid biosynthesis off the beaten track: Unconventional cyclases and their impact on biomimetic synthesis. Angew. Chem. Int. Ed. 2015, 54, 2604–2626. [Google Scholar] [CrossRef]
- Pham, J.V.; Yilma, M.A.; Feliz, A.; Majid, M.T.; Maffetone, N.; Walker, J.R.; Kim, E.; Cho, H.J.; Reynolds, J.M.; Song, M.C. A review of the microbial production of bioactive natural products and biologics. Front. Microbiol. 2019, 10, 1404. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [PubMed]
- Osbourn, A. Secondary metabolic gene clusters: Evolutionary toolkits for chemical innovation. Trends Genet. 2010, 26, 449–457. [Google Scholar] [CrossRef]
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations. 2016. Available online: https://lordslibrary.parliament (accessed on 31 August 2021).
- Miethke, M.; Pieroni, M.; Weber, T.; Brönstrup, M.; Hammann, P.; Halby, L.; Arimondo, P.B.; Glaser, P.; Aigle, B.; Bode, H.B. Towards the sustainable discovery and development of new antibiotics. Nat. Rev. Chem. 2021, 5, 726–749. [Google Scholar] [CrossRef]
- Katz, L.; Baltz, R.H. Natural product discovery: Past, present, and future. J. Ind. Microbiol. Biotechnol. 2016, 43, 155–176. [Google Scholar] [CrossRef]
- Doroghazi, J.R.; Albright, J.C.; Goering, A.W.; Ju, K.-S.; Haines, R.R.; Tchalukov, K.A.; Labeda, D.P.; Kelleher, N.L.; Metcalf, W.W. A roadmap for natural product discovery based on large-scale genomics and metabolomics. Nat. Chem. Biol. 2014, 10, 963–968. [Google Scholar] [CrossRef] [PubMed]
- Cimermancic, P.; Medema, M.H.; Claesen, J.; Kurita, K.; Brown, L.C.W.; Mavrommatis, K.; Pati, A.; Godfrey, P.A.; Koehrsen, M.; Clardy, J.; et al. Insights into Secondary Metabolism from a Global Analysis of Prokaryotic Biosynthetic Gene Clusters. Cell 2010, 158, 412–421. [Google Scholar] [CrossRef]
- Medema, M.H.; Fischbach, M.A. Computational approaches to natural product discovery. Nat. Chem. Biol. 2015, 11, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Ziemert, N.; Alanjary, M.; Weber, T. The evolution of genome mining in microbes—A review. Nat. Prod. Rep. 2016, 33, 988–1005. [Google Scholar] [CrossRef]
- Medema, M.H.; Cimermancic, P.; Sali, A.; Takano, E.; Fischbach, M.A. A systematic computational analysis of biosynthetic gene cluster evolution: Lessons for engineering biosynthesis. PLoS Comput. Biol. 2014, 10, e1004016. [Google Scholar] [CrossRef]
- Ziemert, N.; Lechner, A.; Wietz, M.; Millán-Aguiñaga, N.; Chavarria, K.L.; Jensen, P.R. Diversity and evolution of secondary metabolism in the marine actinomycete genus Salinispora. Proc. Natl. Acad. Sci. USA 2014, 111, E1130–E1139. [Google Scholar] [CrossRef] [PubMed]
