
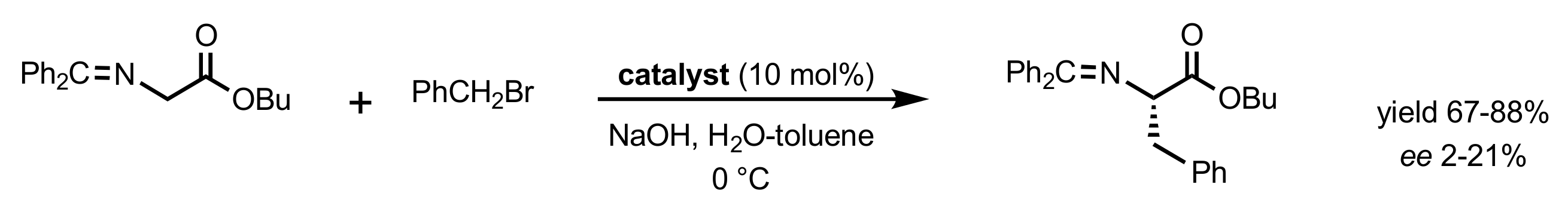
Synthesis and Applications of Carbohydrate-Based Organocatalysts

 , , ,
, , , 
Abstract
:1. Introduction
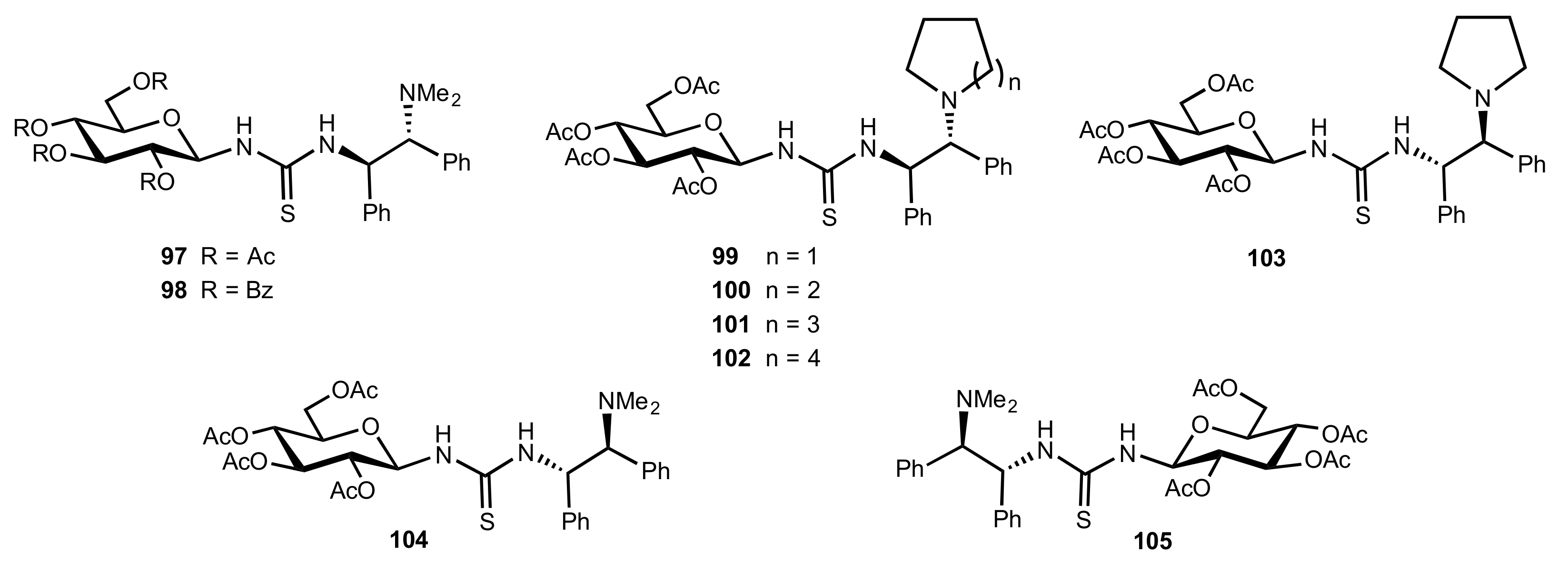
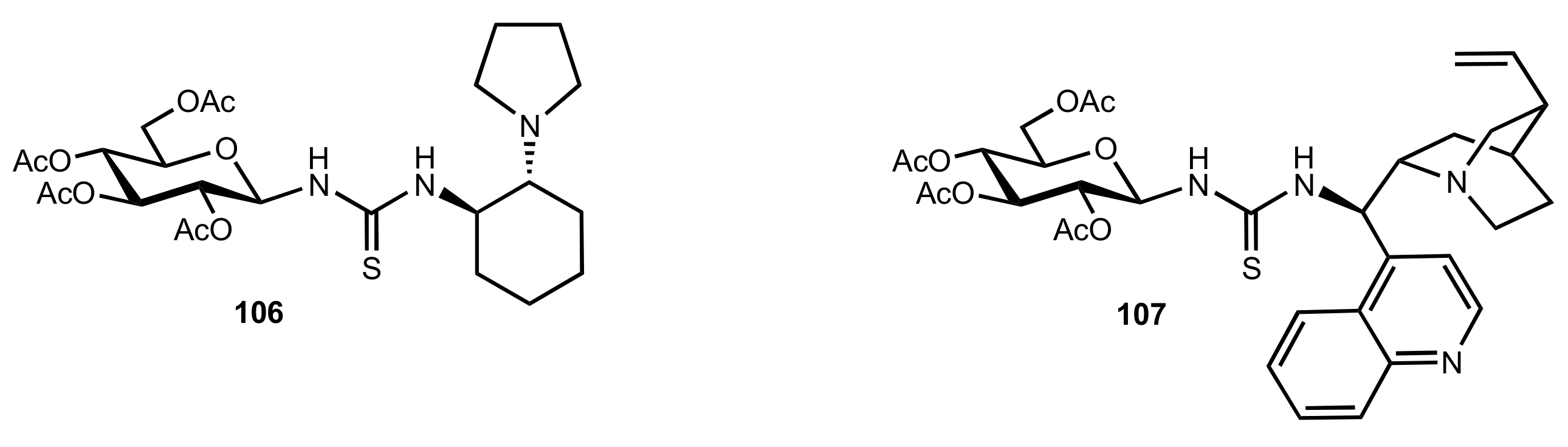
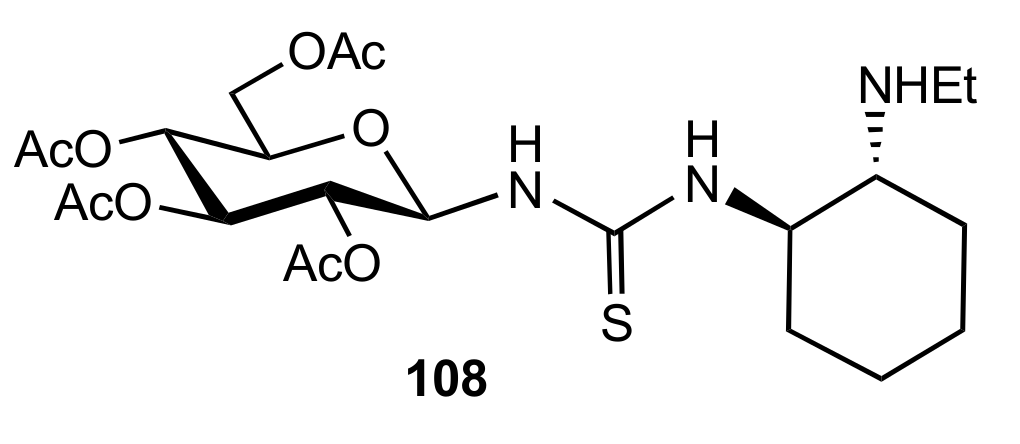
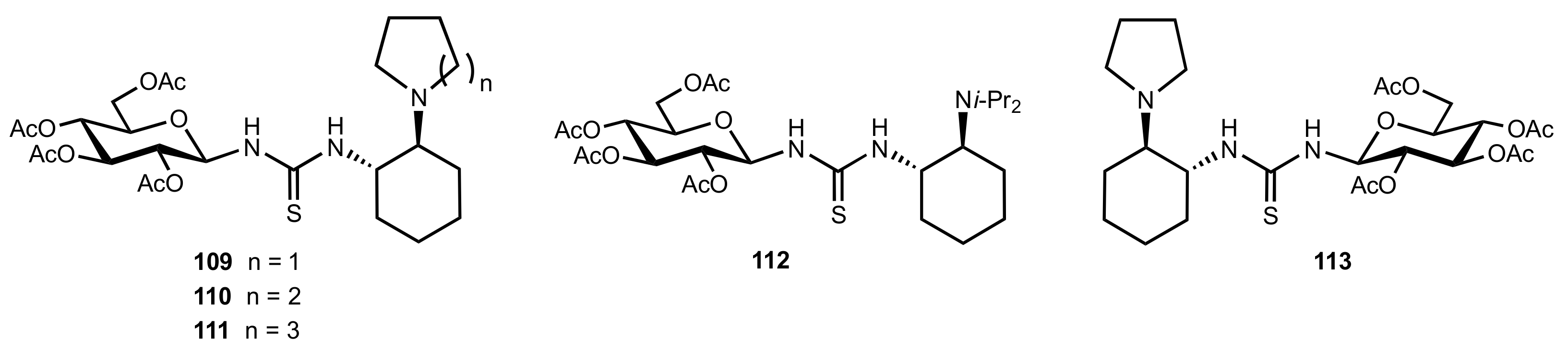
2. Sugar Ureas and Thioureas
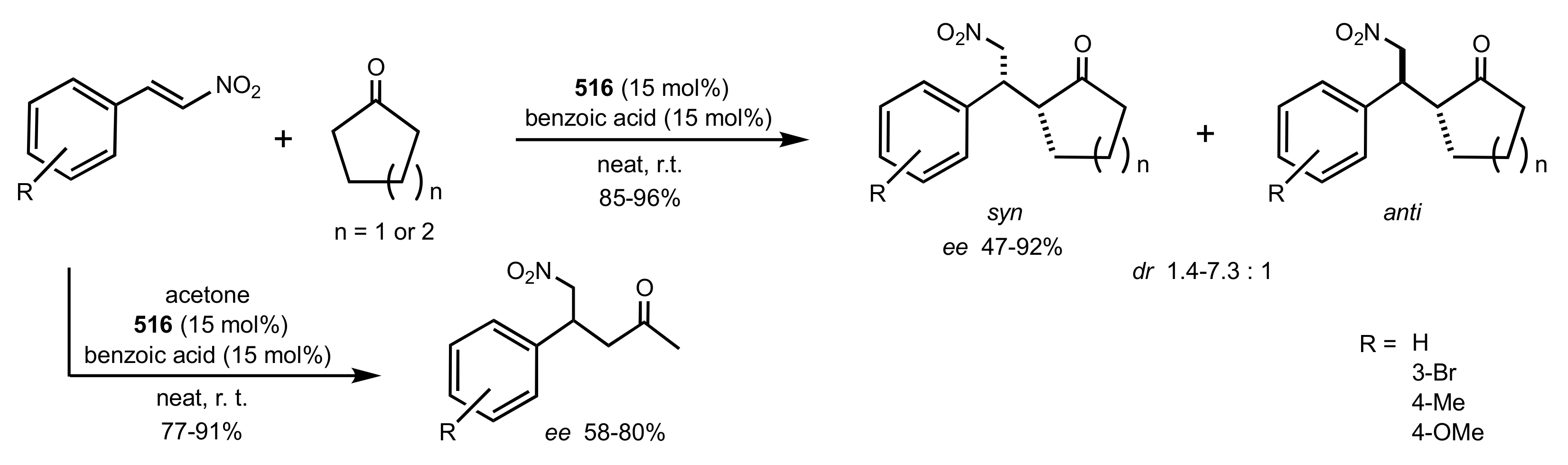
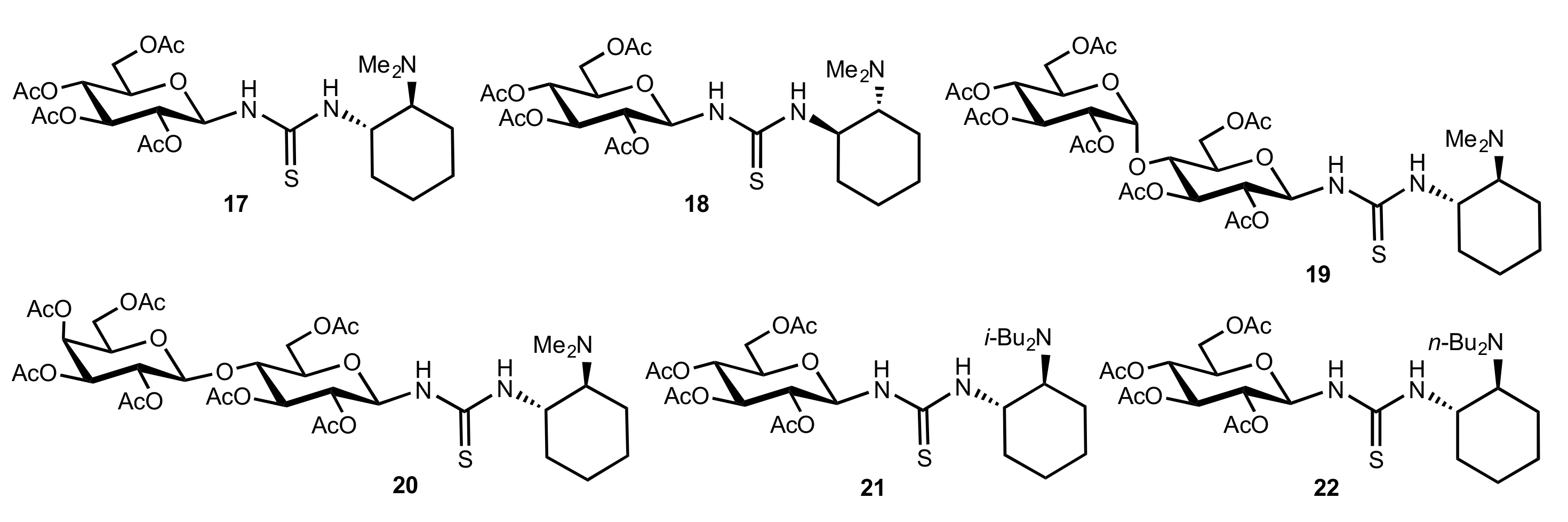
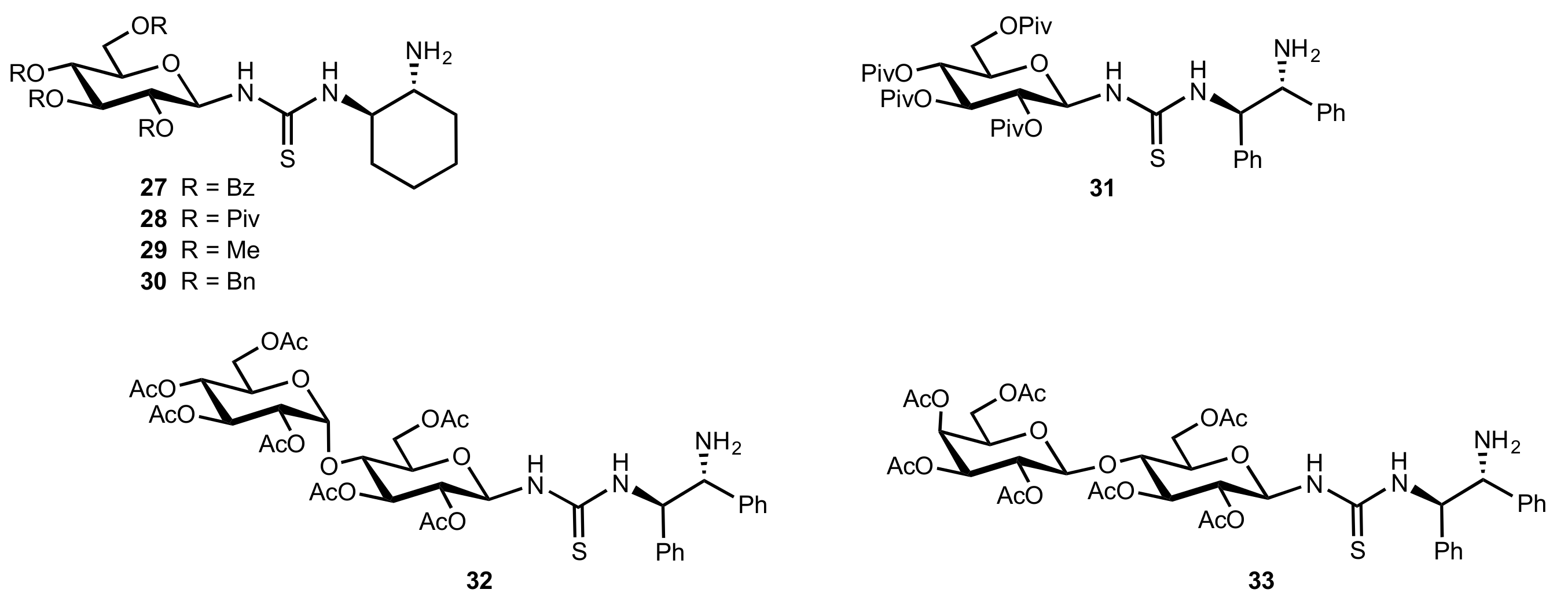
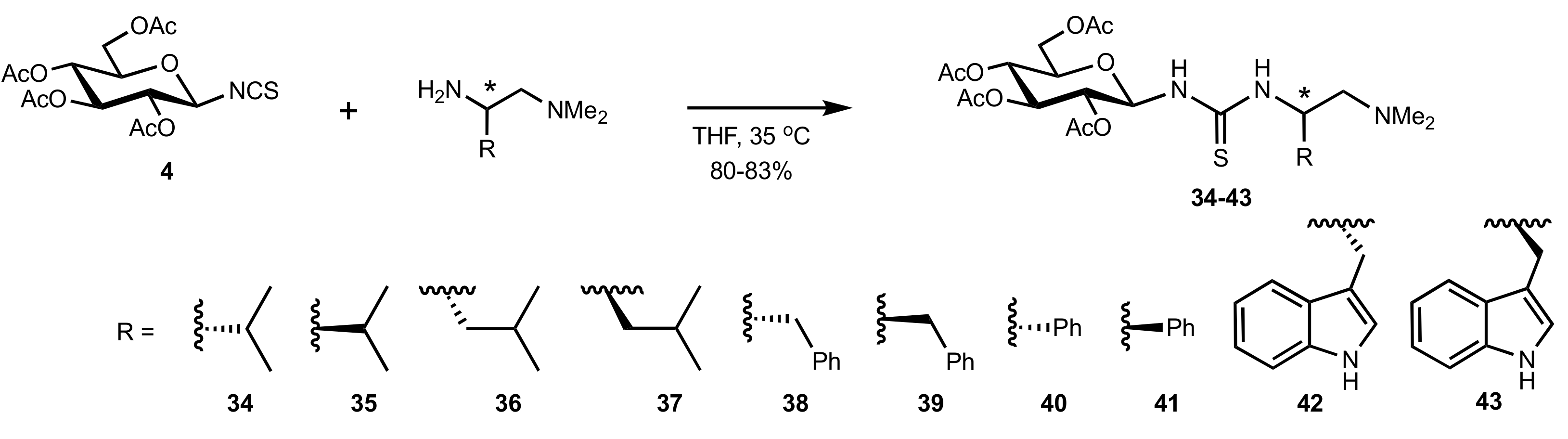
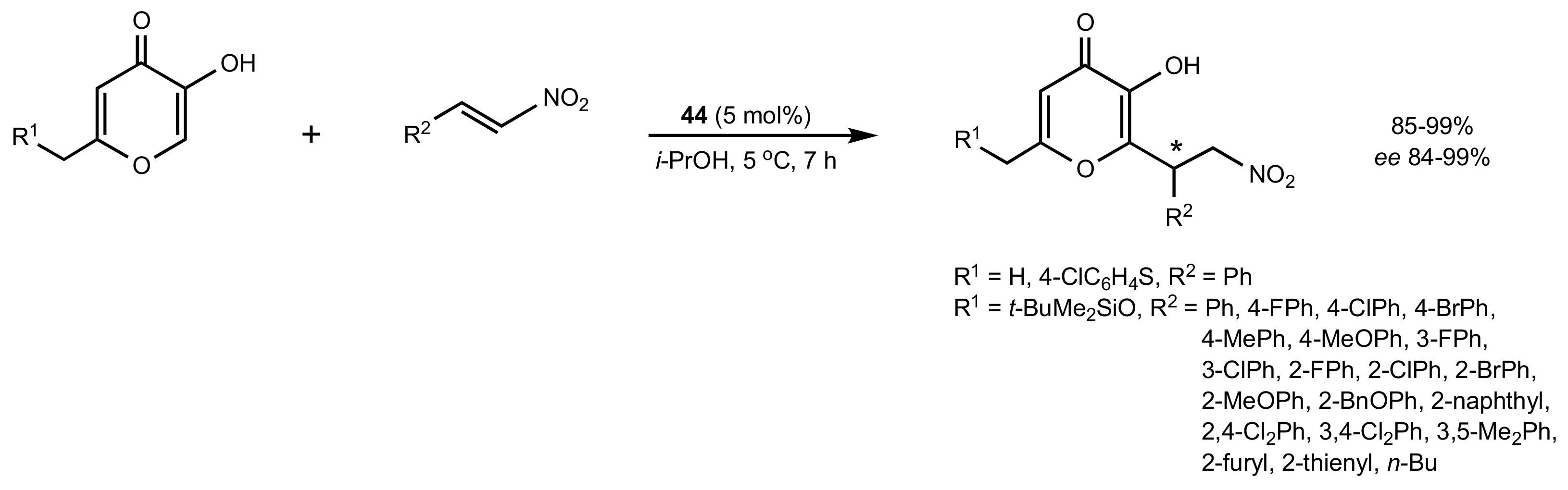
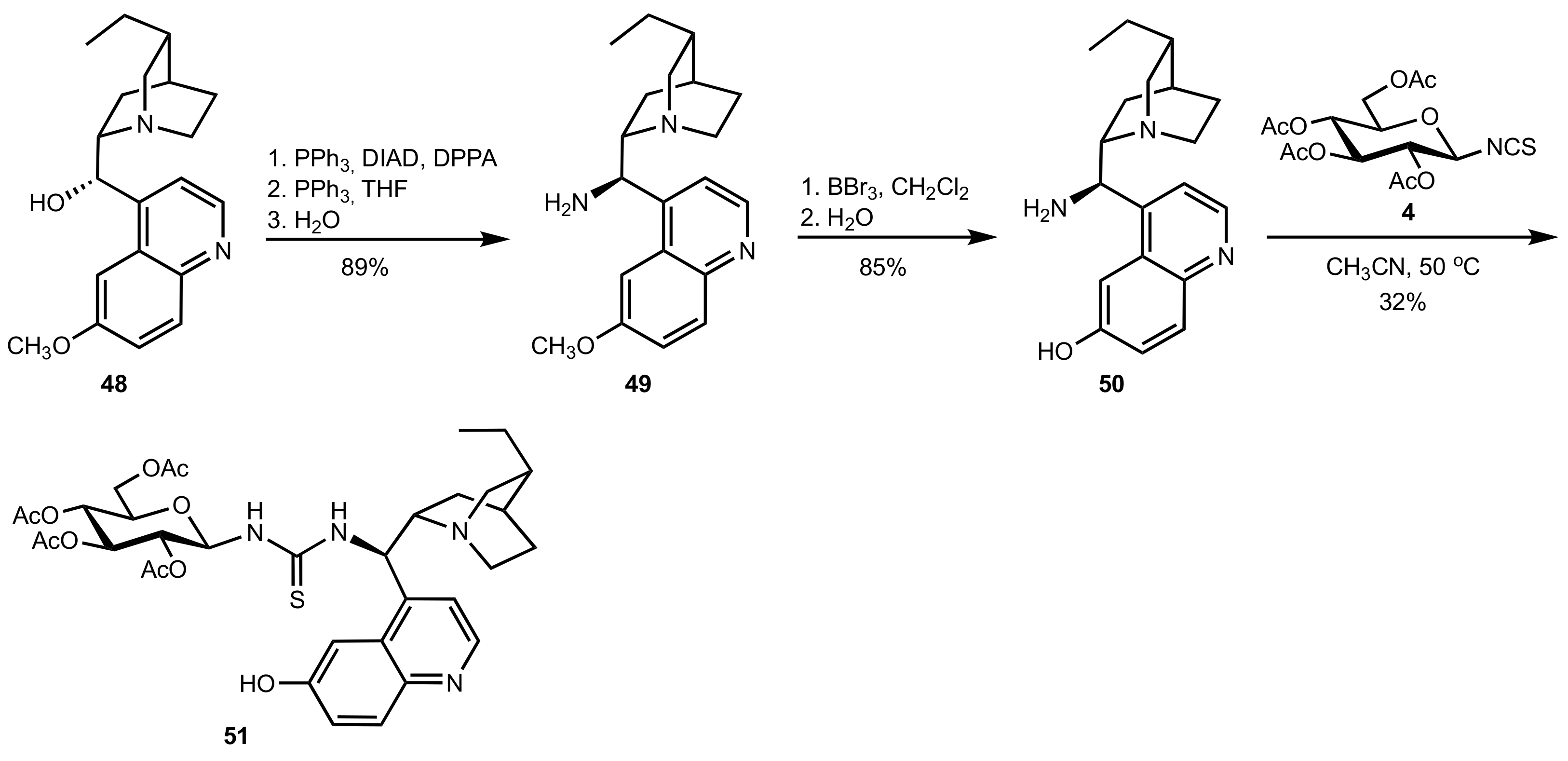
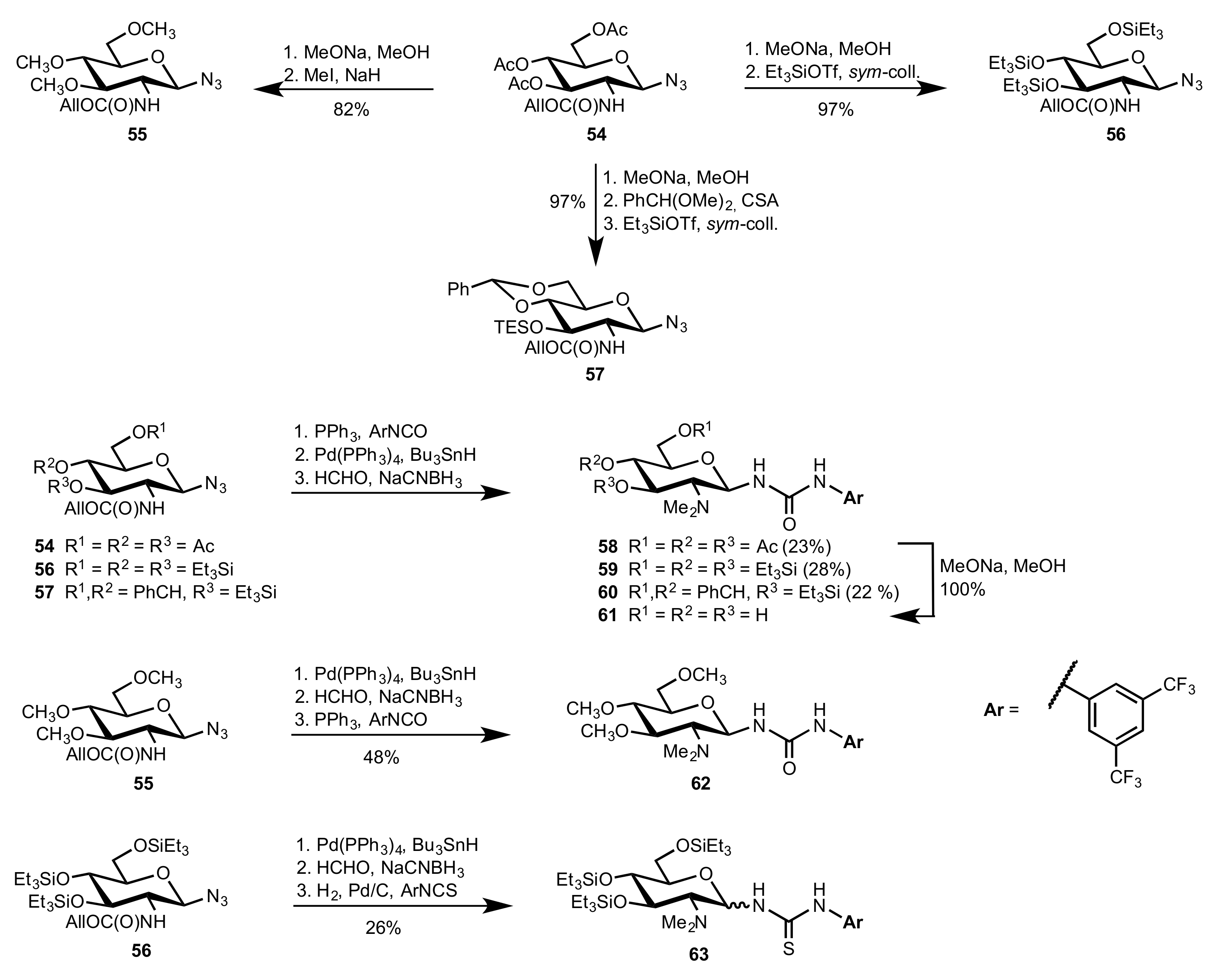
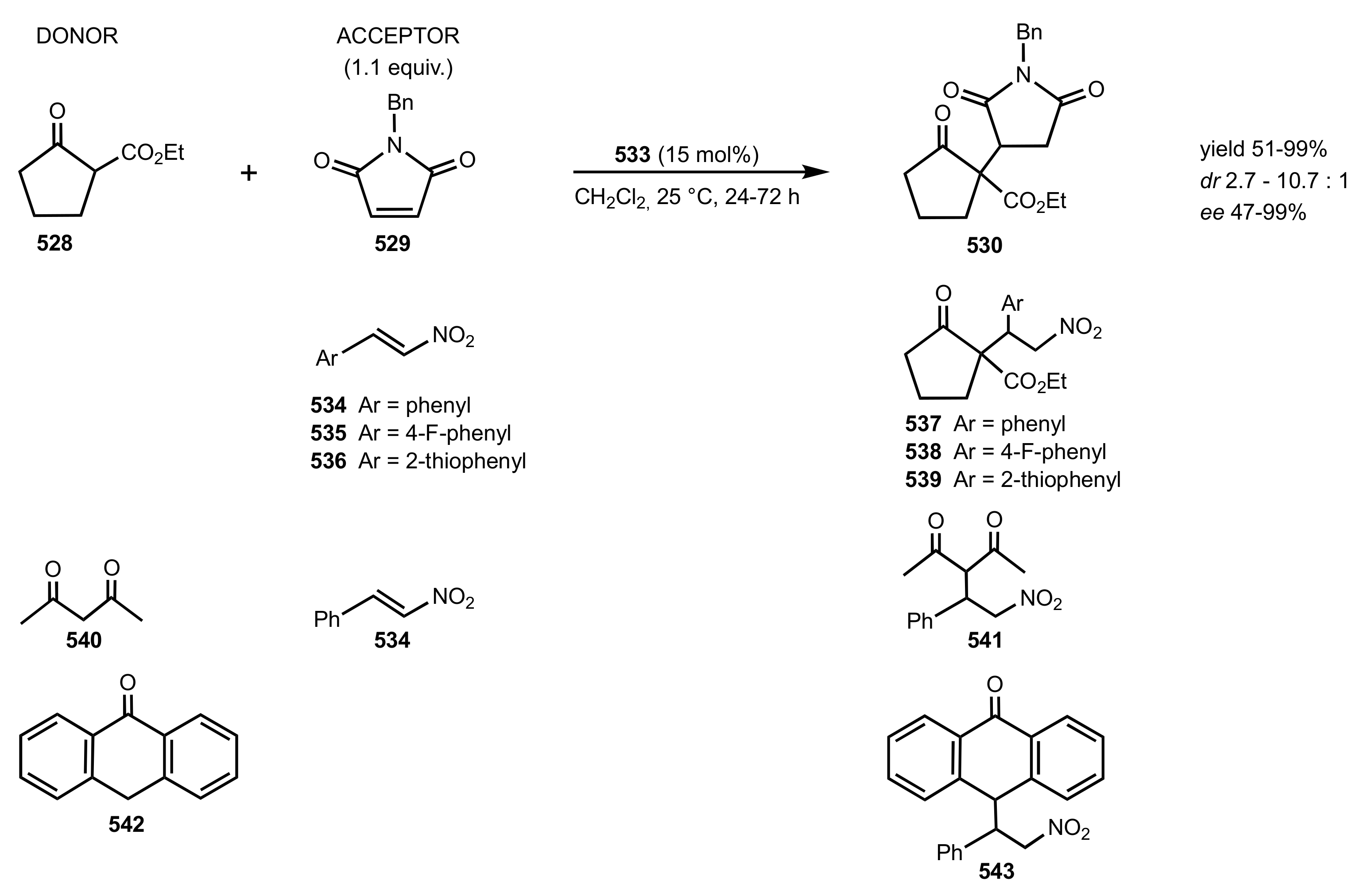
2.1. Michael Addition
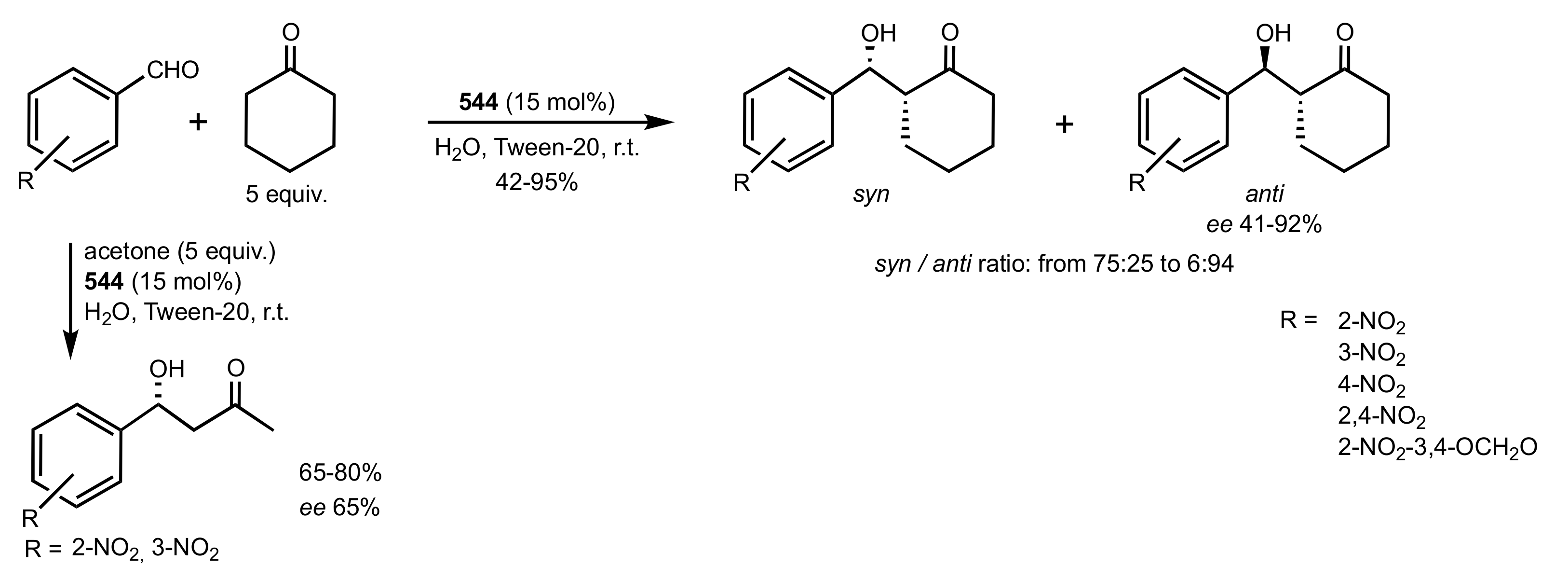
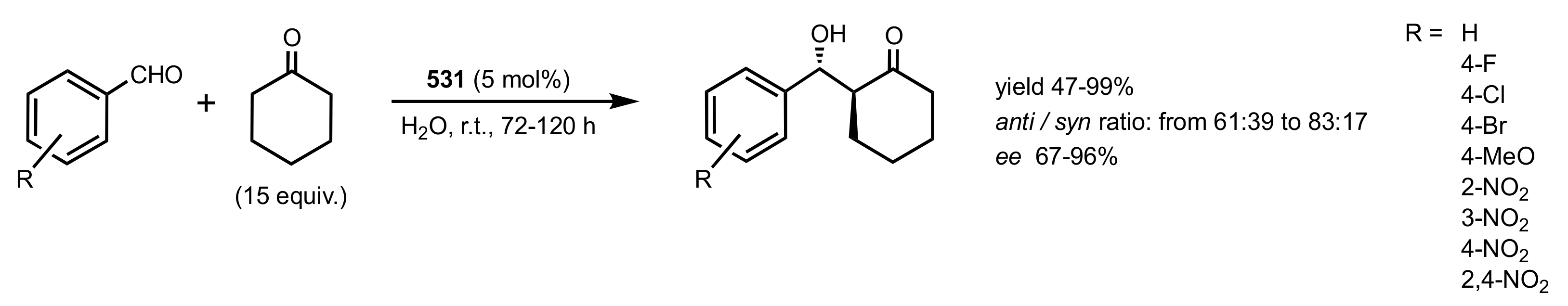
2.2. Aldol Reaction
2.3. Mannich Reaction
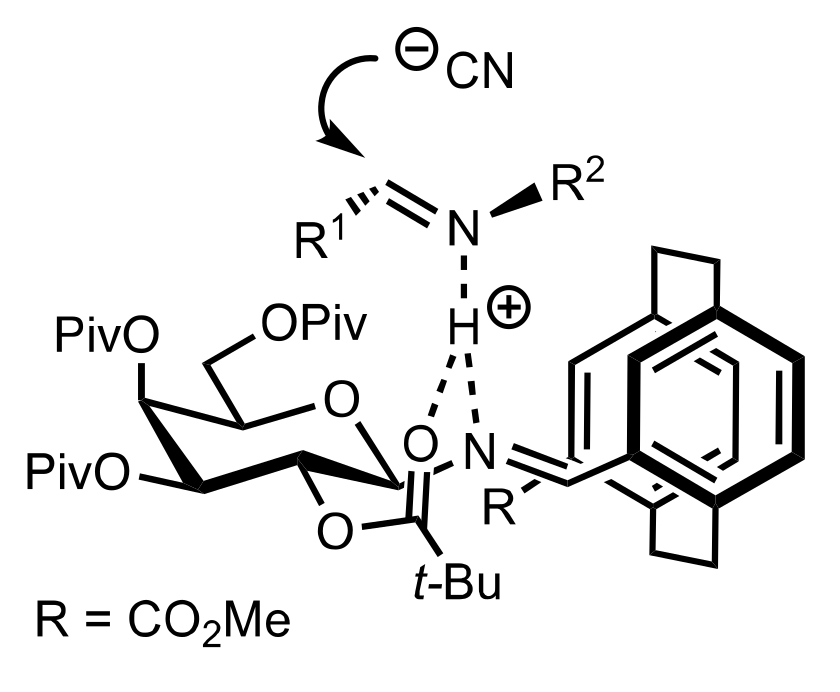
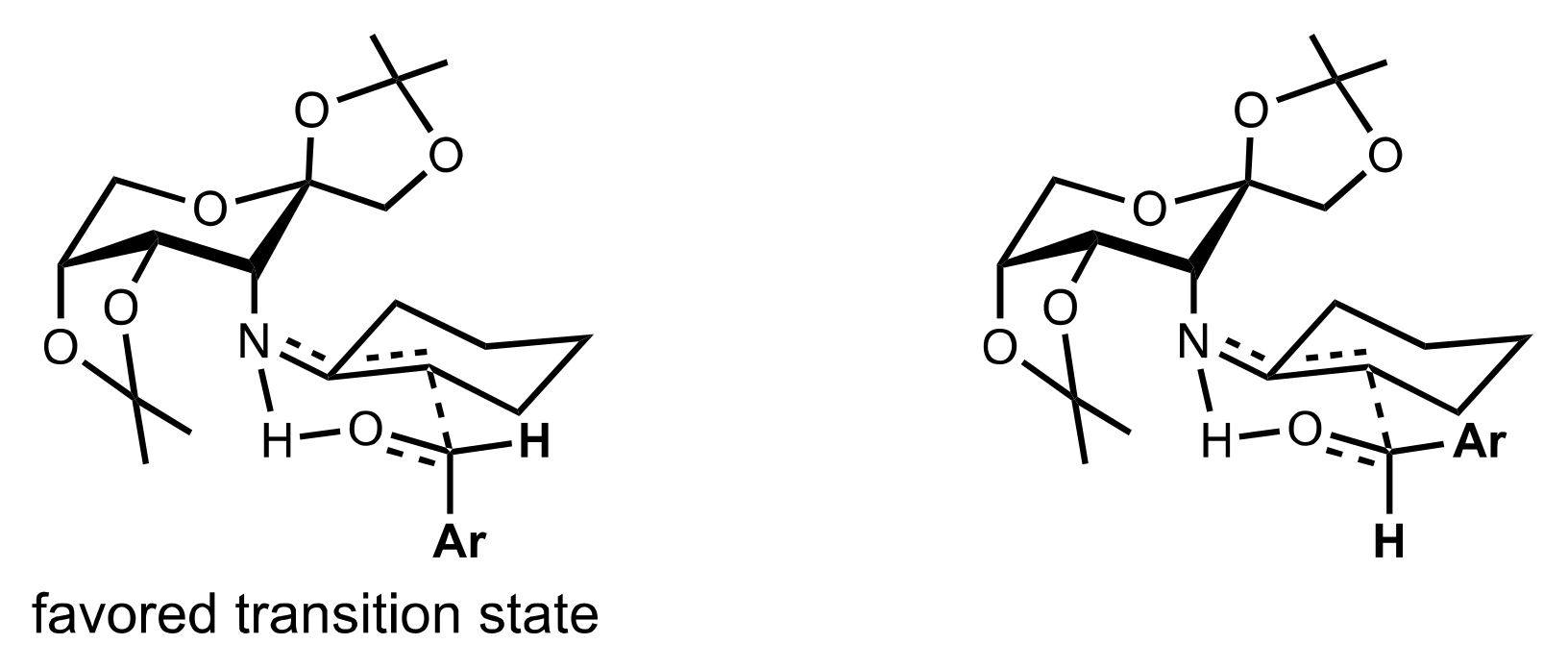
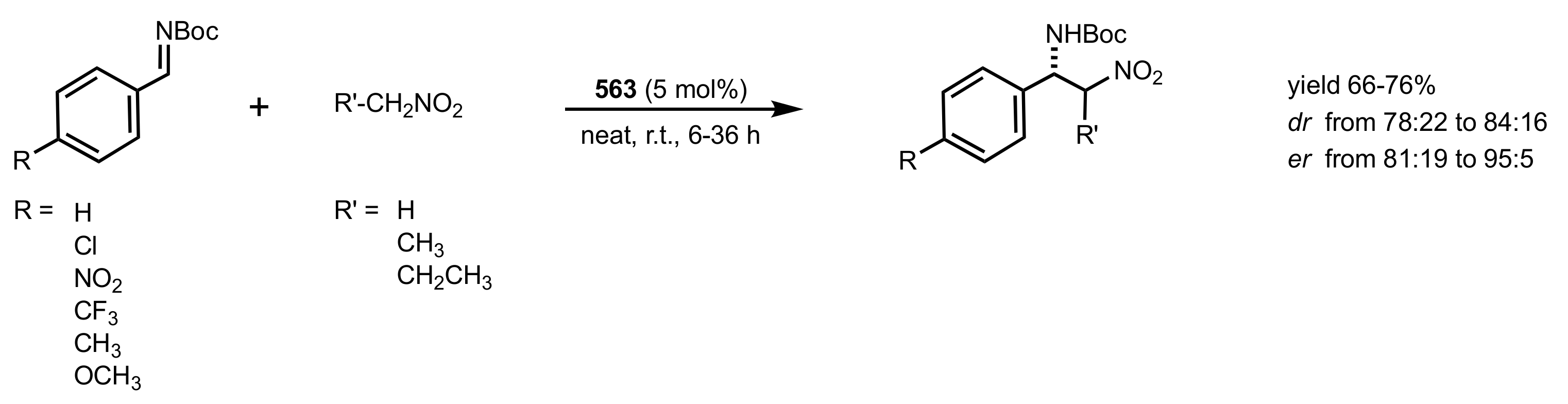
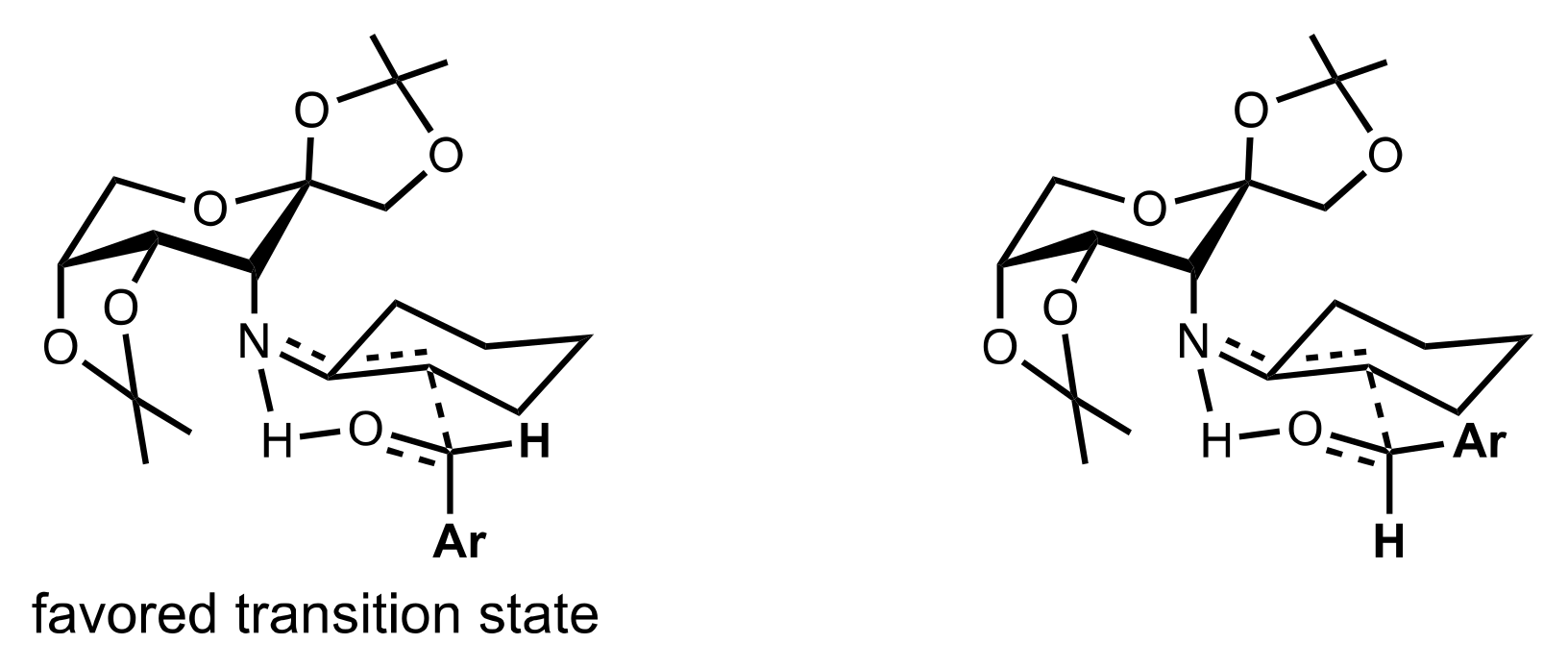
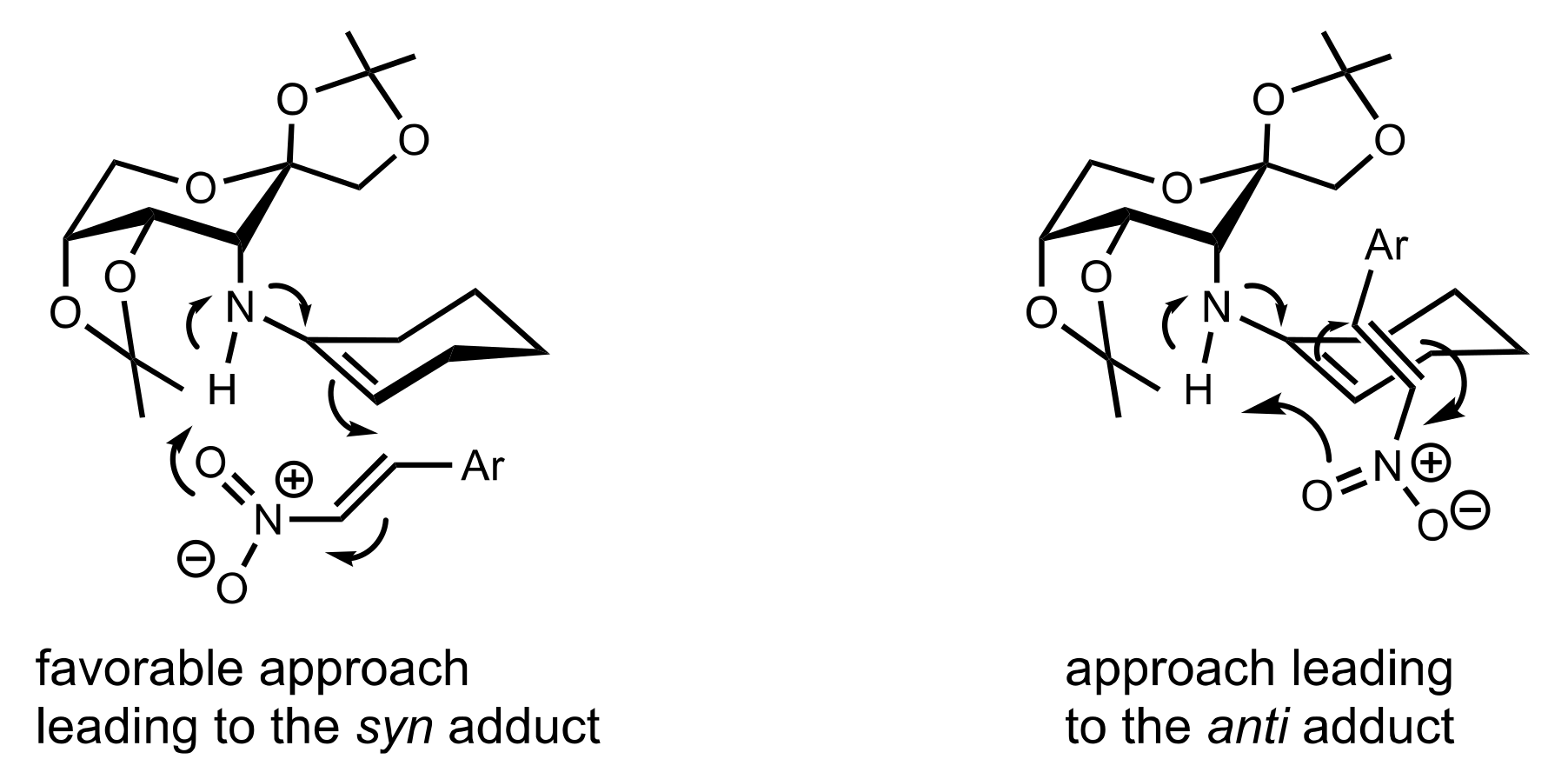
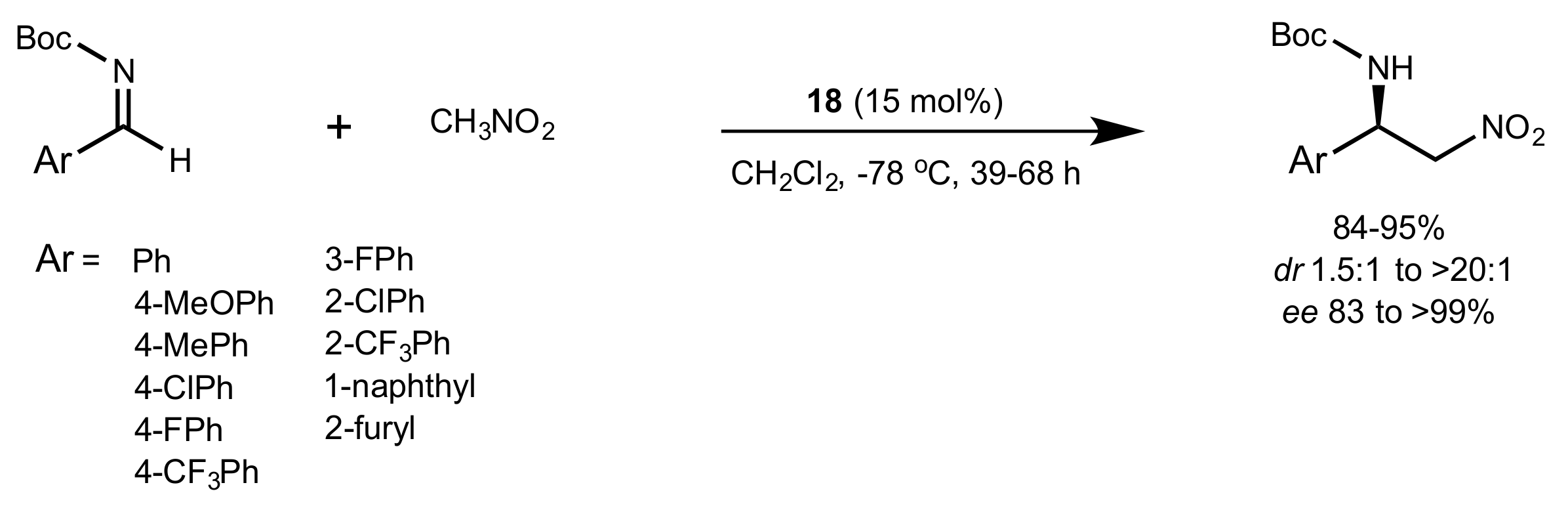
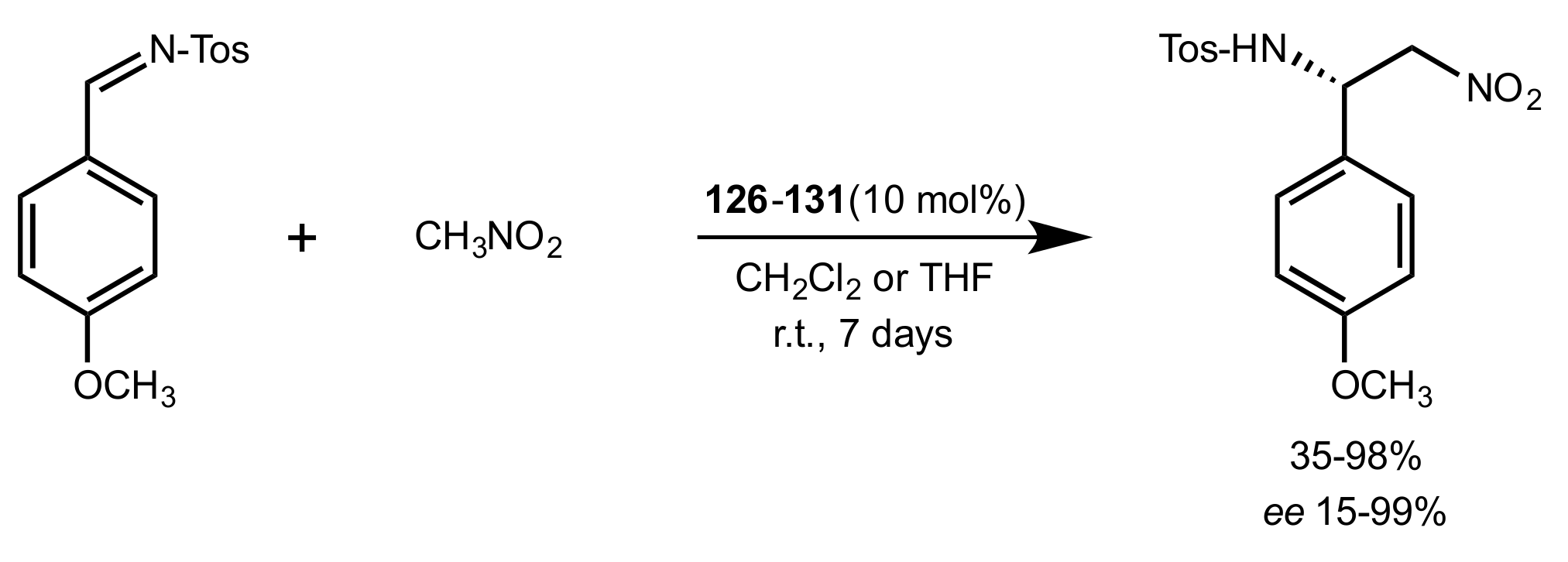
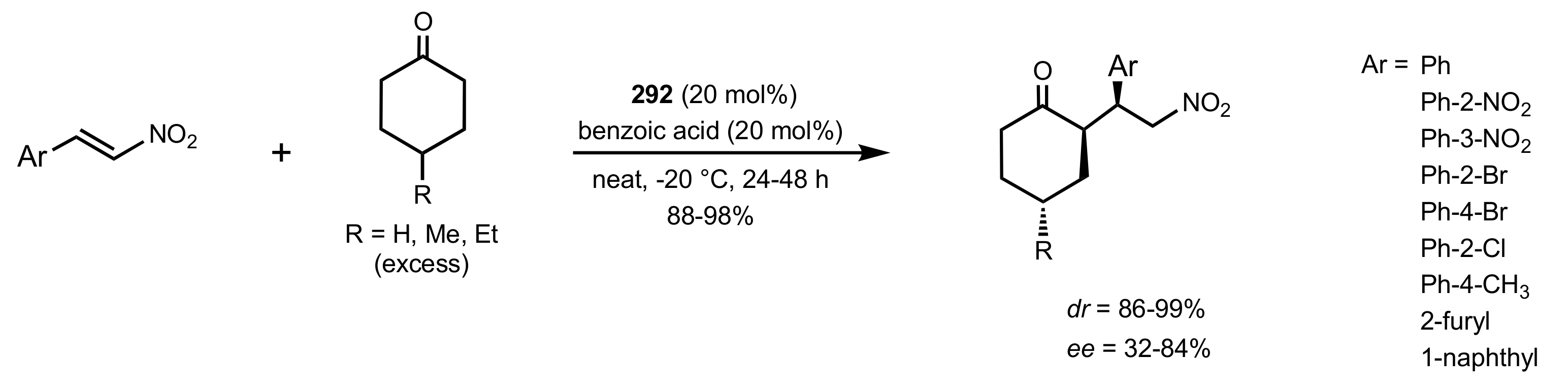
2.4. Aza–Henry (Nitro-Mannich) Reaction
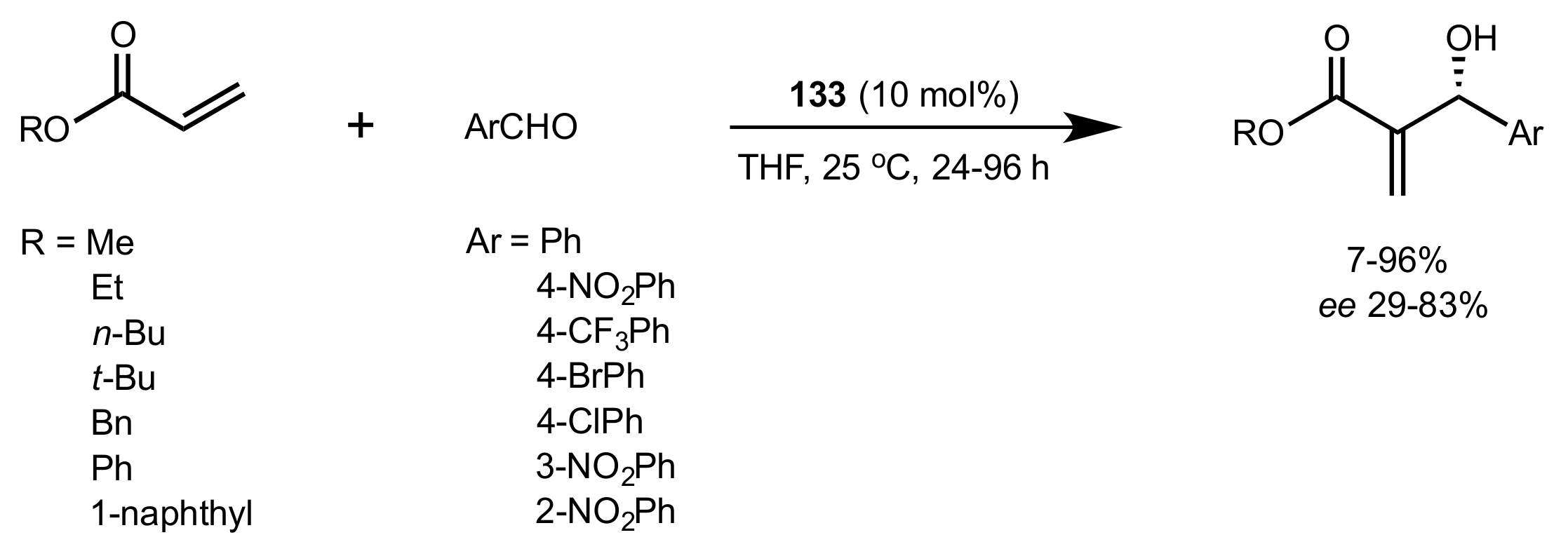
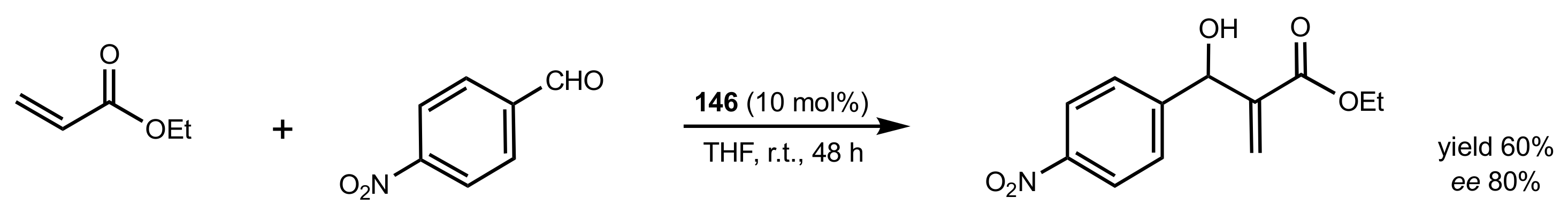
2.5. Morita–Baylis–Hillman Reaction
2.6. Other Asymmetric Transformations
3. Sugar Ketones
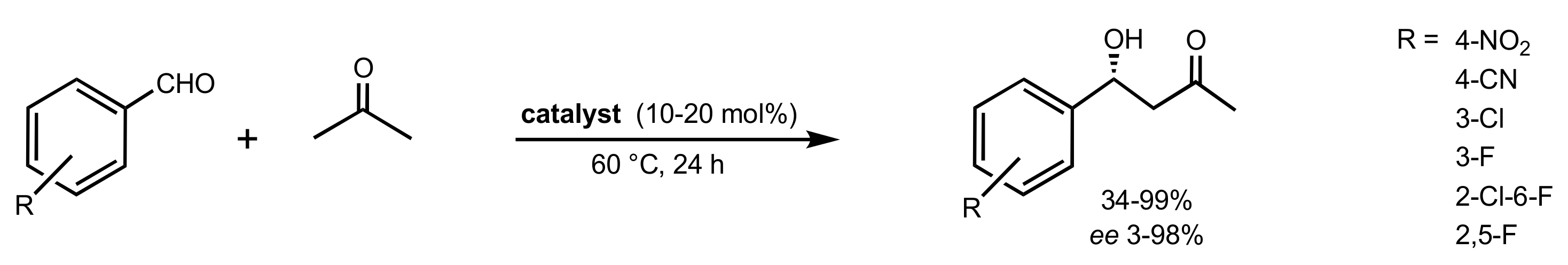
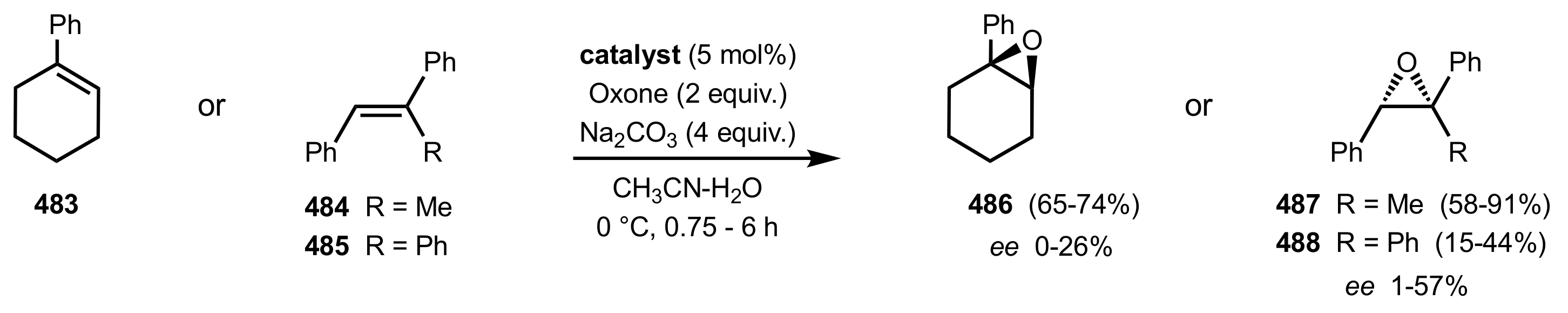
3.1. Asymmetric Epoxidation
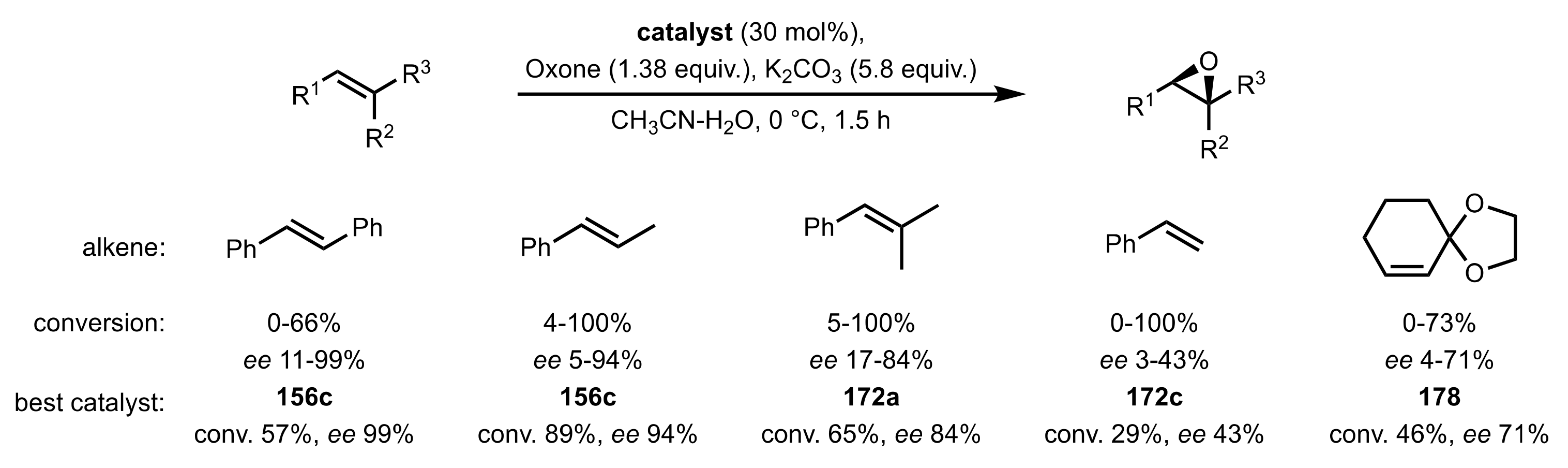
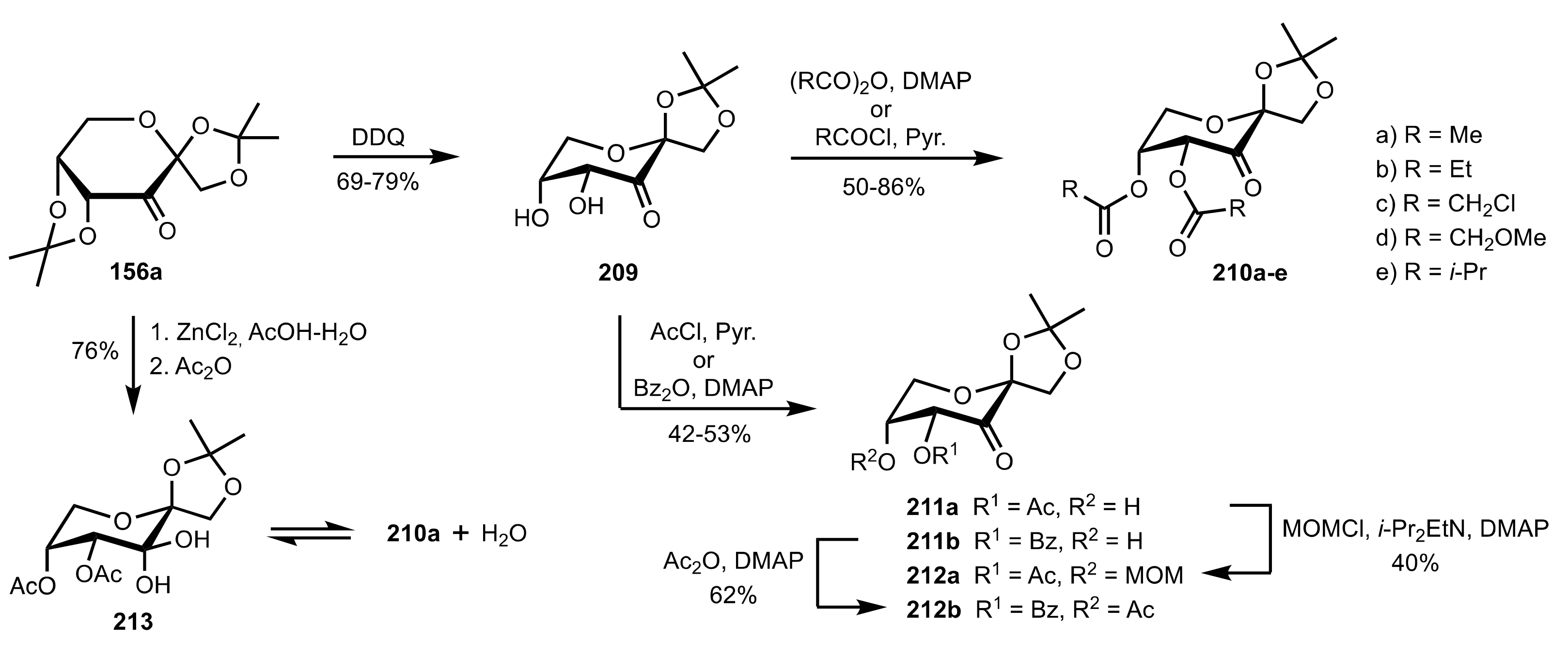
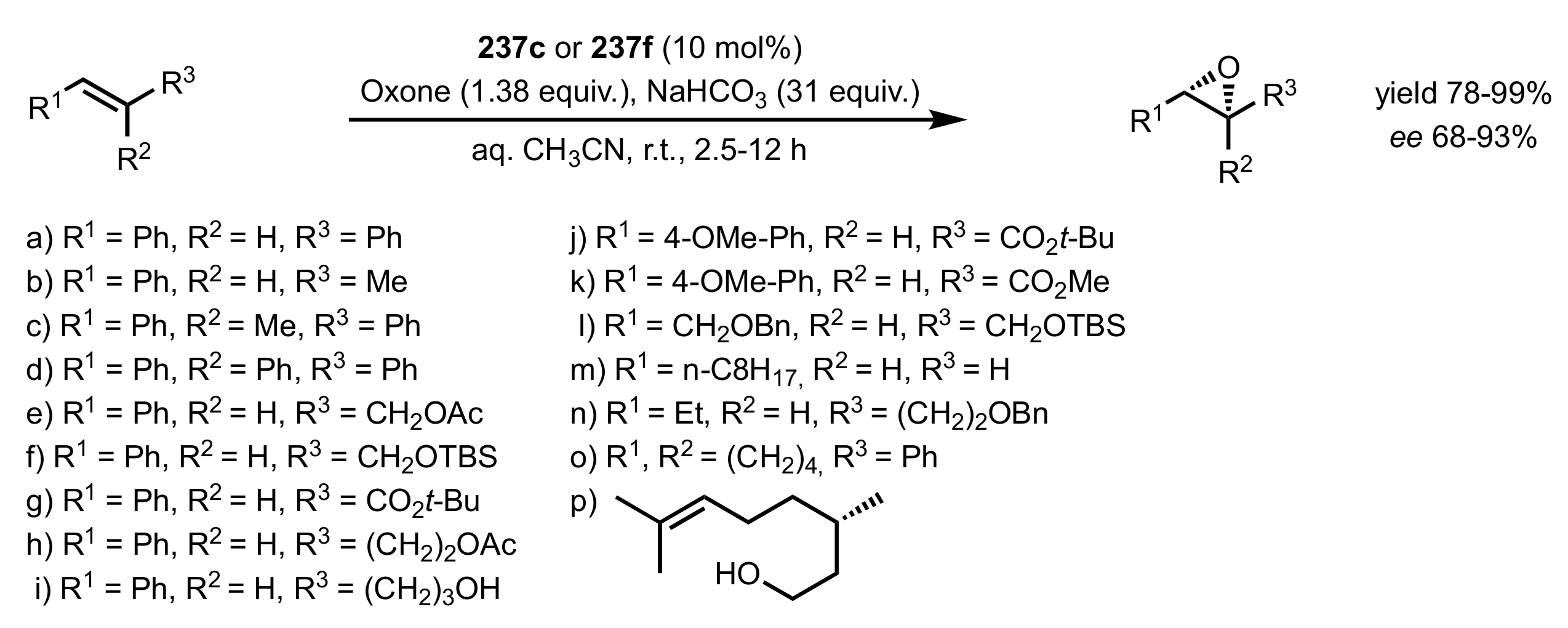
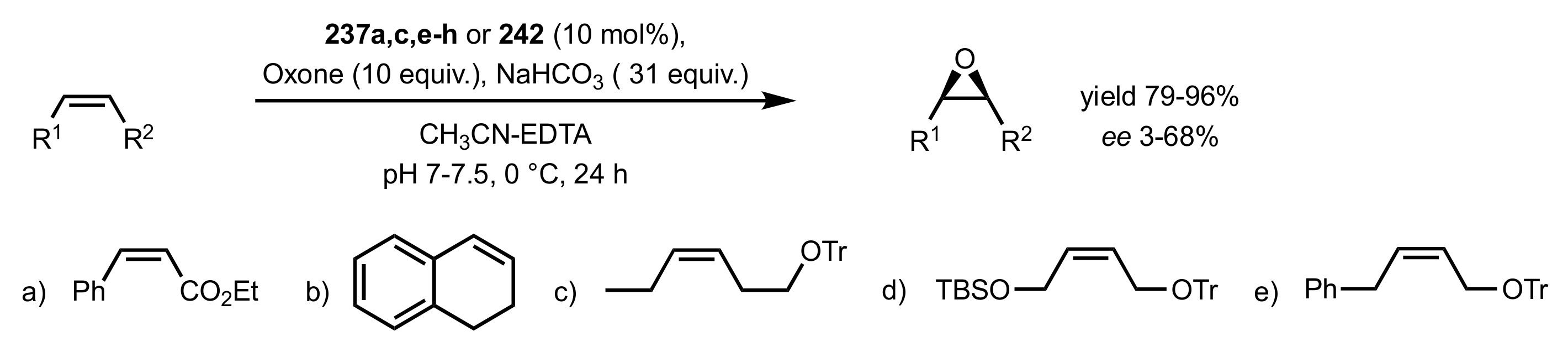
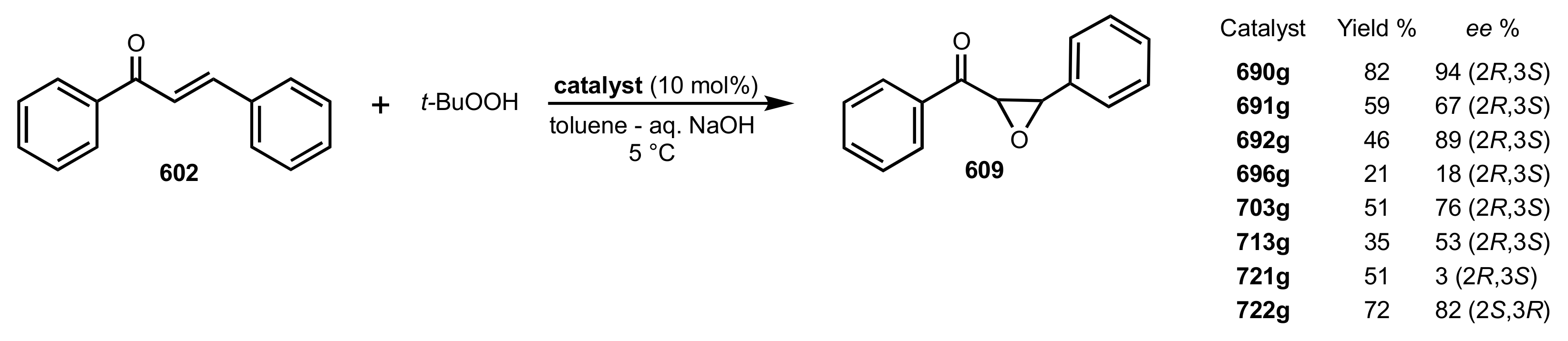
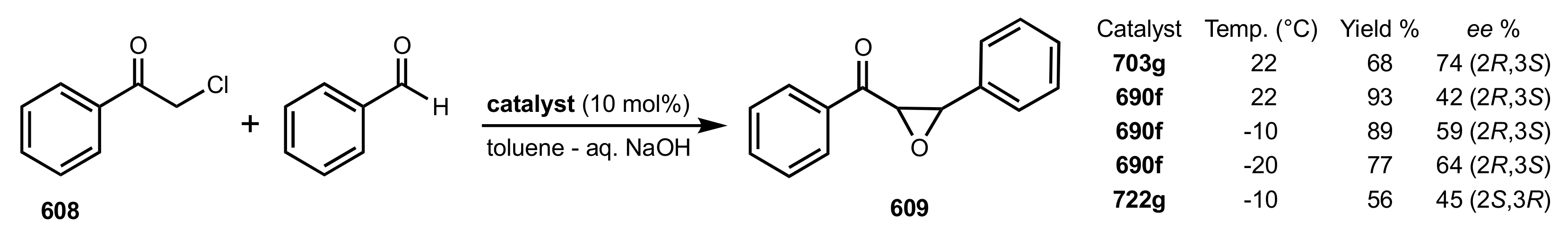
3.1.1. Achievements Reported by Shi and Co-Workers
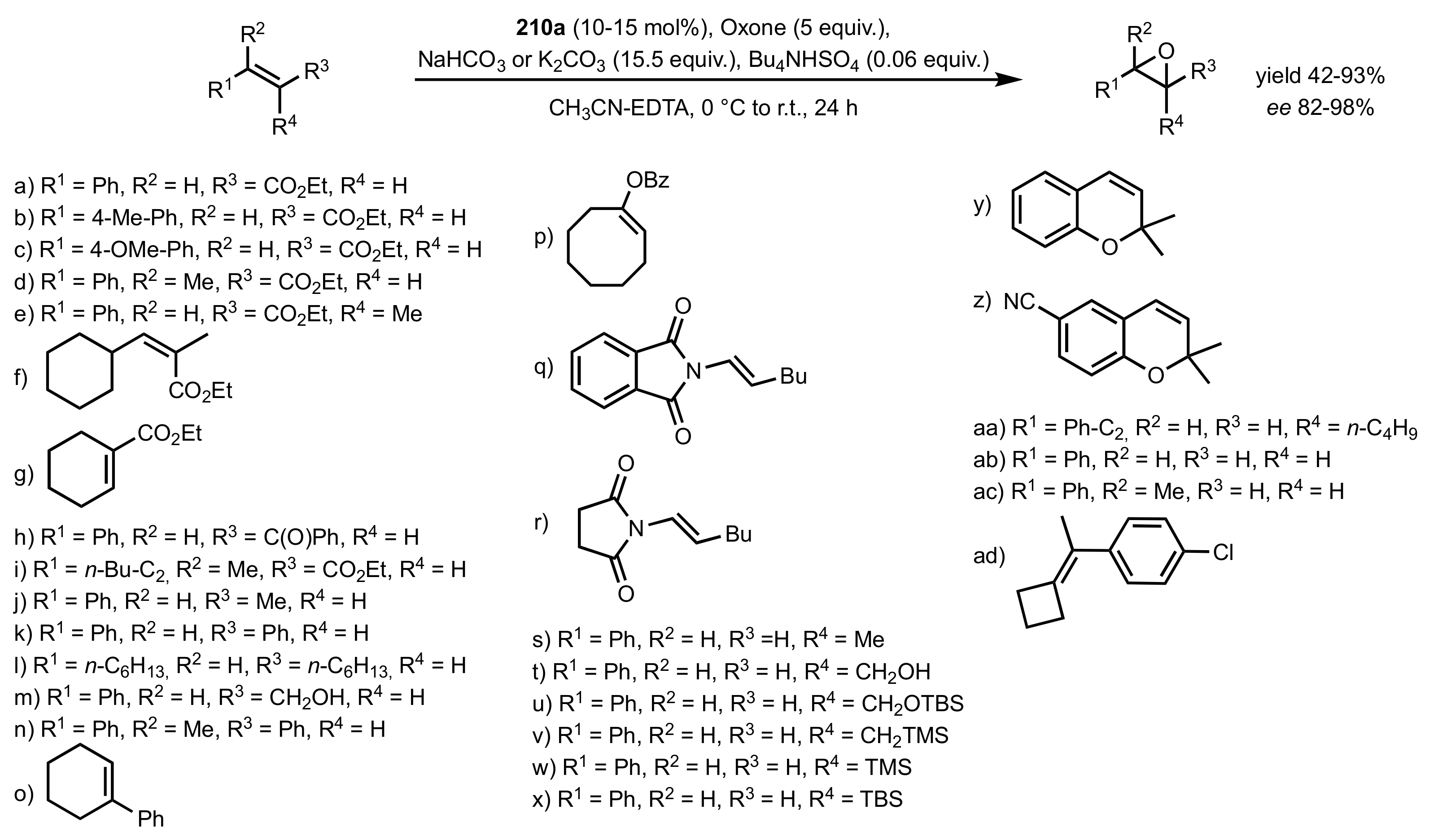
3.1.1.1. Epoxidation of Trans-Alkenes
- Epoxidation of simple trans-alkenes
- New organocatalysts for the epoxidation of trans-alkenes
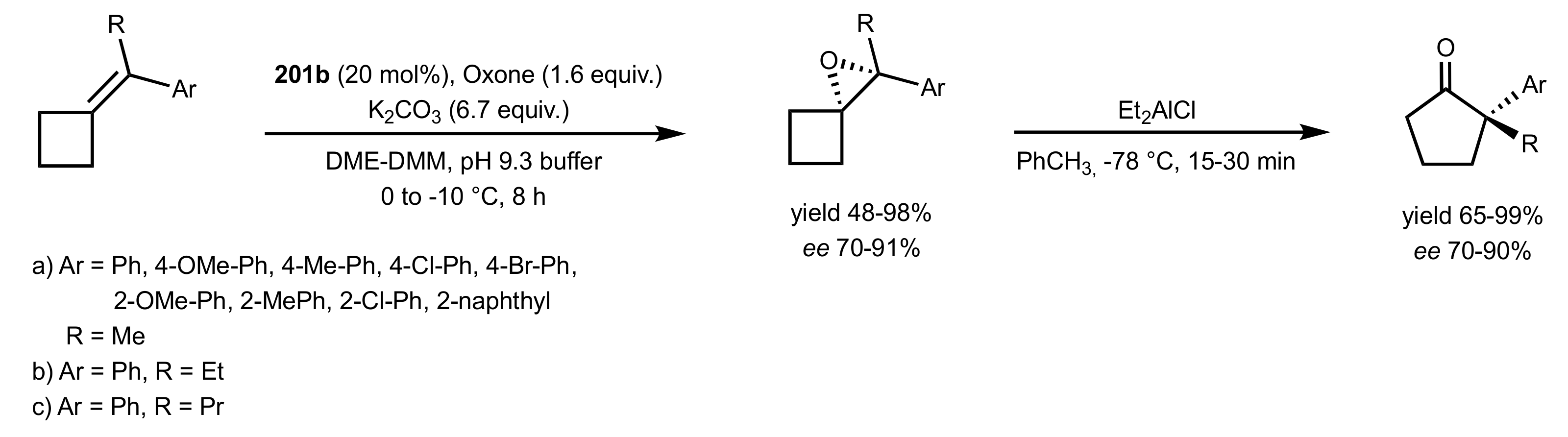
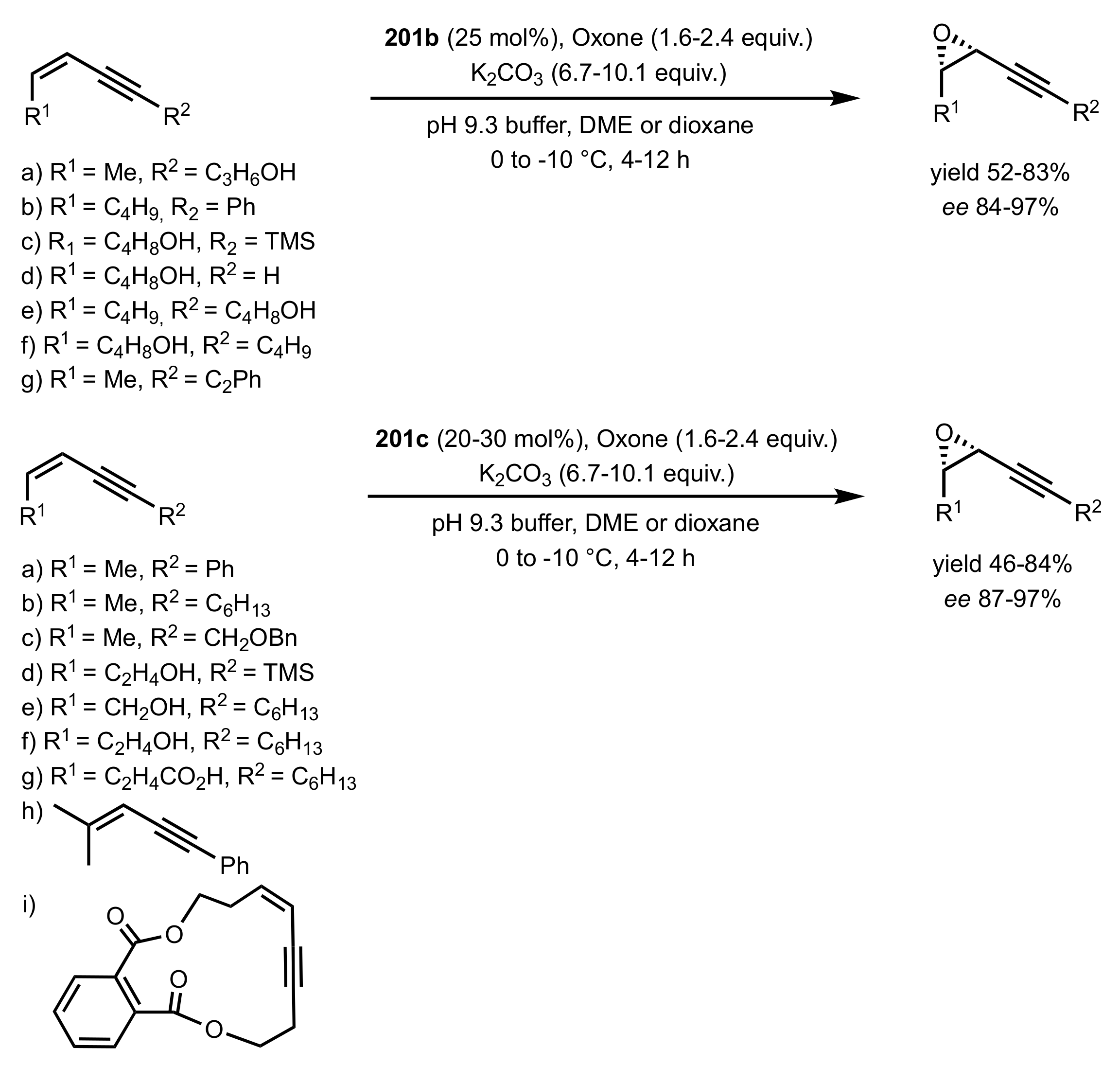
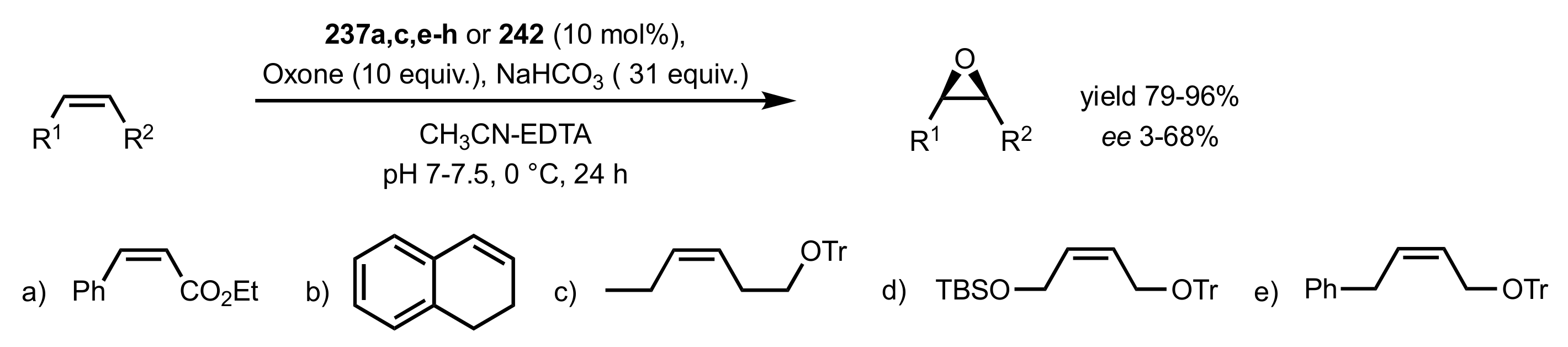
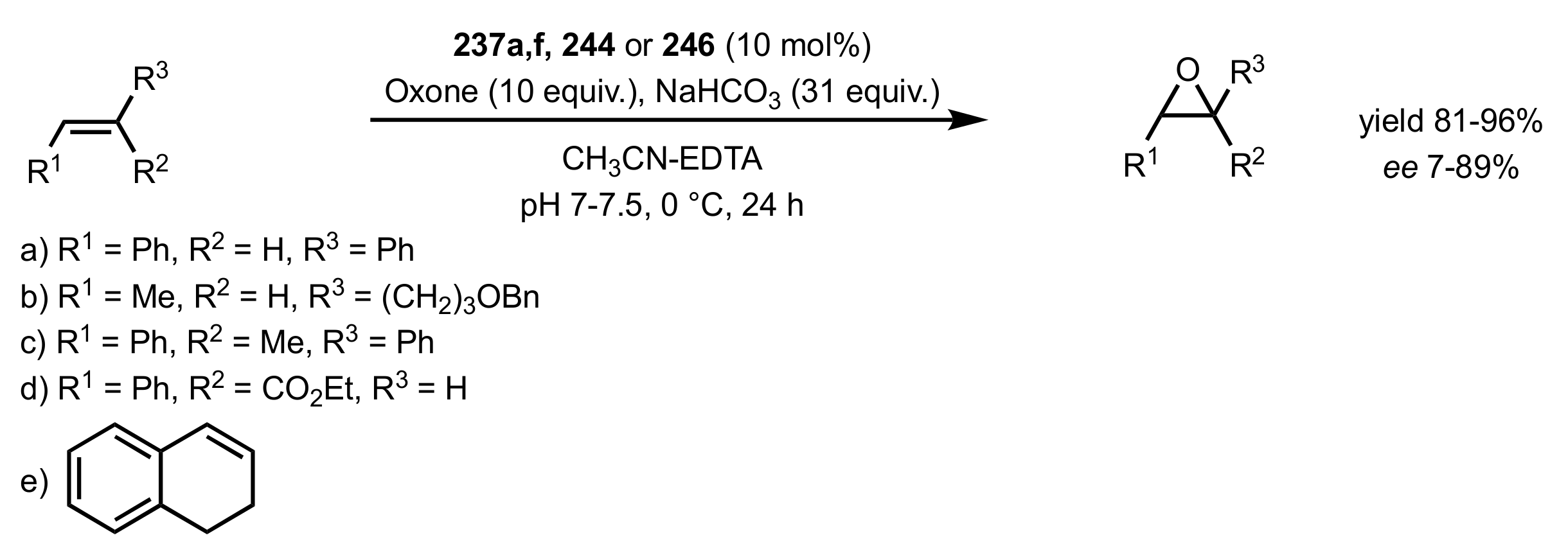
3.1.1.2. Epoxidation of Cis-Alkenes
- Synthesis of nitrogen containing sugar ketones
- Asymmetric epoxidation of cis-alkenes
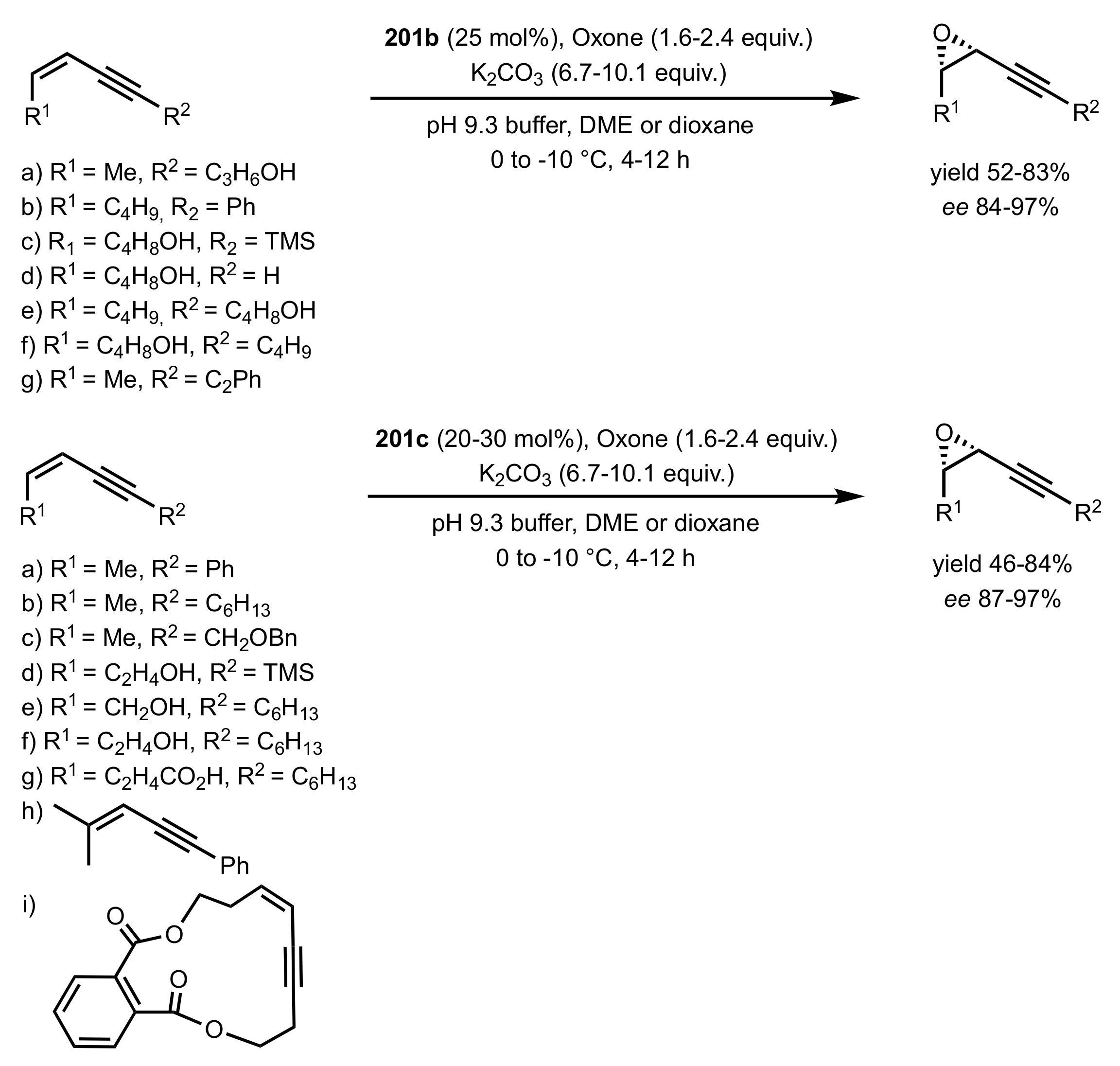
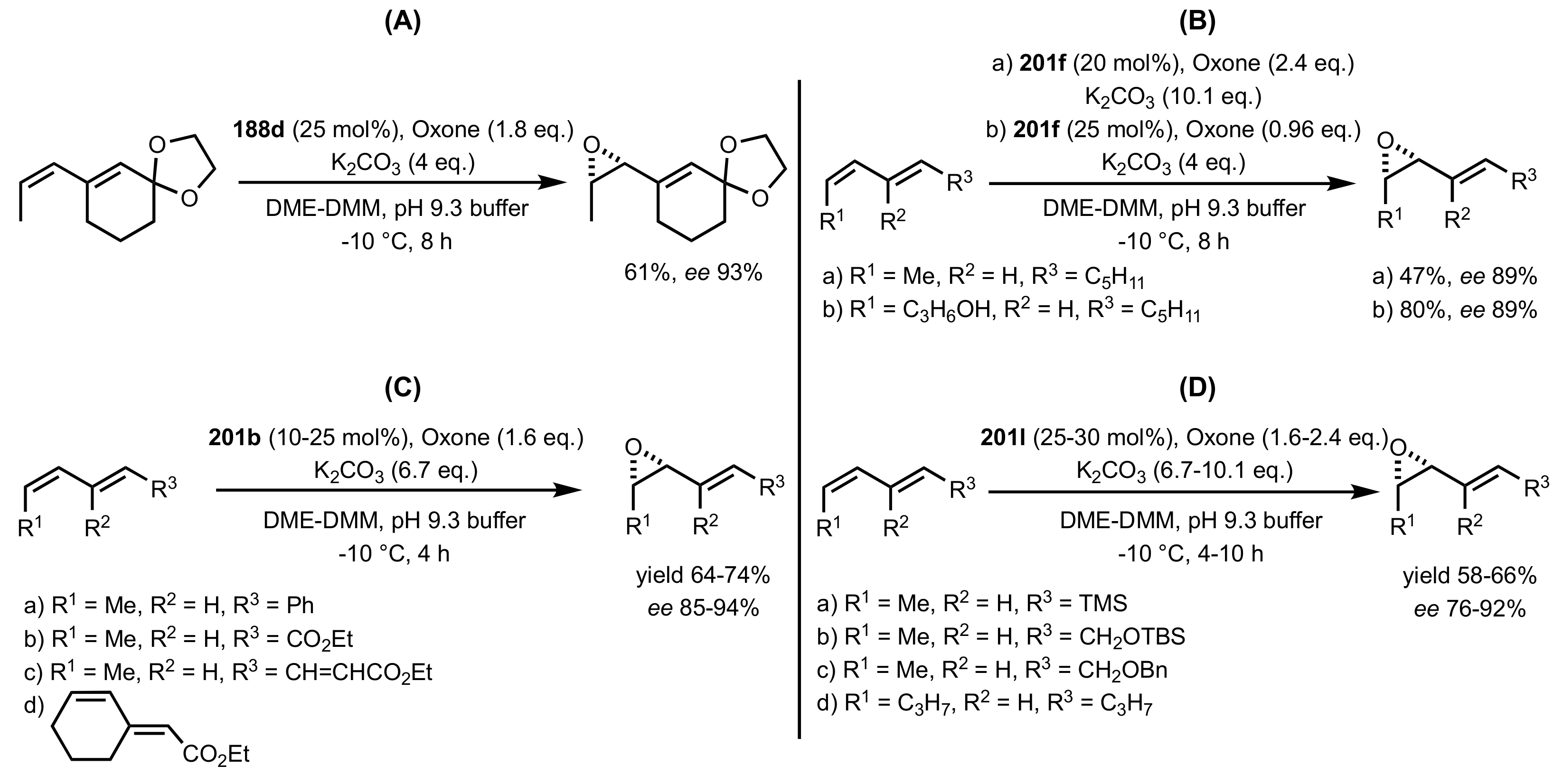
- Epoxidation of conjugated cis-dienes
- Epoxidation of cis-enynes
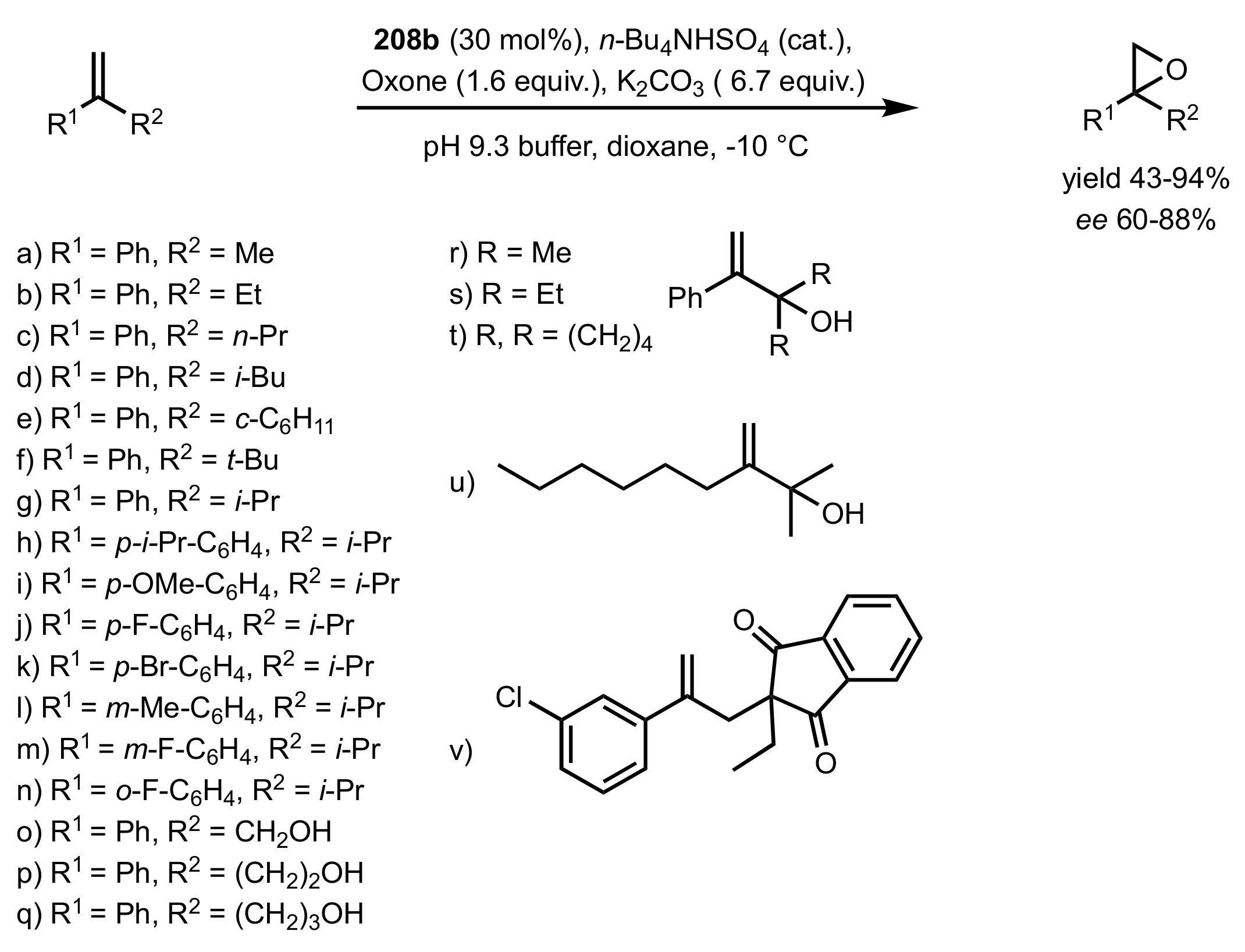
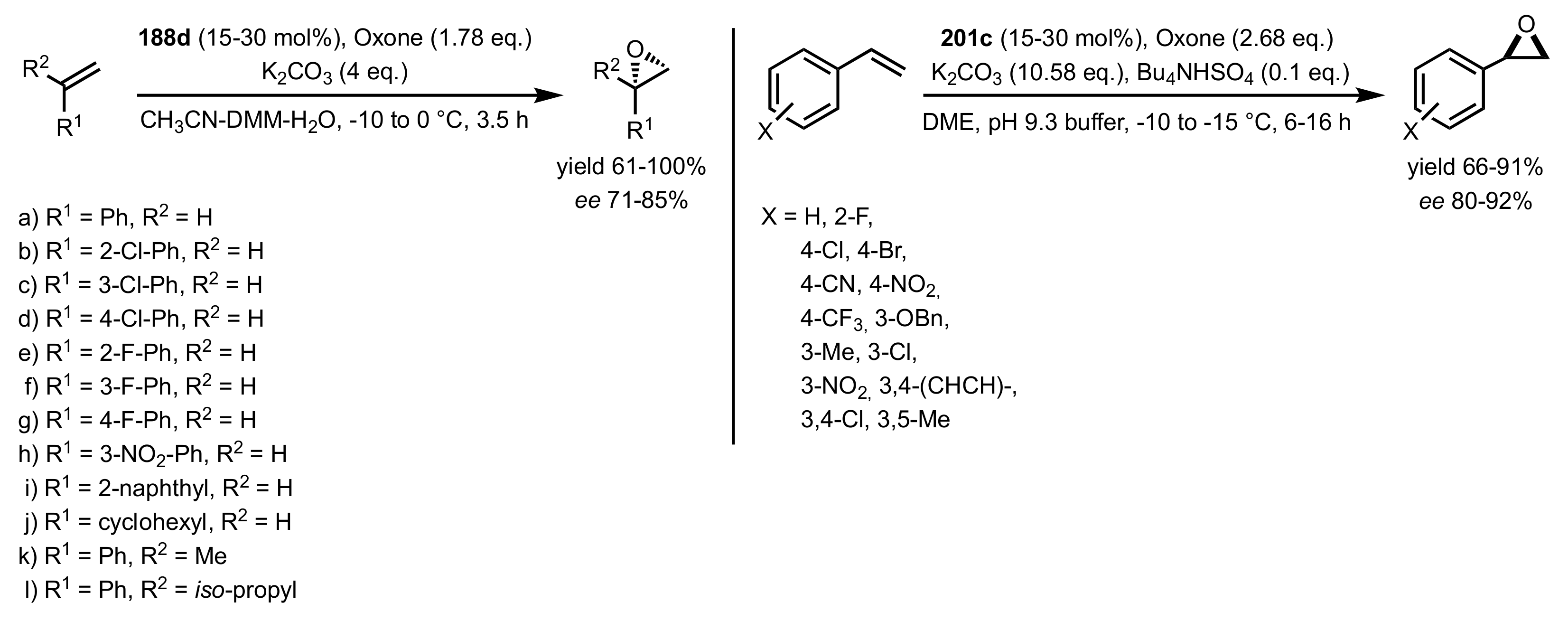
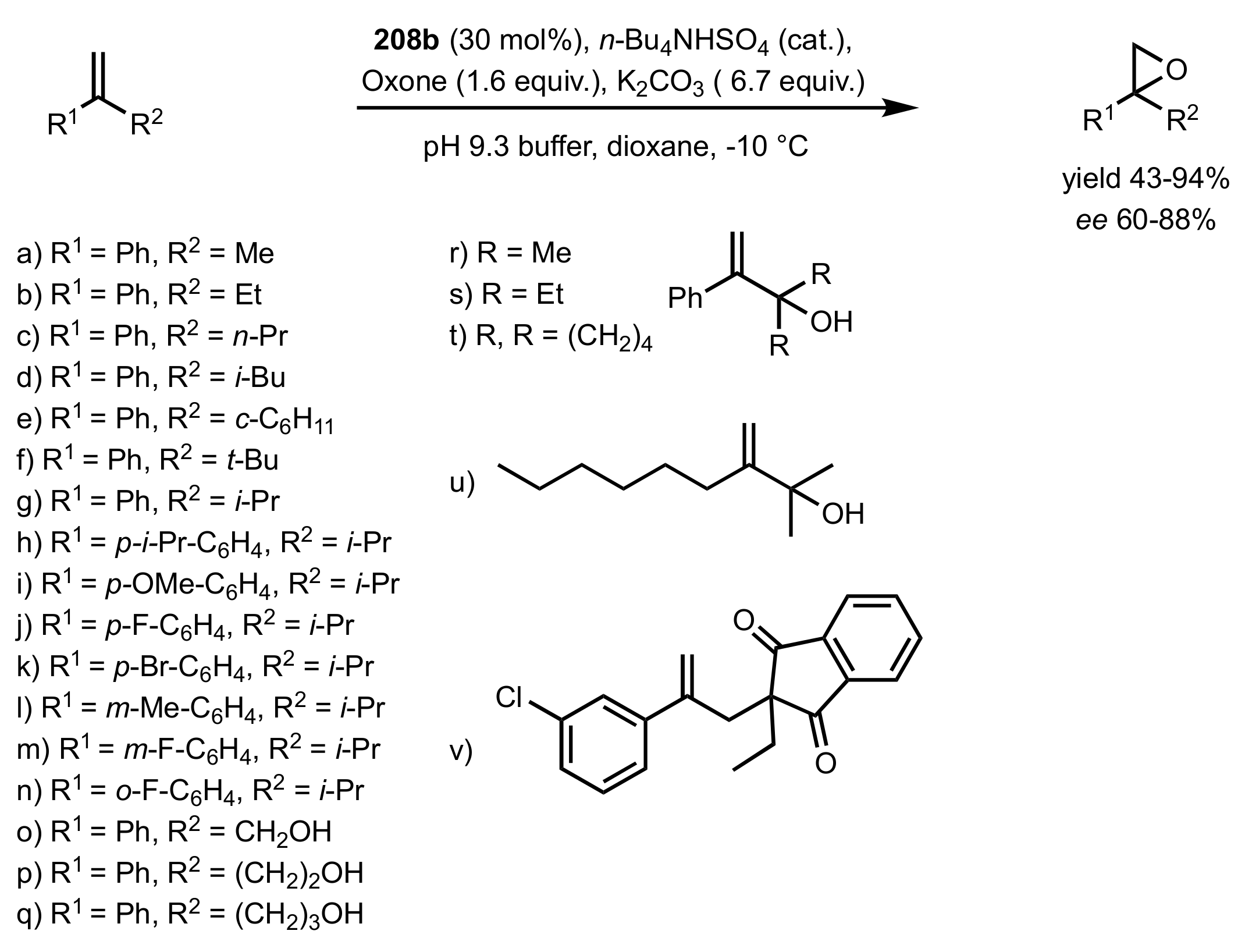
3.1.1.3. Epoxidation of Terminal Alkenes
3.1.1.4. Organocatalysts for the Epoxidation of Tri- and Tetra-Substituted Alkenes
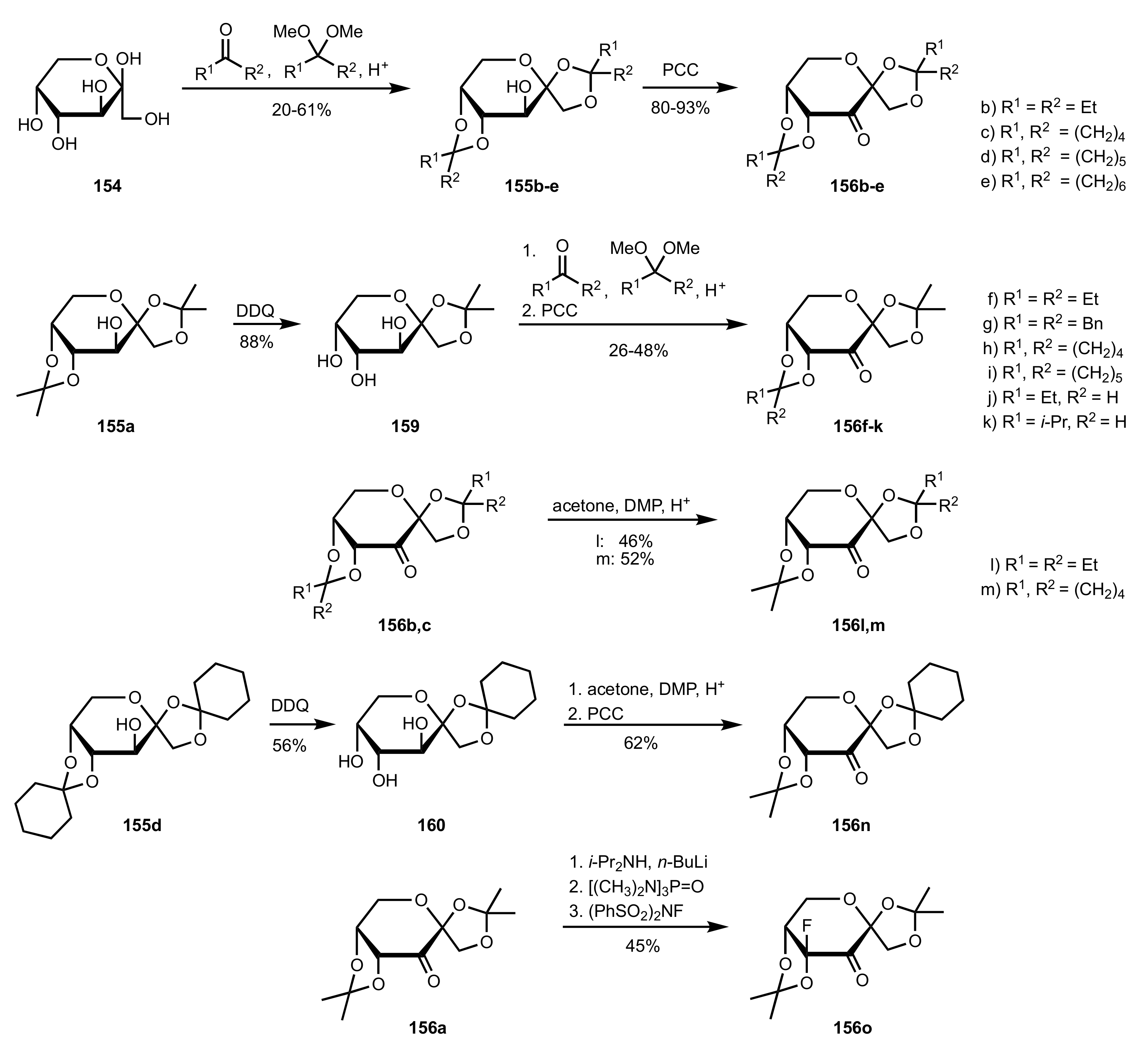
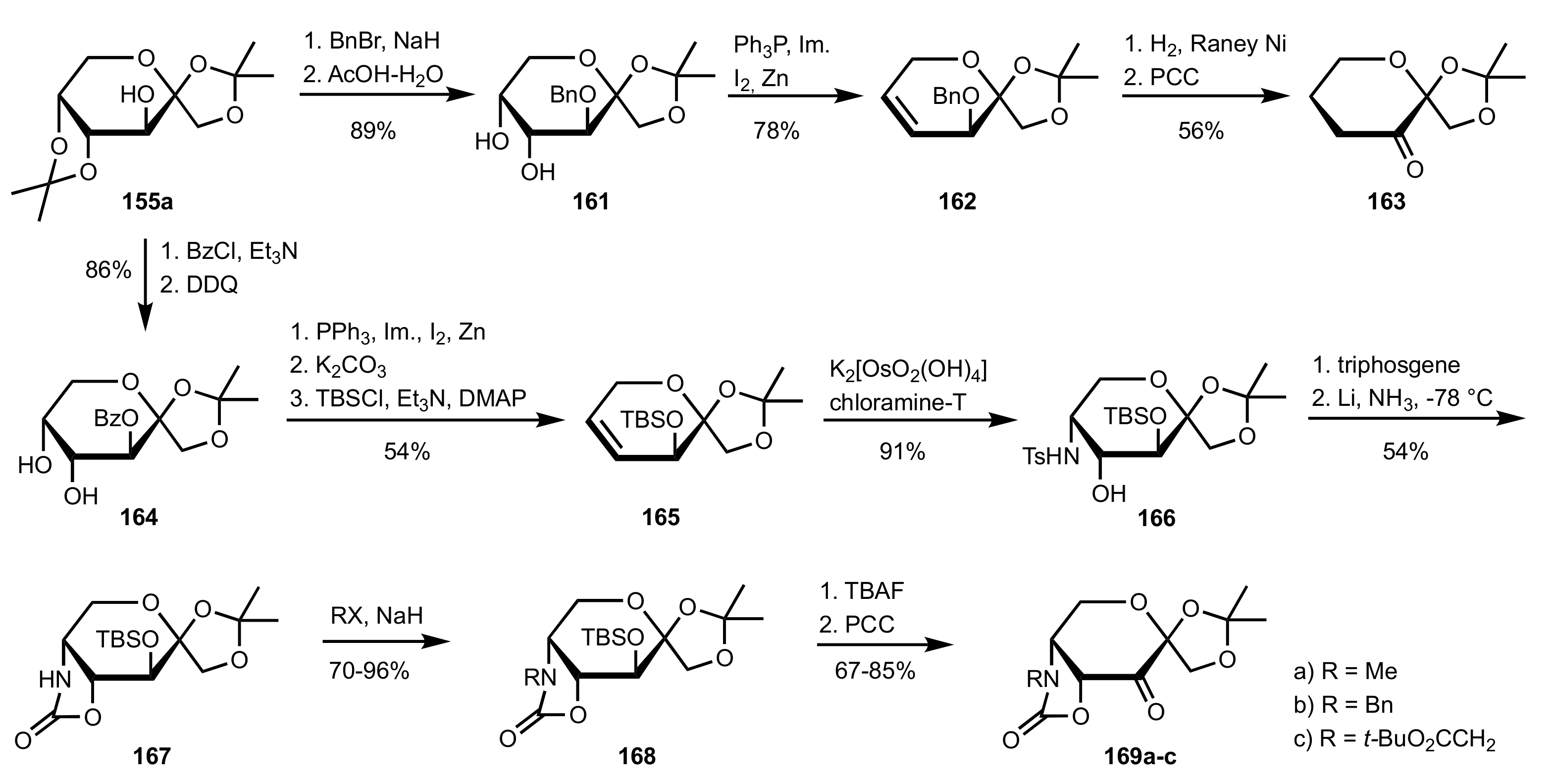
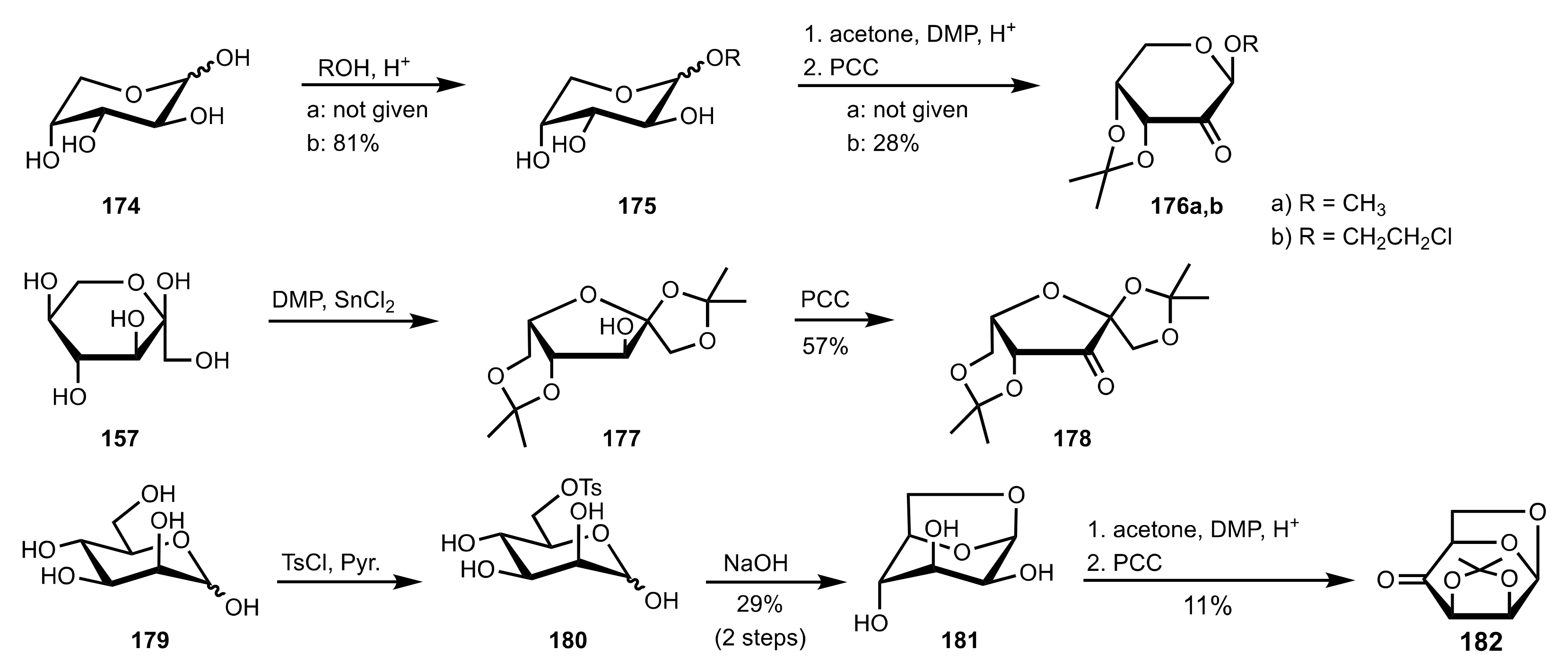
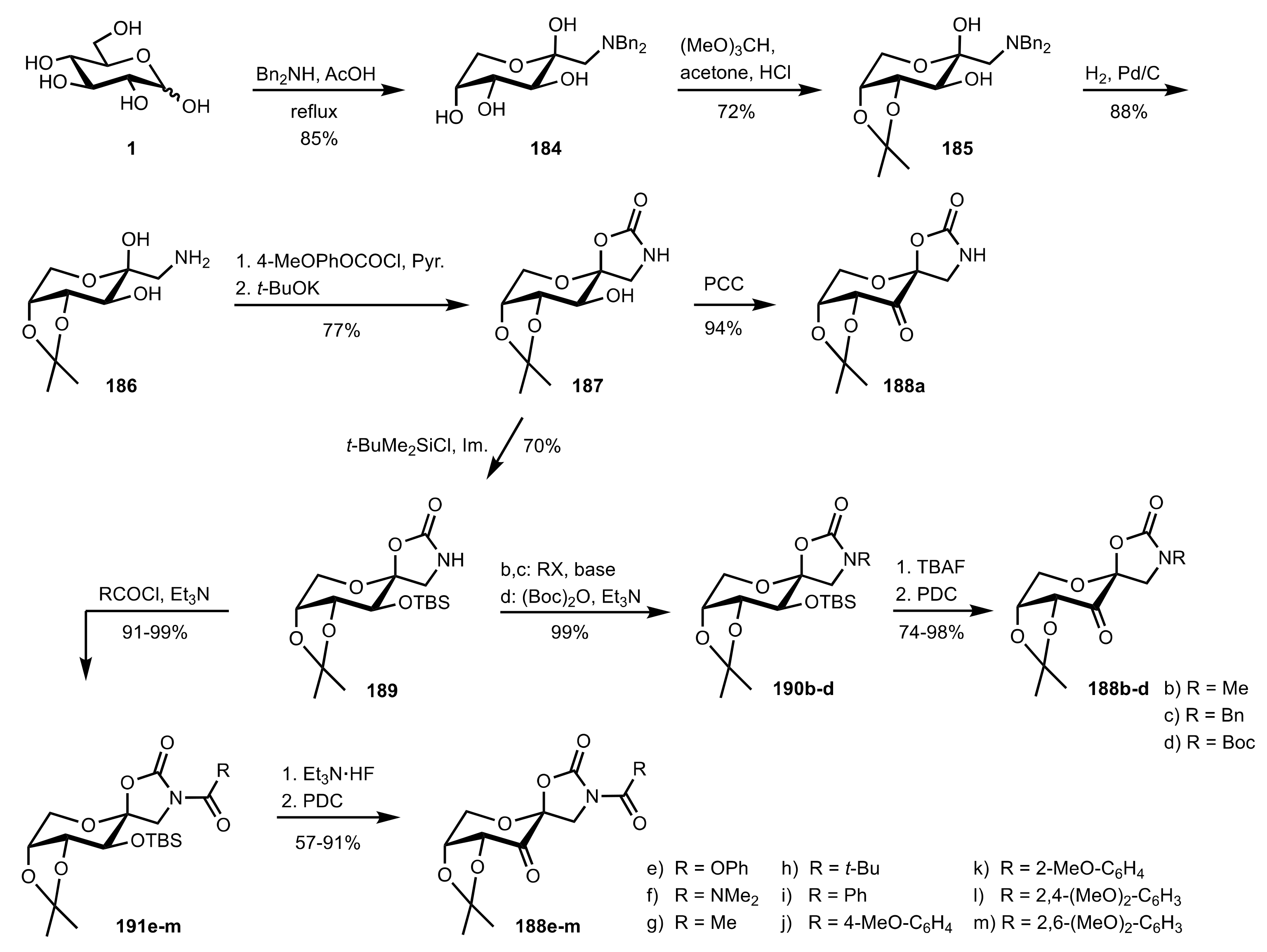
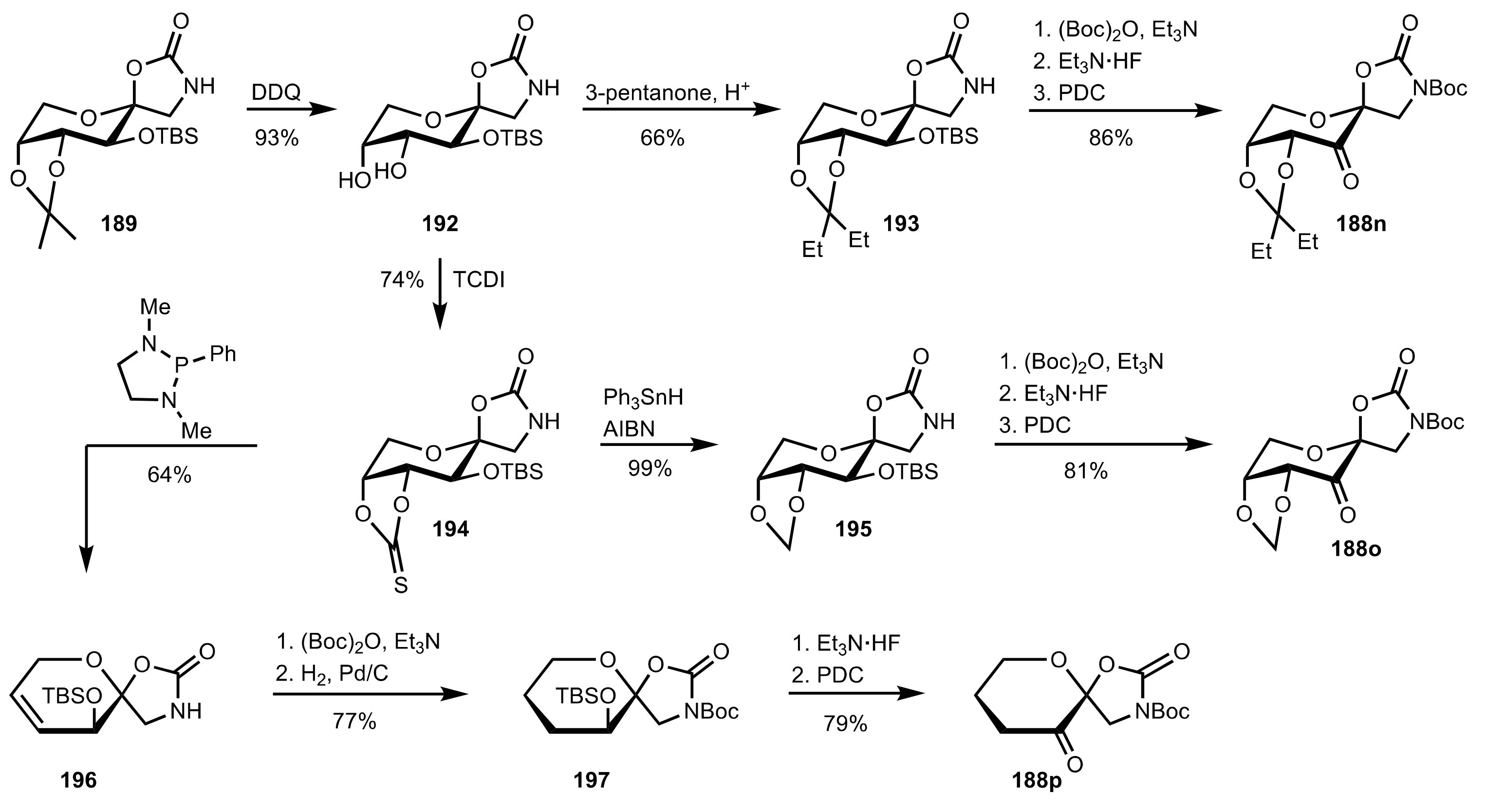
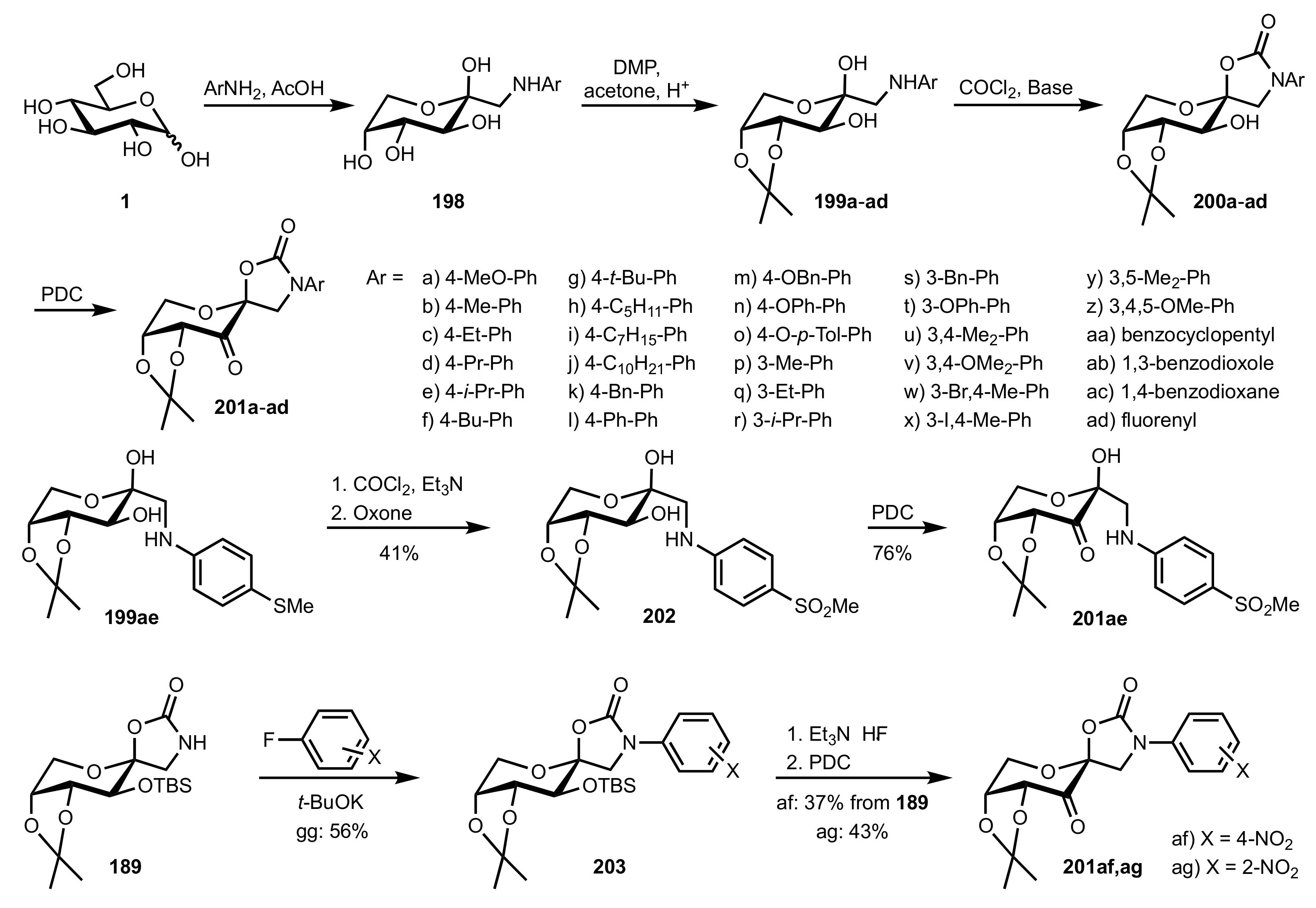
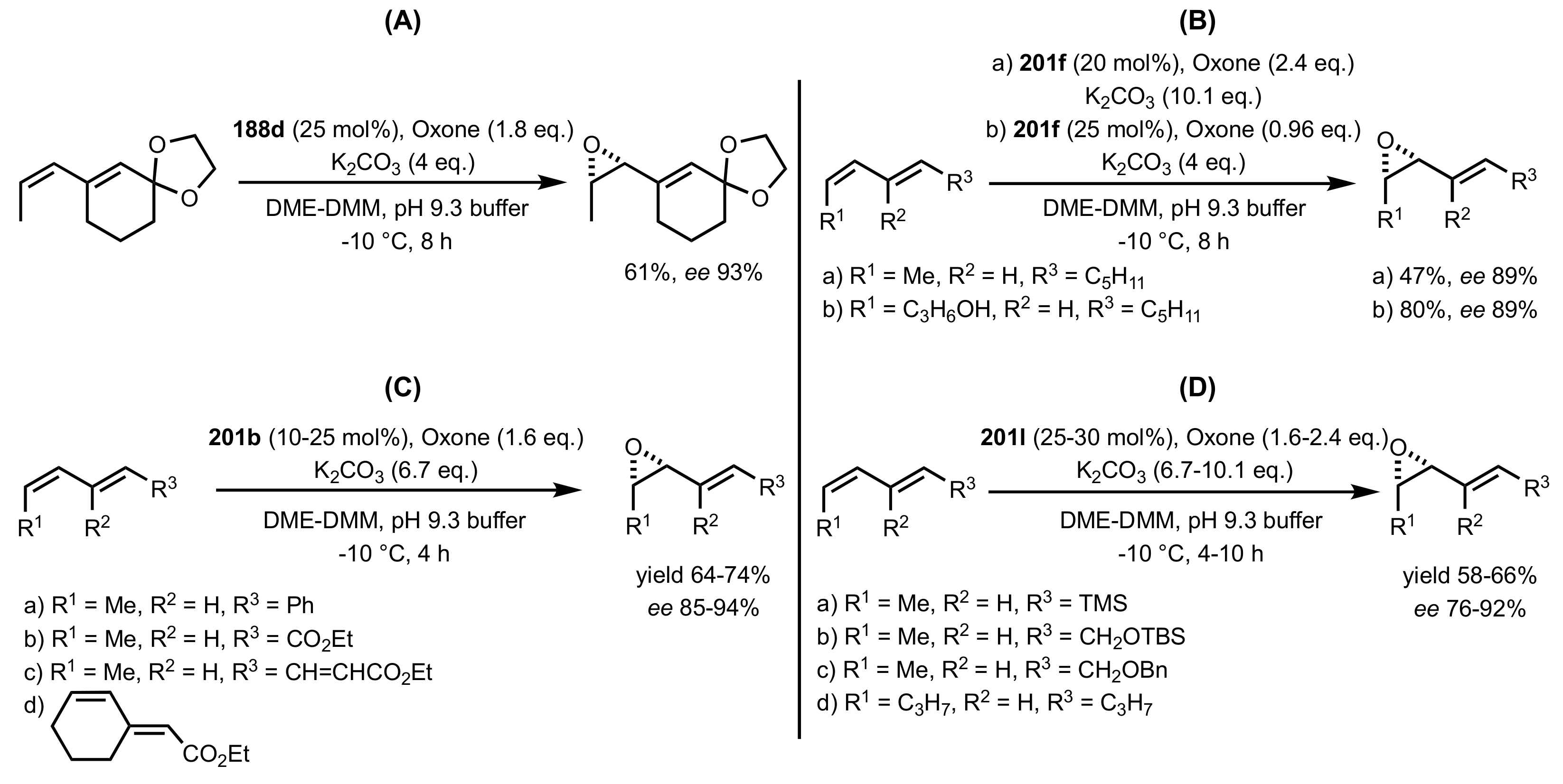
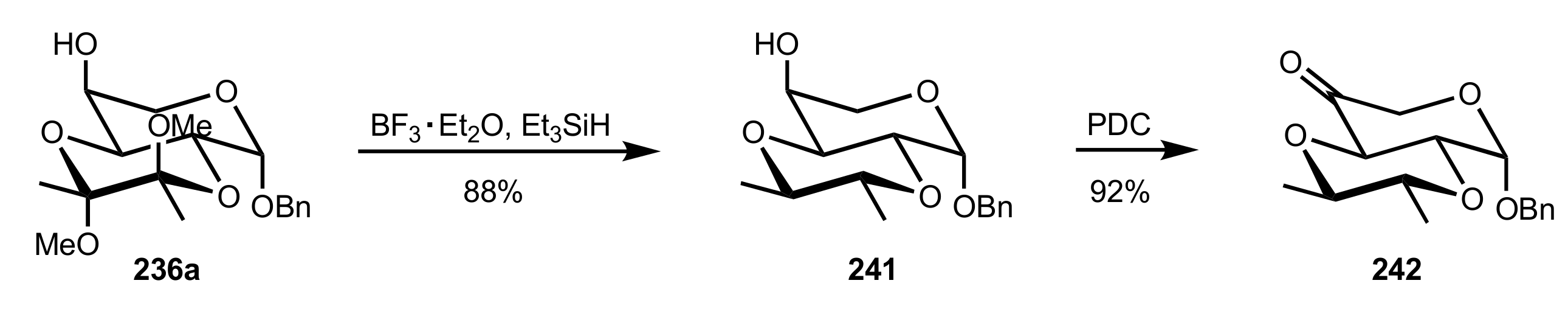
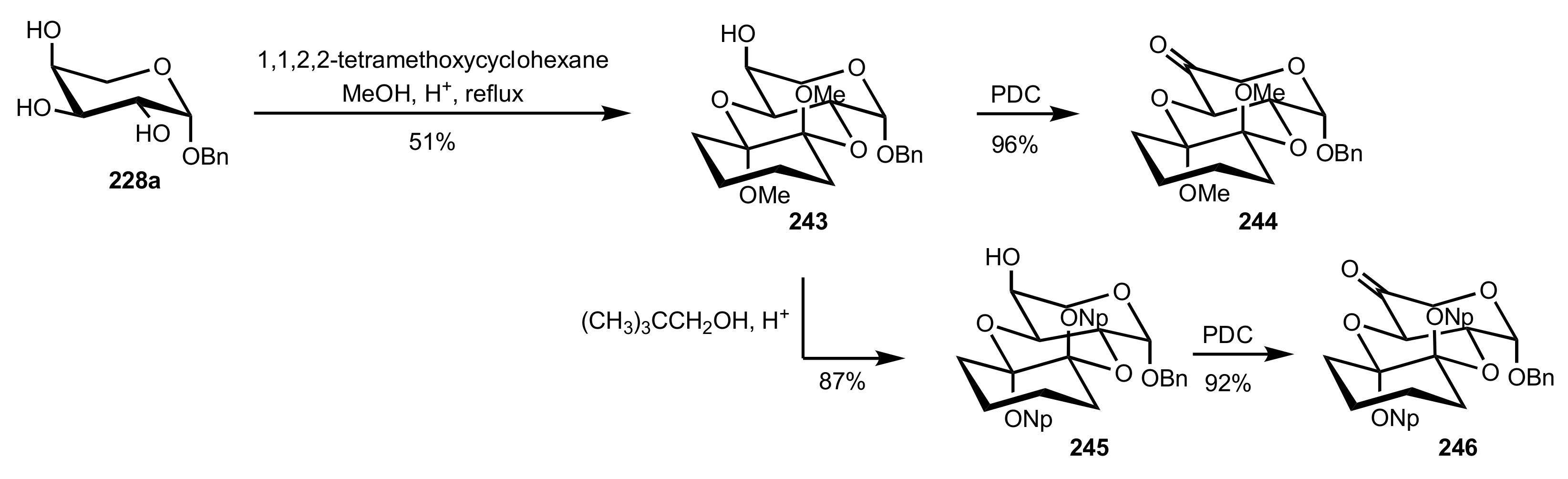
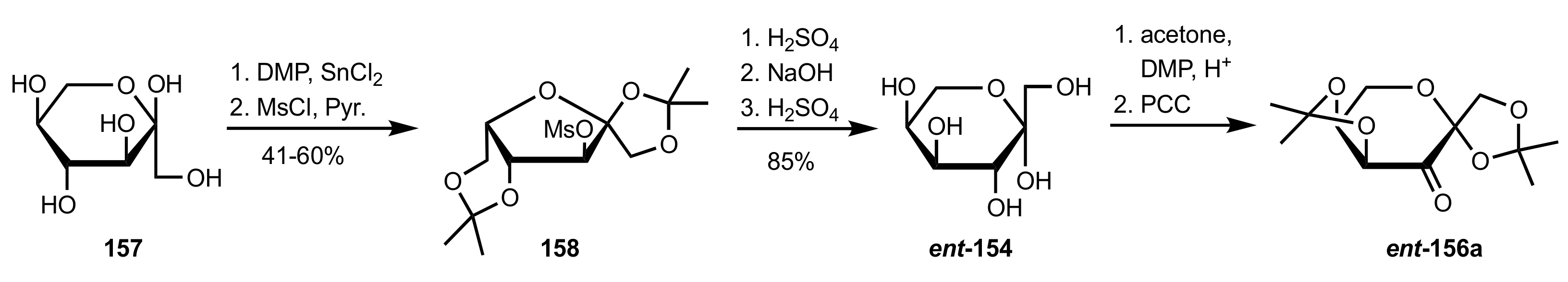
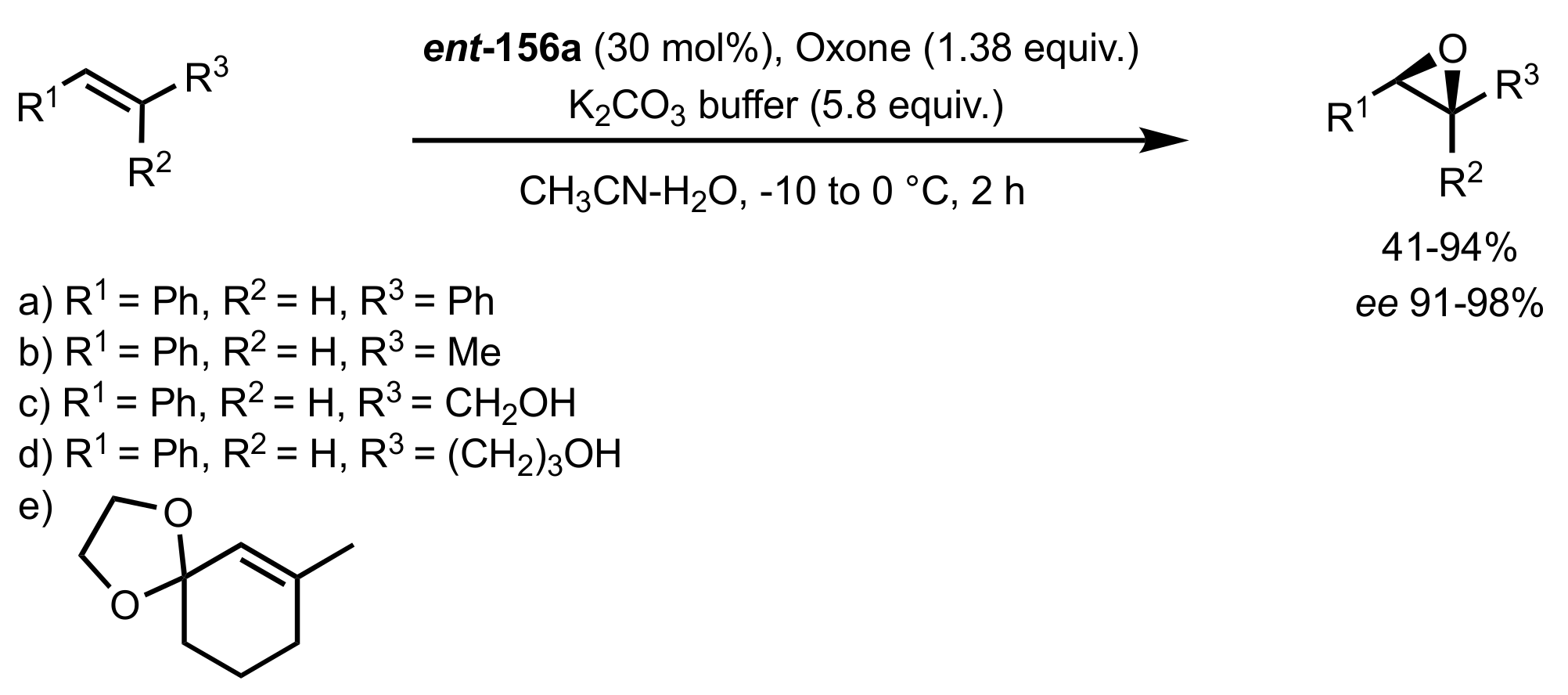
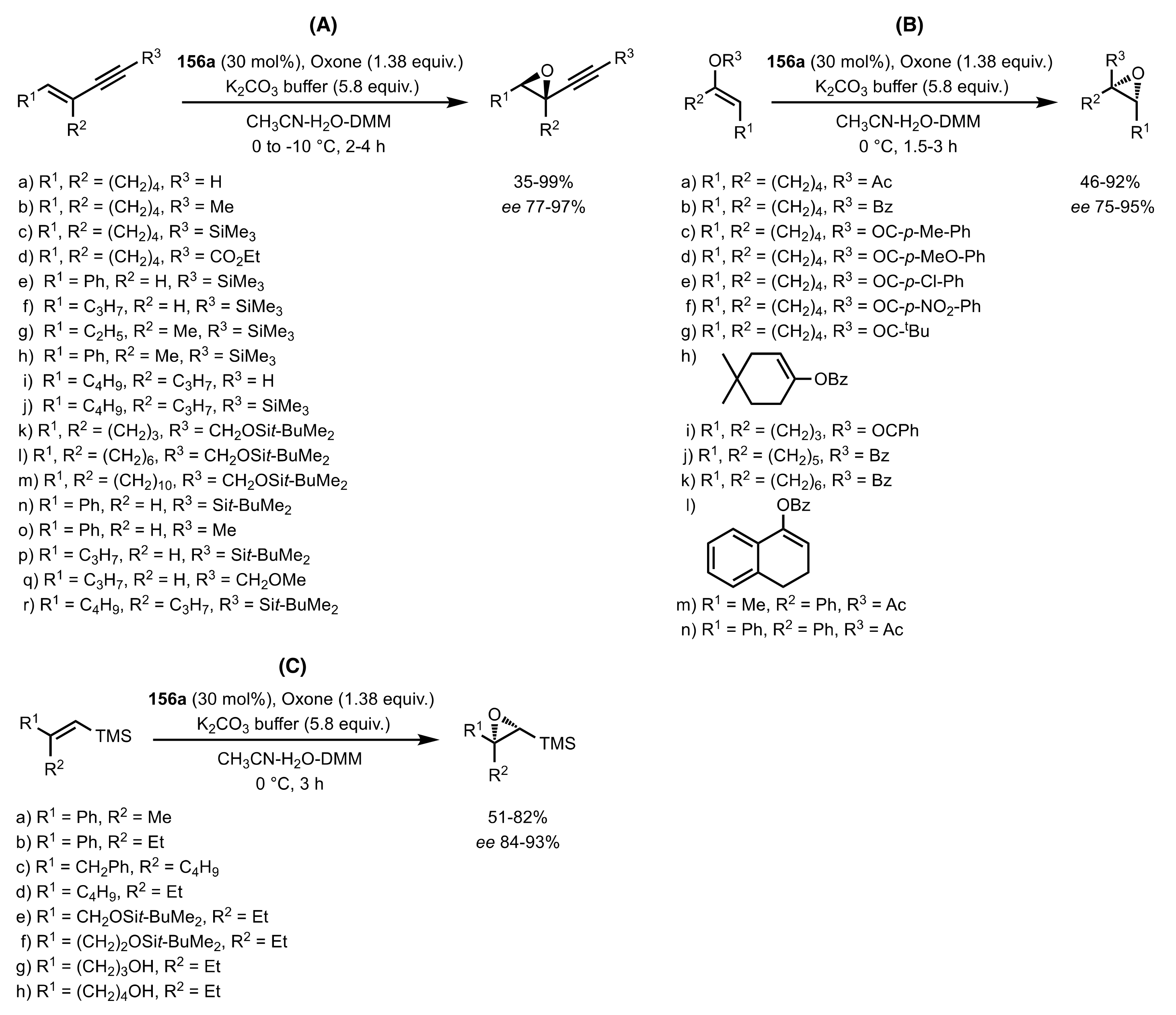
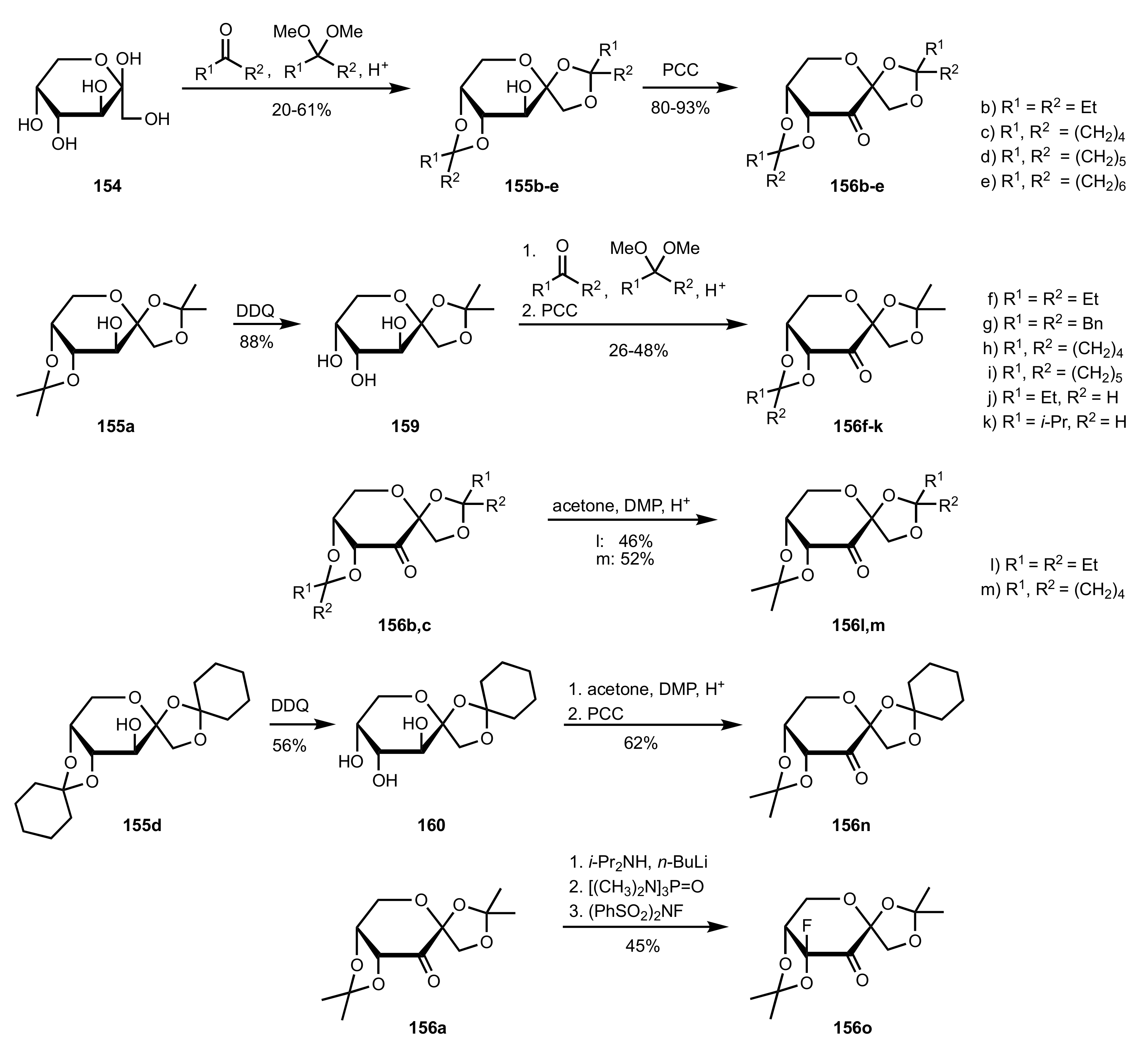
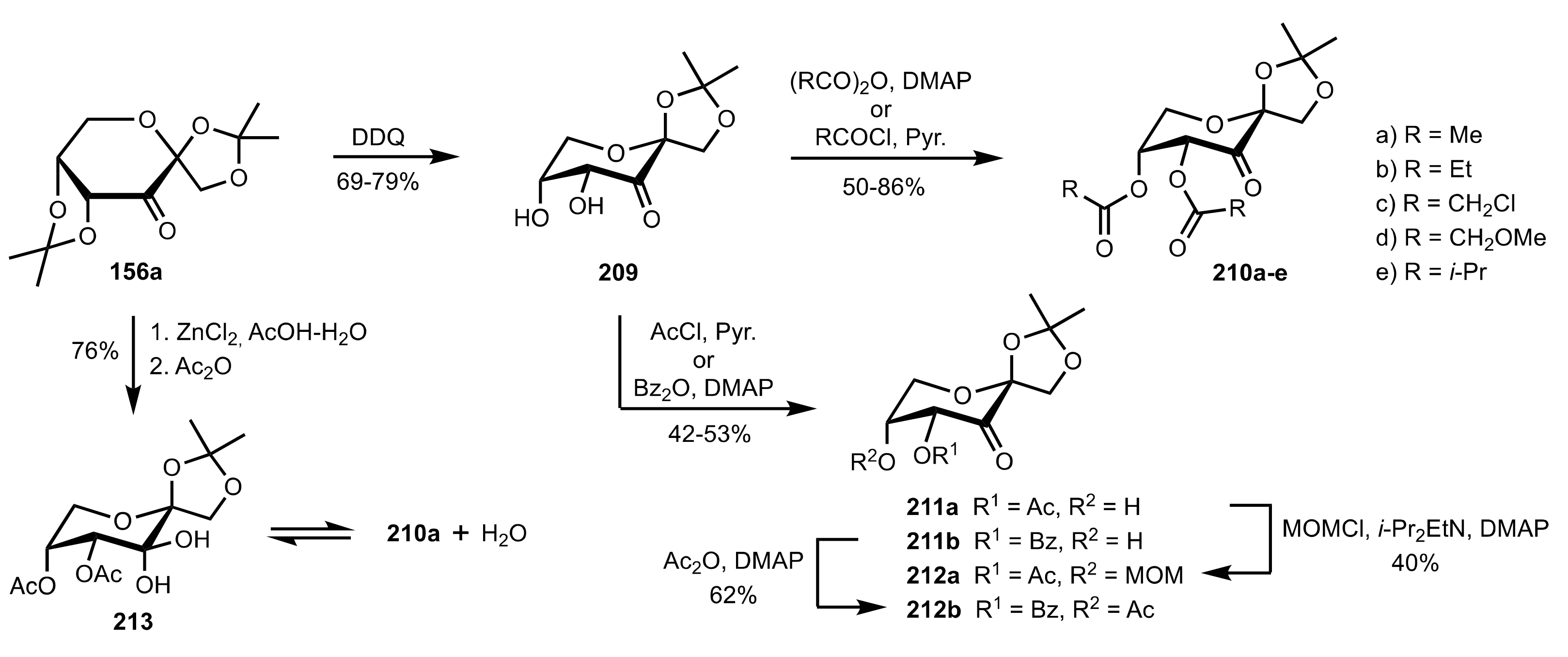
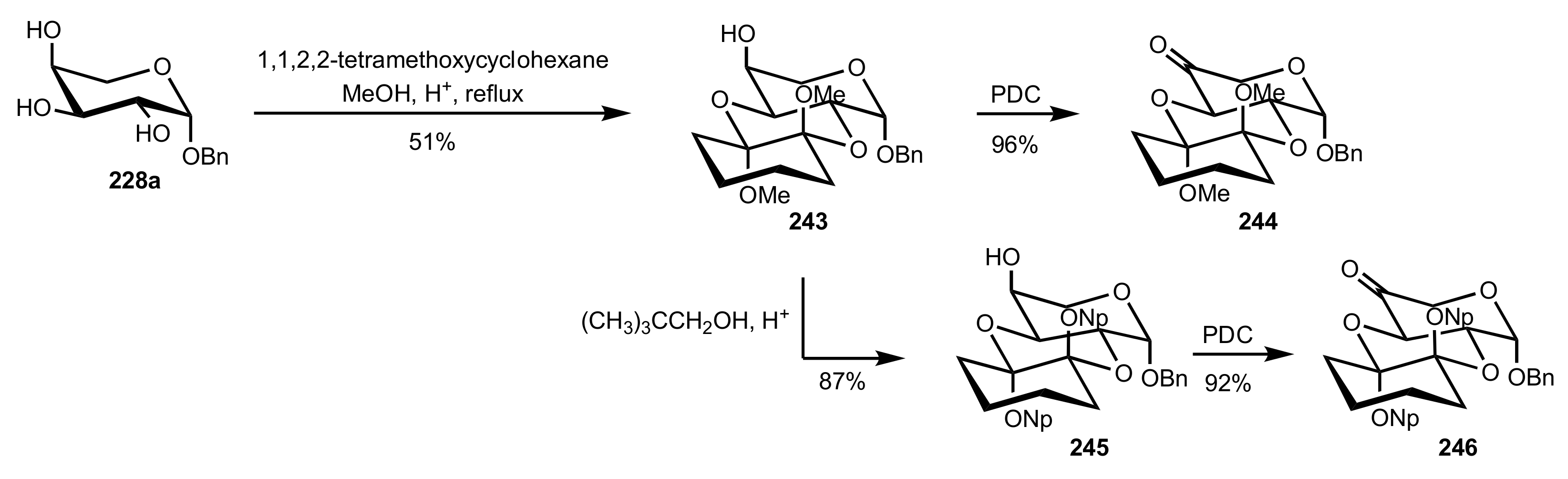
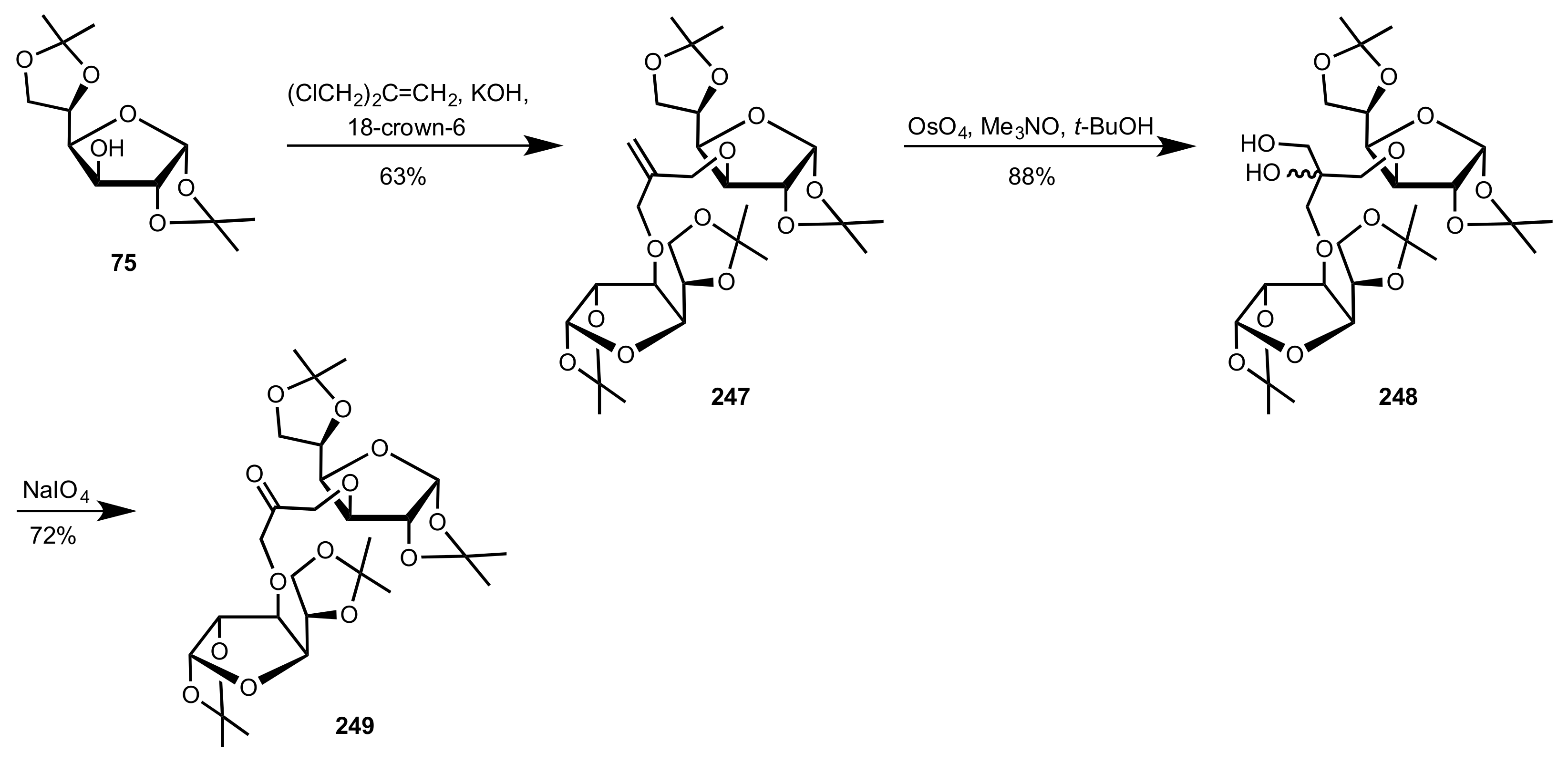
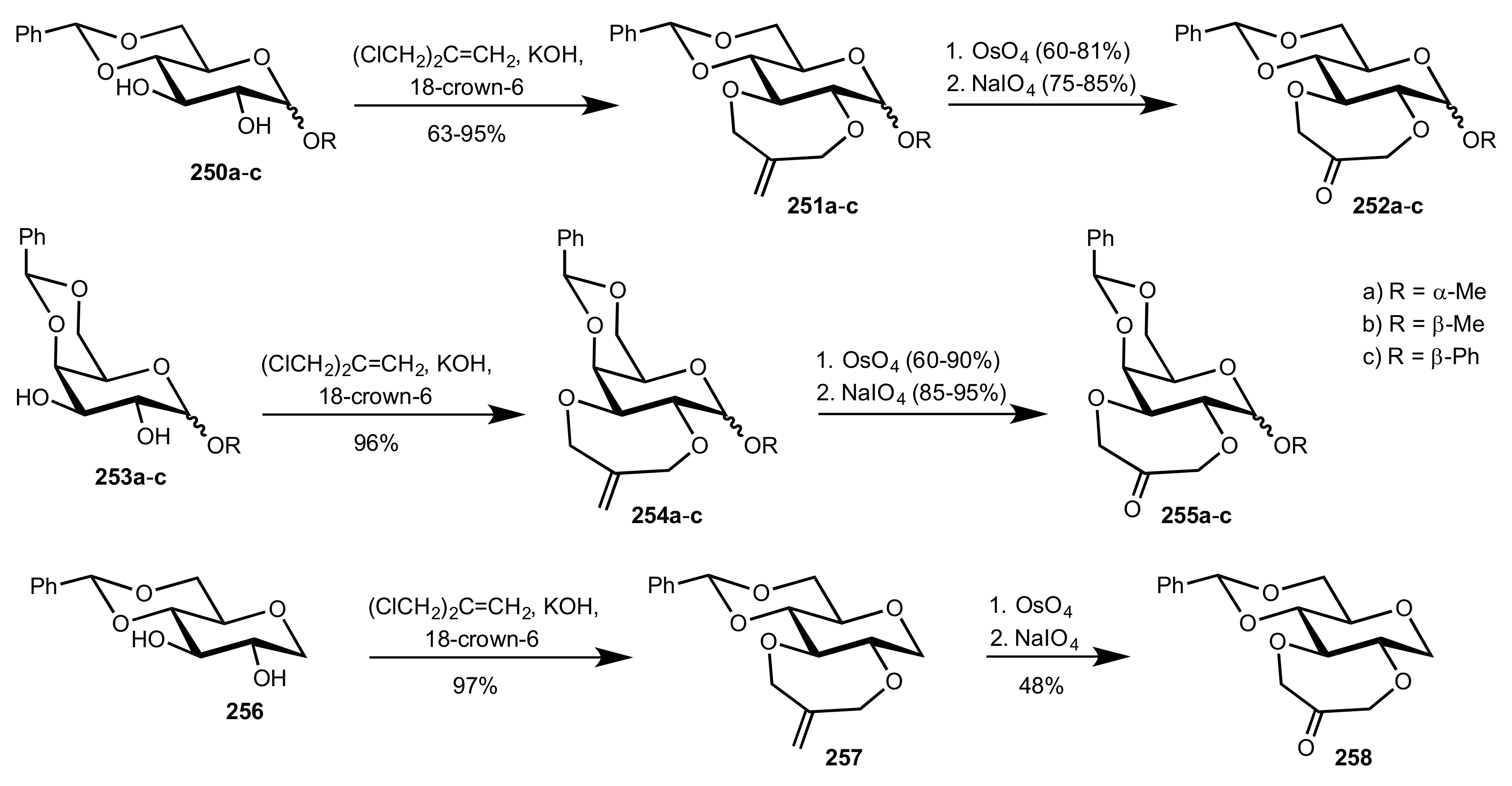
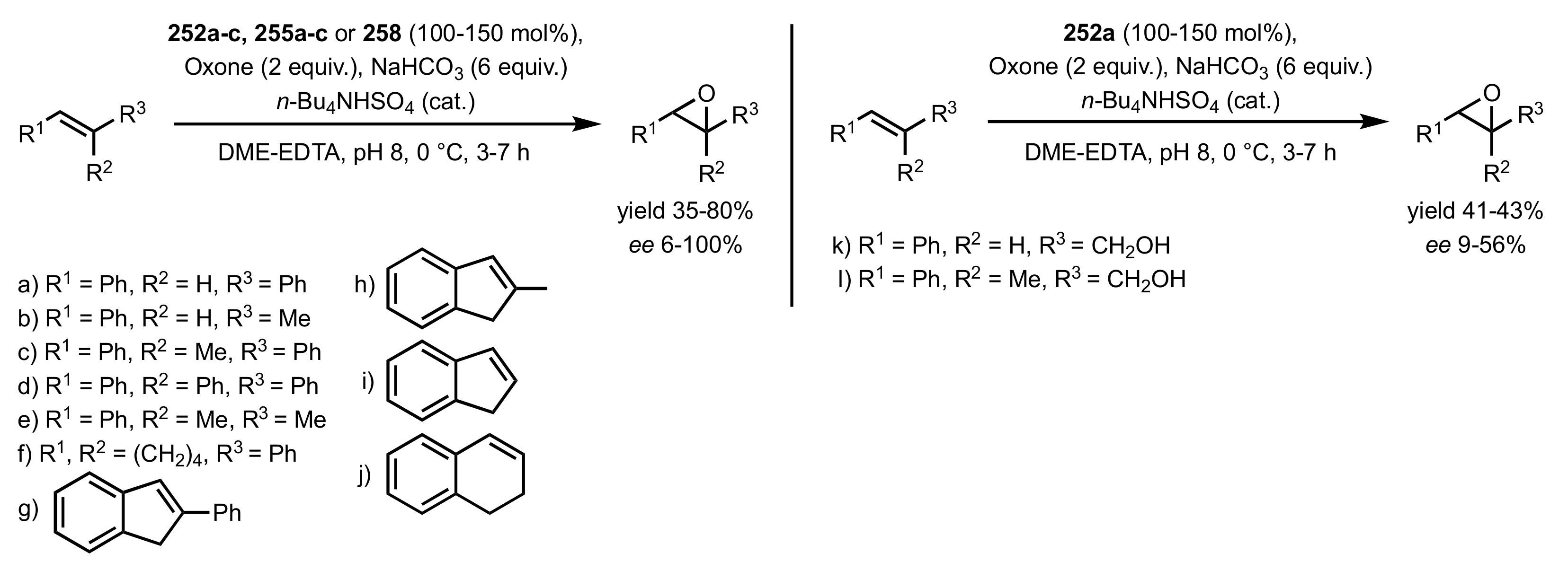
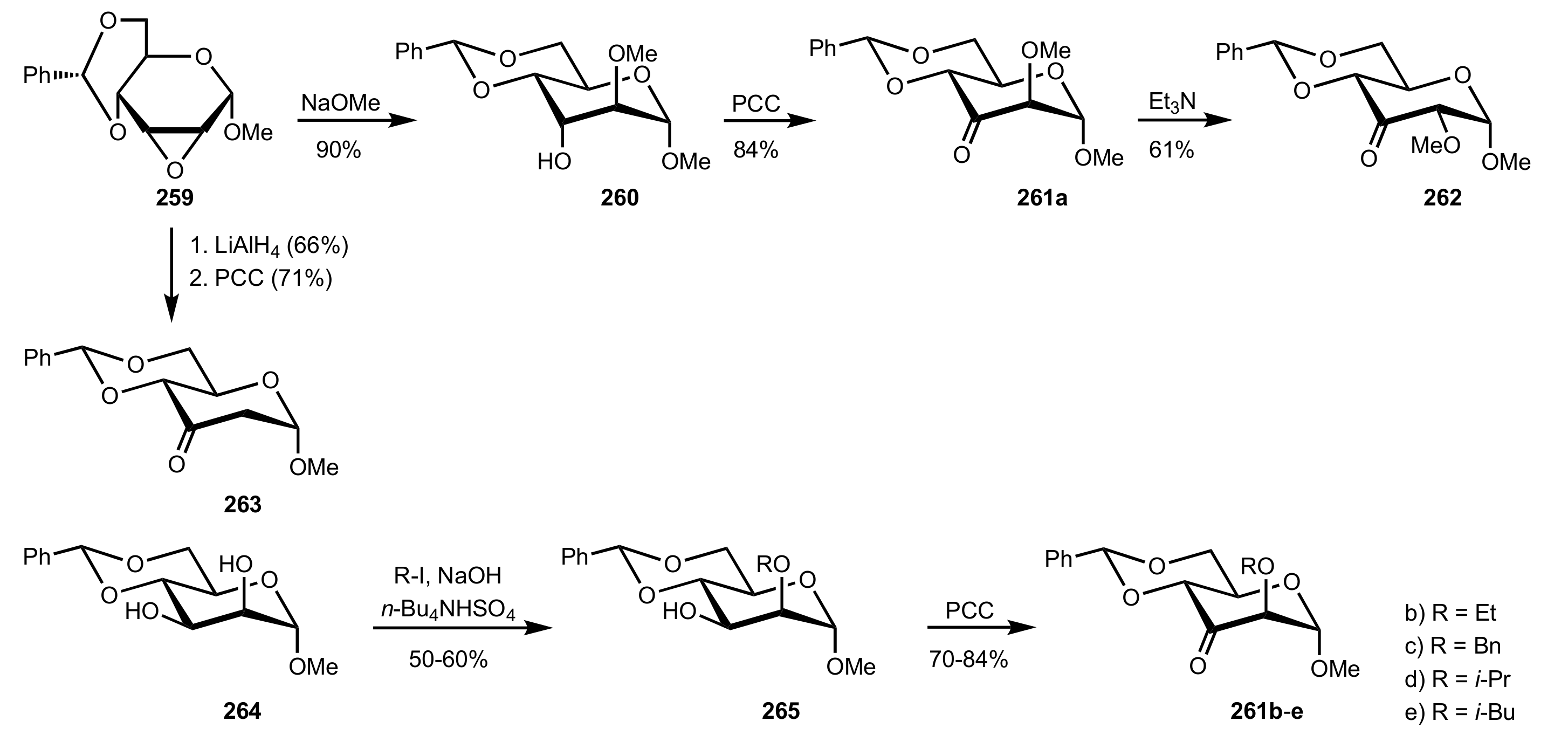
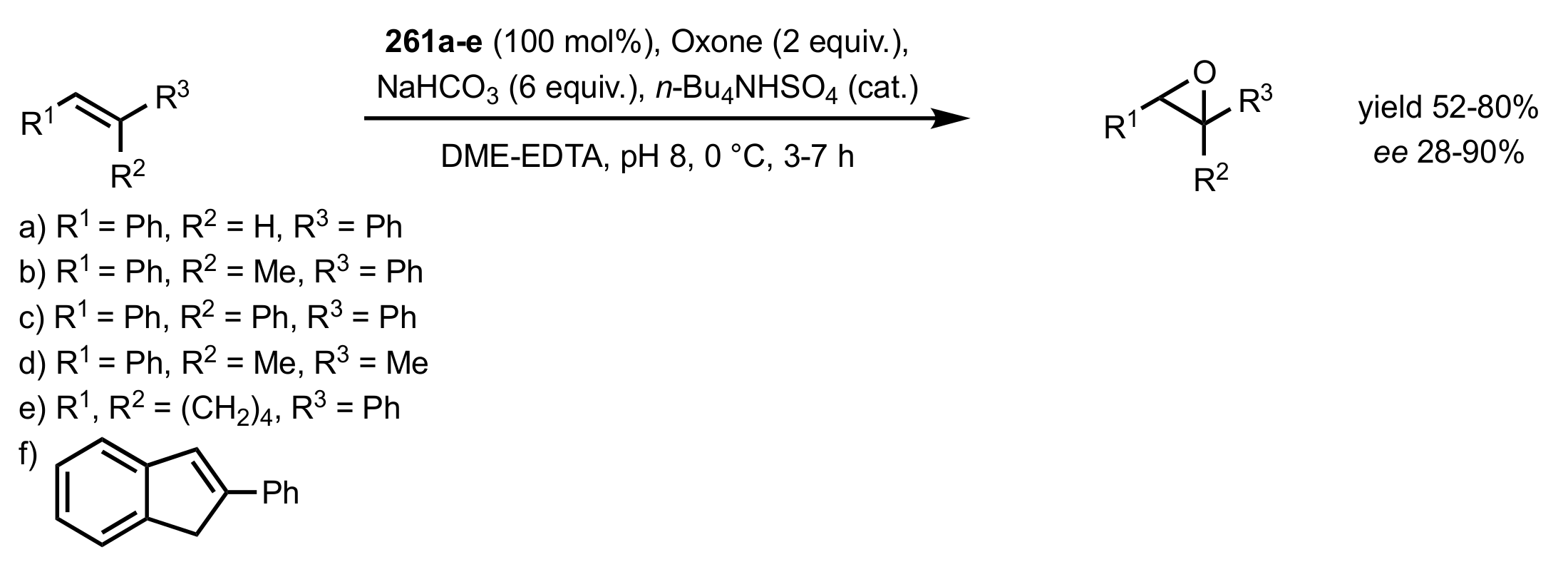
3.1.2. Achievements Reported by Shing and Co-Workers
3.1.3. Achievements Reported by Vega-Pérez, Iglesias-Guerra and Their Co-Workers
3.2. Enantioselective Oxidation of Disulfides
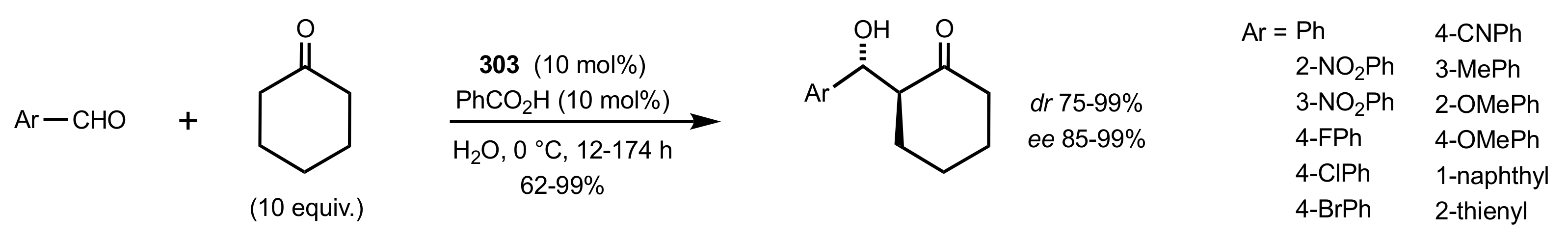
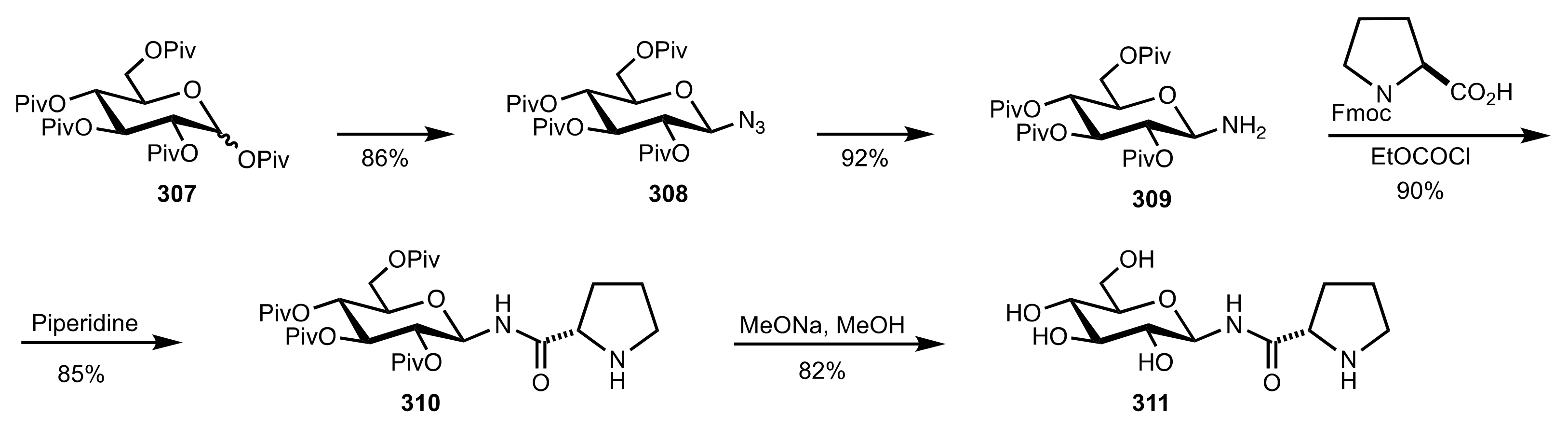
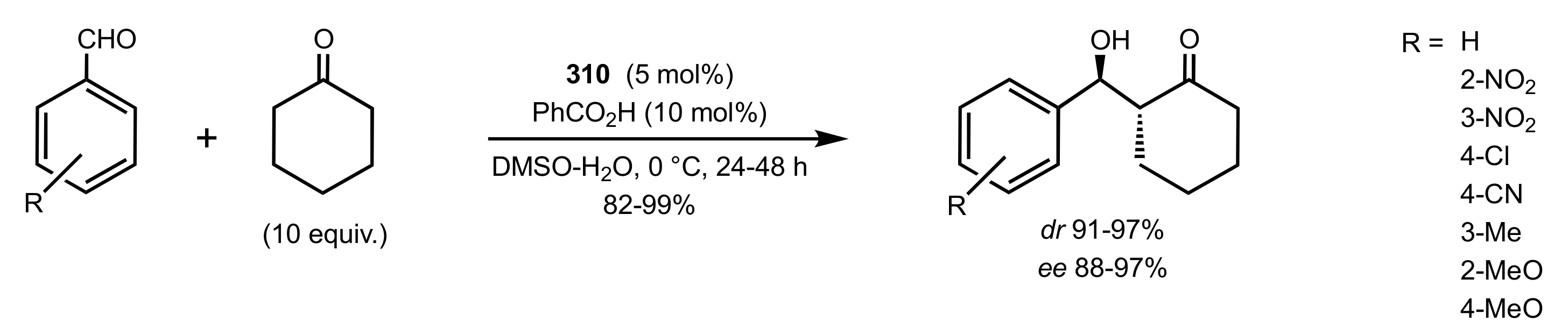
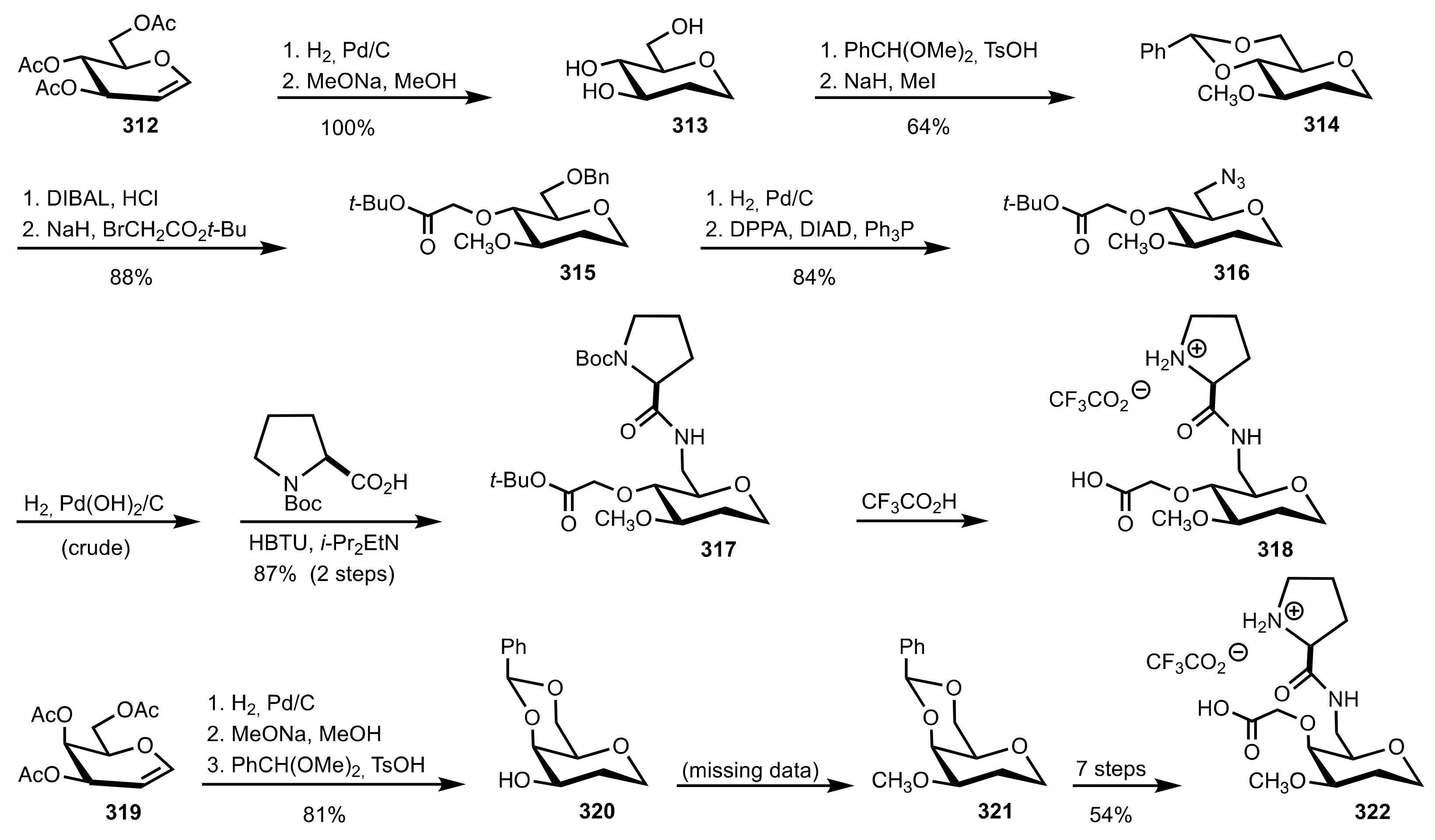
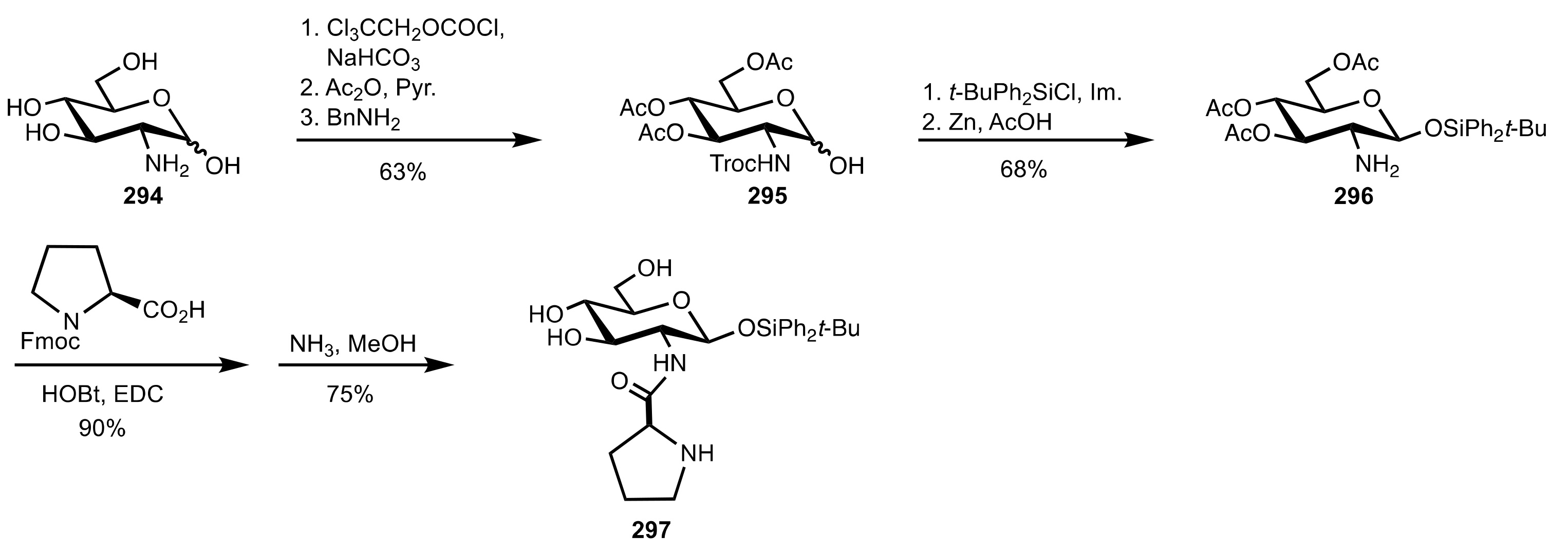
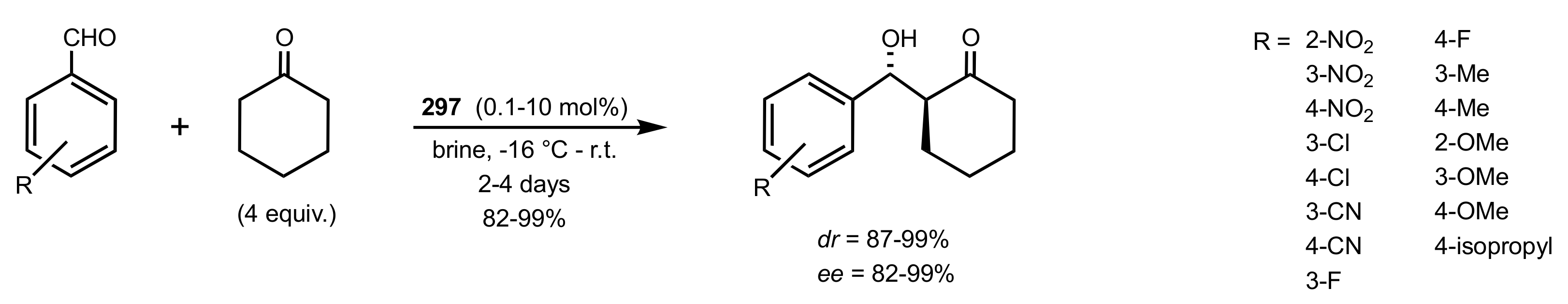
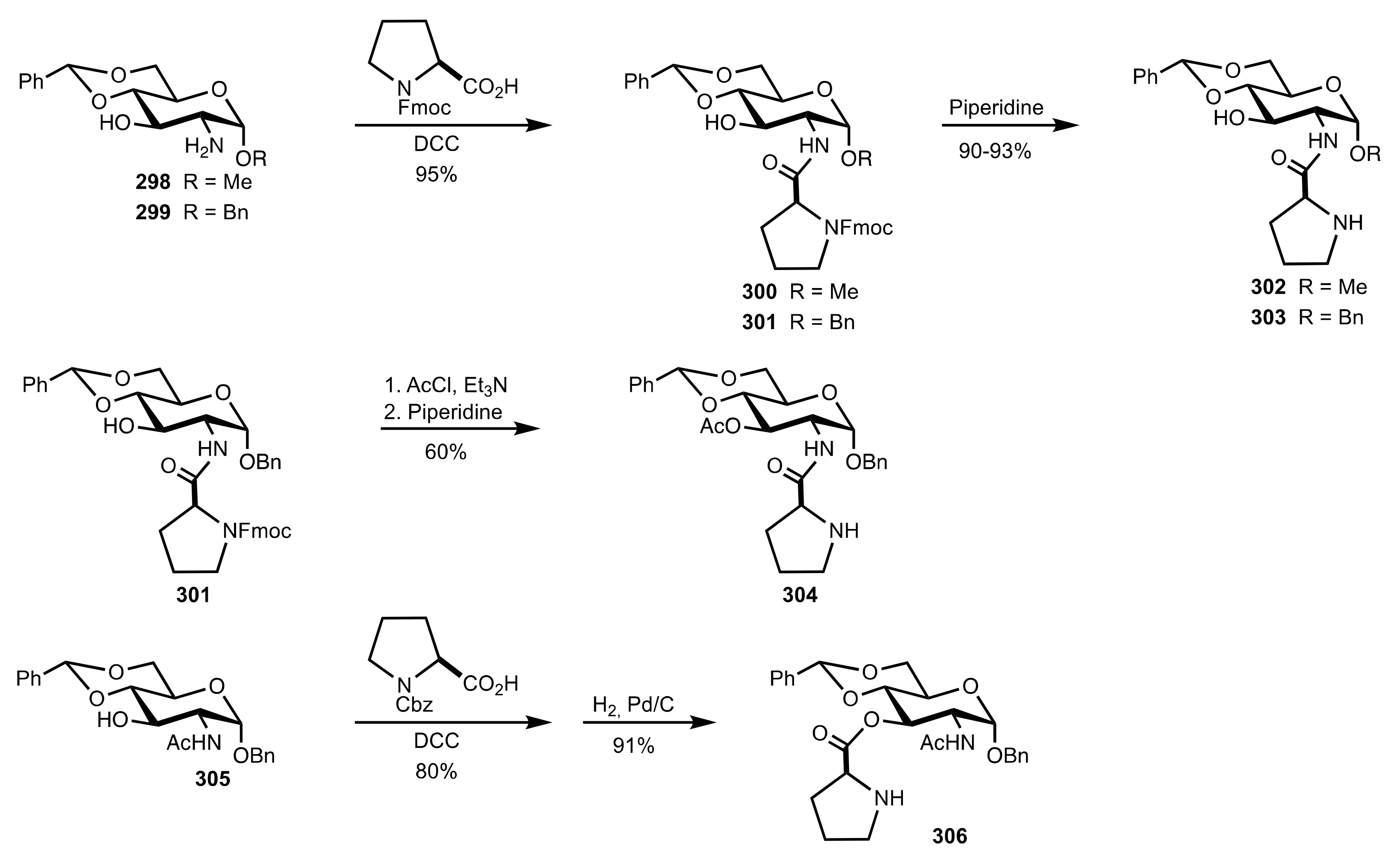
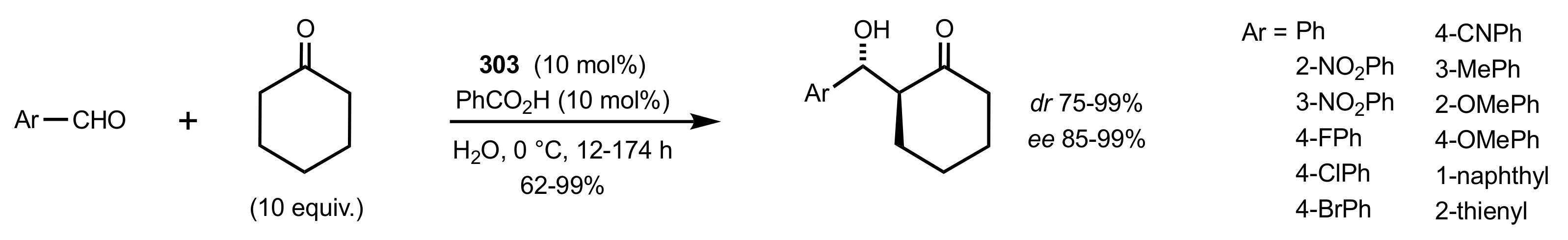
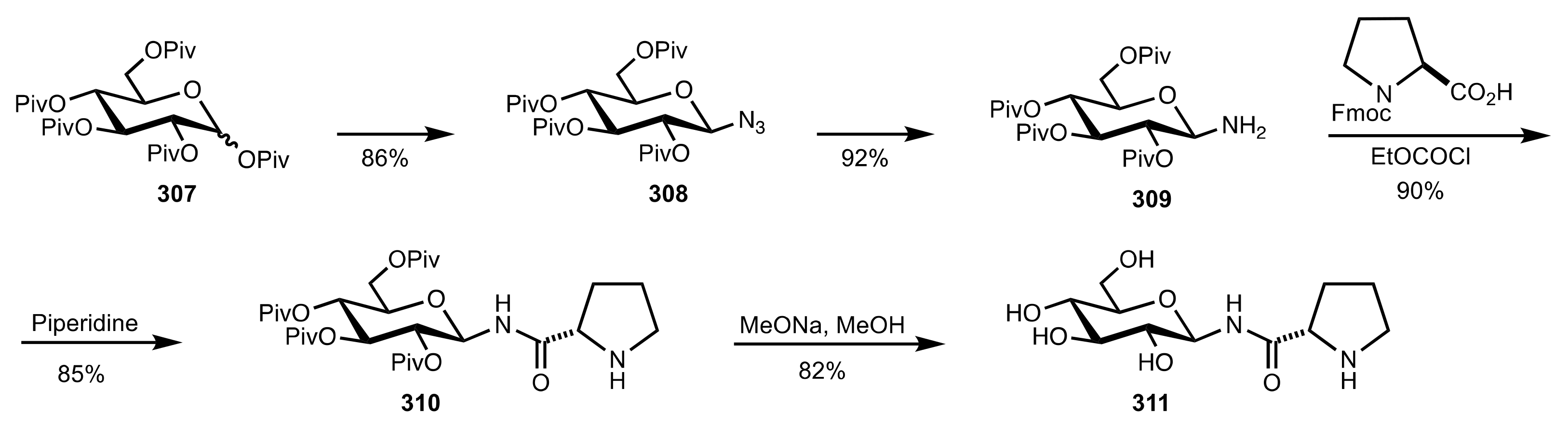
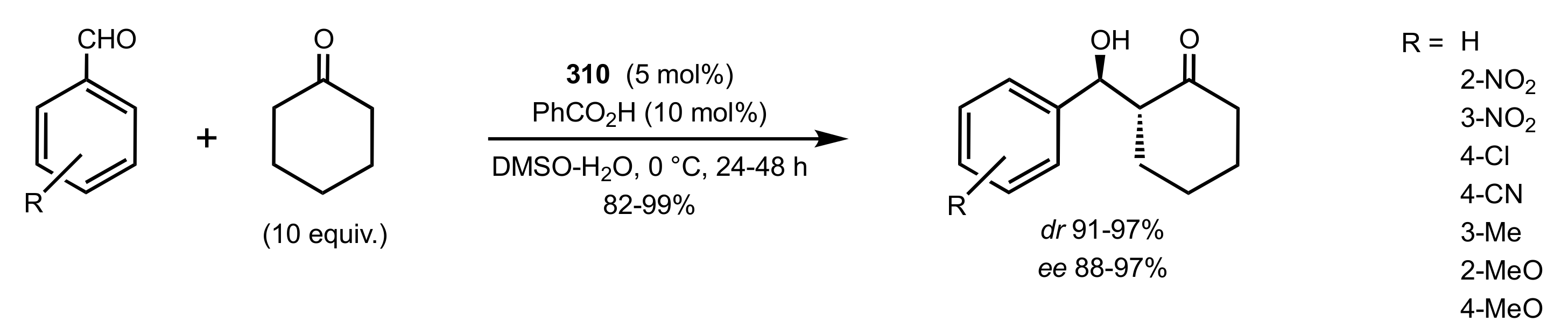
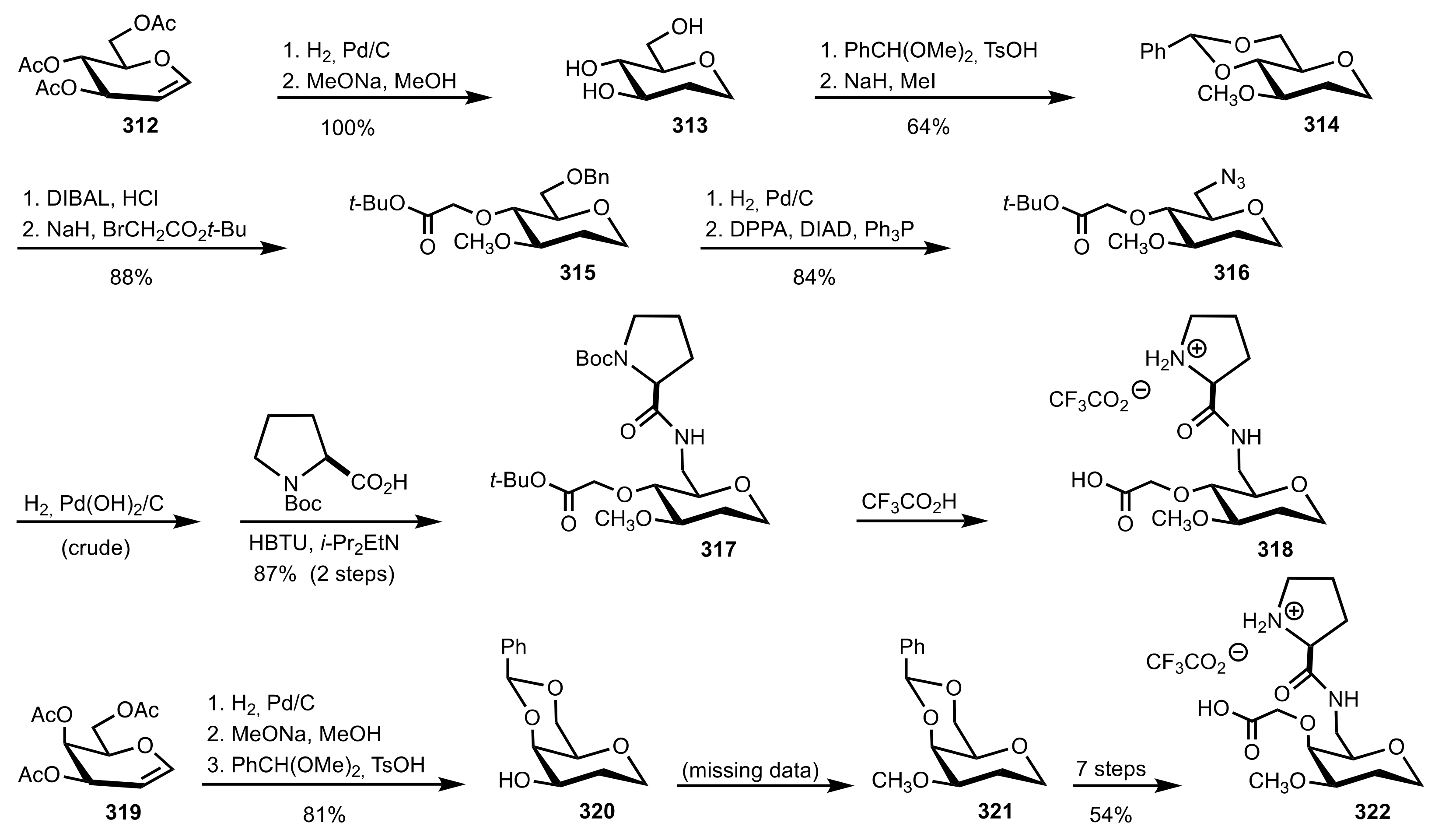
4. Sugar Prolinamides
5. Variously Functionalized Carbohydrates
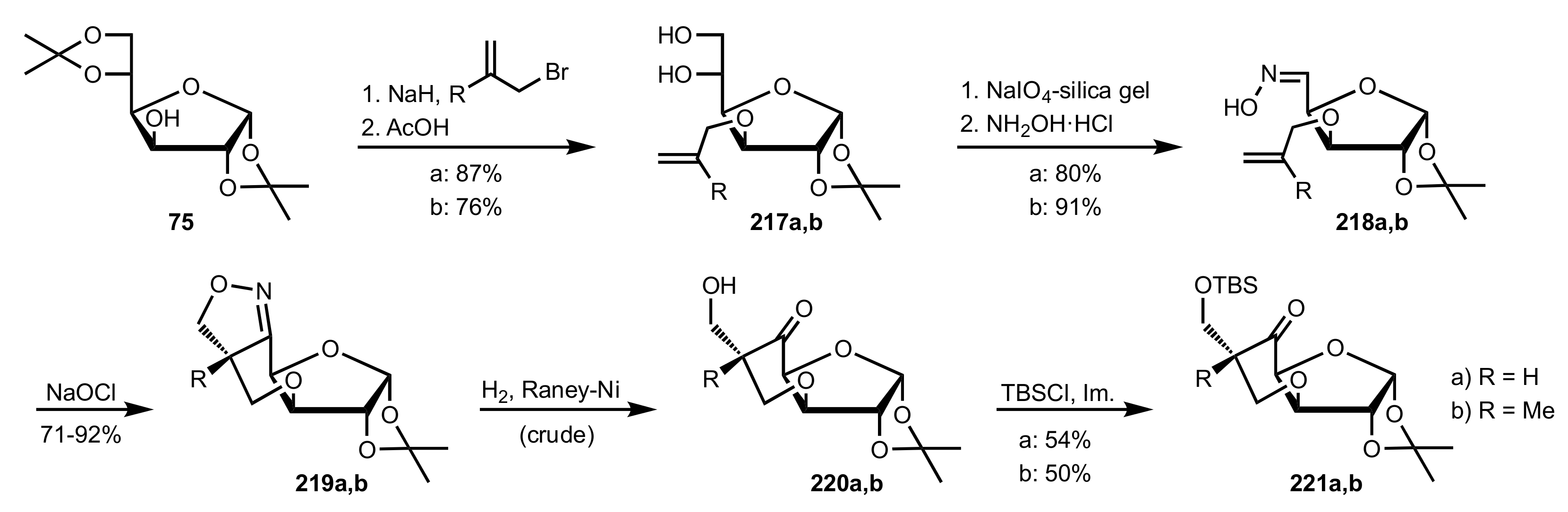
5.1. Sugar Pyrrolidines
5.2. Sugar Pyridines
5.3. Sugar Pyrimidines
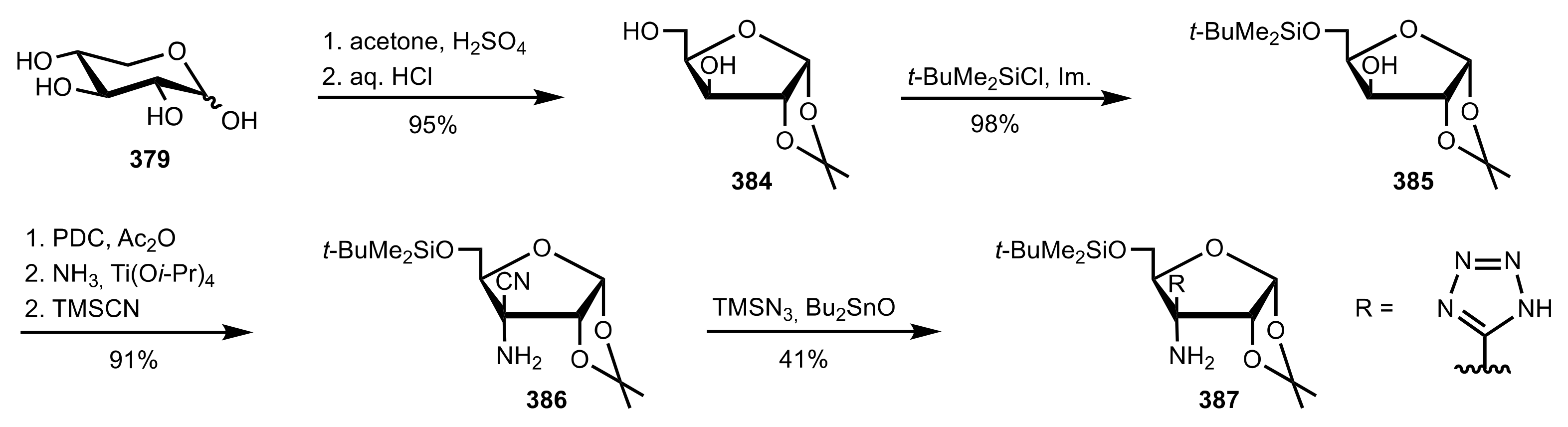
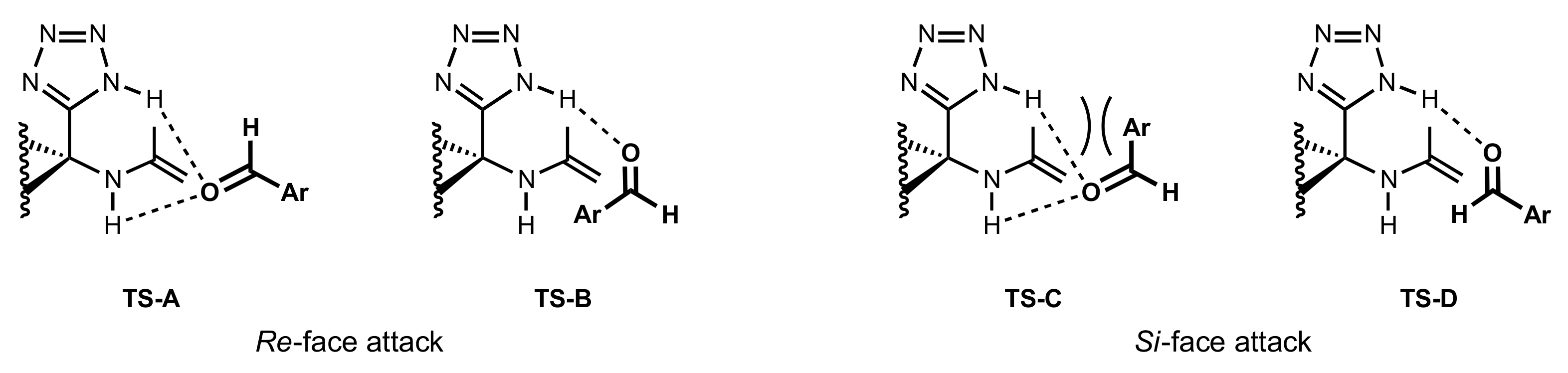
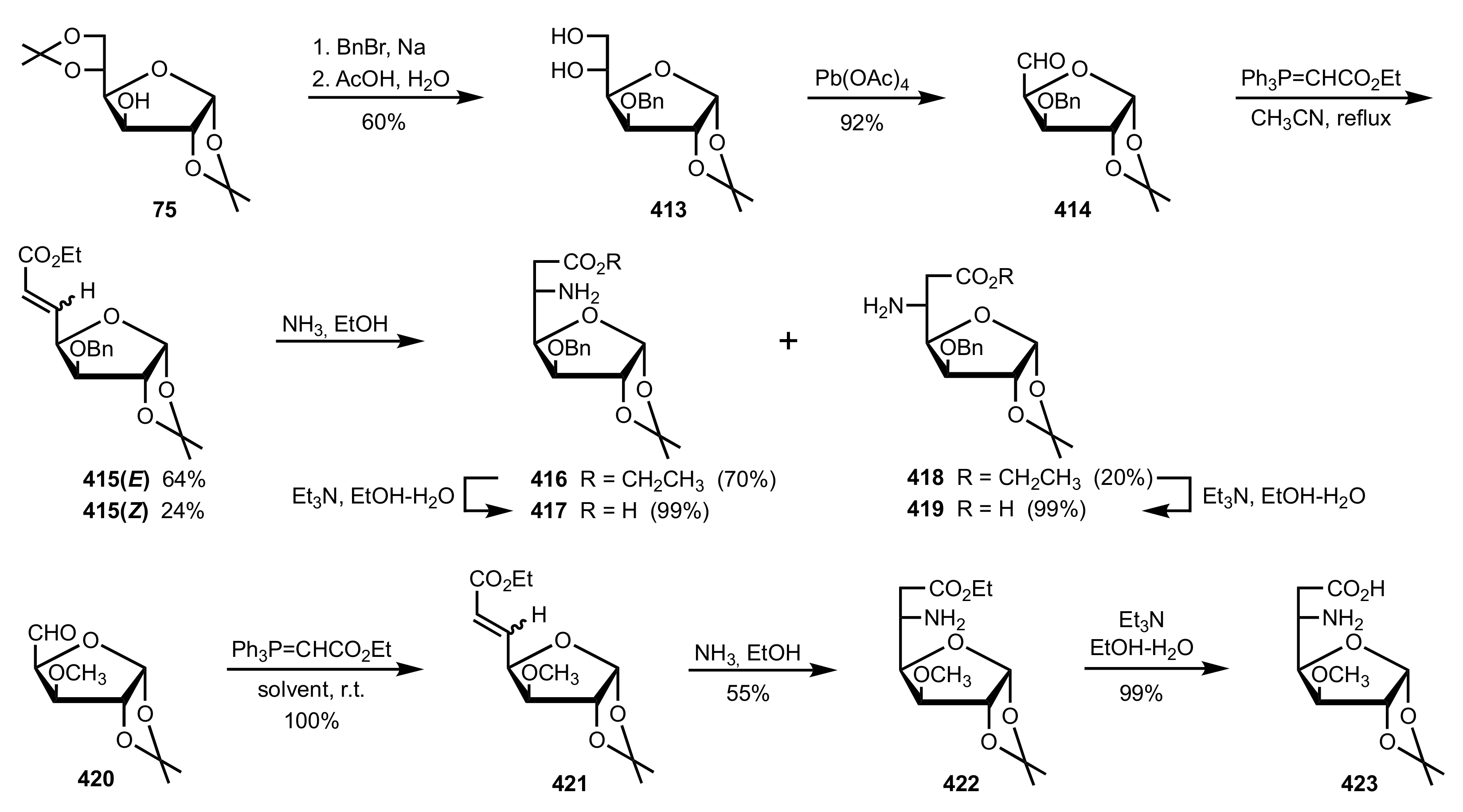
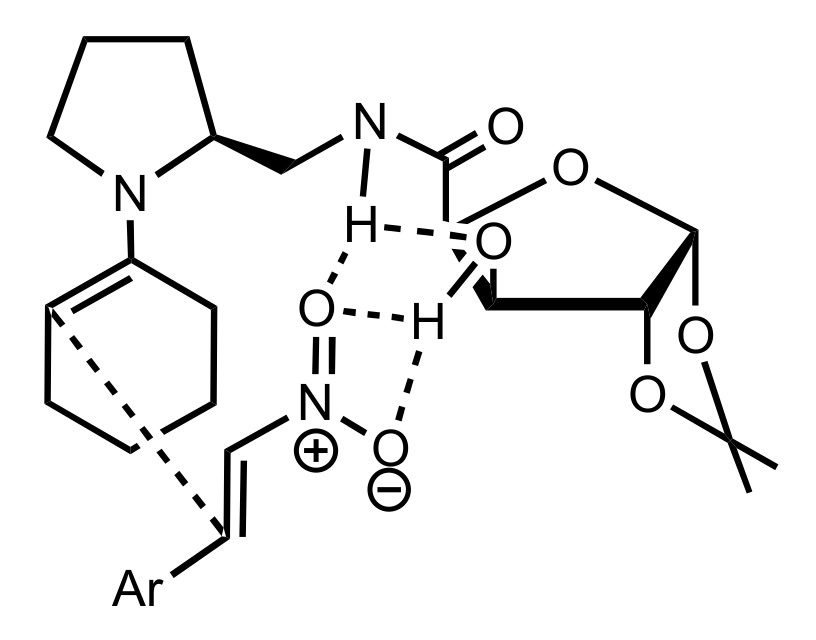
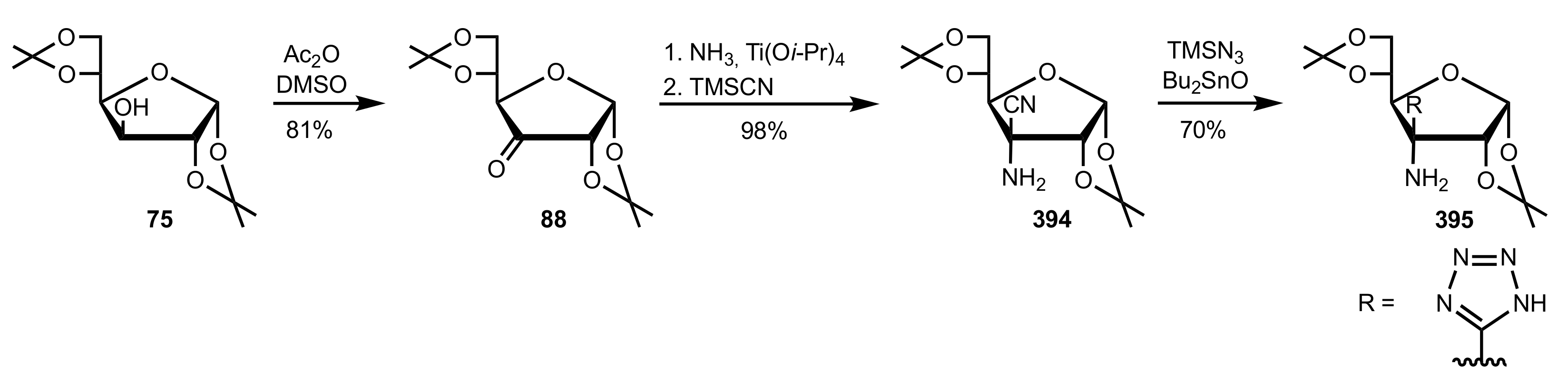
5.4. Sugar Tetrazoles
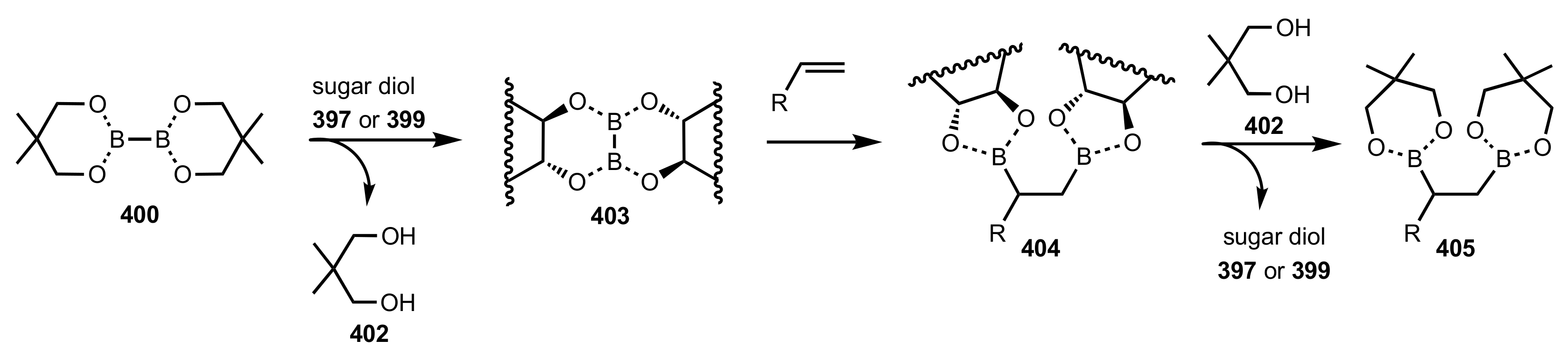
5.5. Sugar Diols
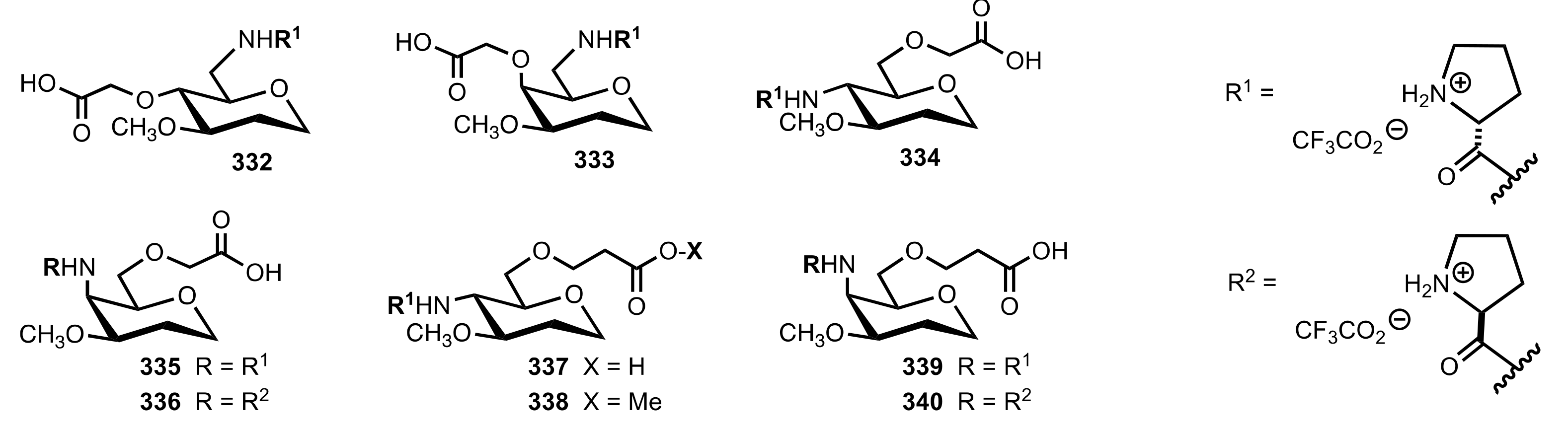
5.6. Sugar Carboxylic Acids
5.7. Sugar Aminoacids
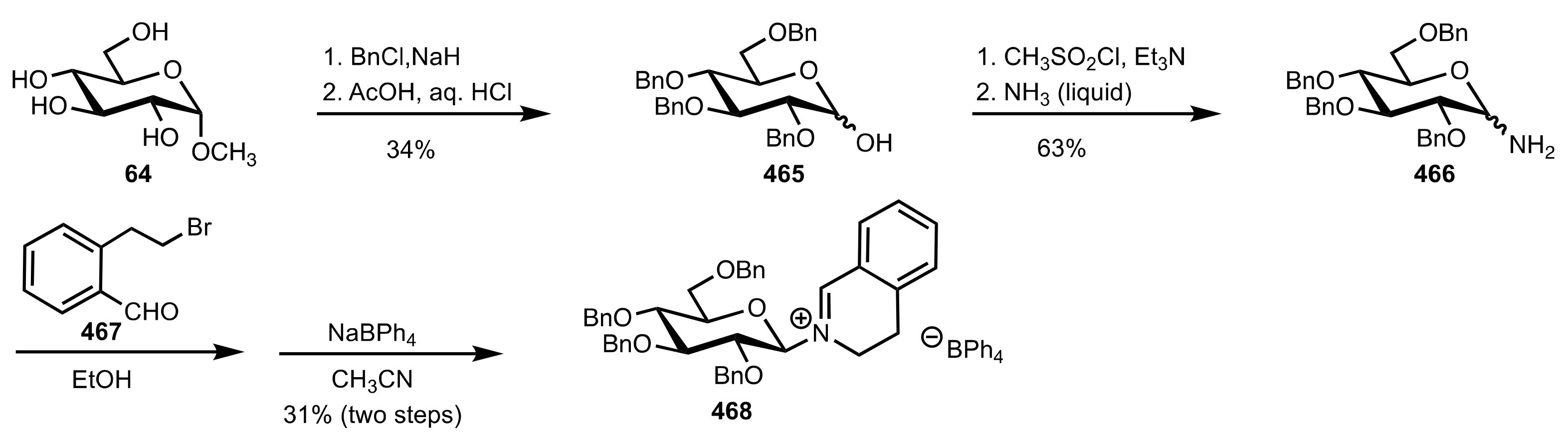
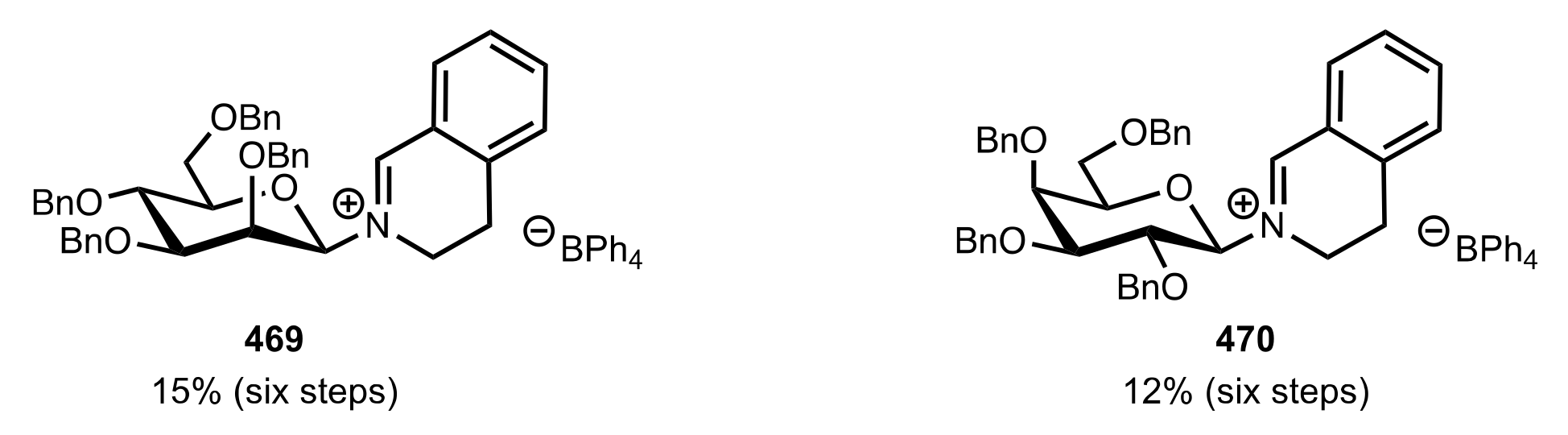
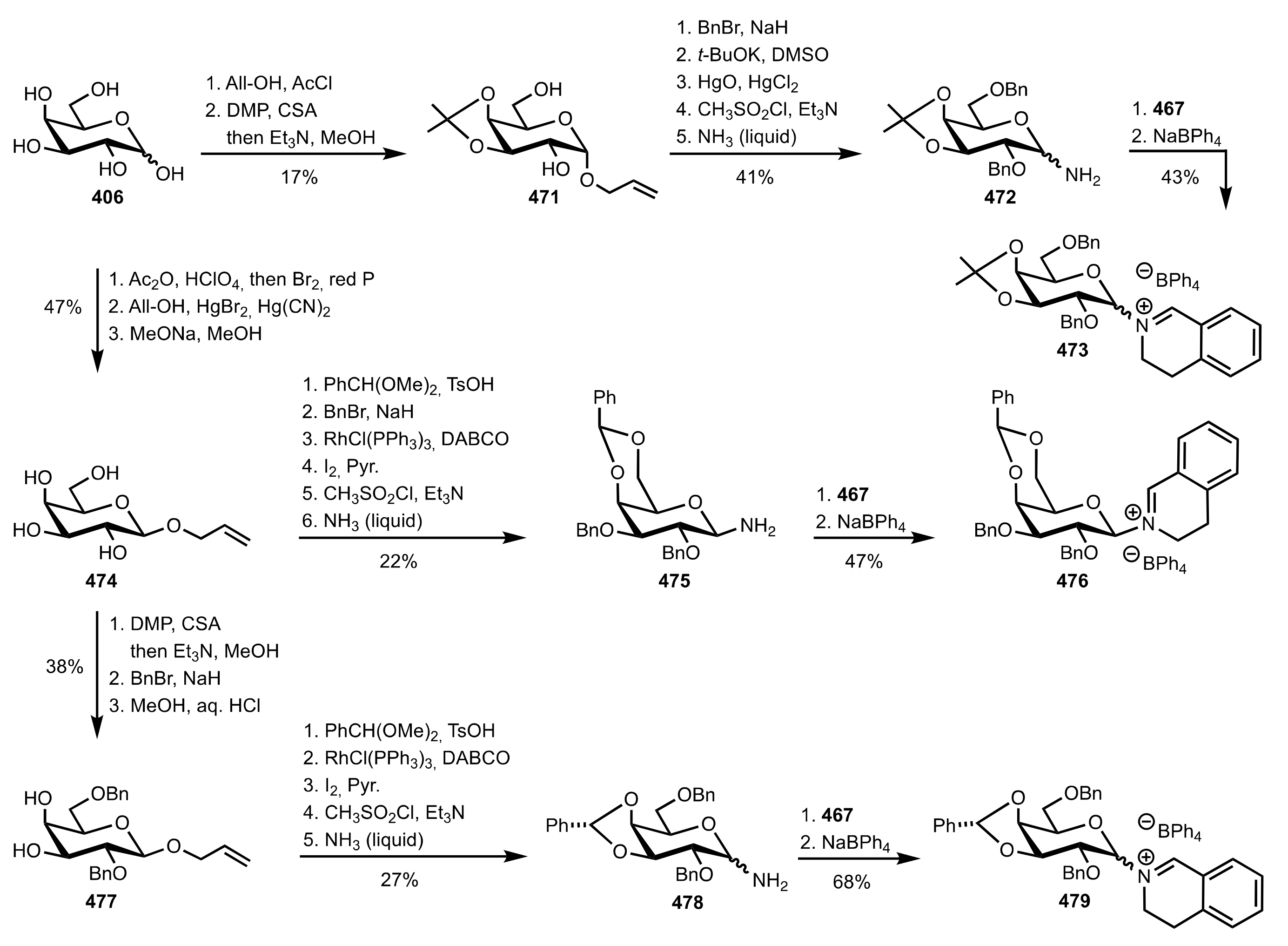
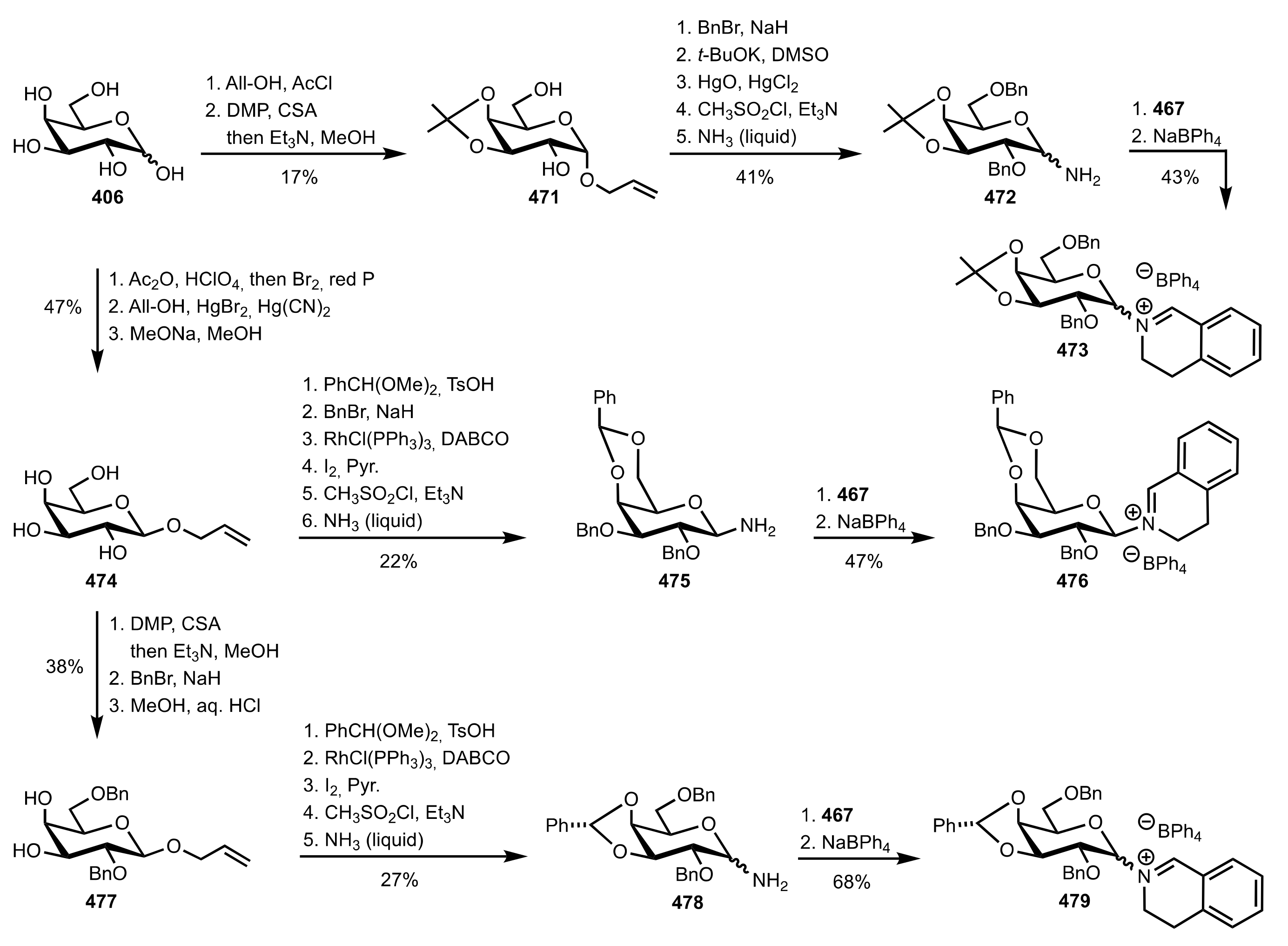
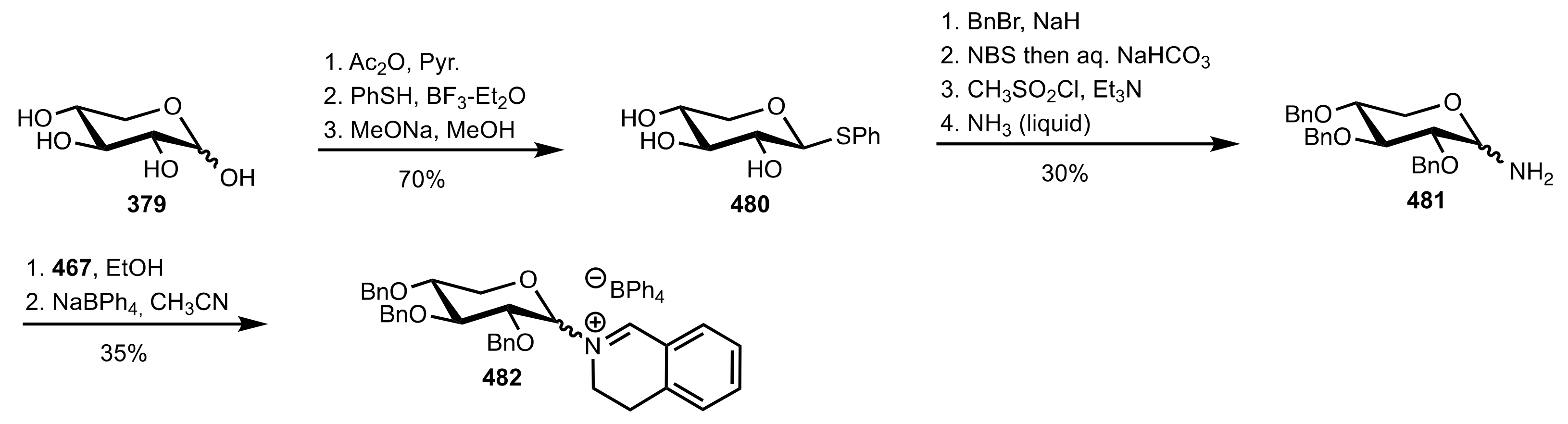
5.8. Sugar Imines and Iminium Salts
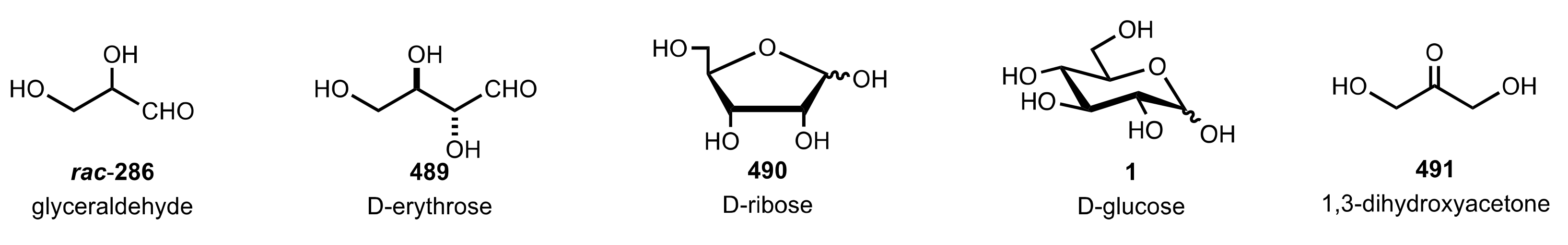
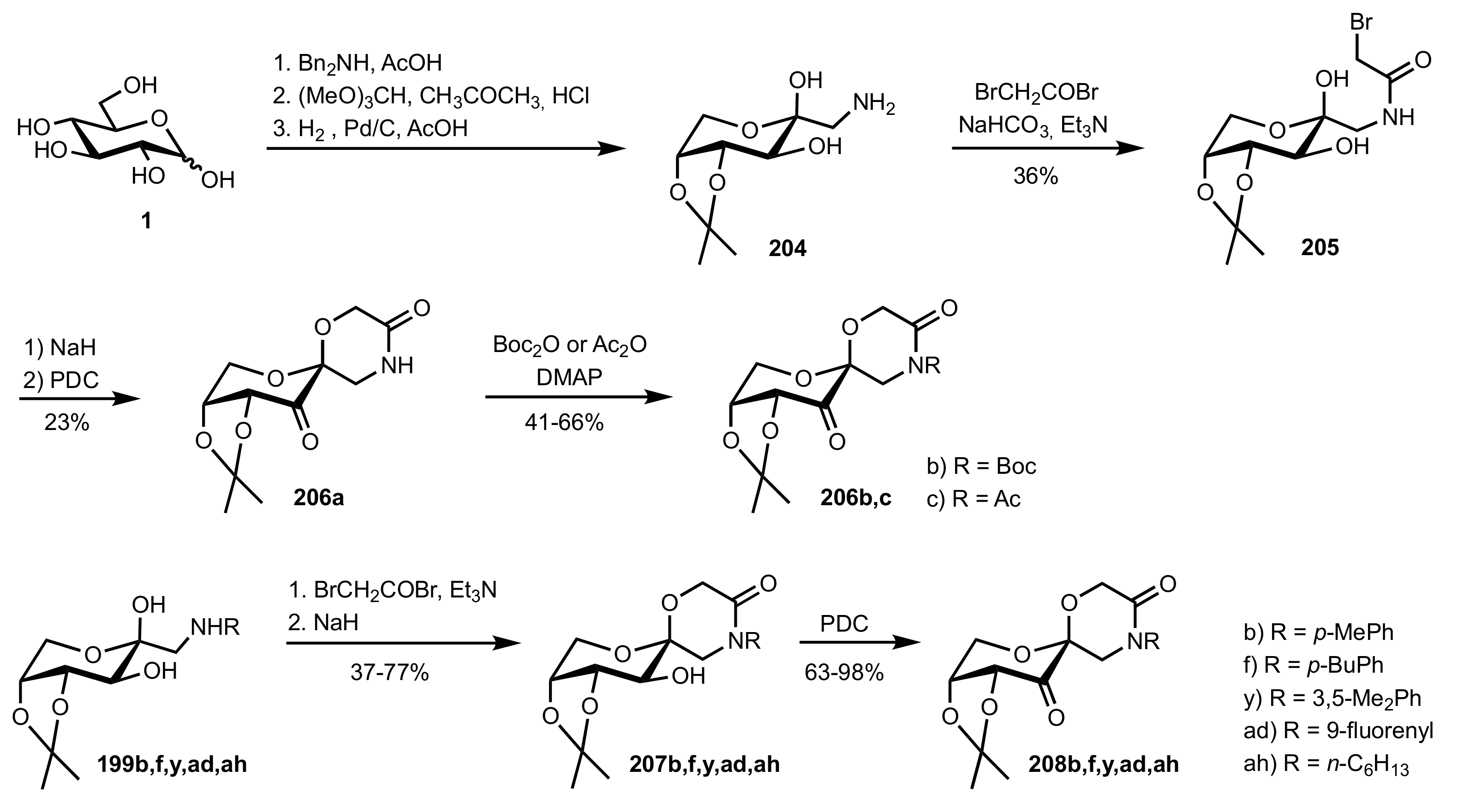
6. Deprotected Monosaccharides
6.1. Neutral Sugars
6.2. Aminosugars
7. Polysaccharides
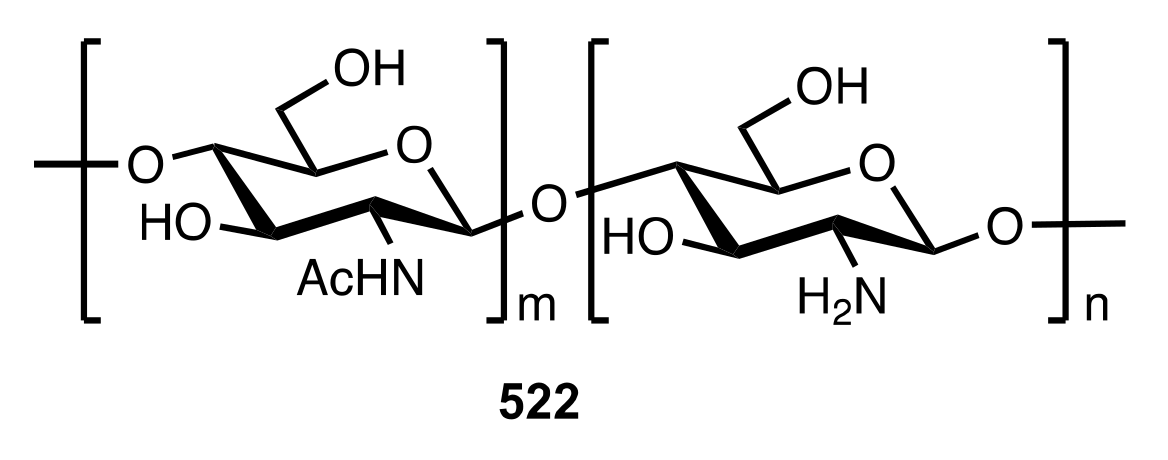
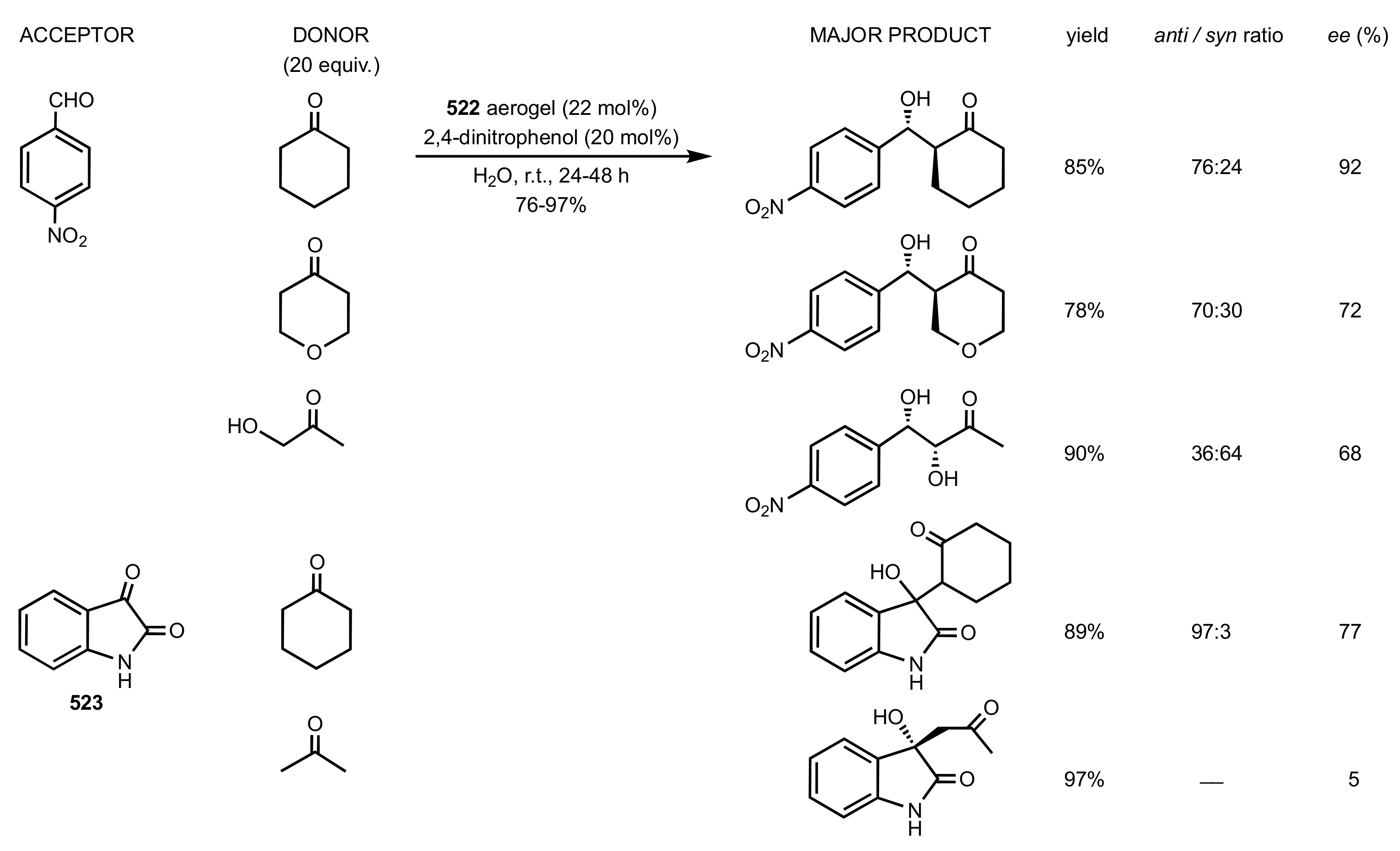
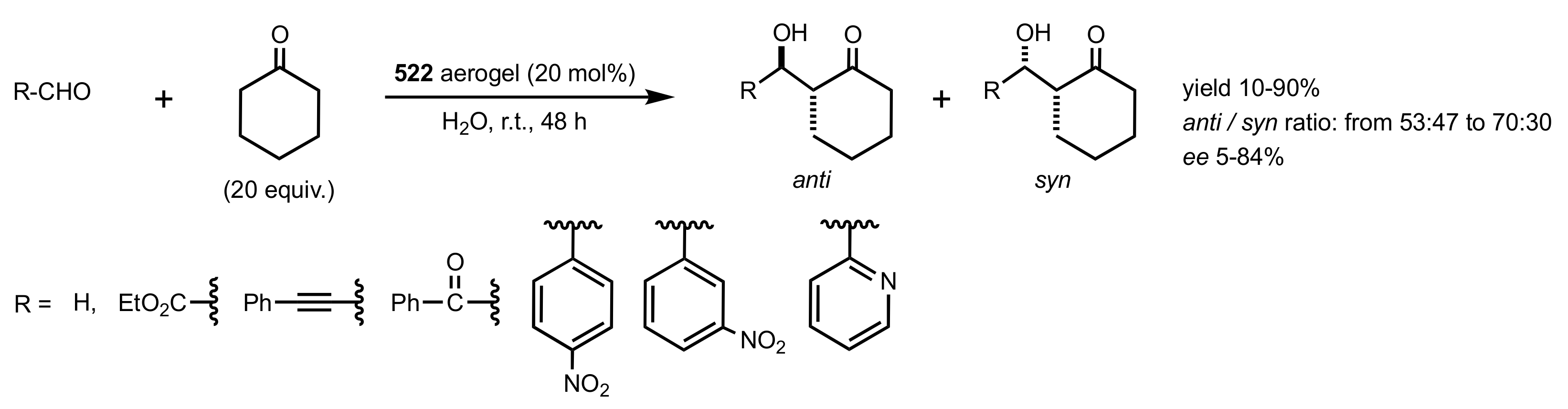
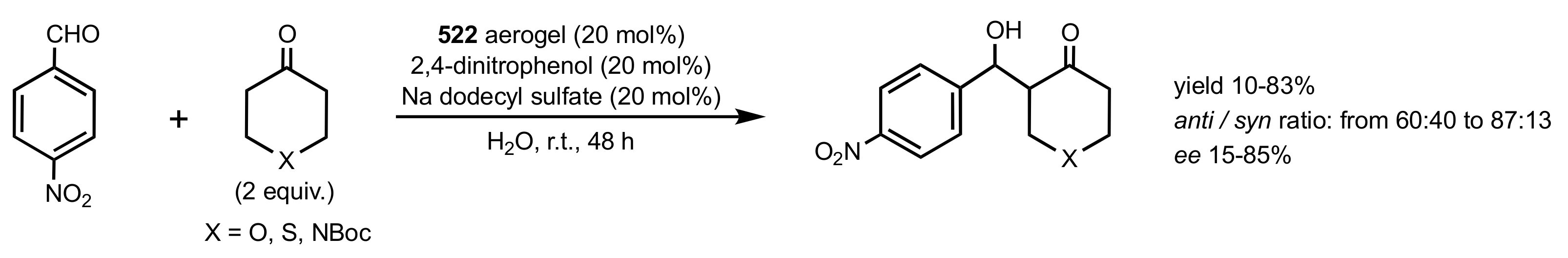
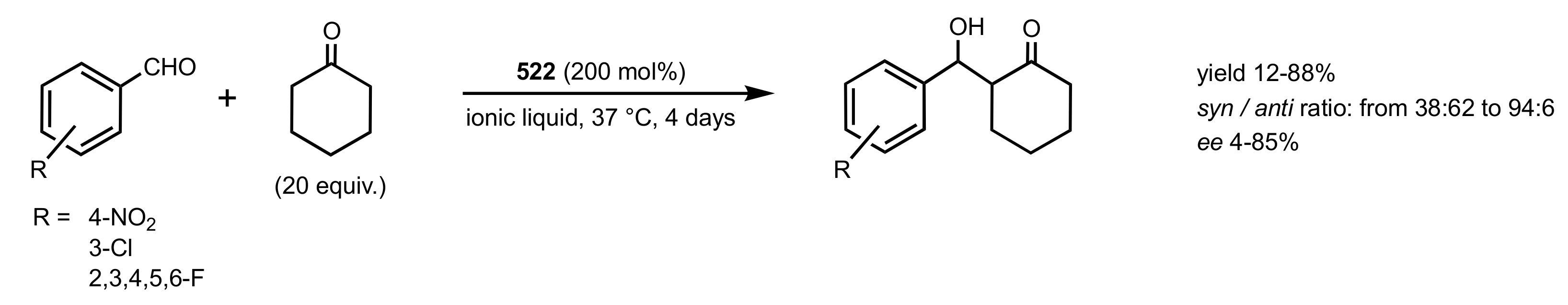
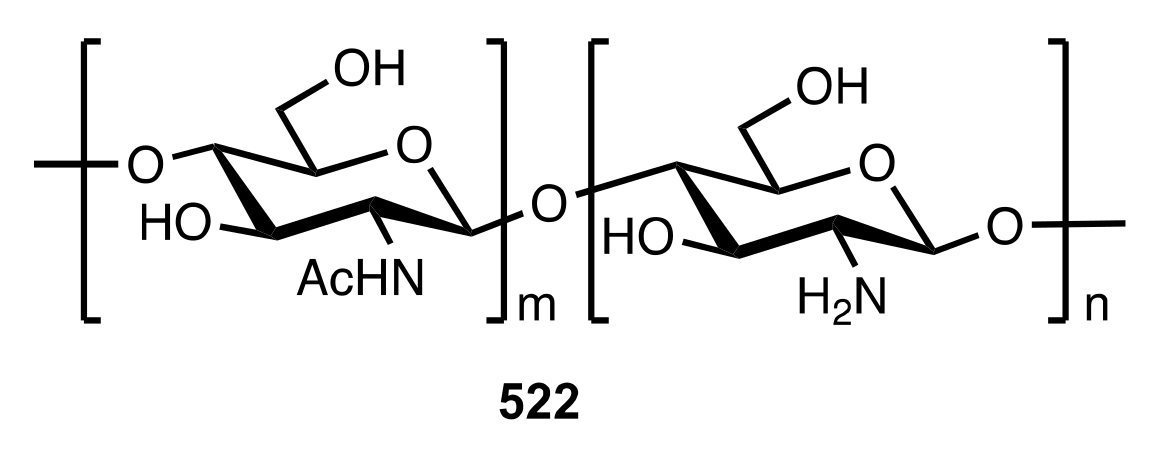
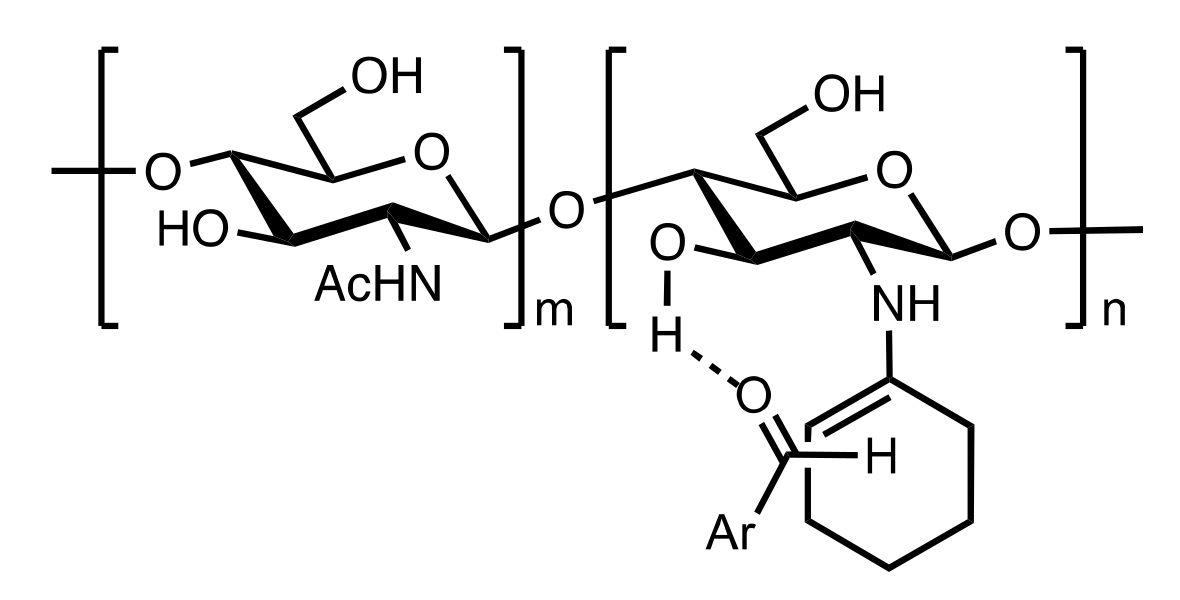
7.1. Chitosan
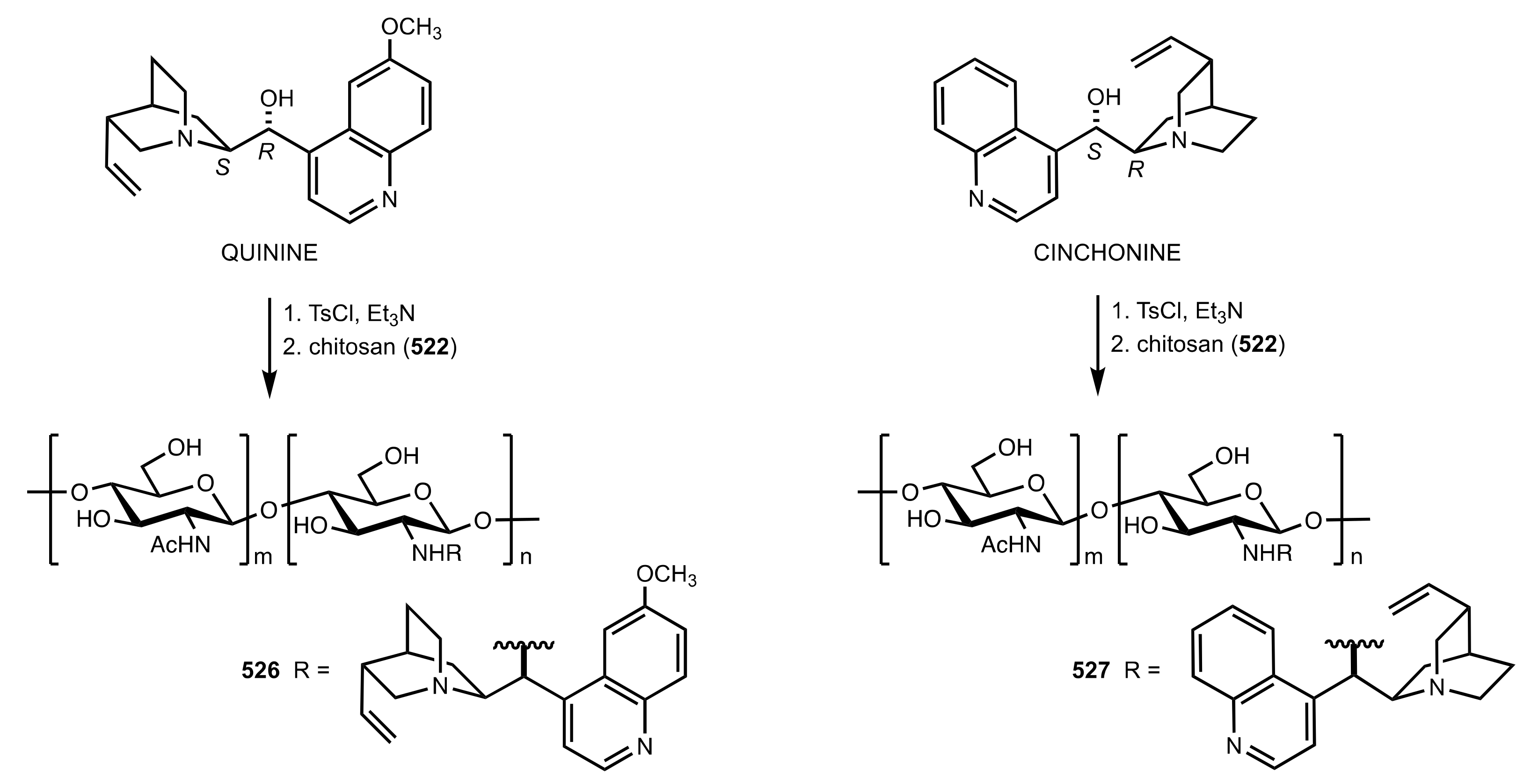
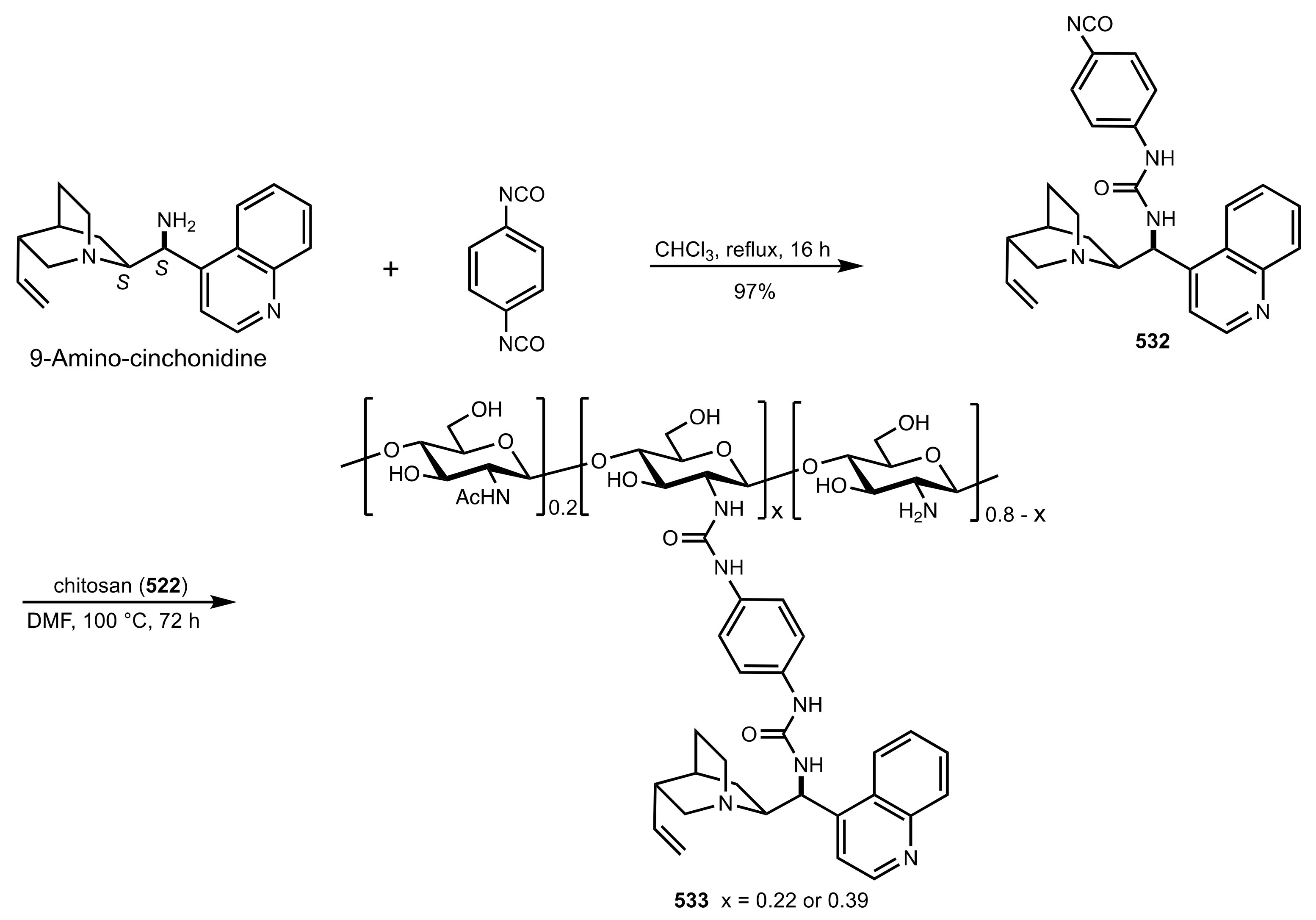
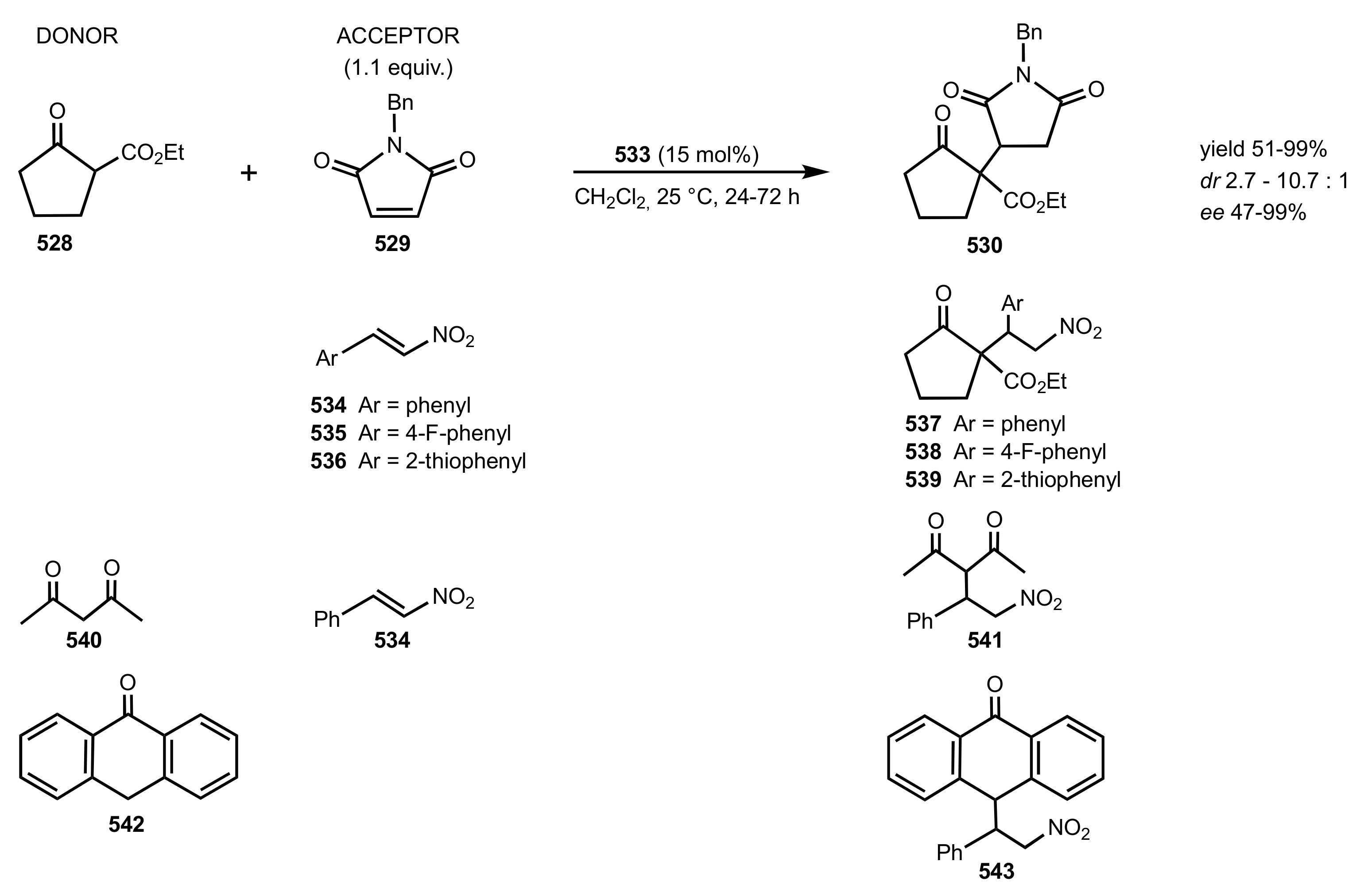
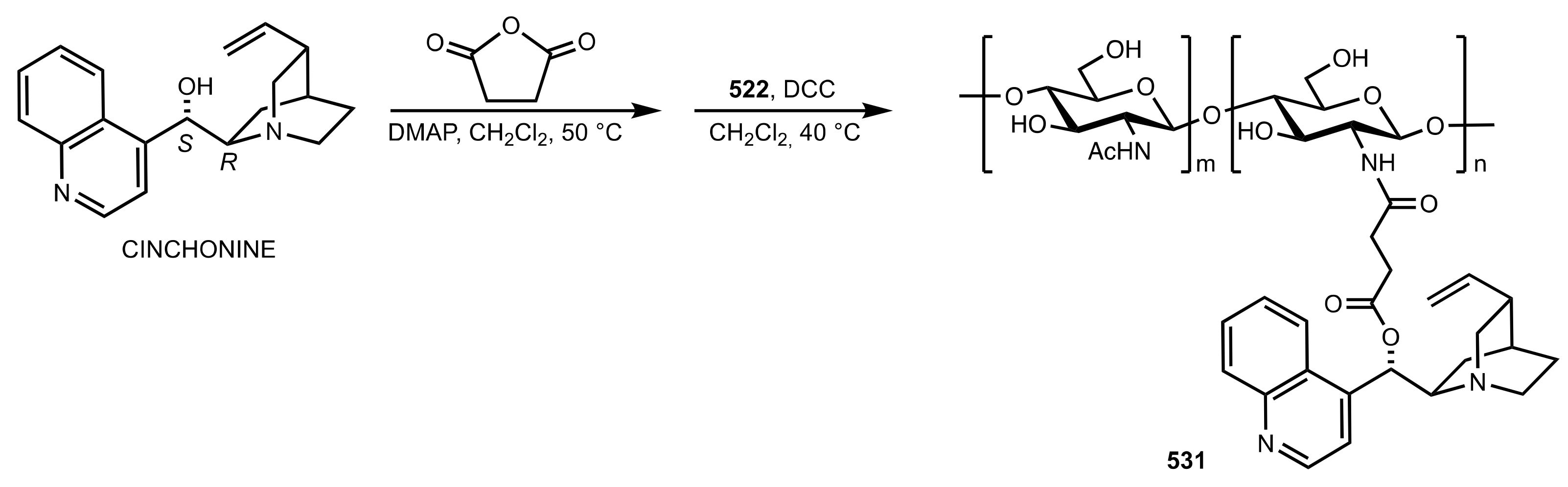
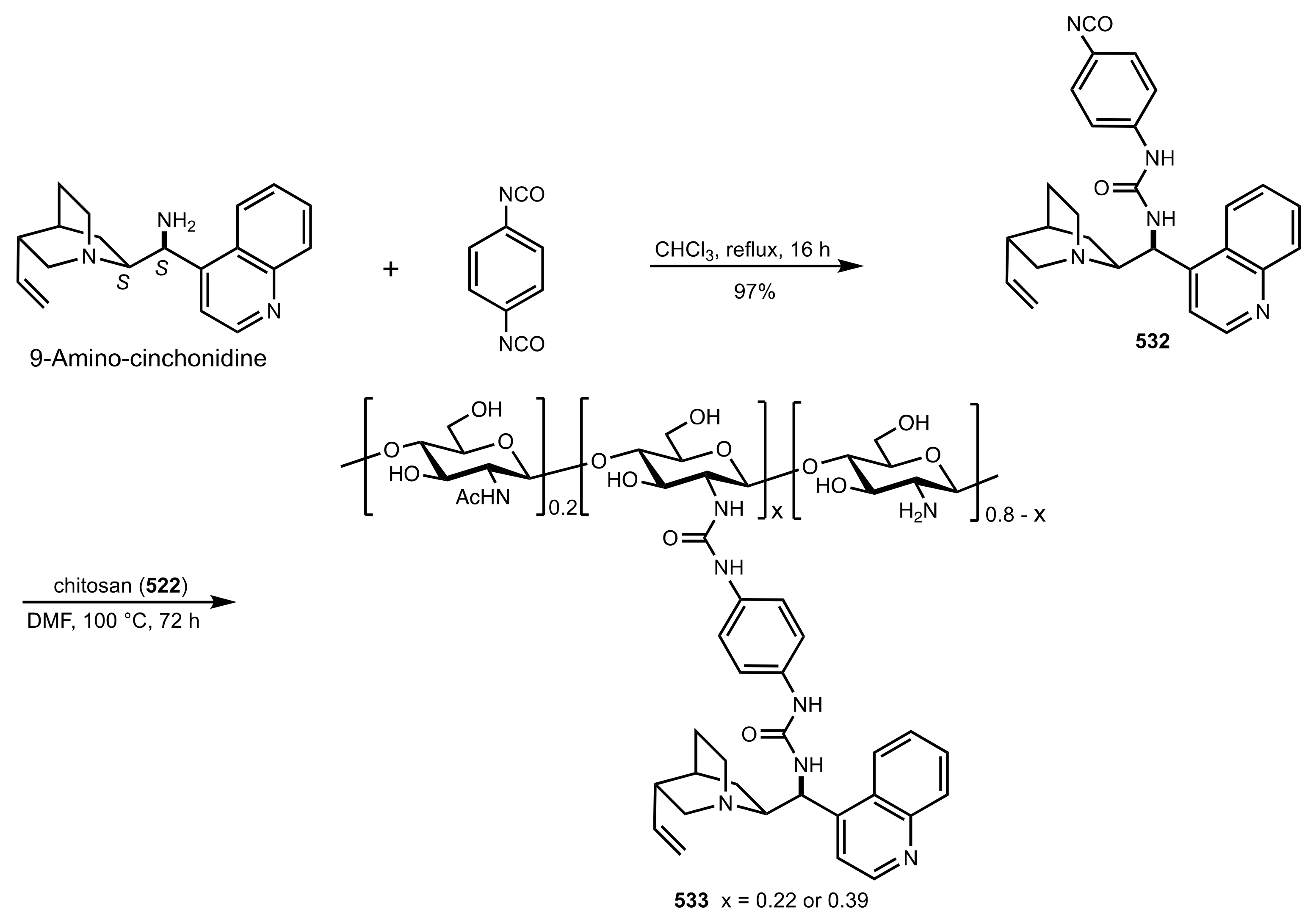
7.2. Chitosan-Cinchona Alkaloids Conjugates
7.3. Functionalized Chitosan
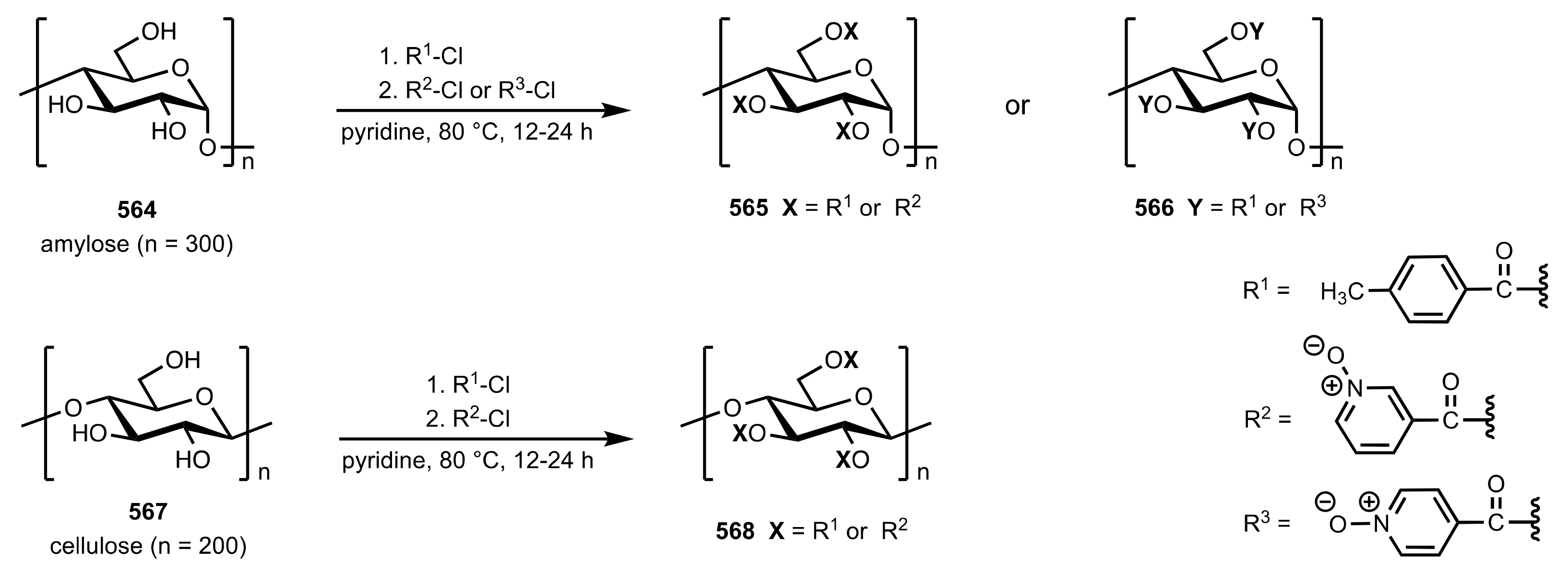
7.4. Functionalized Amylose and Cellulose
8. Phase-Transfer Catalysts
8.1. Sugar Ammonium and Triazolium Salts
8.2. Sugar Crown Ethers
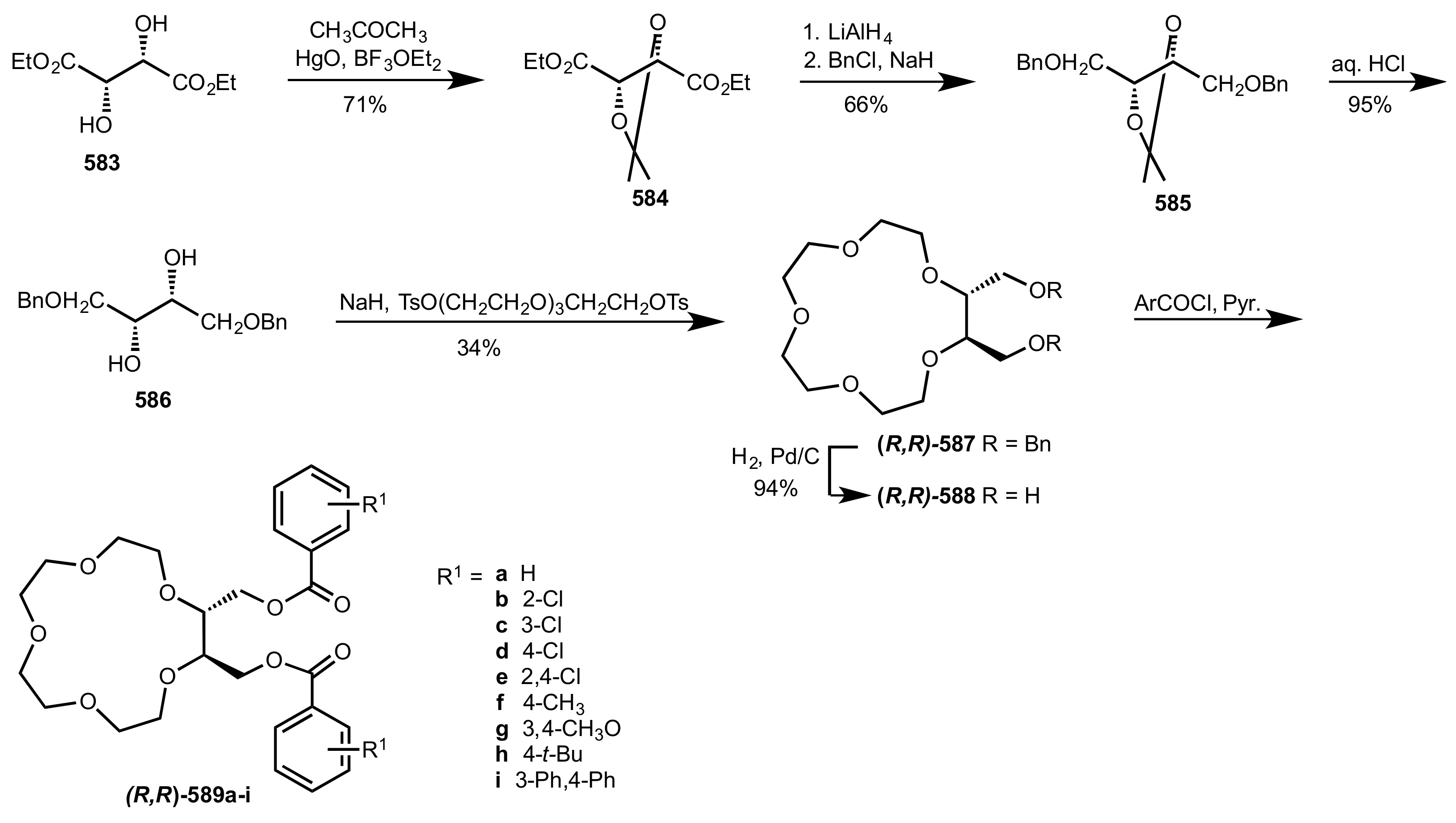
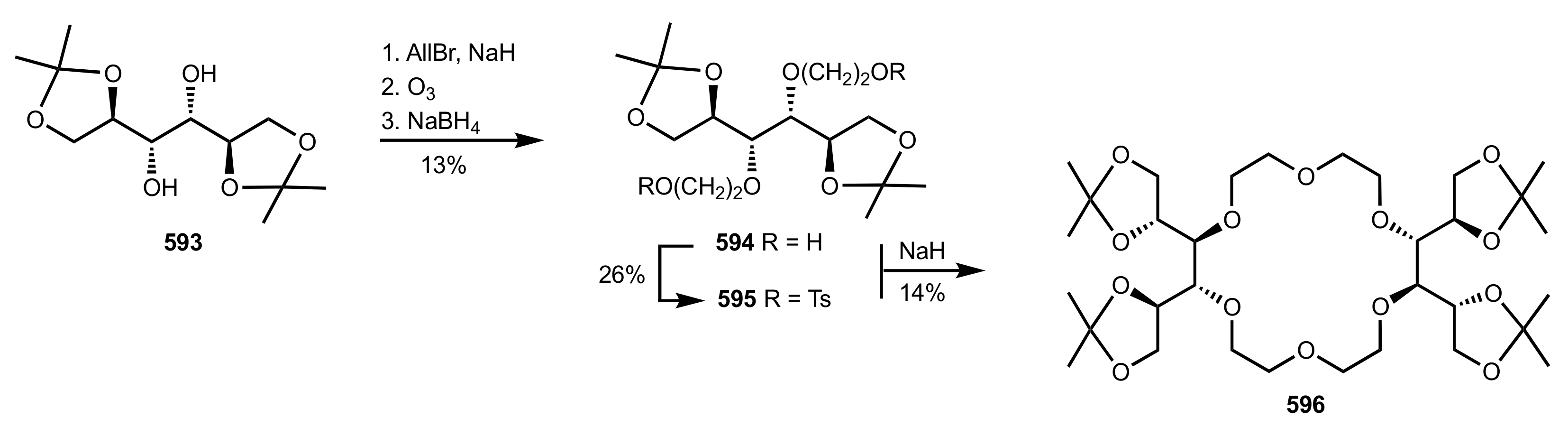
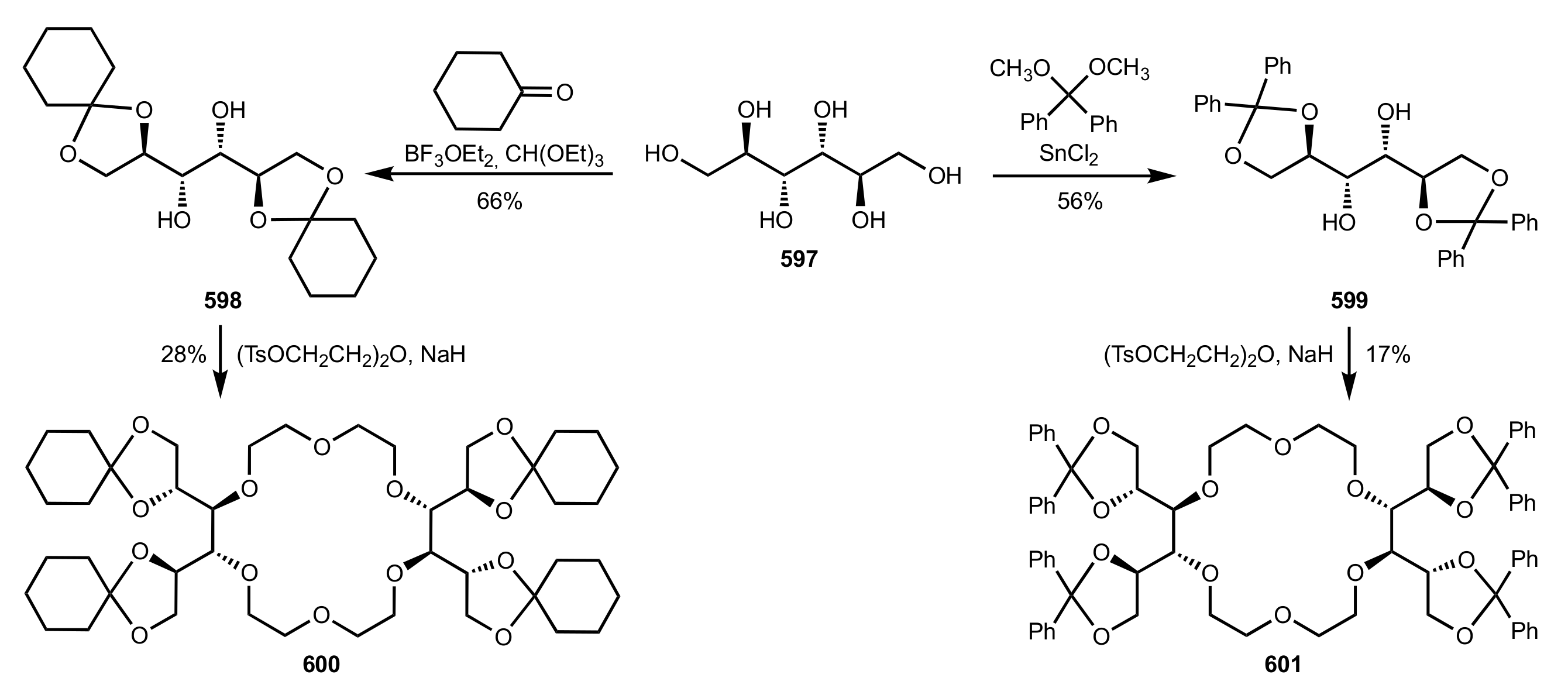
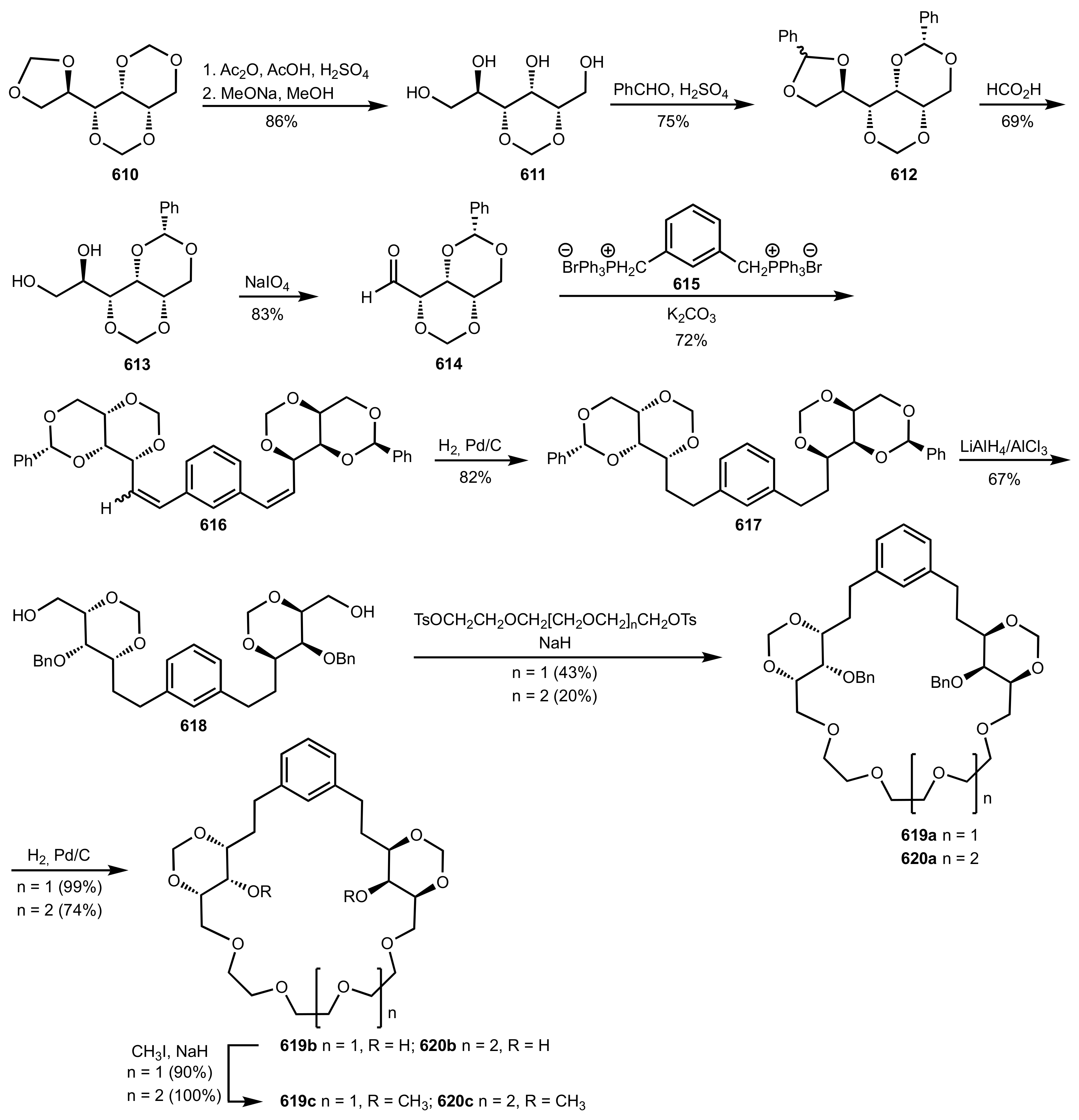
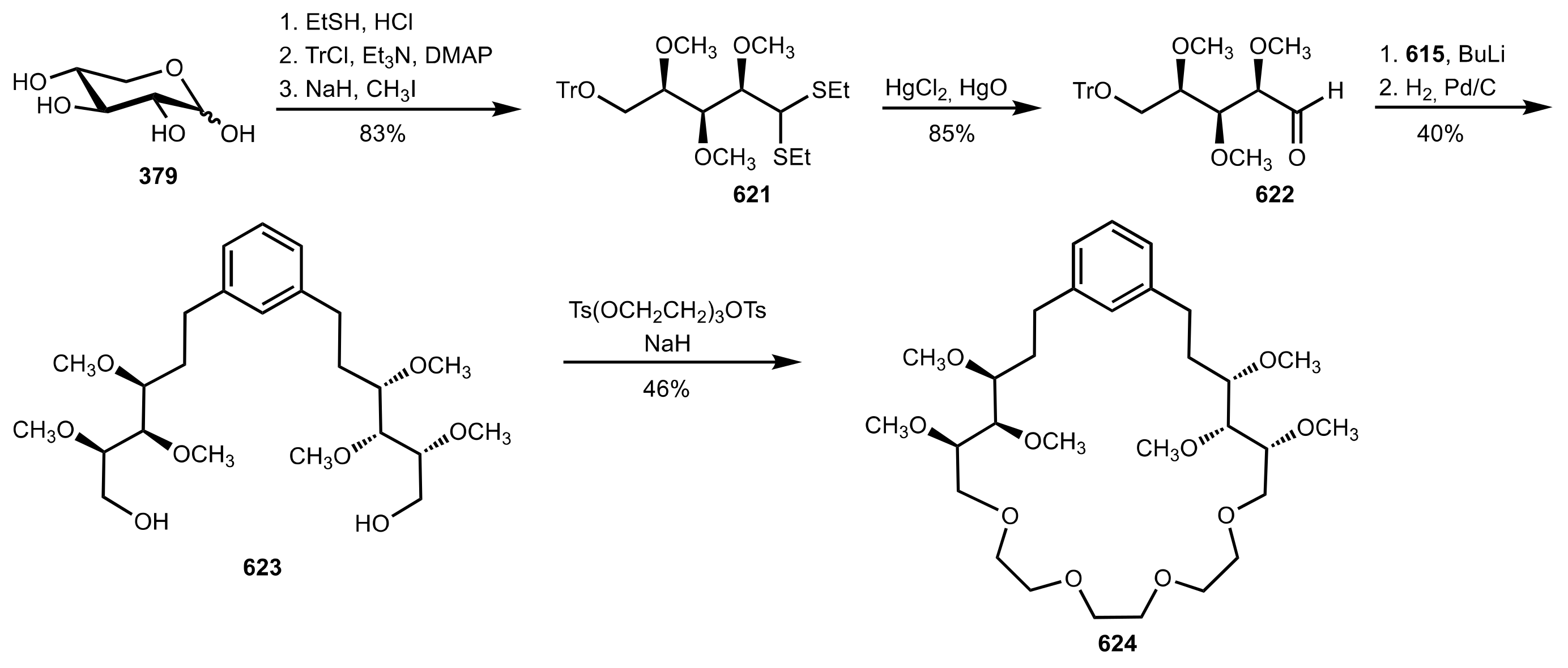
8.2.1. Alditol-Based Crown Ethers
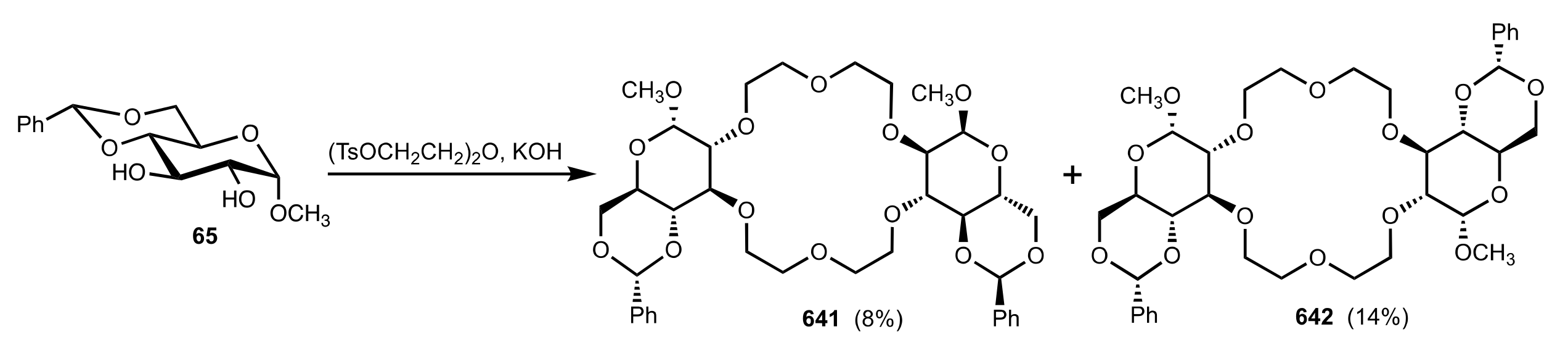
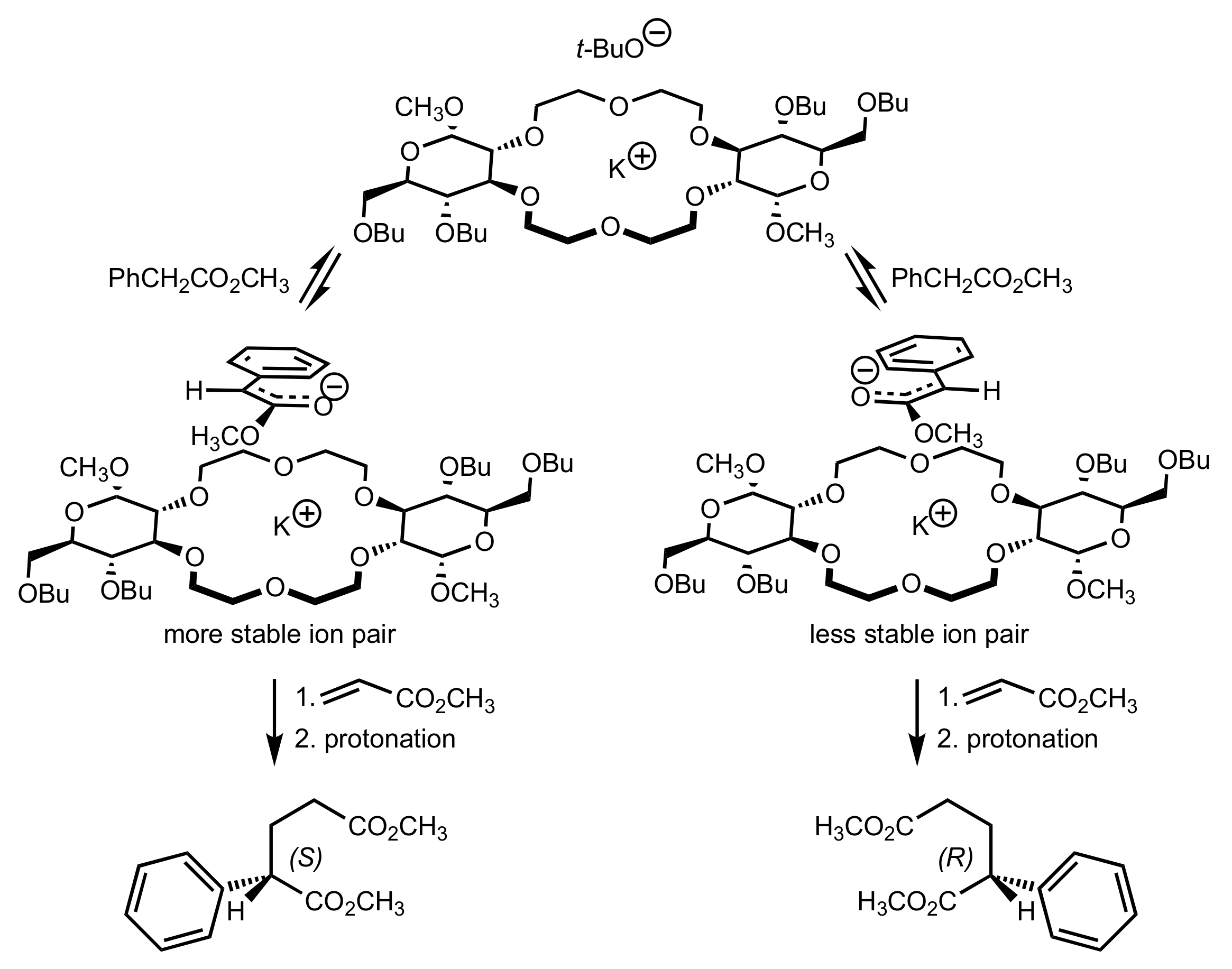
8.2.2. Aldose-Based Crown Ethers
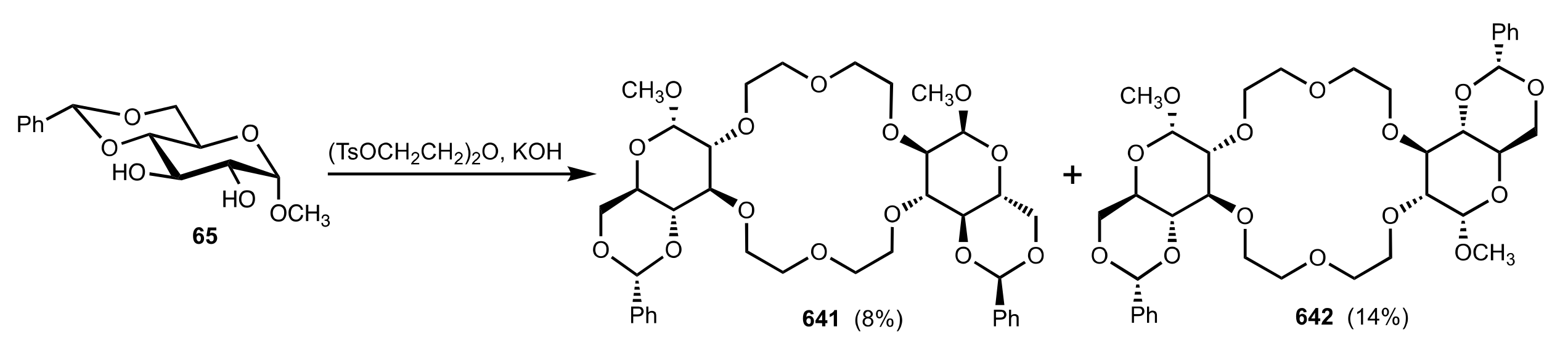
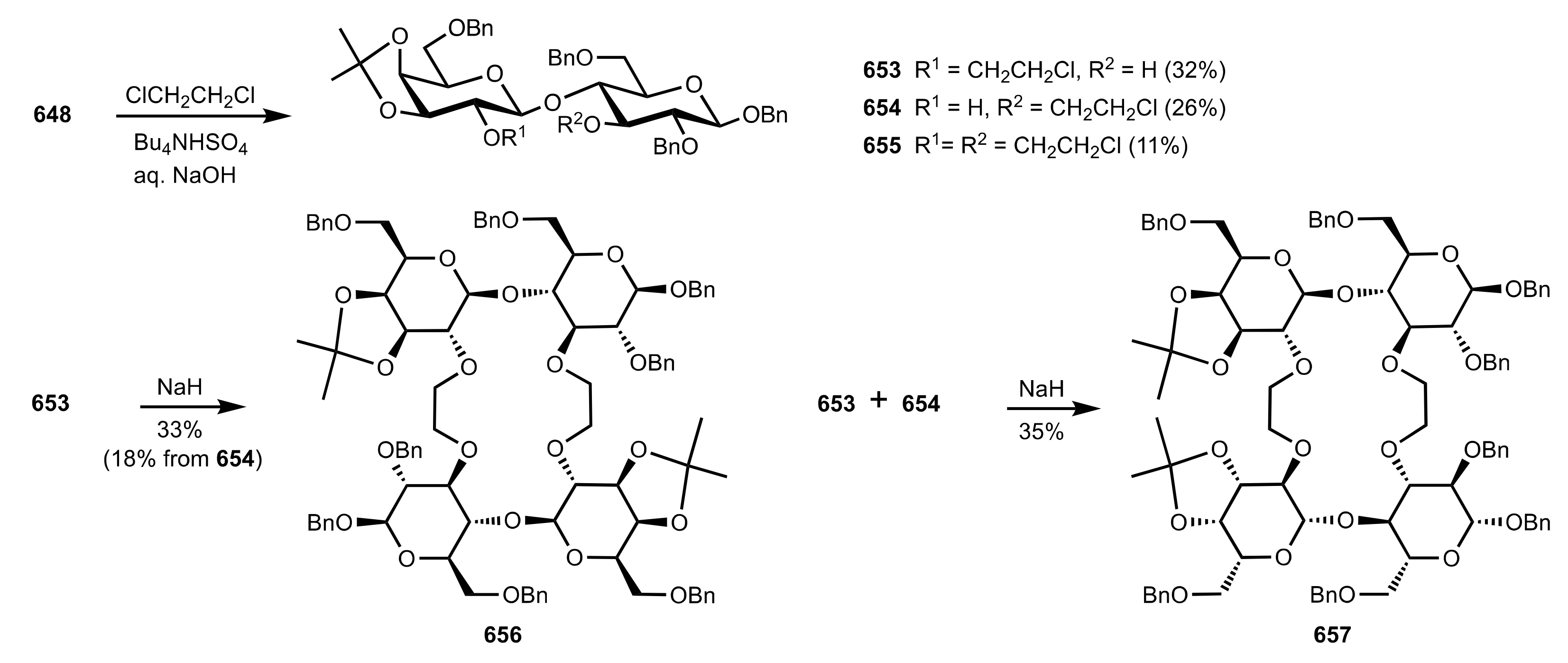
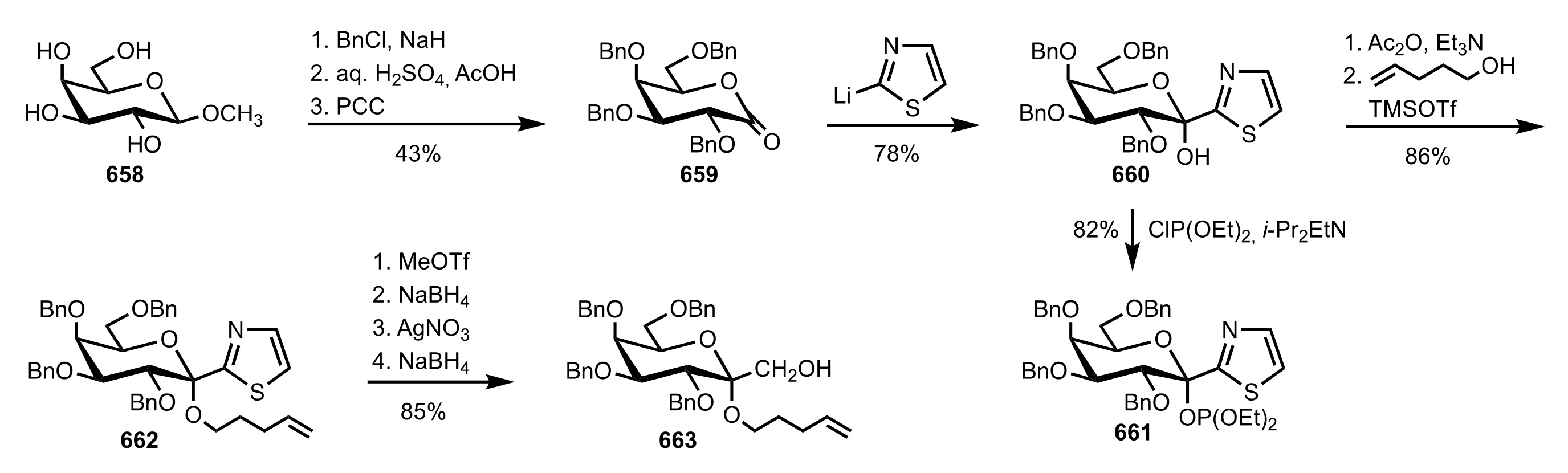
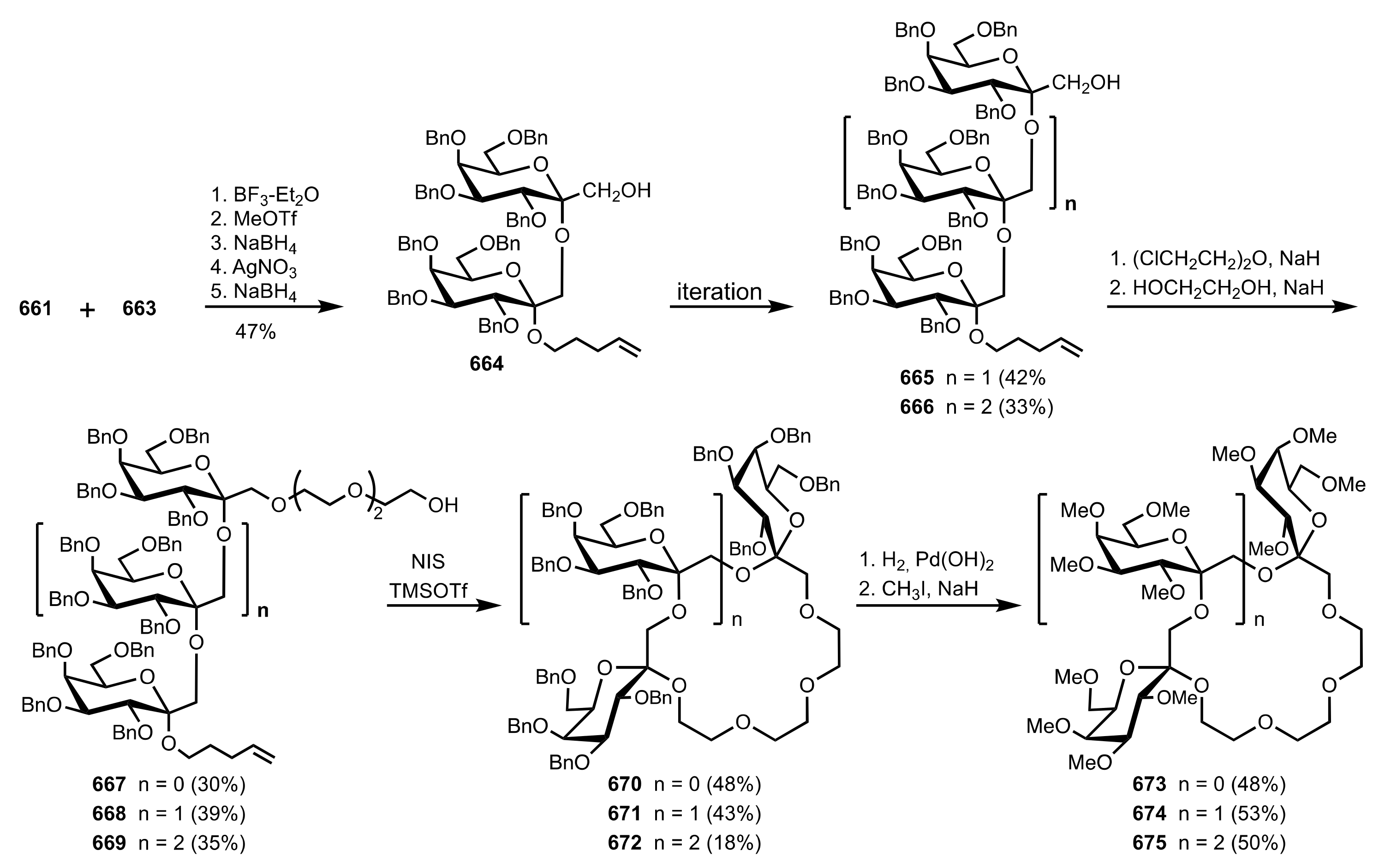
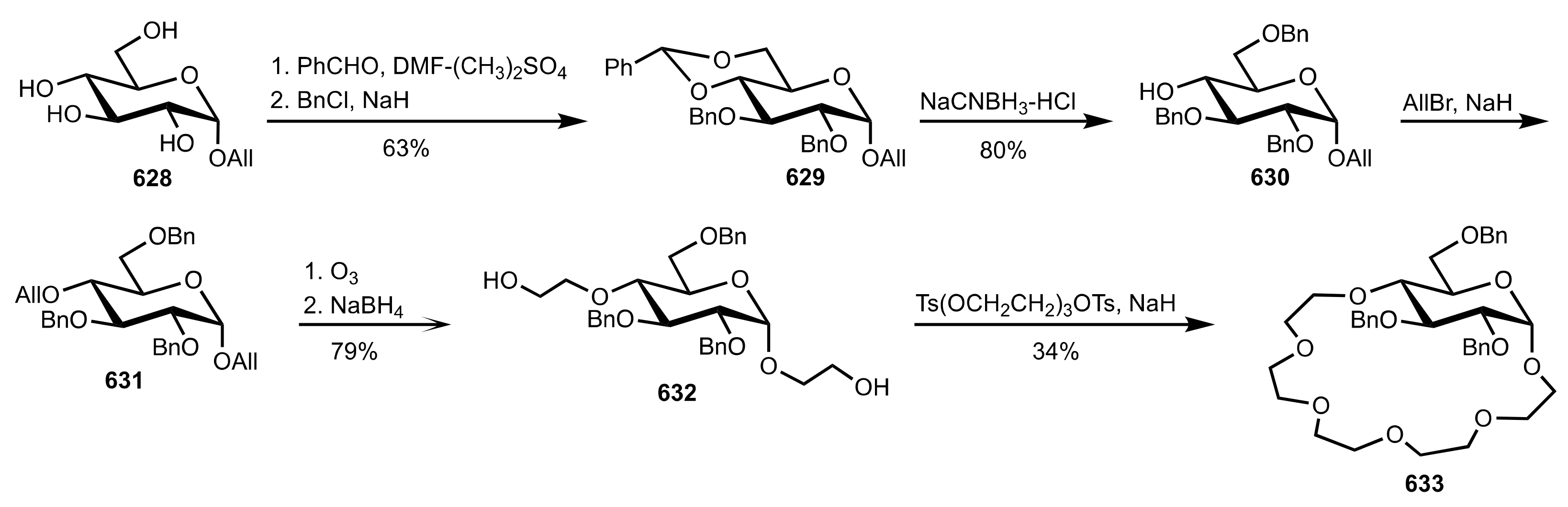
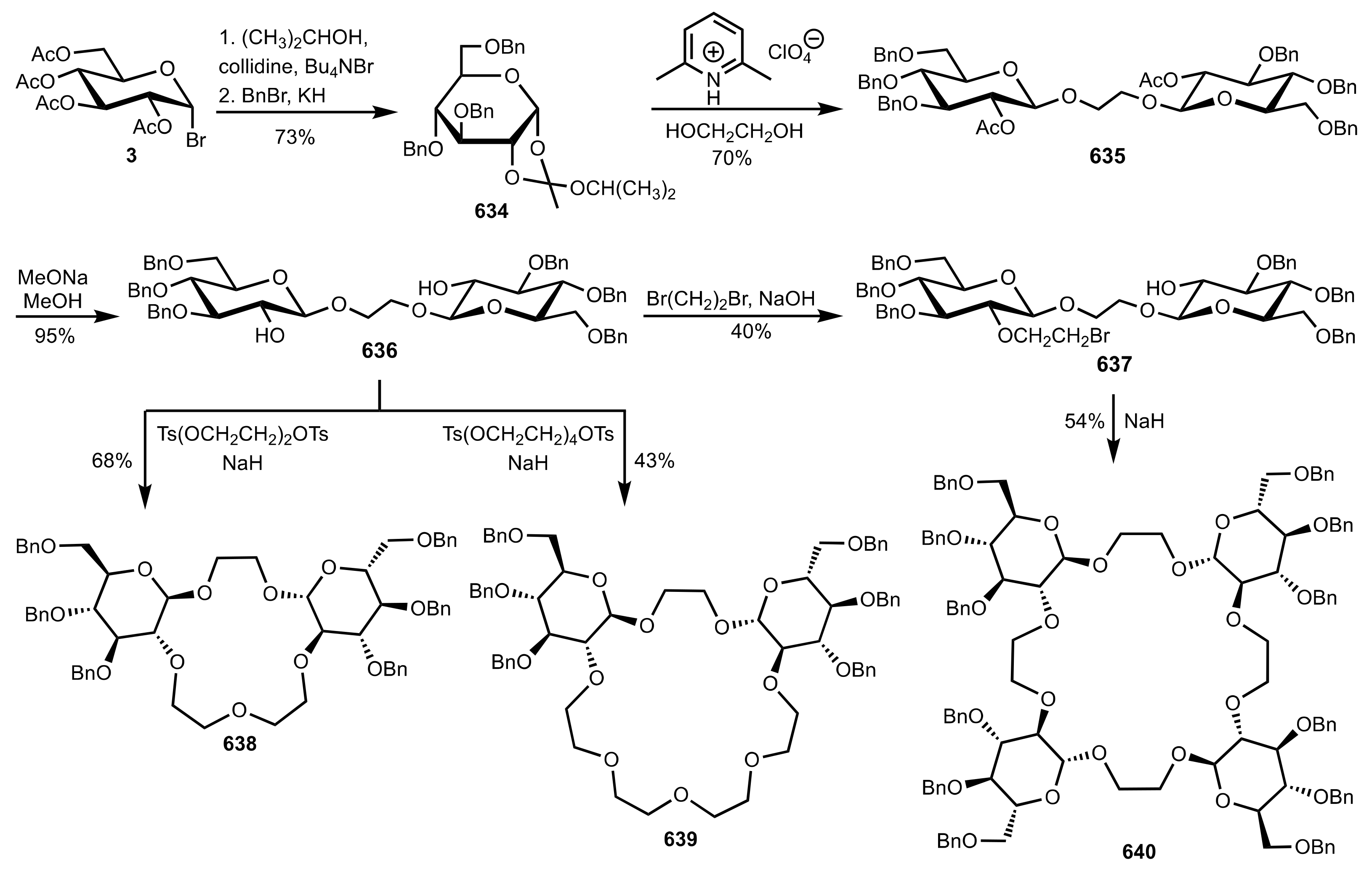
8.2.3. Disaccharide and Oligosaccharide-Based Crown Ethers
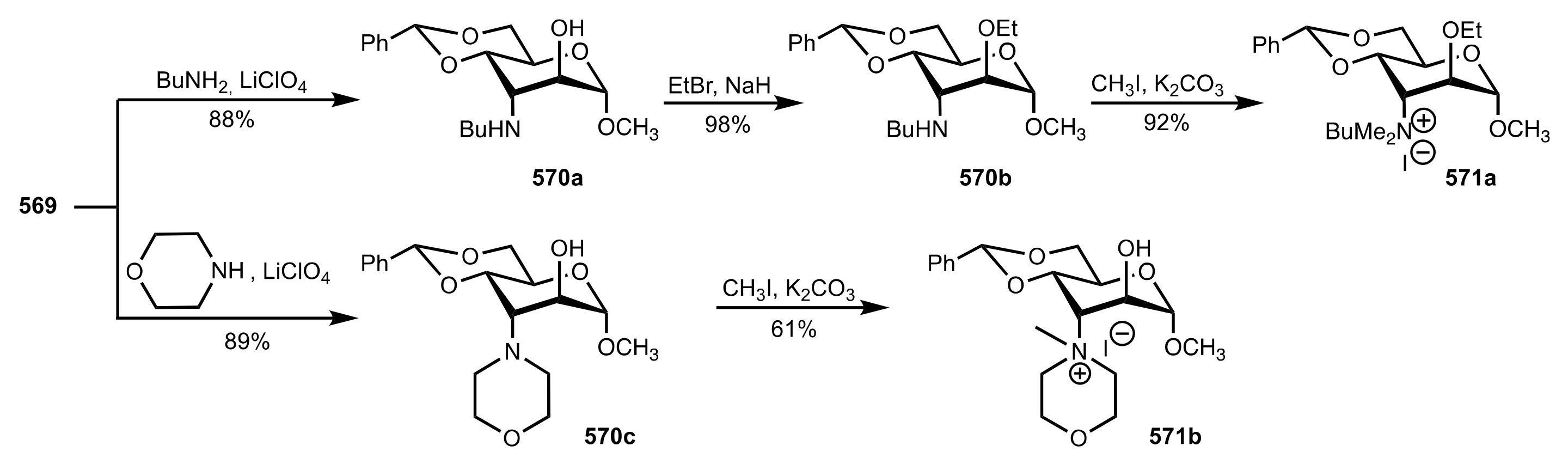
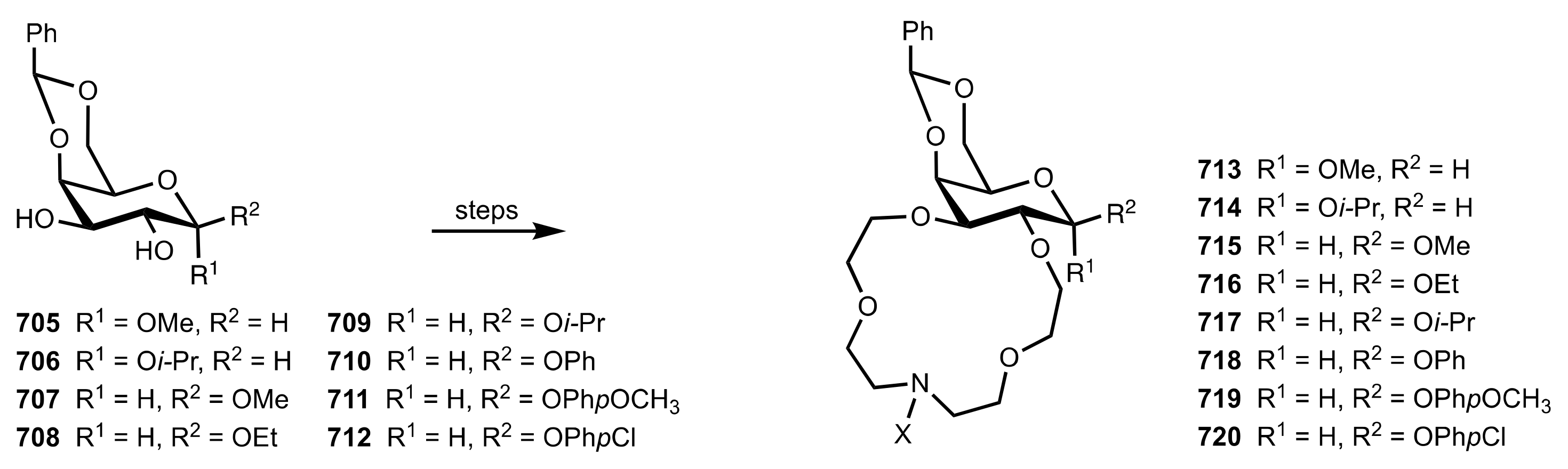
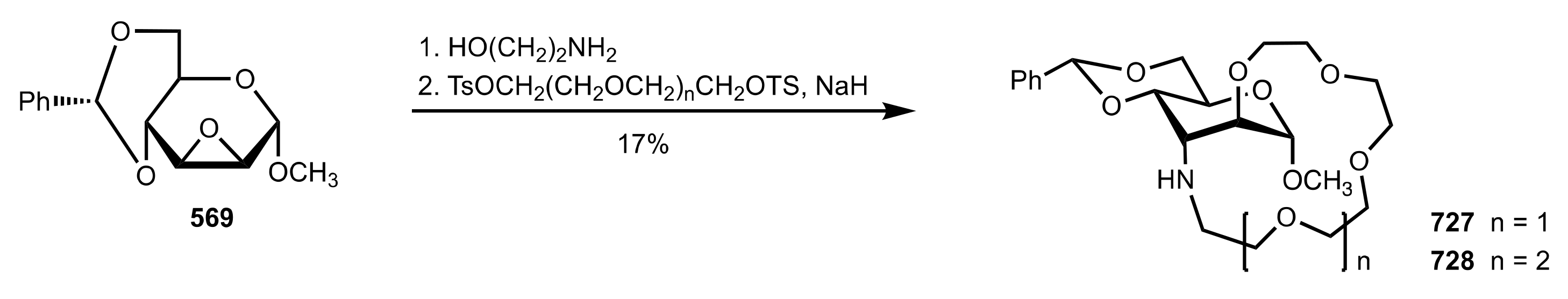
8.3. Sugar Aza-Crown Ethers
8.3.1. Alditol-Based Aza-Crown Ethers
8.3.2. Glycal-Based Aza-Crown Ethers
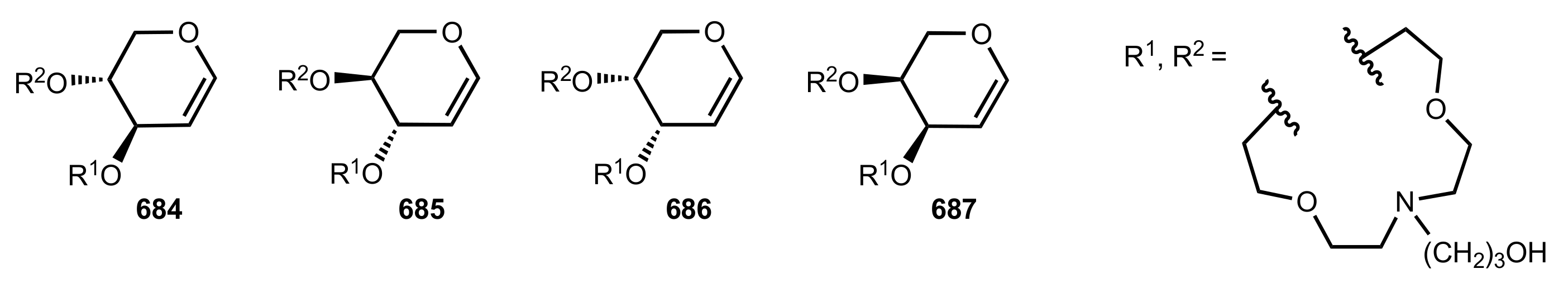
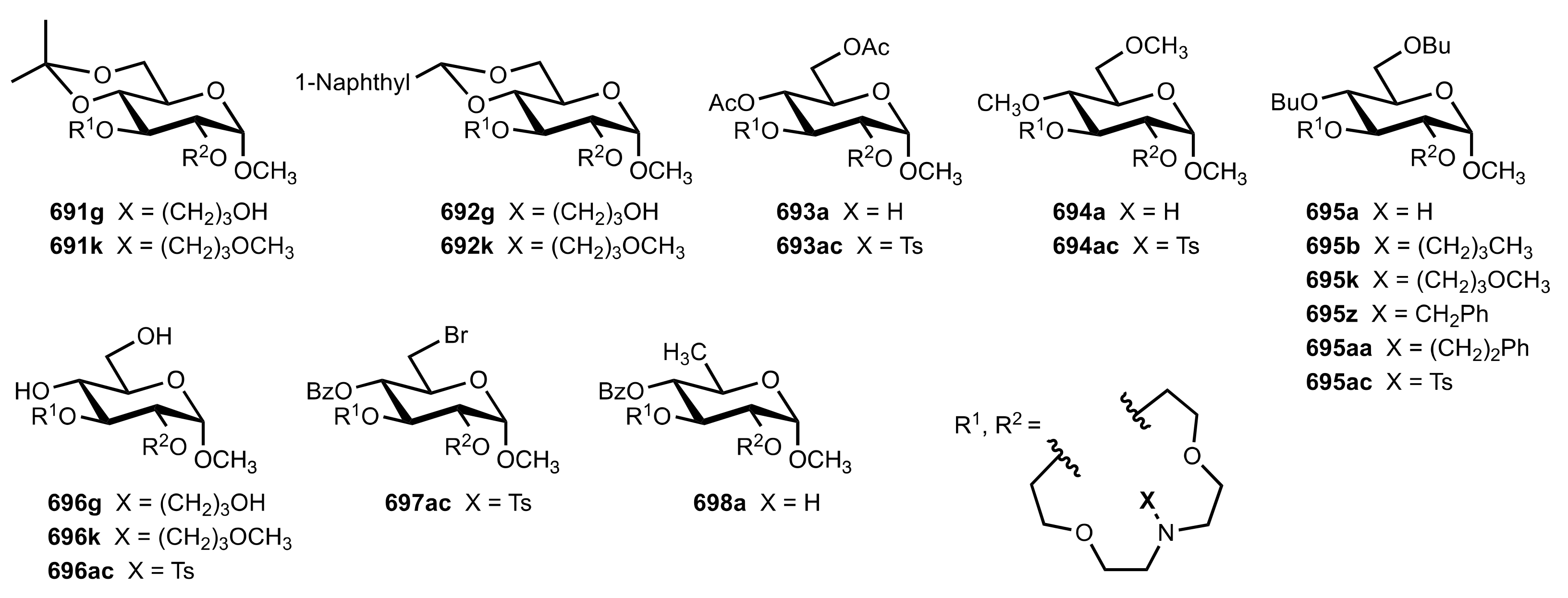
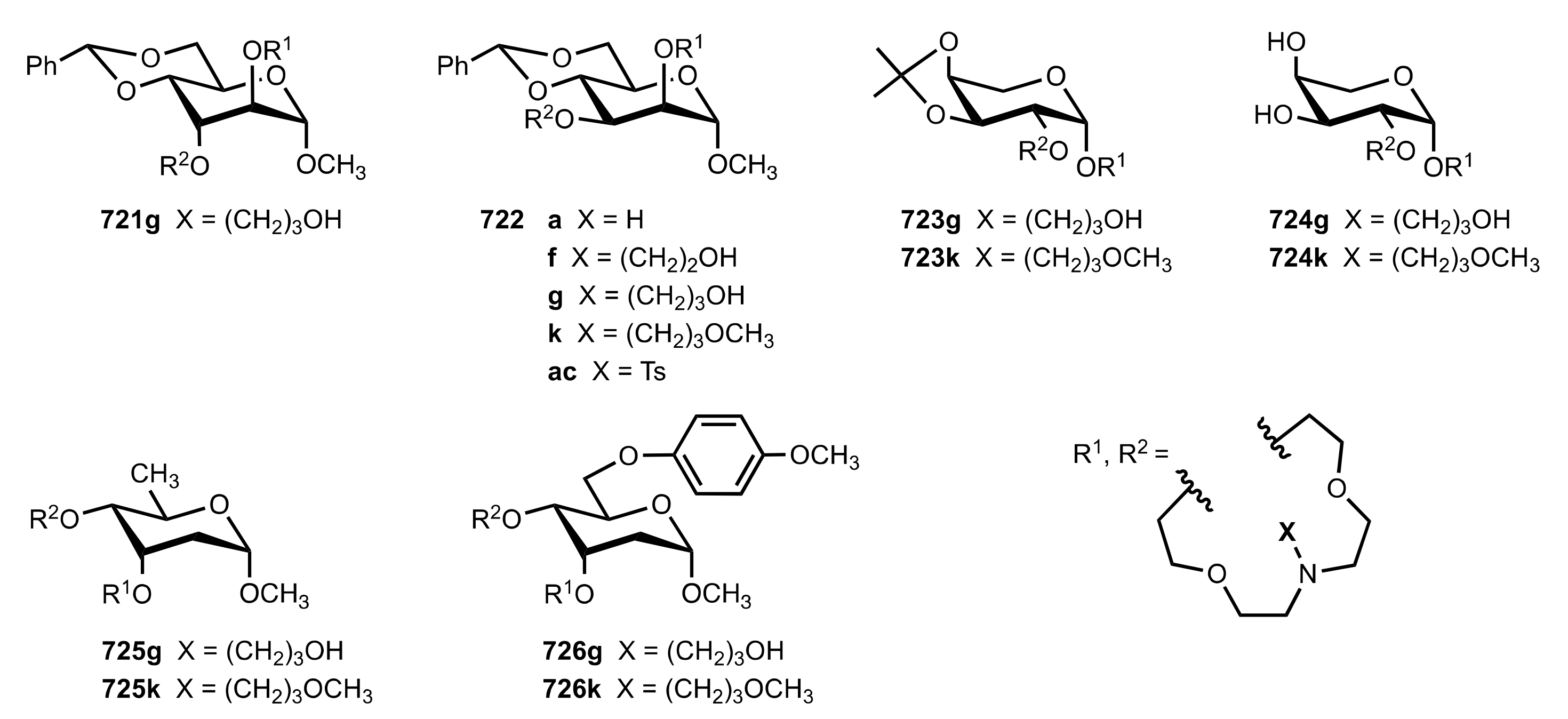
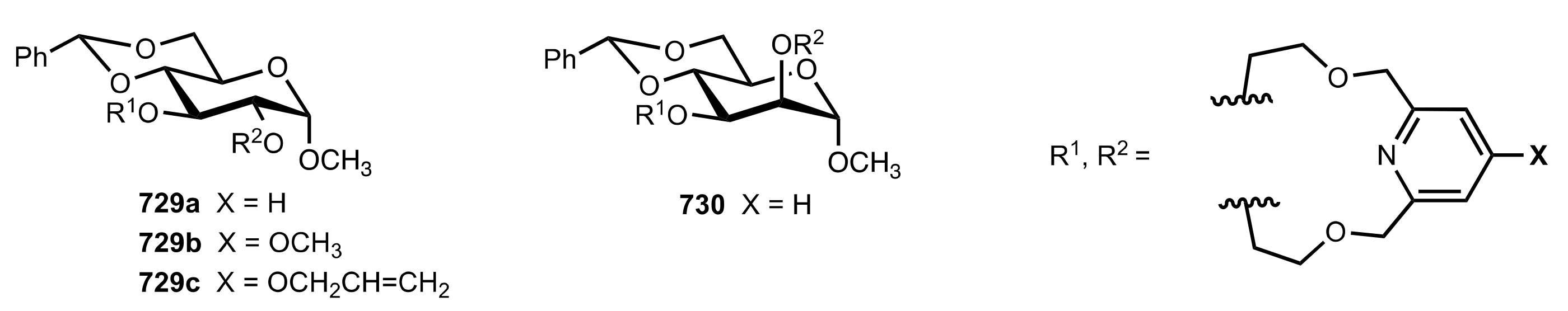
8.3.3. Aldose-Based Aza-Crown Ethers
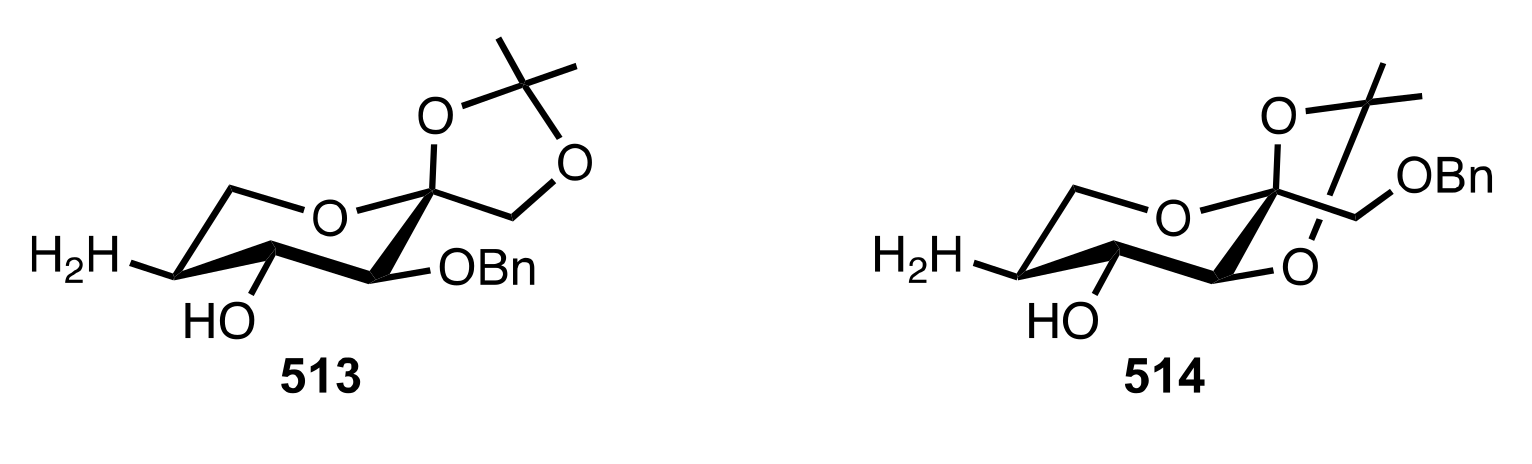
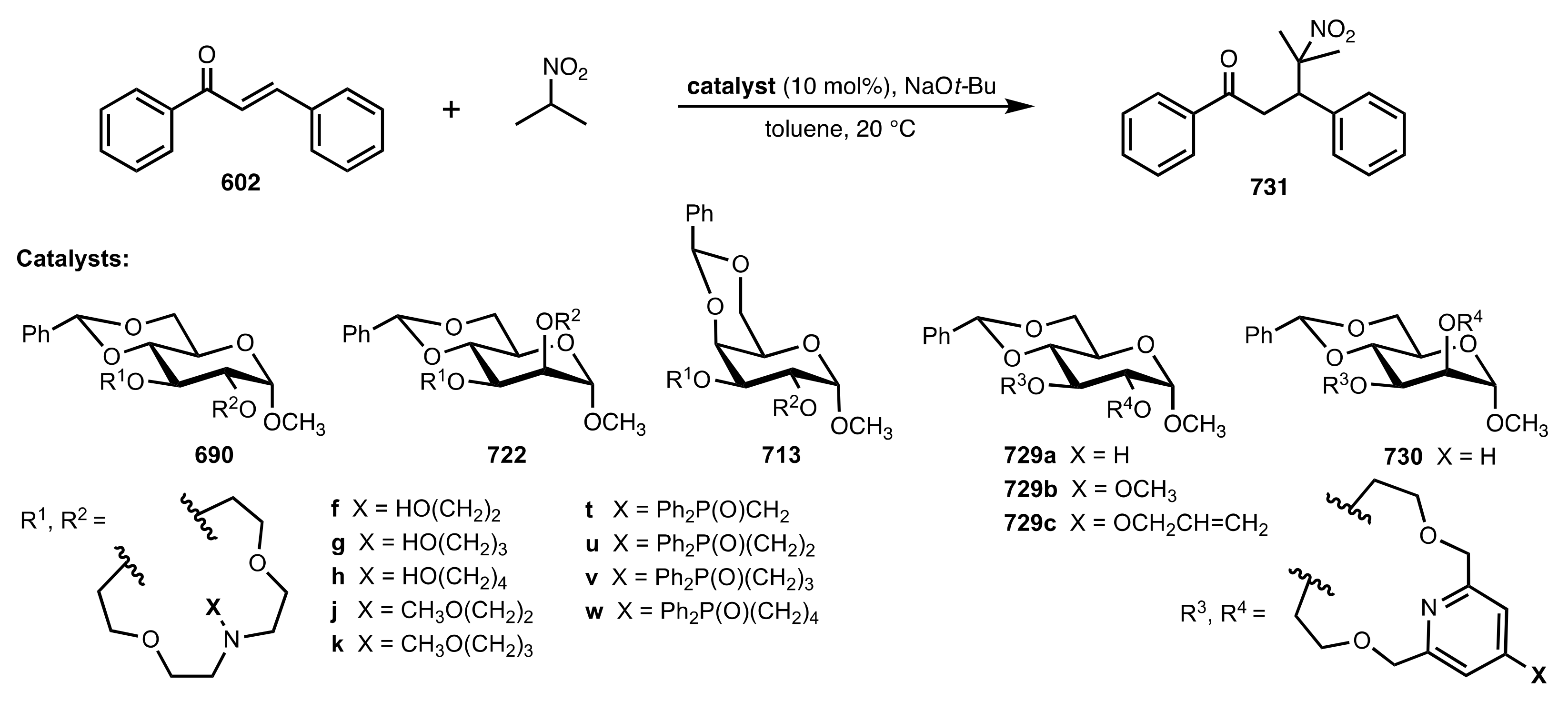
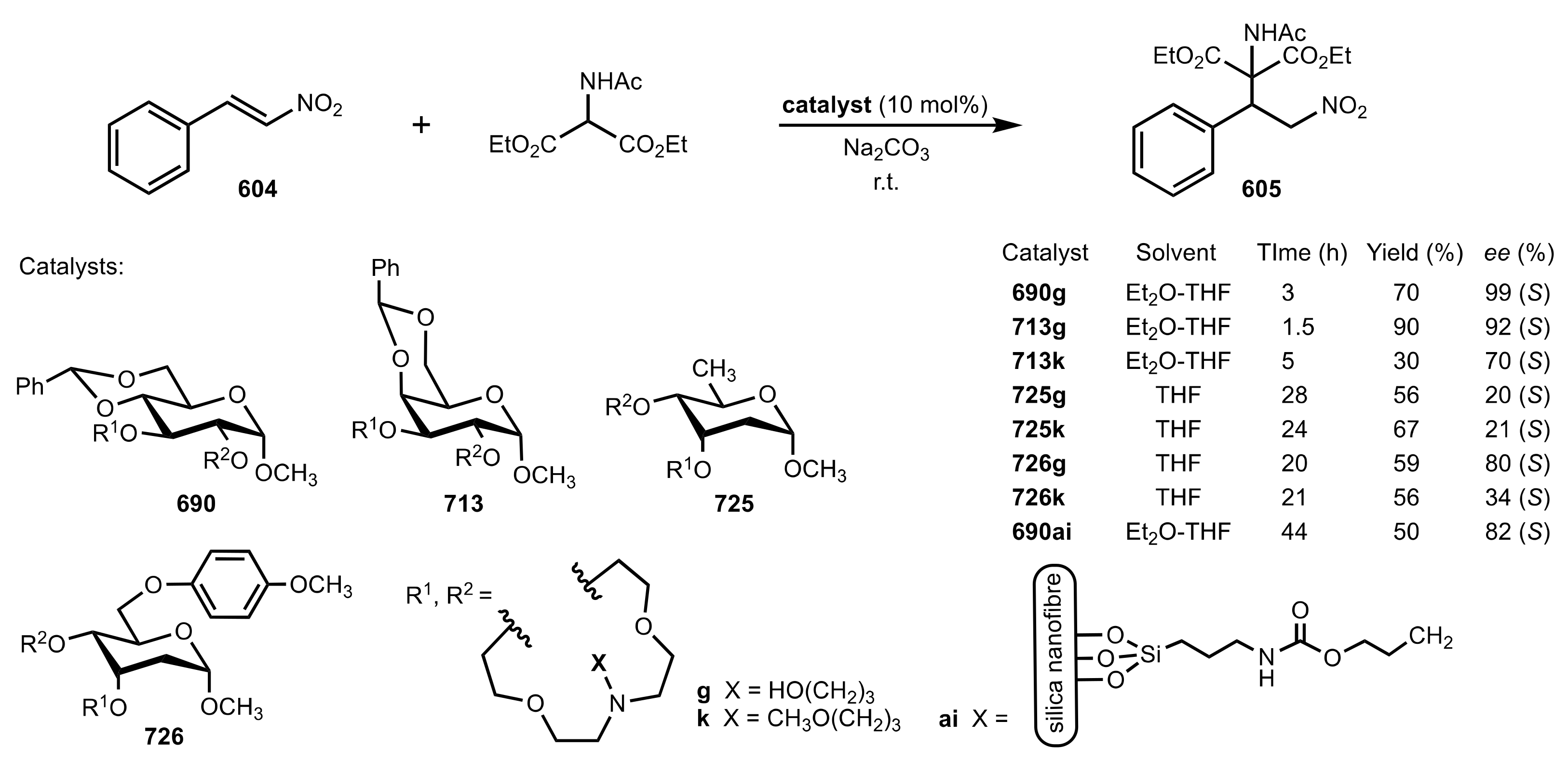
8.3.3.1. Synthesis
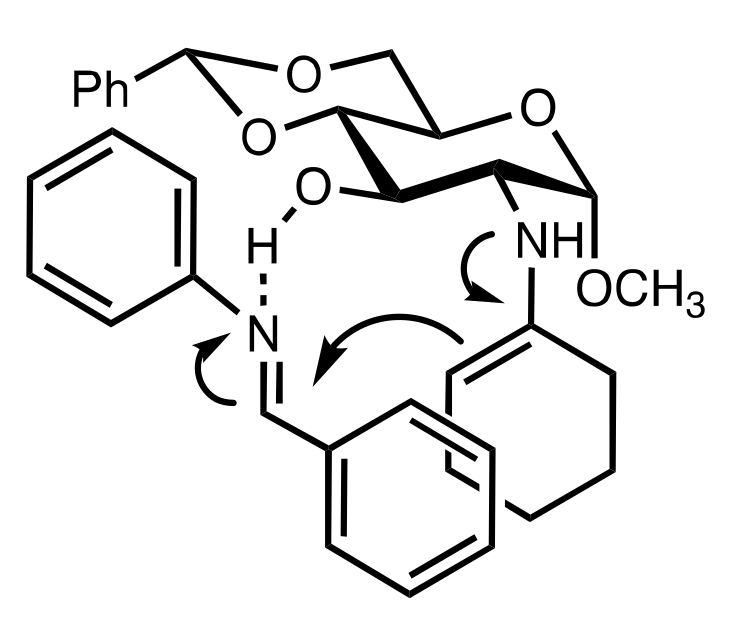
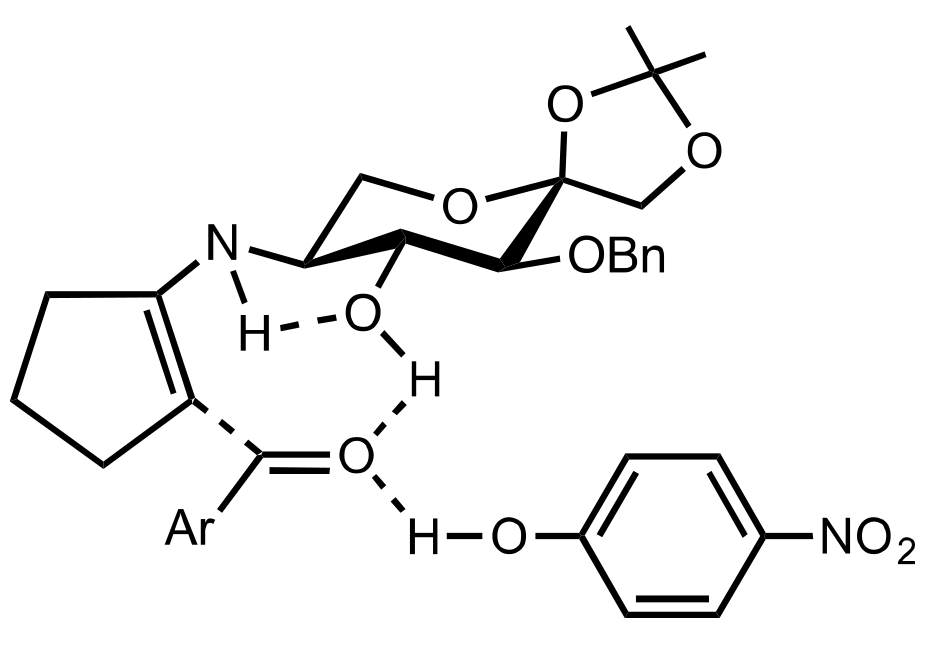
8.3.3.2. Catalysis
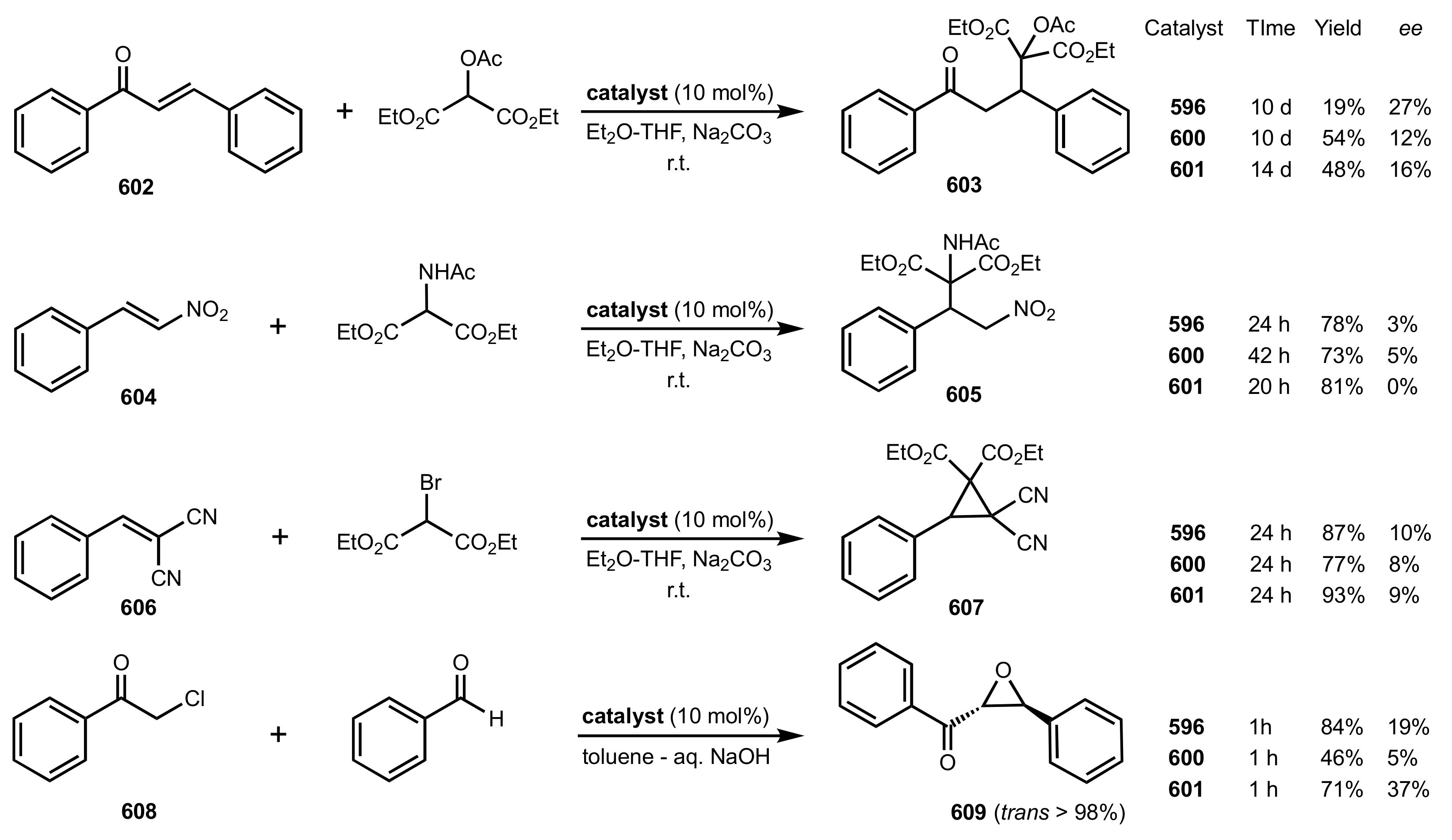
- Michael additions
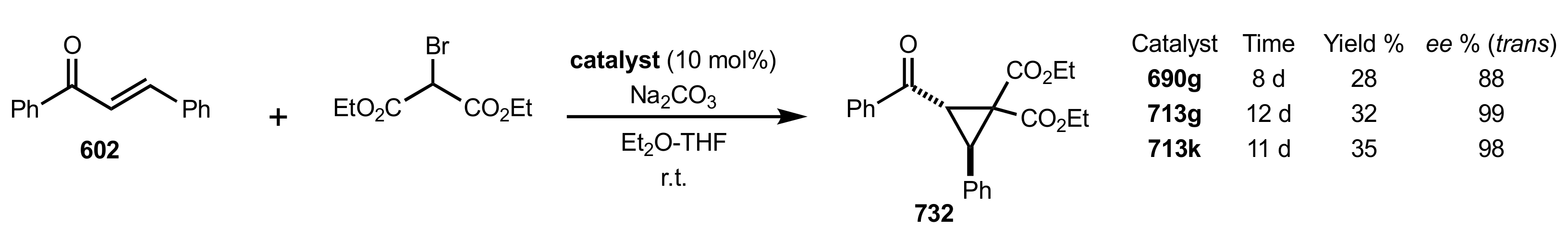
- Cyclopropanation
- Epoxidation of enones
- Darzens condensation
9. Conclusions
Funding
Conflicts of Interest
References and Notes
- Sharpless, K.B. Searching for New Reactivity (Nobel Lecture). Angew. Chem. Int. Ed. 2002, 41, 2024–2032. [Google Scholar] [CrossRef]
- Van der Helm, M.P.; Klemm, B.; Eelkema, R. Organocatalysis in aqueous media. Nat. Rev. Chem. 2019, 3, 491–508. [Google Scholar] [CrossRef]
- Xiang, S.-H.; Tan, B. Advances in asymmetric organocatalysis over the last 10 years. Nat. Commun. 2020, 11, 3786. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; He, X.-H.; Liu, Y.-Q.; He, G.; Peng, C.; Li, J.-L. Asymmetric organocatalysis: An enabling technology for medicinal chemistry. Chem. Soc. Rev. 2021, 50, 1522–1586. [Google Scholar] [CrossRef]
- Jarosz, S.; Listkowski, A. Sugar Derived Crown Ethers and Their Analogs: Synthesis and Properties. Curr. Org. Chem. 2006, 10, 643–662. [Google Scholar] [CrossRef]
- Bakó, P.; Keglevich, G.; Rapi, Z. Asymmetric Phase Transfer Reactions Catalyzed by Chiral Crown Ethers Derived from Monosaccharides. Lett. Org. Chem. 2010, 7, 645–656. [Google Scholar] [CrossRef]
- Bakó, P.; Rapi, Z.; Keglevich, G. Sugar-based Crown Ethers in Enantioselective Syntheses. Period. Polytech. Chem. Eng. 2015, 59, 51–58. [Google Scholar] [CrossRef] [Green Version]
- Schettini, R.; Sicignano, M.; De Riccardis, F.; Izzo, I.; Sala, G.D. Macrocyclic Hosts in Asymmetric Phase-Transfer Catalyzed Reactions. Synthesis 2018, 50, 4777–4795. [Google Scholar]
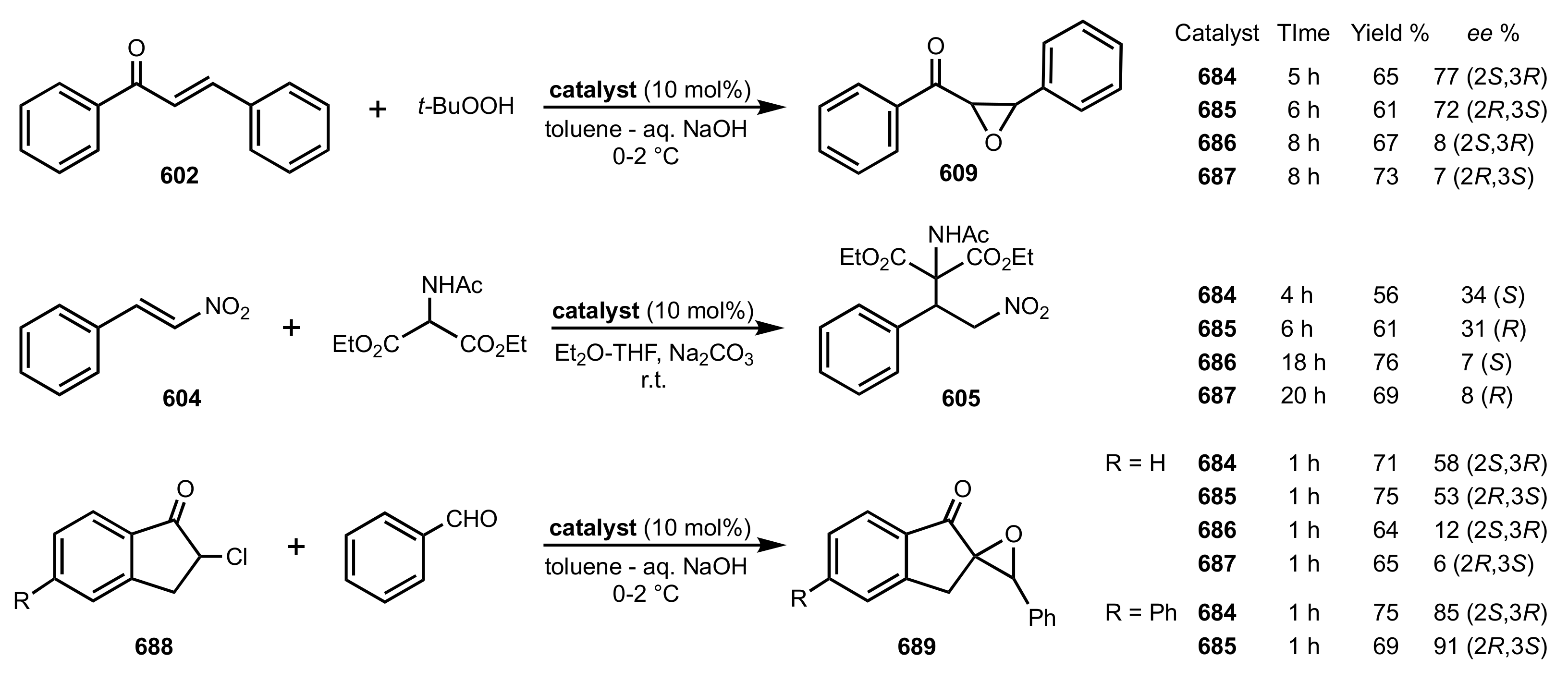
- Frohn, M.; Shi, Y. Chiral Ketone-Catalyzed Asymmetric Epoxidation of Olefins. Synthesis 2000, 2000, 1979–2000. [Google Scholar] [CrossRef] [Green Version]
- Wong, O.A.; Shi, Y. Organocatalytic oxidation. Asymmetric epoxidation of olefins catalyzed by chiral ketones and iminium salts. Chem. Rev. 2008, 108, 3958–3987. [Google Scholar] [CrossRef]
- Shi, Y. Organocatalytic asymmetric epoxidation of olefins by chiral ketones. Acc. Chem. Res. 2004, 37, 488–496. [Google Scholar] [CrossRef]
- Yang, D. Ketone-Catalyzed Asymmetric Epoxidation Reactions. Acc. Chem. Res. 2004, 37, 497–505. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, Q.; Cornwall, R.G.; Shi, Y. Organocatalytic asymmetric epoxidation and aziridination of olefins and their synthetic applications. Chem. Rev. 2014, 114, 8199–8256. [Google Scholar] [CrossRef]
- Boysen, M.M.K. Carbohydrates as synthetic tools in organic chemistry. Chem. A Eur. J. 2007, 13, 8648–8659. [Google Scholar] [CrossRef]
- Lehnert, T.; Özüduru, G.; Grugel, H.; Albrecht, F.; Telligmann, S.M.; Boysen, M.M.K. More than just sweet—Sugar-derived stereodifferentiating agents for asymmetric synthesis. Synthesis 2011, 2011, 2685–2708. [Google Scholar]
- Ma, J.A.; Zhang, G.-W. Enantioselective Addition Reactions Catalyzed by Carbohydrate-Derived Organocatalysts. In Carbohydrates—Tools for Stereoselective Synthesis, 1st ed.; Boysen, M.M.K., Ed.; Wiley-VCH: Weinheim, Germany, 2013; pp. 351–370. [Google Scholar]
- Phillips, A.M.F. Applications of Carbohydrate-Based Organocatalysts in Enantioselective Synthesis. Eur. J. Org. Chem. 2014, 2014, 7291–7303. [Google Scholar] [CrossRef]
- Henderson, A.S.; Bower, J.F.; Galan, M.C. Carbohydrates as enantioinduction components in stereoselective catalysis. Org. Biomol. Chem. 2016, 14, 4008–4017. [Google Scholar] [CrossRef] [Green Version]
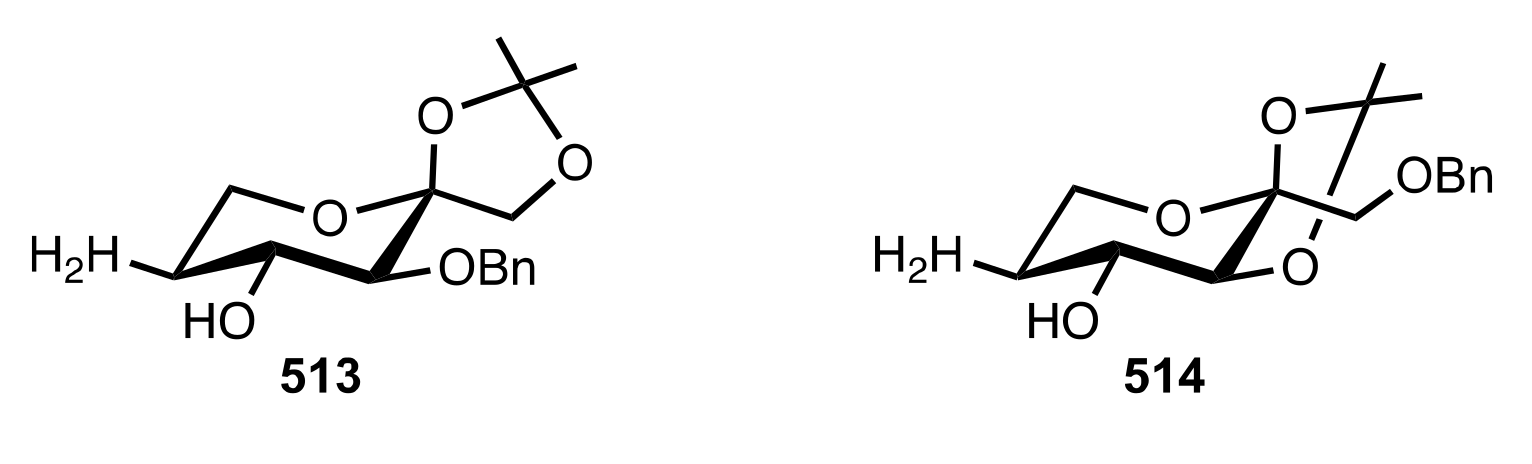
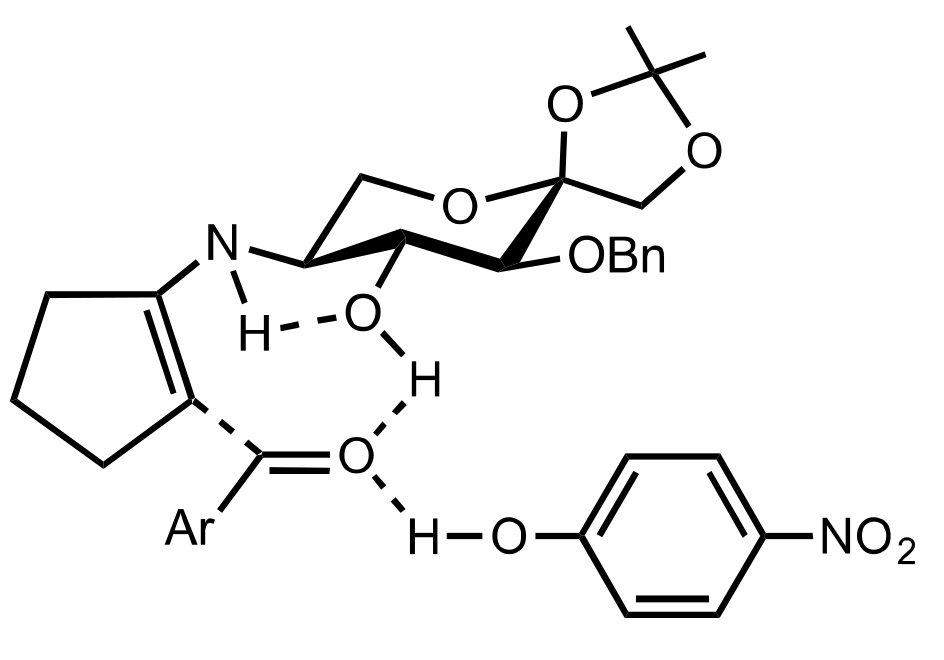
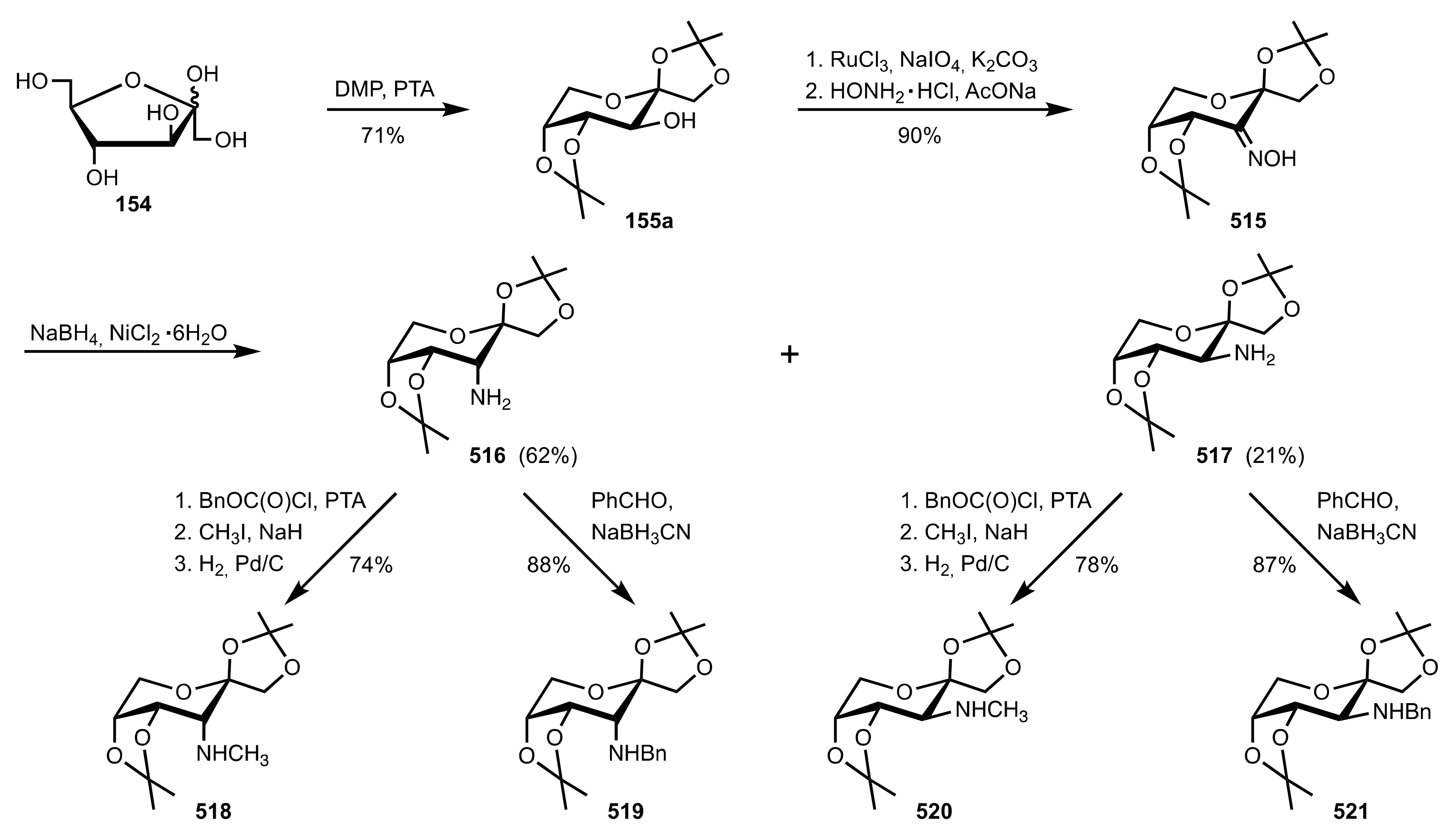
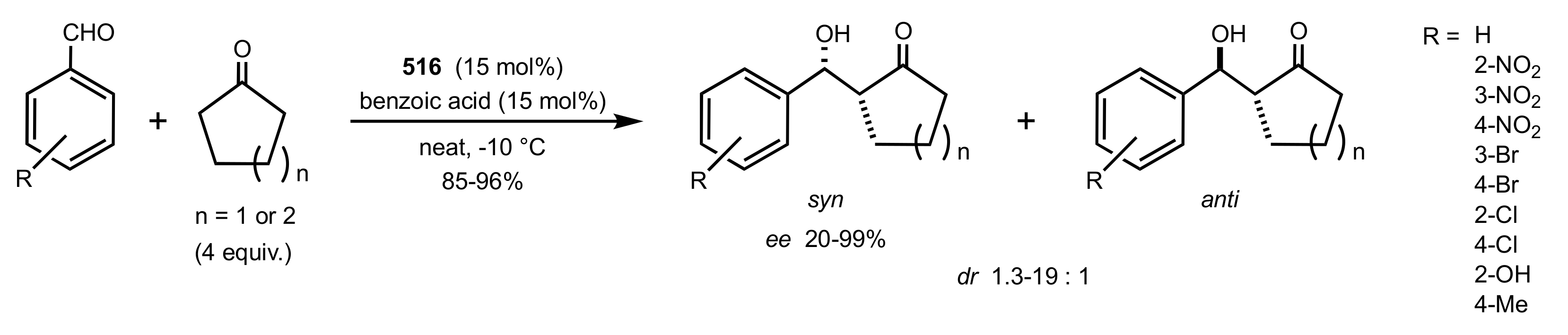
- Agarwal, J. Progress in aminosugar derived asymmetric organocatalysis. Org. Biomol. Chem. 2016, 14, 10747–10762. [Google Scholar] [CrossRef]
- Mondal, M.A.; Mandal, D. Carbohydrate-based organocatalysts in direct asymmetric aqueous aldol reaction. J. Carbohydr. Chem. 2016, 35, 181–200. [Google Scholar] [CrossRef]
- Ikai, T. The dawn of chiral material development using saccharide-based helical polymers. Polym. J. 2017, 49, 355–362. [Google Scholar] [CrossRef]
- Mahé, O.; Brière, J.-F.; Dez, I. Chitosan: An Upgraded Polysaccharide Waste for Organocatalysis. Eur. J. Org. Chem. 2015, 2015, 2559–2578. [Google Scholar] [CrossRef]
- El Kadib, A. Chitosan as a Sustainable Organocatalyst: A Concise Overview. ChemSusChem 2015, 8, 217–244. [Google Scholar] [CrossRef]
- Pan, D.; Ganguly, J. Assessment of Chitosan Based Catalyst and Their Mode of Action. Curr. Organocatal. 2019, 6, 106–138. [Google Scholar] [CrossRef]
- Meninno, S. Valorization of Waste: Sustainable Organocatalysts from Renewable Resources. ChemSusChem 2020, 13, 439–468. [Google Scholar] [CrossRef]
- Sigman, M.S.; Jacobsen, E.N. Schiff base catalysts for the asymmetric Strecker reaction identified and optimized from parallel synthetic libraries. J. Am. Chem. Soc. 1998, 120, 4901–4902. [Google Scholar] [CrossRef]
- Sigman, M.S.; Vachal, P.; Jacobsen, E.N. A general catalyst for the asymmetric Strecker reaction. Angew. Chem. Int. Ed. 2000, 39, 1279–1281. [Google Scholar] [CrossRef]
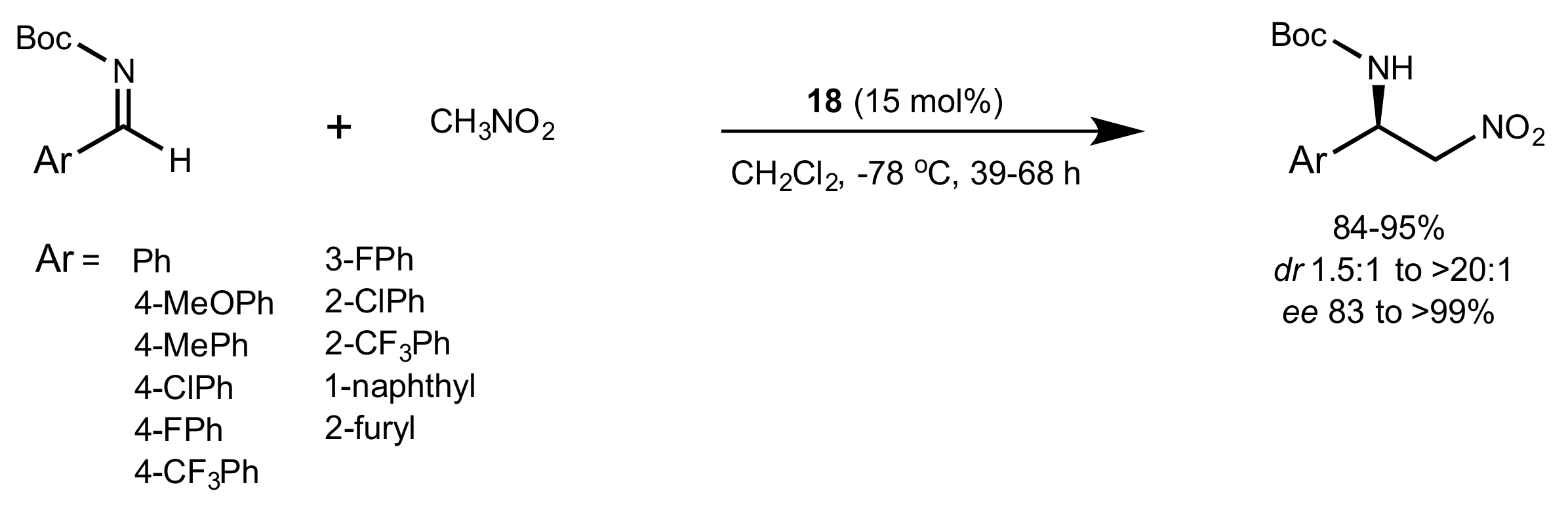
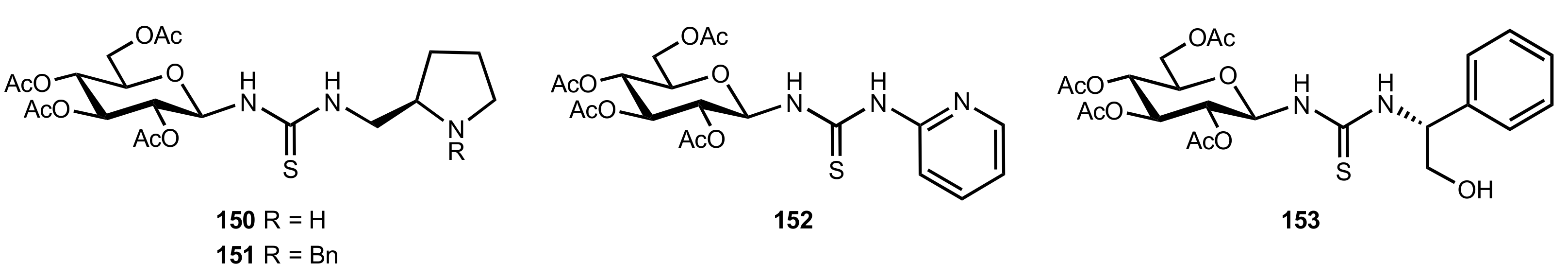
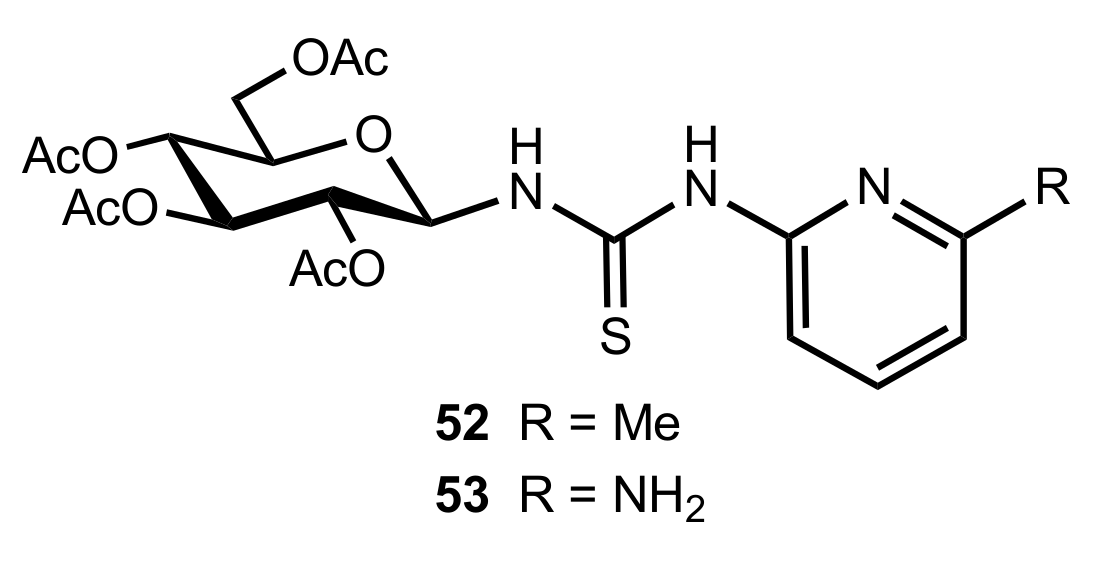
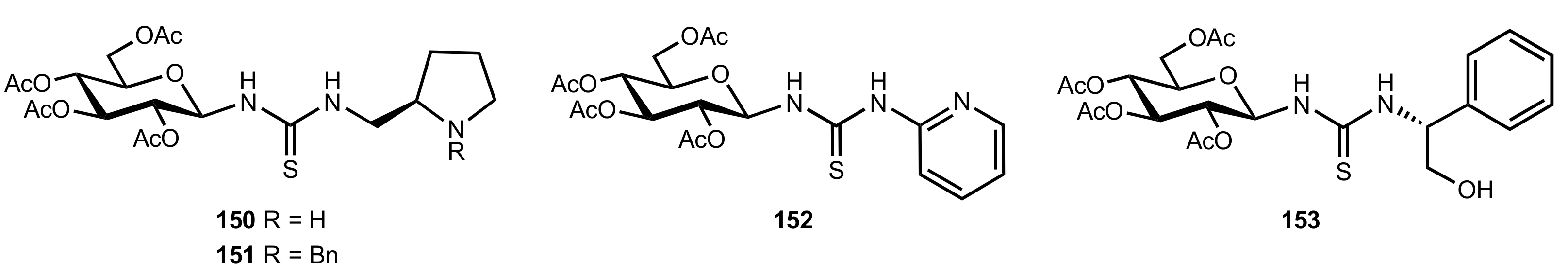
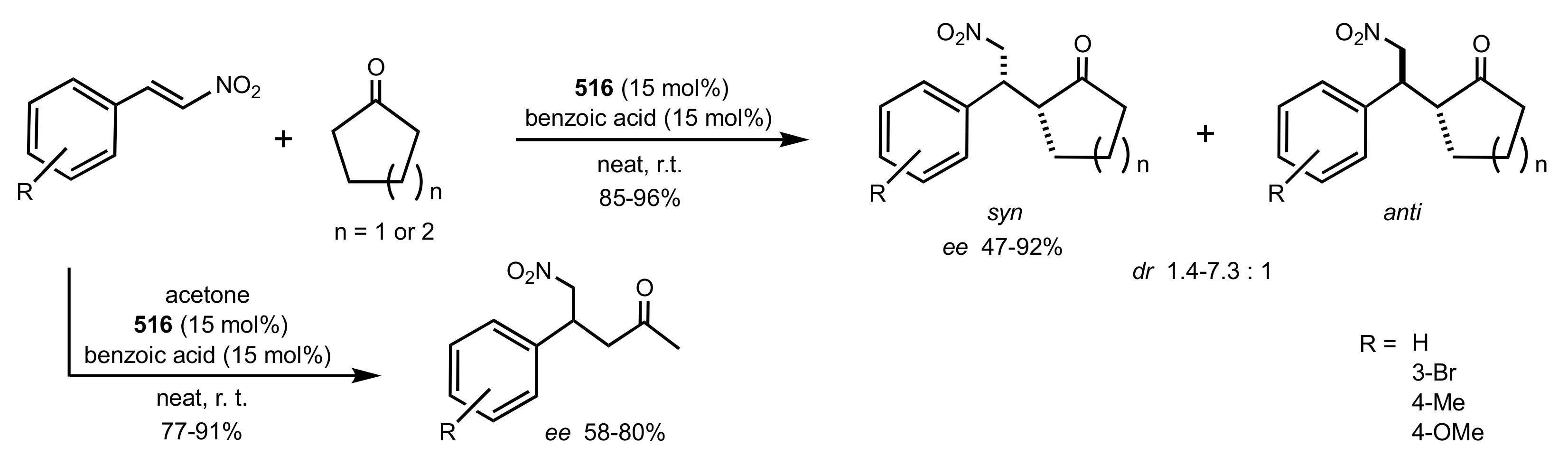
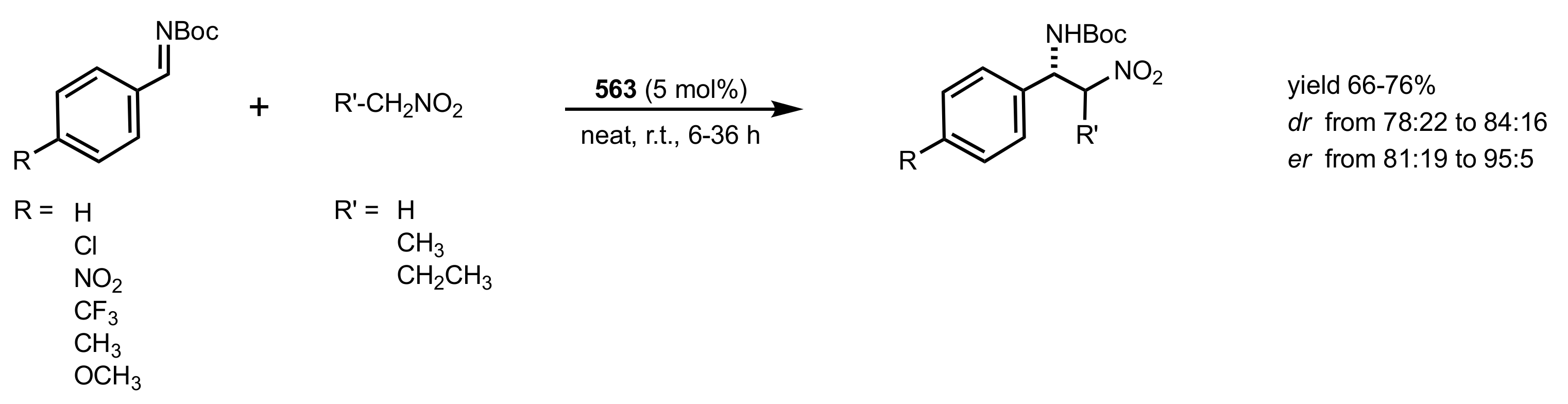
- Okino, T.; Nakamura, S.; Furukawa, T.; Takemoto, Y. Enantioselective aza-Henry reaction catalyzed by a bifunctional organocatalyst. Org. Lett. 2004, 6, 625–627. [Google Scholar] [CrossRef]
- Sohtome, Y.; Tanatani, A.; Hashimoto, Y.; Nagasawa, K. Development of bis-thiourea-type organocatalyst for asymmetric Baylis–Hillman reaction. Tetrahedron Lett. 2004, 45, 5589–5592. [Google Scholar] [CrossRef]
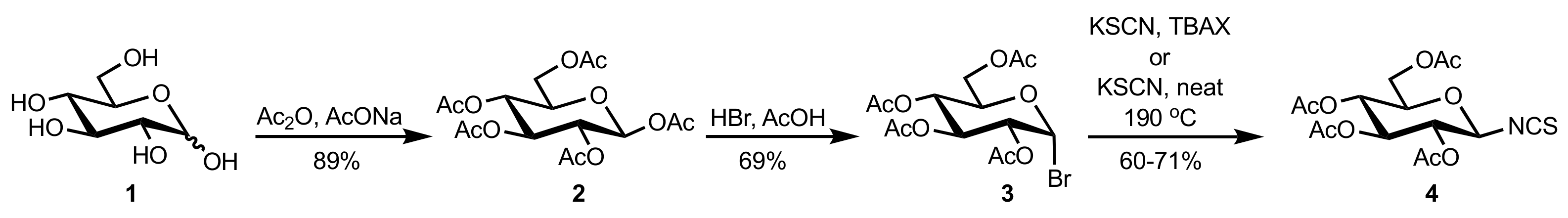
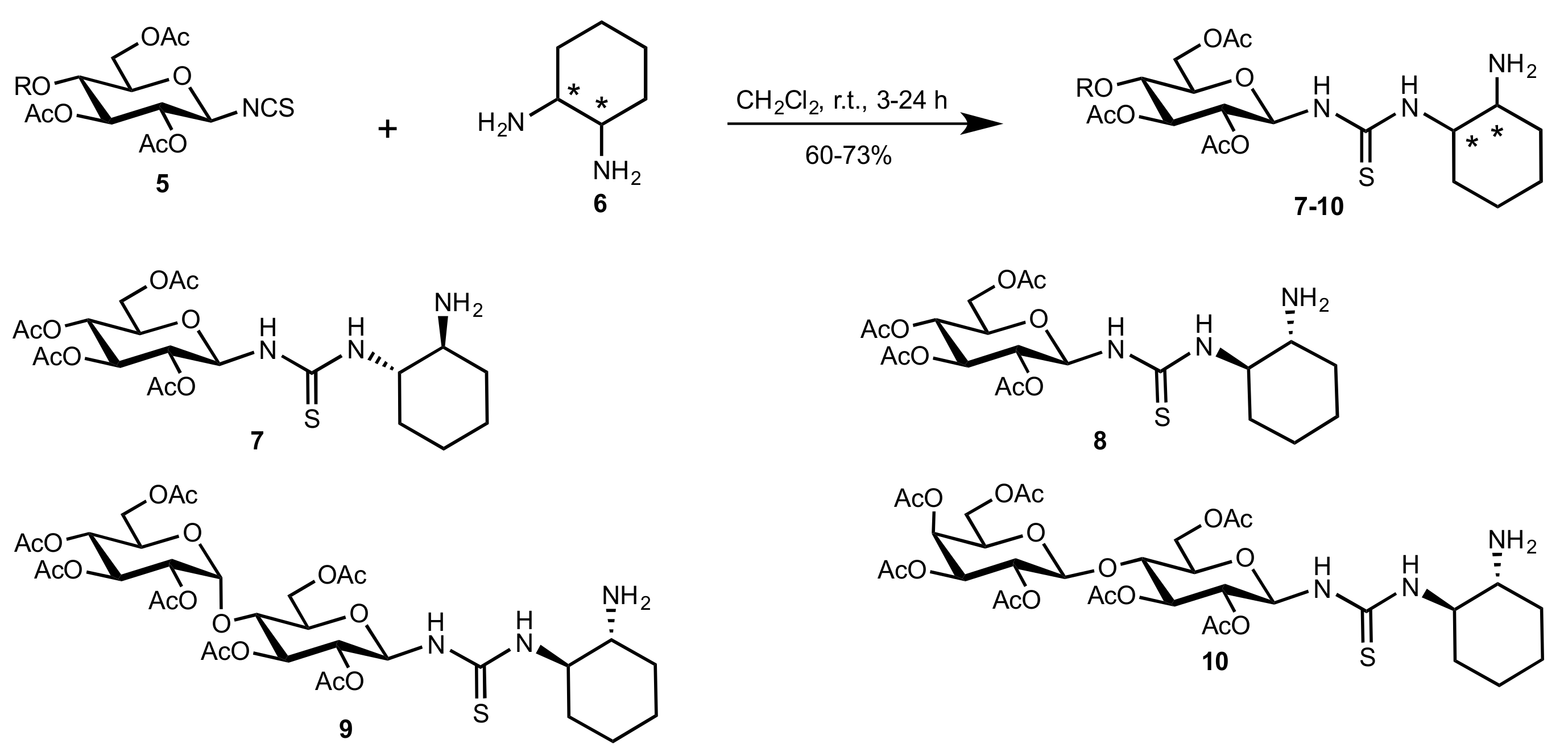
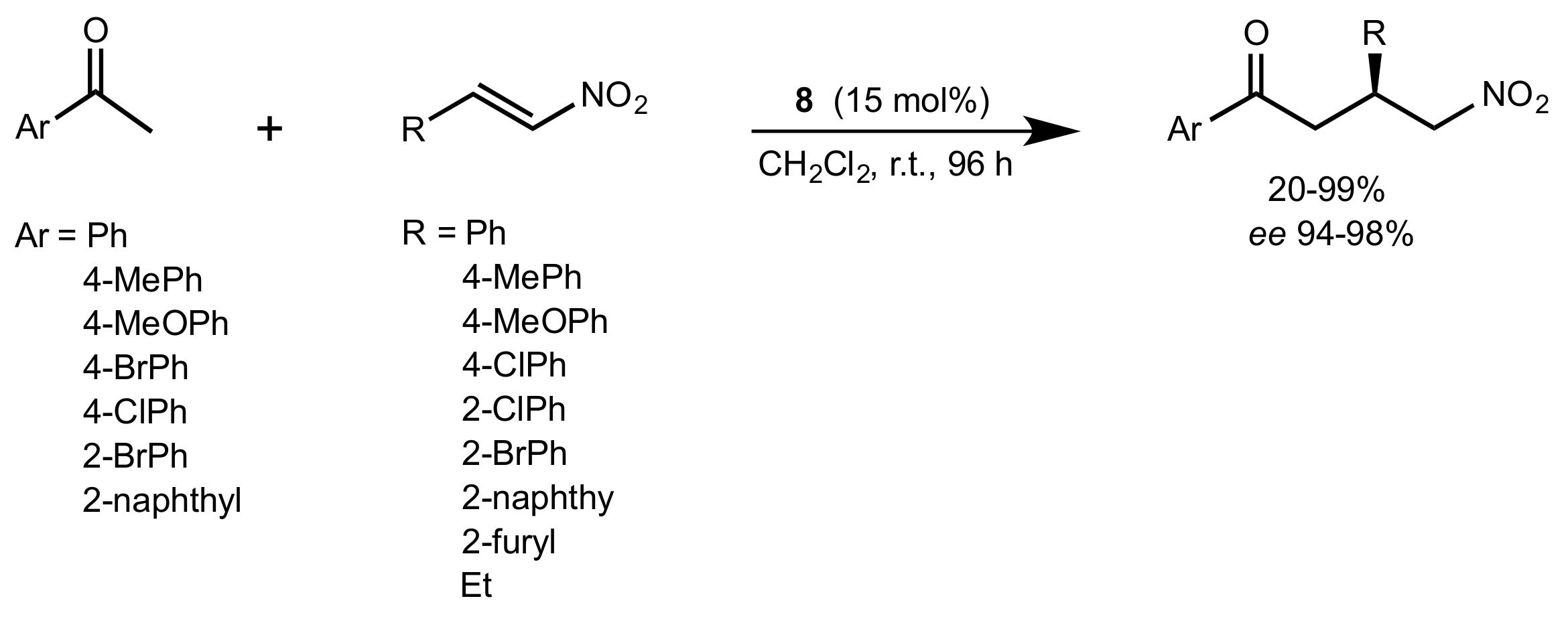
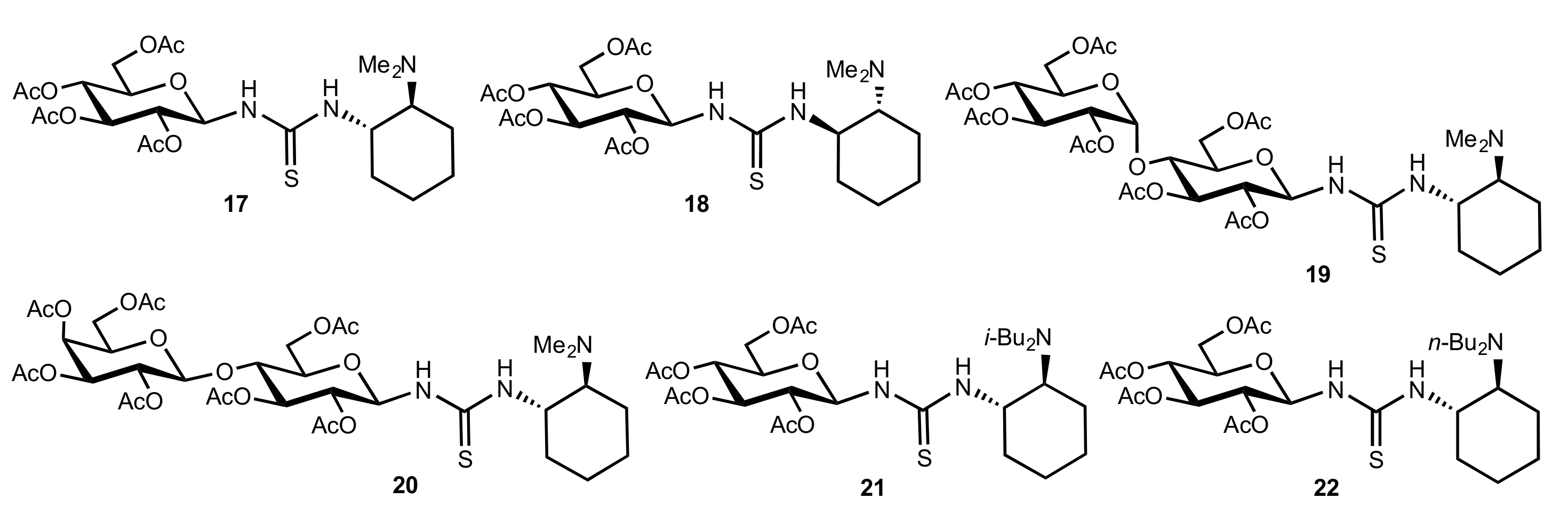
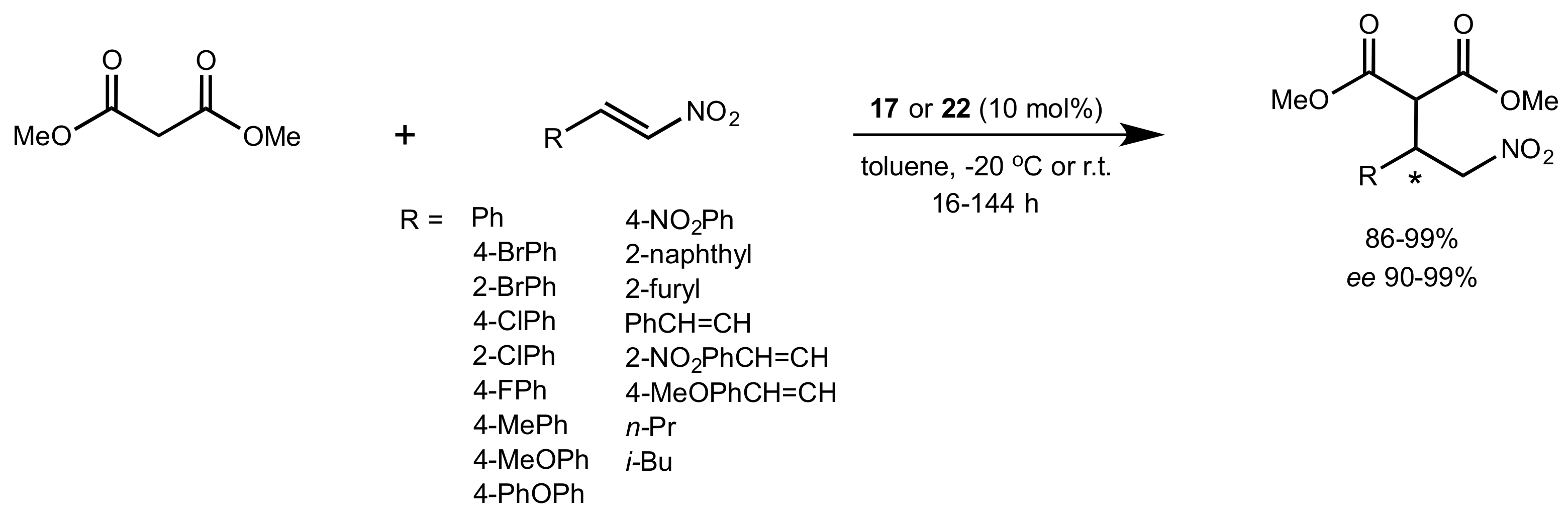
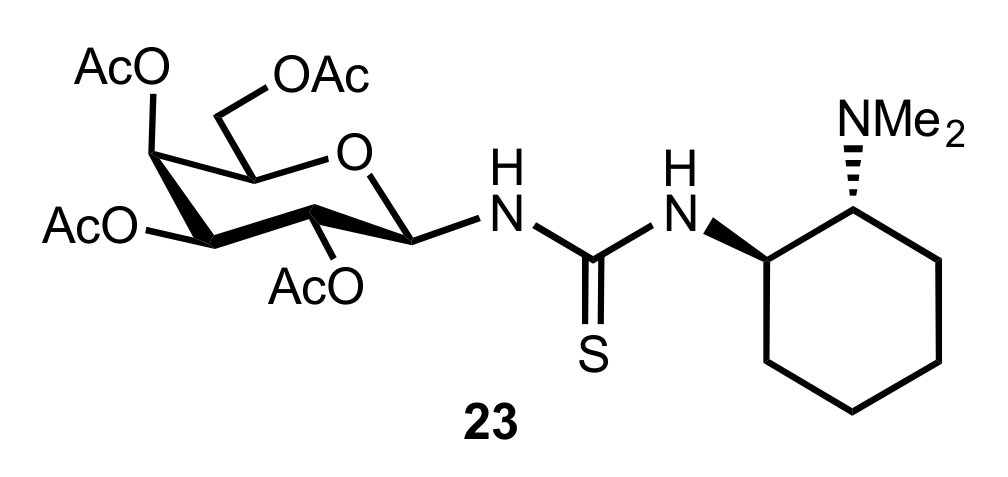
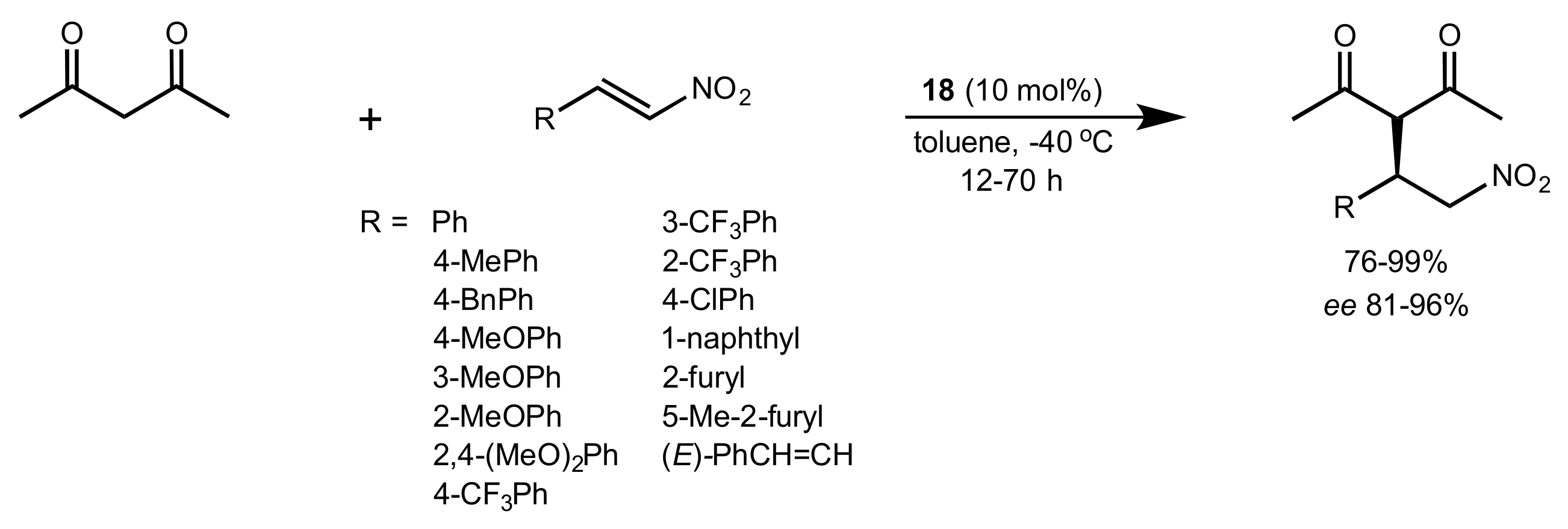
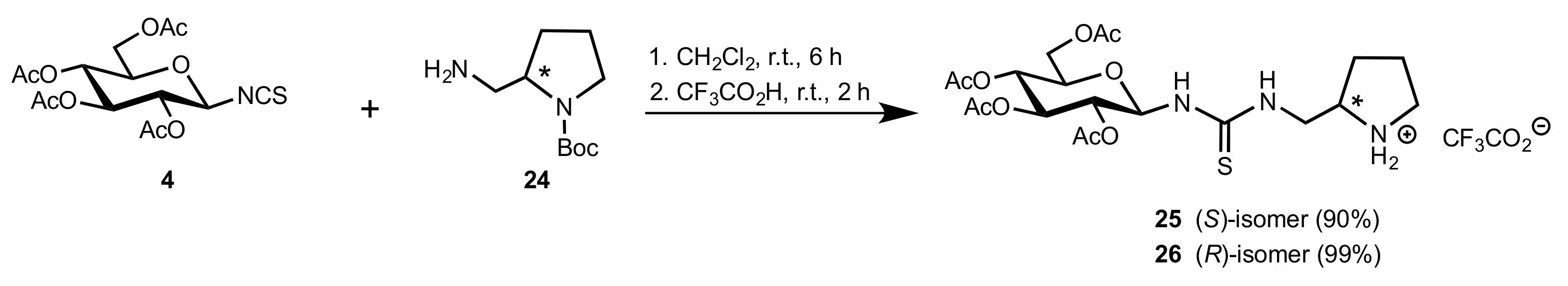
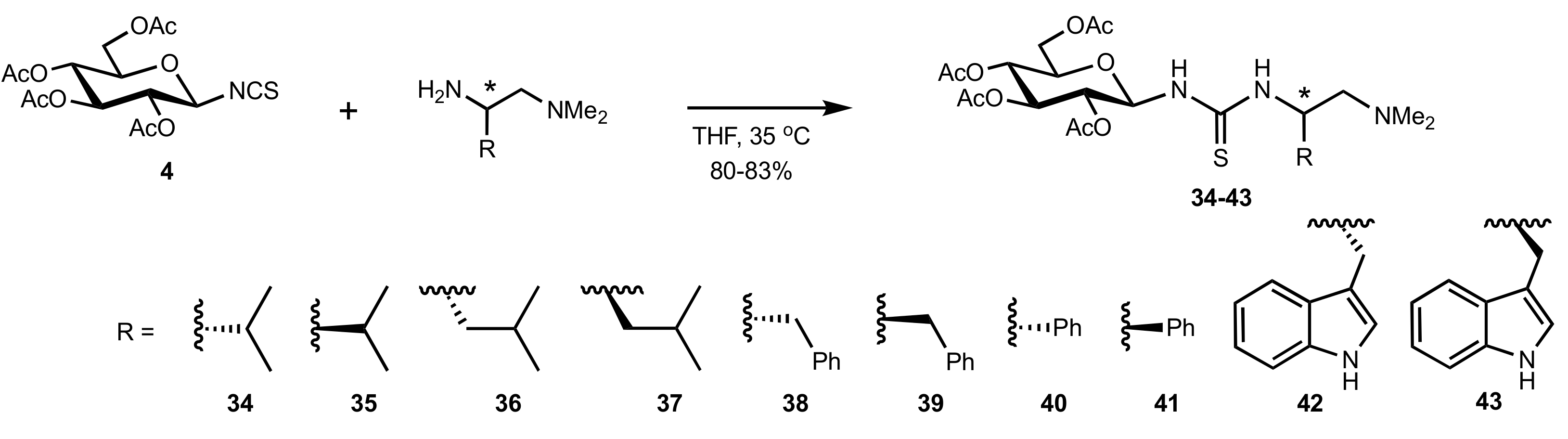
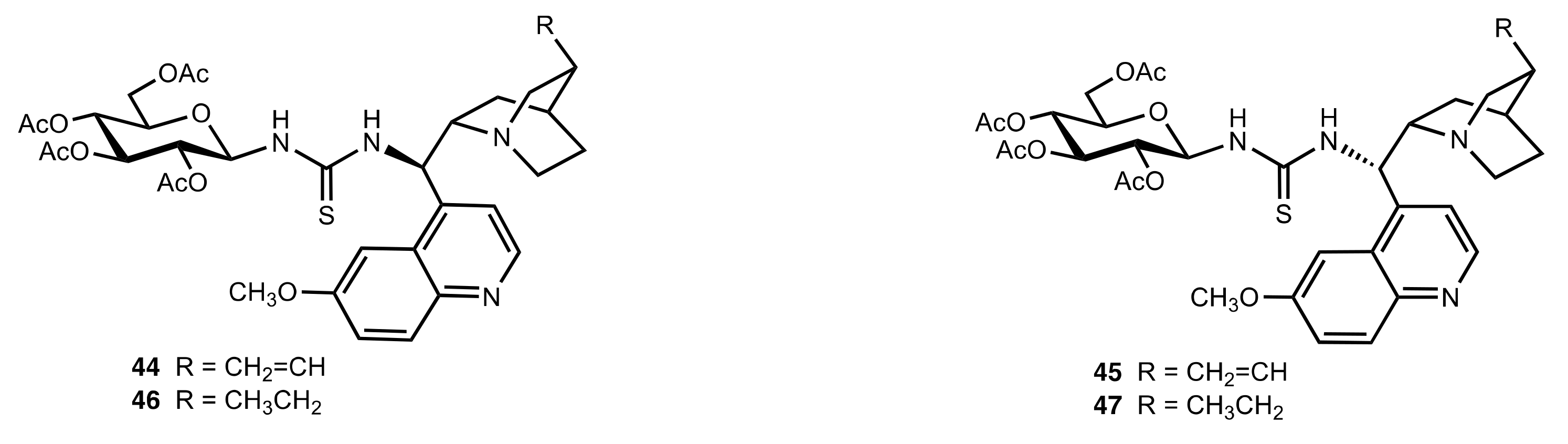
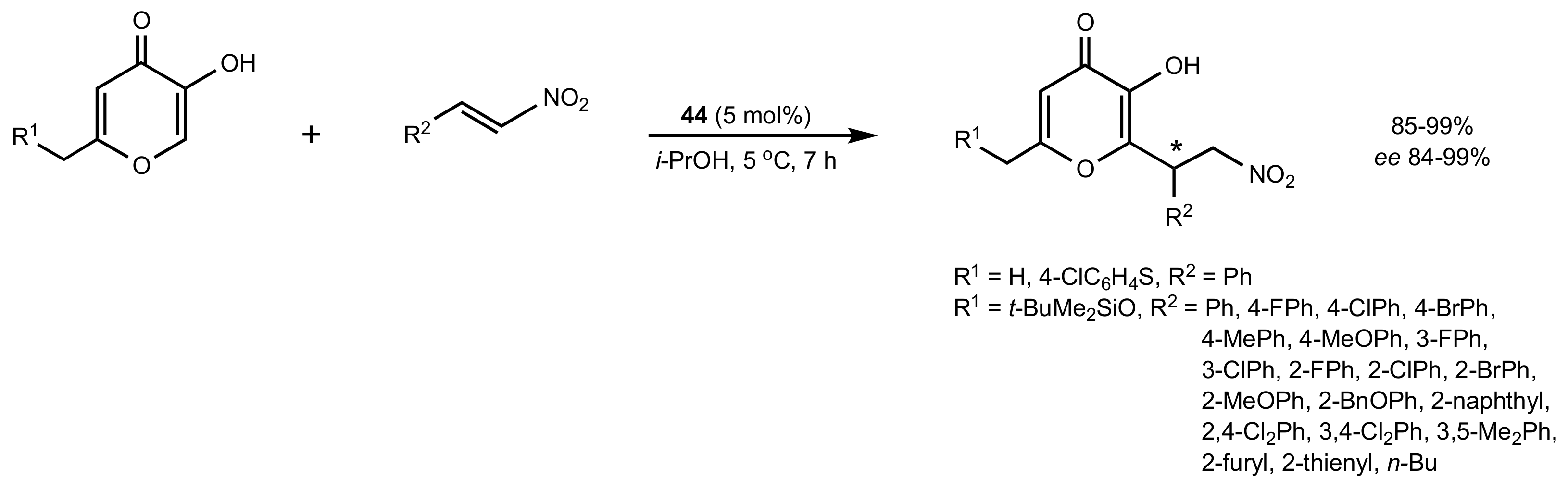
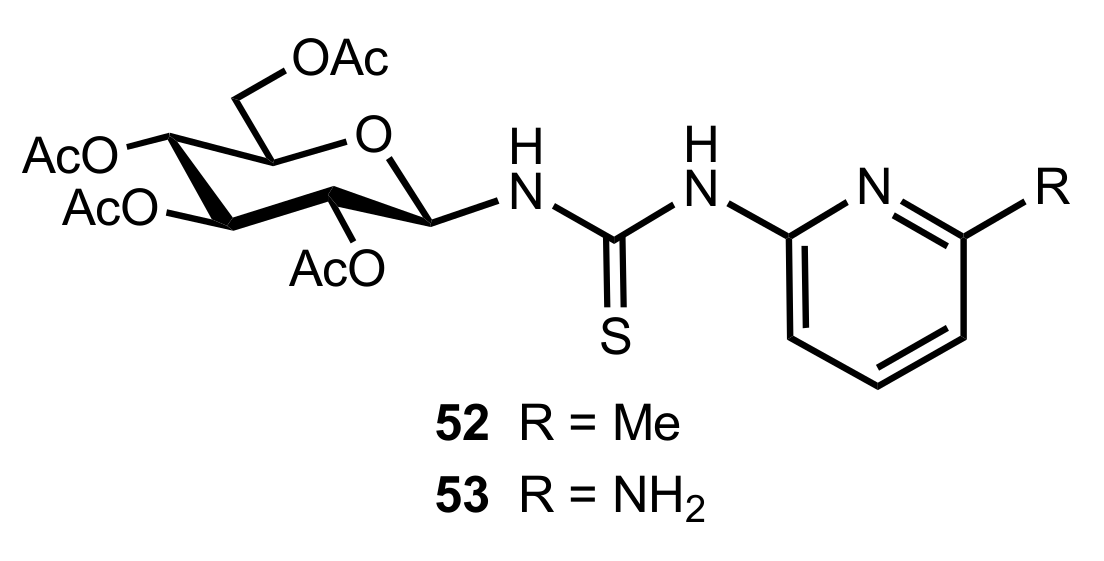
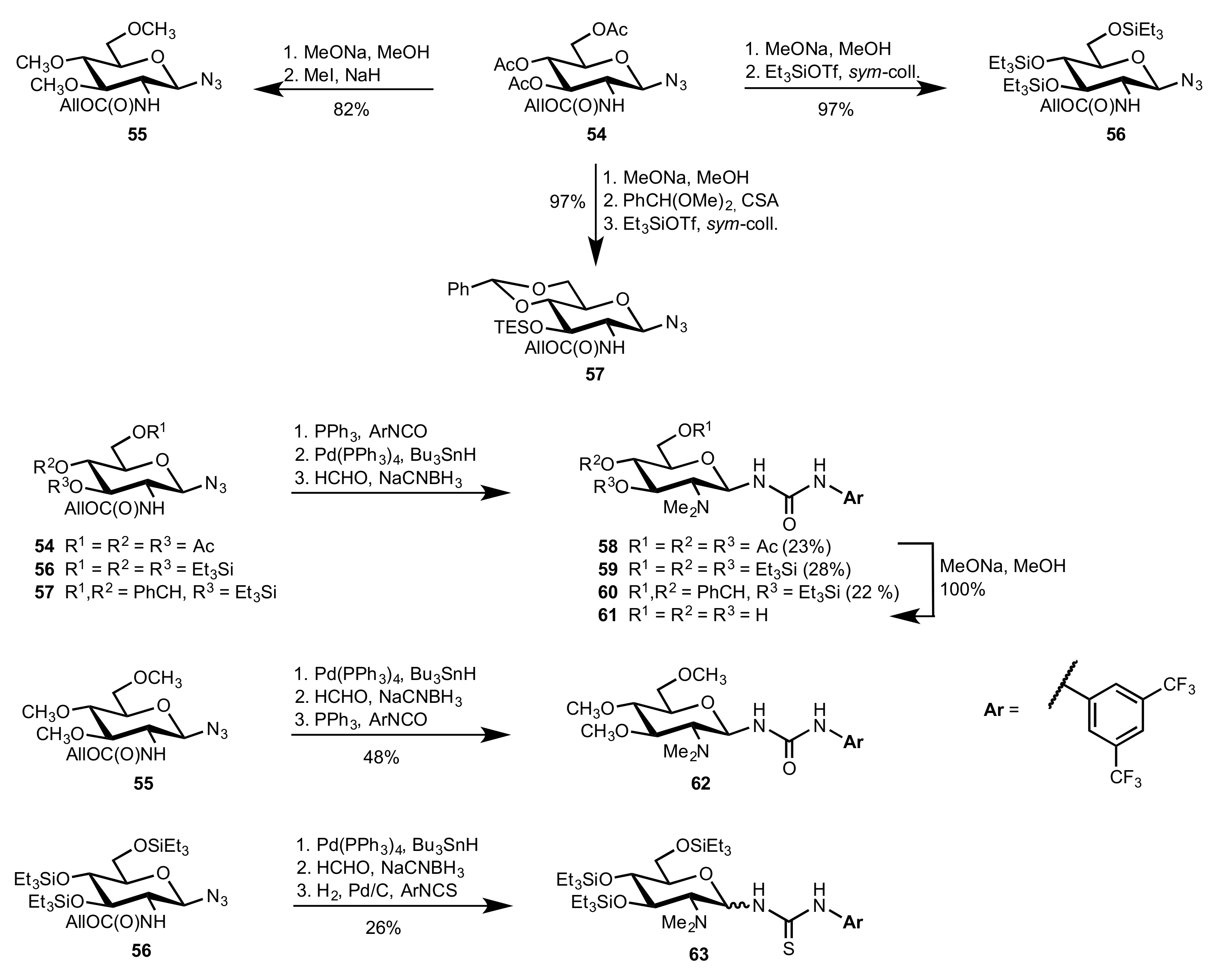
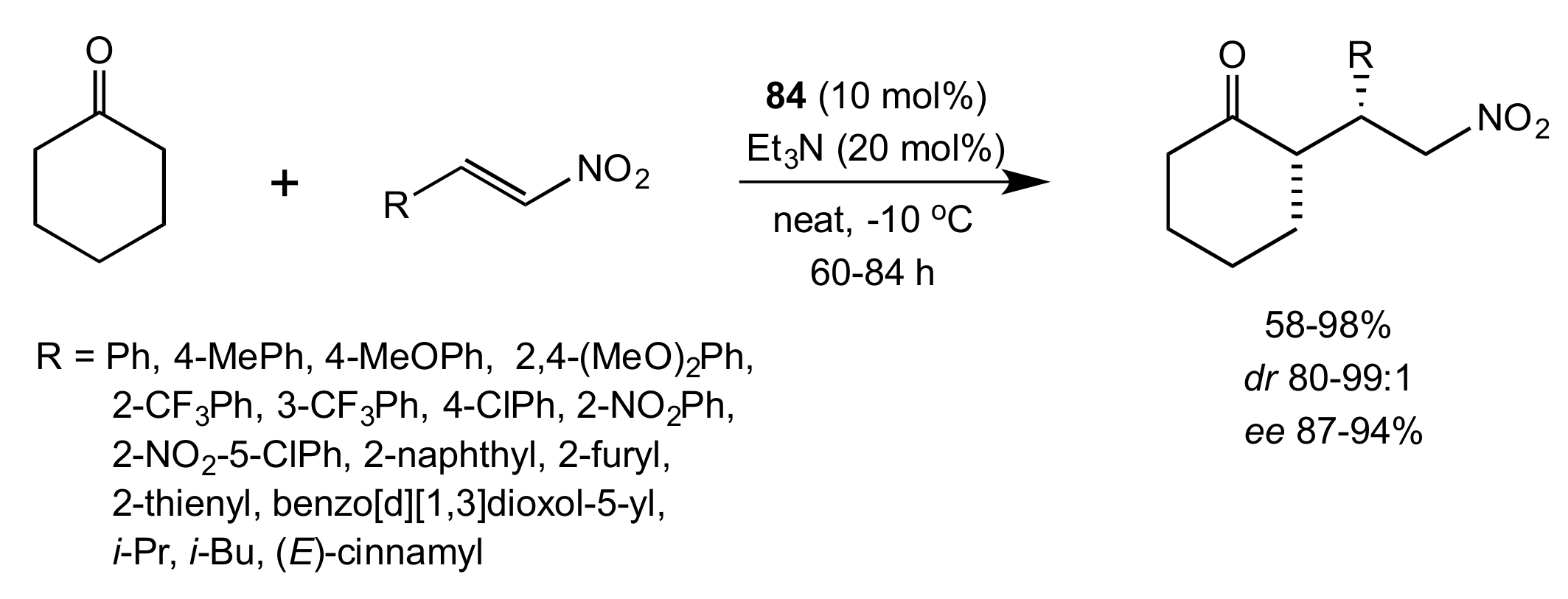
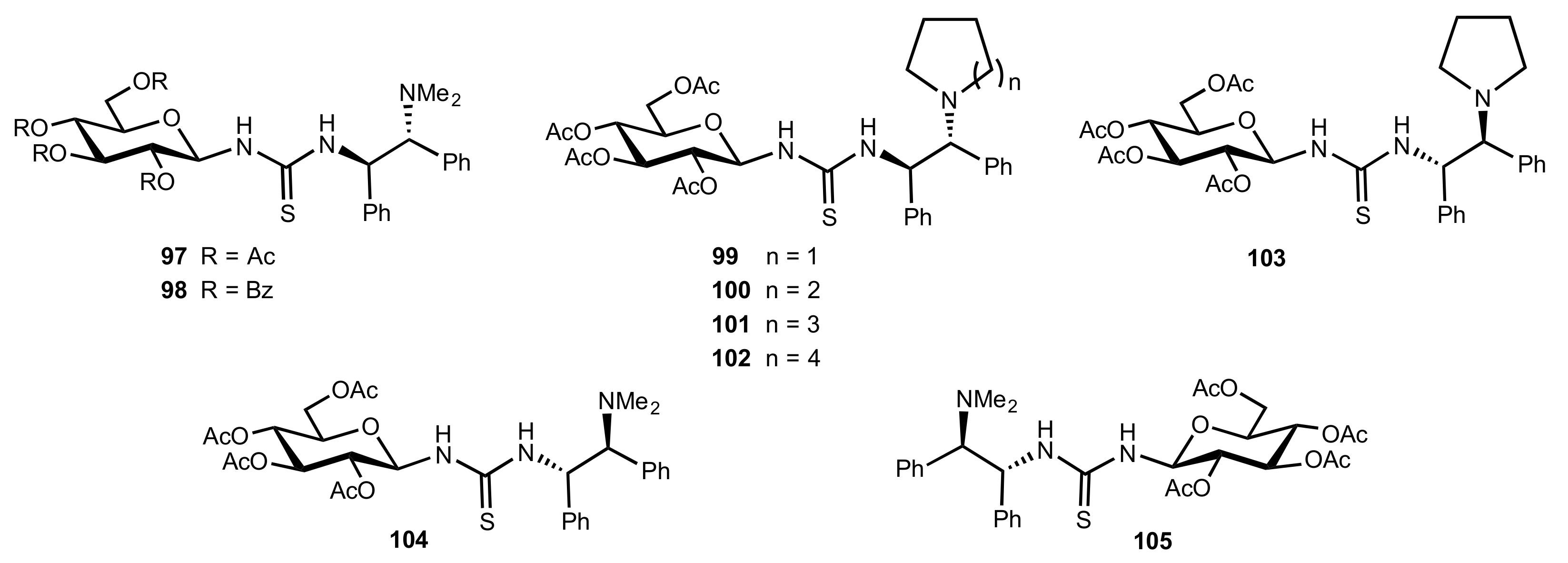
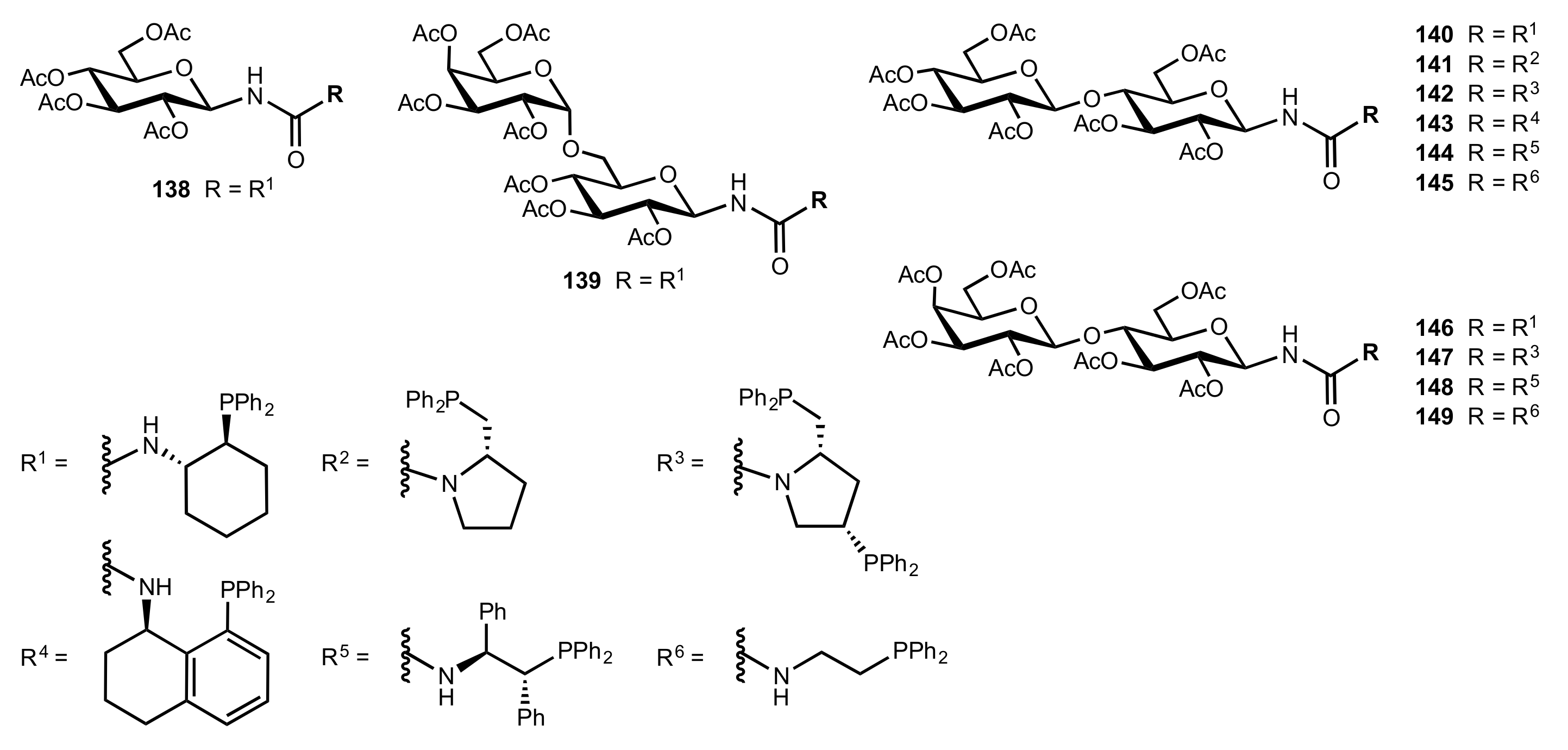
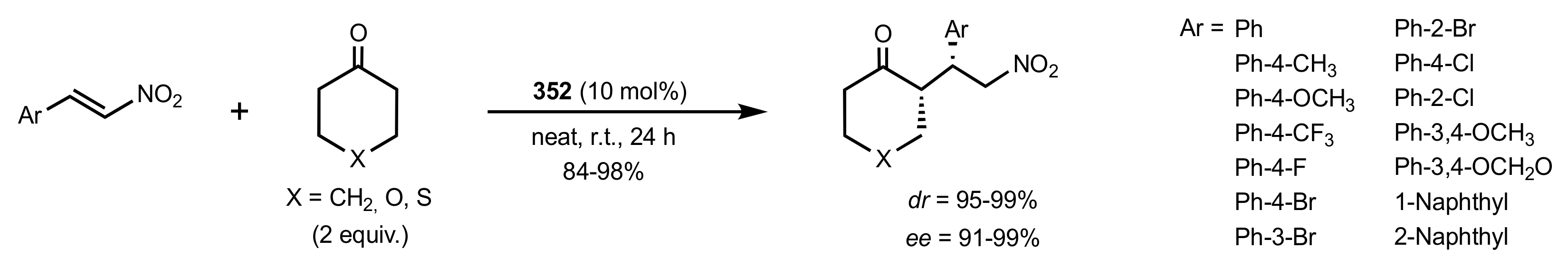
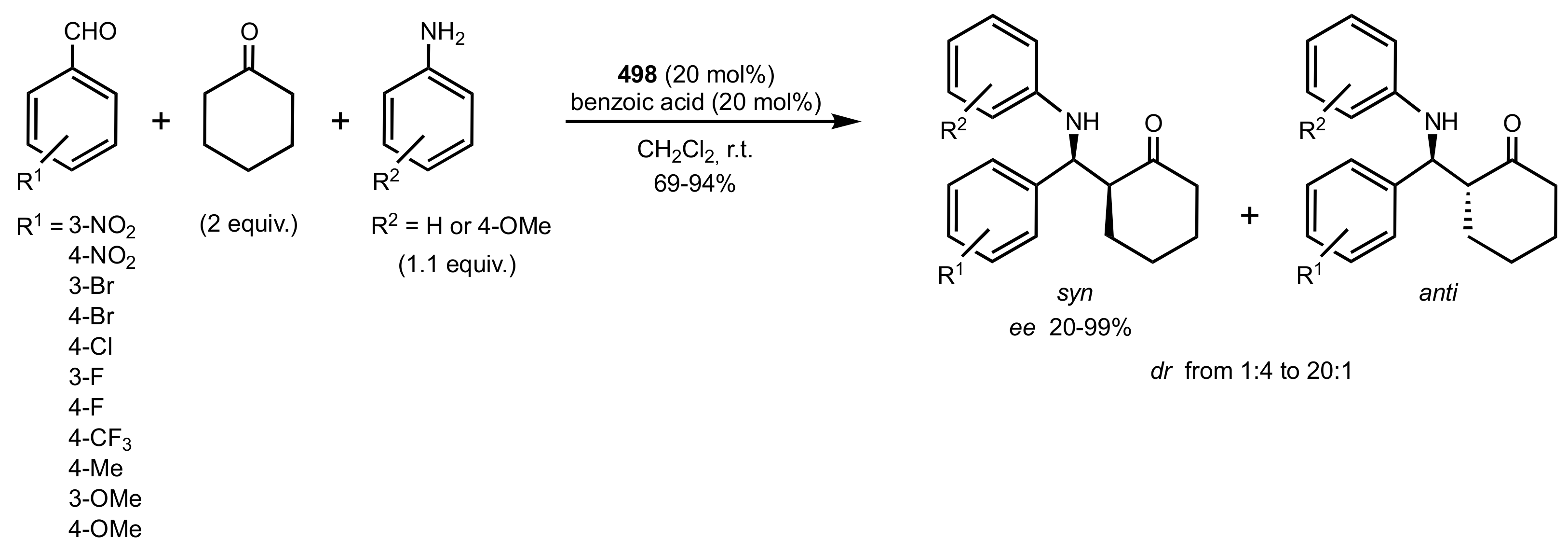
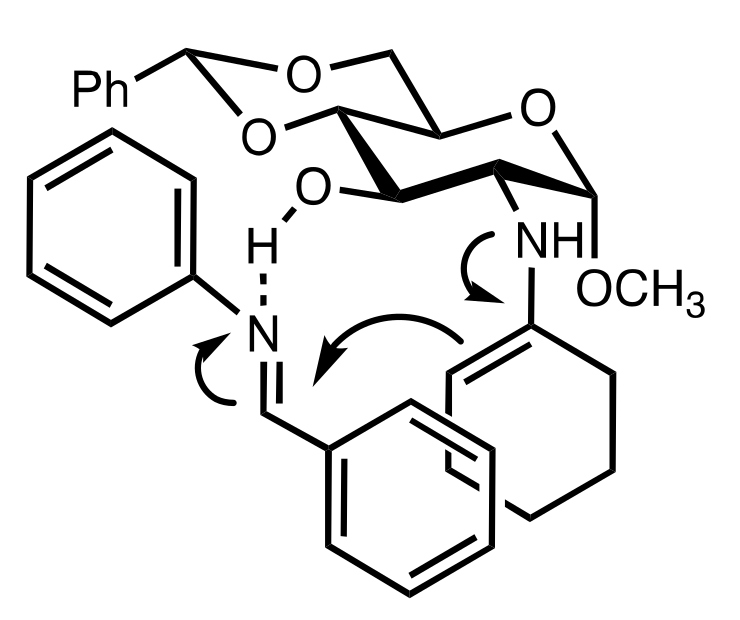
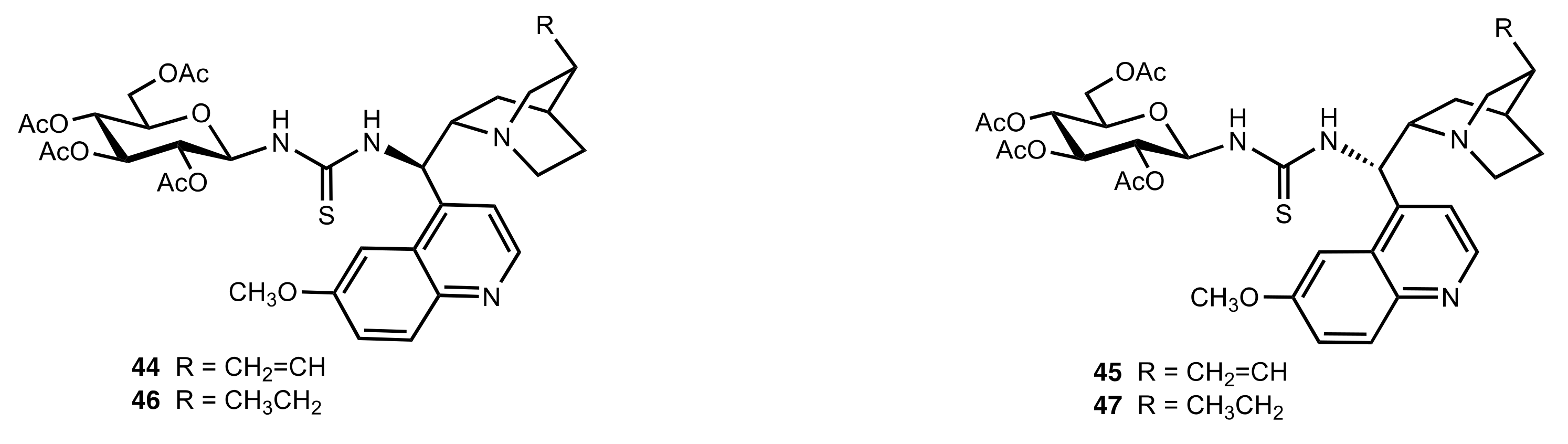
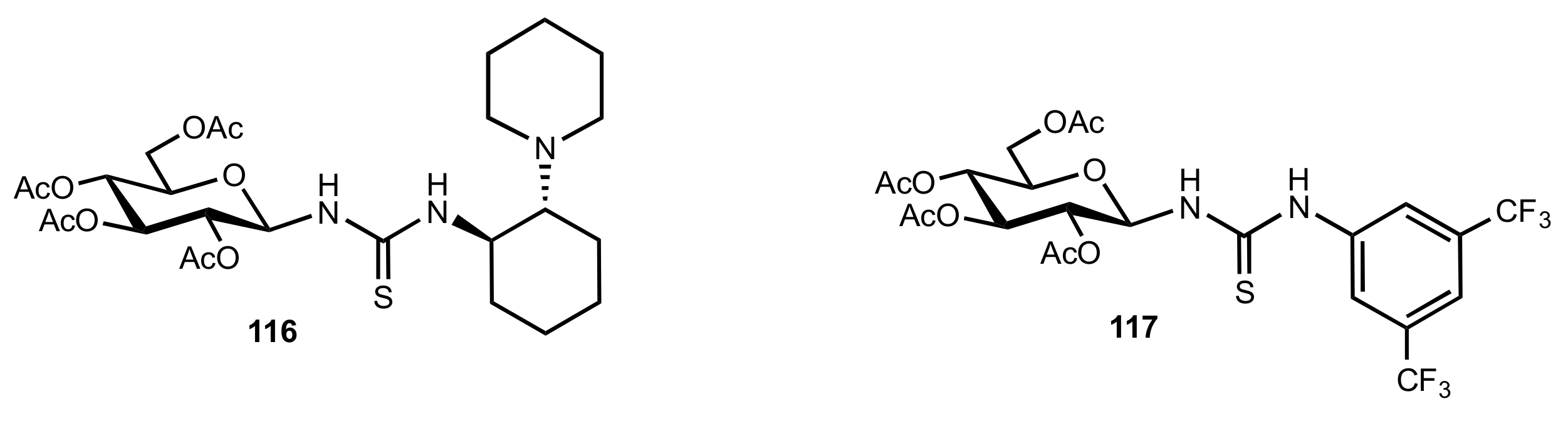
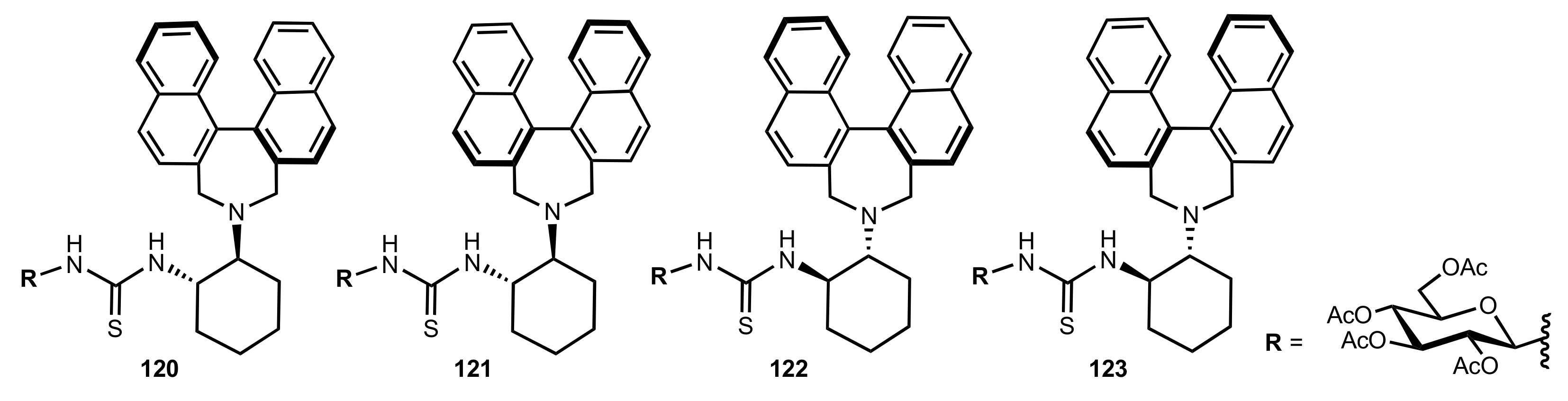
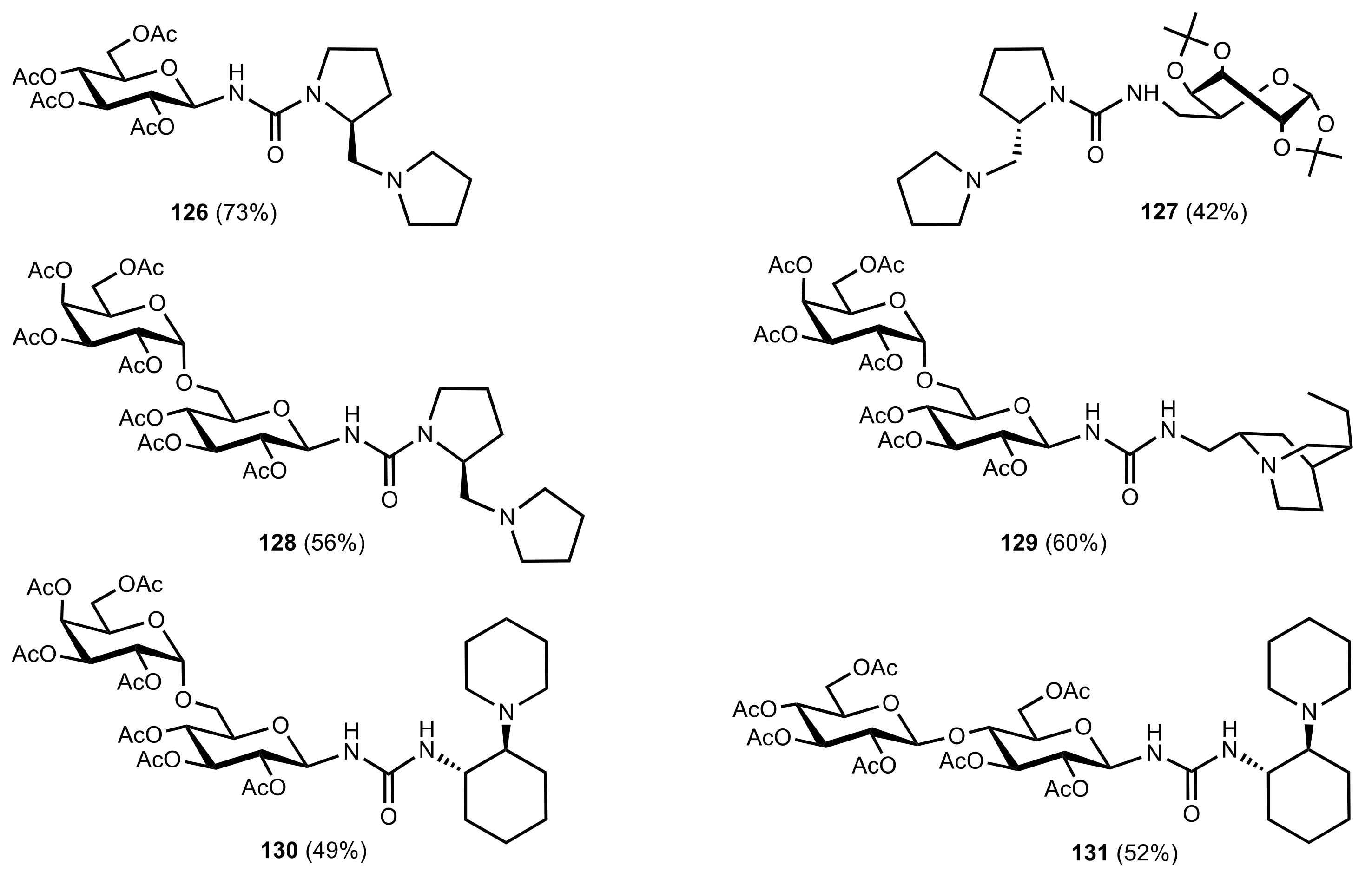
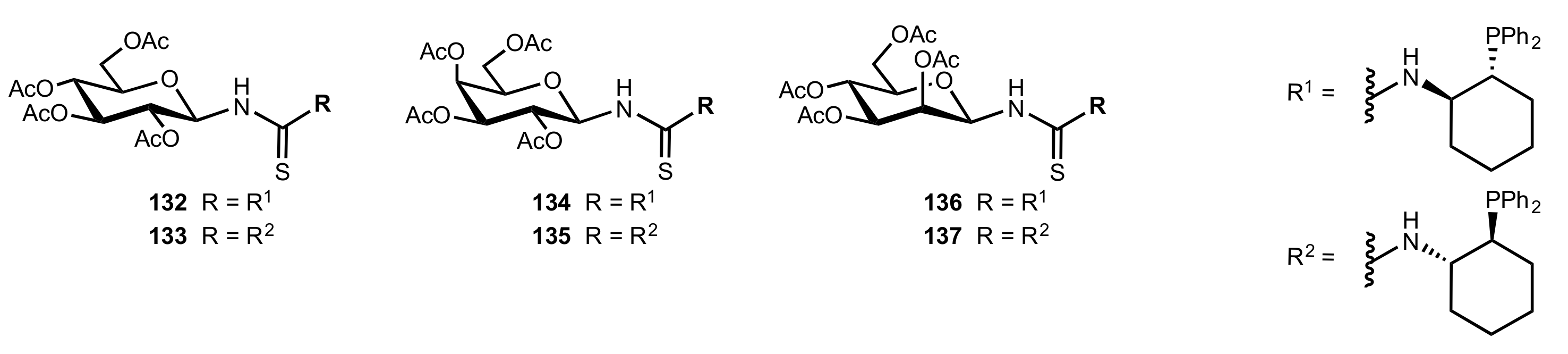
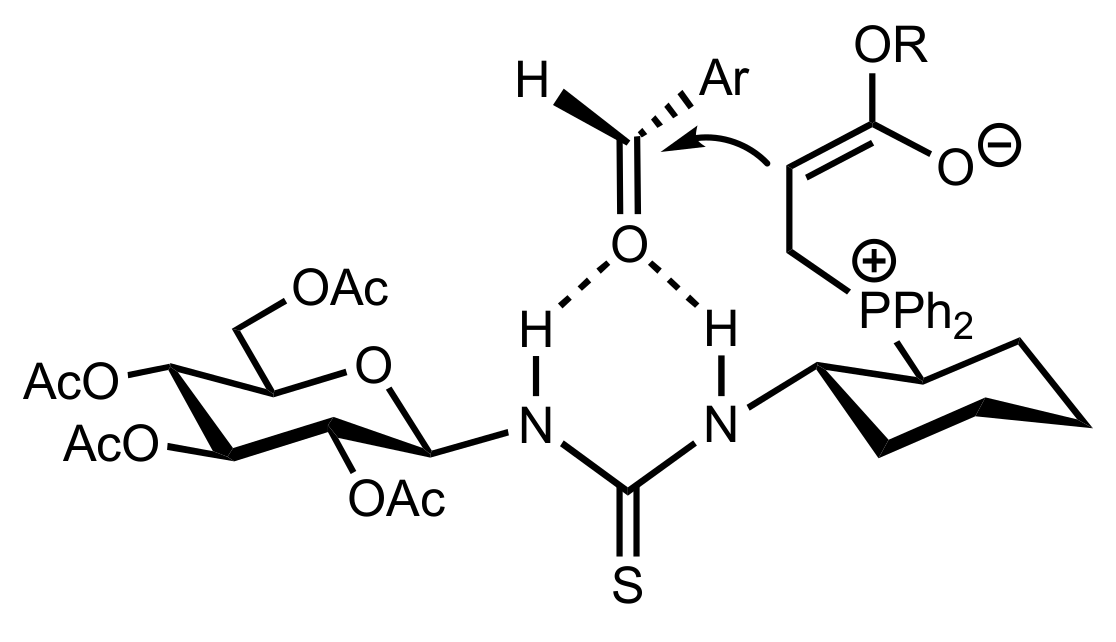
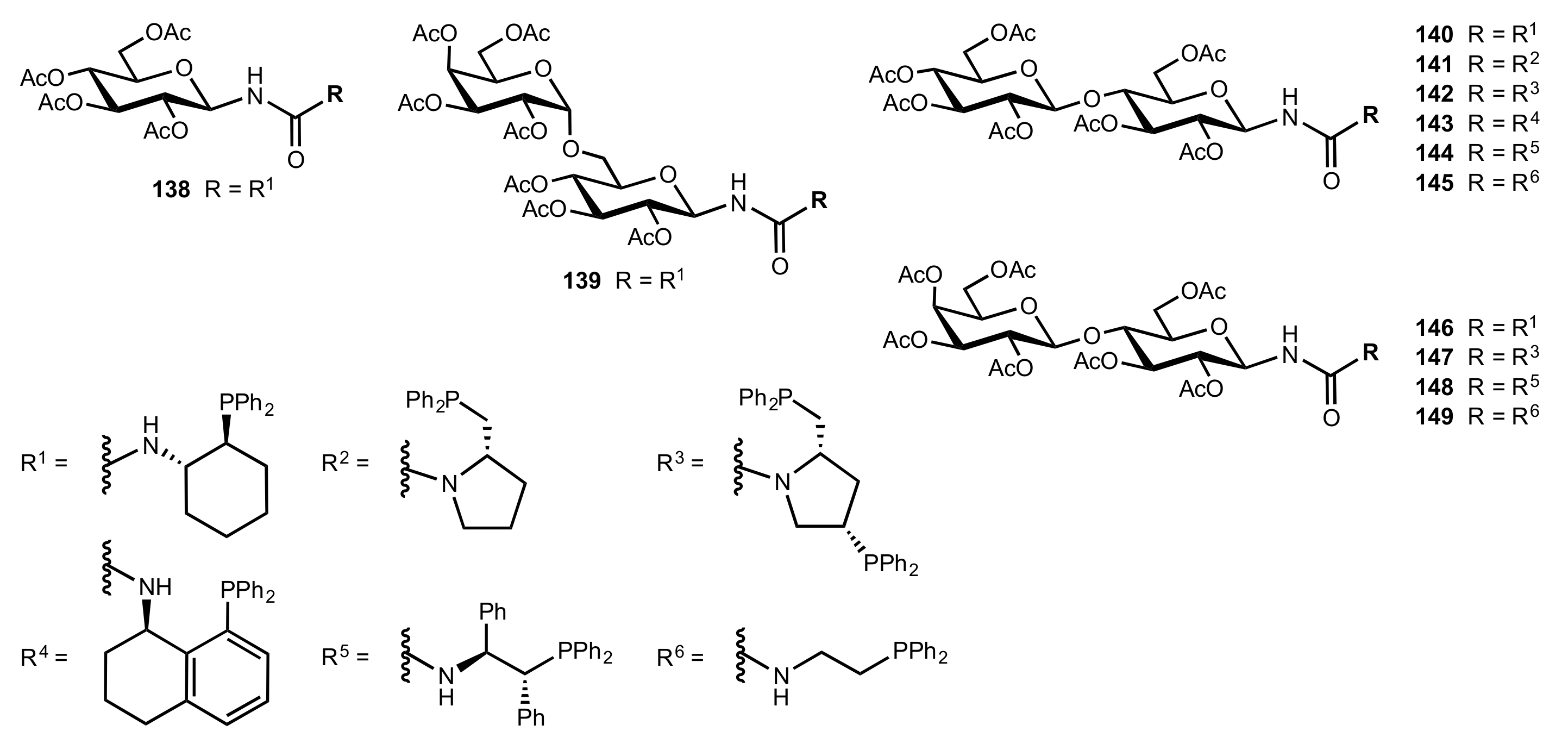
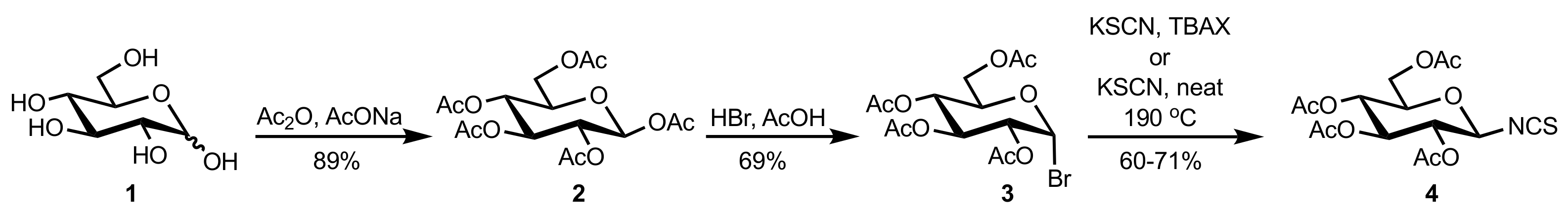
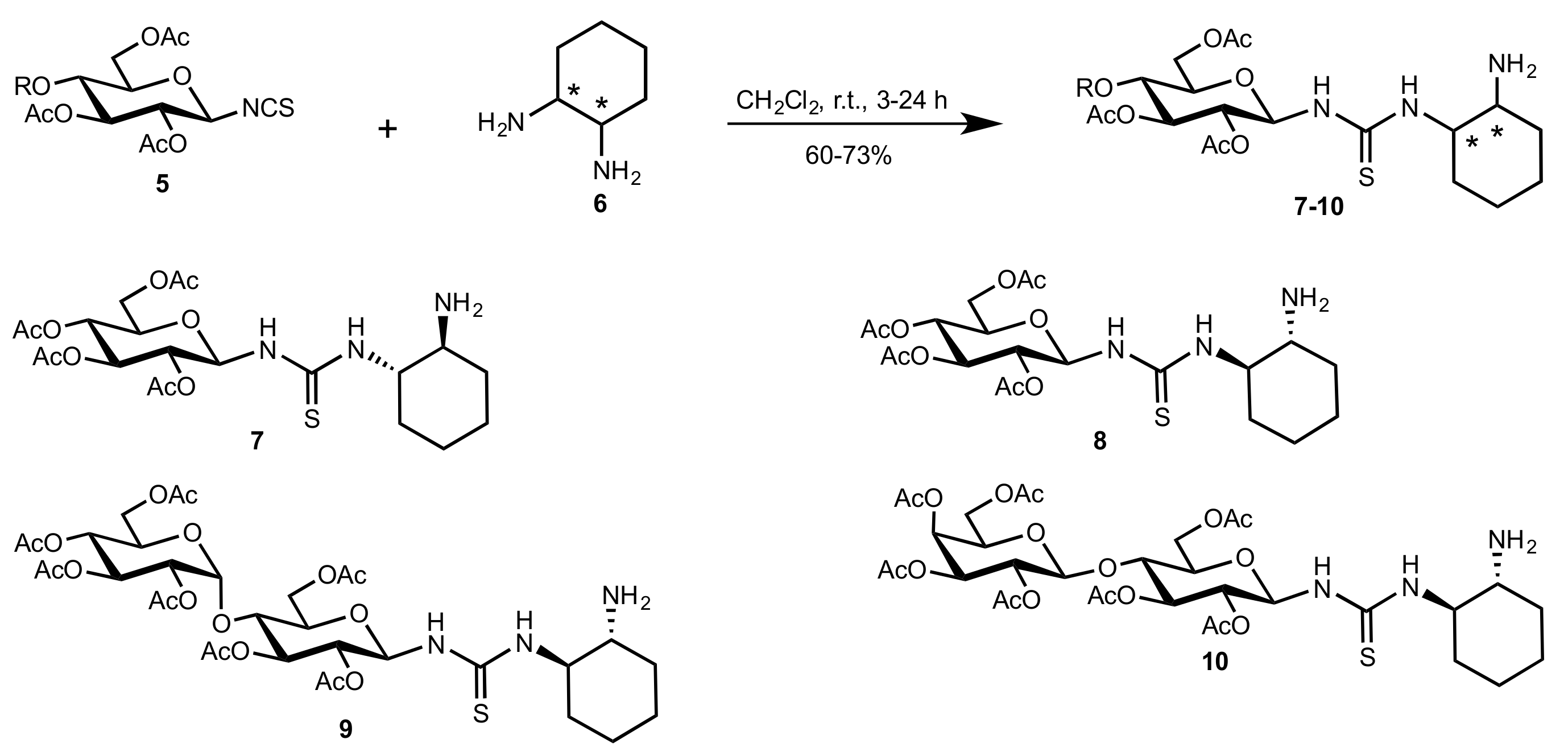
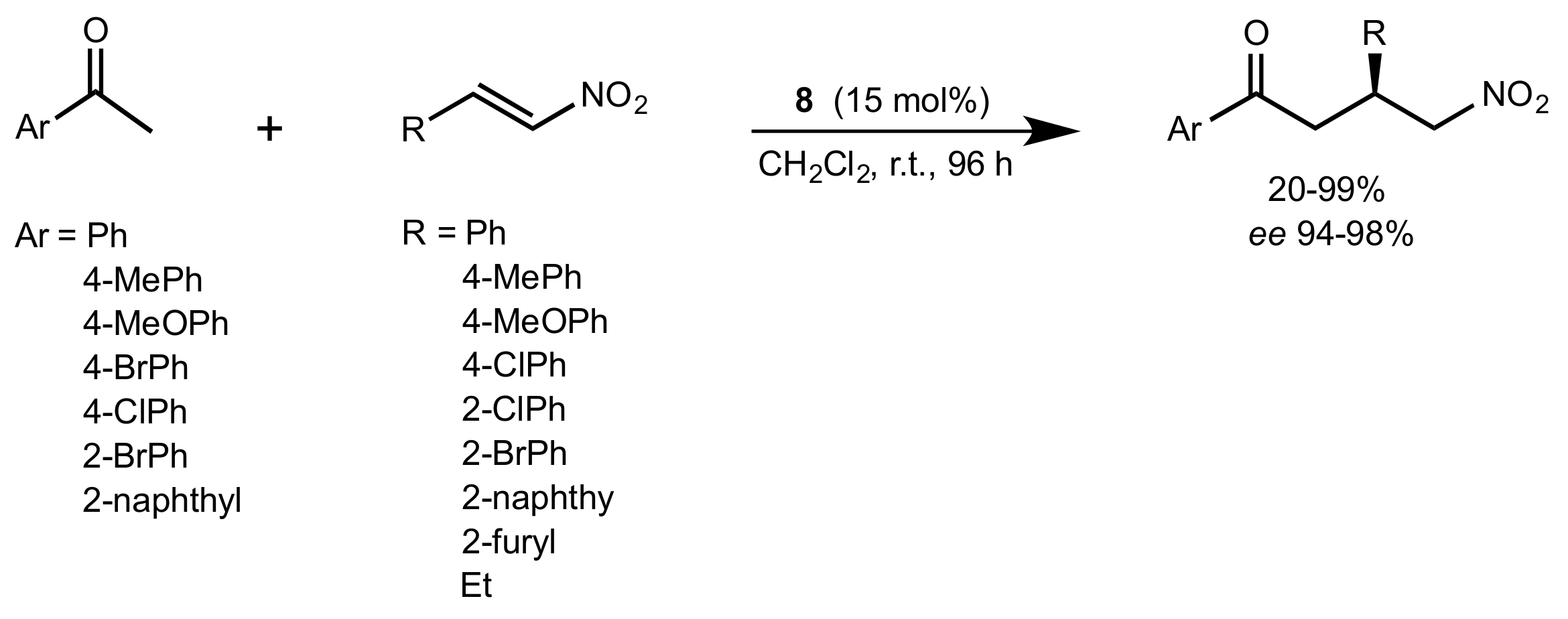
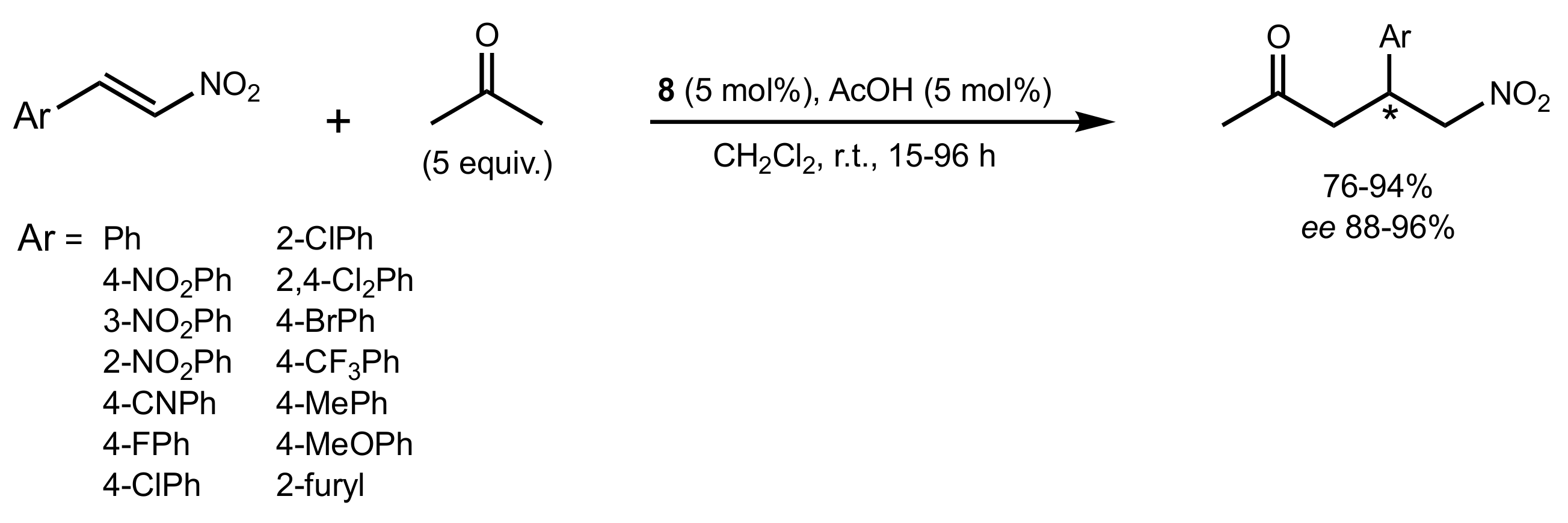
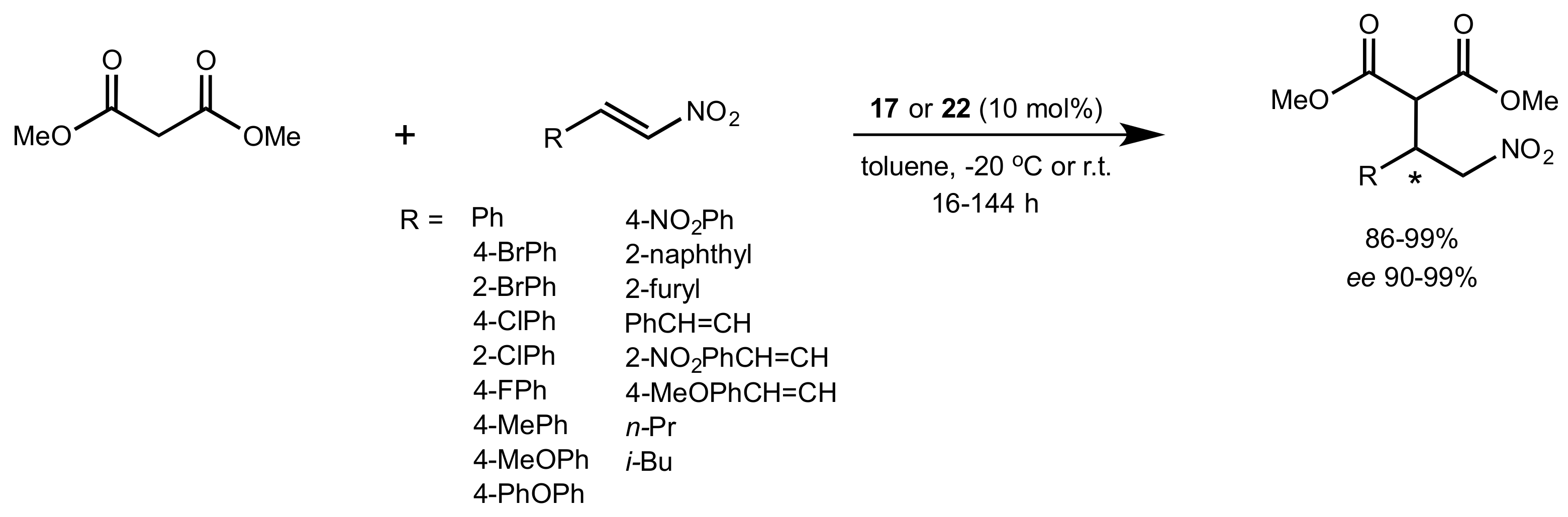
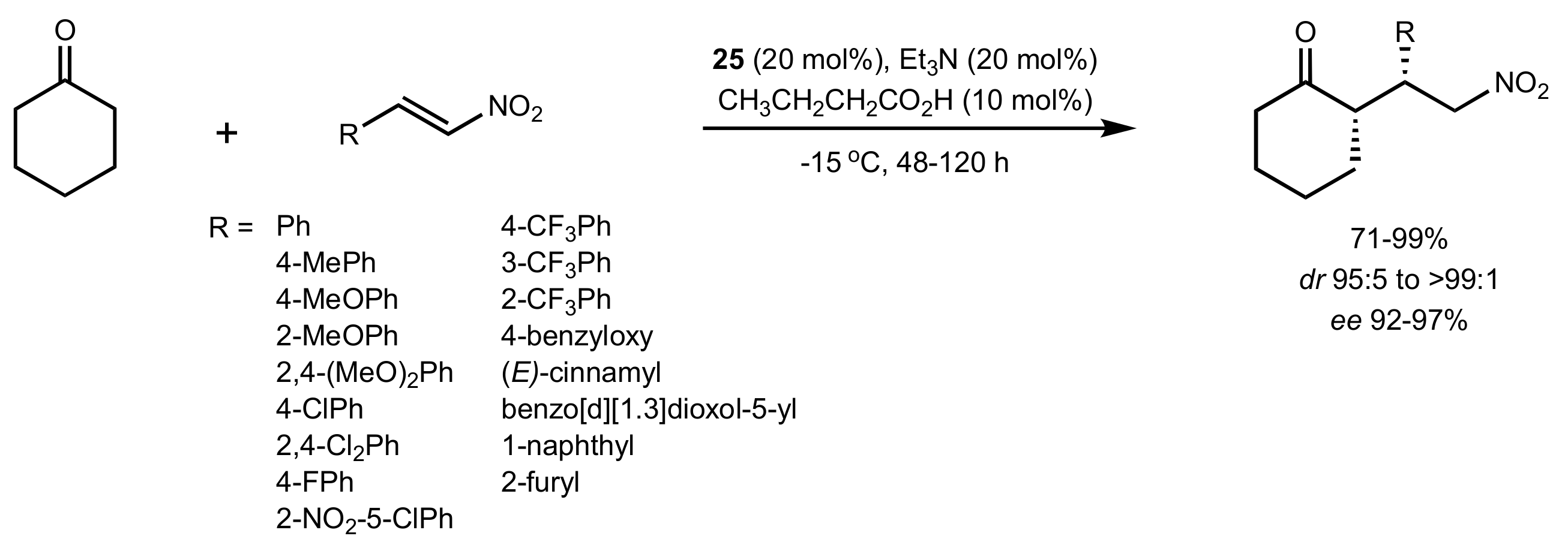
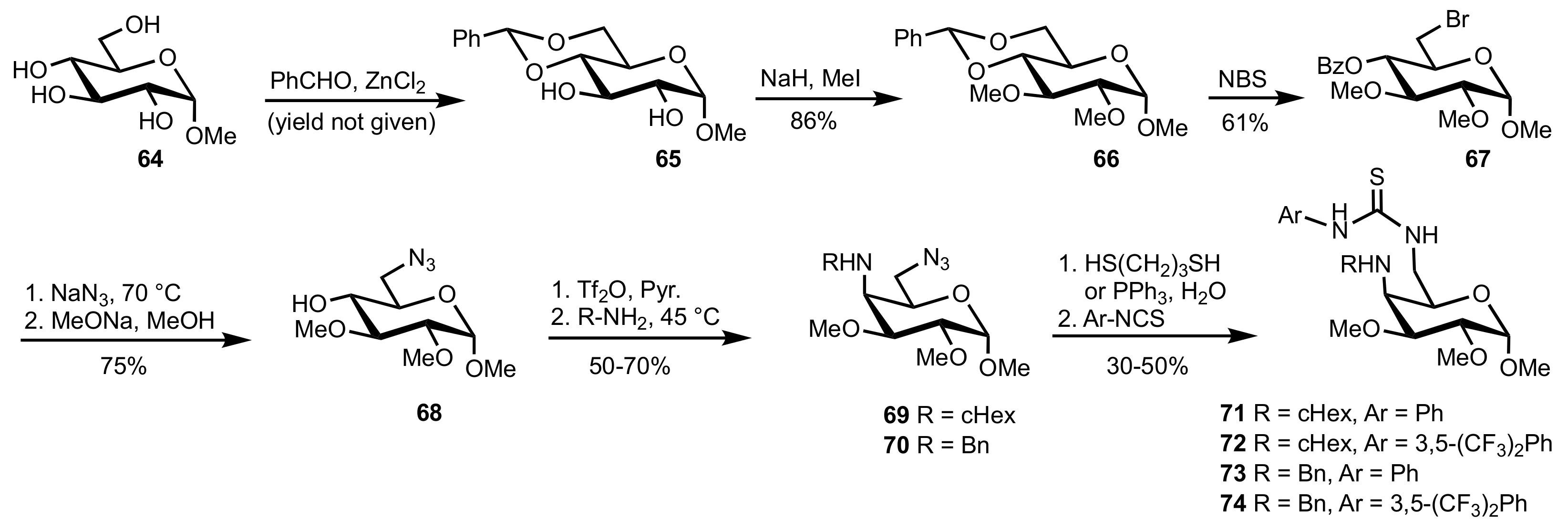
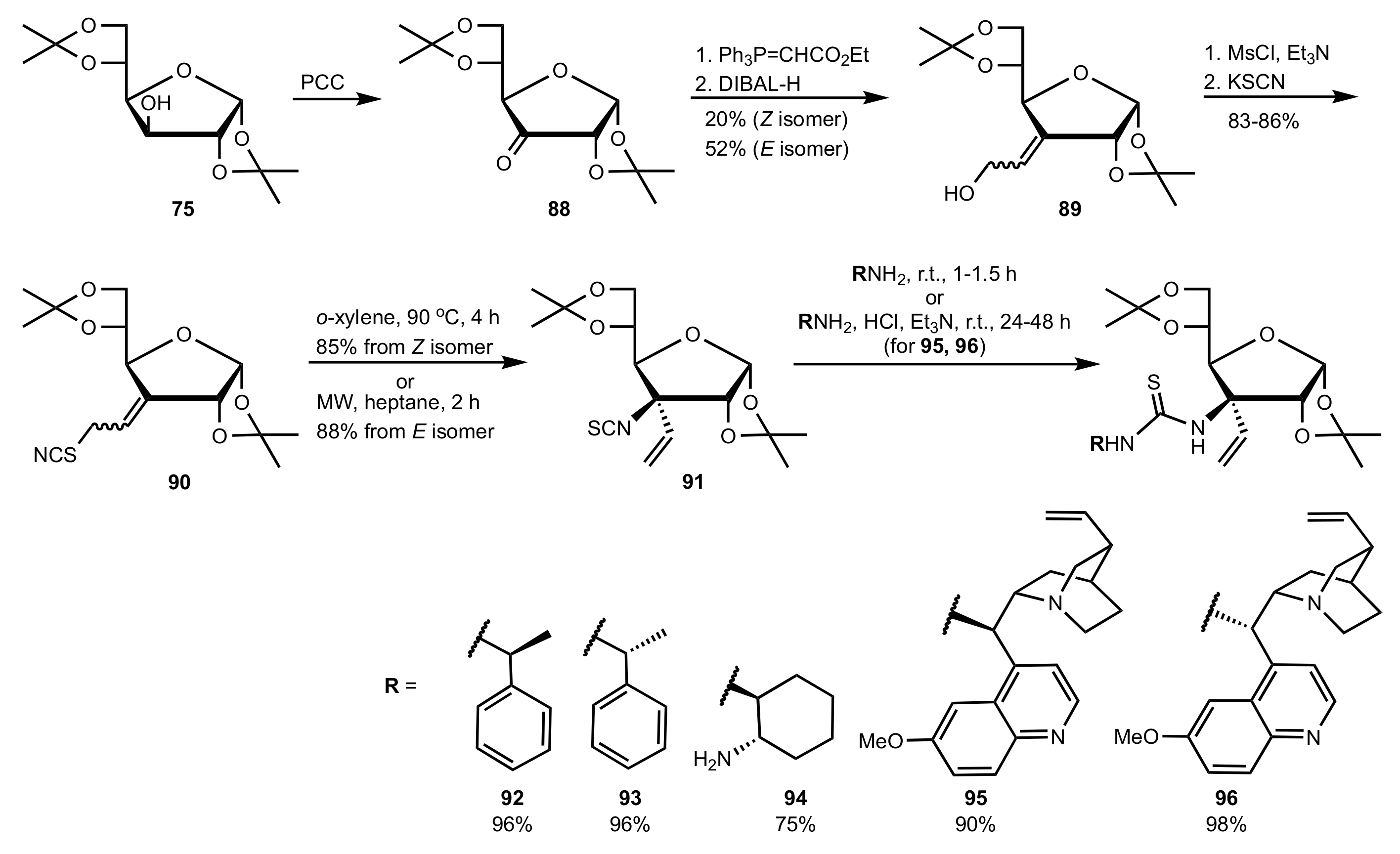
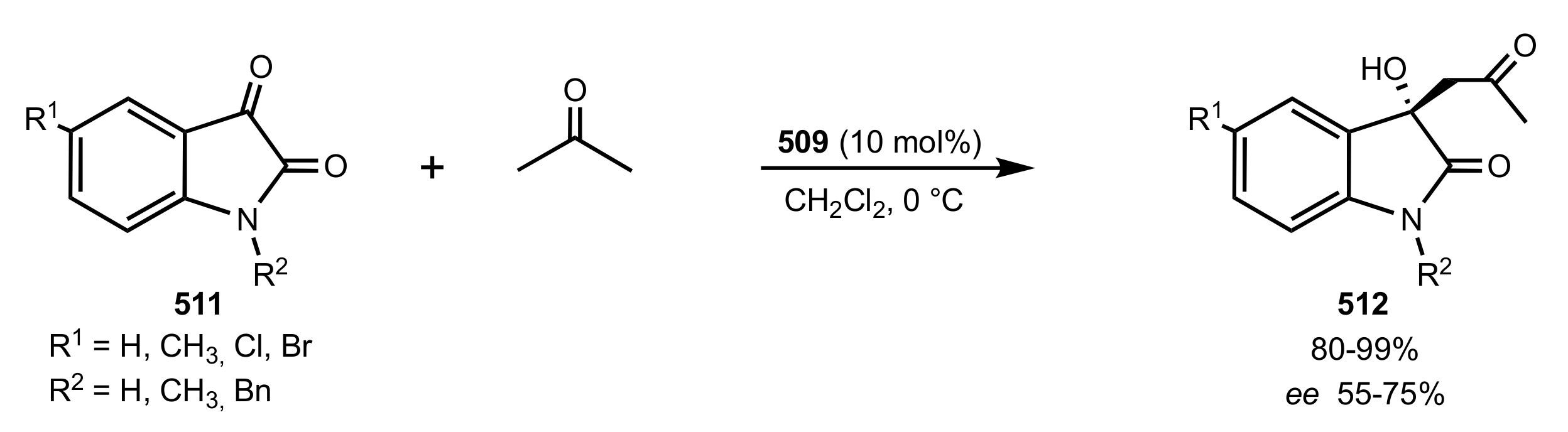
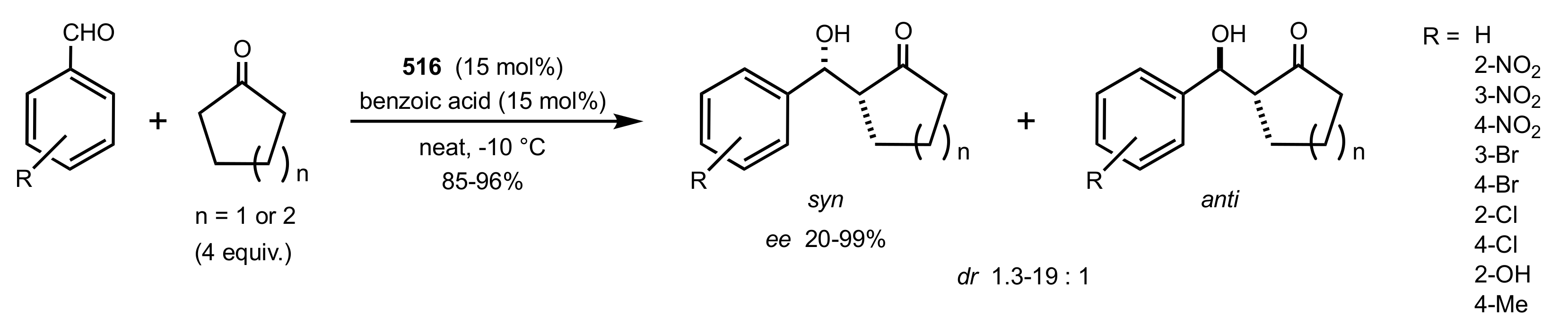
- Liu, K.; Cui, H.-F.; Nie, J.; Dong, K.-Y.; Li, X.-J.; Ma, J.-A. Highly Enantioselective Michael Addition of Aromatic Ketones to Nitroolefins Promoted by Chiral Bifunctional Primary Amine-thiourea Catalysts Based on Saccharides. Org. Lett. 2007, 9, 923–925. [Google Scholar] [CrossRef]
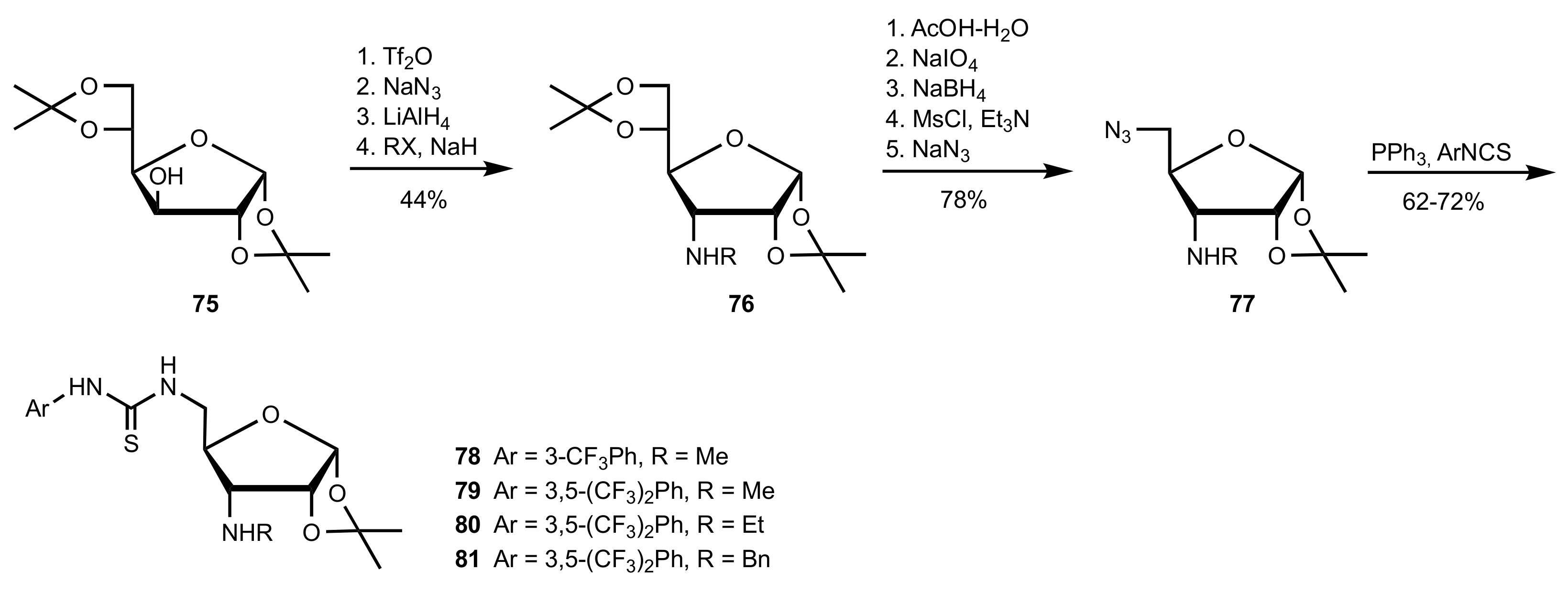
- Timmons, S.C.; Jakeman, D.L. Stereoselective Chemical Synthesis of Sugar Nucleotides via Direct Displacement of Acylated Glycosyl Bromides. Org. Lett. 2007, 9, 1227–1230. [Google Scholar] [CrossRef]
- Zhang, S.; Moussodia, R.-O.; Sun, H.-J.; Leowanawat, P.; Muncan, A.; Nusbaum, C.D.; Chelling, K.M.; Heiney, P.A.; Klein, M.L.; André, S.; et al. Mimicking Biological Membranes with Programmable Glycan Ligands Self-Assembled from Amphiphilic Janus Glycodendrimers. Angew. Chem. Int. Ed. 2014, 53, 10899–10903. [Google Scholar] [CrossRef] [PubMed]
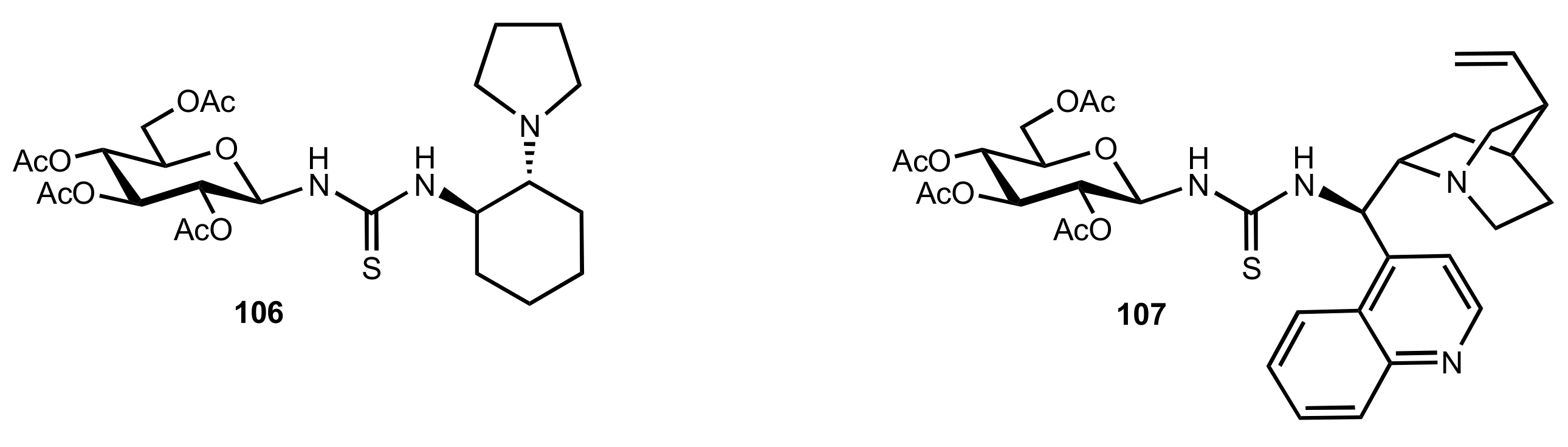
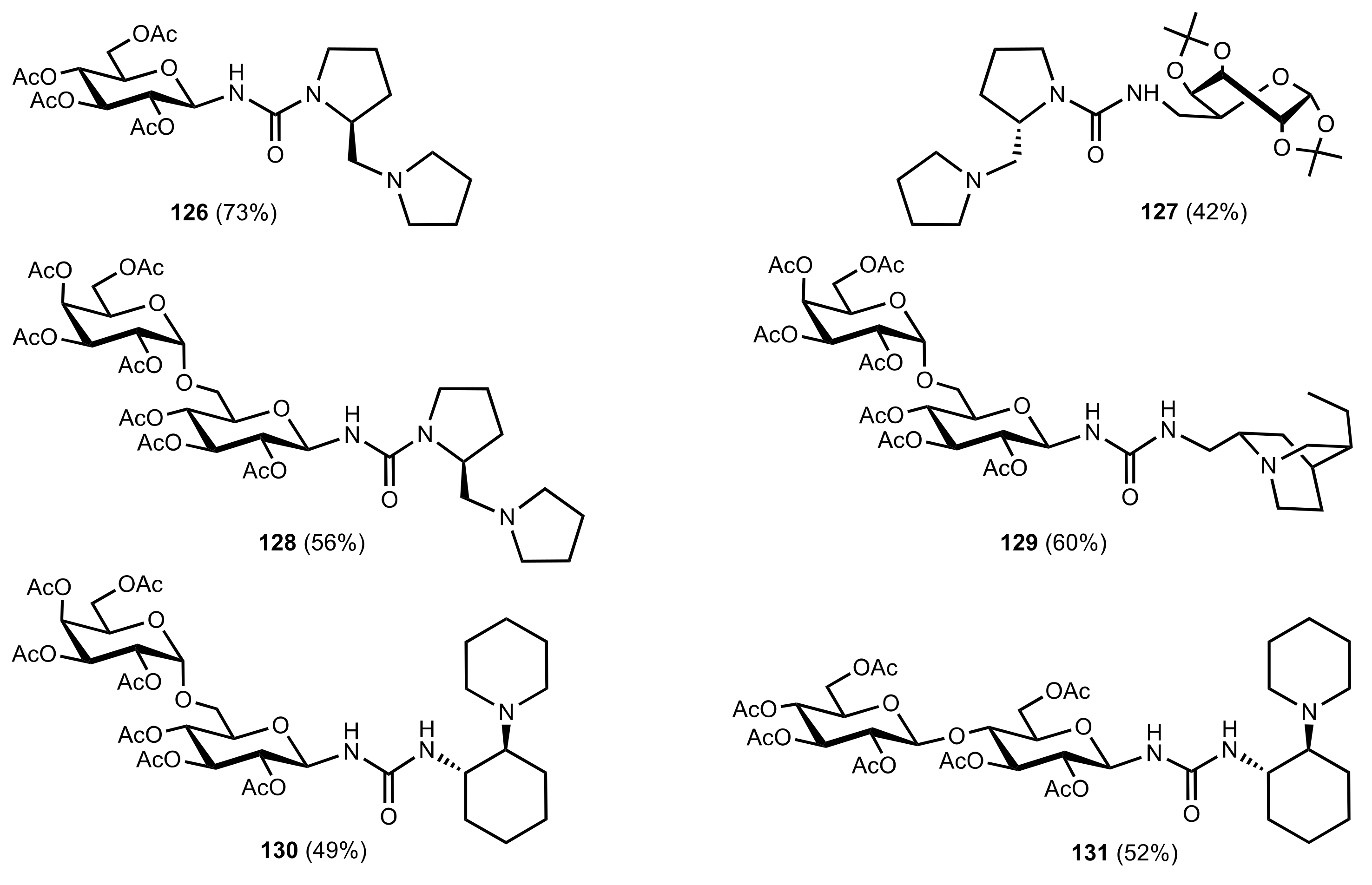
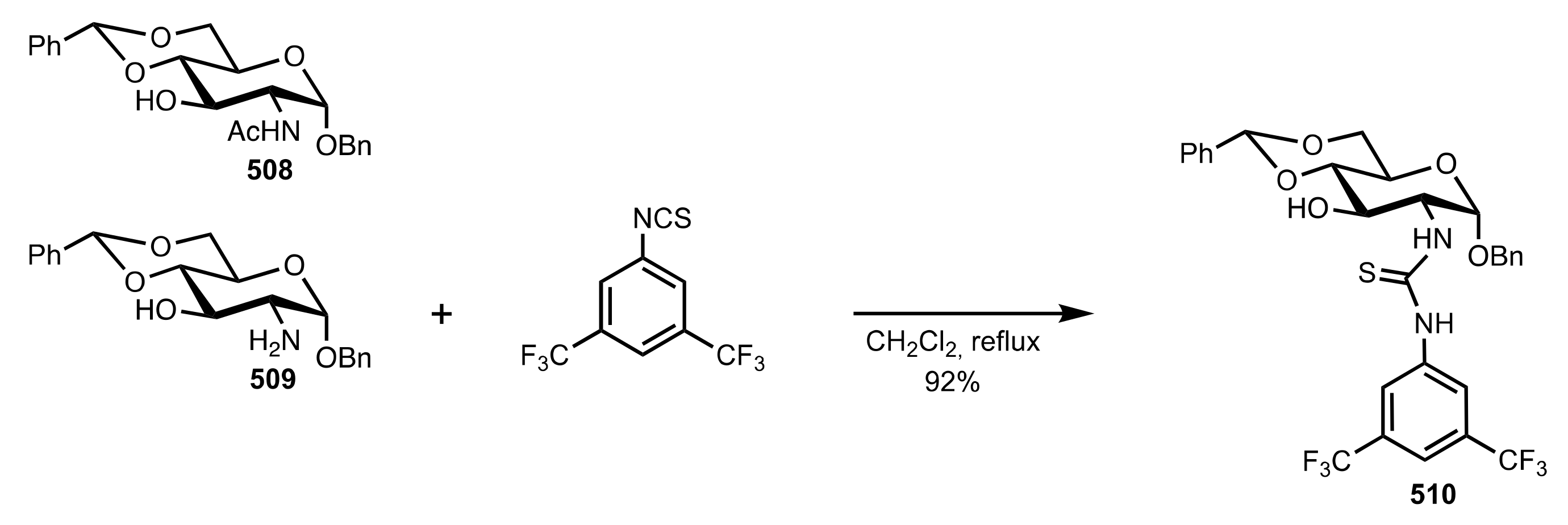
- Nagy, S.; Kozma, P.; Kisszékelyi, P.; Bezzegh, D.; Huszthy, P.; Kupai, J. Synthesis of Three New Bifunctional Glucose-Thiourea Organocatalysts and Their Application in Asymmetric Michael Addition. Studia Ubb. Chem. 2017, 62, 183–194. [Google Scholar] [CrossRef]
- Pearson, M.S.M.; Robin, A.; Bourgougnon, N.; Meslin, J.C.; Deniaud, D. An Efficient Route to Pyrimidine Nucleoside Analogues by [4 + 2] Cycloaddition Reaction. J. Org. Chem. 2003, 68, 8583–8587. [Google Scholar] [CrossRef] [PubMed]
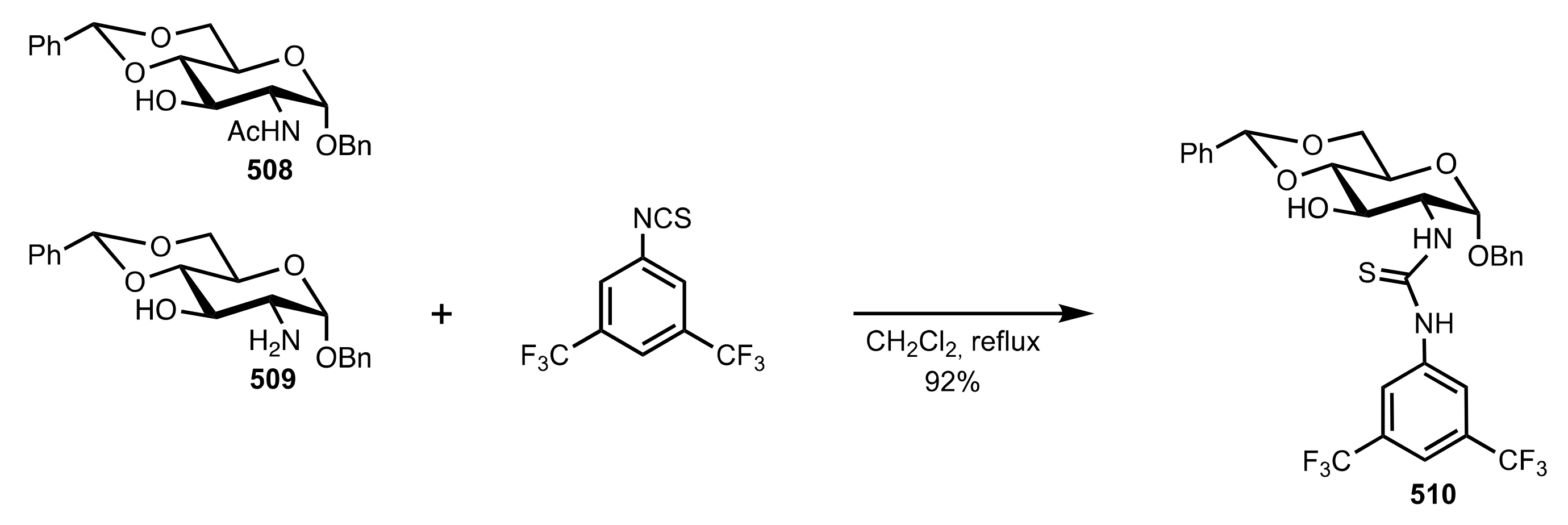
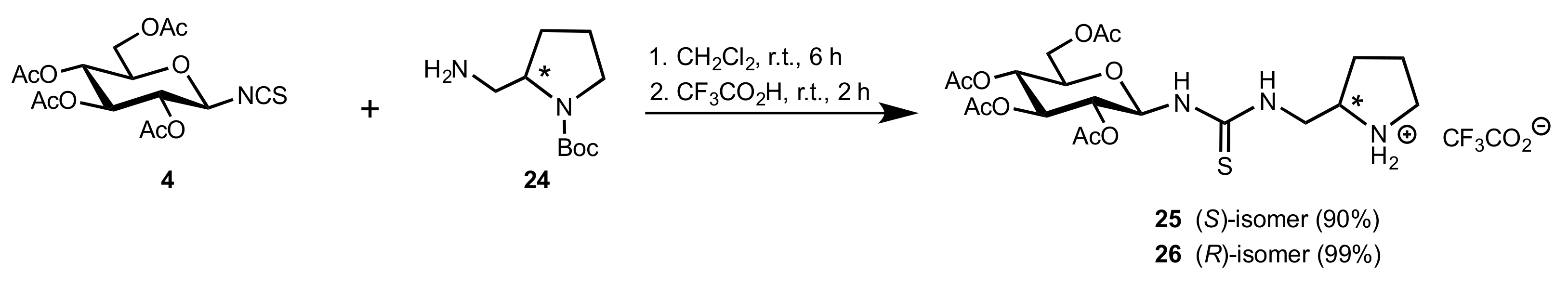
- Camarasa, M.J.; Fernández-Resa, P.; García-López, M.T.; de las Heras, F.G.; Méndez-Castrillón, P.P.; Felix, A.S. A New Procedure for the Synthesis of Glycosyl Isothiocyanates. Synthesis 1984, 6, 509–510. [Google Scholar] [CrossRef]
- Lindhorst, T.K.; Kieburg, C. Solvent-Free Preparation of Glycosyl Isothiocyanates. Synthesis 1995, 1995, 1228–1230. [Google Scholar] [CrossRef]
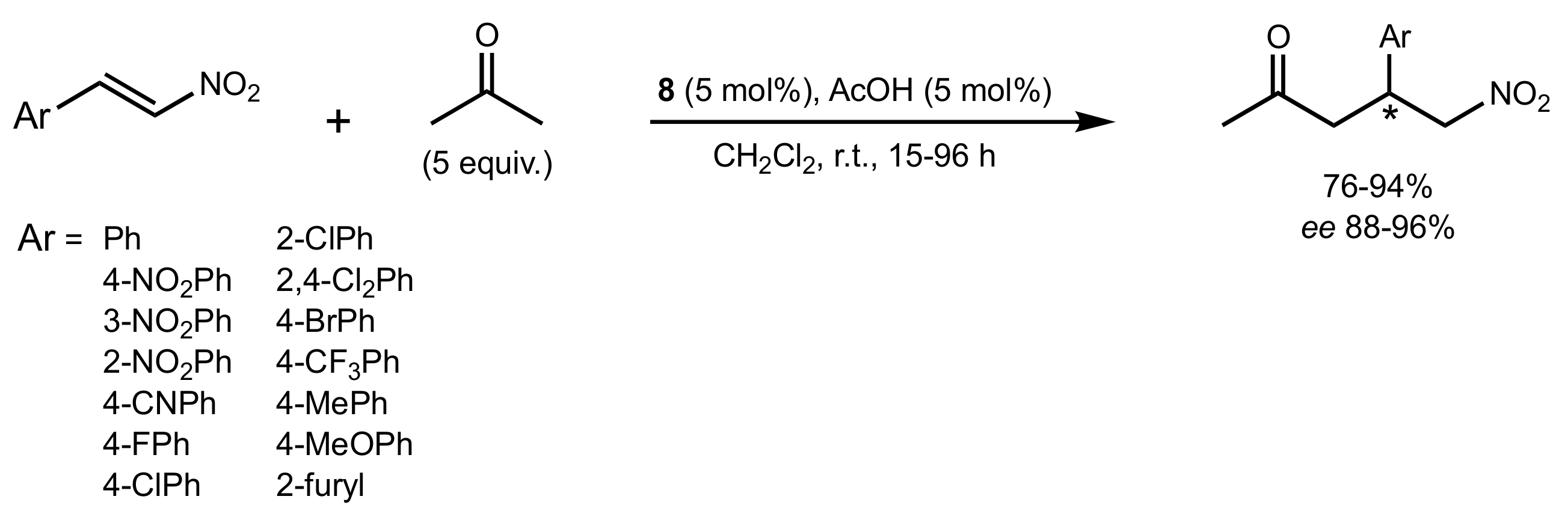
- Gu, X.-T.; Guo, Q.; Wu, X.-Y. Highly enantioselective Michael addition of acetone to nitroolefins catalyzed by chiral bifunctional primary amine-thiourea catalysts with acetic acid. Tetrahedron 2009, 65, 5265–5270. [Google Scholar] [CrossRef]
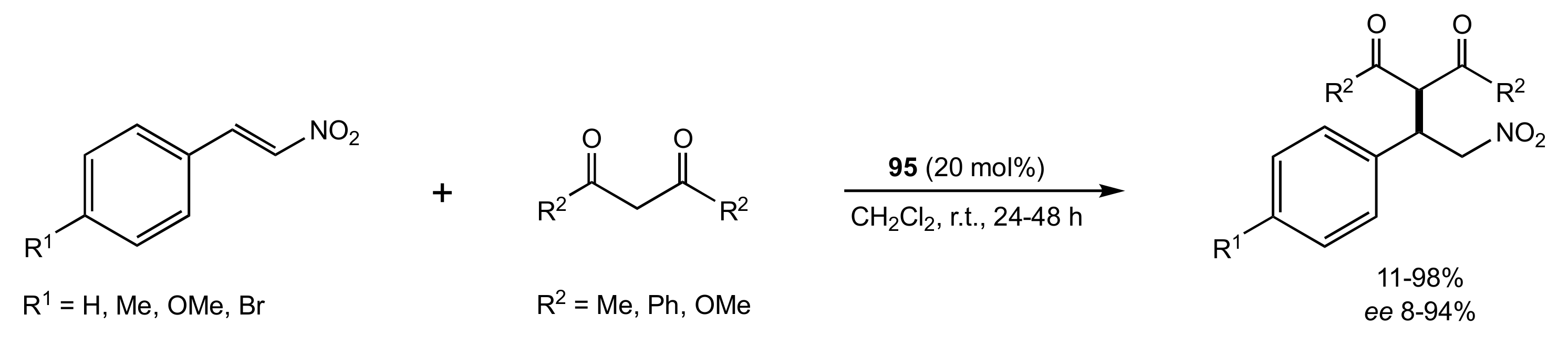
- Li, X.-J.; Liu, K.; Ma, H.; Nie, J.; Ma, J.-A. Highly Enantioselective Michael Addition of Malonates to Nitroolefins Catalyzed by Chiral Bifunctional Tertiary Amine-Thioureas Based on Saccharides. Synlett 2008, 2008, 3242–3246. [Google Scholar] [CrossRef]
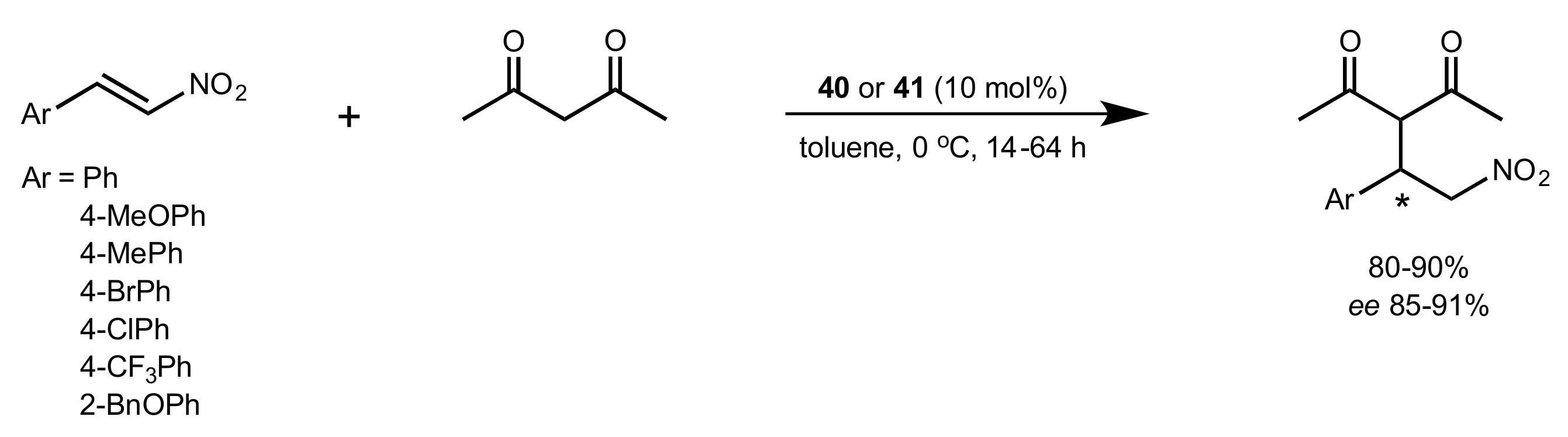
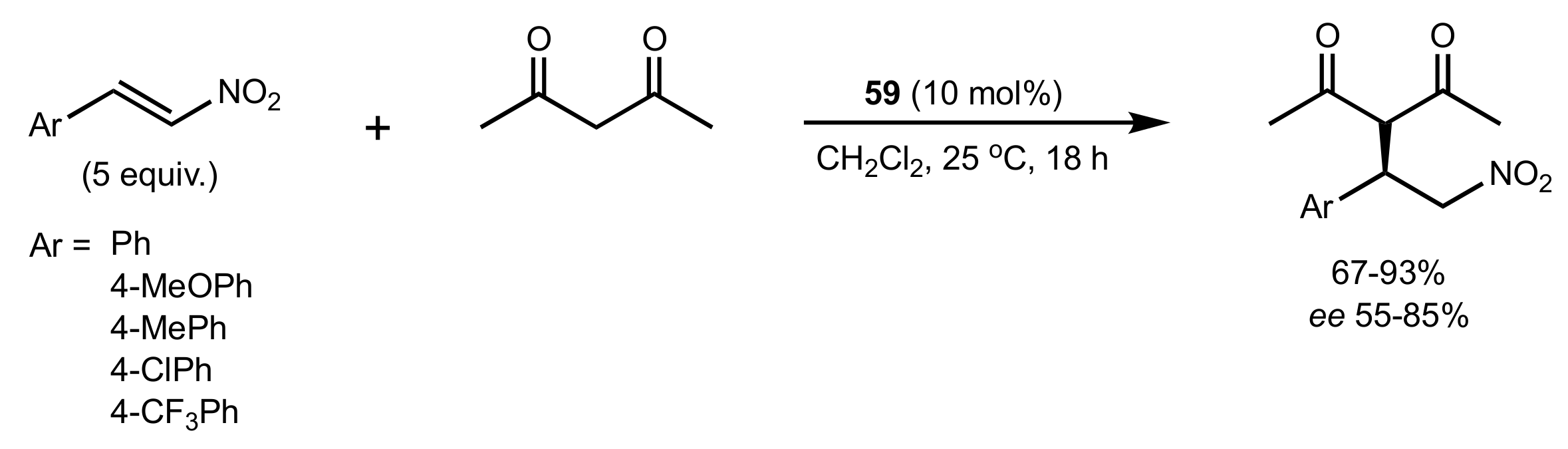
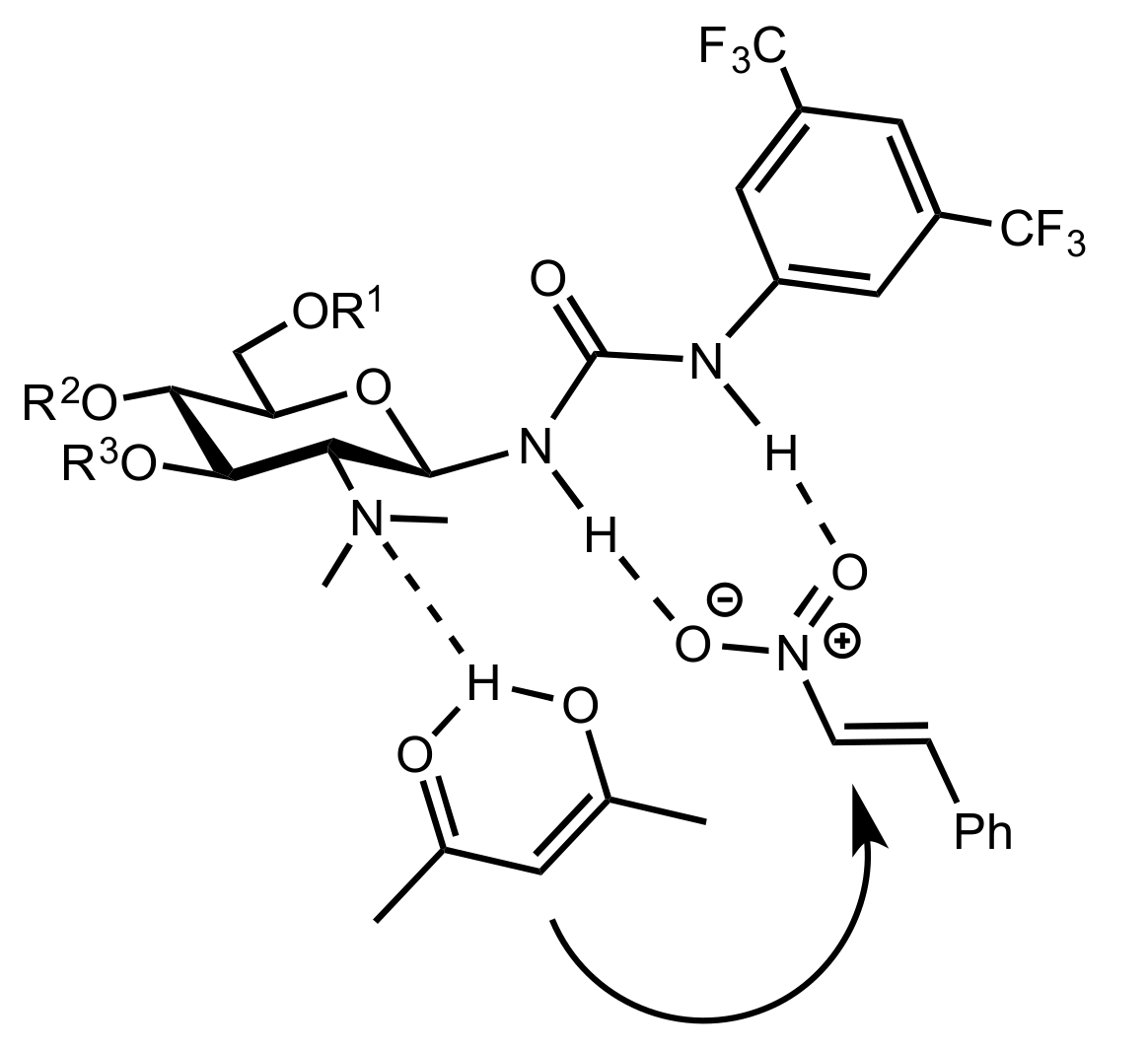
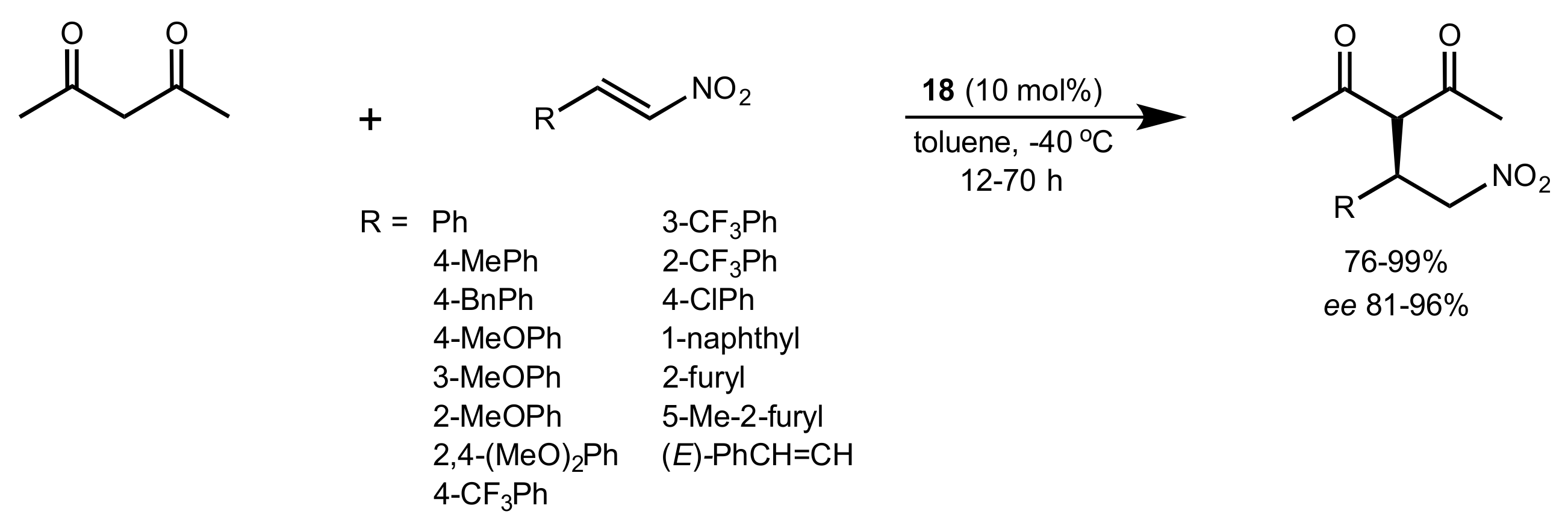
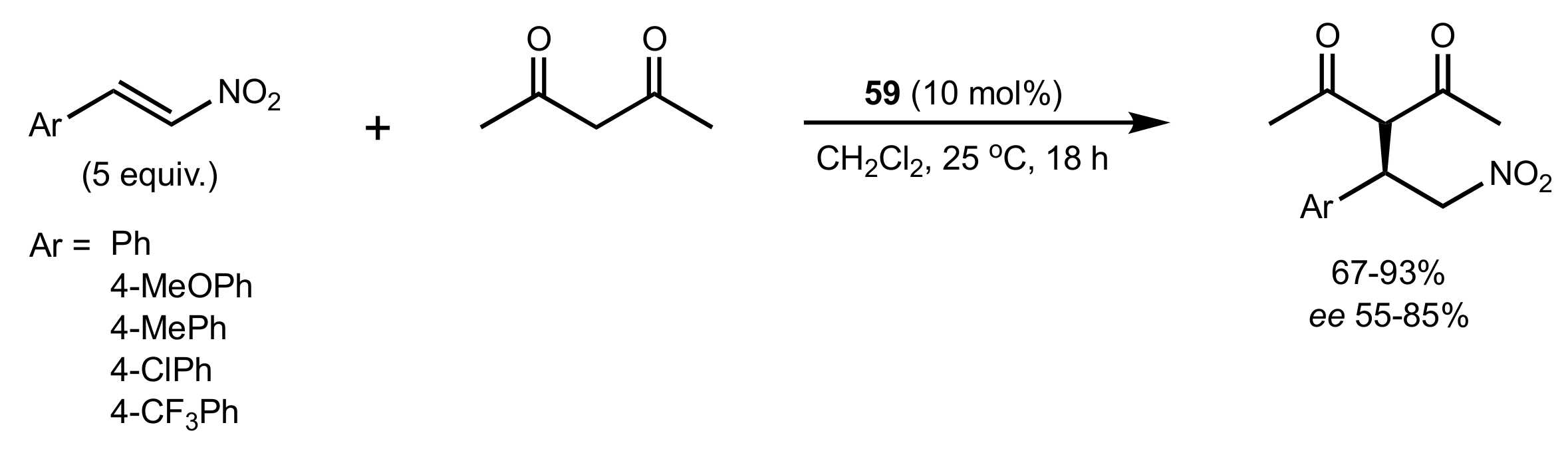
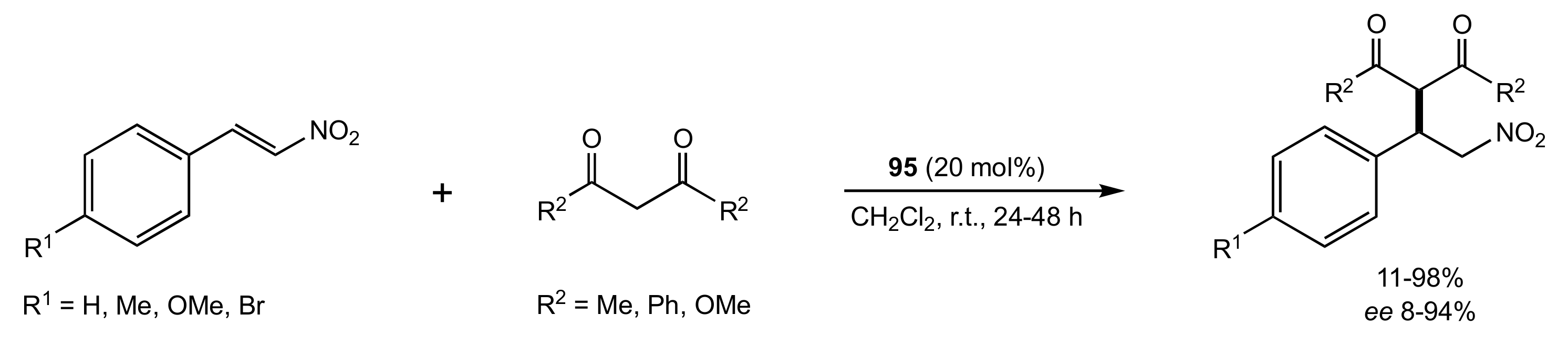
- Gao, P.; Wang, C.; Wu, Y.; Zhou, Z.; Tang, C. Sugar-Derived Bifunctional Thiourea Organocatalyzed Asymmetric Michael Addition of Acetylacetone to Nitroolefins. Eur. J. Org. Chem. 2008, 2008, 4563–4566. [Google Scholar] [CrossRef]
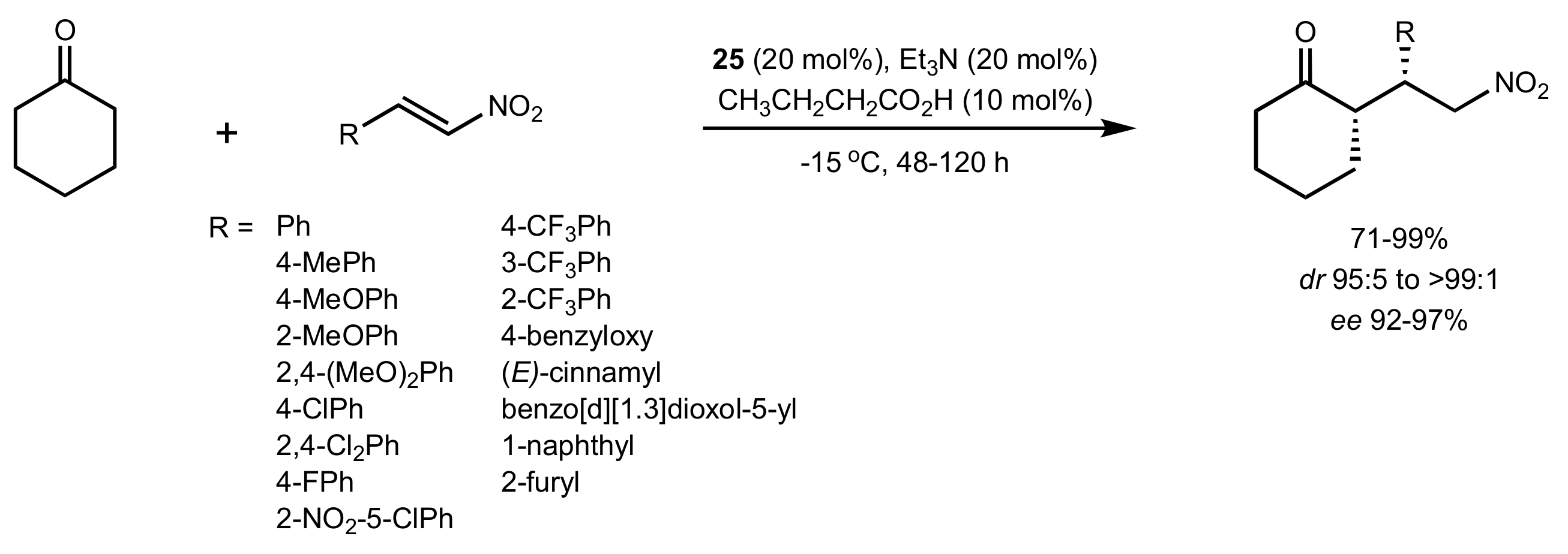
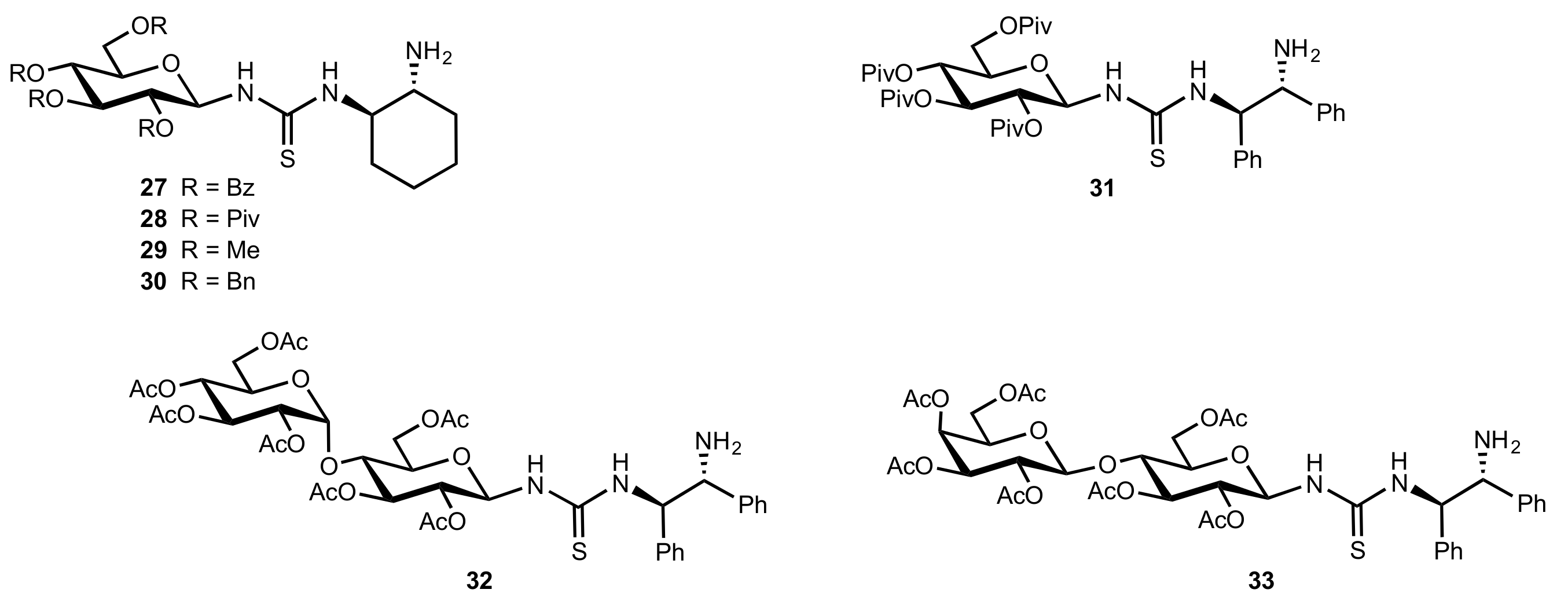
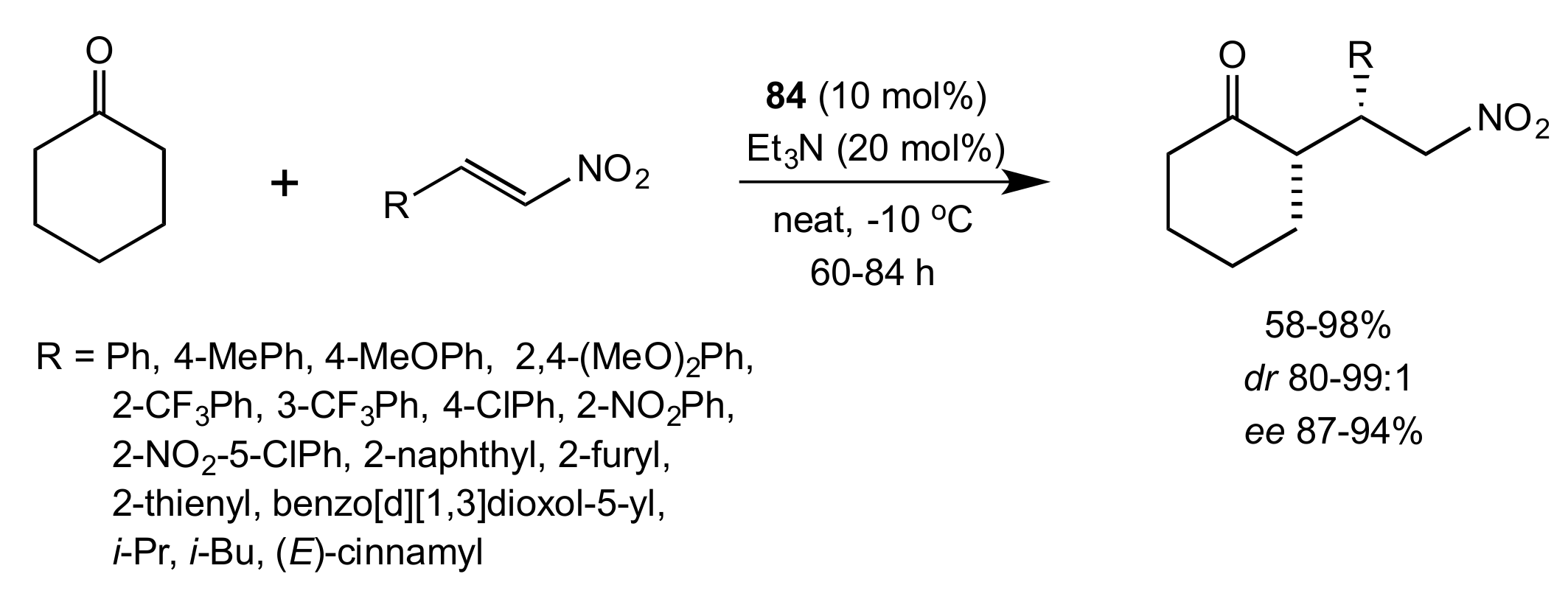
- Lu, A.; Gao, P.; Wu, Y.; Wang, Y.; Zhou, Z.; Tang, C. Highly enantio- and diastereoselective Michael addition of cyclohexanone to nitroolefins catalyzed by a chiral glucose-based bifunctional secondary amine-thiourea catalyst. Org. Biomol. Chem. 2009, 7, 3141–3147. [Google Scholar] [CrossRef]
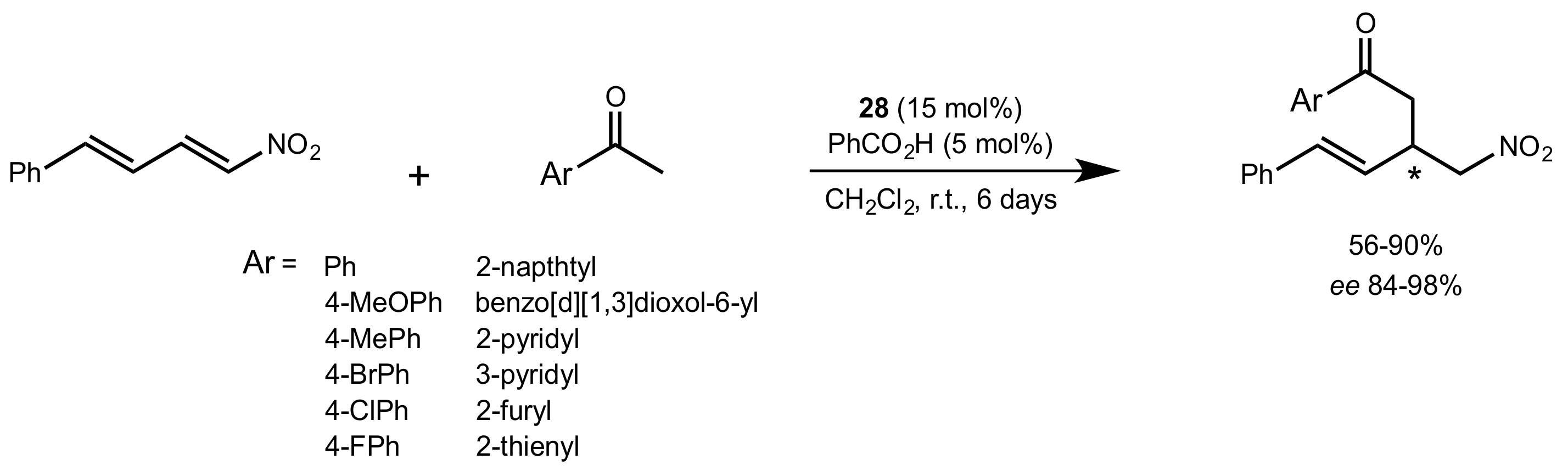
- Ma, H.; Liu, K.; Zhang, F.-G.; Zhu, C.-L.; Nie, J.; Ma, J.-A. Chiral bifunctional thiourea-catalyzed enantioselective Michael addition of ketones to nitrodienes. J. Org. Chem. 2010, 75, 1402–1409. [Google Scholar] [CrossRef]