- Jun, S.-R.; Wassenaar, T.M.; Nookaew, I.; Hauser, L.; Wanchai, V.; Land, M.; Timm, C.M.; Lu, T.-Y.S.; Schadt, C.W.; Doktycz, M.J. Diversity of Pseudomonas genomes, including Populus-associated isolates, as revealed by comparative genome analysis. Appl. Environ. Microbiol. 2016, 82, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Tartor, Y.H.; Gharieb, R.; El-Aziz, A.; Norhan, K.; El Damaty, H.M.; Enany, S.; Khalifa, E.; Attia, A.S.A.; Abdellatif, S.S.; Ramadan, H. Virulence Determinants and Plasmid-Mediated Colistin Resistance mcr Genes in Gram-Negative Bacteria Isolated from Bovine Milk. Front. Cell. Infect. Microbiol. 2021, 11, 761417. [Google Scholar] [CrossRef]
- Moore, E.B.; Tindall, B.; Martins Dos Santos, V.A.P.; Pieper, D.; Ramos, J.-L.; Palleroni, N. Nonmedical: Pseudomonas. Prokaryotes 2006, 6, 646–703. [Google Scholar]
- Weston, D.J.; Pelletier, D.A.; Morrell-Falvey, J.L.; Tschaplinski, T.J.; Jawdy, S.S.; Lu, T.-Y.; Allen, S.M.; Melton, S.J.; Martin, M.Z.; Schadt, C.W. Pseudomonas fluorescens induces strain-dependent and strain-independent host plant responses in defense networks, primary metabolism, photosynthesis, and fitness. Mol. Plant-Microbe Interact. 2012, 25, 765–778. [Google Scholar] [CrossRef]
- Jahanshah, G.; Yan, Q.; Gerhardt, H.; Pataj, Z.; Lämmerhofer, M.; Pianet, I.; Josten, M.; Sahl, H.-G.; Silby, M.W.; Loper, J.E. Discovery of the cyclic lipopeptide gacamide A by genome mining and repair of the defective GacA regulator in Pseudomonas fluorescens Pf0-1. J. Nat. Prod. 2019, 82, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Van Der Voort, M.; Meijer, H.; Schmidt, Y.; Watrous, J.; Dekkers, E.; Mendes, R.; Dorrestein, P.C.; Gross, H.; Raaijmakers, J.M. Genome mining and metabolic profiling of the rhizosphere bacterium Pseudomonas sp. SH-C52 for antimicrobial compounds. Front. Microbiol. 2015, 6, 693. [Google Scholar] [CrossRef]
- Kirchner, N.; Cano-Prieto, C.; Schulz-Fincke, A.-C.; Gütschow, M.; Ortlieb, N.; Moschny, J.; Niedermeyer, T.H.J.; Horak, J.; Lämmerhofer, M.; Van Der Voort, M. Discovery of Thanafactin A, a Linear, Proline-Containing Octalipopeptide from Pseudomonas sp. SH-C52, Motivated by Genome Mining. J. Nat. Prod. 2020, 84, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Chlebek, D.; Pinski, A.; Żur, J.; Michalska, J.; Hupert-Kocurek, K. Genome Mining and Evaluation of the Biocontrol Potential of Pseudomonas fluorescens BRZ63, a New Endophyte of Oilseed Rape (Brassica napus L.) against Fungal Pathogens. Int. J. Mol. Sci. 2020, 21, 8740. [Google Scholar] [CrossRef] [PubMed]
- Loeschcke, A.; Thies, S. Pseudomonas putida—A versatile host for the production of natural products. Appl. Microbiol. Biotechnol. 2015, 99, 6197–6214. [Google Scholar] [CrossRef]
- Aiman, S.; Shehroz, M.; Munir, M.; Gul, S.; Shah, M.; Khan, A. Species-wide genome mining of Pseudomonas putida for potential secondary metabolites and drug-like natural products characterization. J. Proteom. Bioinform 2018, 11, 1–7. [Google Scholar] [CrossRef]
- Zotchev, S.B. Genomics-based insights into the evolution of secondary metabolite biosynthesis in actinomycete bacteria. In Evolutionary Biology: Genome Evolution, Speciation, Coevolution and Origin of Life; Springer: Berlin/Heidelberg, Germany, 2014; pp. 35–45. [Google Scholar]