- Pu, X.; Li, P.; Peng, F.; Li, X.; Zhang, H.; Shao, Z. Asymmetric Conjugate Addition of Acetylacetone to Nitroolefins with Chiral Organocatalysts Derived from Both α-Amino-Acids and Carbohydrates. Eur. J. Org. Chem. 2009, 2009, 4622–4626. [Google Scholar] [CrossRef]
- Pu, X.-W.; Peng, F.-Z.; Zhang, H.-B.; Shao, Z.-H. Doubly stereocontrolled asymmetric conjugate addition of acetylacetone to nitroolefins catalyzed by bifunctional tertiary amine–thiourea catalysts derived from both acyclic α-amino acids and carbohydrates. Tetrahedron 2010, 66, 3655–3661. [Google Scholar] [CrossRef]
- Reddy, B.V.S.; Reddy, S.M.; Swain, M.; Dudem, S.; Kalivendi, S.V.; Reddy, C.S. Enantioselective 1,4-addition of kojic acid derivatives to β-nitroolefins catalyzed by a cinchonine derived sugar thiourea. RSC Adv. 2014, 4, 9107–9111. [Google Scholar] [CrossRef]
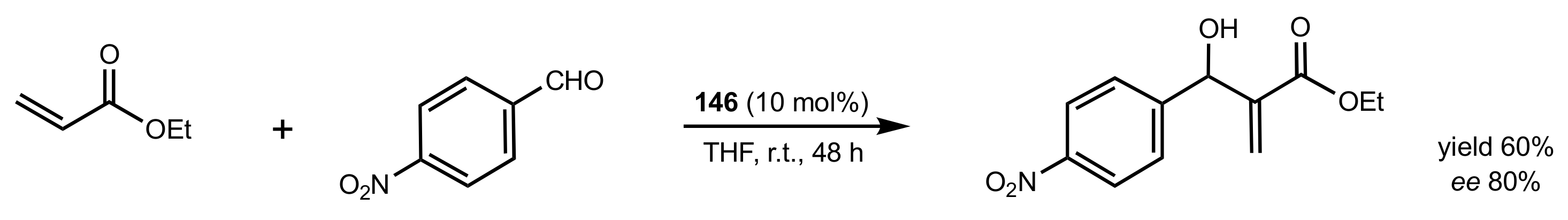
- Puglisi, A.; Benaglia, M.; Raimondi, L.; Lay, L.; Poletti, L. Novel carbohydrate-based bifunctional organocatalysts for nucleophilic addition to nitroolefins and imines. Org. Biomol. Chem. 2011, 9, 3295–3302. [Google Scholar] [CrossRef] [Green Version]
- Ágoston, K.; Fügedi, P. Preparation of new type of organocatalysts having a carbohydrate scaffold. Carbohydr. Res. 2014, 389, 50–56. [Google Scholar] [CrossRef] [Green Version]
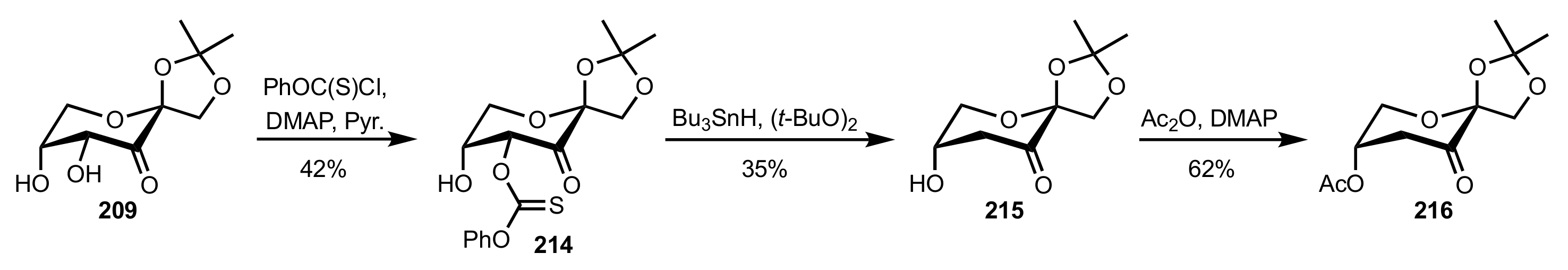
- Fuller, T.S.; Stick, R.V. Further Stereoselective Reductions of 3-O-Hexofuranosyl S-Methyl Dithiocarbonates with Tributyltin Deuteride. A Comment on Mechanism. Austr. J. Chem. 1980, 33, 2509–2515. [Google Scholar] [CrossRef]
- Azad, C.S.; Khan, I.A.; Narula, A.K. Organocatalyzed asymmetric Michael addition by an efficient bifunctional carbohydrate–thiourea hybrid with mechanistic DFT analysis. Org. Biomol. Chem. 2016, 14, 11454–11461. [Google Scholar] [CrossRef]
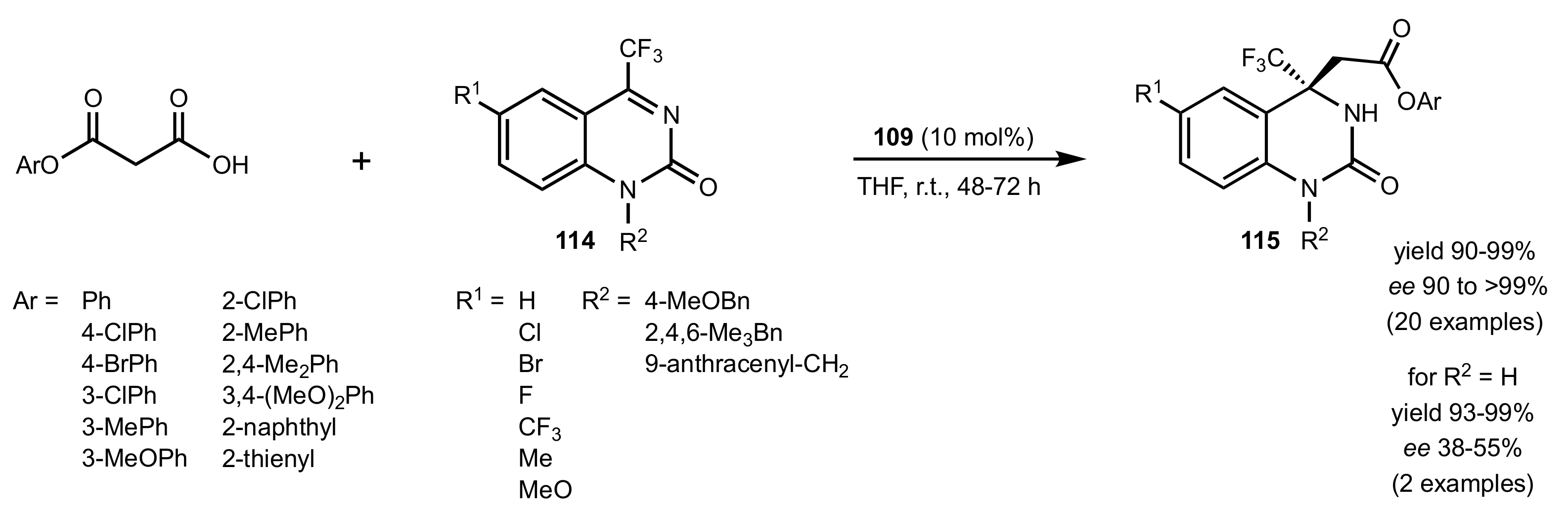
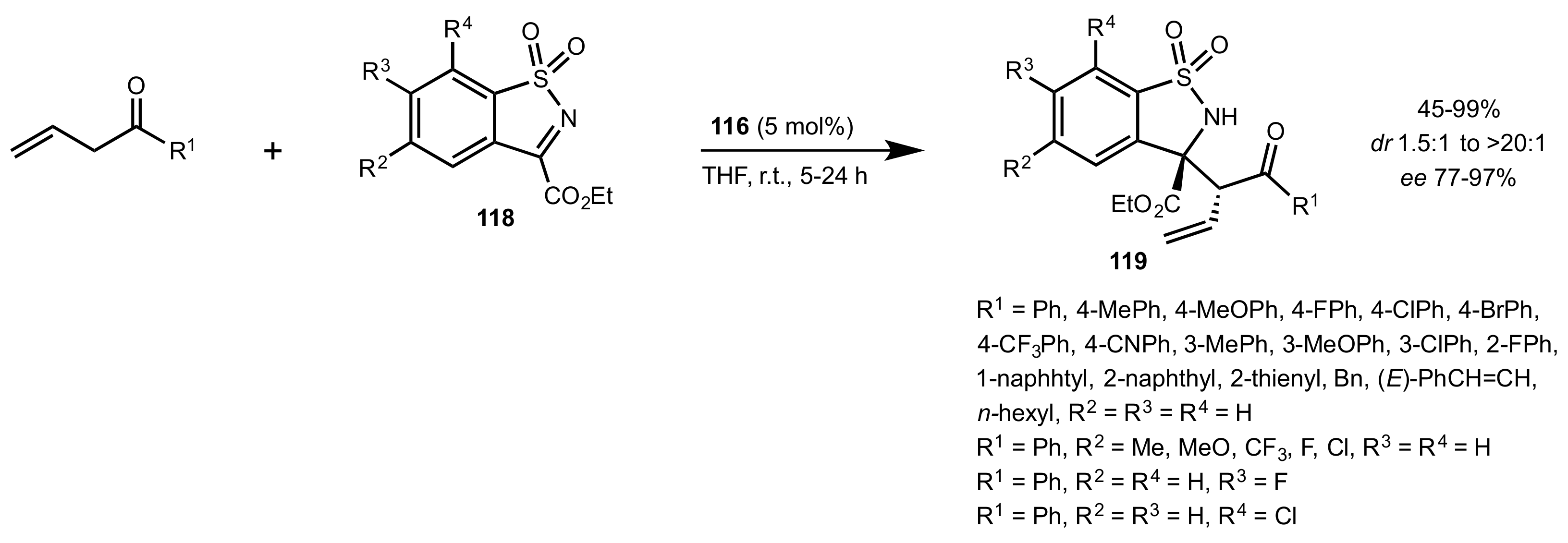
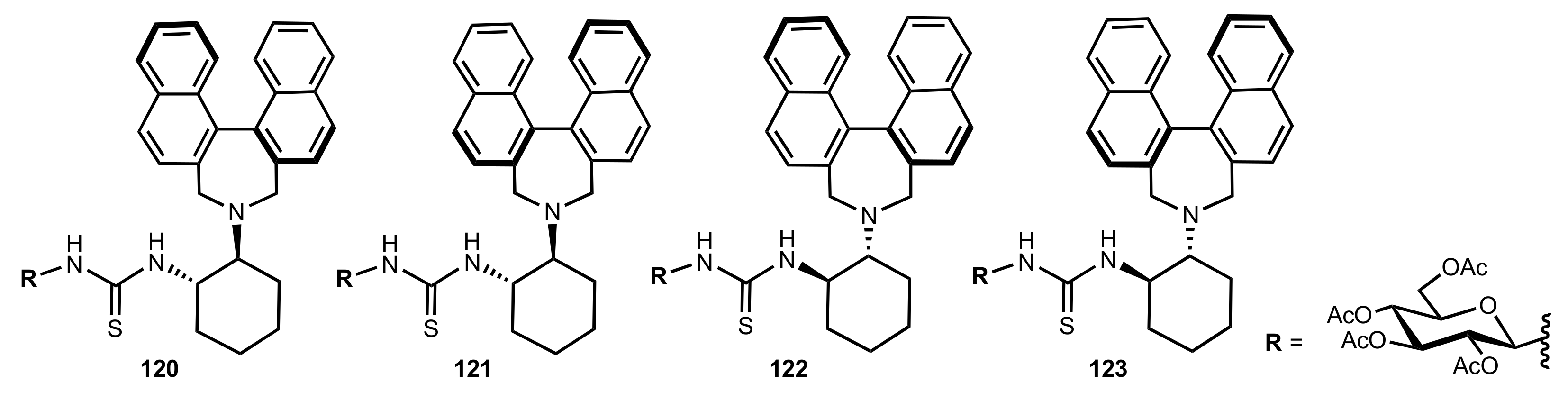
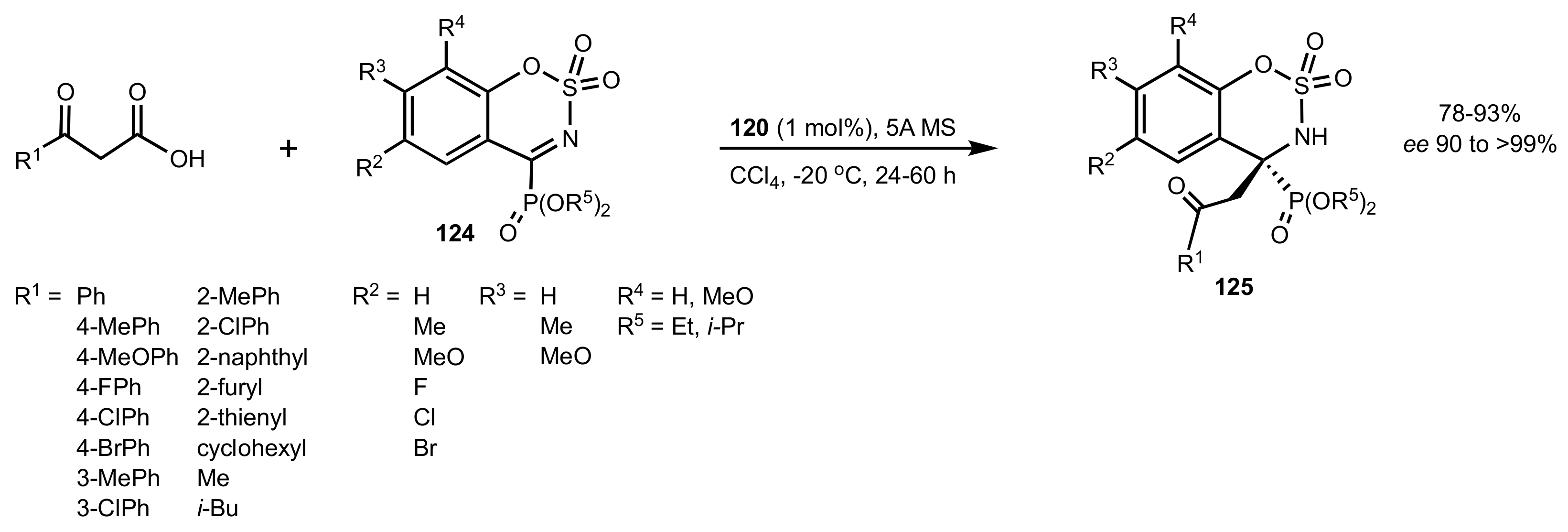
- Rončák, R.; Tvrdoňová, M.; Gonda, J.; Elečko, J. Novel carbohydrate-based thioureas as organocatalysts for asymmetric Michael addition of 1,3-dicarbonyl compounds to nitroolefins. Tetrahedron 2020, 76, 131339. [Google Scholar] [CrossRef]
- Gonda, J.; Martinková, M.; Raschmanová, J.; Balentová, E. Creation of quarternary stereocentres via [3,3]-sigmatropic rearrangement of allylic thiocyanates. A synthetic approach to (+)-myriocin. Tetrahedron Asymmetry 2006, 17, 1875–1882. [Google Scholar] [CrossRef]
- Tronchet, J.M.J.; Gentile, B. 3-C-(Acylméthylène)-3-désoxy-1,2:5,6-di-O-isopropylidène-α-d-ribo- et -xylo-hexofuranoses. Carbohydr. Res. 1975, 44, 23–35. [Google Scholar] [CrossRef]
- Tadano, K.; Idogaki, Y.; Yamada, H.; Suami, T. Introduction of gem-dialkyl group to hexofuranose by ortho ester Claisen rearrangement. Chem. Lett. 1985, 14, 1925–1928. [Google Scholar] [CrossRef]
- Tadano, K.; Idogaki, Y.; Yamada, H.; Suami, T. Ortho Ester Claisen Rearrangements of Three 3-C-(Hydroxymethyl)methylene Derivatives of Hexofuranose: Stereoselective Introduction of a Quaternary Center on C-3 of d-ribo-, l-lyxo-, and d-arabino-Hexofuranoses. J. Org. Chem. 1987, 52, 1201–1210. [Google Scholar] [CrossRef]
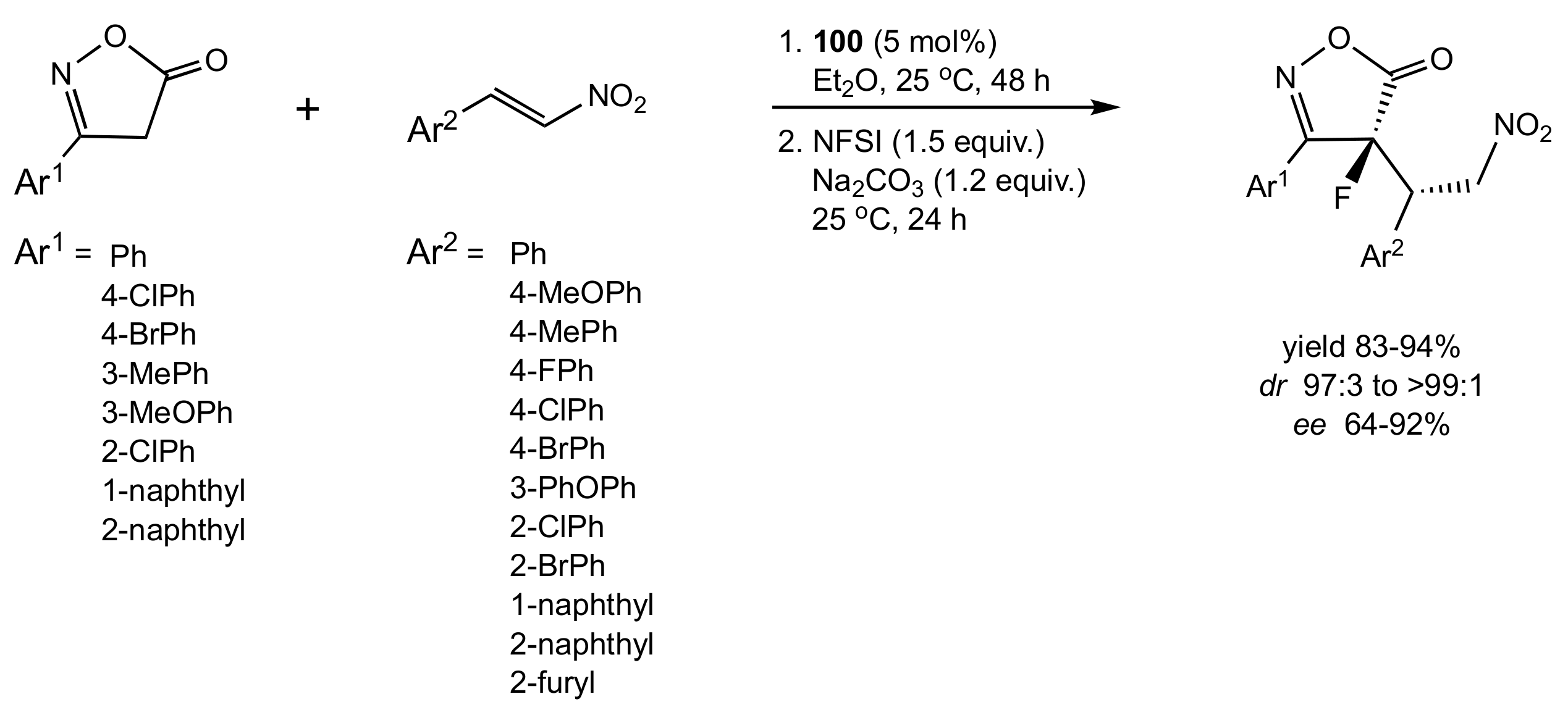
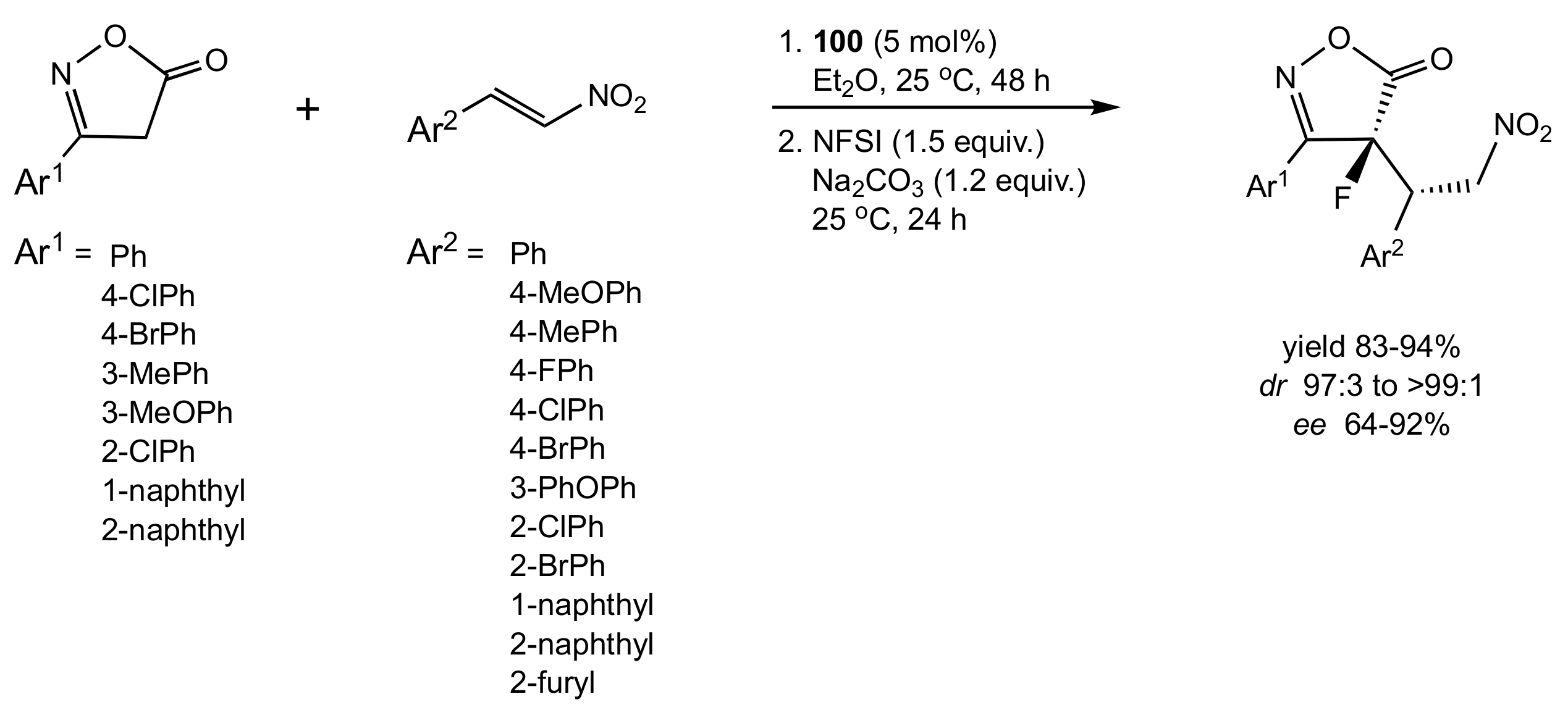
- Li, F.; Sun, L.; Teng, Y.; Yu, P.; Zhao, J.C.-G.; Ma, J.-A. Highly Diastereo- and Enantioselective Organocatalytic One-Pot Sequential 1,4-Addition/Dearomative-Fluorination Transformation. Chem. Eur. J. 2012, 18, 14255–14260. [Google Scholar] [CrossRef]
- Meng, W.-T.; Zheng, Y.; Nie, J.; Xiong, H.-Y.; Ma, J.-A. Organocatalytic Asymmetric One-Pot Sequential Conjugate Addition/Dearomative Fluorination: Synthesis of Chiral Fluorinated Isoxazol-5(4H)-ones. J. Org. Chem. 2013, 78, 559–567. [Google Scholar] [CrossRef]
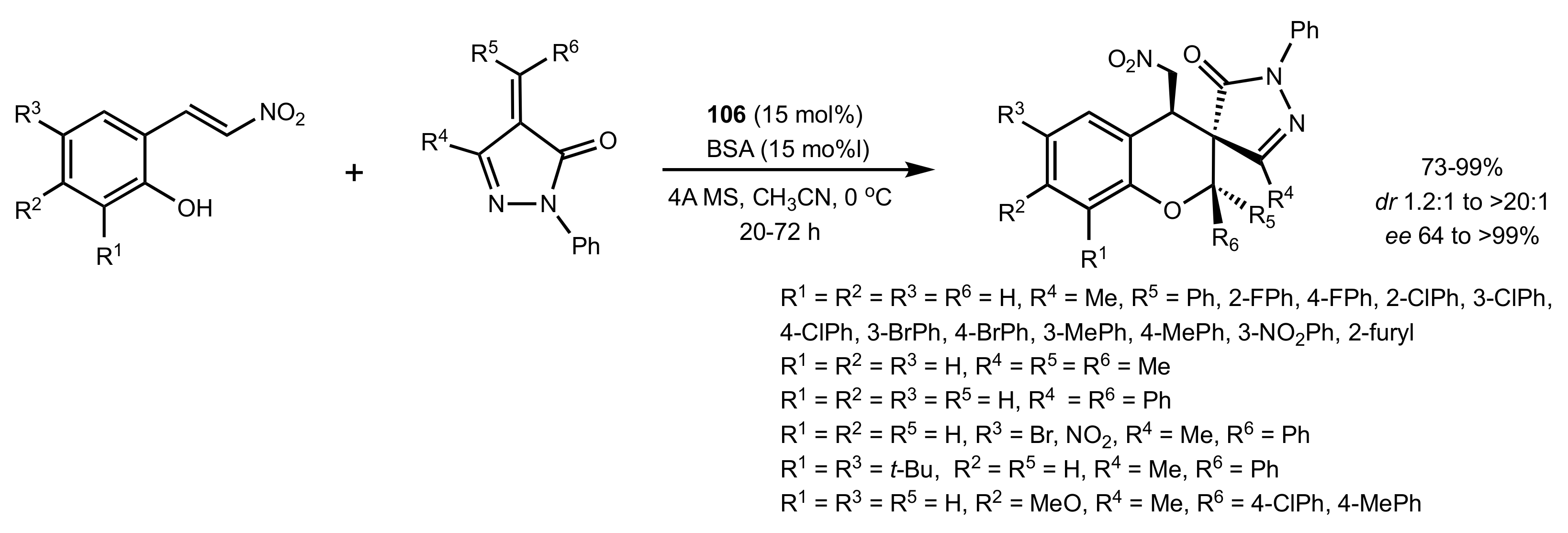
- Zheng, W.; Zhang, J.; Liu, S.; Yu, C.; Miao, Z. Asymmetric synthesis of spiro[chroman-3,3′-pyrazol] scaffolds with an all-carbon quaternary stereocenter via a oxa-Michael–Michael cascade strategy with bifunctional amine-thiourea organocatalysts. RSC Adv. 2015, 5, 91108–91113. [Google Scholar] [CrossRef]
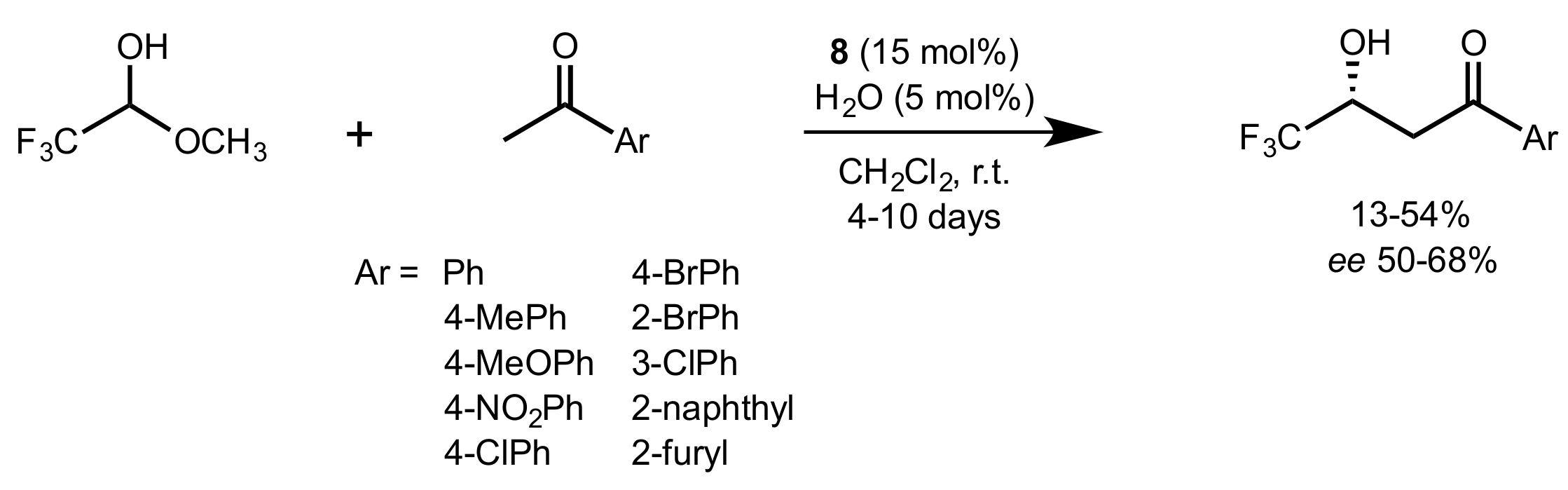
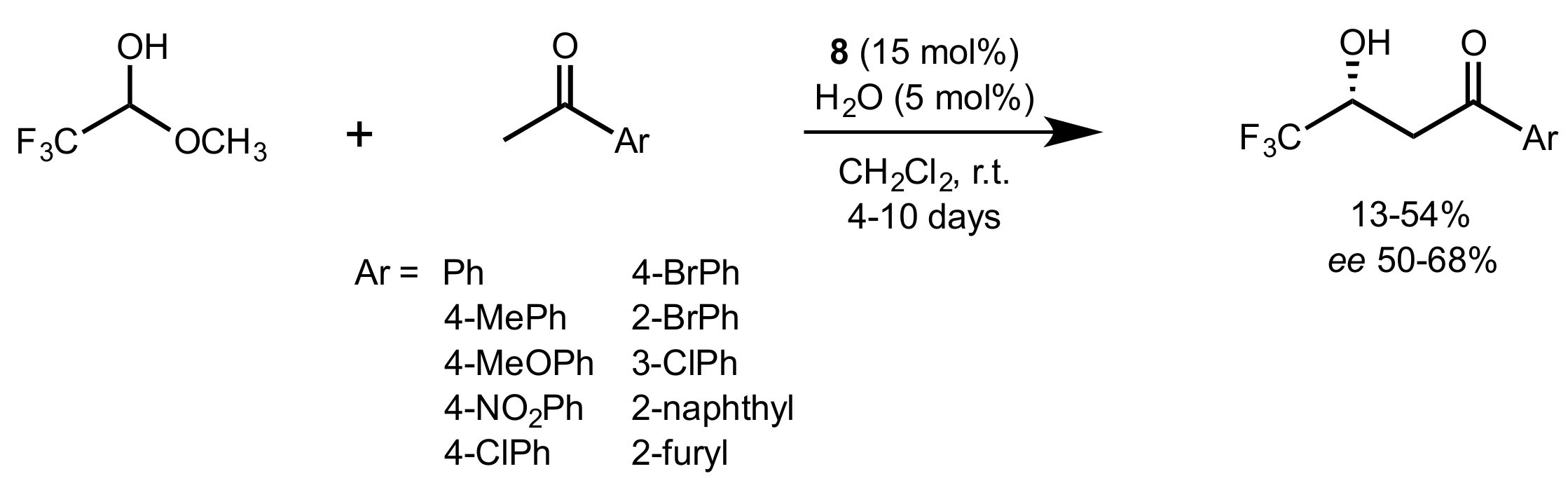
- Nie, J.; Li, X.-J.; Zheng, D.-H.; Zhang, F.-G.; Cui, S.; Ma, J.-A. Chiral bifunctional thiourea-catalyzed enantioselective aldol reaction of trifluoroacetaldehyde hemiacetal with aromatic ketones. J. Fluor. Chem. 2011, 132, 468–473. [Google Scholar] [CrossRef]
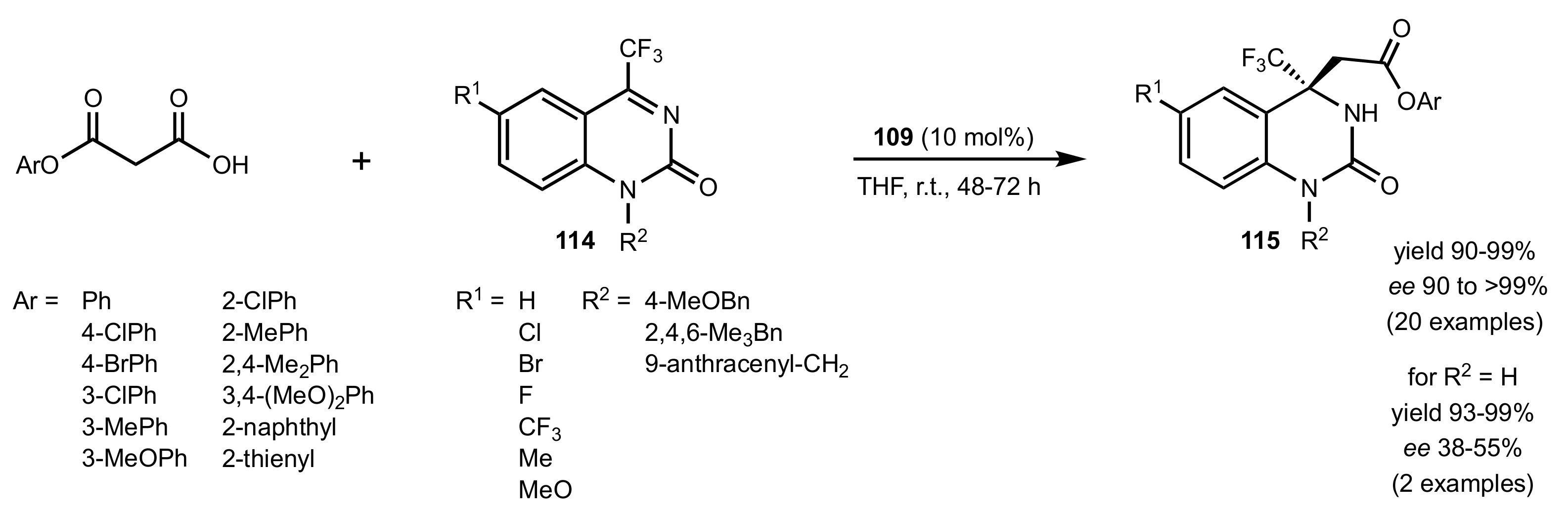
- Yuan, H.-N.; Wang, S.; Nie, J.; Meng, W.; Yao, Q.; Ma, J.-A. Hydrogen-Bond-Directed Enantioselective Decarboxylative Mannich Reaction of β-Ketoacids with Ketimines: Application to the Synthesis of Anti-HIV Drug DPC 083. Angew. Chem. Int. Ed. 2013, 52, 3869–3873. [Google Scholar] [CrossRef]
- Yuan, H.-N.; Li, S.; Nie, J.; Zheng, Y.; Ma, J.-A. Highly Enantioselective Decarboxylative Mannich Reaction of Malonic Acid Half Oxyesters with Cyclic Trifluoromethyl Ketimines: Synthesis of β-Amino Esters and Anti-HIV Drug DPC 083. Chem. Eur. J. 2013, 19, 15856–15860. [Google Scholar] [CrossRef]
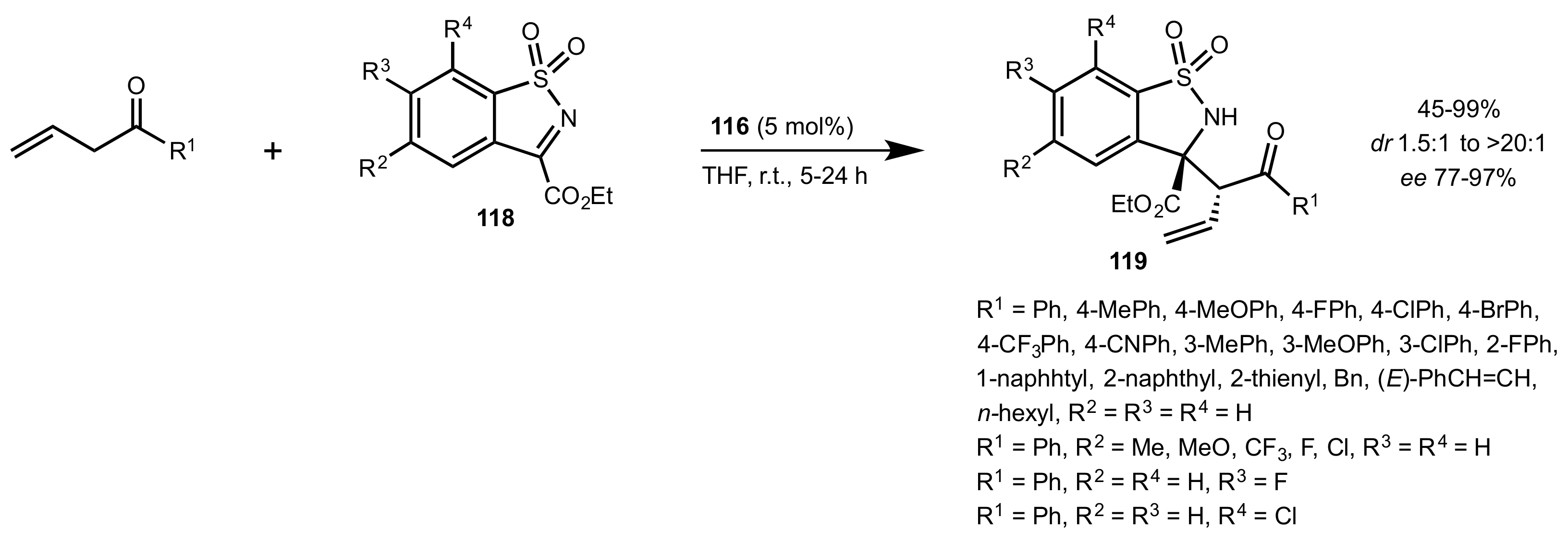
- Qiao, B.; Huang, Y.-J.; Nie, J.; Ma, J.-A. Highly Regio-, Diastereo-, and Enantioselective Mannich Reaction of Allylic Ketones and Cyclic Ketimines: Access to Chiral Benzosultam. Org. Lett. 2015, 17, 4608–4611. [Google Scholar] [CrossRef]
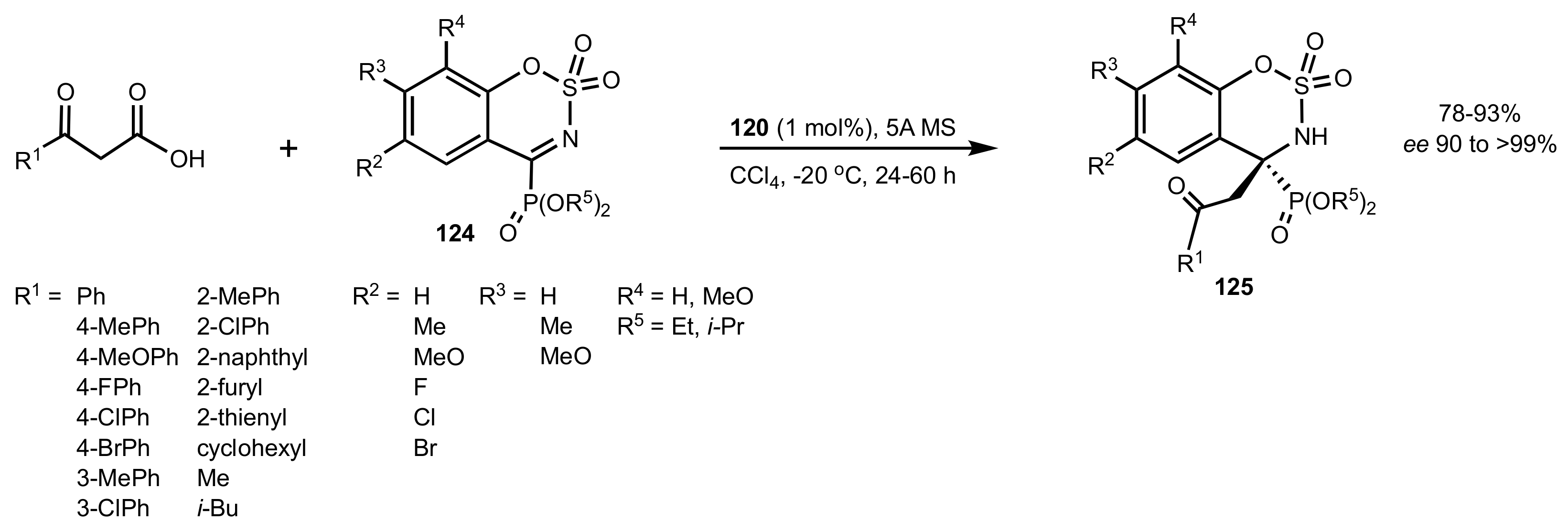
- Liu, Y.-J.; Li, J.-S.; Nie, J.; Ma, J.-A. Organocatalytic Asymmetric Decarboxylative Mannich Reaction of β-Keto Acids with Cyclic α-Ketiminophosphonates: Access to Quaternary α-Aminophosphonates. Org. Lett. 2018, 20, 3643–3646. [Google Scholar] [CrossRef]
- Wang, C.; Zhou, Z.; Tang, C. Novel Bifunctional Chiral Thiourea Catalyzed Highly Enantioselective aza-Henry Reaction. Org. Lett. 2008, 10, 1707–1710. [Google Scholar] [CrossRef]
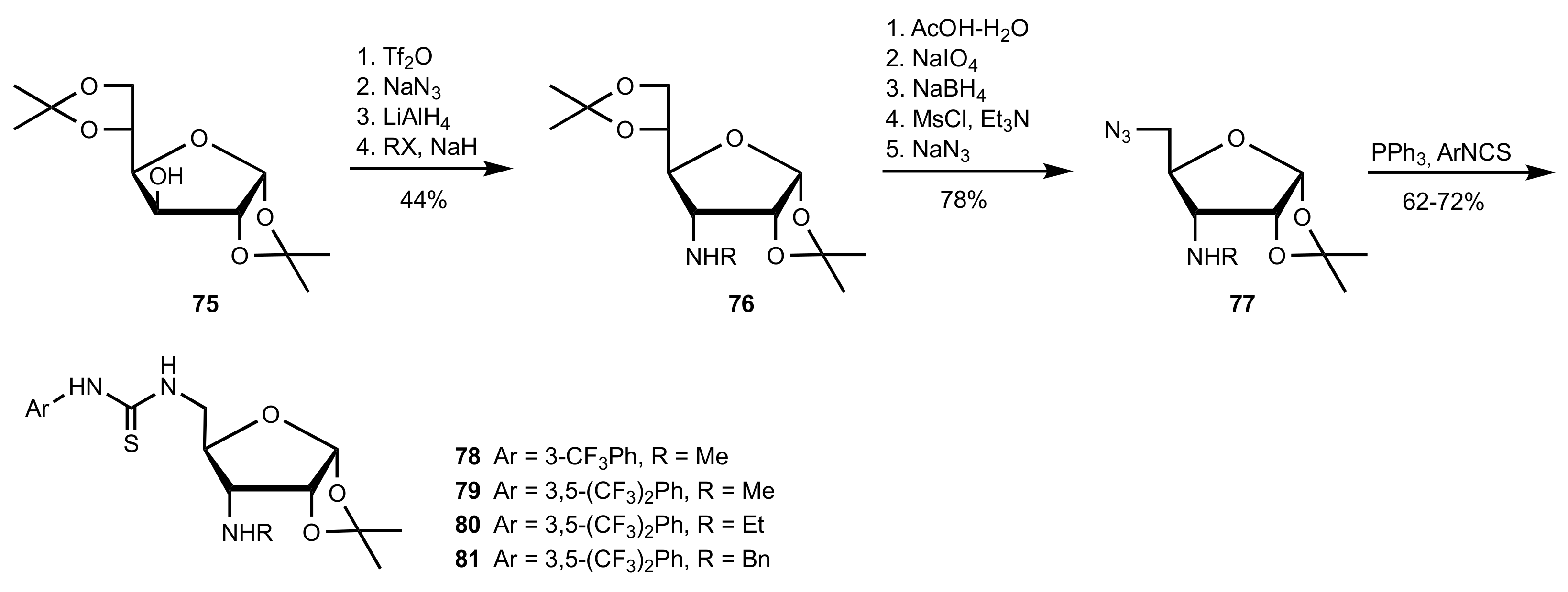
- Robak, J.; Kryczka, B.; Świerczyńska, B.; Zawisza, A.; Porwański, S. New sugar-derived bifunctional chiral ureas as highly effective organocatalysts in asymmetric aza-Henry reaction. Carbohydr. Res. 2015, 404, 83–86. [Google Scholar] [CrossRef] [PubMed]
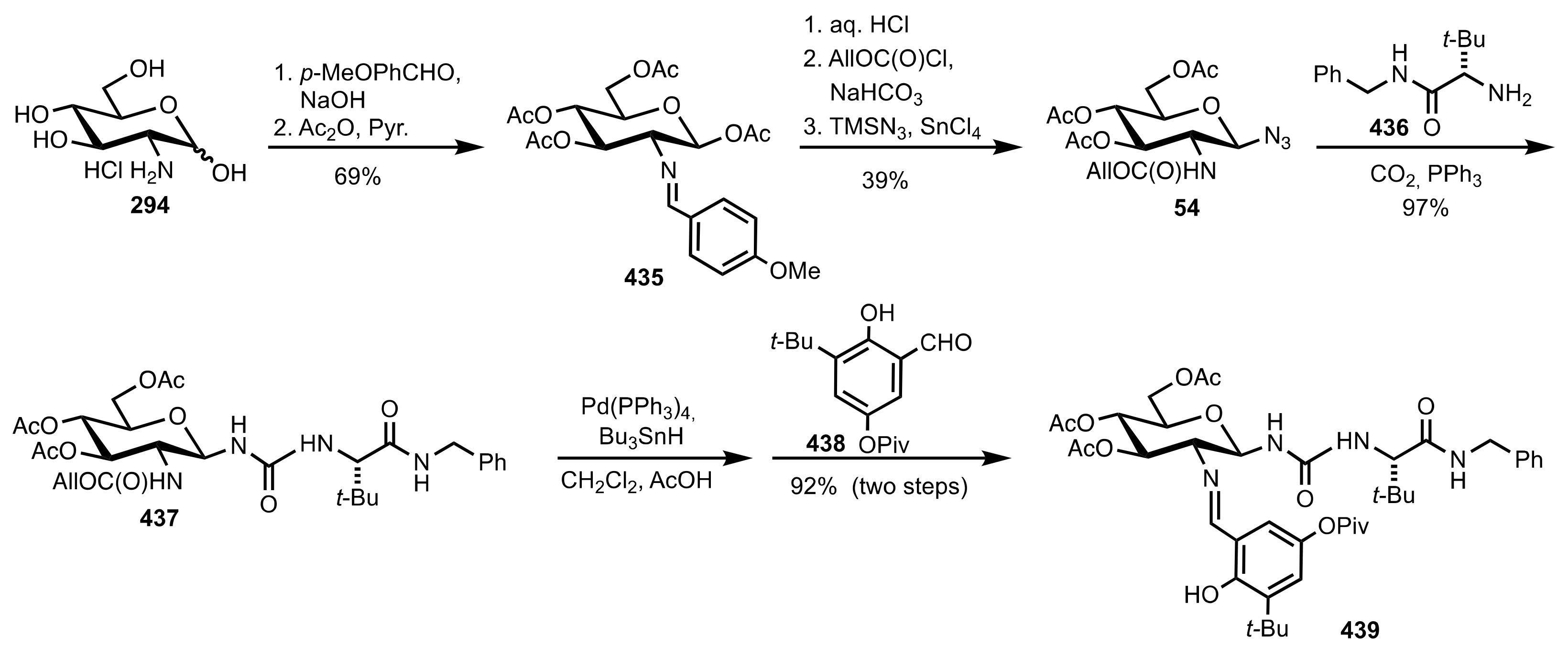
- Deras, I.L.; Takegawa, K.; Kondo, A.; Kato, I.; Lee, Y.C. Synthesis of a high-mannose-type glycopeptide analog containing a glucose-asparagine linkage. Bioorg. Med. Chem. Lett. 1998, 8, 1763–1766. [Google Scholar] [CrossRef]
- Szarek, W.A.; Jones, J.K.N. Carbohydrate containing nitrogen in a five-membered ring and an attempted synthesis of a carbohydrate with nitrogen in a seven-membered ring. Can. J. Chem. 1965, 43, 2345–2356. [Google Scholar] [CrossRef] [Green Version]
- Pető, C.; Batta, G.; Györgydeák, Z.; Sztaricskai, F. Zur Darstellung der Anomeren des Hepta-O-acetylcellobiosyl-, -lactosyl-, -maltosyl- und -melibiosylazids. Liebigs Ann. Chem. 1991, 1991, 505–507. [Google Scholar] [CrossRef]
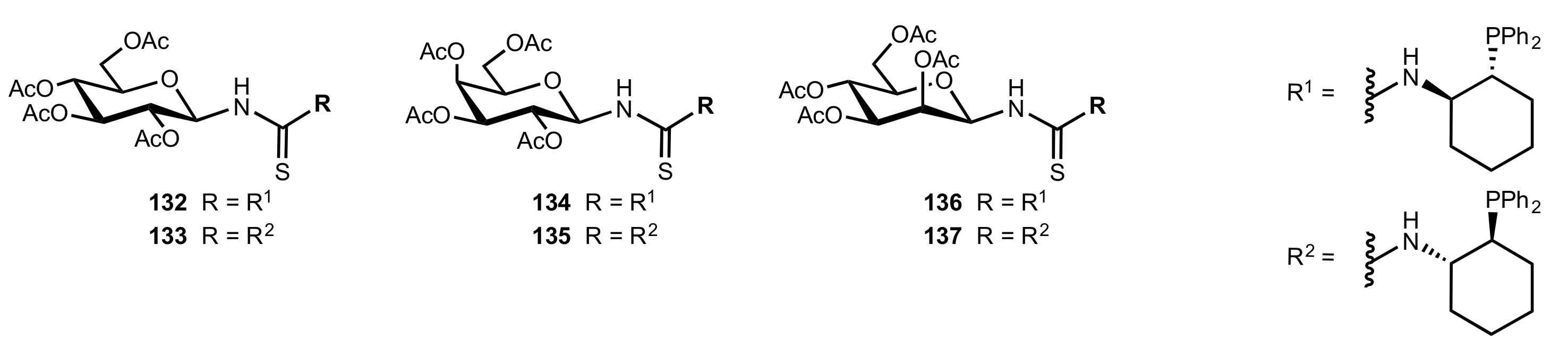
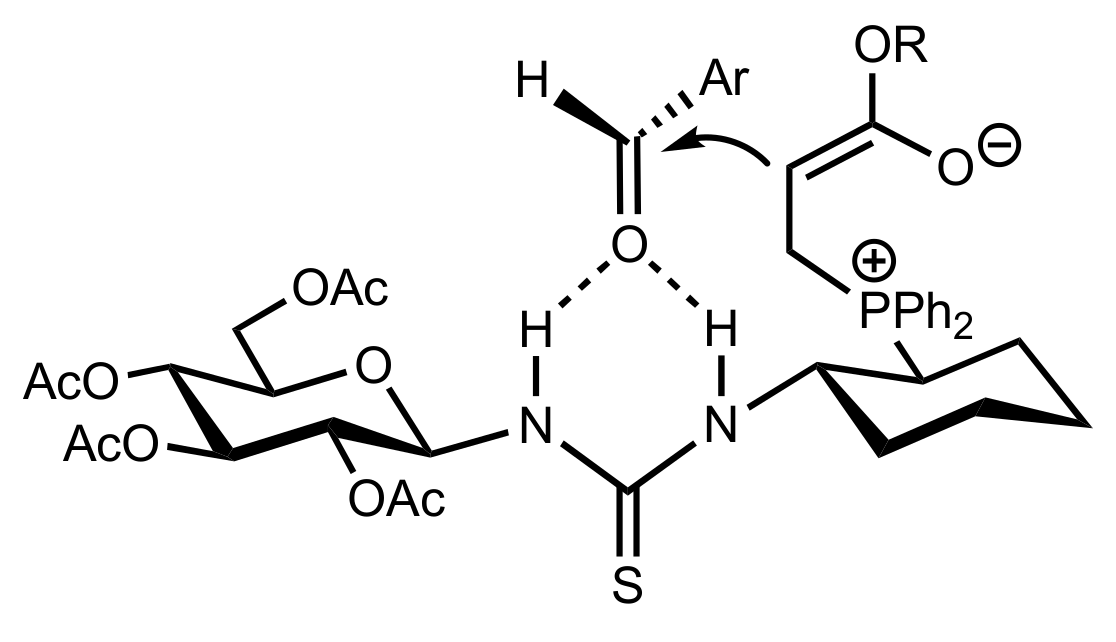
- Yang, W.; Sha, F.; Zhang, X.; Yuan, K.; Wu, X. Enantioselective Morita-Baylis-Hillman Reaction Organocatalyzed by Glucose-based Phosphinothiourea. Chin. J. Chem. 2012, 30, 2652–2656. [Google Scholar] [CrossRef]
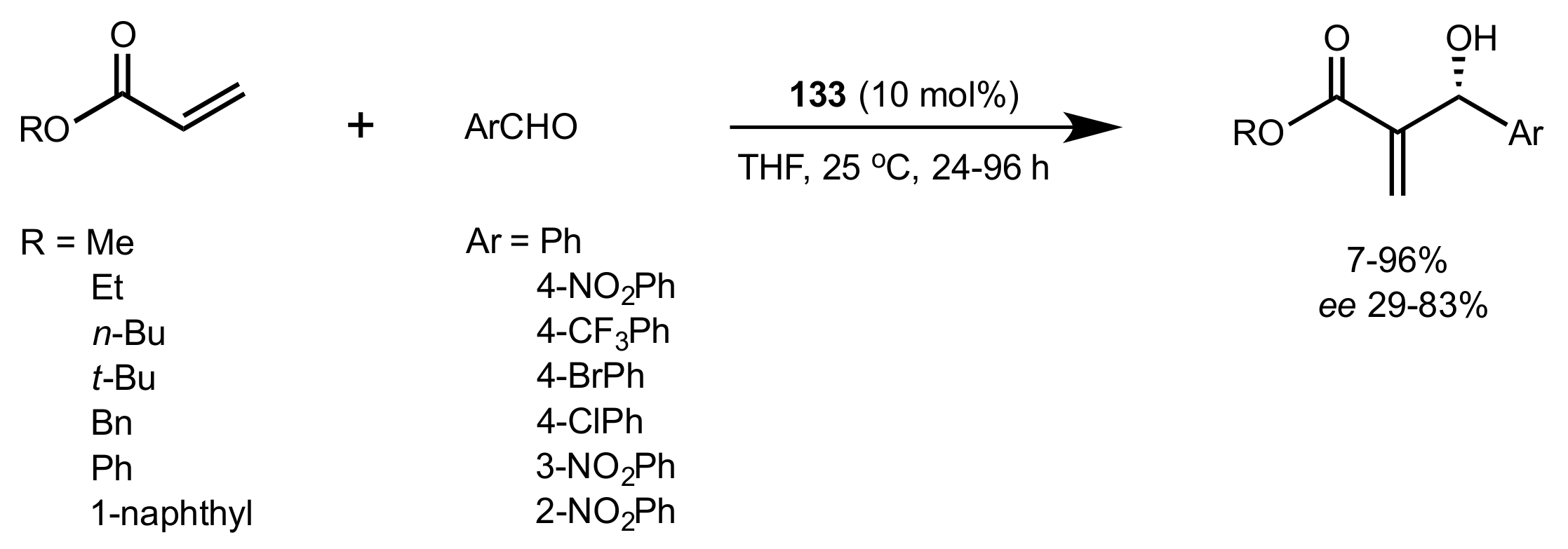
- Porwański, S. New ureas containing glycosyl and diphenylphosphinyl scaffolds: Synthesis and the first attempts to use them in asymmetric synthesis. Carbohydr. Res. 2014, 394, 7–12. [Google Scholar] [CrossRef]
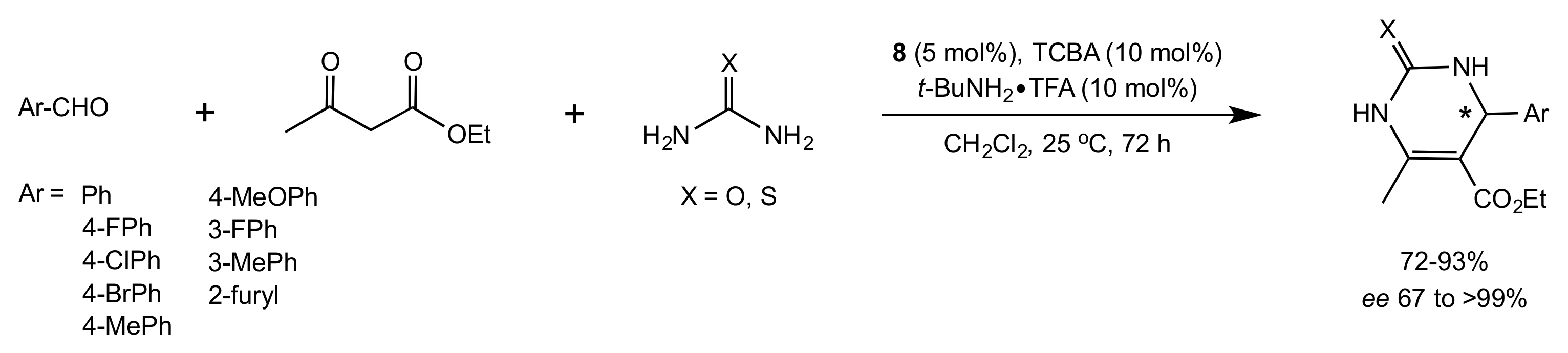
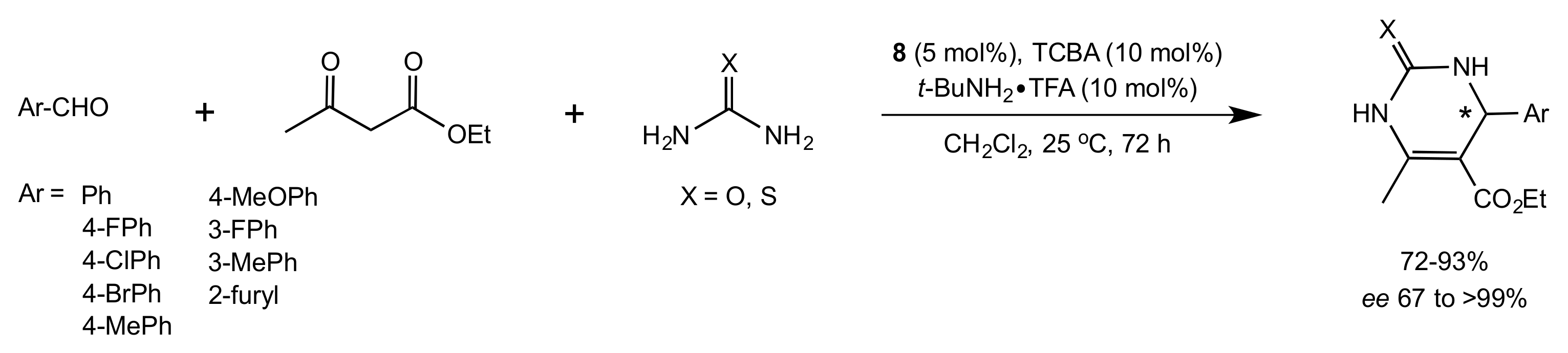
- Wang, Y.; Yang, H.; Yu, J.; Miao, Z.; Chen, R. Highly Enantioselective Biginelli Reaction Promoted by Chiral Bifunctional Primary Amine-Thiourea Catalysts: Asymmetric Synthesis of Dihydropyrimidines. Adv. Synth. Catal. 2009, 351, 3057–3062. [Google Scholar] [CrossRef]
- Kong, S.; Fan, W.; Wu, G.; Miao, Z. Enantioselective Synthesis of Tertiary α-Hydroxy Phosphonates Catalyzed by Carbohydrate/Cinchona Alkaloid Thiourea Organocatalysts. Angew. Chem. Int. Ed. 2012, 51, 8864–8867. [Google Scholar] [CrossRef]
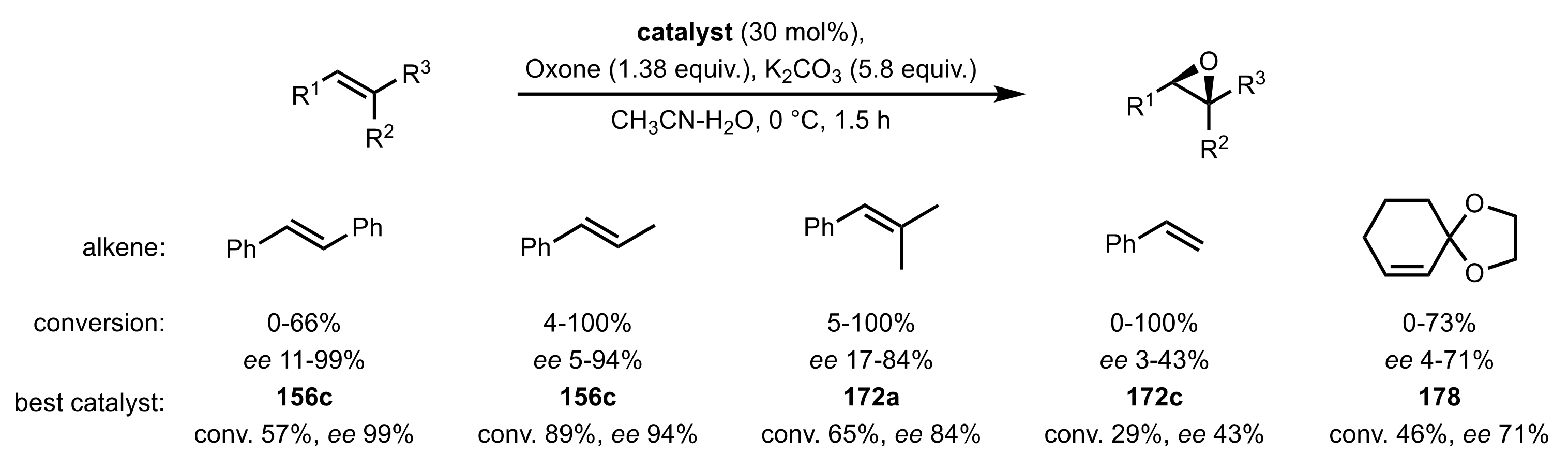
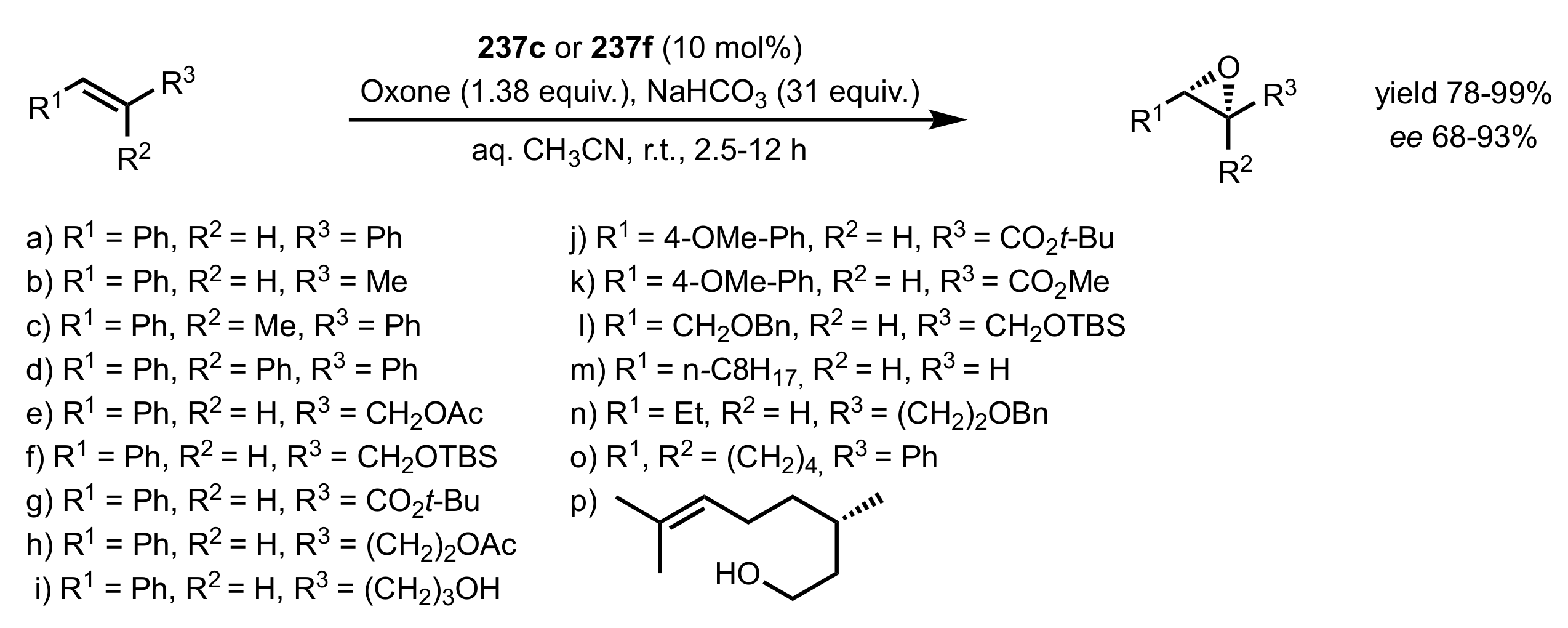
- Tu, Y.; Wang, Z.X.; Shi, Y. An efficient asymmetric epoxidation method for trans-olefins mediated by a fructose-derived ketone. J. Am. Chem. Soc. 1996, 118, 9806–9807. [Google Scholar] [CrossRef]
- Mio, S.; Kumagawa, Y.; Sugai, S. Synthetic studies on (+)-hydantocidin (3): A new synthetic method for construction of the spiro-hydantoin ring at the anomeric position of d-ribofuranose. Tetrahedron 1991, 47, 2133–2144. [Google Scholar] [CrossRef]
- Wang, Z.X.; Tu, Y.; Frohn, M.; Shi, Y. A Dramatic pH Effect Leads to a Catalytic Asymmetric Epoxidation. J. Org. Chem. 1997, 62, 2328–2329. [Google Scholar] [CrossRef]
- Wang, Z.X.; Tu, Y.; Frohn, M.; Zhang, J.R.; Shi, Y. An efficient catalytic asymmetric epoxidation method. J. Am. Chem. Soc. 1997, 119, 11224–11235. [Google Scholar] [CrossRef]
- Chen, C.C.; Whistler, R.L. Synthesis of l-fructose. Carbohydr. Res. 1988, 175, 265–271. [Google Scholar] [CrossRef]
- Wang, Z.X.; Shi, Y. A pH Study on the Chiral Ketone Catalyzed Asymmetric Epoxidation of Hydroxyalkenes. J. Org. Chem. 1998, 63, 3099–3104. [Google Scholar] [CrossRef]
- Cao, G.A.; Wang, Z.X.; Tu, Y.; Shi, Y. Chemo- and enantioselective epoxidation of enynes. Tetrahedron Lett. 1998, 39, 4425–4428. [Google Scholar] [CrossRef]
- Wang, Z.X.; Cao, G.A.; Shi, Y. Chiral ketone catalyzed highly chemo- and enantioselective epoxidation of conjugated enynes. J. Org. Chem. 1999, 64, 7646–7650. [Google Scholar] [CrossRef]
- Zhu, Y.; Tu, Y.; Yu, H.; Shi, Y. Highly enantioselective epoxidation of enol silyl ethers and esters. Tetrahedron Lett. 1998, 39, 7819–7822. [Google Scholar] [CrossRef]
- Zhu, Y.; Shu, L.; Tu, Y.; Shi, Y. Enantioselective synthesis and stereoselective rearrangements of enol ester epoxides. J. Org. Chem. 2001, 66, 1818–1826. [Google Scholar] [CrossRef]
- Warren, J.D.; Shi, Y. Chiral ketone-catalyzed asymmetric epoxidation of 2,2-disubstituted vinylsilanes. J. Org. Chem. 1999, 64, 7675–7677. [Google Scholar] [CrossRef]
- Frohn, M.; Zhou, X.; Zhang, J.R.; Tang, Y.; Shi, Y. Kinetic resolution of racemic cyclic olefins via chiral dioxirane. J. Am. Chem. Soc. 1999, 121, 7718–7719. [Google Scholar] [CrossRef]
- Shu, L.; Shi, Y. Asymmetric epoxidation using hydrogen peroxide (H2O2) as primary oxidant. Tetrahedron Lett. 1999, 40, 8721–8724. [Google Scholar] [CrossRef]
- Shu, L.; Shi, Y. An efficient ketone-catalyzed asymmetric epoxidation using hydrogen peroxide (H2O2) as primary oxidant. Tetrahedron 2001, 57, 5213–5218. [Google Scholar] [CrossRef]
- Tu, Y.; Wang, Z.X.; Frohn, M.; He, M.; Yu, H.; Tang, Y.; Shi, Y. Structural probing of ketone catalysts for asymmetric epoxidation. J. Org. Chem. 1998, 63, 8475–8485. [Google Scholar] [CrossRef]
- Fernández, J.M.G.; Mellet, C.O.; Marín, A.M.; Fuentes, J. A mild and efficient procedure to remove acetal and dithioacetal protecting groups in carbohydrate derivatives using 2,3-dichloro-5,6-dicyano-1,4-benzoquinone. Carbohydr. Res. 1995, 274, 263–268. [Google Scholar] [CrossRef]
- Differding, E.; Ofner, H. N-Fluorobenzenesulfonimide: A Practical Reagent for Electrophilic Fluorinations. Synlett 1991, 1991, 187–189. [Google Scholar] [CrossRef]
- Kang, J.; Lim, G.J.; Yoon, S.K.; Kim, M.Y. Asymmetric Cyclopropanation Using New Chiral Auxiliaries Derived from d-Fructose. J. Org. Chem. 1995, 60, 564–577. [Google Scholar] [CrossRef]
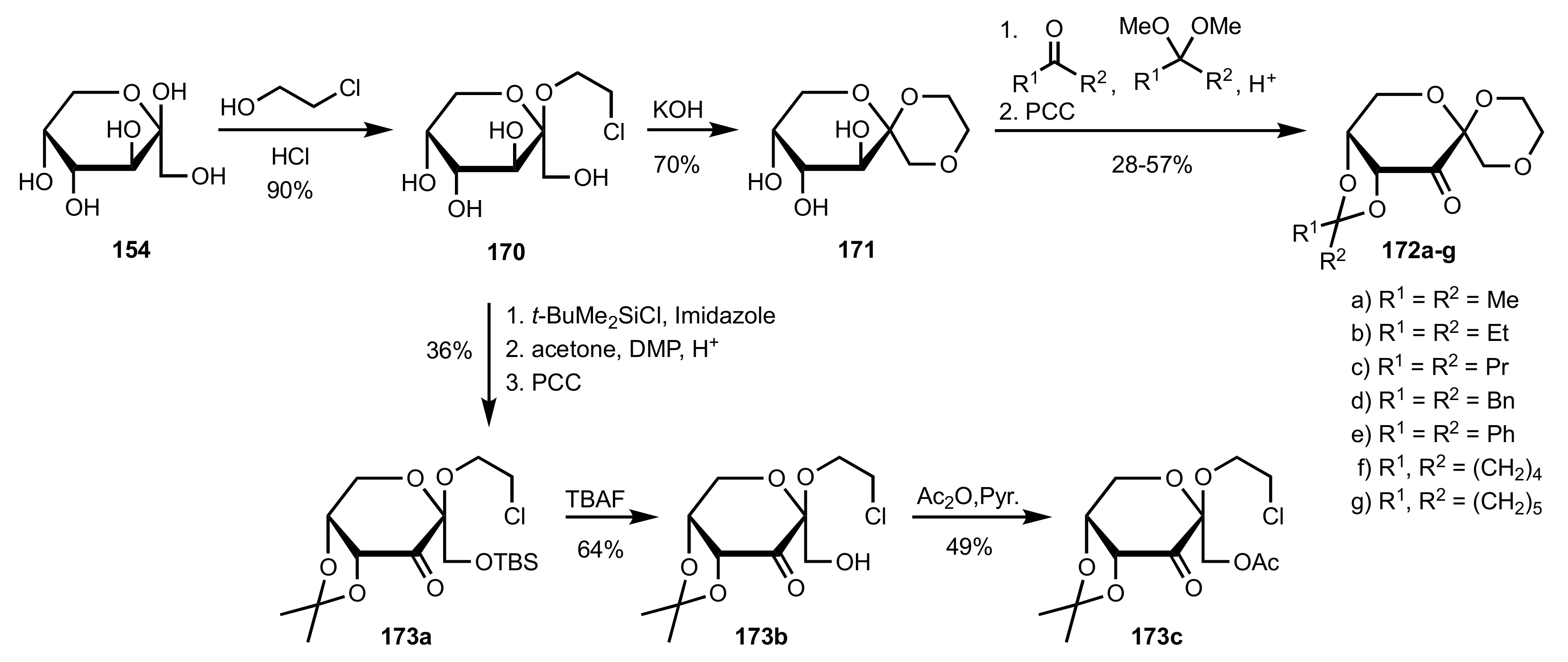
- Garegg, P.J.; Samuelsson, B. One-step conversion of vicinal diols into olefins, using a novel reagent system. Synthesis 1979, 1979, 469–470. [Google Scholar] [CrossRef]
- Lichtenthaler, F.W.; Doleschal, W.; Hahn, S. Spirocyclic Dihydropyranones from d-Fructose. Liebigs Ann. Chem. 1985, 1985, 2454–2464. [Google Scholar] [CrossRef]
- Matsumoto, K.; Ebata, T.; Matsushita, H. Novel synthesis of 3-acetamido-3-deoxy- and 4-acetamido-4-deoxy-d-altrose from levoglucosenone using regioselective cis-oxyamination. Carbohydr. Res. 1995, 267, 187–201. [Google Scholar] [CrossRef]
- Rubin, A.E.; Sharpless, K.B. A Highly Efficient Aminohydroxylation Process. Angew. Chem. Int. Ed. Engl. 1997, 36, 2637–2640. [Google Scholar] [CrossRef]
- Tian, H.; She, X.; Shi, Y. Electronic probing of ketone catalysts for asymmetric epoxidation. Search for more robust catalysts. Org. Lett. 2001, 3, 715–718. [Google Scholar] [CrossRef]
- Chan, J.Y.C.; Cheong, P.P.L.; Hough, L.; Richardson, A.C. The preparation and reactions of a new glycoside: 2′-Chloroethyl β-d-fructopyranoside. J. Chem. Soc. Perkin Trans. 1985, 4, 1447–1455. [Google Scholar] [CrossRef]
- Aamlid, K.H.; Hough, L.; Richardson, A.C.; Hendry, D. An enantiospecific synthesis of (R)-1,4,7-trioxaspiro[5.5]undecane from d-fructose. Carbohydr. Res. 1987, 164, 373–390. [Google Scholar] [CrossRef]
- Bennett, M.; Gill, G.B.; Pattenden, G.; Shuker, A.J.; Stapleton, A. Ylidenebutenolide mycotoxins. Concise syntheses of patulin and neopatulin from carbohydrate precursors. J. Chem. Soc. Perkin Trans. 1991, 1991, 929–937. [Google Scholar] [CrossRef]
- Zottola, M.A.; Alonso, R.; Vite, G.D.; Fraser-Reid, B. A Practical, Efficient Large-Scale Synthesis of 1,6-Anhydrohexopyranoses. J. Org. Chem. 1989, 54, 6123–6125. [Google Scholar] [CrossRef]
- Tian, H.; She, X.; Shu, L.; Yu, H.; Shi, Y. Highly enantioselective epoxidation of cis-olefins by chiral dioxirane. J. Am. Chem. Soc. 2000, 122, 11551–11552. [Google Scholar] [CrossRef]
- Tian, H.; She, X.; Xu, J.; Shi, Y. Enantioselective epoxidation of terminal olefins by chiral dioxirane. Org. Lett. 2001, 3, 1929–1931. [Google Scholar] [CrossRef]
- Tian, H.; She, X.; Yu, H.; Shu, L.; Shi, Y. Designing new chiral ketone catalysts. Asymmetric epoxidation of cis-olefins and terminal olefins. J. Org. Chem. 2002, 67, 2435–2446. [Google Scholar] [CrossRef]
- Shu, L.; Shen, Y.M.; Burke, C.; Goeddel, D.; Shi, Y. An improved synthesis of a ketone catalyst for asymmetric epoxidation of olefins. J. Org. Chem. 2003, 68, 4963–4965. [Google Scholar] [CrossRef]
- Corey, E.J.; Hopkins, B. A mild procedure for the conversion of 1,2-diols to olefins. Tetrahedron Lett. 1982, 23, 1979–1982. [Google Scholar] [CrossRef]
- Shu, L.; Wang, P.; Gan, Y.; Shi, Y. Asymmetric epoxidation catalyzed by N-aryl-substituted oxazolidinone-containing ketones: Further evidence for electronic effects. Org. Lett. 2003, 5, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Goeddel, D.; Shu, L.; Yuan, Y.; Wong, O.A.; Wang, B.; Shi, Y. Effective asymmetric epoxidation of styrenes by chiral dioxirane. J. Org. Chem. 2006, 71, 1715–1717. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.X.; Goeddel, D.; Li, K.; Shi, Y. A practical synthesis of N-aryl-substituted oxazolidinone-containing ketone catalysts for asymmetric epoxidation. Tetrahedron 2006, 62, 8064–8068. [Google Scholar] [CrossRef]
- Shu, L.; Shi, Y. Asymmetric epoxidation of cis-β-methylstyrenes catalyzed by N-aryl substituted oxazolidinone-containing ketones. A beneficial substituent effect. Tetrahedron Lett. 2004, 45, 8115–8117. [Google Scholar] [CrossRef]
- Burke, C.P.; Shi, Y. Enantioselective epoxidation of nonconjugated cis-olefins by chiral dioxirane. Org. Lett. 2009, 11, 5150–5153. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Ling, J.; Chiu, P. Vinyl epoxides in organic synthesis. Chem. Rev. 2014, 114, 8037–8128. [Google Scholar] [CrossRef]
- Marshall, J.A. SN2’ Additions of organocopper reagents to vinyloxiranes. Chem. Rev. 1989, 89, 1503–1511. [Google Scholar] [CrossRef]
- Burke, C.P.; Shi, Y. Regio- and Enantioselective Epoxidation of Dienes by a Chiral Dioxirane: Synthesis of Optically Active Vinylcis-Epoxides. Angew. Chem. Int. Ed. 2006, 45, 4475–4478. [Google Scholar] [CrossRef]
- Burke, C.P.; Shi, Y. Enantioselective epoxidation of conjugated cis-enynes by chiral dioxirane. J. Org. Chem. 2007, 72, 4093–4097. [Google Scholar] [CrossRef]
- Shen, Y.M.; Wang, B.; Shi, Y. Enantioselective synthesis of 2-alkyl-2-aryl cyclopentanones by asymmetric epoxidation of tetrasubstituted cyclobutylidene olefins and epoxide rearrangement. Tetrahedron Lett. 2006, 47, 5455–5458. [Google Scholar] [CrossRef]
- Wang, B.; Wong, O.A.; Zhao, M.X.; Shi, Y. Asymmetric epoxidation of 1,1-disubstituted terminal olefins by chiral dioxirane via a planar-like transition state. J. Org. Chem. 2008, 73, 9539–9543. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.Y.; She, X.; Shi, Y. Highly enantioselective epoxidation of α,β-unsaturated esters by chiral dioxirane. J. Am. Chem. Soc. 2002, 124, 8792–8793. [Google Scholar] [CrossRef]
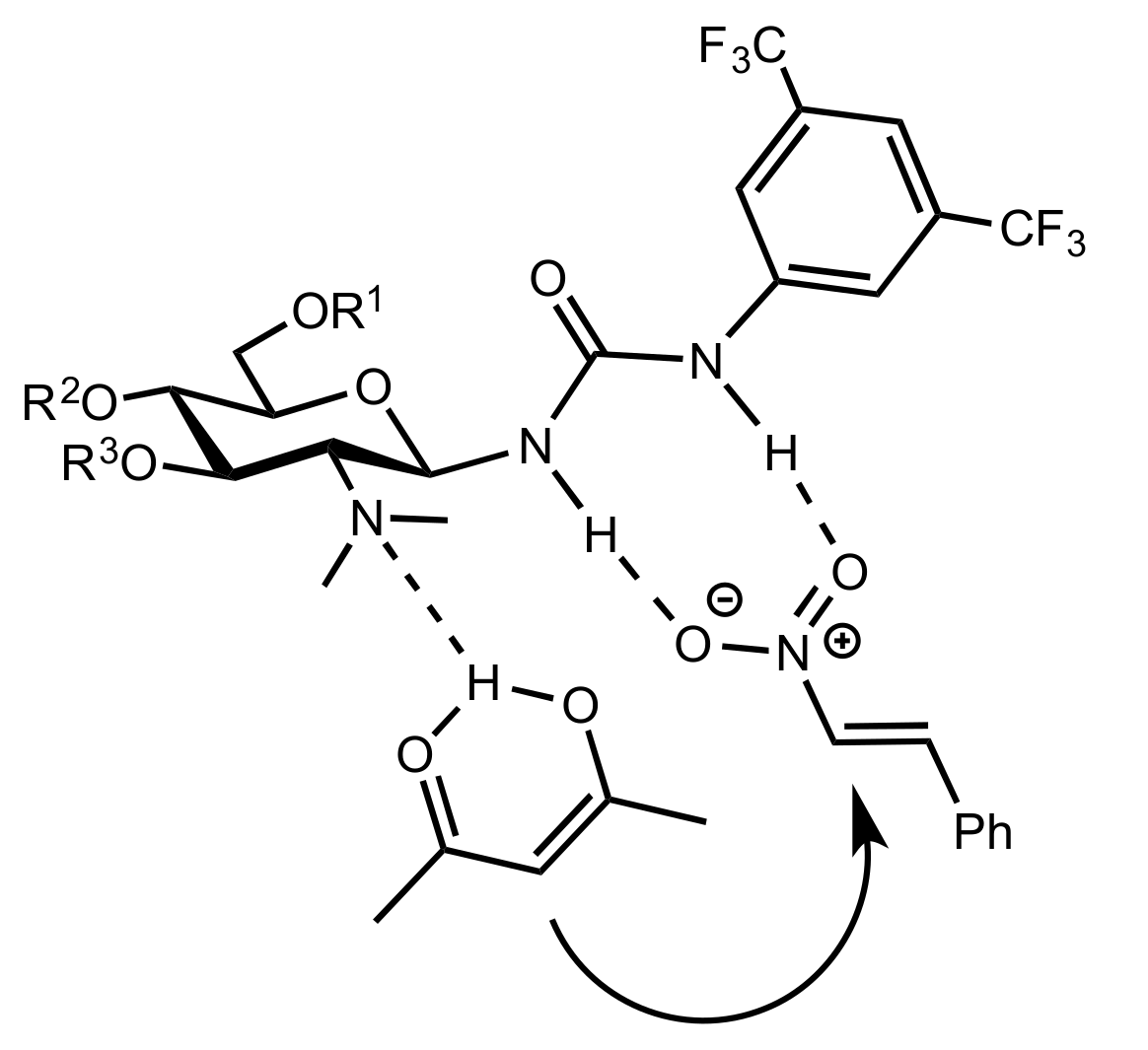
- Wang, B.; Wu, X.Y.; Wong, O.A.; Nettles, B.; Zhao, M.X.; Chen, D.; Shi, Y. A diacetate ketone-catalyzed asymmetric epoxidation of olefins. J. Org. Chem. 2009, 74, 3986–3989. [Google Scholar] [CrossRef] [Green Version]
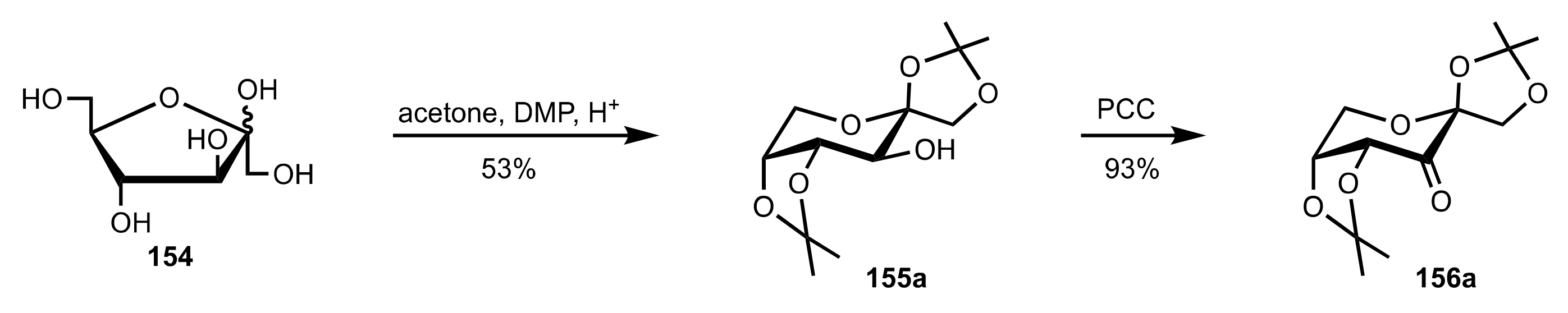
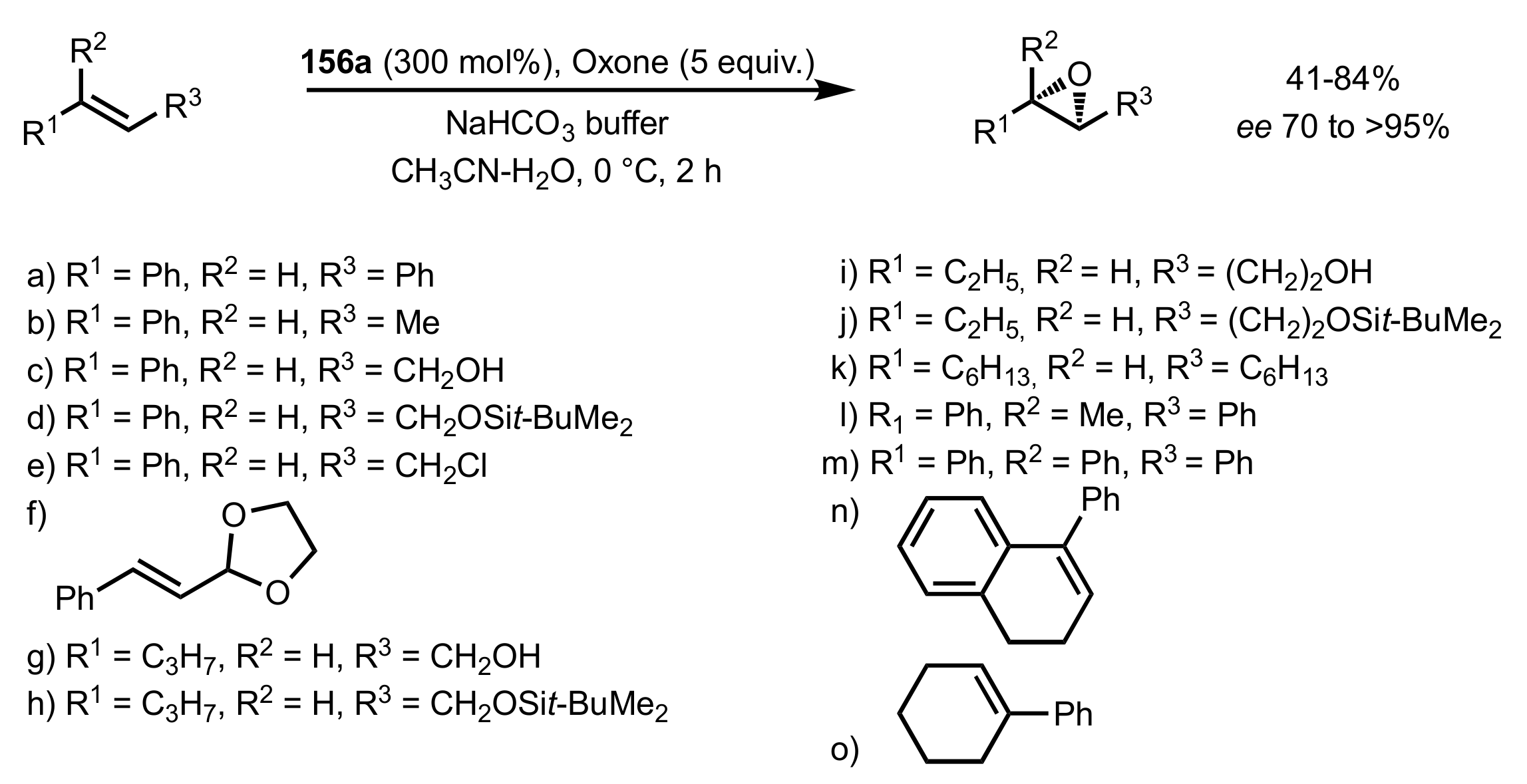
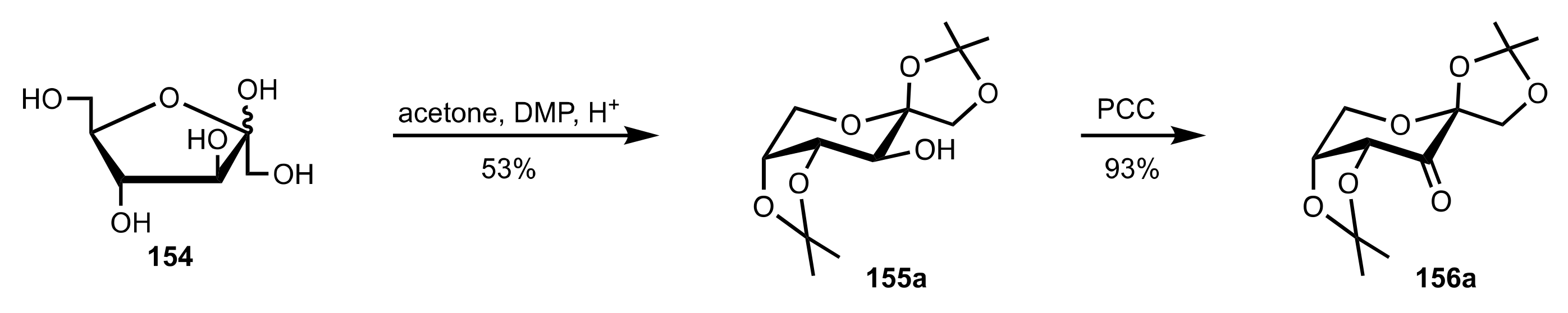
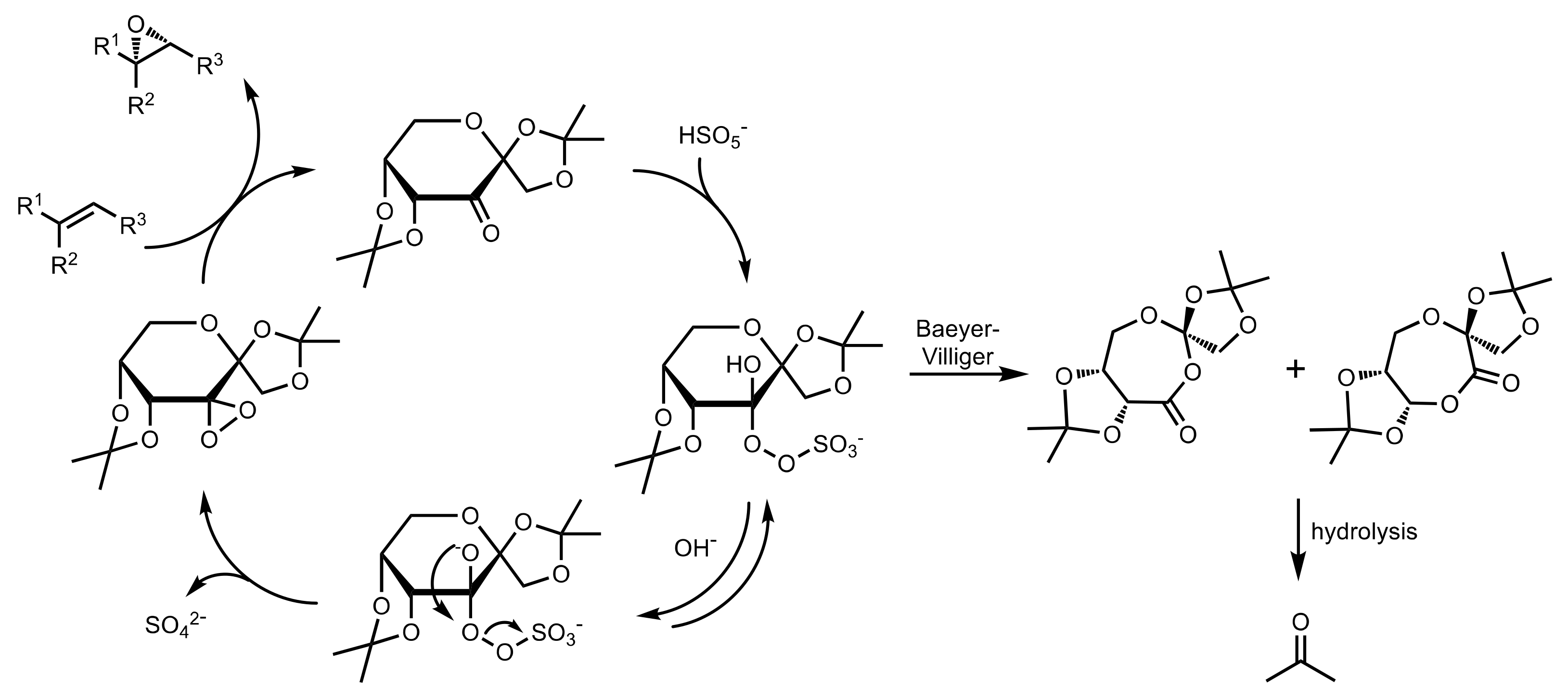
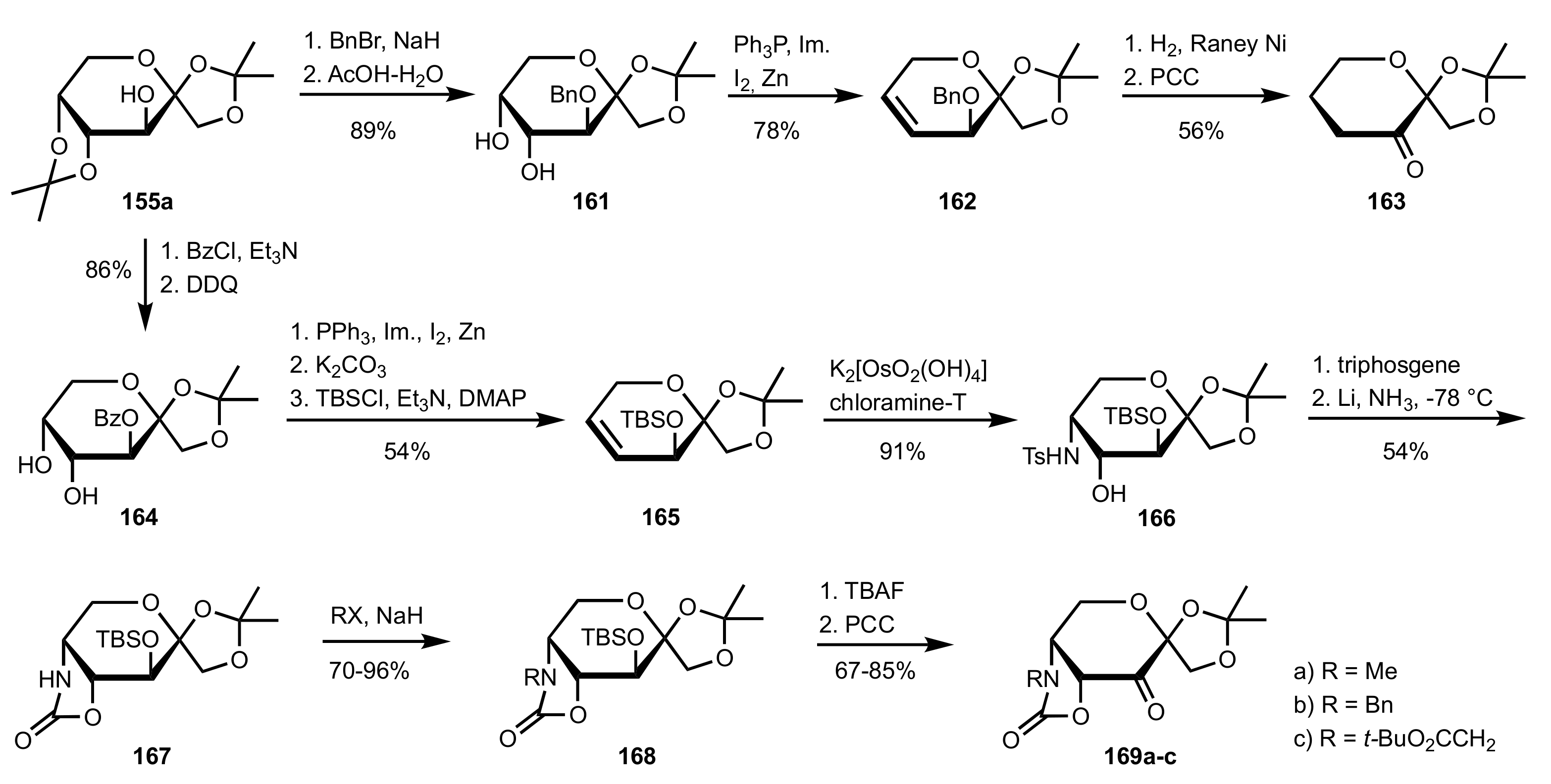
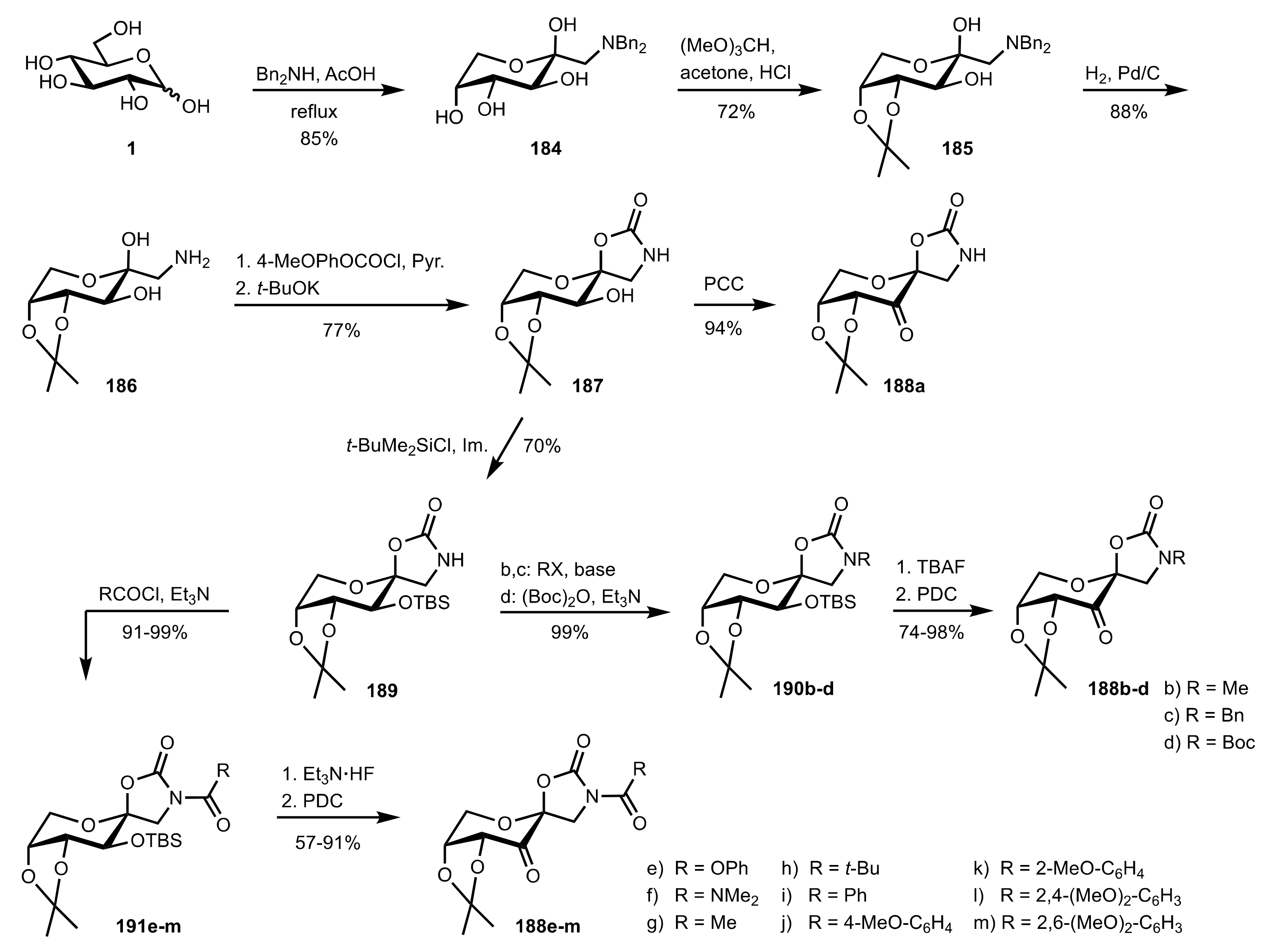
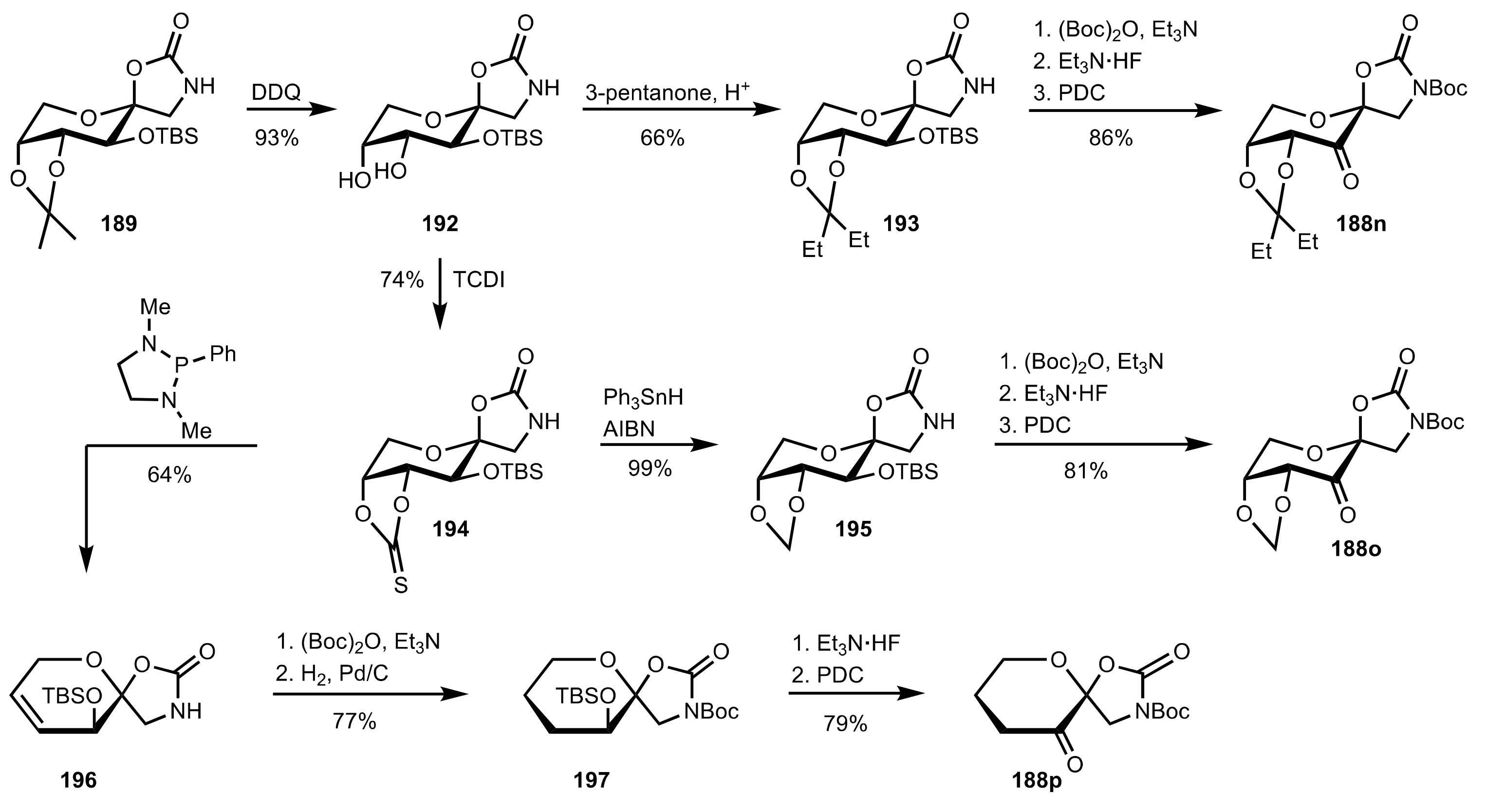
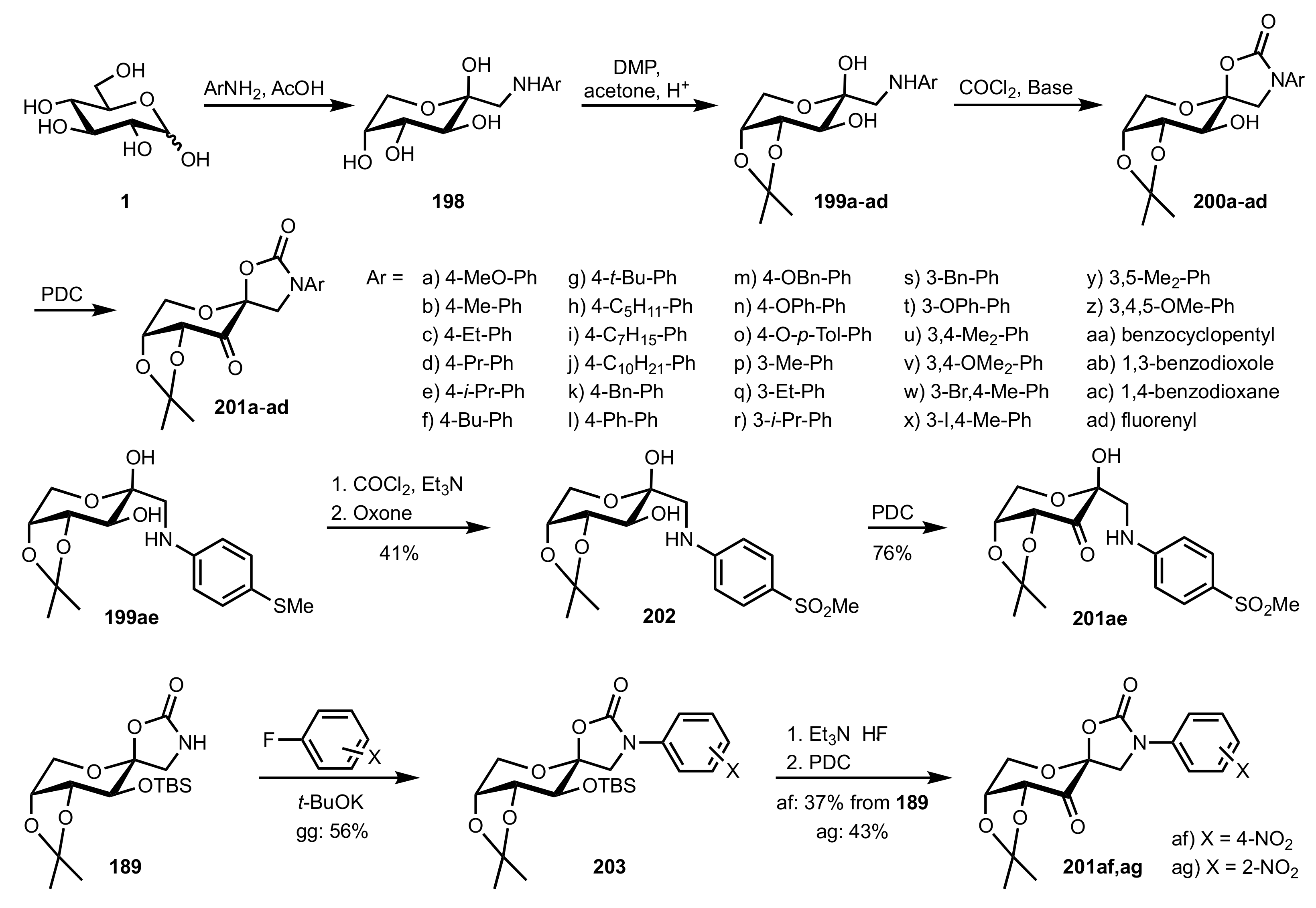
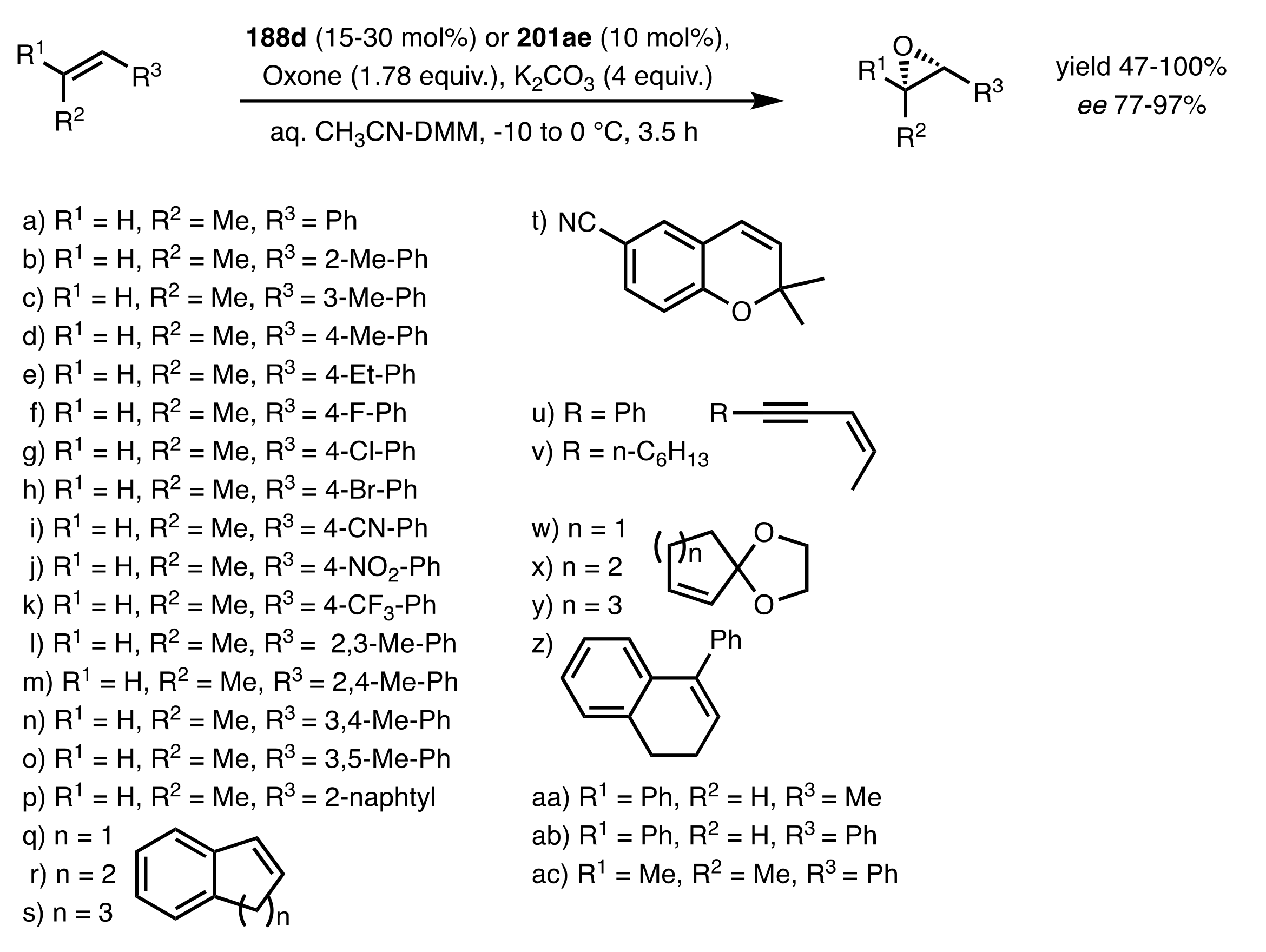
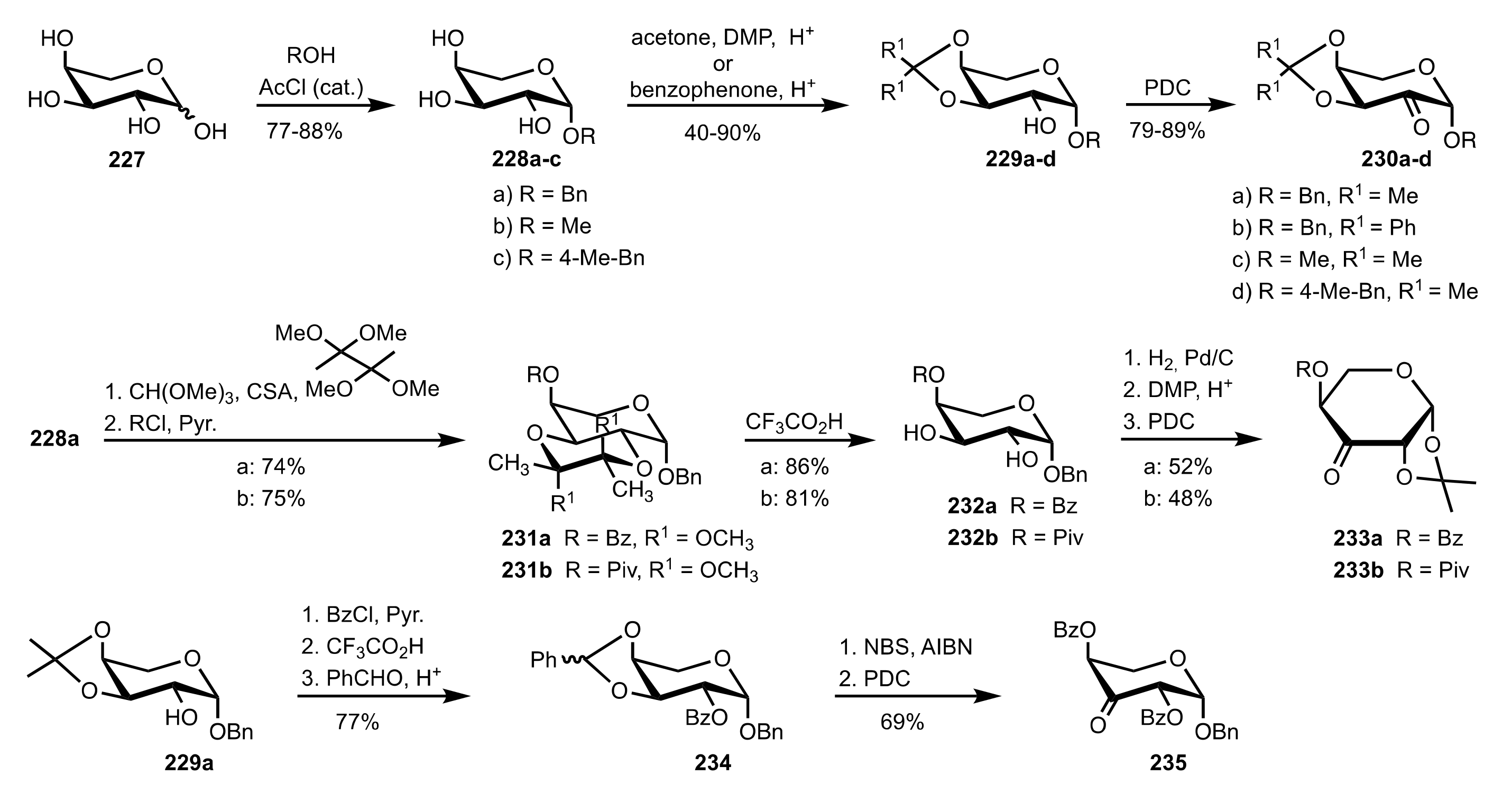
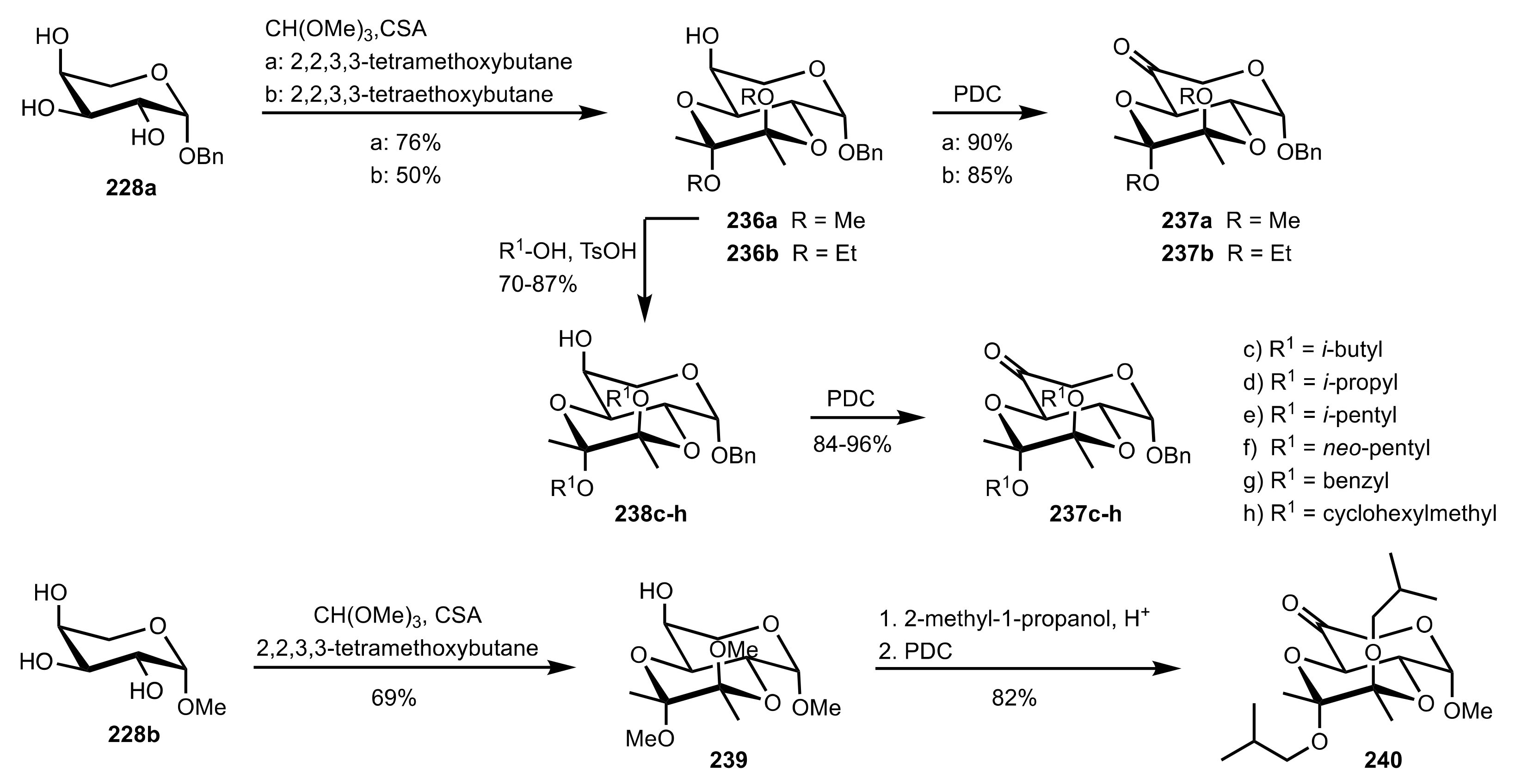
- Shing, T.K.M.; Leung, G.Y.C. Asymmetric epoxidation catalyzed by d-glucose-derived uloses. Tetrahedron 2002, 58, 7545–7552. [Google Scholar] [CrossRef]
- Shing, T.K.M.; Leung, G.Y.C.; Yeung, K.W. Catalytic enantioselective epoxidation with arabinose-derived uloses containing tunable steric sensors. Tetrahedron Lett. 2003, 44, 9225–9228. [Google Scholar] [CrossRef]
- Shing, T.K.M.; Leung, Y.C.; Yeung, K.W. Catalytic asymmetric epoxidation of alkenes with arabinose-derived uloses. Tetrahedron 2003, 59, 2159–2168. [Google Scholar] [CrossRef]
- Shing, T.K.M.; Leung, G.Y.C.; Luk, T. Arabinose-derived ketones as catalysts for asymmetric epoxidation of alkenes. J. Org. Chem. 2005, 70, 7279–7289. [Google Scholar] [CrossRef]
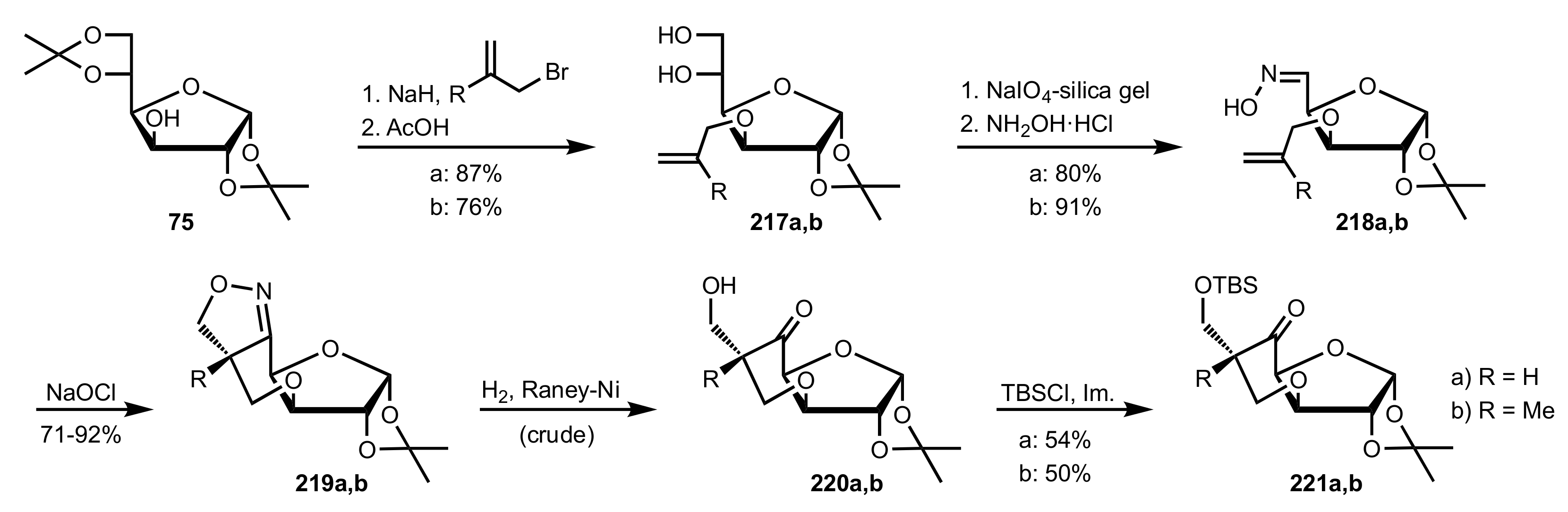
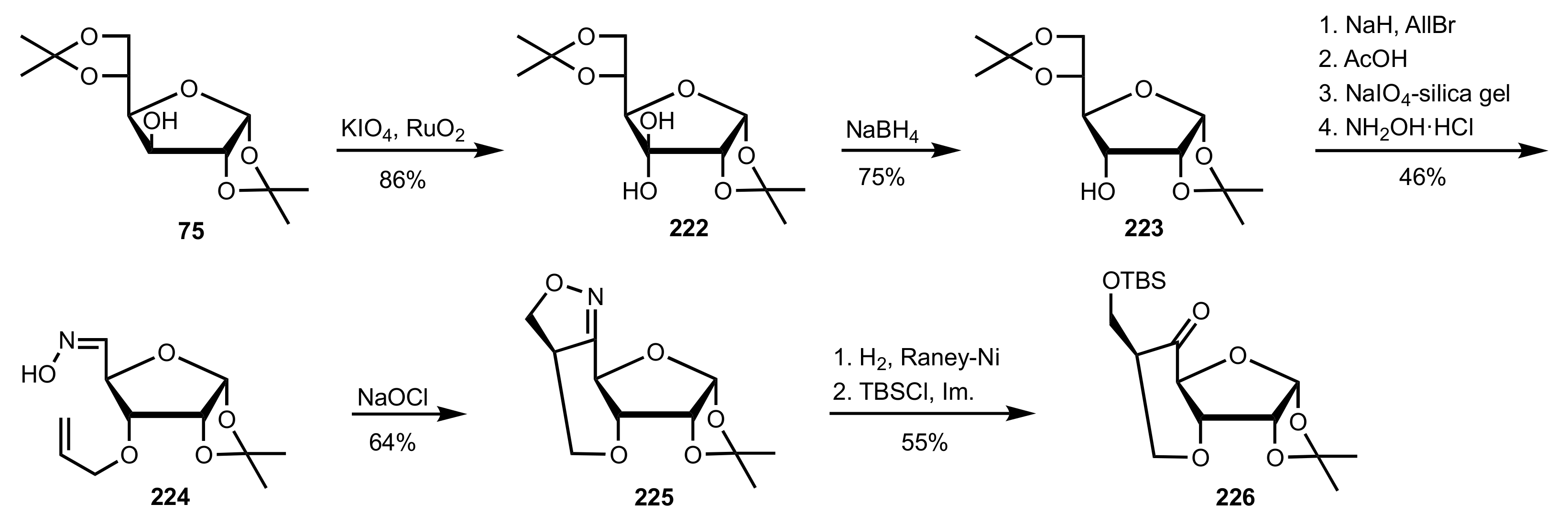
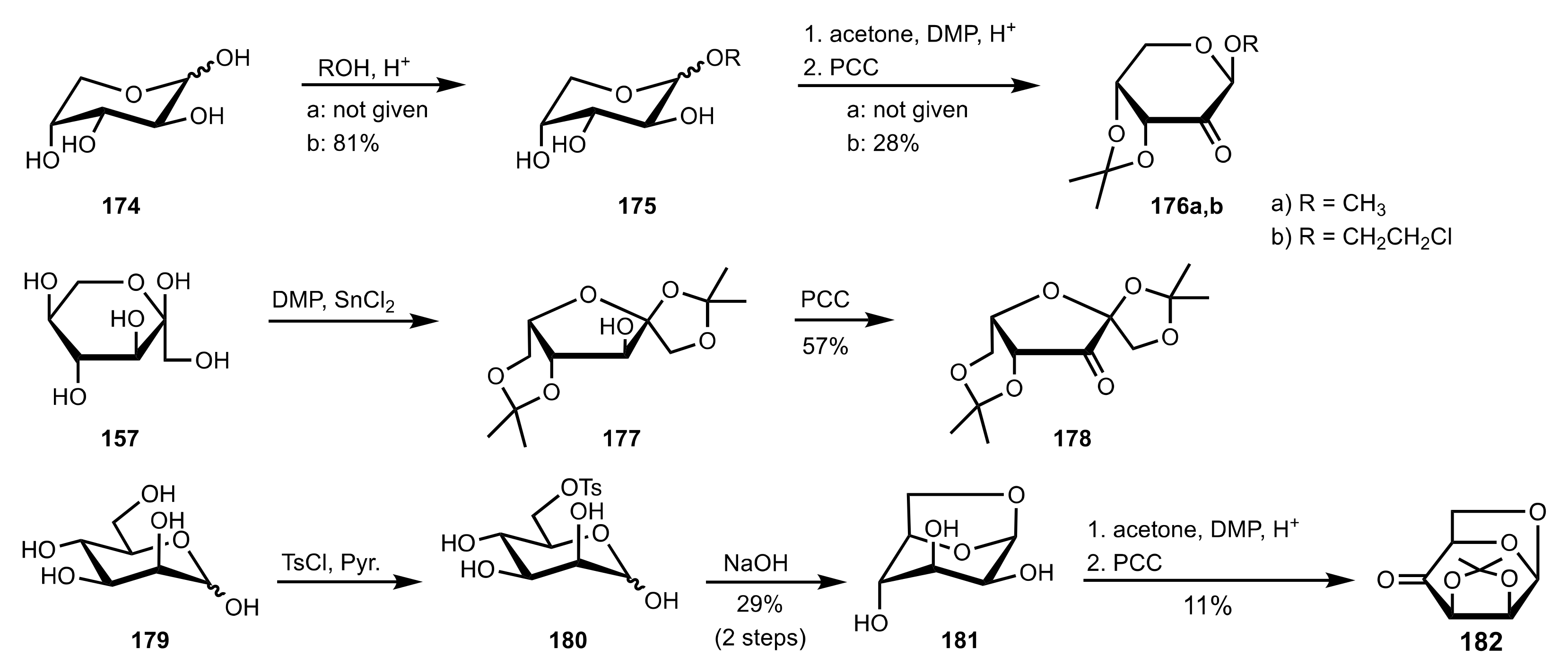
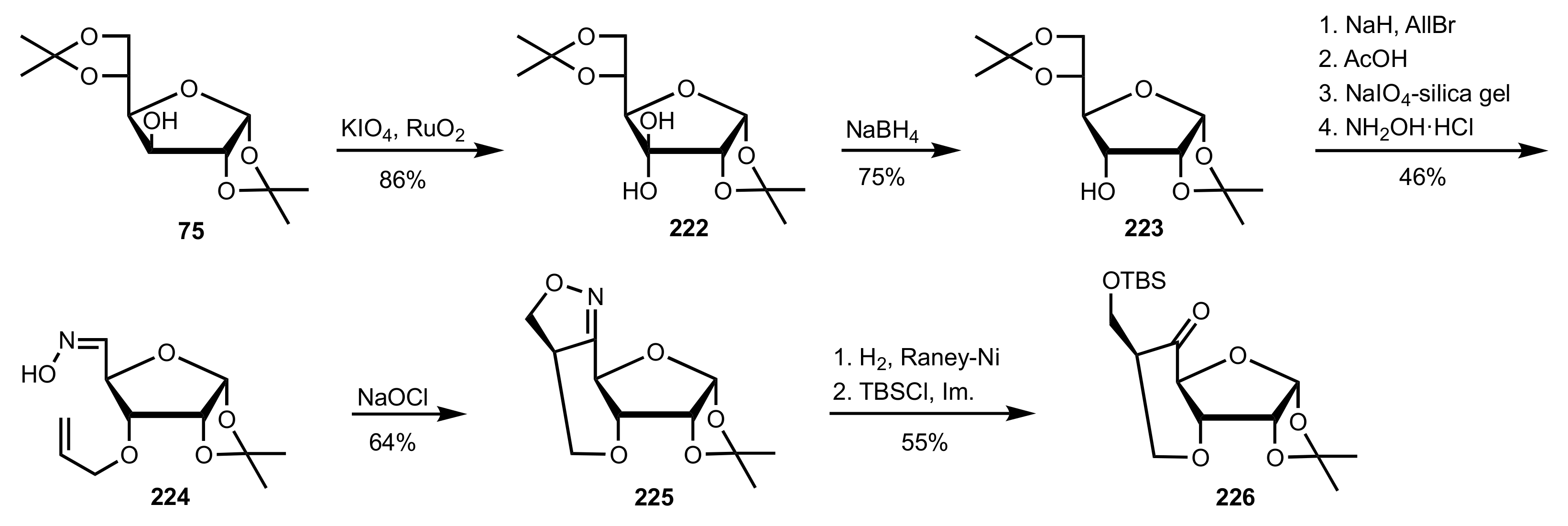