- Gallagher, K.A.; Jensen, P.R. Genomic insights into the evolution of hybrid isoprenoid biosynthetic gene clusters in the MAR4 marine streptomycete clade. BMC Genom. 2015, 16, 960. [Google Scholar] [CrossRef] [PubMed]
- Khaldi, N.; Collemare, J.; Lebrun, M.-H.; Wolfe, K.H. Evidence for horizontal transfer of a secondary metabolite gene cluster between fungi. Genome Biol. 2008, 9, R18. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, A.T.; Elrefaiy, M.A.; Arakawa, K. Computational prediction of the mode of binding of antitumor lankacidin C to tubulin. ACS Omega 2019, 4, 4461–4471. [Google Scholar] [CrossRef] [PubMed]
- Fischbach, M.A.; Walsh, C.T.; Clardy, J. The evolution of gene collectives: How natural selection drives chemical innovation. Proc. Natl. Acad. Sci. USA 2008, 105, 4601–4608. [Google Scholar] [CrossRef] [PubMed]
- Jenke-Kodama, H.; Sandmann, A.; Müller, R.; Dittmann, E. Evolutionary implications of bacterial polyketide synthases. Mol. Biol. Evol. 2005, 22, 2027–2039. [Google Scholar] [CrossRef]
- Kroken, S.; Glass, N.L.; Taylor, J.W.; Yoder, O.C.; Turgeon, B.G. Phylogenomic analysis of type I polyketide synthase genes in pathogenic and saprobic ascomycetes. Proc. Natl. Acad. Sci. USA 2003, 100, 15670–15675. [Google Scholar] [CrossRef]
- Ziemert, N.; Jensen, P.R. Phylogenetic approaches to natural product structure prediction. Methods Enzymol. 2012, 517, 161–182. [Google Scholar] [PubMed]
- Rausch, C.; Hoof, I.; Weber, T.; Wohlleben, W.; Huson, D.H. Phylogenetic analysis of condensation domains in NRPS sheds light on their functional evolution. BMC Evol. Biol. 2007, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Ginolhac, A.; Jarrin, C.; Robe, P.; Perrière, G.; Vogel, T.M.; Simonet, P.; Nalin, R. Type I polyketide synthases may have evolved through horizontal gene transfer. J. Mol. Evol. 2005, 60, 716–725. [Google Scholar] [CrossRef] [PubMed]
- Jenke-Kodama, H.; Dittmann, E. Evolution of metabolic diversity: Insights from microbial polyketide synthases. Phytochemistry 2009, 70, 1858–1866. [Google Scholar] [CrossRef] [PubMed]
- Metsä-Ketelä, M.; Halo, L.; Munukka, E.; Hakala, J.; Mäntsälä, P.; Ylihonko, K. Molecular evolution of aromatic polyketides and comparative sequence analysis of polyketide ketosynthase and 16S ribosomal DNA genes from various Streptomyces species. Appl. Environ. Microbiol. 2002, 68, 4472–4479. [Google Scholar] [CrossRef]
- Doroghazi, J.R.; Metcalf, W.W. Comparative genomics of actinomycetes with a focus on natural product biosynthetic genes. BMC Genom. 2013, 14, 611. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547. [Google Scholar] [CrossRef]
- Tavaré, S. Some probabilistic and statistical problems in the analysis of DNA sequences. Lect. Math. Life Sci. 1986, 17, 57–86. [Google Scholar]