- Shing, T.K.M.; Fung, W.-C.; Wong, C.-H. Ring-selective Syntheses of Homochiral Oxepanes and Tetrahydropyrans from Carbohydrates via Intramolecular Nitrone or Nitrile Oxide Cycloadditions. J. Chem. Soc. Chem. Commun. 1994, 1994, 449–450. [Google Scholar] [CrossRef]
- Zhong, Y.-L.; Shing, T.K.M. Efficient and Facile Glycol Cleavage Oxidation Using Improved Silica Gel-Supported Sodium Metaperiodate. J. Org. Chem. 1997, 62, 2622–2624. [Google Scholar] [CrossRef]
- Baker, D.C.; Horton, D.; Tindall, C.G. Large-scale preparation of d-allose: Observations on the stereoselectivity of the reduction of 1,2:5,6-di-O-isopropylidene-α-d-ribo-hexofuranos-3-ulose hydrate. Carbohydr. Res. 1972, 24, 192–197. [Google Scholar] [CrossRef]
- Shing, T.K.M.; Luk, T.; Lee, C.M. Asymmetric epoxidation of cis-alkenes with arabinose-derived ketones: Enantioselective synthesis of the side chain of Taxol. Tetrahedron 2006, 62, 6621–6629. [Google Scholar] [CrossRef]
- Shing, T.K.M.; Luk, T. Catalytic asymmetric epoxidation of alkenes with arabinose-derived ketones containing a cyclohexane-1,2-diacetal. Tetrahedron Asymmetry 2009, 20, 883–886. [Google Scholar] [CrossRef]
- Vega-Pérez, J.M.; Candela, J.I.; Blanco, E.; Iglesias-Guerra, F. Stereoselective synthesis of epoxyalkyl glycoside precursors of glycosyl glycerol analogues from alkenyl glycosides of N-acetyl-d-glucosamine derivatives. Tetrahedron Asymmetry 2002, 13, 2471–2483. [Google Scholar] [CrossRef]
- Vega-Pérez, J.M.; Vega, M.; Blanco, E.; Iglesias-Guerra, F. Stereoselective synthesis of oxiranes using oxazolidines derived from 2-amino-2-deoxy-d-allose as chiral auxiliaries. Tetrahedron Asymmetry 2001, 12, 3189–3203. [Google Scholar] [CrossRef]
- Vega-Pérez, J.M.; Vega, M.; Blanco, E.; Iglesias-Guerra, F. Stereoselective epoxidation of alkenylidene acetals derived from carbohydrates with d-allo, d-altro, d-galacto, d-gluco and d-xylo configurations. Tetrahedron Asymmetry 2007, 18, 1850–1867. [Google Scholar] [CrossRef]
- Vega-Pérez, J.M.; Holm, M.V.; Martínez, M.L.; Blanco, E.; Iglesias-Guerra, F. Synthesis of New Chiral Ketones from d-Glucose Derivatives and Their Use in the Enantioselective Epoxidation of Arylalkenes. Eur. J. Org. Chem. 2009, 2009, 6009–6018. [Google Scholar] [CrossRef]
- Vega-Pérez, J.M.; Vega-Holm, M.; Periñán, I.; Palo-Nieto, C.; Iglesias-Guerra, F. Synthesis of new carbohydrate-derived ketones as organocatalysts in the enantioselective epoxidation of arylalkenes. Part 2: Chiral ketones from sugars. Tetrahedron 2011, 67, 364–372. [Google Scholar] [CrossRef]
- Vega-Pérez, J.M.; Periñán, I.; Palo-Nieto, C.; Vega-Holm, M.; Iglesias-Guerra, F. Alkenyl β-d-galactopyranoside derivatives as efficient chiral templates in stereoselective cyclopropanation and epoxidation reactions. Tetrahedron Asymmetry 2010, 21, 81–95. [Google Scholar] [CrossRef]
- Vega-Pérez, J.M.; Periñán, I.; Vega-Holm, M.; Palo-Nieto, C.; Iglesias-Guerra, F. New mannose-derived ketones as organocatalysts for enantioselective dioxirane-mediated epoxidation of arylalkenes. Part 3: Chiral ketones from sugars. Tetrahedron 2011, 67, 7057–7065. [Google Scholar] [CrossRef]
- Yang, D.; Yip, Y.; Tang, M.; Road, P.; Kong, H.; August, R.V. A C2 Symmetric Chiral Ketone for Catalytic Asymmetric Epoxidation of Unfunctionalized Olefins. J. Am. Chem. Soc. 1996, 118, 491–492. [Google Scholar] [CrossRef]
- Adam, W.; Zhao, C.G. Synthesis of optically active C2-symmetric ketones for the asymmetric epoxidation of prochiral olefins by dioxiranes generated in situ with Caroate as a peroxide source. Tetrahedron Asymmetry 1997, 8, 3995–3998. [Google Scholar] [CrossRef]
- Moberg, C. C3 Symmetry in Asymmetric Catalysis and Chiral Recognition. Angew. Chem. Int. Ed. 1998, 37, 248–268. [Google Scholar] [CrossRef]
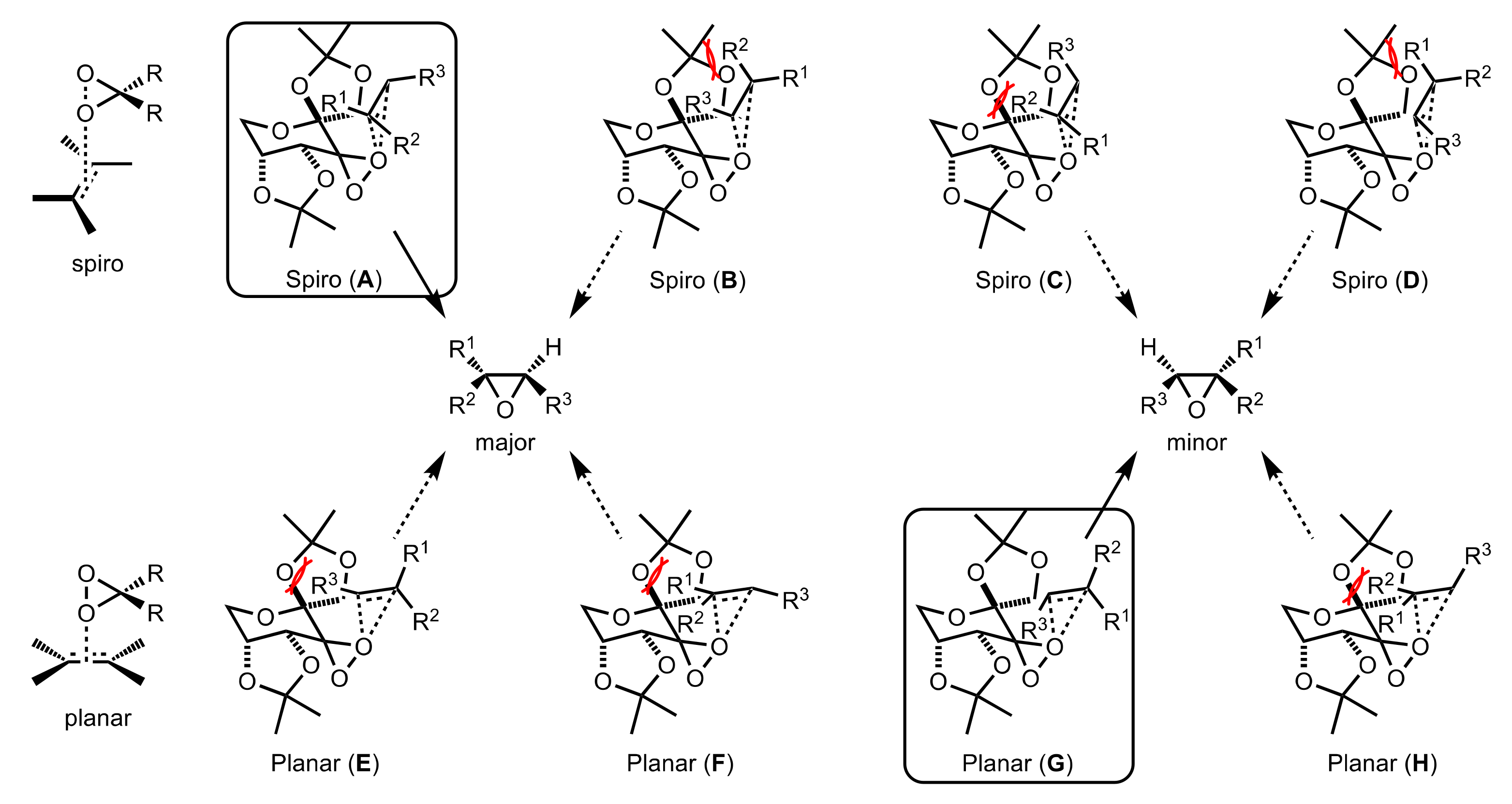
- Baumstark, A.L.; McCloskey, C.J. Epoxidation of alkenes by dimethyldioxirane: Evidence for a spiro transition state. Tetrahedron Lett. 1987, 28, 3311–3314. [Google Scholar] [CrossRef]
- Lee, E.; Mcardle, P.; Browne, P. N.m.r. (1H and 13C) studies of some cis- and trans fused 4,6-O-benzylidene-hexopyranoside derivatives. Carbohydr. Res. 1990, 197, 262–269. [Google Scholar] [CrossRef]
- Jalsa, N.K.; Singh, G. A unique approach to the synthesis of a dengue vaccine and the novel tetrasaccharide that results. Tetrahedron Asymmetry 2009, 20, 867–874. [Google Scholar] [CrossRef]
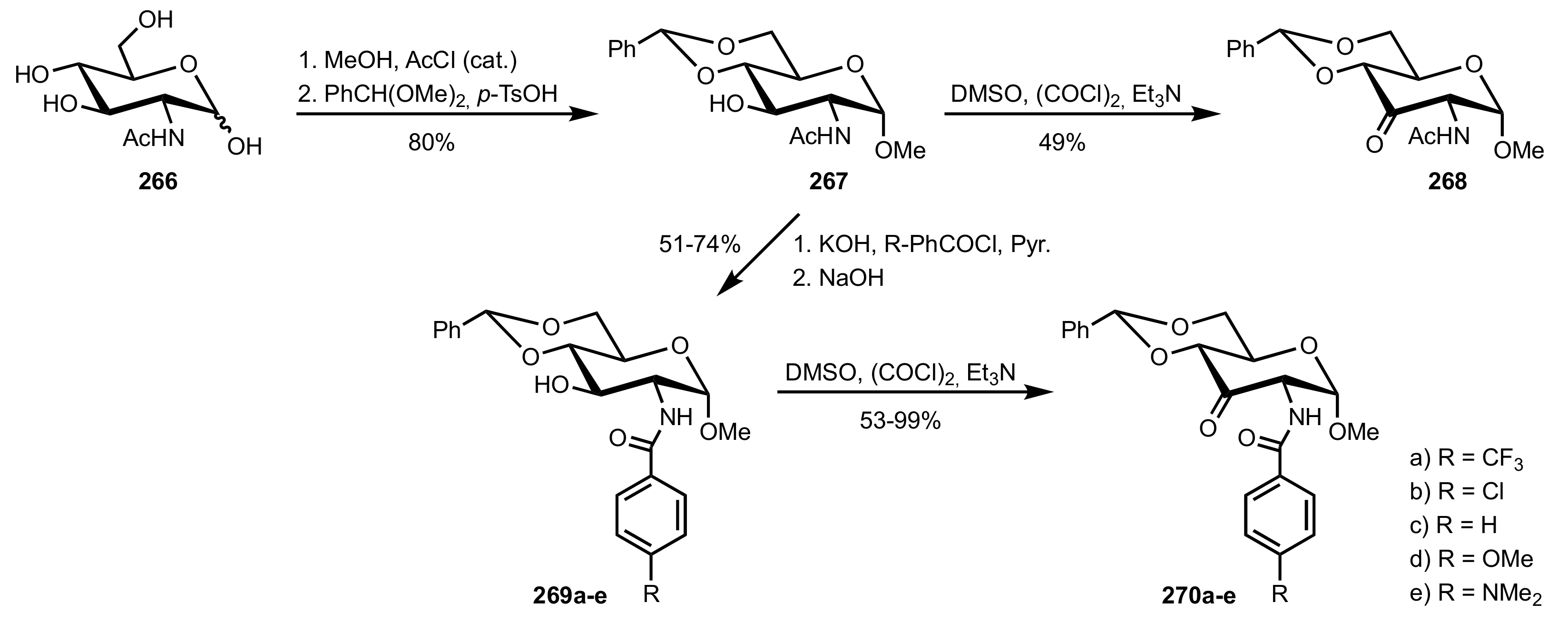
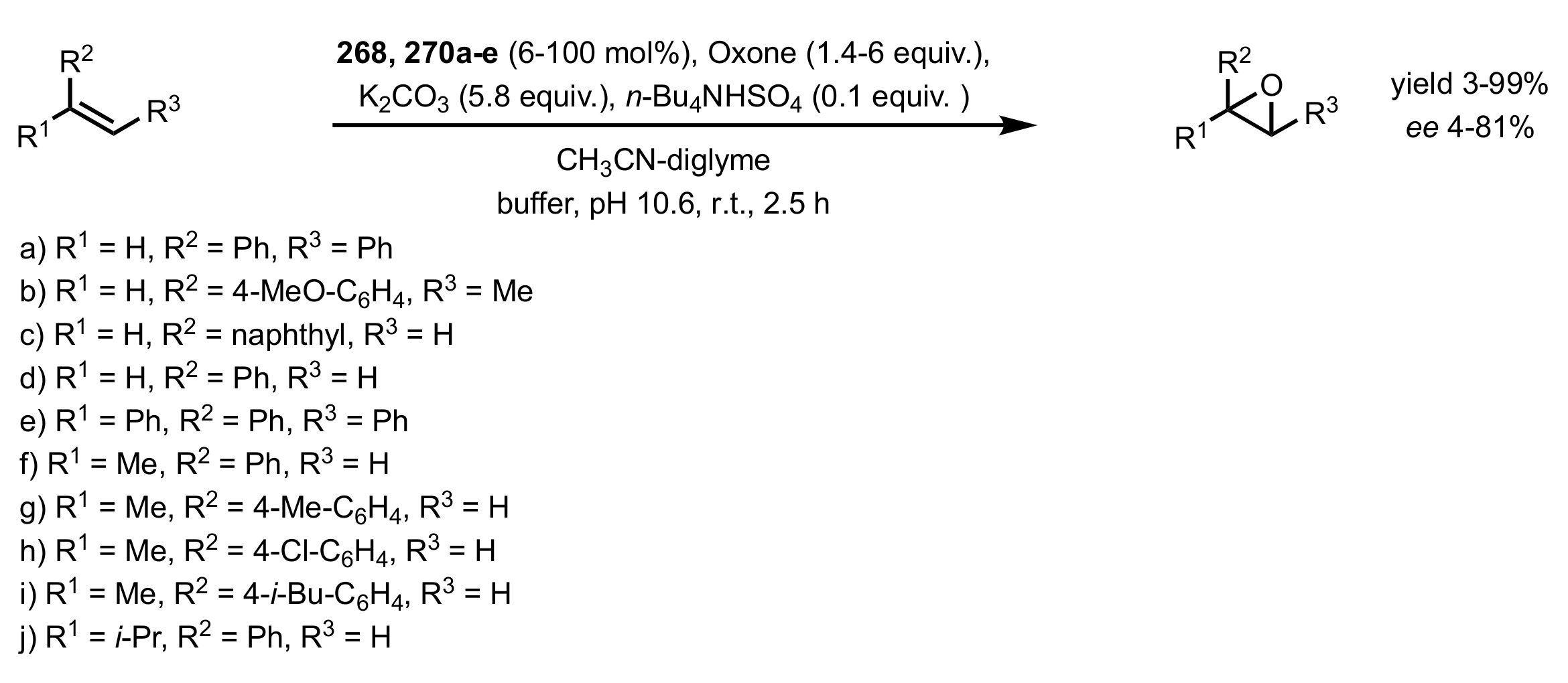
- Boutureira, O.; McGouran, J.F.; Stafford, R.L.; Emmerson, D.P.G.; Davis, B.G. Accessible sugars as asymmetric olefin epoxidation organocatalysts: Glucosaminide ketones in the synthesis of terminal epoxides. Org. Biomol. Chem. 2009, 7, 4285–4288. [Google Scholar] [CrossRef]
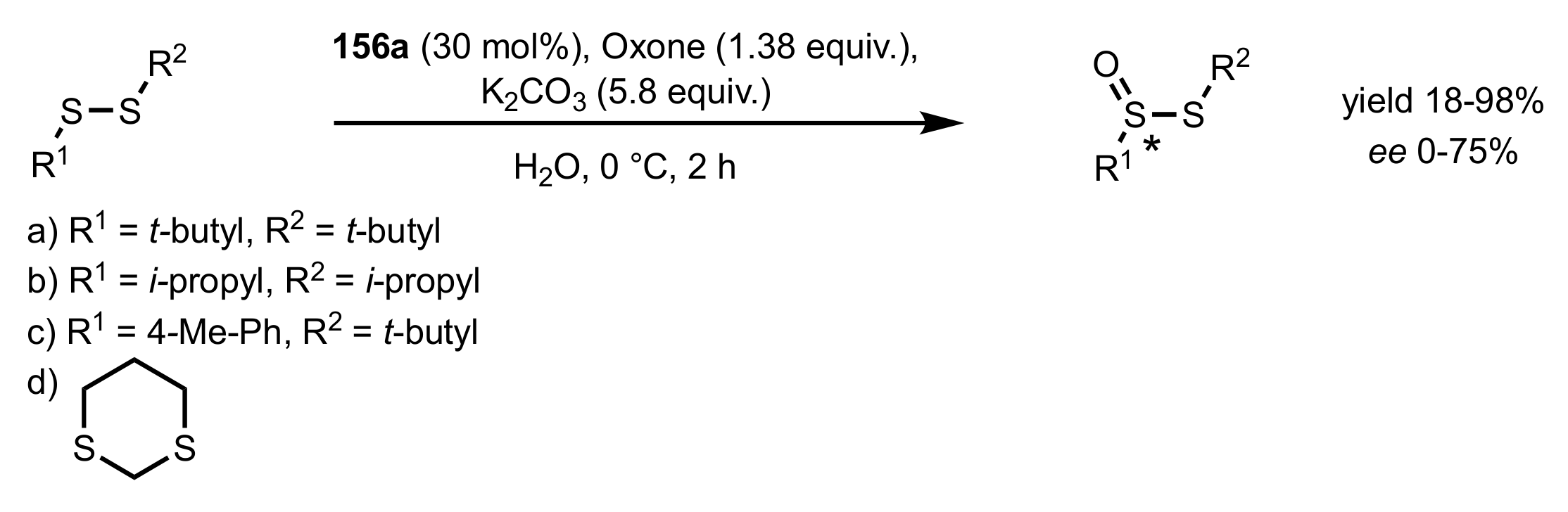
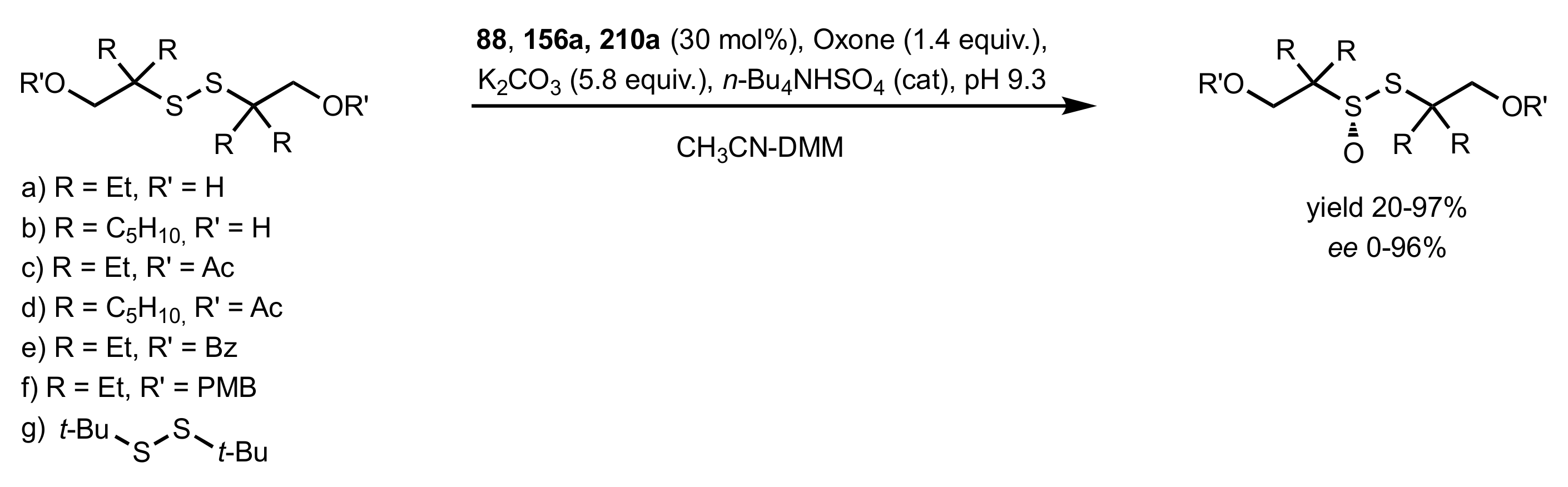
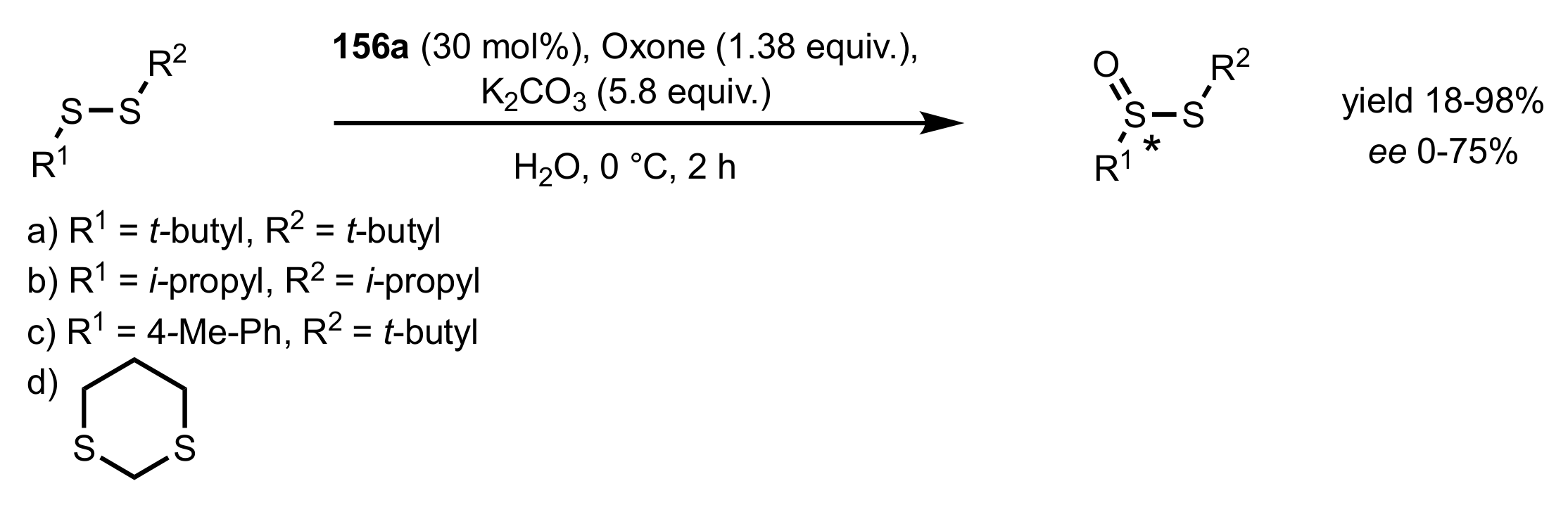
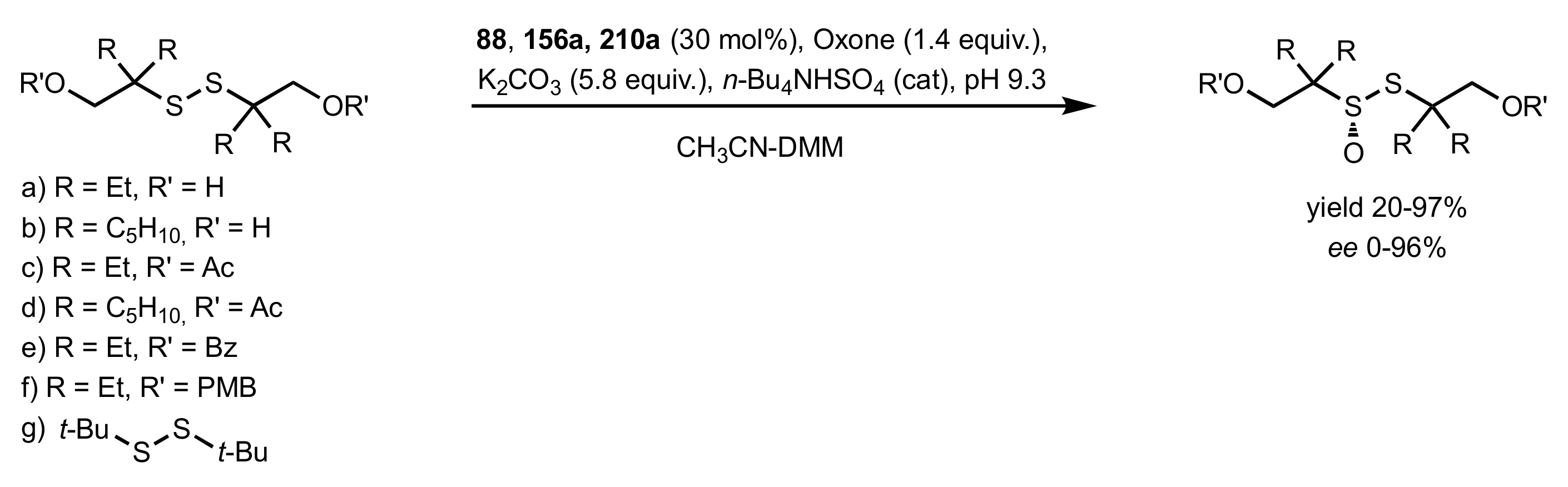
- Colonna, S.; Pironti, V.; Drabowicz, J.; Brebion, F.; Fensterbank, L.; Malacria, M. Enantioselective Synthesis of Thiosulfinates and of Acyclic Alkylidenemethylene Sulfide Sulfoxides. Eur. J. Org. Chem. 2005, 2005, 1727–1730. [Google Scholar] [CrossRef]
- Khiar, N.; Mallouk, S.; Valdivia, V.; Bougrin, K.; Soufiaoui, M.; Fernández, I. Enantioselective organocatalytic oxidation of functionalized sterically hindered disulfides. Org. Lett. 2007, 9, 1255–1258. [Google Scholar] [CrossRef]
- Tsutsui, A.; Takeda, H.; Kimura, M.; Fujimoto, T.; Machinami, T. Novel enantiocontrol system with aminoacyl derivatives of glucoside as enamine-based organocatalysts for aldol reaction in aqueous media. Tetrahedron Lett. 2007, 48, 5213–5217. [Google Scholar] [CrossRef]
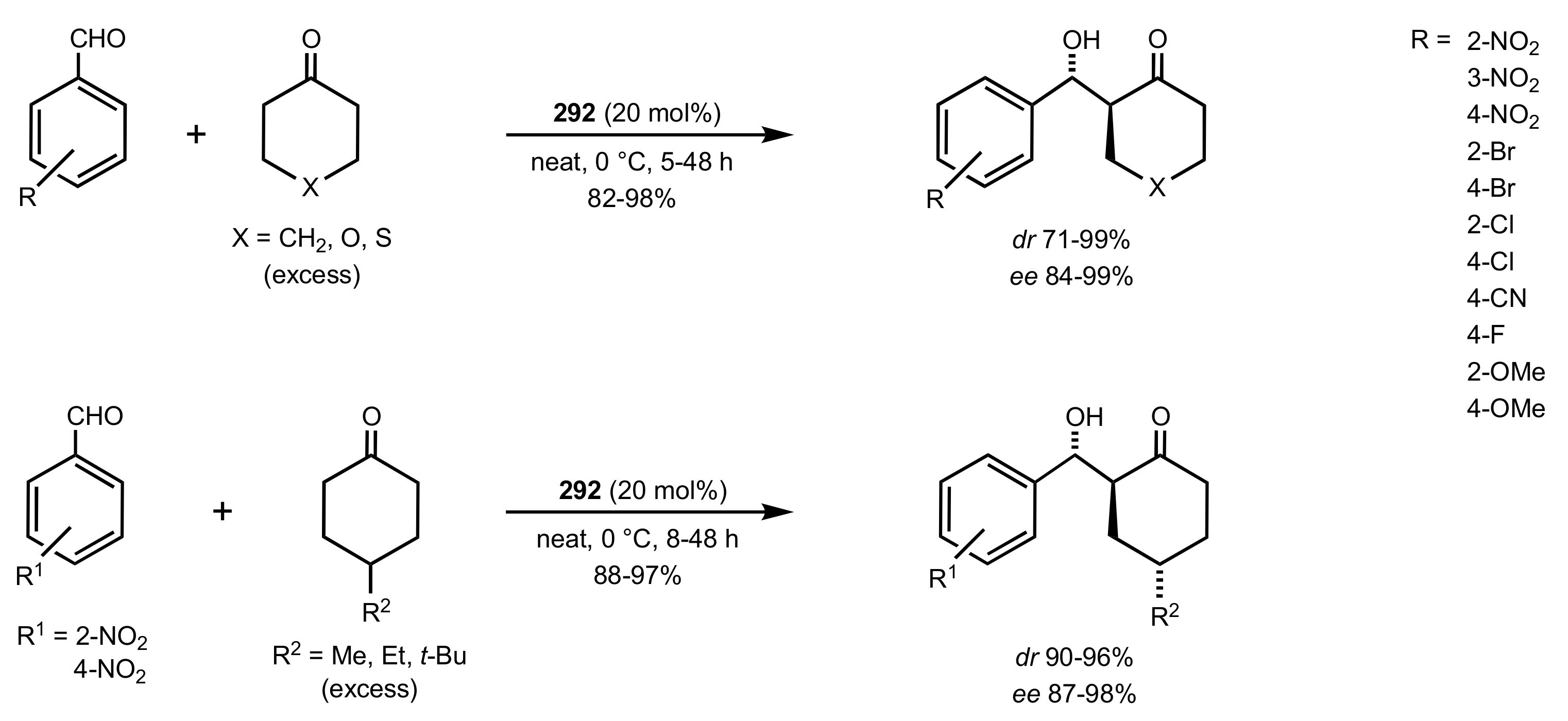
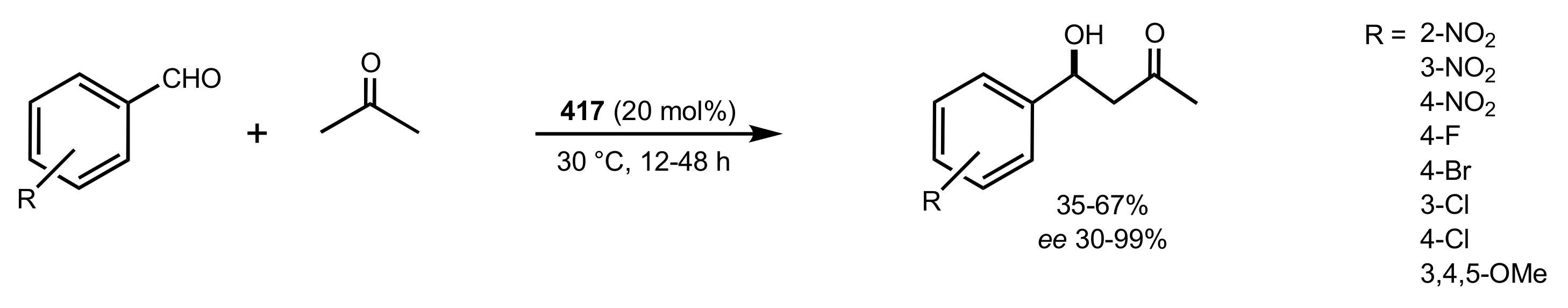
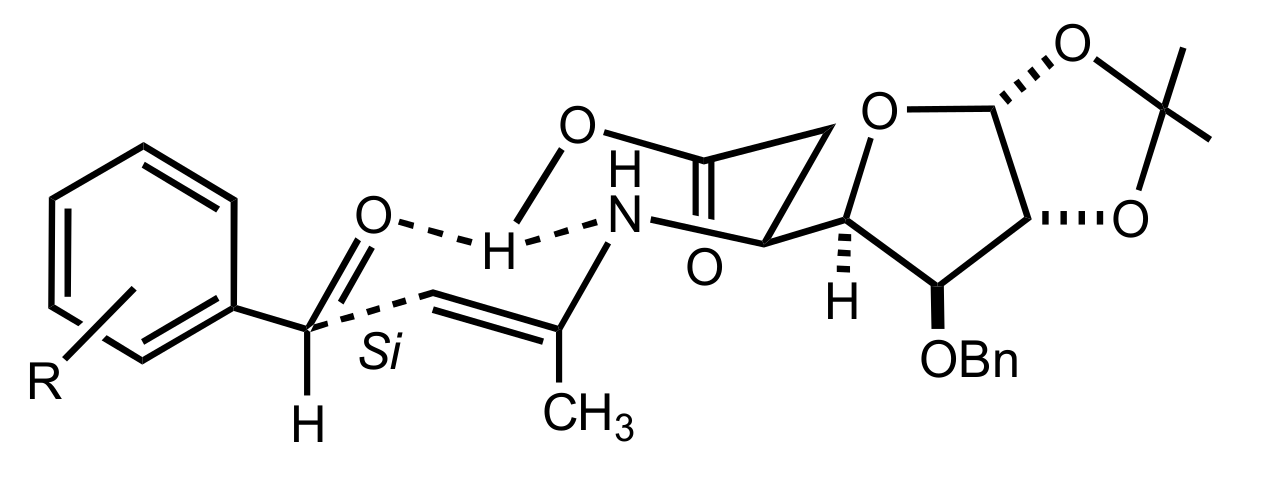
- Miura, D.; Fujimoto, T.; Tsutsui, A.; Machinami, T. Stereoselective Synthesis of Ketoses by Aldol Reaction Using Water-Compatible Prolinamide Catalysts in Aqueous Media. Synlett 2013, 24, 1501–1504. [Google Scholar] [CrossRef]
- Miura, D.; Machinami, T. Stereoselective Aldol Reaction in Aqueous Solution Using Prolinamido-Glycosides as Water-Compatible Organocatalyts. Mod. Res. Catal. 2015, 4, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, J.; Peddinti, R.K. Highly efficient and solvent-free direct aldol reaction catalysed by glucosamine-derived prolinamide. Tetrahedron Asymmetry 2010, 21, 1906–1909. [Google Scholar] [CrossRef]
- Agarwal, J.; Peddinti, R.K. Asymmetric Michael addition catalysed by sugar-based prolinamides in solvent-free conditions. Tetrahedron Lett. 2011, 52, 117–121. [Google Scholar] [CrossRef]
- Agarwal, J.; Peddinti, R.K. Synthesis and Characterization of Monosaccharide Derivatives and Application of Sugar-Based Prolinamides in Asymmetric Synthesis. Eur. J. Org. Chem. 2012, 2012, 6390–6406. [Google Scholar] [CrossRef]
- Jacquinet, J.-C.; Sinaÿ, P. Synthèse du methyl-2-acétamido-2-désoxy-β-d-glucofuranoside et de dérivés. Carbohydr. Res. 1974, 32, 101–114. [Google Scholar] [CrossRef]
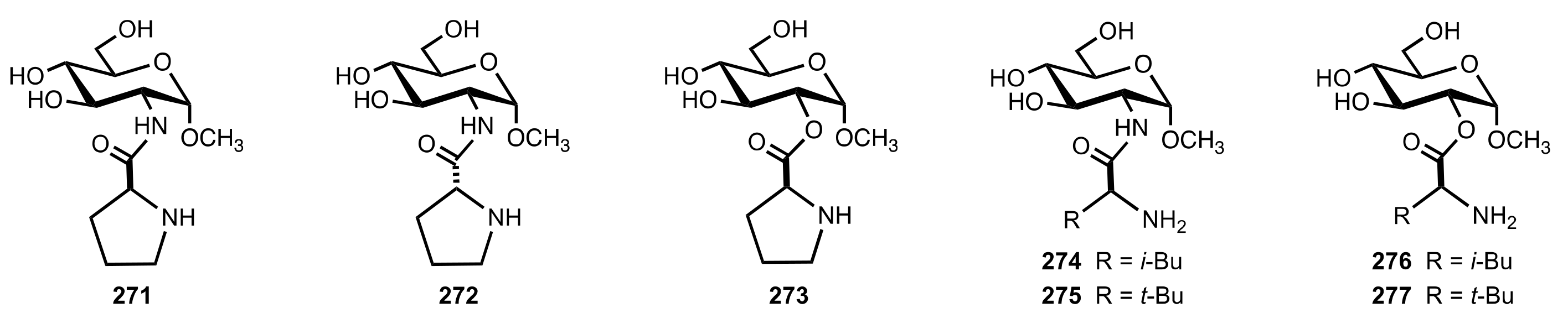
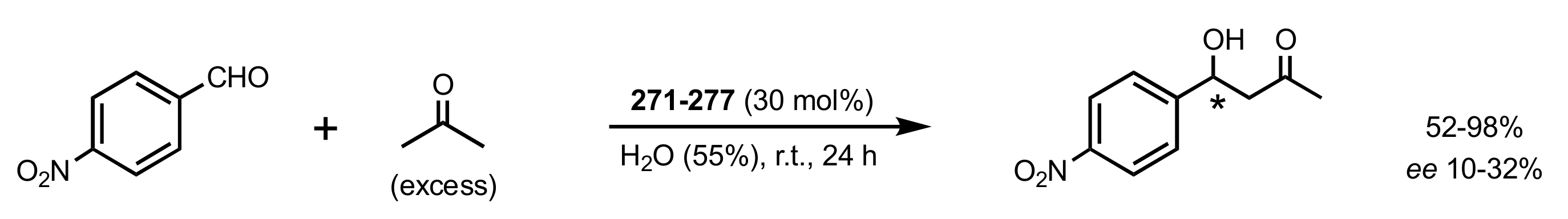
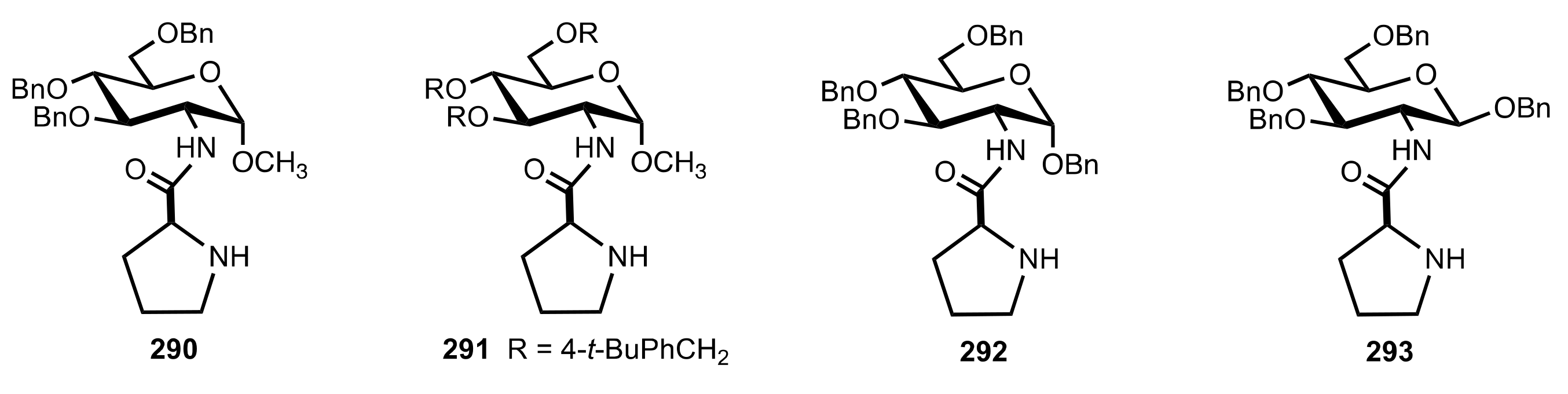
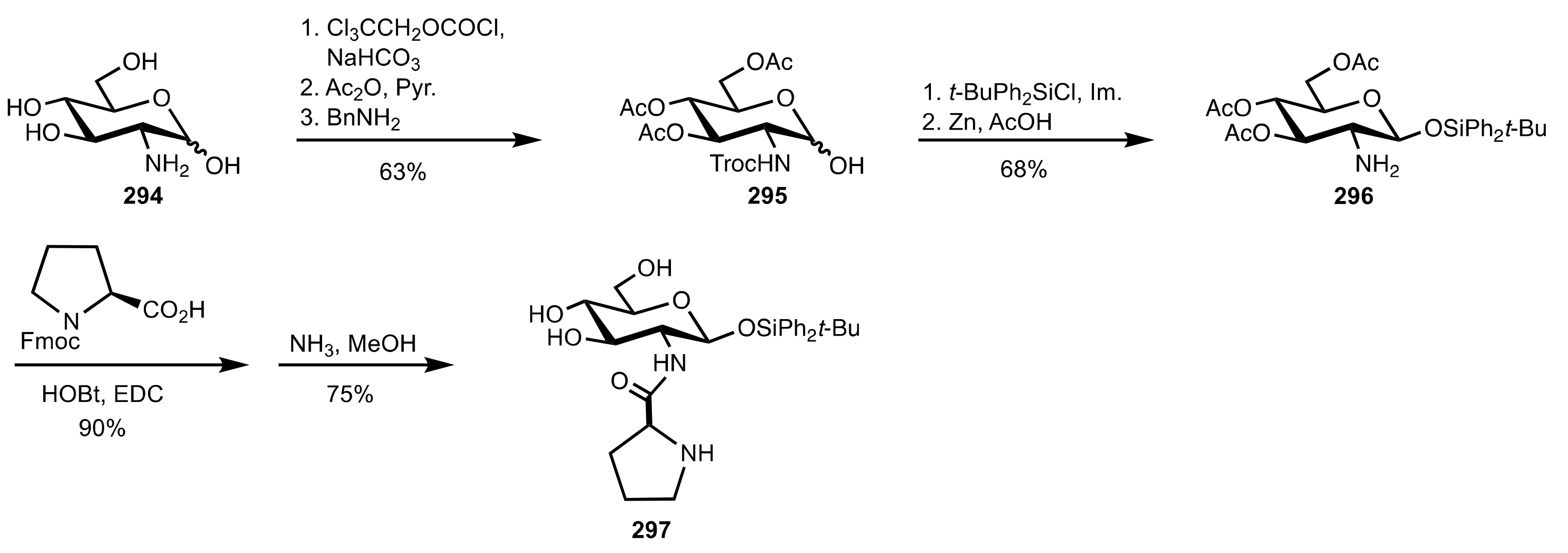
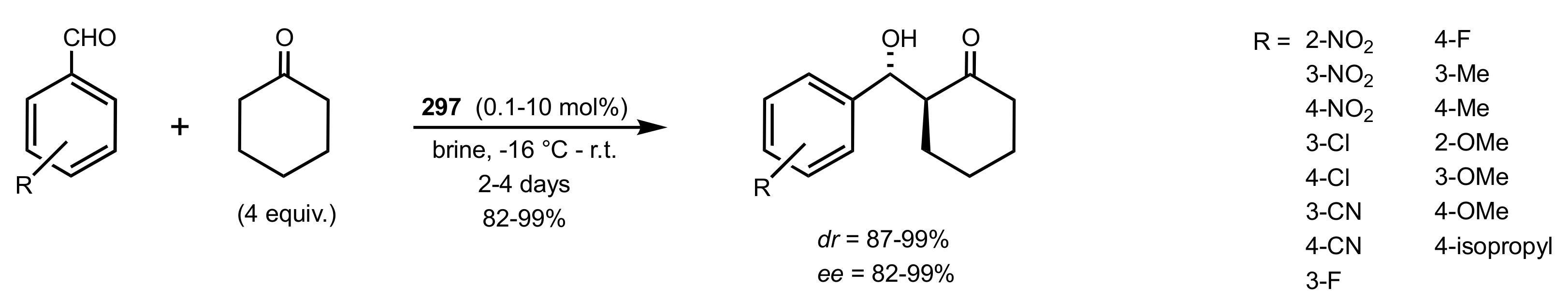
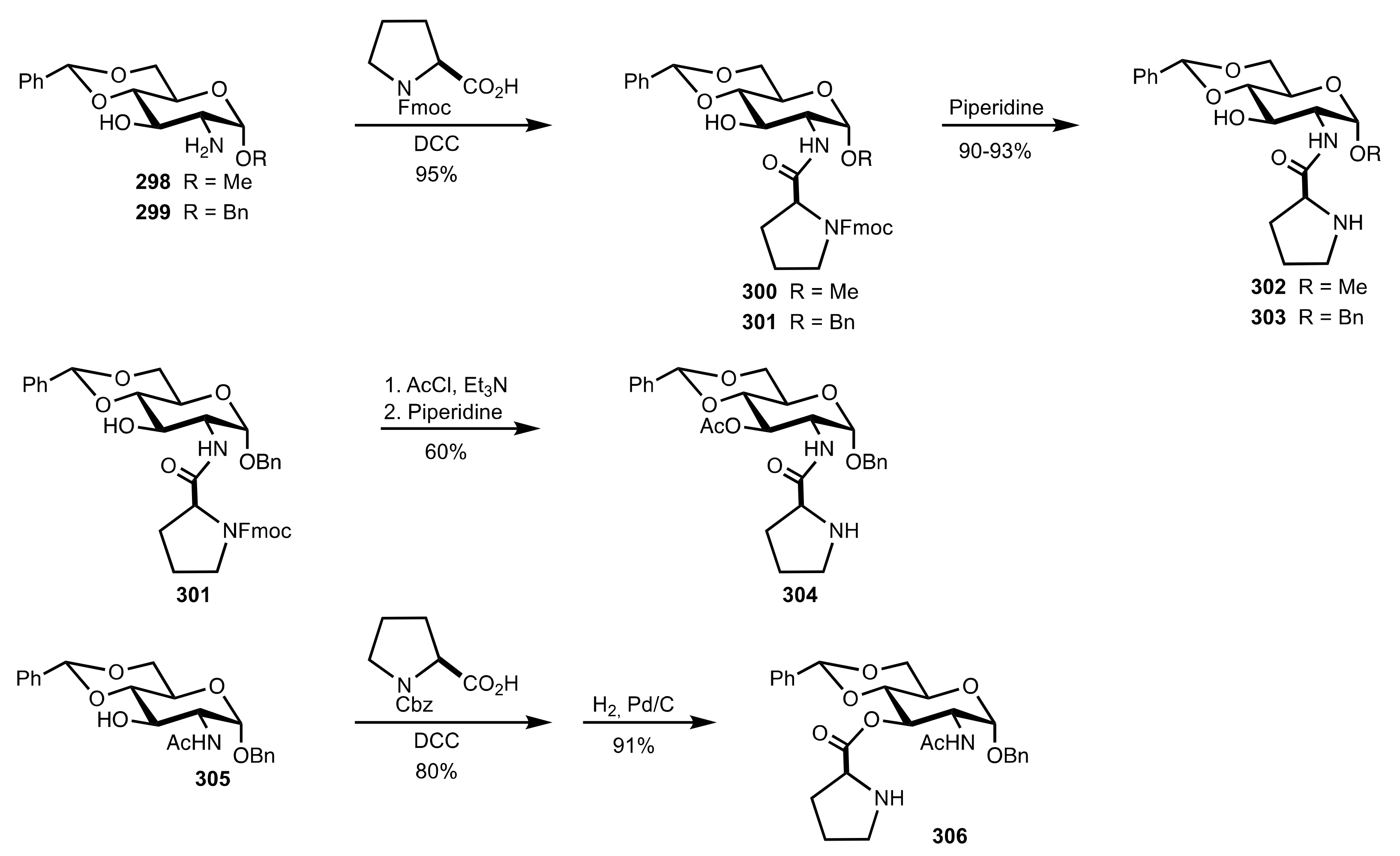
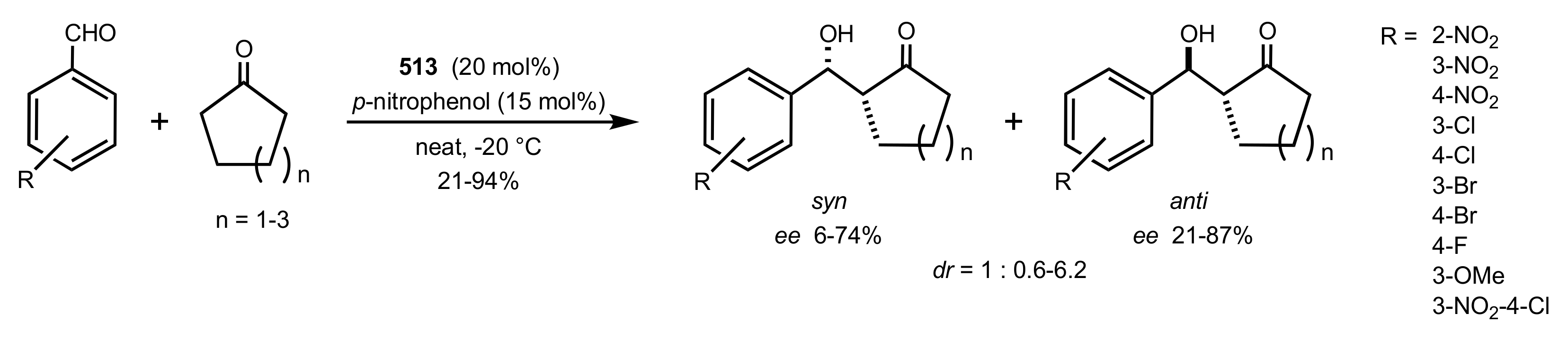
- Pedatella, S.; De Nisco, M.; Mastroianni, D.; Naviglio, D.; Nucci, A.; Caputo, R. Diastereo- and Enantioselective Direct Aldol Reactions in Aqueous Medium: A New Highly Efficient Proline-Sugar Chimeric Catalyst. Adv. Synth. Catal. 2011, 353, 1443–1446. [Google Scholar] [CrossRef]
- De Nisco, M.; Pedatella, S.; Bektaş, S.; Caputo, R. d-Glucosamine in a chimeric prolinamide organocatalyst for direct asymmetric aldol addition. Carbohydr. Res. 2012, 356, 273–277. [Google Scholar] [CrossRef]
- Shen, C.; Shen, F.; Zhou, G.; Xia, H.; Chen, X.; Liu, X.; Zhang, P. Novel carbohydrate-derived prolinamide as a highly efficient, recoverable catalyst for direct aldol reactions in water. Catal. Commun. 2012, 26, 6–10. [Google Scholar] [CrossRef]
- Shen, C.; Liao, H.; Shen, F.; Zhang, P. Novel synthesis of carbohydrate-derived organocatalysts and their application in asymmetric aldol reactions. Catal. Commun. 2013, 41, 106–109. [Google Scholar] [CrossRef] [Green Version]
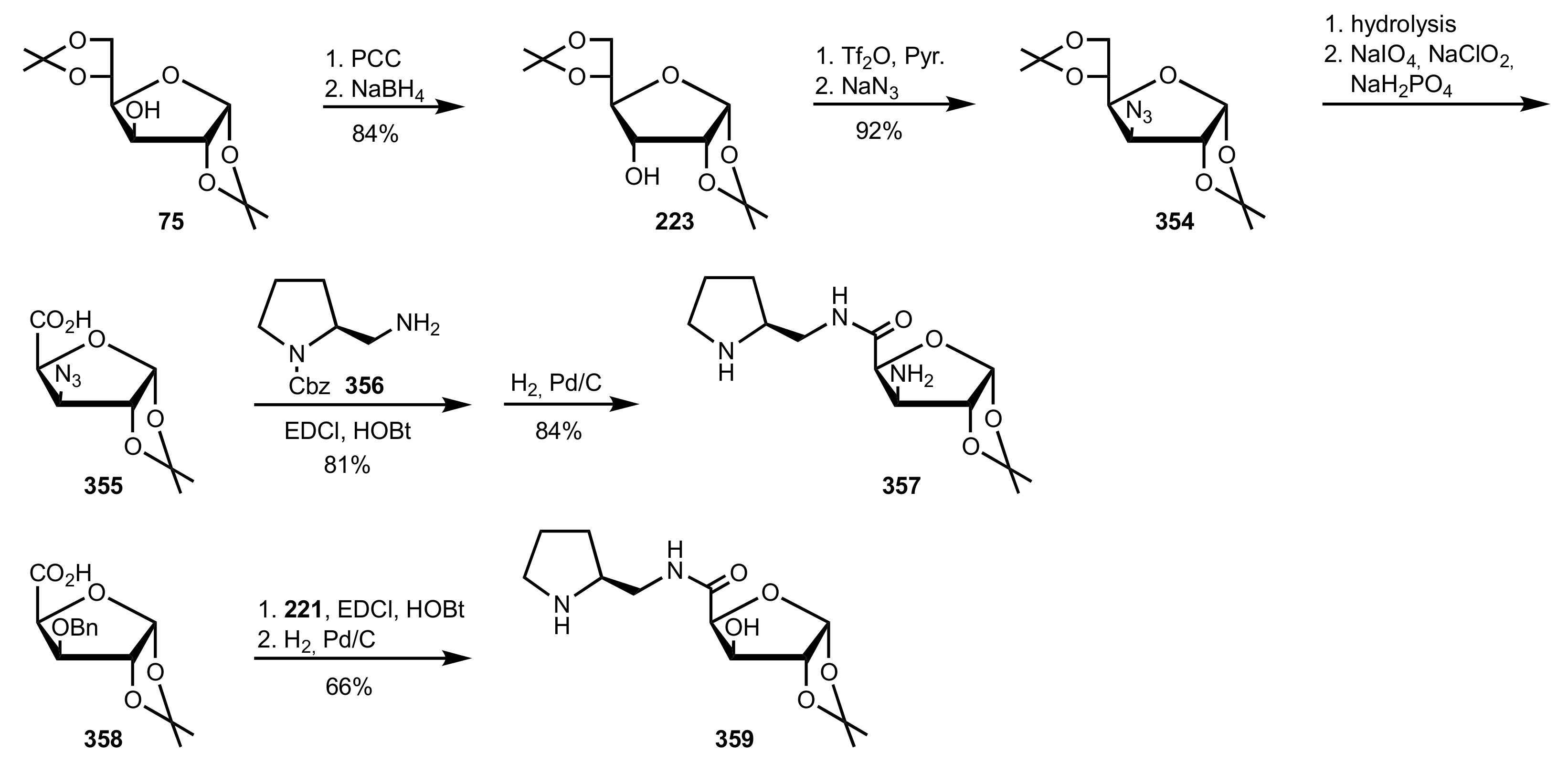
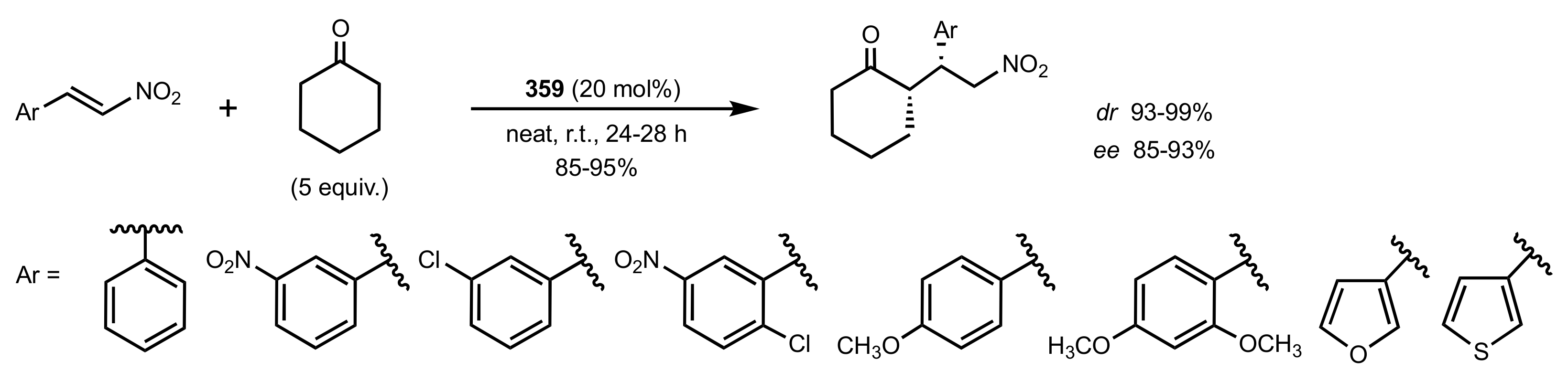
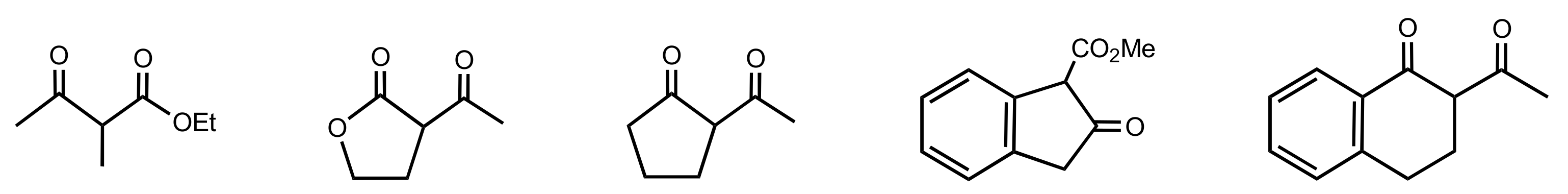
- Borges-Gonzáles, J.; Feher-Voelger, A.; Crisóstomo, F.P.; Morales, E.Q.; Martín, T. Tetrahydropyran-Based Hybrid Dipeptides as Asymmetric Catalysts for Michael Addition of Aldehydes to β-Nitrostyrenes. Adv. Synth. Catal. 2017, 359, 576–583. [Google Scholar] [CrossRef]
- Giuliano, R.M.; Jordan, A.D.; Gauthier, A.D.; Hoogsteen, K. Diastereofacial Selectivity of Diels-Alder Reactions of Carbohydrate-Derived Dienes and Their Carbocyclic Analogs. J. Org. Chem. 1993, 58, 4979–4988. [Google Scholar] [CrossRef]
- Bhunia, A.; Schwardt, O.; Gäthje, H.; Gao, G.-P.; Kelm, S.; Benie, A.J.; Hricovini, M.; Peters, T.; Ernst, B. Consistent Bioactive Conformation of the Neu5Acα(2-3)Gal Epitope Upon Lectin Binding. ChemBioChem 2008, 9, 2941–2945. [Google Scholar] [CrossRef]
- Carrillo, R.; Martín, V.S.; López, M.; Martín, T. Synthesis and cation complexation properties of new macrolides. Tetrahedron 2005, 61, 8177–8191. [Google Scholar] [CrossRef]
- Snatzke, G.; Raza, Z.; Habuš, I.; Šunjic, V. Chiroptical properties of tetrahydropyran-3,4-diols and 2-hydroxymethyltetrahydropyran-3-ols derived from l-arabinose, d-galactose, d-glucose, and d-xylose, and enantioselectivity in reduction with their complexes. Carbohydr. Res. 1988, 182, 179–196. [Google Scholar] [CrossRef]
- Borges-Gonzáles, J.; García-Monzón, I.; Martín, T. Conformational Control of Tetrahydropyran—Based Hybrid Dipeptide Catalysts Improves Activity and Stereoselectivity. Adv. Synth. Catal. 2019, 361, 2141–2147. [Google Scholar] [CrossRef] [Green Version]
- Habuš, I.; Šunjic, V. Preparation of the Chiral Diol (2R,3R)-2-Hydroxymethyl-3-hydroxy-tetrahydropyran from d-Glucose via Reductive Rearrangement of Pseudo-d-glucal Triacetate. Croat. Chem. Acta 1985, 58, 321–330. [Google Scholar]
- Feher-Voelger, A.; Borges-Gonzáles, J.; Morales, E.Q.; Carrillo, R.; Morales, E.Q.; Gonzáles-Platas, J.; Martín, T. Synthesis and Conformational Analysis of Cyclic Homooligomers from Pyranoid ε-Sugar Amino Acids. Chem. Eur. J. 2014, 20, 4007–4022. [Google Scholar] [CrossRef]
- Wang, L.; Liu, J.; Miao, T.; Zhou, W.; Li, P.; Ren, K.; Zhang, X. Sugar-Based Pyrrolidine as a Highly Enantioselective Organocatalyst for Asymmetric Michael Addition of Ketones to Nitrostyrenes. Adv. Synth. Catal. 2010, 352, 2571–2578. [Google Scholar] [CrossRef]
- Mondal, S.; Fan, E. Mild and Efficient Synthesis of Fmoc-Protected Amino Azides from Fmoc-Protected Amino Alcohols. Synlett 2006, 2006, 306–308. [Google Scholar] [CrossRef]
- Kumar, T.P.; Balaji, S.V. Sugar amide-pyrrolidine catalyst for the asymmetric Michael addition of ketones to nitroolefins. Tetrahedron Asymmetry 2014, 25, 473–477. [Google Scholar] [CrossRef]
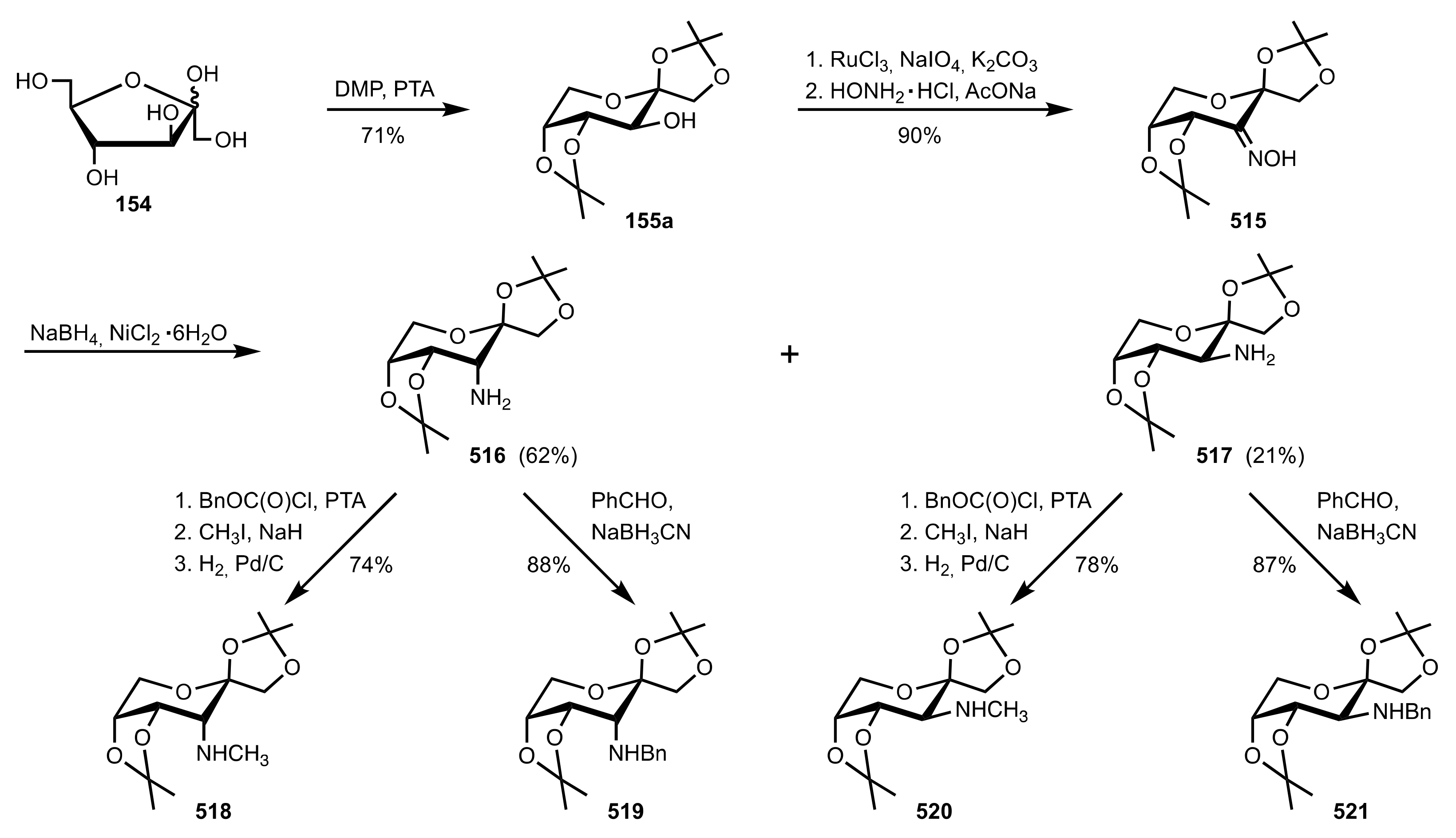
- Austin, G.N.; Baird, P.D.; Fleet, G.W.J.; Peach, J.M.; Smith, P.W.; Watkin, D.J. 3,6-Dideoxy-3,6-imino-1,2-O-isopropylidene-α-d-glucofuranose as a divergent intermediate for the synthesis of hydroxylated pyrrolidines: Synthesis of 1,4-dideoxy-1,4-imino-l-gulitol, 1,4-dideoxy-1,4-imino-l-lyxitol, 2S,3S,4R-3,4-dihydroxyproline and (1S,2R,8S,8aR)-1,2,8-trihydroxyoctahydroindolizine [8-epi-swainsonine]. X-ray crystal structure of (1S,2R,8S,8aR)-1,2,8-trihydroxy-5-oxo-octahydroindolizine. Tetrahedron 1987, 43, 3095–3108. [Google Scholar]
- Chandrasekhar, S.; Reddy, M.S.; Jagadeesh, B.; Prabhakar, A.; Rao, M.H.V.R.; Jagannadh, B. Formation of a Stable 14-Helix in Short Oligomers of Furanoid cis-β-Sugar-Amino Acid. J. Am. Chem. Soc. 2004, 126, 13586–13587. [Google Scholar] [CrossRef] [PubMed]
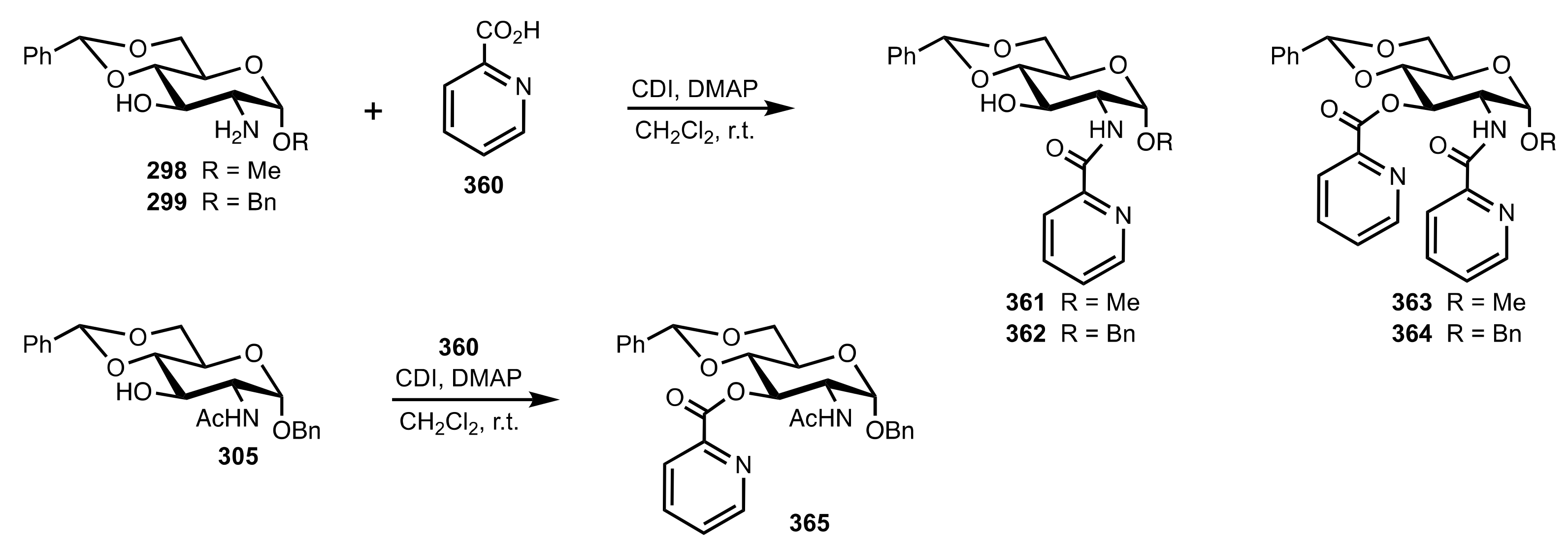
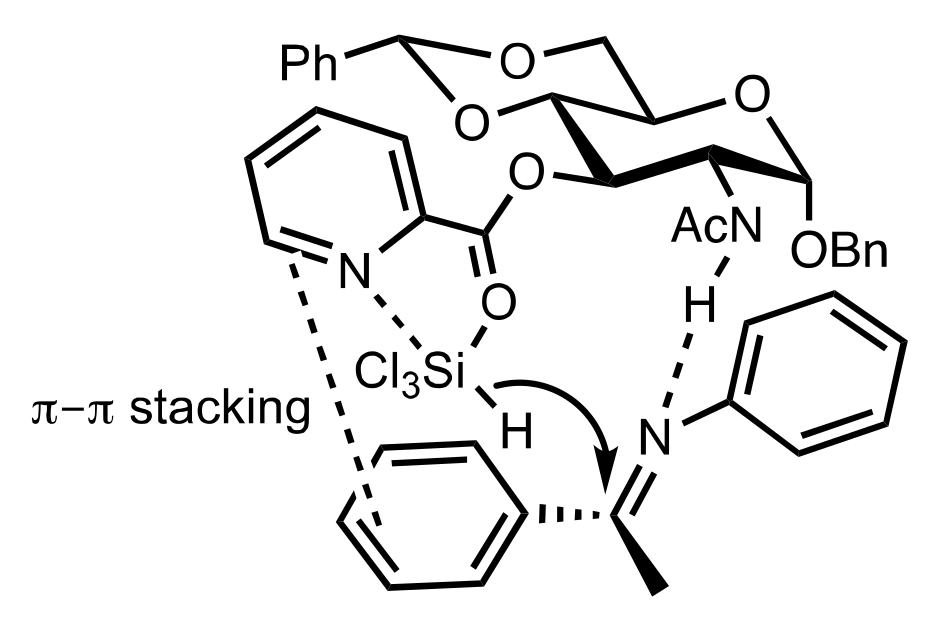
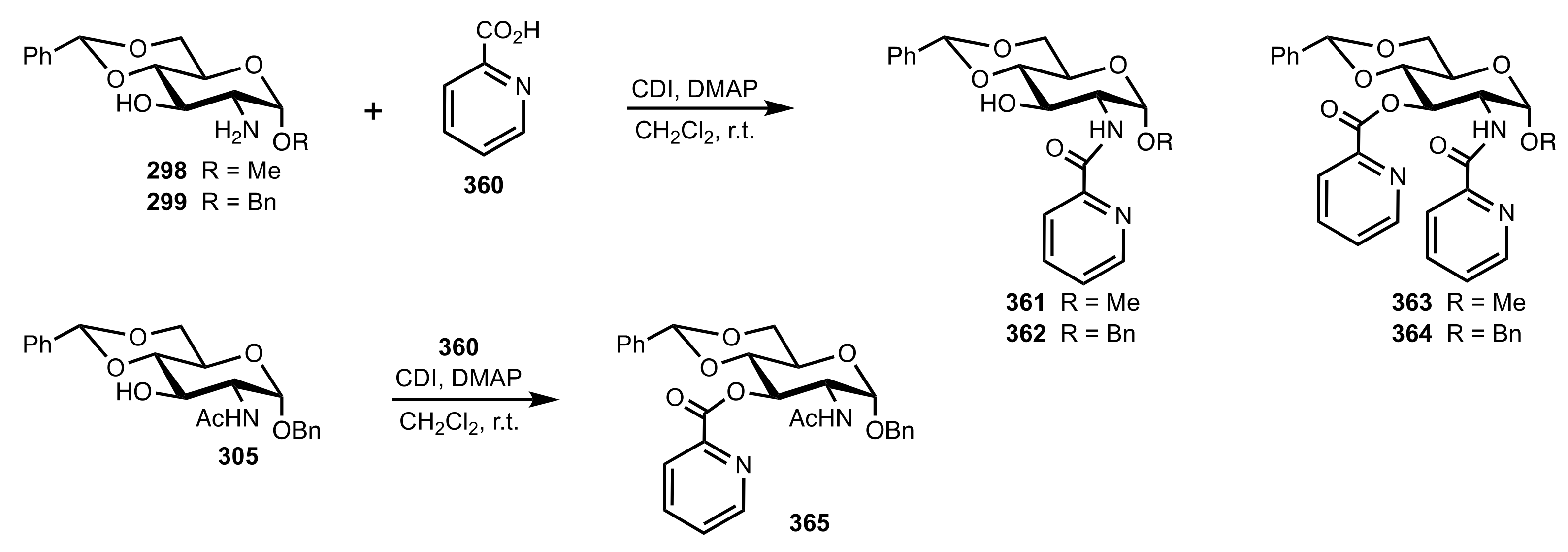
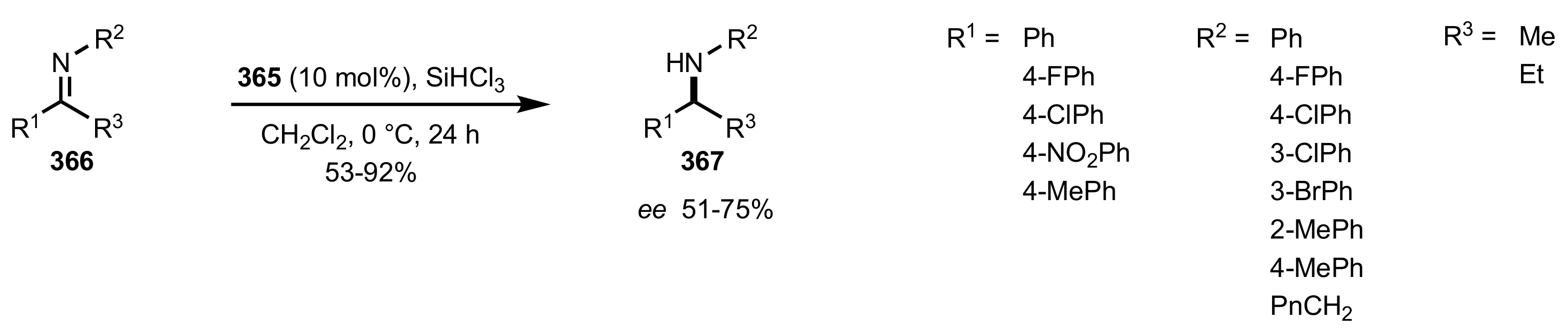
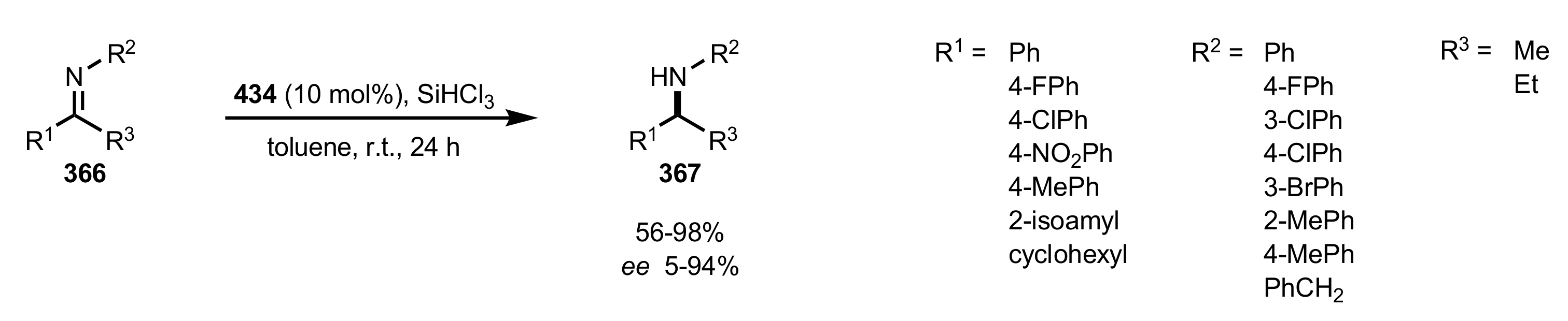
- Ge, X.; Qian, C.; Chen, Y.; Chen, X. Novel carbohydrate-derived pyridinecarboxylic organocatalysts for the enantioselective reduction of imines with trichlorosilane. Tetrahedron Asymmetry 2014, 25, 596–601. [Google Scholar] [CrossRef]
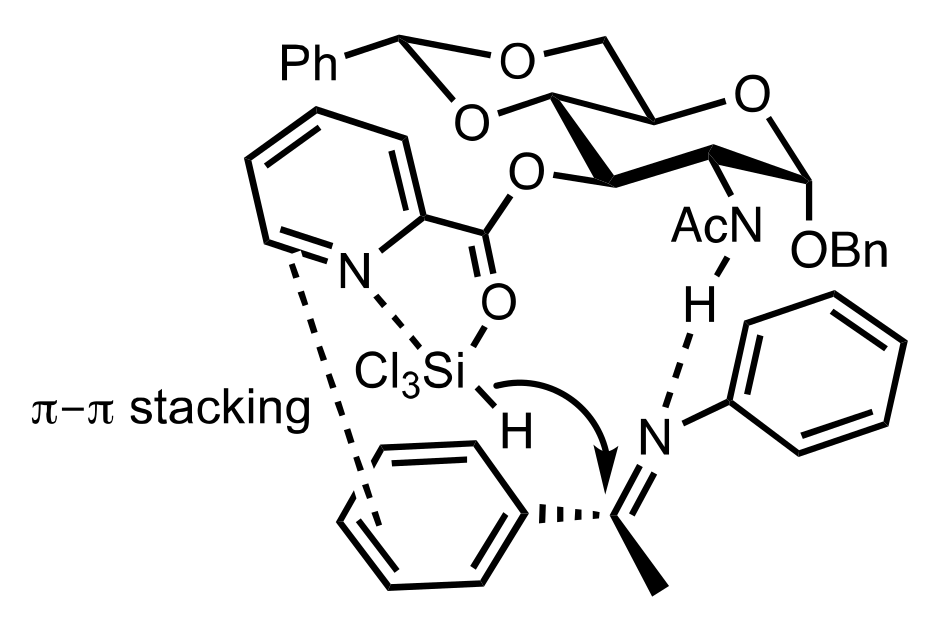
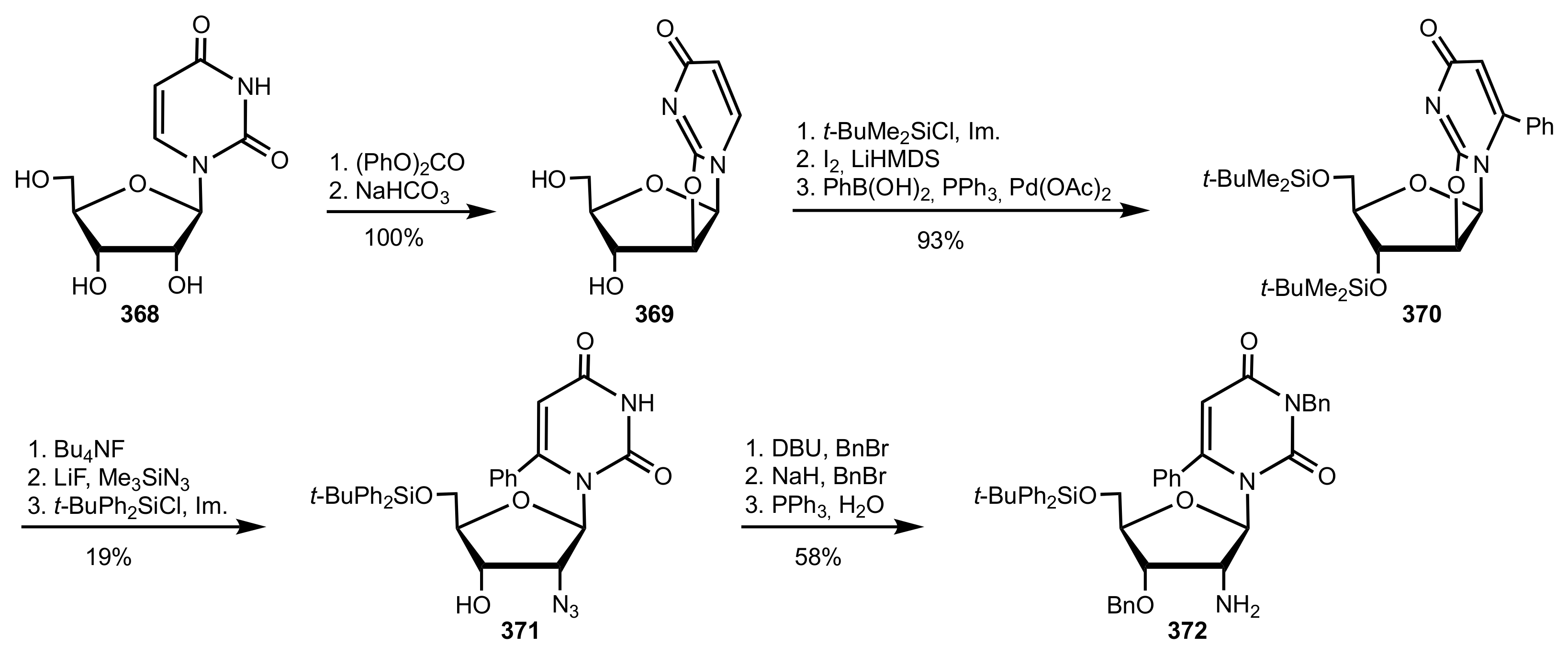
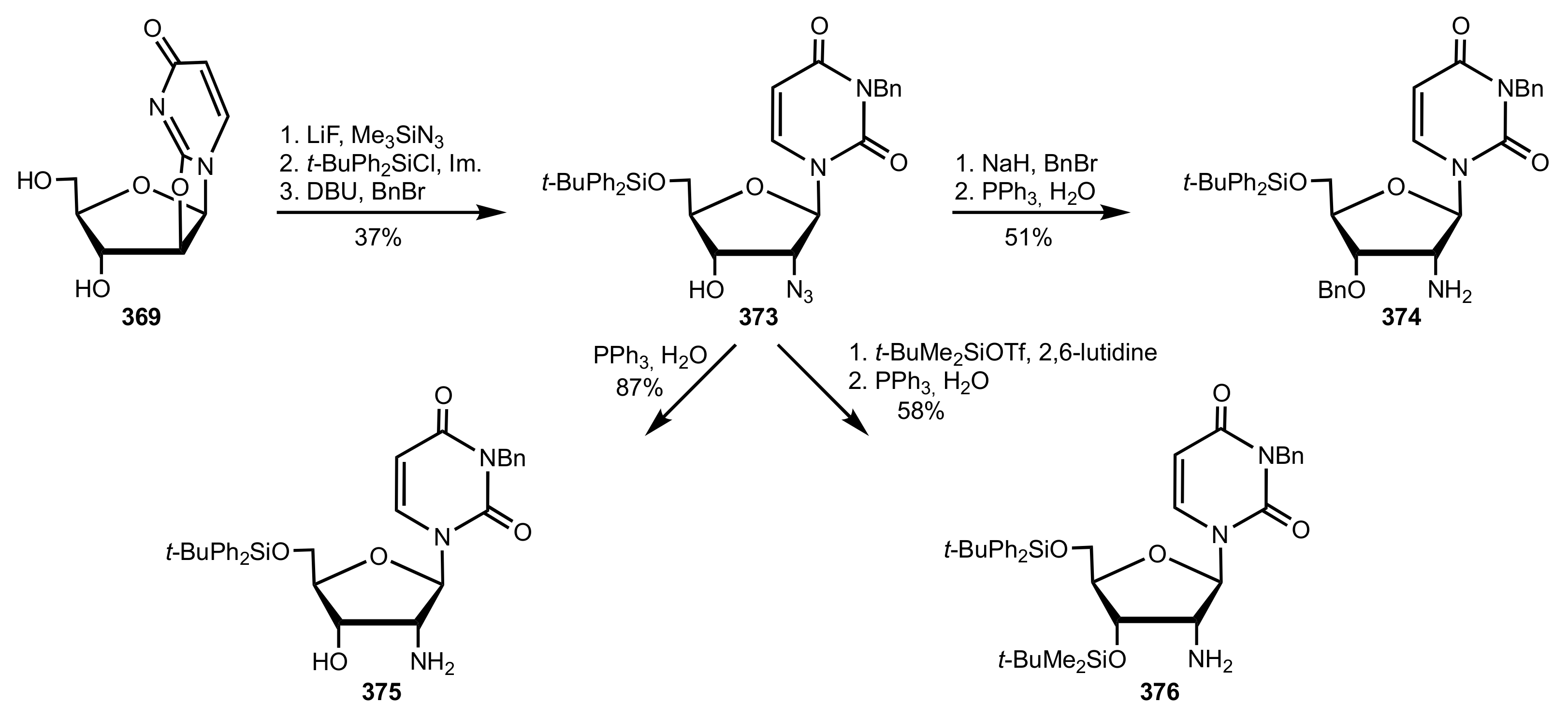
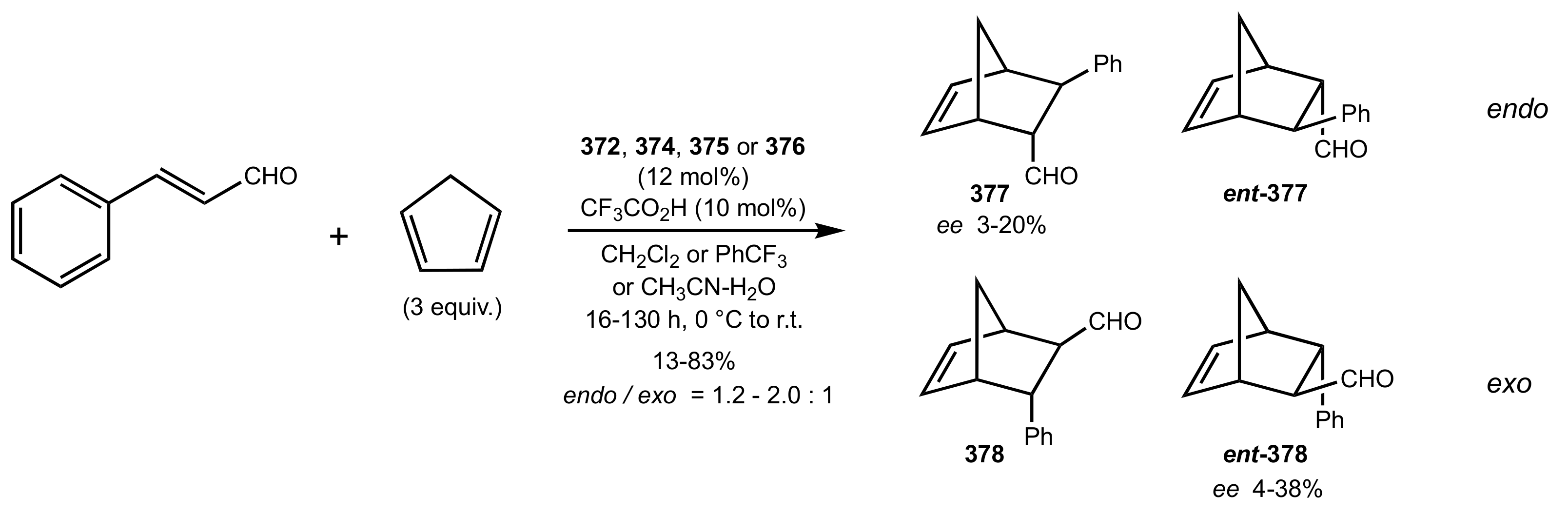
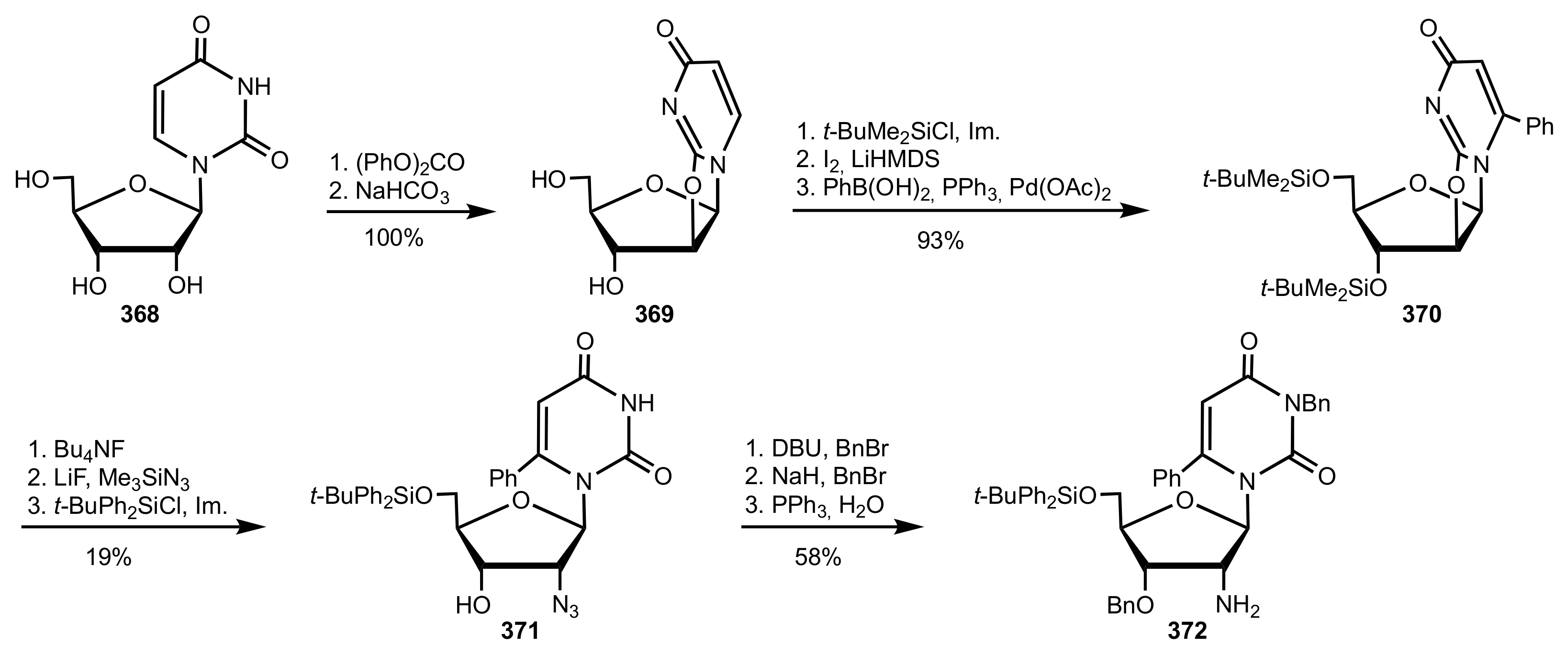
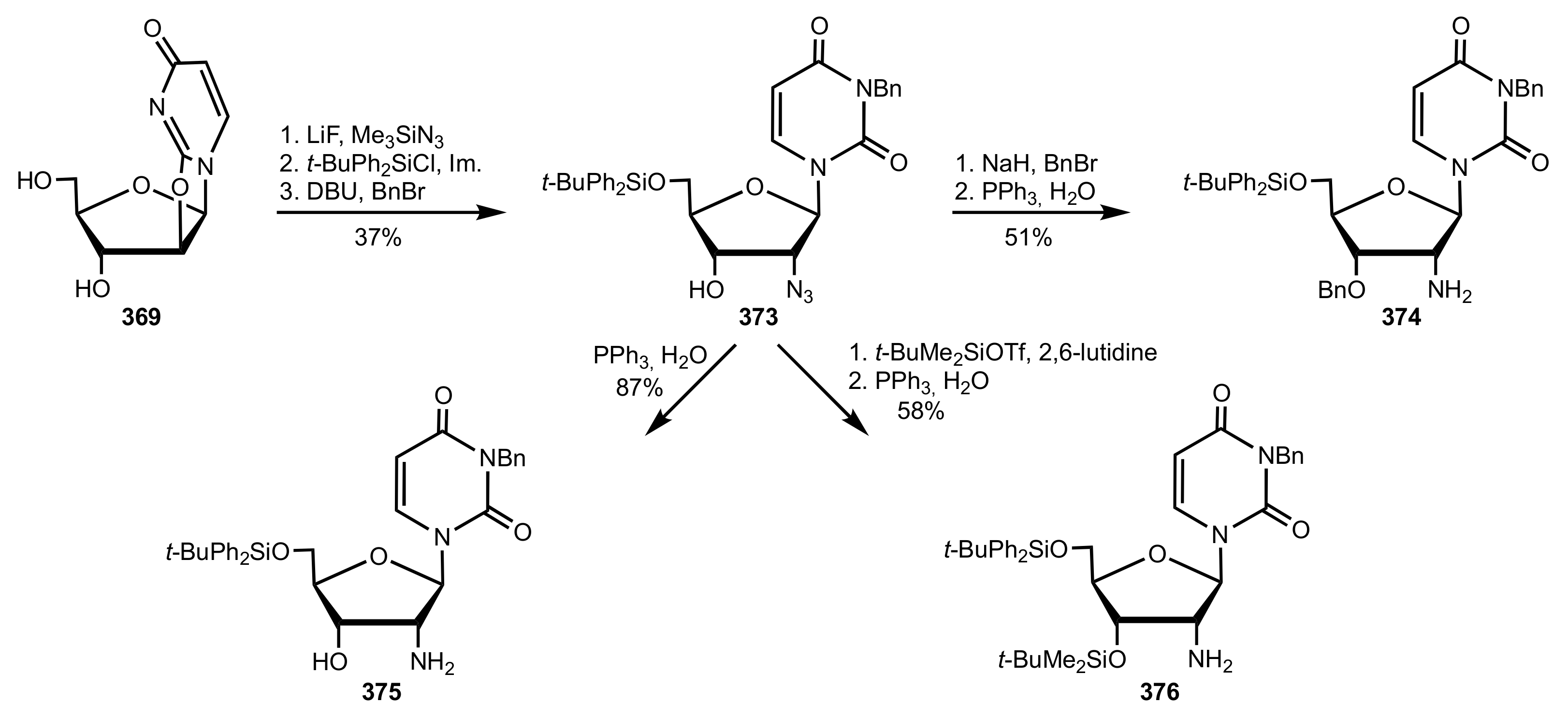
- Wakamatsu, H.; Itoh, M.; Natori, Y.; Yoshimura, Y. Synthesis of 2′-aminouridine derivatives as an organocatalyst for Diels-Alder reaction. Nucleosides Nucleotides Nucleic Acids 2020, 39, 365–383. [Google Scholar] [CrossRef]
- McGee, D.P.C.; Vaughn-Settle, A.; Vargeese, C.; Zhai, Y. 2′-Amino-2′-deoxyuridine via an Intramolecular Cyclization of a Trichloroacetimidate. J. Org. Chem. 1996, 61, 781–785. [Google Scholar] [CrossRef]
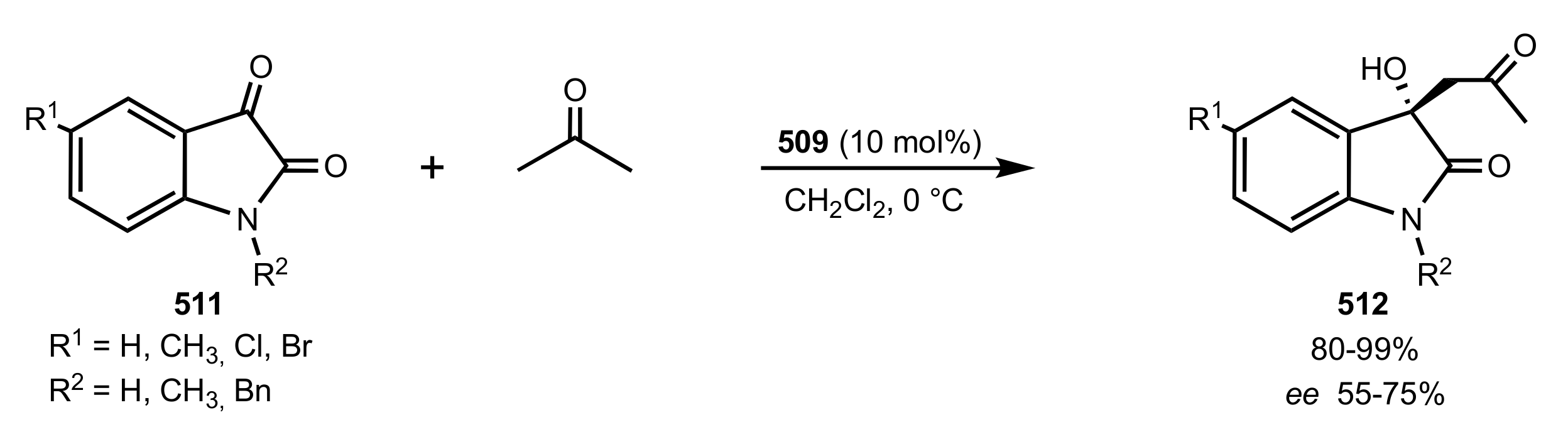
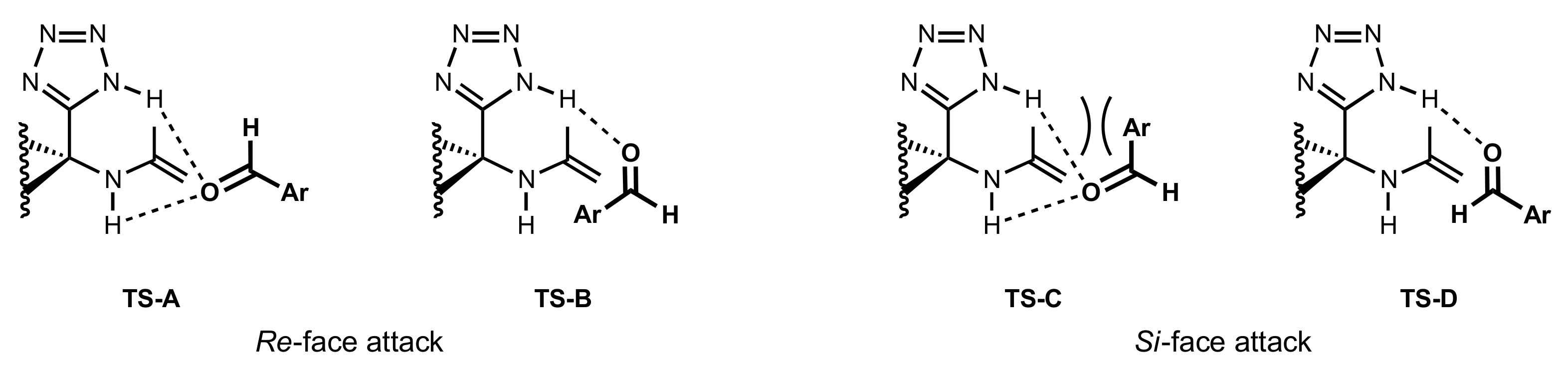
- Wynands, L.; Delacroix, S.; Van Nhien, A.N.; Soriano, E.; Marco-Contelles, J.; Postel, D. New glycosyl-α-aminotetrazole-based catalysts for highly enantioselective aldol reactions. Tetrahedron 2013, 69, 4899–4907. [Google Scholar] [CrossRef] [Green Version]
- Len, C.; Postel, D.; Mackenzie, G.; Villa, P.; Ronco, G. Strategy for the Preparation of 2′ and 3′ Branched Nucleosides. Pharm. Pharmacol. Commun. 1999, 5, 165–168. [Google Scholar] [CrossRef]
- Domínguez, L.; Van Nhien, A.N.; Tomassi, C.; Len, C.; Postel, D.; Marco-Contelles, J. Synthesis of 4-Amino-5-H-2,3-dihydroisothiazole-1,1-dioxide Ring Systems on Sugar Templates via Carbanion-Mediated Sulfonamide Intramolecular Cyclization Reactions (CSIC Protocols) of Glyco-α-sulfonamidonitriles. J. Org. Chem. 2004, 69, 843–856. [Google Scholar] [CrossRef]
- Muraoka, O.; Yoshikai, K.; Takahashi, H.; Minematsu, T.; Lu, G.; Tanabe, G.; Wang, T.; Matsuda, H.; Yoshikawa, M. Synthesis and biological evaluation of deoxy salacinols, the role of polar substituents in the side chain on the α-glucosidase inhibitory activity. Bioorg. Med. Chem. 2006, 14, 500–509. [Google Scholar] [CrossRef]
- Sowa, W. Convenient synthesis of 3-amino-3-deoxy-d-ribose. Can. J. Chem. 1968, 46, 1586–1589. [Google Scholar] [CrossRef]
- Van Nhien, A.N.; Domínguez, L.; Tomassi, C.; Torres, M.R.; Len, C.; Postel, D.; Marco-Contelles, J. Synthesis and transformations of [1,2-O-isopropylidene-α-d-erythro (and α-d-ribo) furanose]-3-spiro-3′-(4′-amino-5′H-2′,3′-dihydroisothiazole -1′,1′-dioxide) derivatives. Tetrahedron 2004, 60, 4709–4727. [Google Scholar] [CrossRef]
- Mazur, A.; Tropp, B.E.; Engel, R. Isosteres of natural phosphates. 11. Synthesis of a phosphonic acid analogue of an oligonucleotide. Tetrahedron 1984, 40, 3949–3956. [Google Scholar] [CrossRef]
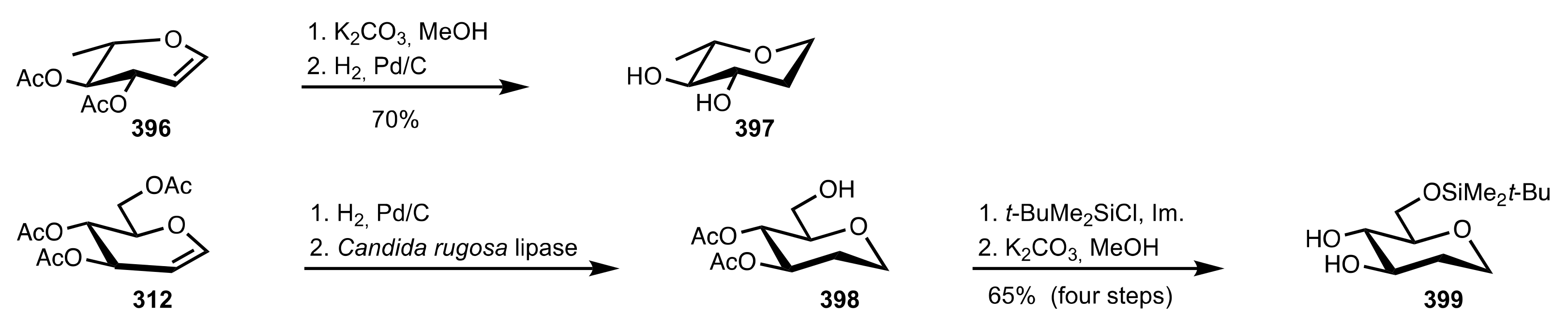
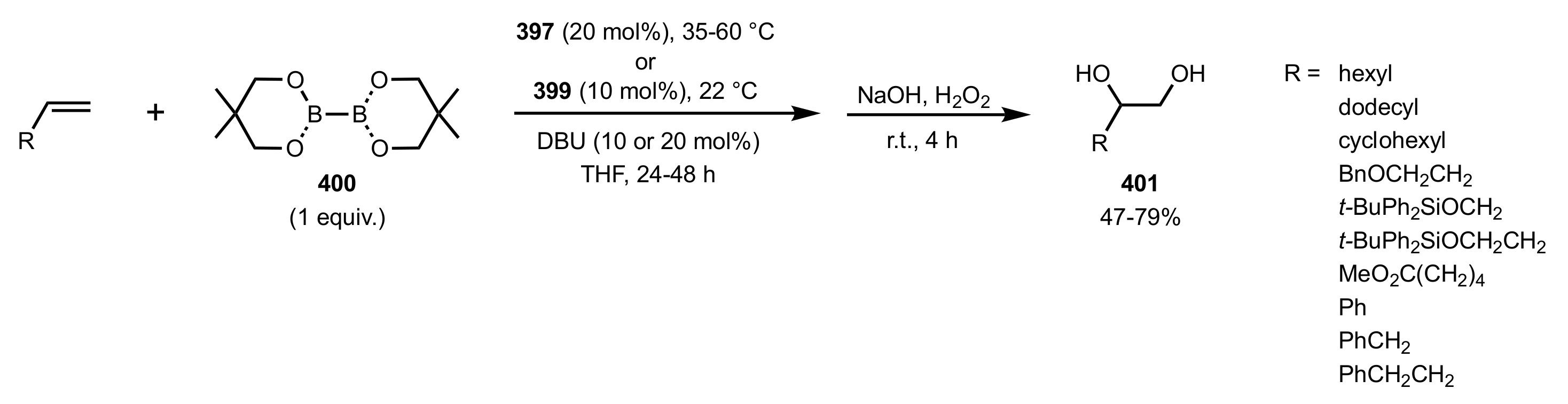
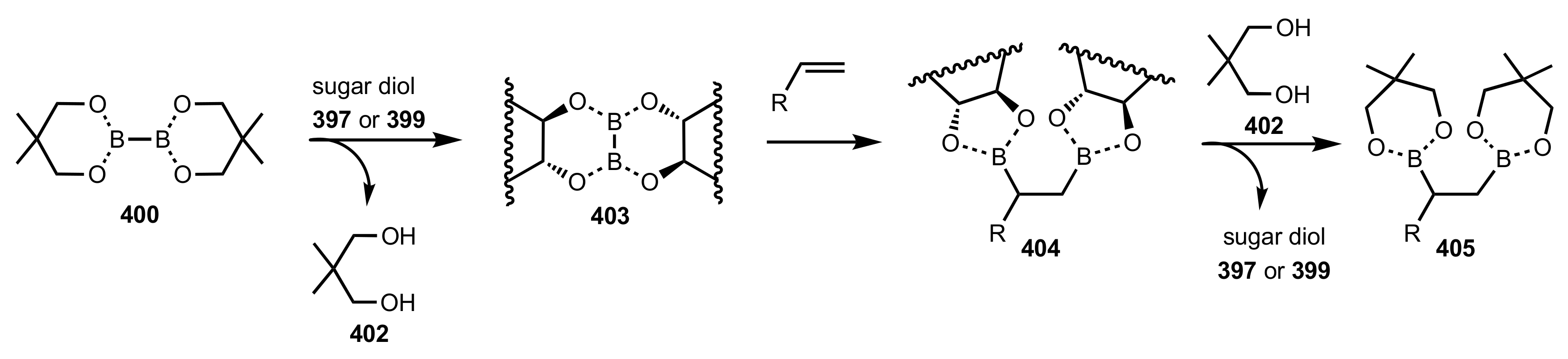
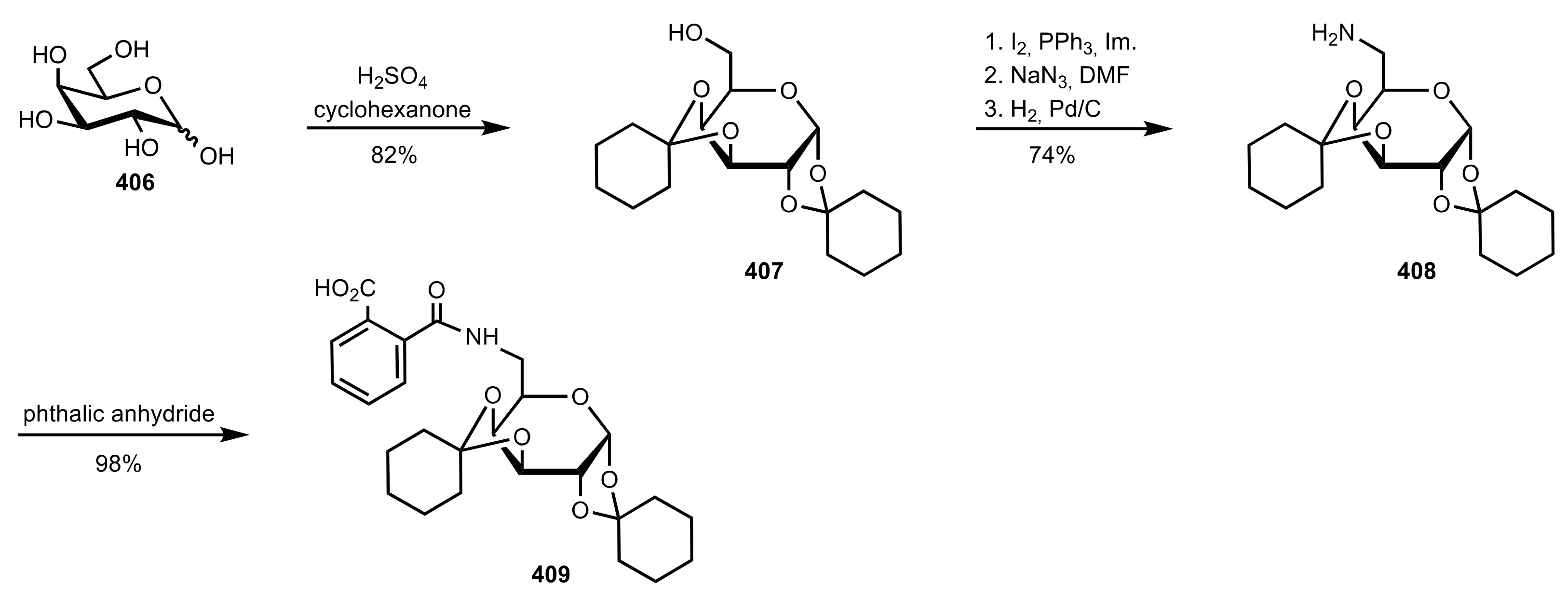
- Fang, L.; Yan, L.; Haeffner, F.; Morken, J.P. Carbohydrate-Catalyzed Enantioselective Alkene Diboration: Enhanced Reactivity of 1,2-Bonded Diboron Complexes. J. Am. Chem. Soc. 2016, 138, 2508–2511. [Google Scholar] [CrossRef] [Green Version]
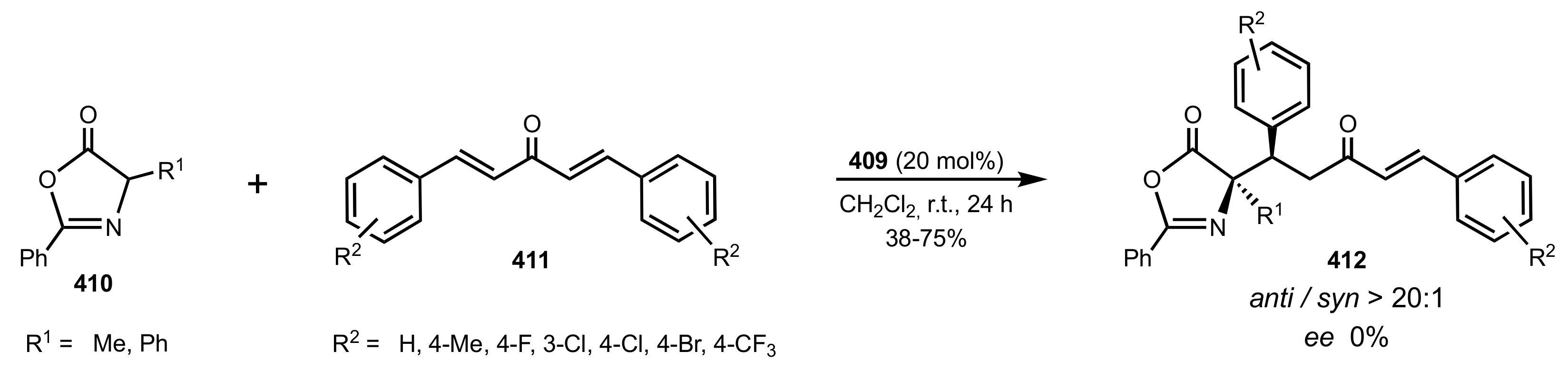
- Pinheiro, D.L.J.; Batista, G.M.F.; Gonçalves, J.R.; Duarte, T.N.; Amarante, G.W. Sugar-Based Organocatalyst for the Diastereoselective Desymmetrization of Dibenzylideneacetones. Eur. J. Org. Chem. 2016, 2016, 459–462. [Google Scholar] [CrossRef]
- Hoppe, I.; Schöllkopf, U.; Tölle, R. Asymmetrische Synthese von α-Methylaminosäure-methylestern unter Verwendung von d-Galactodialdehydals chiralem Hilfsreagens. Synthesis 1983, 1983, 789–791. [Google Scholar] [CrossRef]
- Dwivedi, N.; Bisht, S.S.; Tripathi, R.P. Asymmetric organocatalysis with glycosyl-β-amino acids: Direct asymmetric aldol reaction of acetone with aldehydes. Carbohydr. Res. 2006, 341, 2737–2743. [Google Scholar] [CrossRef]
- Meyer, A.S.; Reichstein, T. l-Idose aus l-glucose, sowie ein neuer Weg zur l-Idomethylose. Helv. Chim. Acta 1946, 29, 152–162. [Google Scholar] [CrossRef]
- Wolfrom, M.L.; Hanessian, S. The Reaction of Free Carbonyl Sugar Derivatives with Organometallic Reagents. I. 6-Deoxy-l-idose and Derivatives. J. Org. Chem. 1962, 27, 1800–1804. [Google Scholar] [CrossRef]
- Desai, V.N.; Saha, N.N.; Dhavale, D.D. Intramolecular Michael addition of benzylamine to sugar derived α,β-unsaturated ester: A new diastereoselective synthesis of a higher homologue of 1-deoxy-l-ido-nojirimycin. Chem. Commun. 1999, 1999, 1719–1720. [Google Scholar] [CrossRef]
- Tronchet, J.M.J.; Gentile, B. Réaction d’ylures stabilisés sur des aldéhydosucres: Influence de divers facteurs dont la structure de l’aldéhydosucres sur le pourcentage d’isomères géométriques obtenu. Helv. Chim. Acta 1979, 62, 2091–2098. [Google Scholar] [CrossRef]
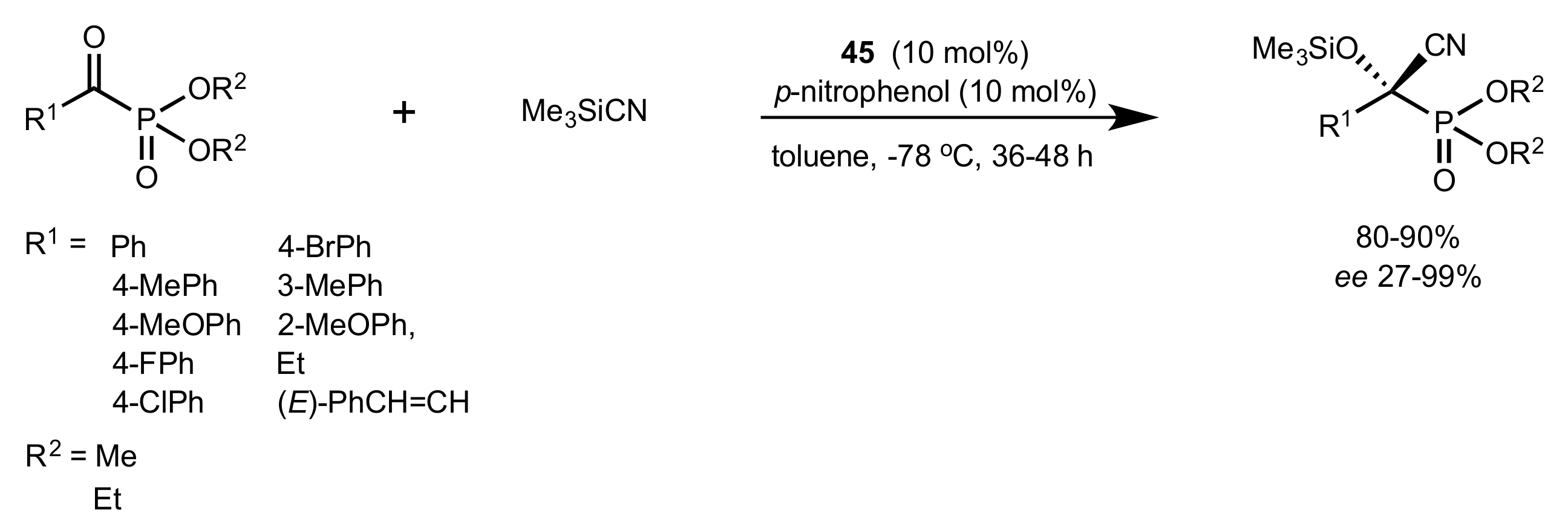
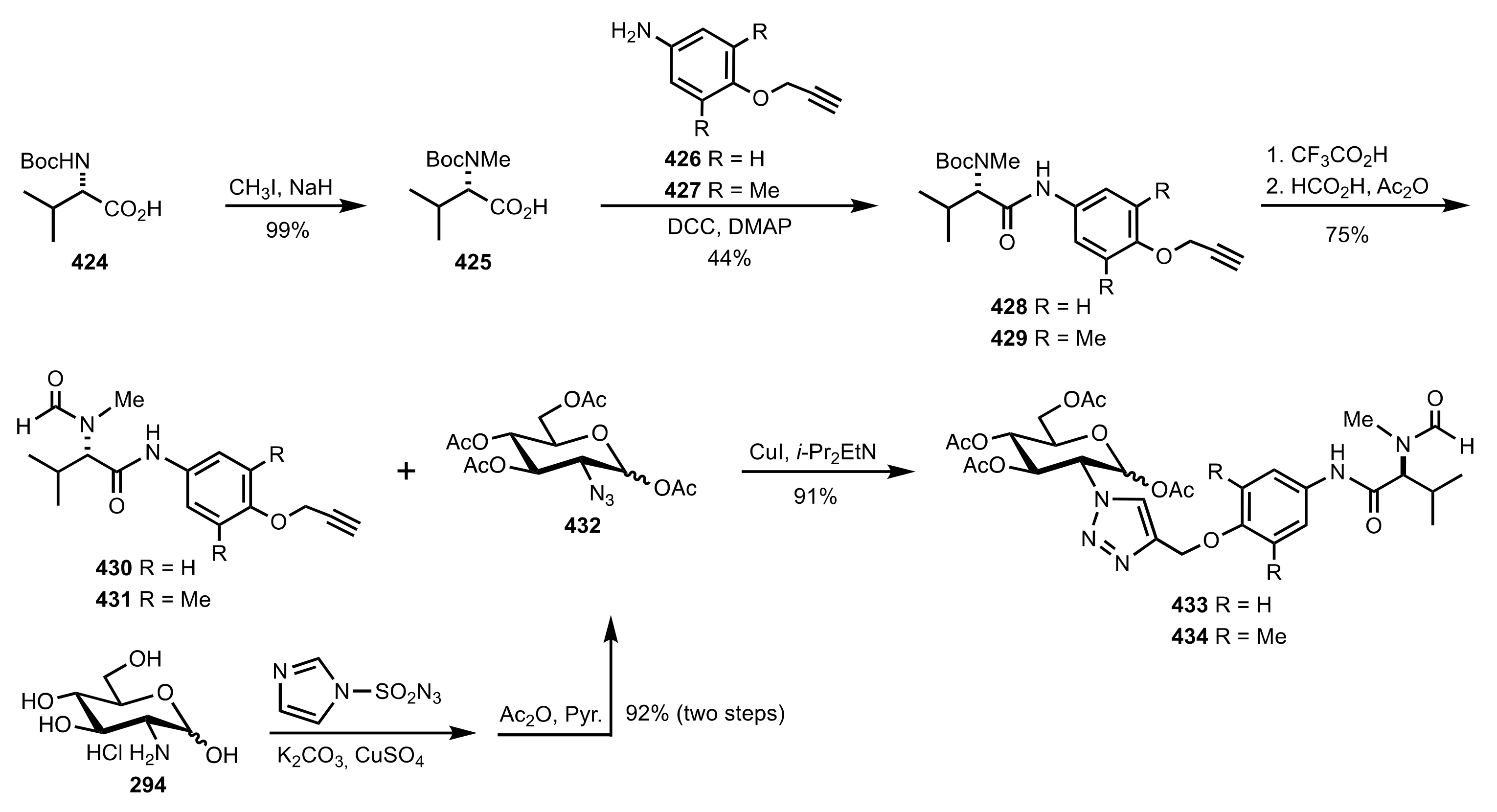
- Ge, X.; Qian, C.; Chen, X. Synthesis of novel carbohydrate-based valine-derived formamide organocatalysts by CuAAC click chemistry and their application in asymmetric reduction of imines with trichlorosilane. Tetrahedron Asymmetry 2014, 25, 1450–1455. [Google Scholar] [CrossRef]
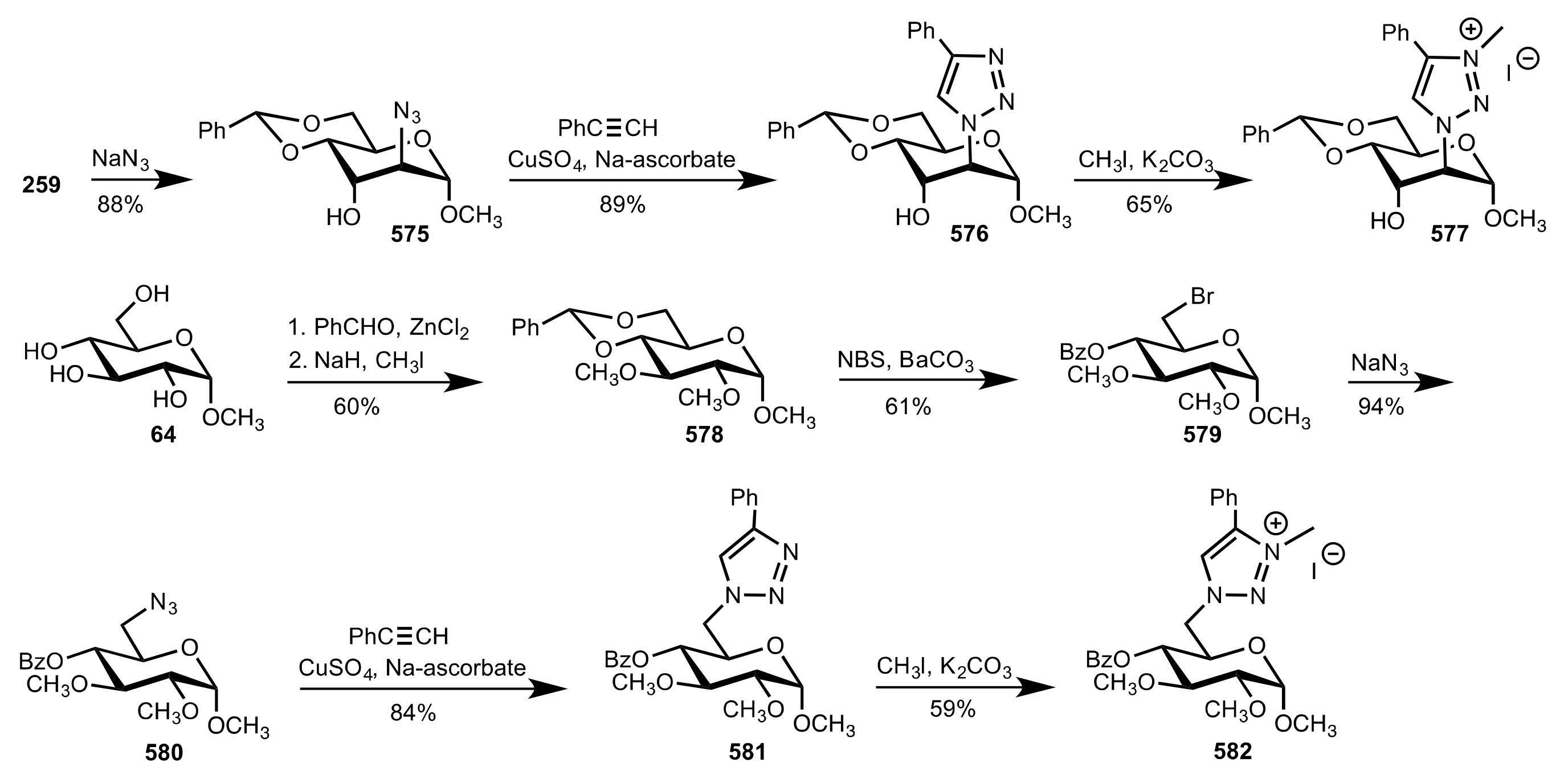
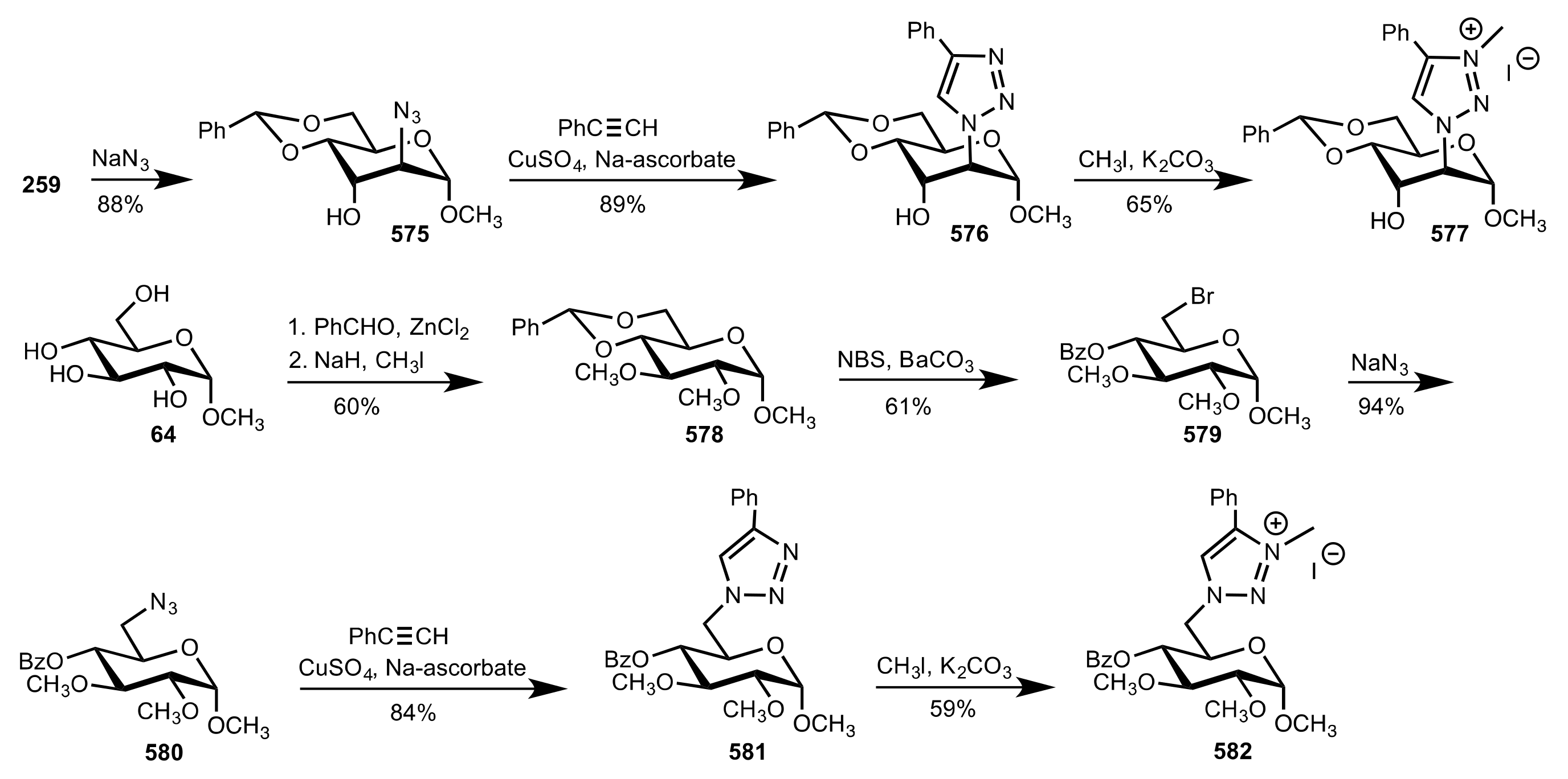
- Goddard-Borger, E.D.; Stick, R.V. An Efficient, Inexpensive, and Shelf-Stable Diazotransfer Reagent: Imidazole-1-sulfonyl Azide Hydrochloride. Org. Lett. 2007, 9, 3797–3800. [Google Scholar] [CrossRef]
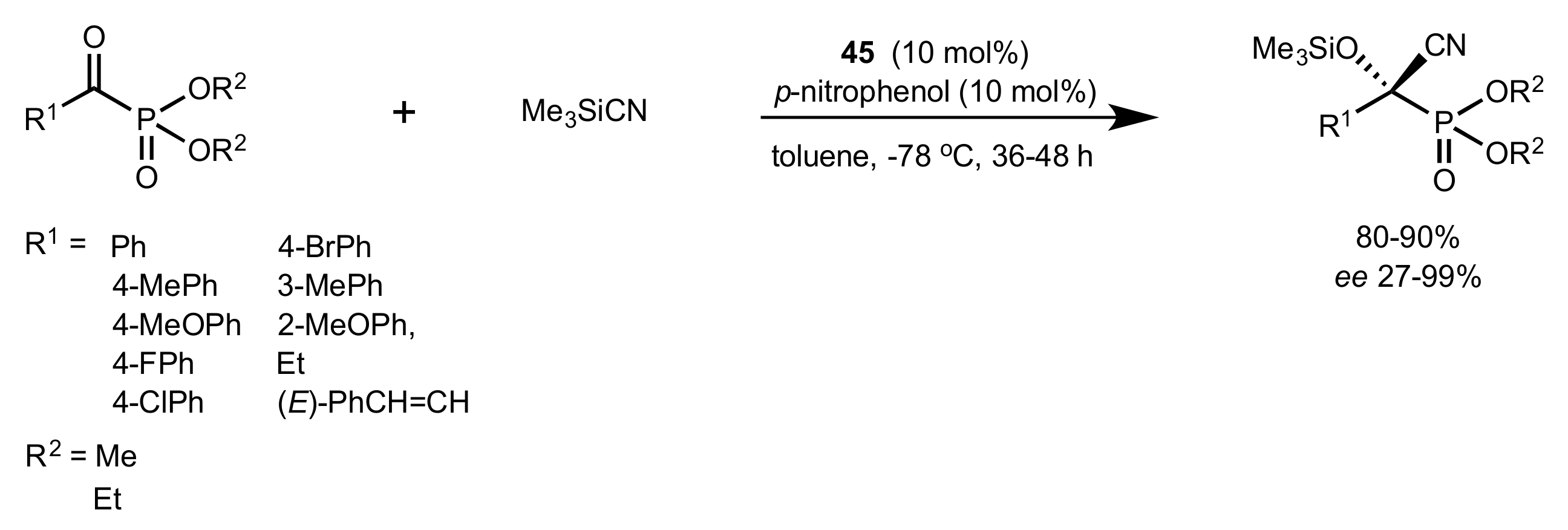
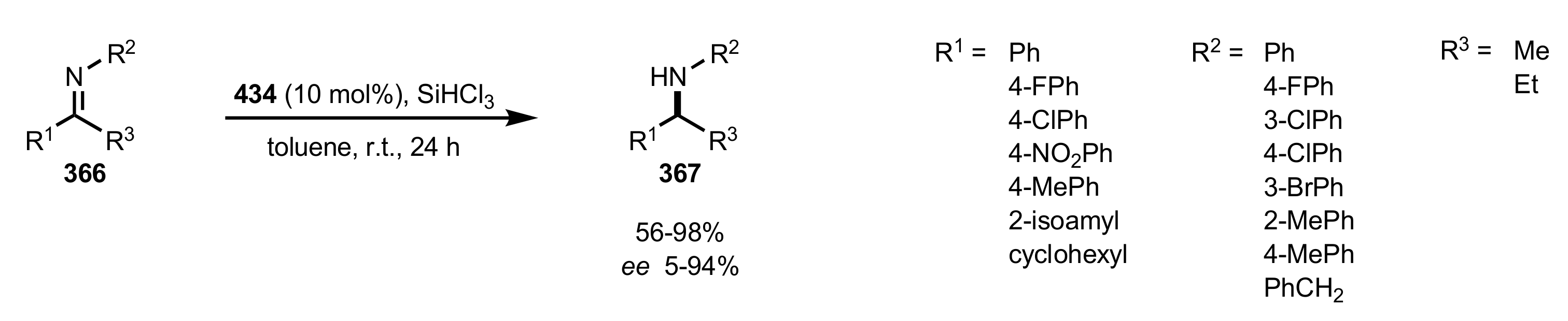
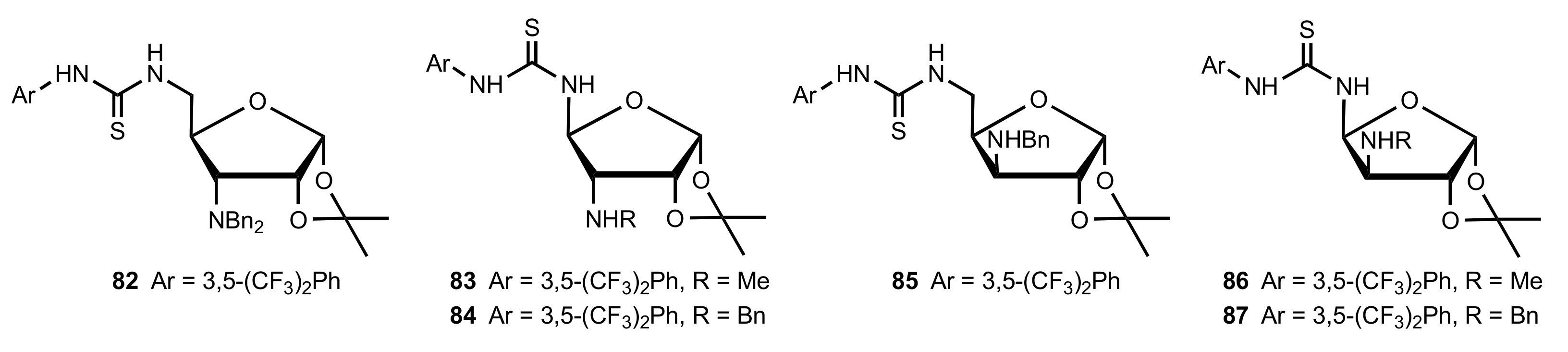
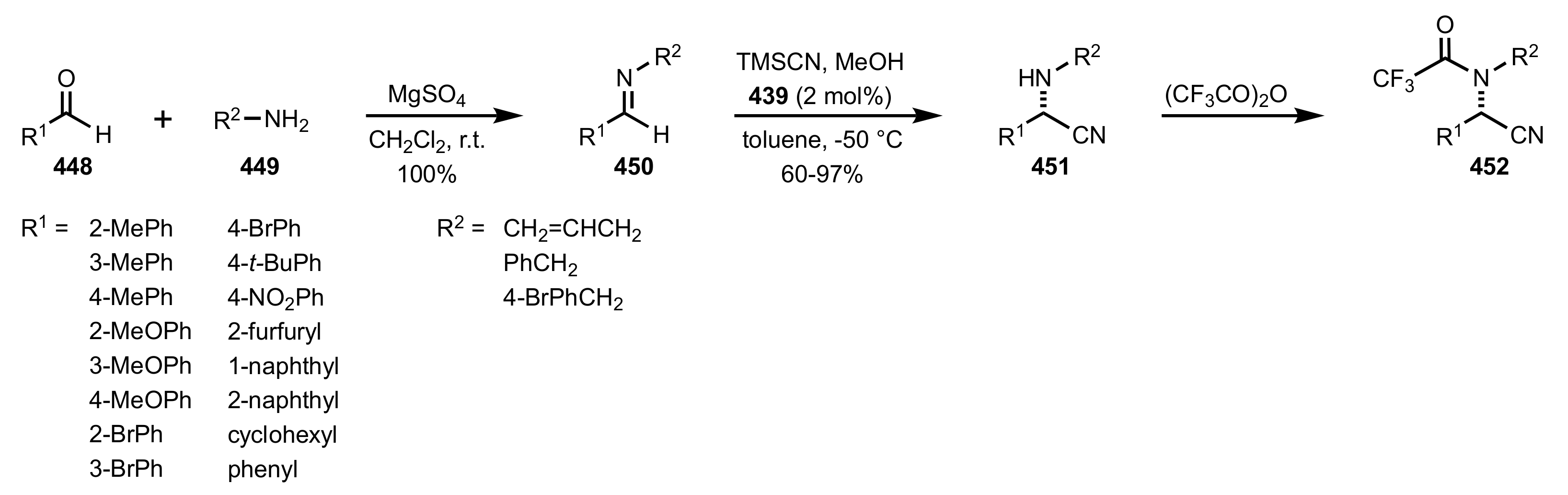
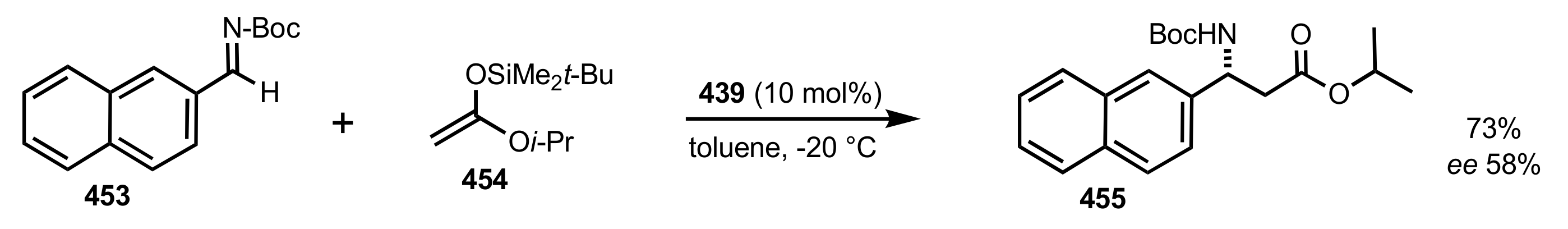
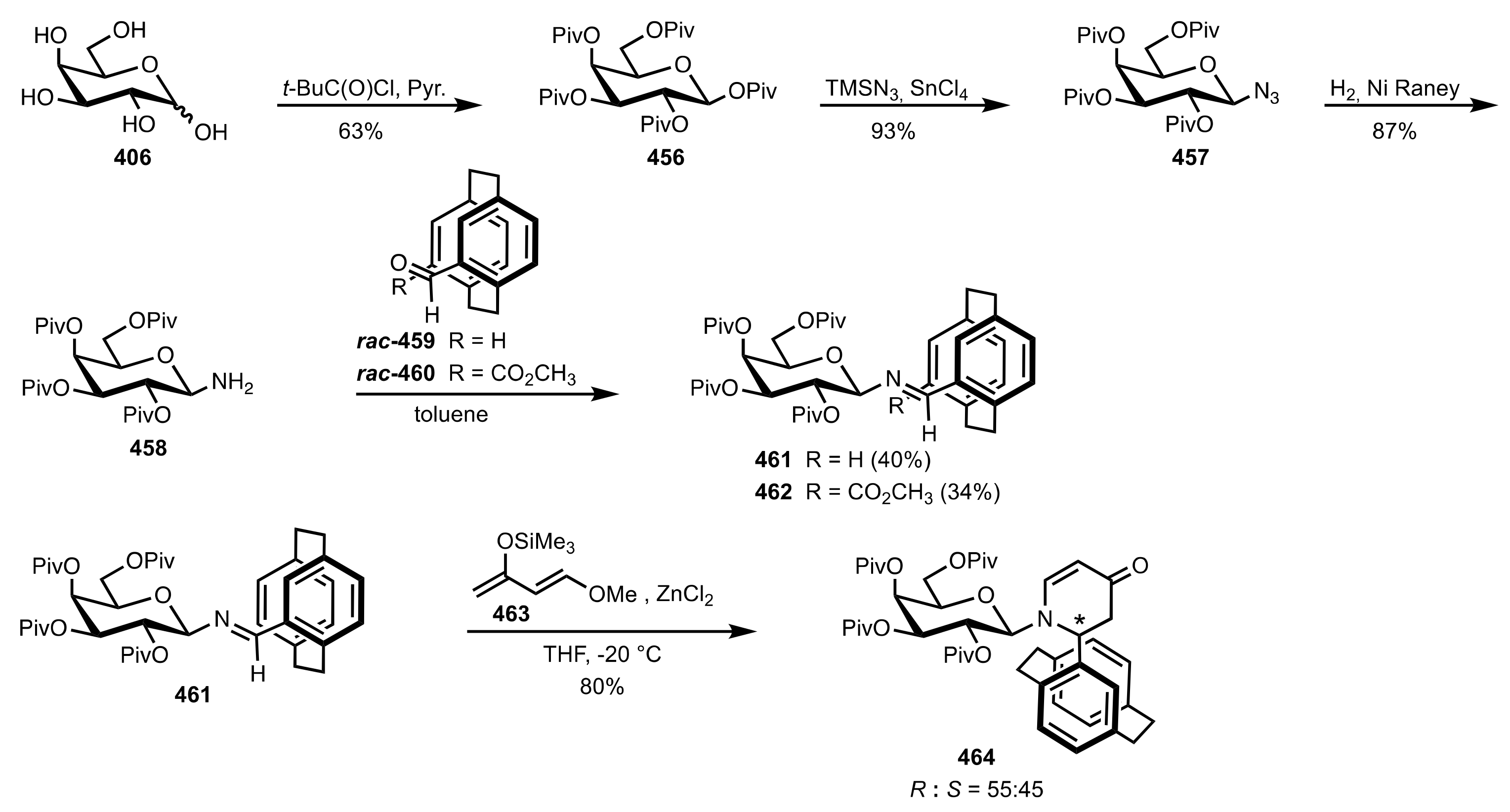
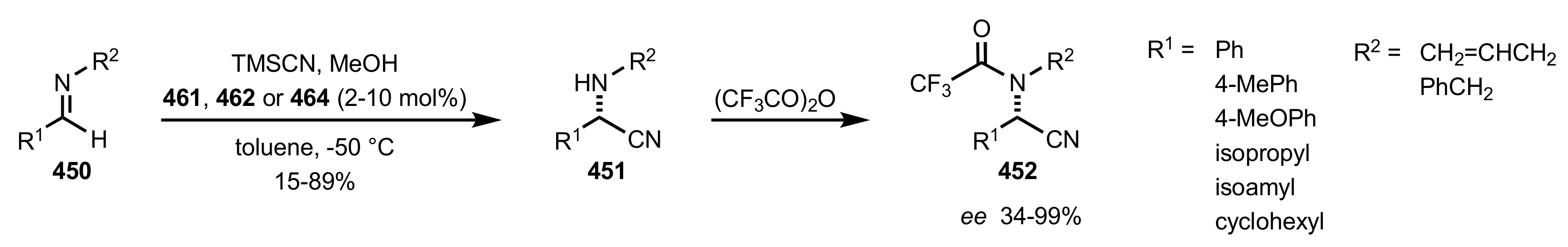
- Becker, C.; Hoben, C.; Kunz, H. Enantioselective Organocatalysis of Strecker and Mannich Reactions Based on Carbohydrates. Adv. Synth. Catal. 2007, 349, 417–424. [Google Scholar] [CrossRef]
- Negru, M.; Schollmeyer, D.; Kunz, H. Enantioselective Strecker Reaction Catalyzed by an Organocatalyst Lacking a Hydrogen-Bond-Donor Function. Angew. Chem. Int. Ed. 2007, 46, 9339–9341. [Google Scholar] [CrossRef]