- Yoon, S.-H.; Ha, S.-M.; Lim, J.; Kwon, S.; Chun, J. A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek 2017, 110, 1281–1286. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema, M.H.; Weber, T. antiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef] [PubMed]
- Skinnider, M.A.; Johnston, C.W.; Gunabalasingam, M.; Merwin, N.J.; Kieliszek, A.M.; MacLellan, R.J.; Li, H.; Ranieri, M.R.M.; Webster, A.L.H.; Cao, M.P.T.; et al. Comprehensive prediction of secondary metabolite structure and biological activity from microbial genome sequences. Nat. Commun. 2020, 11, 6058. [Google Scholar] [CrossRef]
- Van Heel, A.J.; de Jong, A.; Song, C.; Viel, J.H.; Kok, J.; Kuipers, O.P. BAGEL4: A user-friendly web server to thoroughly mine RiPPs and bacteriocins. Nucleic Acids Res. 2018, 46, W278–W281. [Google Scholar] [CrossRef]
- Machado, H.; Sonnenschein, E.C.; Melchiorsen, J.; Gram, L. Genome mining reveals unlocked bioactive potential of marine Gram-negative bacteria. BMC Genom. 2015, 16, 158. [Google Scholar] [CrossRef] [PubMed]
- Churchill, G.A. Stochastic models for heterogeneous DNA sequences. Bull. Math. Biol. 1989, 51, 79–94. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2013, 41, D36–D42. [Google Scholar] [CrossRef] [PubMed]
- Consortium, U. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar] [CrossRef]
- Hammami, R.; Zouhir, A.; Le Lay, C.; Hamida, J.B.; Fliss, I. BACTIBASE second release: A database and tool platform for bacteriocin characterization. BMC Microbiol. 2010, 10, 22. [Google Scholar] [CrossRef]
- Waghu, F.H.; Barai, R.S.; Gurung, P.; Idicula-Thomas, S. CAMPR3: A database on sequences, structures and signatures of antimicrobial peptides. Nucleic Acids Res. 2016, 44, D1094–D1097. [Google Scholar] [CrossRef] [PubMed]
- Medema, M.H.; Kottmann, R.; Yilmaz, P.; Cummings, M.; Biggins, J.B.; Blin, K.; De Bruijn, I.; Chooi, Y.H.; Claesen, J.; Coates, R.C. Minimum information about a biosynthetic gene cluster. Nat. Chem. Biol. 2015, 11, 625–631. [Google Scholar] [CrossRef]
- Ziemert, N.; Podell, S.; Penn, K.; Badger, J.H.; Allen, E.; Jensen, P.R. The natural product domain seeker NaPDoS: A phylogeny based bioinformatic tool to classify secondary metabolite gene diversity. PLoS ONE 2012, 7, e34064. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Profile hidden Markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Bloudoff, K.; Schmeing, T.M. Structural and functional aspects of the nonribosomal peptide synthetase condensation domain superfamily: Discovery, dissection and diversity. Biochim. Biophys. Acta (BBA)—Proteins Proteom. 2017, 1865, 1587–1604. [Google Scholar] [CrossRef] [PubMed]
- Scherlach, K.; Hertweck, C. Triggering cryptic natural product biosynthesis in microorganisms. Org. Biomol. Chem. 2009, 7, 1753–1760. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Isolation Source | Size (mb) | Genes | antiSMASH | PRISM | BAGEL | KS Domains | C Domain |
|---|---|---|---|---|---|---|---|---|
| Hit | Hit | Hit | ||||||
| P. bijieensis L22-9 | N/A | 6.730 | 5984 | 15 | 7 | 5 | 8 | 37 |
| P. brassicacearum 3Re2-7 | Endorhiza of potato | 6.739 | 6014 | 11 | 4 | 3 | 8 | 27 |
| P. viciae 11K1 | Rhizosphere | 6.705 | 5868 | 13 | 9 | 2 | 7 | 66 |
| P. corrugata RM1-1-4 | Rhizosphere | 6.124 | 5394 | 10 | 7 | 3 | 8 | 45 |
| P. chlororaphis qlu-1 | Rhizosphere | 6.828 | 6093 | 16 | 9 | 1 | 11 | 26 |
| P. protegens CHA0 | N/A | 6.868 | 6252 | 15 | 9 | 1 | 10 | 28 |
| P. umsongensis CY-1 | Soil | 6.690 | 6060 | 11 | 3 | 1 | 8 | 12 |
| P. atacamensis SM1 | Rhizospheric soil | 5.991 | 5436 | 9 | 3 | 3 | 7 | 13 |
| P. glycinae MS586 | Cotton field | 6.397 | 5818 | 11 | 7 | 3 | 11 | 26 |
| P. mandelii JR-1 | N/A | 7.189 | 6604 | 11 | 3 | 1 | 10 | 12 |
| P. silesiensis A3 | Wastewater | 6.824 | 6166 | 12 | 5 | 1 | 9 | 12 |
| P. rhodesiae NL2019 | Soil | 5.779 | 5262 | 10 | 4 | 0 | 7 | 17 |
| P. lurida MYb11 | Rotting apple | 6.101 | 5549 | 13 | 6 | 1 | 7 | 24 |
| P. simiae PCL1751 | Soil | 6.144 | 5643 | 12 | 4 | 1 | 7 | 16 |
| P. lundensis 2T.2.5.2 | Meltwater pond | 4.934 | 4563 | 7 | 4 | 2 | 7 | 12 |
| P. psychrophila KM02 | Food | 5.314 | 4813 | 6 | 2 | 1 | 7 | 0 |
| P. versuta L10.10 | Soil | 5.15 | 4671 | 6 | 2 | 0 | 6 | 10 |
| P. amygdali pv. tabaci str. ATCC 11528 | N/A | 6.202 | 5489 | 10 | 9 | 2 | 6 | 26 |
| P. syringae BIM B-268 | Ribes nigrum leaves | 6.019 | 5165 | 10 | 8 | 0 | 6 | 70 |
| P. cannabina pv. alisalensis MAFF 301419 | Radish | 6.145 | 5486 | 7 | 5 | 0 | 6 | 27 |
| P. syringae pv. tomato str. DC3000 | Tomato | 6.538 | 5891 | 10 | 8 | 3 | 9 | 32 |
| P. eucalypticola NP-1 | Plant leaf | 6.402 | 5782 | 8 | 7 | 0 | 4 | 29 |
| P. rhizosphaerae DSM 16299 | Rhizospheric soil | 4.689 | 4214 | 7 | 4 | 0 | 6 | 3 |
| P. alkylphenolica Neo | Soil | 5.612 | 5092 | 9 | 3 | 1 | 4 | 23 |
| P. monteilii B5 | Soil | 6.079 | 5661 | 6 | 2 | 1 | 5 | 17 |
| P. putida NBRC 14164 | N/A | 6.157 | 5539 | 7 | 3 | 0 | 5 | 17 |
| P. plecoglossicida XSDHY-P | Fish spleen | 5.526 | 5067 | 7 | 2 | 2 | 2 | 11 |
| P. entomophila L48 | N/A | 5.889 | 5199 | 14 | 11 | 2 | 10 | 44 |
| P. soli SJ10 | N/A | 6.248 | 5798 | 12 | 6 | 1 | 7 | 32 |
| P. sediminis B10D7D | N/A | 4.934 | 4612 | 7 | 3 | 0 | 9 | 9 |
| P. toyotomiensis SM2 | Rhizospheric soil | 5.235 | 4857 | 8 | 3 | 0 | 8 | 11 |
| P. mendocina S5.2 | N/A | 5.372 | 5081 | 7 | 3 | 0 | 10 | 9 |
| P. lalkuanensis PE08 | Soil | 6.057 | 5558 | 7 | 3 | 0 | 7 | 12 |
| P. otitidis MrB4 DNA | Water | 6.089 | 5615 | 10 | 4 | 0 | 9 | 13 |
| P. aeruginosa PAO1 | N/A | 6.264 | 5697 | 14 | 14 | 4 | 6 | 21 |
| P. citronellolis P3B5 | Basil | 6.951 | 6219 | 8 | 3 | 1 | 8 | 14 |
| P. multiresinivorans populi | Rhizosphere soil | 6.518 | 5974 | 7 | 2 | 1 | 9 | 7 |
| Species Name | Source | Size | Genes | AntiSMASH | BAGEL | PRISM | KS Domain | C Domain |
|---|---|---|---|---|---|---|---|---|
| (Mbp) | Hit | Hit | Hit | |||||