- Kunz, H.; Sager, W.; Schanzenbach, D.; Decker, M. Carbohydrates as Chiral Templates: Stereoselective Strecker Synthesis of d-α-Amino Nitriles and Acids Using O-Pivaloylated d-Galactosylamine as the Auxiliary. Liebigs Ann. Chem. 1991, 1991, 649–654. [Google Scholar] [CrossRef]
- Armstrong, A.; Ahmed, G.; Garnett, I.; Goacolou, K.; Wailes, J.S. Exocyclic iminium salts as catalysts for alkene epoxidation by Oxone. Tetrahedron 1999, 55, 2341–2352. [Google Scholar] [CrossRef]
- Page, P.C.B.; Buckley, B.R.; Appleby, L.F.; Alsters, P.A. Highly Efficient Catalysts for Epoxidation Mediated by Iminium Salts. Synthesis 2005, 2005, 3405–3411. [Google Scholar] [CrossRef]
- Page, P.C.B.; Chan, Y.; Liddle, J.; Elsegood, M.R.J. Carbohydrate-derived iminium salt organocatalysts for the asymmetric epoxidation of alkenes. Tetrahedron 2014, 70, 7283–7305. [Google Scholar] [CrossRef] [Green Version]
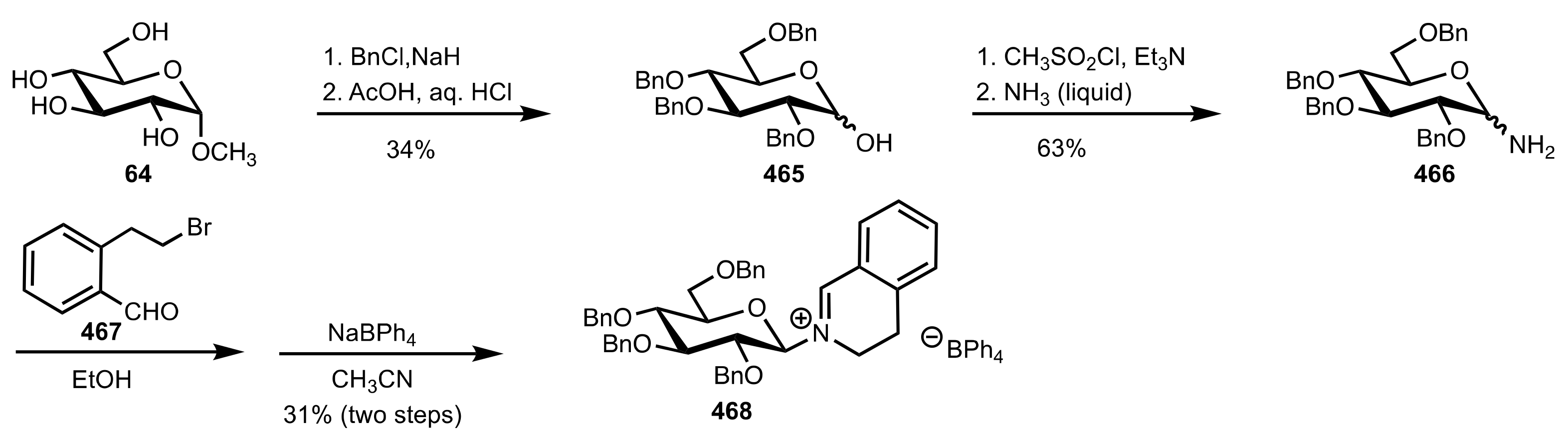
- Dorsey, A.D.; Barbarow, J.E.; Trauner, D. Reductive Cyclization of δ-Hydroxy Nitriles: A New Synthesis of Glycosylamines. Org. Lett. 2003, 5, 3237–3239. [Google Scholar] [CrossRef] [PubMed]
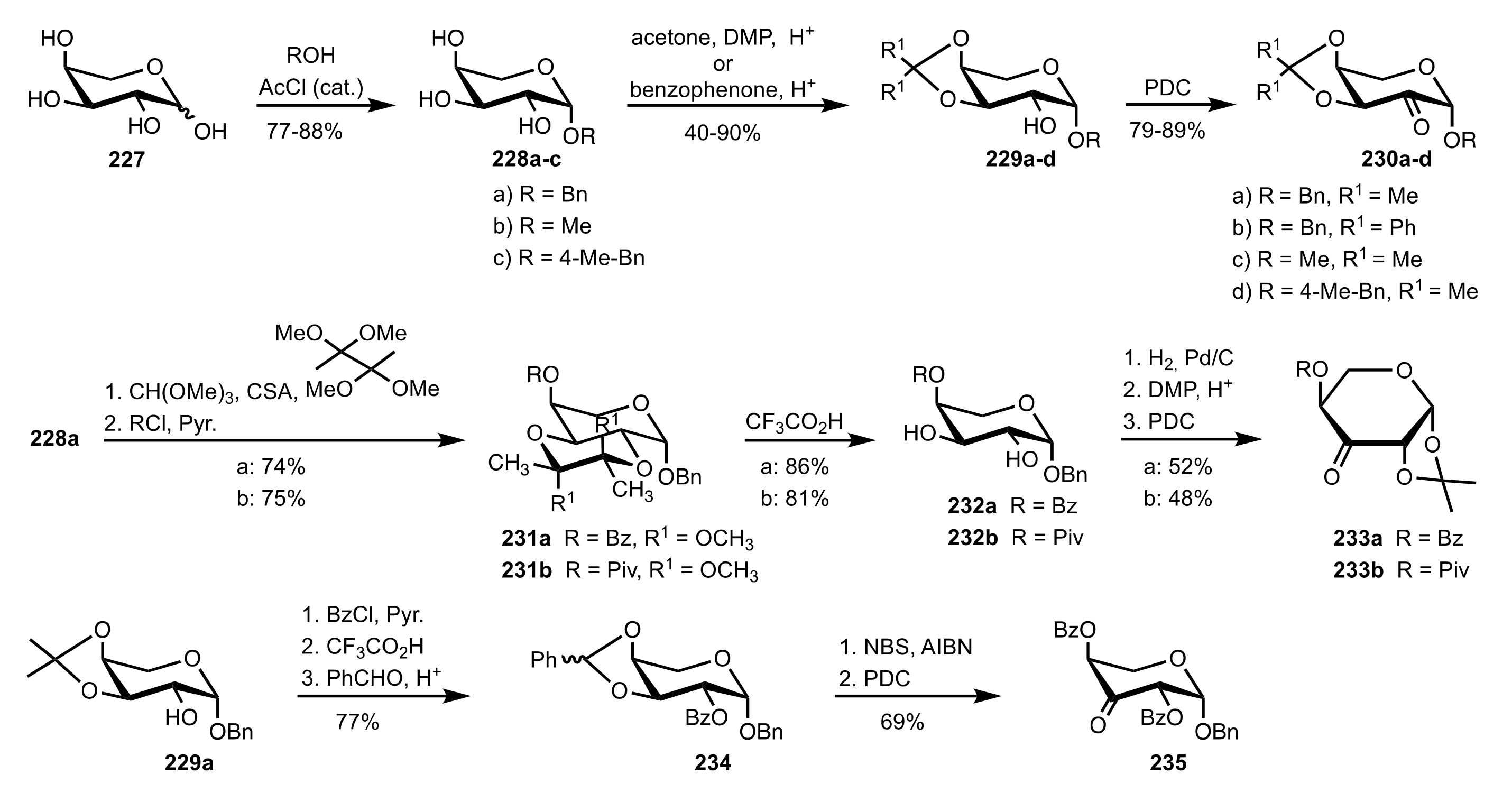
- Catelani, G.; Colonna, F.; Marra, A. An improved preparation of 3,4-O-isopropylidene derivatives of α- and β-d-galactopyranosides. Carbohydr. Res. 1988, 182, 297–300. [Google Scholar] [CrossRef]
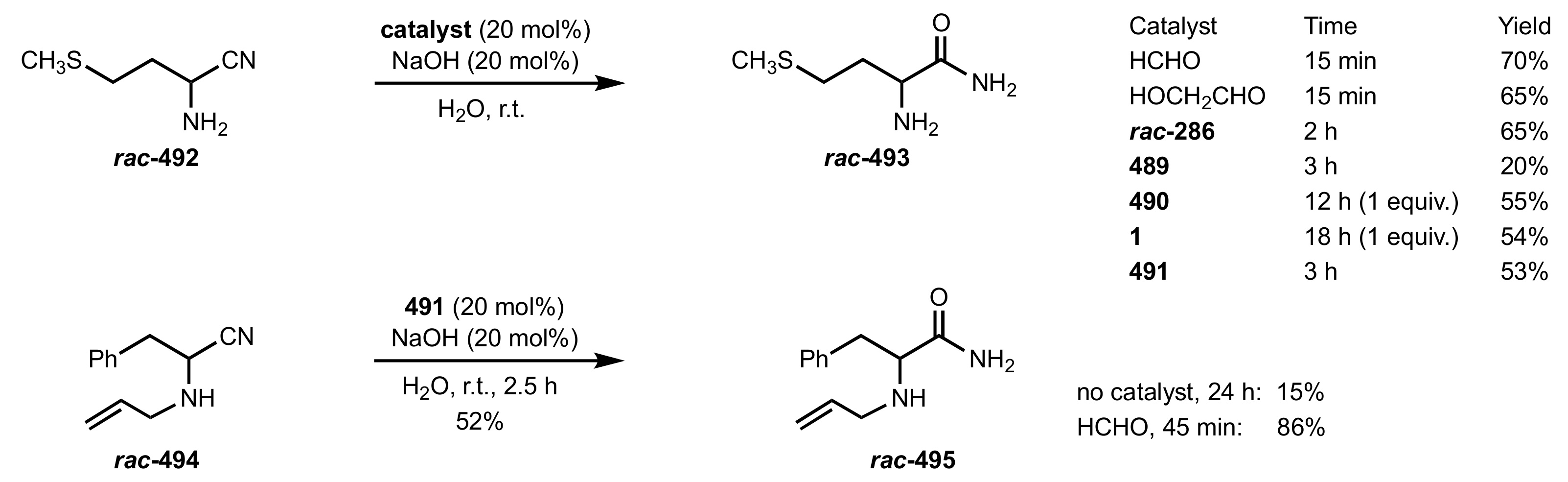
- Chitale, S.; Derasp, J.S.; Hussain, B.; Tanveer, K.; Beauchemin, A.M. Carbohydrates as efficient catalysts for the hydration of α-amino nitriles. Chem. Commun. 2016, 52, 13147–13150. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Pandey, J.; Tripathi, R.P. d-Glucosamine, a natural aminosugar as organocatalyst for an ecofriendly direct aldol reaction of ketones with aromatic aldehydes in water. Catal. Commun. 2008, 9, 743–746. [Google Scholar] [CrossRef]
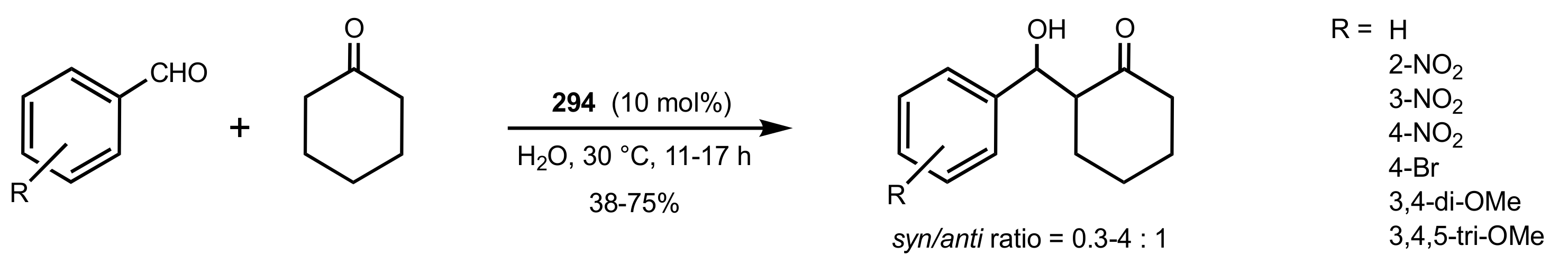
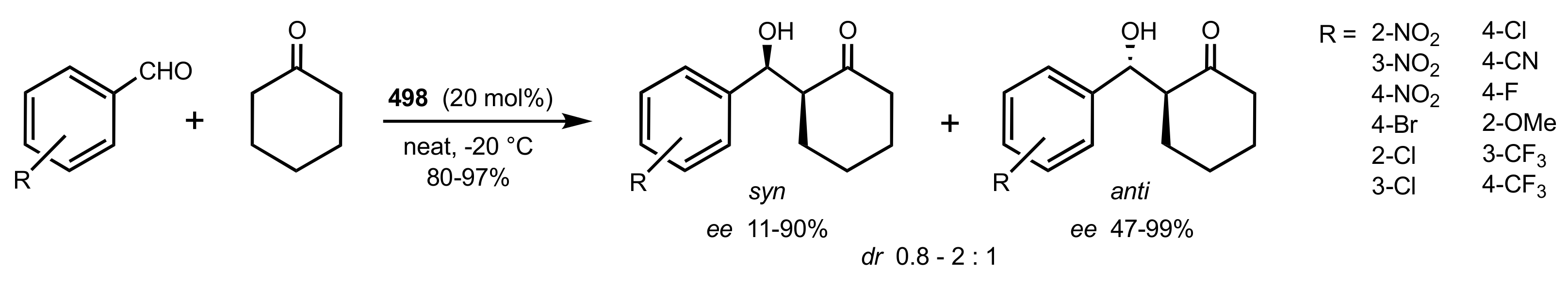
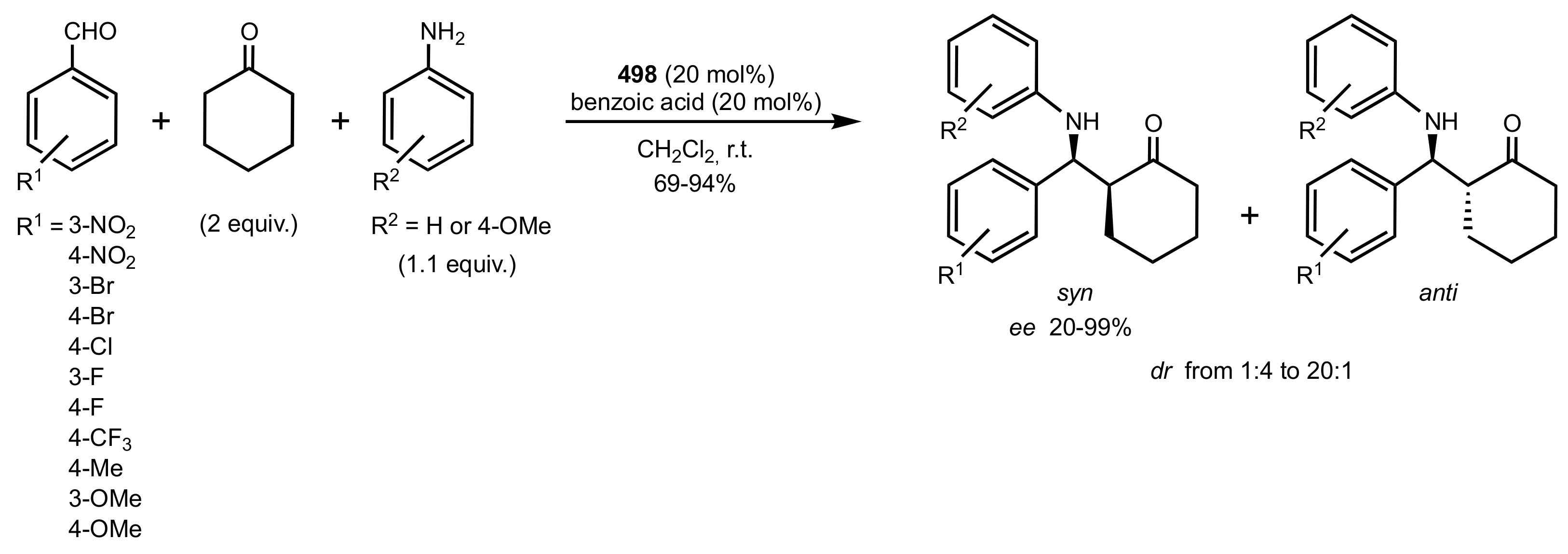
- Agarwal, J.; Peddinti, R.K. Glucosamine-Based Primary Amines as Organocatalysts for the Asymmetric Aldol Reaction. J. Org. Chem. 2011, 76, 3502–3505. [Google Scholar] [CrossRef]
- Sharma, A.; Peddinti, R.K. Direct Asymmetric Mannich Reaction Catalyzed by a d-Glucosamine-Derived Organocatalyst. Synlett 2018, 29, 630–634. [Google Scholar]
- Shen, C.; Shen, F.; Xia, H.; Zhang, P.; Chen, X. Carbohydrate-derived alcohols as organocatalysts in enantioselective aldol reactions of isatins with ketones. Tetrahedron Asymmetry 2011, 22, 708–712. [Google Scholar] [CrossRef]
- Shen, C.; Xia, H.J.; Zheng, H.; Zhang, P.; Chen, X. Synthesis of novel carbohydrate-based iminophosphinite ligands in Pd-catalyzed asymmetric allylic alkylations. Tetrahedron Asymmetry 2010, 21, 1936–1941. [Google Scholar] [CrossRef]
- Li, L.; Fang, Z.; Fang, J.; Zhoub, J.; Xiang, Y. d-Fructose-derived β-amino alcohol catalyzed direct asymmetric aldol reaction in the presence of p-nitrophenol. RSC Adv. 2013, 3, 21084–21091. [Google Scholar] [CrossRef]
- Li, L.; Fang, Z. Synthesis of novel β-amino-alcohols from d-fructose. Chem. Res. Appl. 2012, 24, 146–150. [Google Scholar]
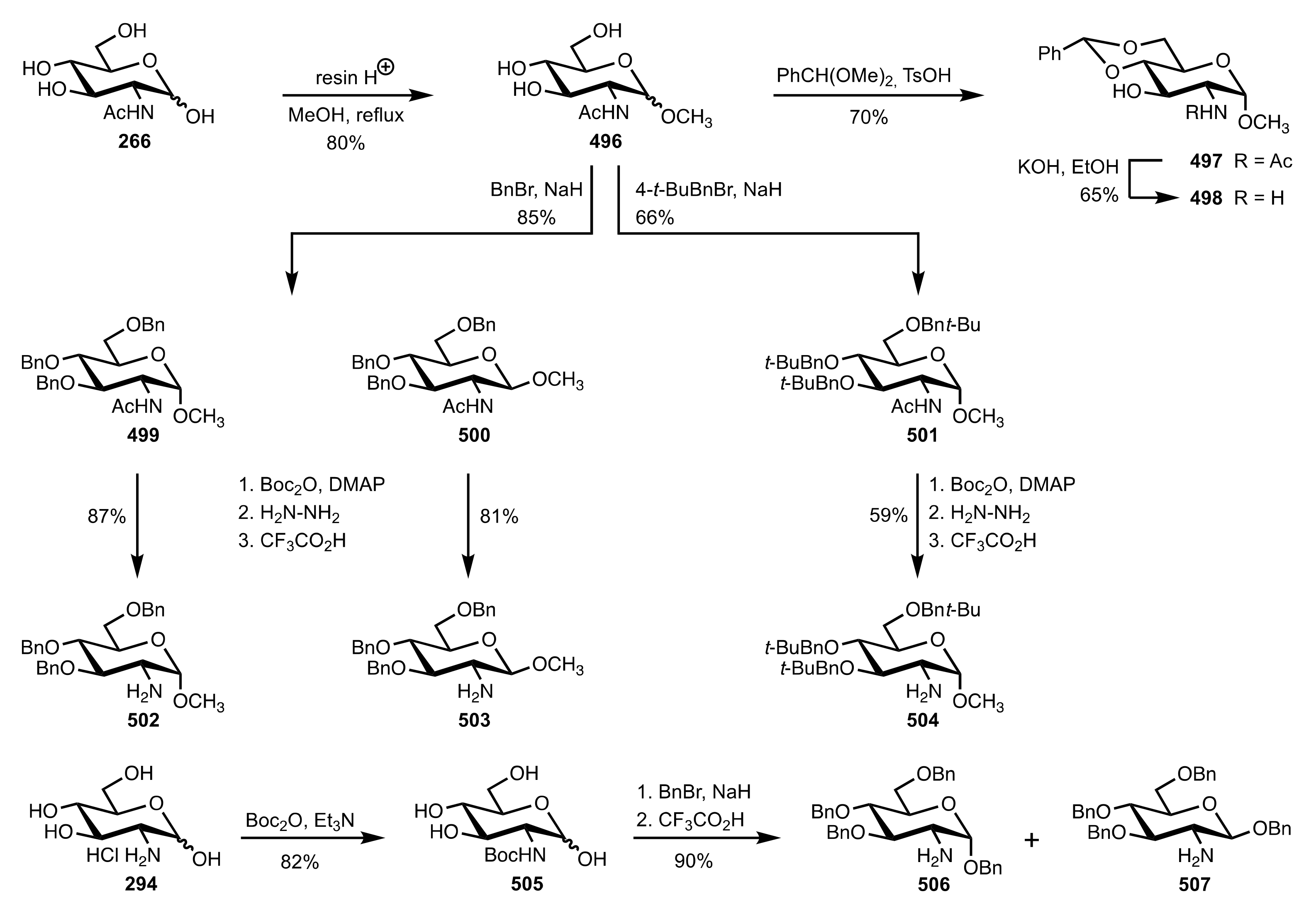
- Vanlaldinpuia, K.; Bora, P.; Bez, G. Monofunctional primary amine: A new class of organocatalyst for asymmetric Aldol reaction. J. Chem. Sci. 2017, 129, 301–312. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Frost, J.W. Biosynthesis of 1-Deoxy-1-imino-d-erythrose 4-Phosphate: A Defining Metabolite in the Aminoshikimate Pathway. J. Am. Chem. Soc. 2002, 124, 528–529. [Google Scholar] [CrossRef]
- Vanlaldinpuia, K.; Bora, P.; Basumatary, G.; Mohanta, R.; Bez, G. Enantioselective aminocatalysis: Michael addition of unactivated ketones to nitroolefins catalyzed by d-fructose derived monofunctional primary amine. J. Chem. Sci. 2017, 129, 1603–1610. [Google Scholar] [CrossRef]
- Reddy, K.R.; Rajgopal, K.; Maheswari, C.U.; Kantam, M.L. Chitosan hydrogel: A green and recyclable biopolymer catalyst for aldol and Knoevenagel reactions. New J. Chem. 2006, 30, 1549–1552. [Google Scholar] [CrossRef]
- Kühbeck, D.; Saidulu, G.; Reddy, K.R.; Díaz, D.D. Critical assessment of the efficiency of chitosan biohydrogel beads as recyclable and heterogeneous organocatalyst for C–C bond formation. Green Chem. 2012, 14, 378–392. [Google Scholar] [CrossRef] [Green Version]
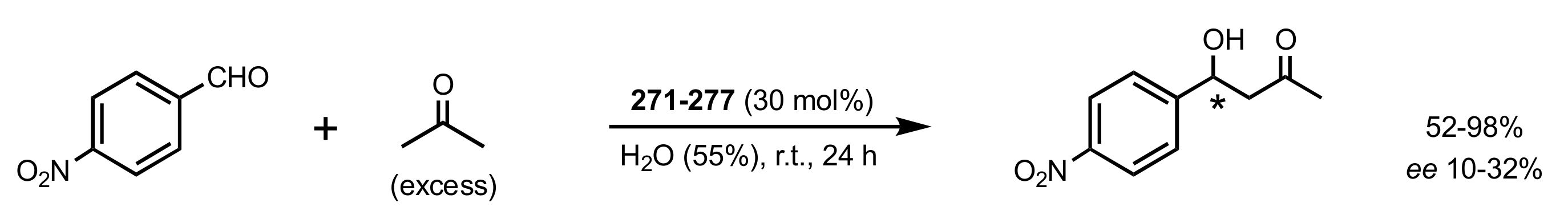
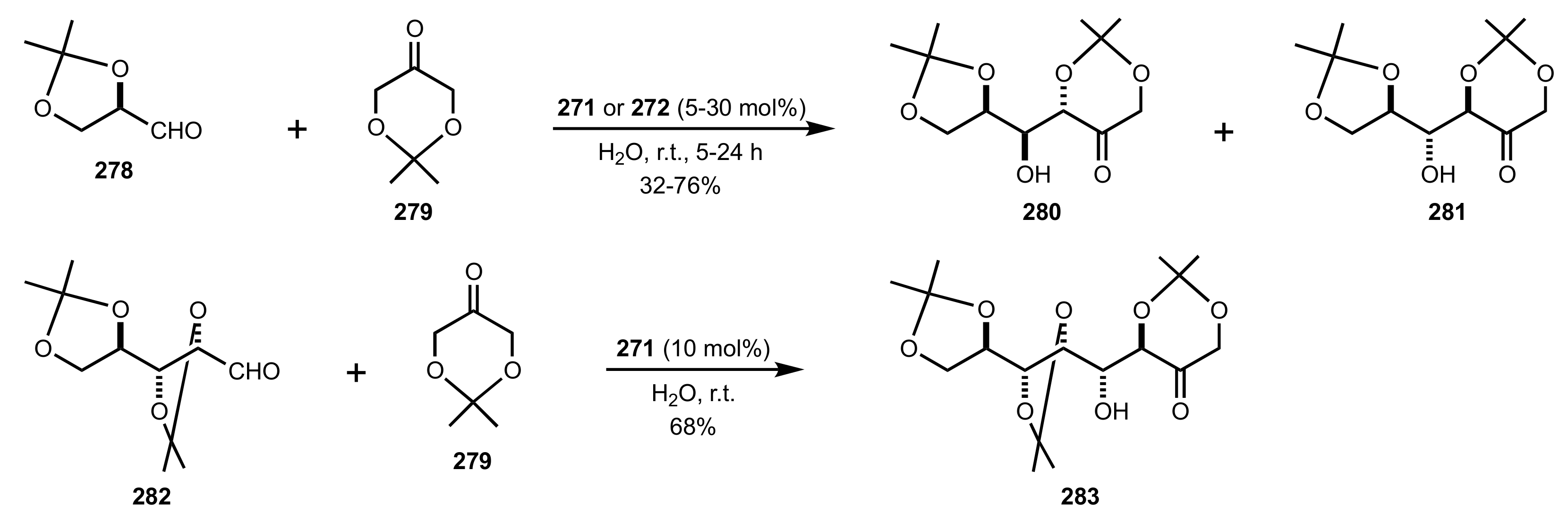
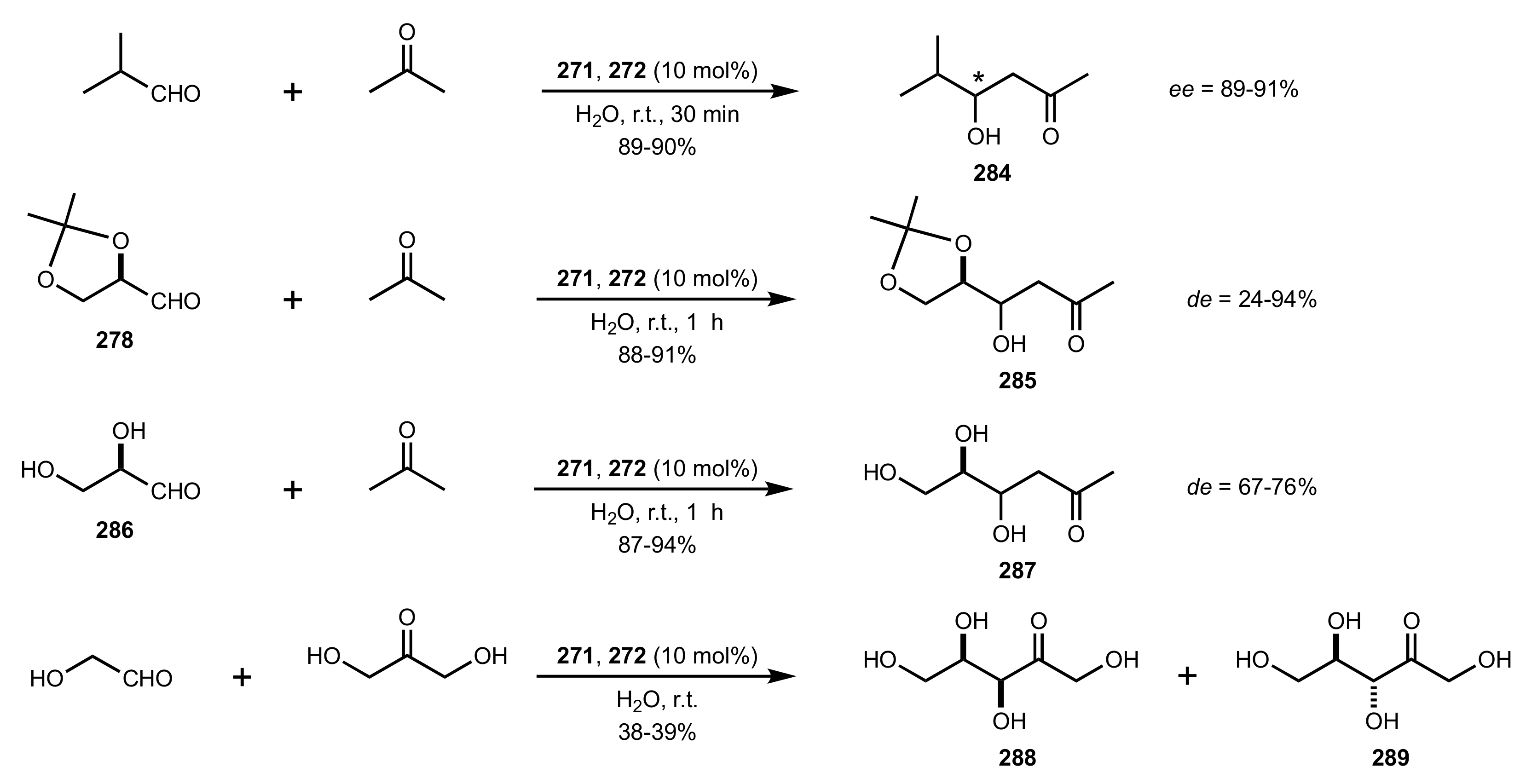
- Rani, D.; Singla, P.; Agarwal, J. ‘Chitosan in water’ as an eco-friendly and efficient catalytic system for Knoevenagel condensation reaction. Carbohydr. Polym. 2018, 202, 355–364. [Google Scholar] [CrossRef]
- Anbu, N.; Hariharan, S.; Dhakshinamoorthy, A. Knoevenagel-Doebner condensation promoted by chitosan as a reusable solid base catalyst. Mol. Catal. 2020, 484, 110744. [Google Scholar] [CrossRef]
- Anbu, N.; Maheswari, R.; Elamathi, V.; Varalakshmi, P.; Dhakshinamoorthy, A. Chitosan as a biodegradable heterogeneous catalyst for Knoevenagel condensation between benzaldehydes and cyanoacetamide. Catal. Commun. 2020, 138, 105954. [Google Scholar] [CrossRef]
- Hirayama, Y.; Kanomata, K.; Hatakeyama, M.; Kitaoka, T. Chitosan nanofiber-catalyzed highly selective Knoevenagel condensation in aqueous methanol. RSC Adv. 2020, 10, 26771–26776. [Google Scholar] [CrossRef]
- Kayser, H.; Müller, C.R.; García-González, C.A.; Smirnova, I.; Leitner, W.; de María, P.D. Dried chitosan-gels as organocatalysts for the production of biomass-derived platform chemicals. Appl. Catal. A Gen. 2012, 445–446, 180–186. [Google Scholar] [CrossRef]
- De Vylder, A.; Lauwaert, J.; De Clercq, J.; Van Der Voort, P.; Stevens, C.V.; Thybaut, J.W. Kinetic evaluation of chitosan-derived catalysts for the aldol reaction in water. React. Chem. Eng. 2019, 4, 1948–1956. [Google Scholar] [CrossRef]
- Siddiqui, I.R.; Rai, P.; Rahila; Srivastava, A. Chitosan: An efficient promoter for the synthesis of 2-aminopyrimidine-5-carbonitrile derivatives in solvent free conditions. New J. Chem. 2014, 38, 3791–3795. [Google Scholar] [CrossRef]
- Sahu, P.K.; Gupta, S.K.; Agarwal, D.D. Chitosan: An Efficient, Reusable, and Biodegradable Catalyst for Green Synthesis of Heterocycles. Ind. Eng. Chem. Res. 2014, 53, 2085–2091. [Google Scholar] [CrossRef]
- Shivhare, K.N.; Siddiqui, I.R. Chitosan: A Natural and Sustainable Polymeric Organocatalyst for C-C Bond Formation During the Synthesis of 5-amino-2,3-dihydrobenzo[d]thiazole-4,6-dicarbonitriles. Curr. Organocatal. 2019, 6, 69–76. [Google Scholar] [CrossRef]
- Jose, T.; Sudheesh, N.; Shukla, R.S. Amino functionalized chitosan as a catalyst for selective solvent-free self-condensation of linear aldehydes. J. Mol. Catal. A Chem. 2010, 333, 158–166. [Google Scholar] [CrossRef]
- Han, X.; Li, Y.; An, H.; Zhao, X.; Wang, Y. Chitosan-catalyzed n-butyraldehyde self-condensation reaction mechanism and kinetics. Chin. J. Chem. Eng. 2019, 27, 2447–2454. [Google Scholar] [CrossRef]
- Valentin, R.; Molvinger, K.; Quignard, F.; Brunel, D. Supercritical CO2 dried chitosan: An efficient intrinsic heterogeneous catalyst in fine chemistry. New J. Chem. 2003, 27, 1690–1692. [Google Scholar] [CrossRef]
- Zhao, Y.; Tian, J.-S.; Qi, X.-H.; Han, Z.-N.; Zhuang, Y.-Y.; He, L.-N. Quaternary ammonium salt-functionalized chitosan: An easily recyclable catalyst for efficient synthesis of cyclic carbonates from epoxides and carbon dioxide. J. Mol. Catal. A Chem. 2007, 271, 284–289. [Google Scholar] [CrossRef]
- Dekamin, M.G.; Azimoshan, M.; Ramezani, L. Chitosan: A highly efficient renewable and recoverable bio-polymer catalyst for the expeditious synthesis of α-amino nitriles and imines under mild conditions. Green Chem. 2013, 15, 811–820. [Google Scholar] [CrossRef]
- Rao, S.N.; Mohan, D.C.; Adimurthy, S. Chitosan: An efficient recyclable catalyst for transamidation of carboxamides with amines under neat conditions. Green Chem. 2014, 16, 4122–4126. [Google Scholar]
- Reddy, S.R.S.; Reddy, B.R.P.; Reddy, P.V.G. Chitosan: Highly efficient, green, and reusable biopolymer catalyst for the synthesis of alkylaminophenols via Petasis borono–Mannich reaction. Tetrahedron Lett. 2015, 56, 4984–4989. [Google Scholar] [CrossRef]
- Zhaleh, S.; Hazeri, N.; Faghihi, M.R.; Maghsoodlou, M.T. Chitosan: A sustainable, reusable and biodegradable organocatalyst for green synthesis of 1,4-dihydropyridine derivatives under solvent-free condition. Res. Chem. Intermed. 2016, 42, 8069–8081. [Google Scholar] [CrossRef]
- Asghari-Haji, F.; Rad-Moghadam, K.; Mahmoodi, N.O. An efficient approach to bis-benzoquinonylmethanes on water under catalysis of the bioderived O-carboxymethyl chitosan. RSC Adv. 2016, 6, 27388–27394. [Google Scholar] [CrossRef]
- Shelke, P.B.; Mali, S.N.; Chaudhari, H.K.; Pratap, A.P. Chitosan hydrochloride mediated efficient, green catalysis for the synthesis of perimidine derivatives. J. Heterocycl. Chem. 2019, 56, 3048–3054. [Google Scholar] [CrossRef]