| P. fluorescens Pf275 | Soil | 6.61 | 5884 | 15 | 5 | 6 | 8 | 37 |
| P. fluorescens DR133 | Rhizosphere | 6.848 | 6102 | 16 | 4 | 6 | 8 | 33 |
| P. fluorescens 2P24 | Soil | 6.611 | 5803 | 14 | 3 | 7 | 19 | 34 |
| P. fluorescens FW300-N2C3 | Ground water | 7.119 | 6149 | 18 | 1 | 13 | 10 | 80 |
| P. fluorescens F113 | N/A | 6.846 | 6093 | 12 | 2 | 5 | 13 | 17 |
| P. fluorescens FW300-N2E3 | Ground water | 6.392 | 5951 | 12 | 0 | 4 | 10 | 2 |
| P. fluorescens G20-18 | Arctic grass | 6.481 | 6001 | 11 | 3 | 4 | 8 | 13 |
| P. fluorescens NCIMB 11764 | N/A | 6.998 | 6404 | 11 | 3 | 3 | 8 | 13 |
| P. fluorescens NCTC9428 | N/A | 6.034 | 5413 | 7 | 2 | 4 | 6 | 16 |
| P. fluorescens LBUM677 | Rhizosphere | 6.14 | 5487 | 12 | 1 | 6 | 6 | 25 |
| P. fluorescens G7 | Soil | 6.336 | 5804 | 13 | 0 | 7 | 8 | 26 |
| P. fluorescens MS82 | Rhizosphere | 6.208 | 5690 | 12 | 4 | 9 | 11 | 26 |
| P. fluorescens YK-310 | Soil | 6.499 | 5825 | 15 | 0 | 8 | 9 | 41 |
| P. fluorescens Pf0-1 | N/A | 6.438 | 5852 | 12 | 0 | 6 | 9 | 33 |
| P. fluorescens UK4 | Drinking water | 6.064 | 5513 | 13 | 0 | 6 | 6 | 19 |
| P. fluorescens PF08 | Scophthalmus maximus | 6.031 | 5518 | 12 | 0 | 6 | 7 | 0 |
| P. fluorescens KF1 | Kumarahou flower | 6.957 | 6306 | 13 | 0 | 6 | 8 | 15 |
| P. fluorescens SBW25 | N/A | 6.723 | 6123 | 11 | 2 | 7 | 8 | 33 |
| P. fluorescens SIK_W1 | Soil | 6.791 | 6058 | 15 | 0 | 6 | 7 | 24 |
| P. fluorescens JNU01 | N/A | 6.79 | 6058 | 15 | 0 | 6 | 7 | 24 |
| P. fluorescens NCTC10038 | N/A | 6.515 | 5965 | 14 | 2 | 6 | 6 | 22 |
| P. fluorescens FDAARGOS_1088 | N/A | 6.135 | 5585 | 13 | 0 | 9 | 7 | 16 |
| P. fluorescens A506 | Tree leaf | 6.02 | 5493 | 12 | 2 | 9 | 7 | 15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alam, K.; Islam, M.M.; Li, C.; Sultana, S.; Zhong, L.; Shen, Q.; Yu, G.; Hao, J.; Zhang, Y.; Li, R.; et al. Genome Mining of Pseudomonas Species: Diversity and Evolution of Metabolic and Biosynthetic Potential. Molecules 2021, 26, 7524. https://doi.org/10.3390/molecules26247524
Alam K, Islam MM, Li C, Sultana S, Zhong L, Shen Q, Yu G, Hao J, Zhang Y, Li R, et al. Genome Mining of Pseudomonas Species: Diversity and Evolution of Metabolic and Biosynthetic Potential. Molecules. 2021; 26(24):7524. https://doi.org/10.3390/molecules26247524
Chicago/Turabian StyleAlam, Khorshed, Md. Mahmudul Islam, Caiyun Li, Sharmin Sultana, Lin Zhong, Qiyao Shen, Guangle Yu, Jinfang Hao, Youming Zhang, Ruijuan Li, and et al. 2021. "Genome Mining of Pseudomonas Species: Diversity and Evolution of Metabolic and Biosynthetic Potential" Molecules 26, no. 24: 7524. https://doi.org/10.3390/molecules26247524
APA StyleAlam, K., Islam, M. M., Li, C., Sultana, S., Zhong, L., Shen, Q., Yu, G., Hao, J., Zhang, Y., Li, R., & Li, A. (2021). Genome Mining of Pseudomonas Species: Diversity and Evolution of Metabolic and Biosynthetic Potential. Molecules, 26(24), 7524. https://doi.org/10.3390/molecules26247524