- Tsutsumi, Y.; Koga, H.; Qi, Z.-D.; Saito, T.; Isogai, A. Nanofibrillar Chitin Aerogels as Renewable Base Catalysts. Biomacromolecules 2014, 15, 4314–4319. [Google Scholar] [CrossRef]
- Koga, H.; Kitaoka, T.; Isogai, A. In situ modification of cellulose paper with amino groups for catalytic applications. J. Mater. Chem. 2011, 21, 9356–9361. [Google Scholar] [CrossRef]
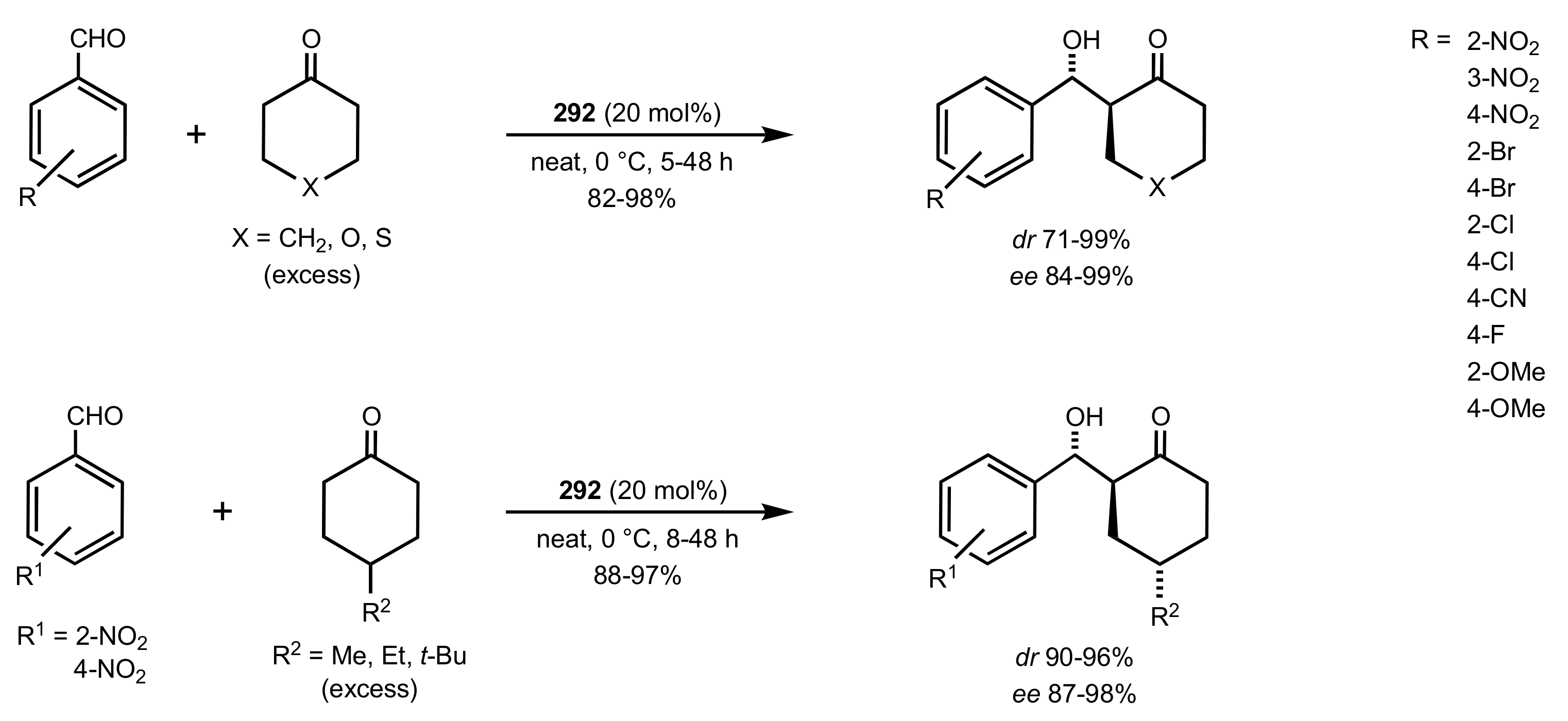
- Kumar, K.S.; Krishna, B.S.; Reddy, C.B.; Reddy, M.V.N.; Reddy, C.S. Solvent-free synthesis of α-aminophosphonates: Cellulose-SO3H as an efficient catalyst. Arab. J. Chem. 2017, 10, S368–S375. [Google Scholar] [CrossRef]
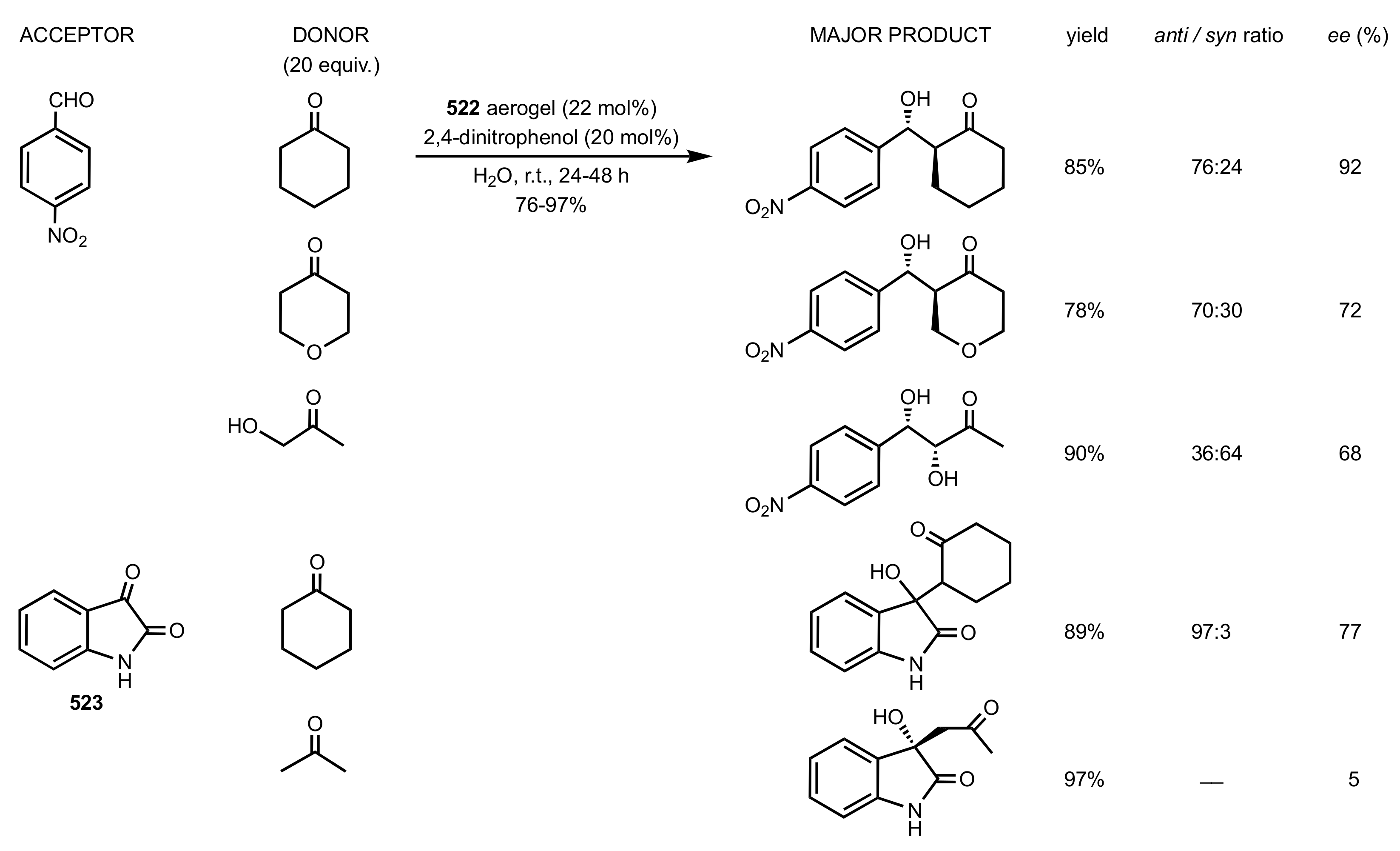
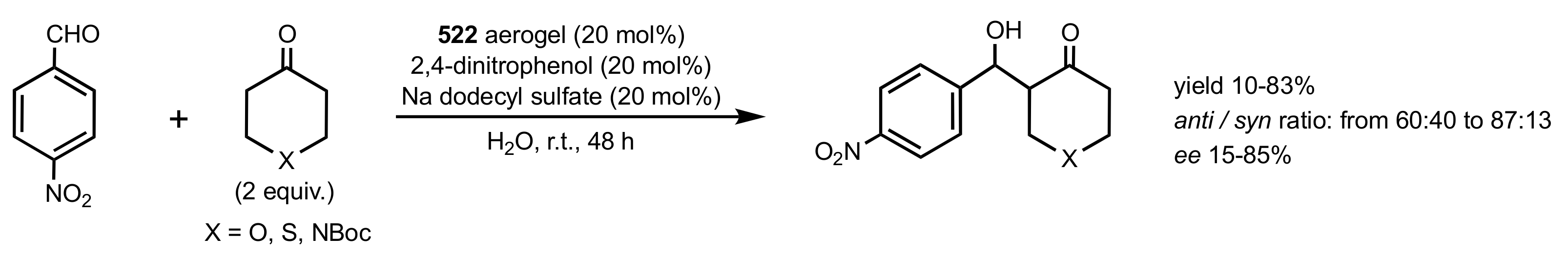
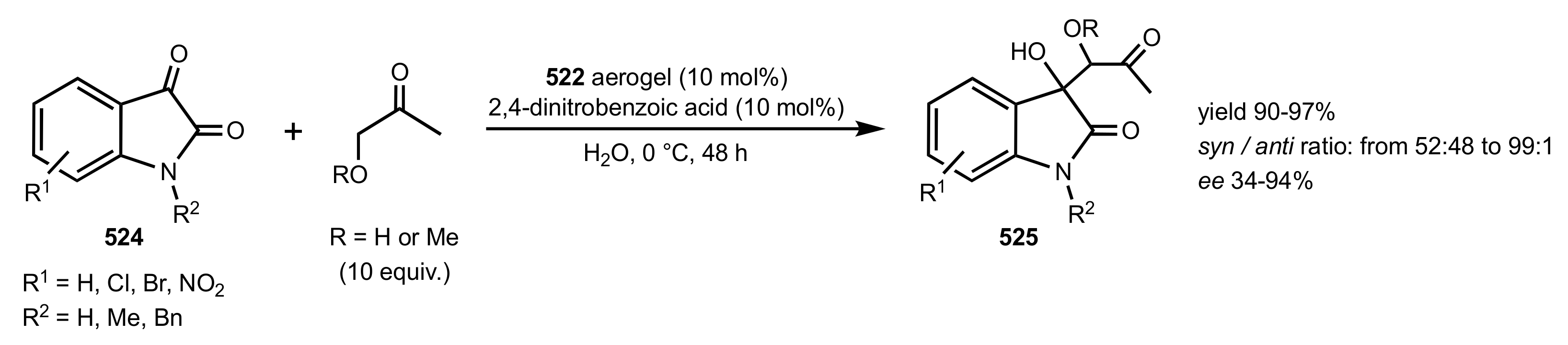
- Ricci, A.; Bernardi, L.; Gioia, C.; Vierucci, S.; Robitzer, M.; Quignard, F. Chitosan aerogel: A recyclable, heterogeneous organocatalyst for the asymmetric direct aldol reaction in water. Chem. Commun. 2010, 46, 6288–6290. [Google Scholar] [CrossRef]
- Gioia, C.; Ricci, A.; Bernardi, L.; Bourahla, K.; Tanchoux, N.; Robitzer, M.; Quignard, F. Chitosan Aerogel Beads as a Heterogeneous Organocatalyst for the Asymmetric Aldol Reaction in the Presence of Water: An Assessment of the Effect of Additives. Eur. J. Org. Chem. 2013, 2013, 588–594. [Google Scholar] [CrossRef]
- Dong, H.; Liu, J.; Ma, L.; Ouyang, L. Chitosan Aerogel Catalyzed Asymmetric Aldol Reaction in Water: Highly Enantioselective Construction of 3-Substituted-3-hydroxy-2-oxindoles. Catalyst 2016, 6, 186. [Google Scholar] [CrossRef] [Green Version]
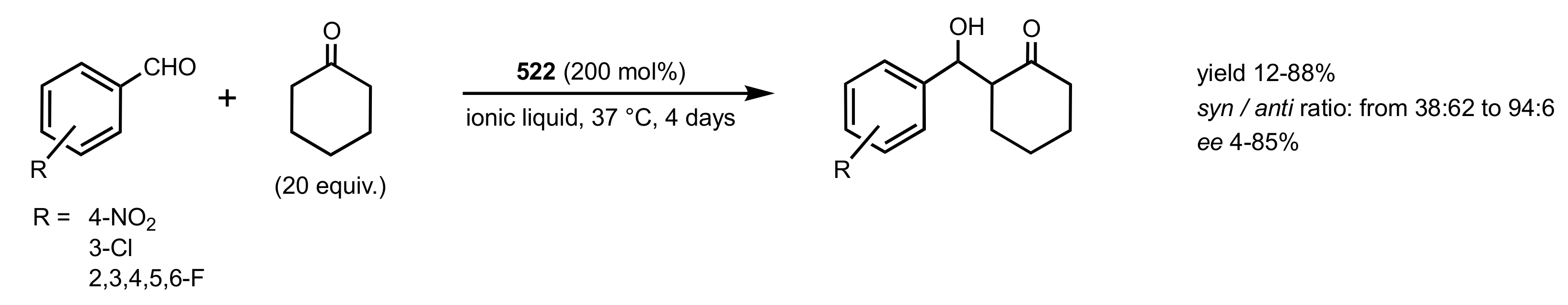
- Heckel, T.; Konieczna, D.D.; Wilhelm, R. An Ionic Liquid Solution of Chitosan as Organocatalyst. Catalyst 2013, 3, 914–921. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.; Zhao, W.; Yang, L.; Zhang, X.; Cui, Y. Chitosan-Based Heterogeneous Catalysts for Enantioselective Michael Reaction. Chirality 2012, 24, 640–645. [Google Scholar] [CrossRef]
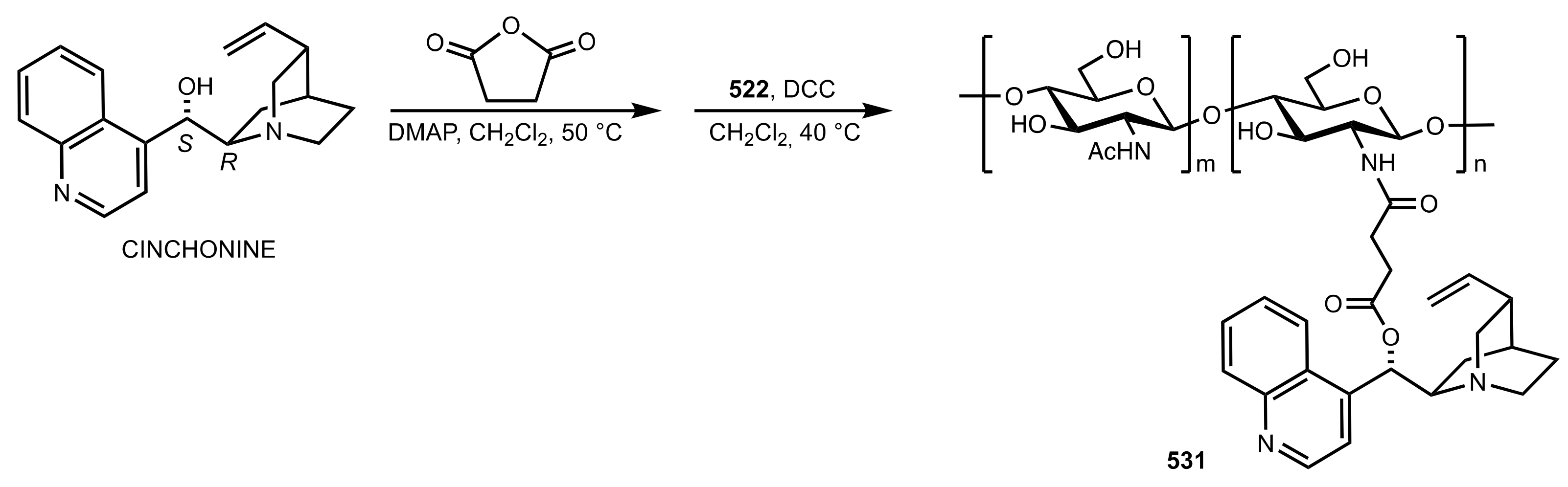
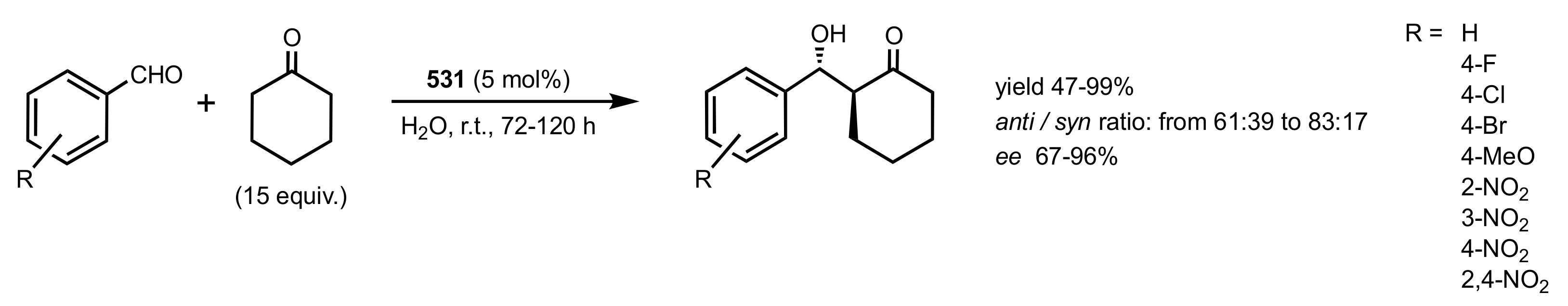
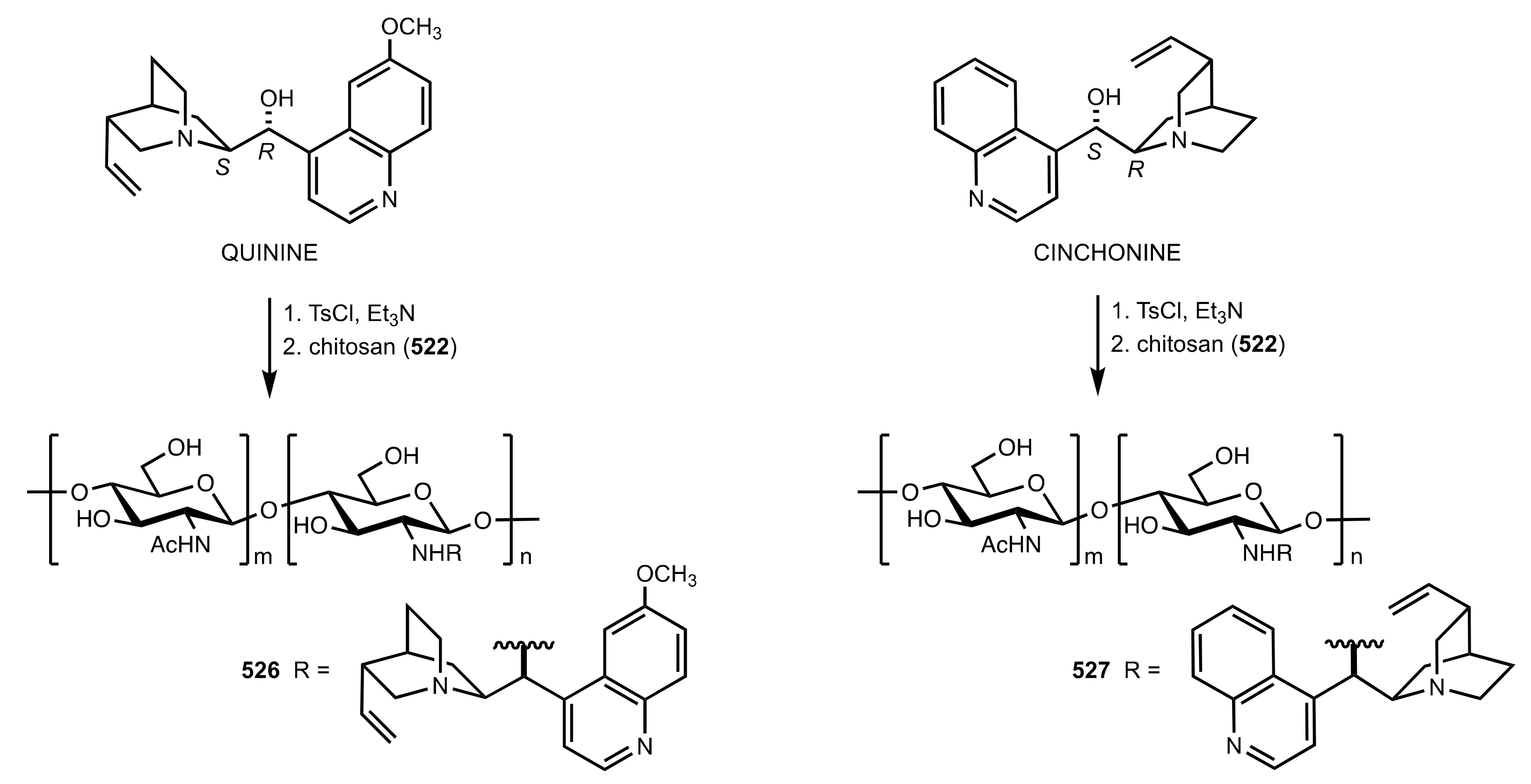
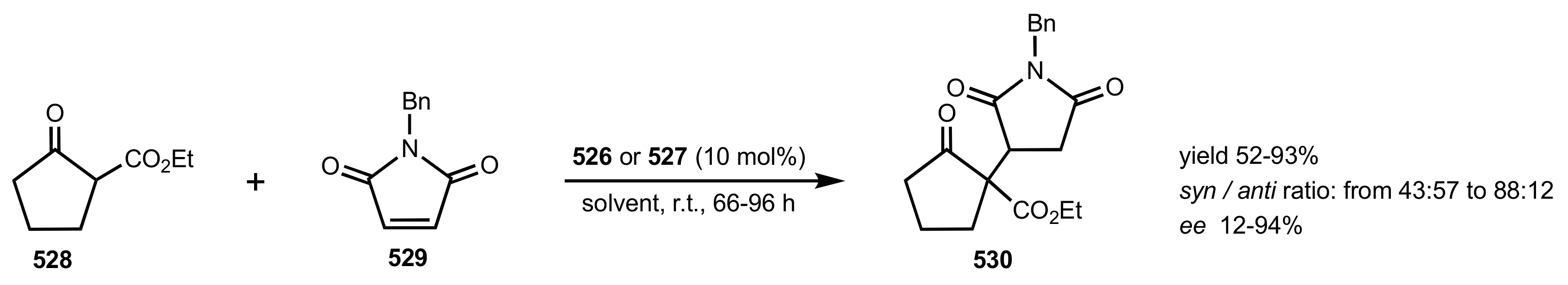
- Zhao, W.; Qu, C.; Yang, L.; Cui, Y. Chitosan-supported cinchonine as an efficient organocatalyst for direct asymmetric aldol reaction in water. Chin. J. Catal. 2015, 36, 367–371. [Google Scholar] [CrossRef]
- Abdelkawy, M.A.; Aly, E.A.; El-Badawi, M.A.; Itsuno, S. Chitosan-supported cinchona urea: Sustainable organocatalyst for asymmetric Michael reaction. Catal. Commun. 2020, 146, 106132. [Google Scholar] [CrossRef]
- Zhang, H.; Zhao, W.; Zou, J.; Liu, Y.; Li, R.; Cui, Y. Aldol Reaction Catalyzed by a Hydrophilic Catalyst in Aqueous Micelle as an Enzyme Mimic System. Chirality 2009, 21, 492–496. [Google Scholar] [CrossRef]
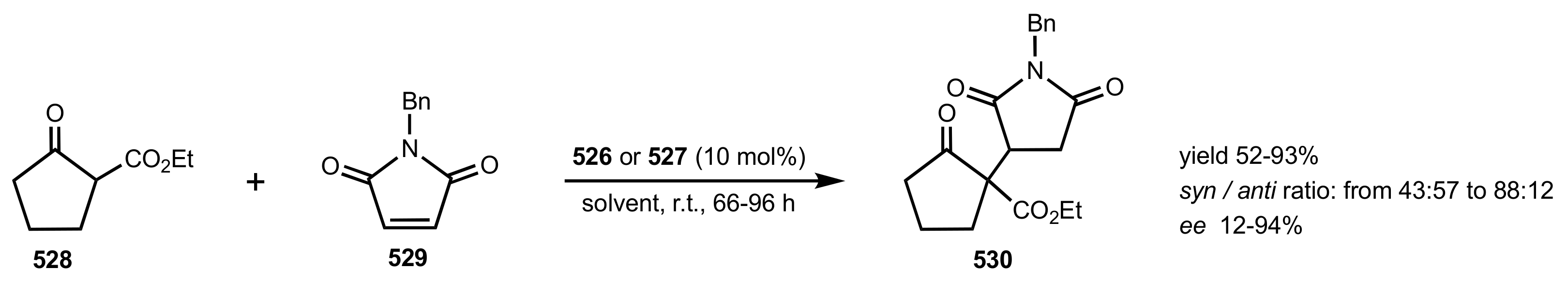
- Andrés, J.M.; González, F.; Maestro, A.; Pedrosa, R.; Valle, M. Biodegradable Chitosan-Derived Thioureas as Recoverable Supported Organocatalysts—Application to the Stereoselective Aza-Henry Reaction. Eur. J. Org. Chem. 2017, 2017, 3658–3665. [Google Scholar] [CrossRef]
- Andrés, J.M.; Maestro, A.; Rodríguez-Ferrer, P.; Simón, I.; Pedrosa, R. Short Synthesis of Novel Recyclable Chiral Bifunctional Thioureas from Aminoalkyl Polystyrene and their use as Organocatalysts in Stereoselective aza-Henry Reaction. ChemistrySelect 2016, 1, 5057–5061. [Google Scholar] [CrossRef] [Green Version]
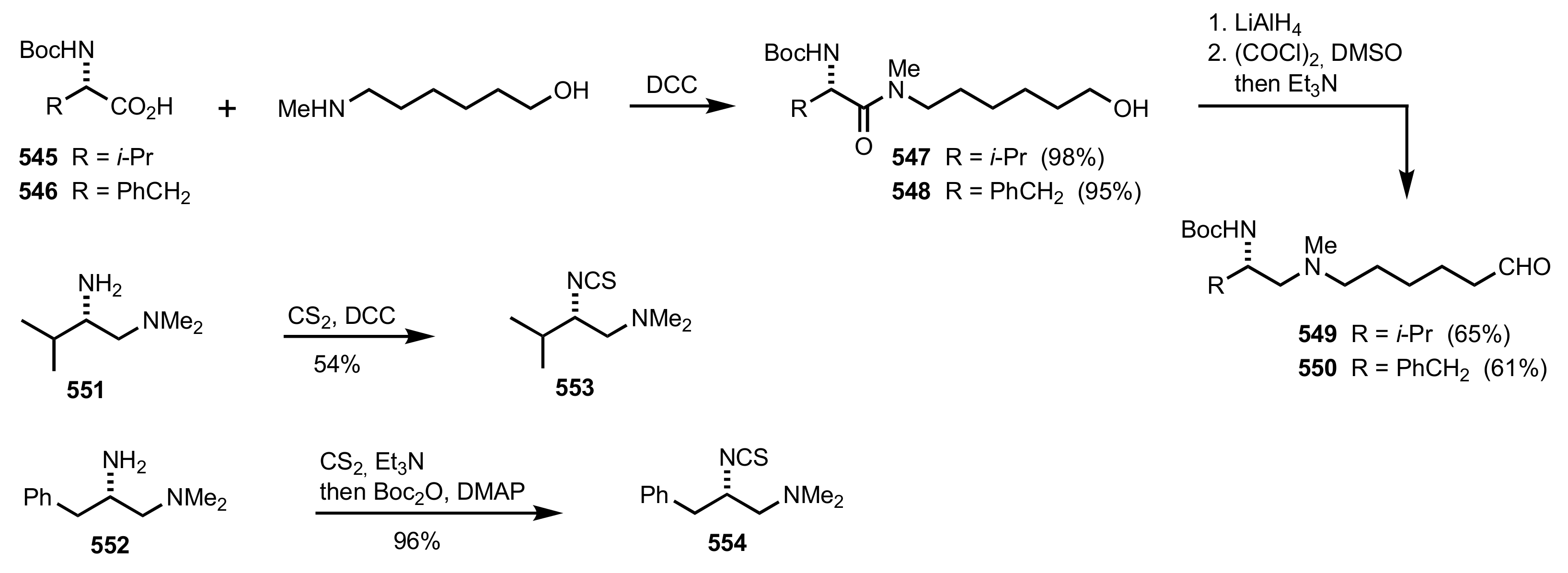
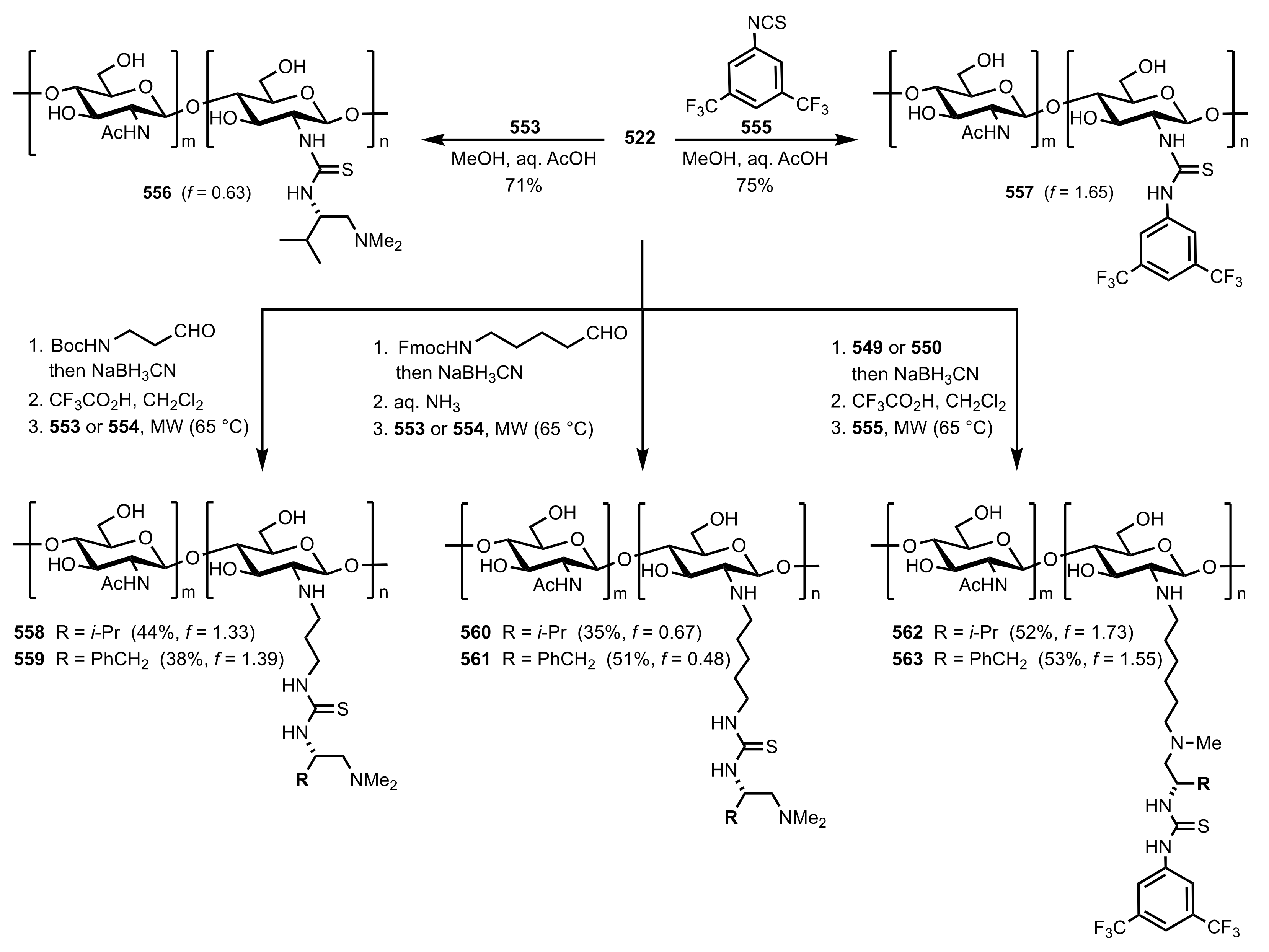
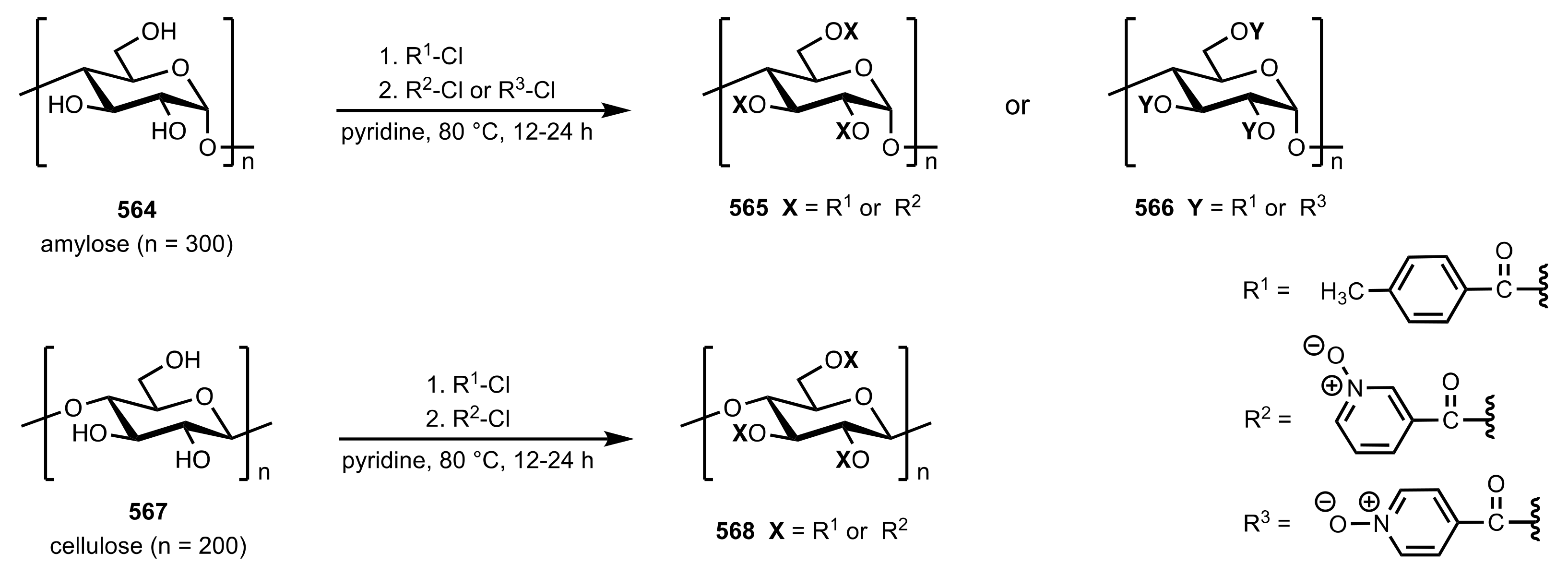
- Ikai, T.; Moro, M.; Maeda, K.; Kanoh, S. Synthesis of polysaccharide derivatives bearing pyridine N-oxide groups and their use as asymmetric organocatalysts. Eur. React. Funct. Polym. 2011, 71, 1055–1058. [Google Scholar] [CrossRef]
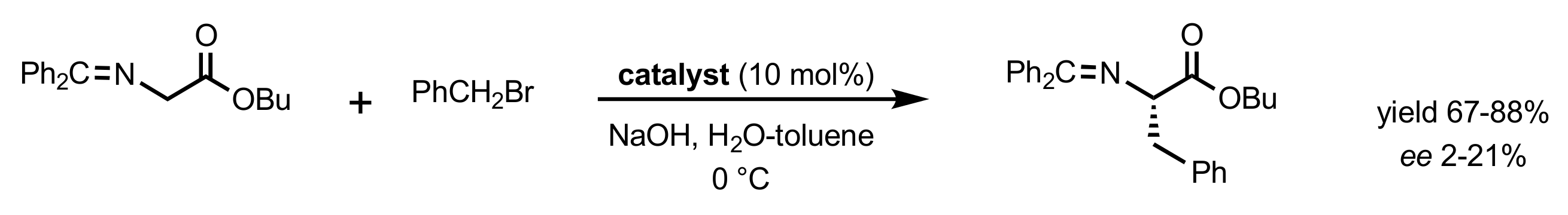
- For a review, see: Mąkosza, M.; Fedoryński, M. Interfacial Processes—The Key Steps of Phase Transfer Catalyzed Reactions. Catalysts 2020, 10, 1436. [Google Scholar] [CrossRef]
- Starks, C.M. Phase-transfer catalysis. I. Heterogeneous reactions involving anion transfer by quaternary ammonium and phosphonium salts. J. Am. Chem. Soc. 1971, 93, 195–199. [Google Scholar]
- For a review, see: Nakamura, T.; Okuno, K.; Nishiyori, R.; Shirakawa, S. Hydrogen-Bonding Catalysis of Alkyl-Onium Salts. Chem. Asian J. 2020, 15, 463–472. [Google Scholar] [CrossRef]
- For a review, see: Ooi, T.; Maruoka, K. Recent advances in asymmetric phase-transfer catalysis. Angew. Chem. Int. Ed. 2007, 46, 4222–4266. [Google Scholar] [CrossRef]
- For a review, see: Wang, H. Chiral Phase-Transfer Catalysts with Hydrogen Bond: A Powerful Tool in the Asymmetric Synthesis. Catalysts 2019, 9, 244. [Google Scholar] [CrossRef] [Green Version]
- For a review, see: Kaneko, S.; Kumatabara, Y.; Shirakawa, S. A new generation of chiral phase-transfer catalysts. Org. Biomol. Chem. 2016, 14, 5367–5376. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, C.J.; Frensdorff, H.K. Macrocyclic Polyethers and Their Complexes. Angew. Chem. Int. Ed. Engl. 1972, 11, 16–25. [Google Scholar] [CrossRef]
- Liotta, C.L.; Harris, H.P.J. The Chemistry of “Naked” Anions. I. Reactions of the 18-Crown-6 Complex of Potassium Fluoride with Organic Substrates in Aprotic Organic Solvents. J. Am. Chem. Soc. 1974, 96, 2250–2252. [Google Scholar] [CrossRef]
- Cram, D.; Sogah, G.D.Y. Chiral crown complexes catalyse Michael addition reactions to give adducts in high optical yields. J. Chem. Soc. Chem. Commun. 1981, 1981, 625–628. [Google Scholar] [CrossRef]
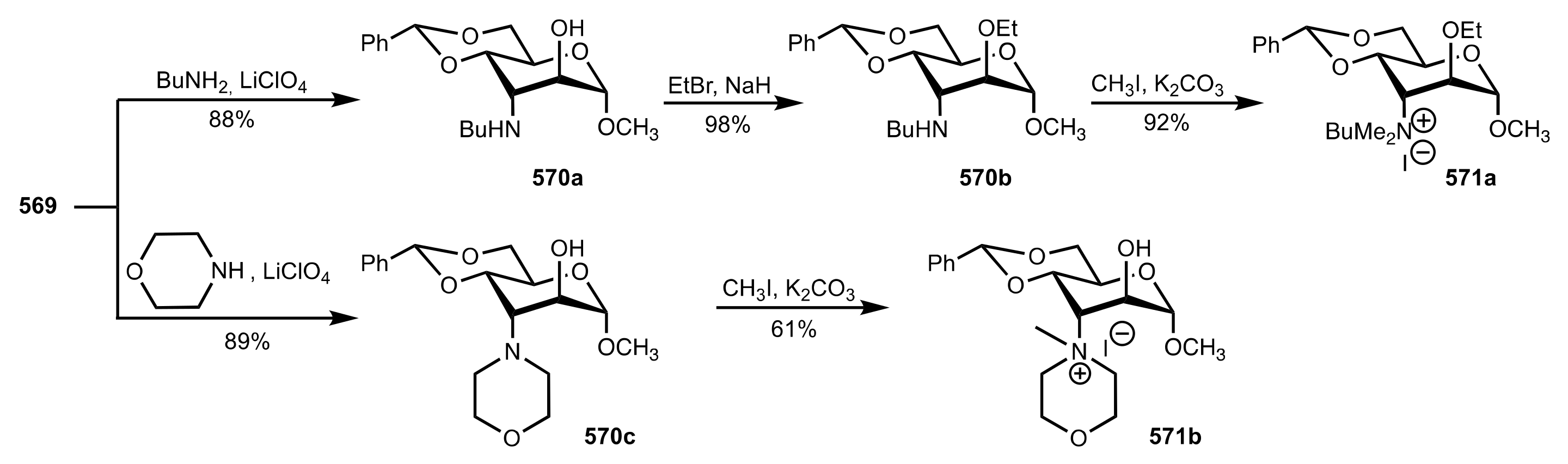
- Nemcsok, T.; Rapi, Z.; Bagi, P.; Bakó, P. Synthesis and application of novel carbohydrate-based ammonium and triazolium salts. Synth. Commun. 2019, 49, 2388–2400. [Google Scholar] [CrossRef] [Green Version]
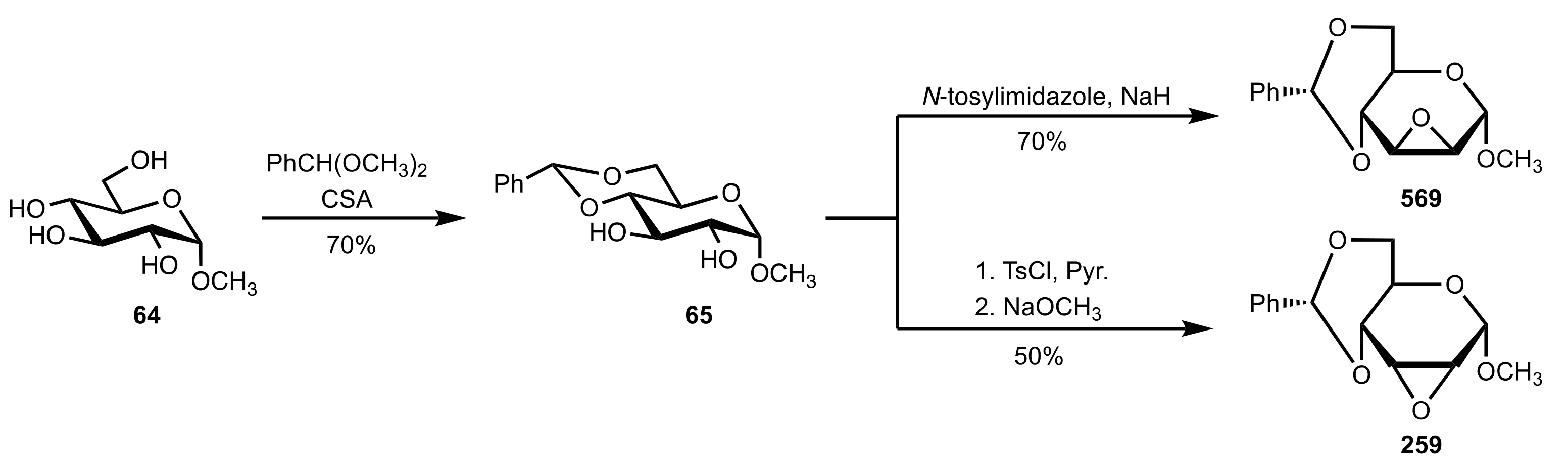
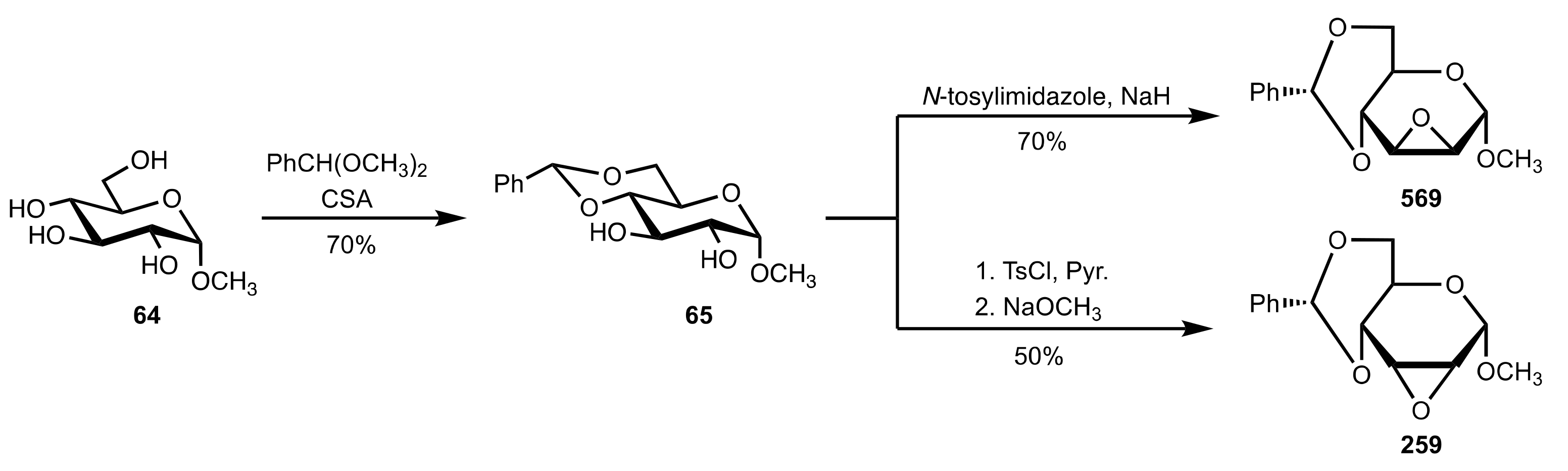
- Hicks, D.R.; Fraser-Reid, B. Selective sulphonylation with N-Tosylimidazole. A one-Step Preparation of Methyl 2,3-Anhydro-4,6-O-benzylidene-α-d-mannopyranoside. Synthesis 1974, 1974, 203–204. [Google Scholar] [CrossRef]
- Comba, M.B.; Mangione, M.I.; Suárez, A.G.; Sarotti, A.M.; Spanevello, R.A. A Domino Epoxide Ring-Opening Xanthate Migration Reaction: An Alternative Entry to Thiosugars. Eur. J. Org. Chem. 2018, 2018, 6848–6856. [Google Scholar] [CrossRef]
- Goyal, N.; Cheuk, S.; Wang, G. Synthesis and characterization of d-glucosamine-derived low molecular weight gelators. Tetrahedron 2010, 66, 5962–5971. [Google Scholar] [CrossRef]
- Dehmlow, E.V.; Sauerbier, C. Enantioselektive Phasen transfer-Kata1yse durch optisch aktive Kronenether. Liebigs Ann. Chem. 1989, 1989, 181–185. [Google Scholar] [CrossRef]
- Curtis, W.D.; Laidler, D.A.; Stoddart, J.F.; Jones, G.H. Synthesis of configurationally chiral cryptands and cryptates from carbohydrate precursors. J. Chem. Soc. Chem. Commun. 1975, 1975, 833–835. [Google Scholar] [CrossRef]
- Nemcsok, T.; Rapi, Z.; Keglevich, G.; Grün, A.; Bakó, P. Synthesis of d-mannitol-based crown ethers and their application as catalyst in asymmetric phase transfer reactions. Chirality 2018, 30, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, T.; Sugawara, H.; Watanabe, M.; Yamashita, K. Facile synthesis of 1,2:5,6-Di-O-cyclohexylidene-d-mannitol and 2,3-O-cyclohexylidene-d-glyceraldehyde. Agric. Biol. Chem. 1984, 48, 1841–1844. [Google Scholar]
- Banerjee, A.; Schepmann, D.; Köhler, J.; Würthwein, E.U.; Wünsch, B. Synthesis and SAR studies of chiral non-racemic dexoxadrol analogues as uncompetitive NMDA receptor antagonists. Bioorg. Med. Chem. 2010, 18, 7855–7867. [Google Scholar] [CrossRef] [PubMed]
- Ness, A.T.; Hahn, R.M.; Hudson, C.S. The Acetolysis of Trimethylene-d-sorbitol. 2,4-Methylene-d-sorbitol and 1,3:2,4-Dimethylene-d-sorbitol. J. Am. Chem. Soc. 1944, 66, 665–670. [Google Scholar] [CrossRef]
- Van Maarschalkerwaart, D.A.H.; Willard, N.P.; Pandit, U.K. Synthesis of Carbohydrate containing Crown Ethers and their application as Catalysts in Asymmetric Michael Additions. Tetrahedron 1992, 48, 8825–8840. [Google Scholar] [CrossRef]
- Nakaya, T.; Imoto, M. Plastic Scintillators. II. The Synthesis of Some Distyrylbenzene Derivatives as Wavelength Shifters in Plastic Scintillators. Bull. Chem. Soc. Jpn. 1966, 39, 1547–1551. [Google Scholar] [CrossRef]
- Kanakamma, P.P.; Mani, N.S.; Maitra, U.; Nair, V. Chiral crown ethers incorporating d-glucose. J. Chem. Soc. Perkin Trans. 1 1995, 1995, 2339–2344. [Google Scholar] [CrossRef]
- Vicent, C.; Martin-Lomas, M.; Penadés, S. New synthetic strategy to highly symmetric chiral macrocycles from carbohydrate derivatives. Tetrahedron 1989, 45, 3605–3612. [Google Scholar] [CrossRef]
- Kaji, E.; Hosokawa, Y.; Watanabe, Y.; Kobayashi, M.; Yamakawa, M. Stereospecific access to β-mannosides from glucose-derived 1,2-orthoesters as glycosyl donors. Heterocycles 2003, 61, 459–470. [Google Scholar] [CrossRef]
- Bakó, P.; Fenichel, L.; Tőke, L.; Czugler, M. Synthese und Komplexbildung von Kronenethern aus Methyl-4,6-O-benzyliden-α-d-glucopyranosid. Liebigs Ann. Chem. 1981, 1981, 1163–1171. [Google Scholar] [CrossRef]
- Bakó, P.; Fenichel, L.; Tőke, L.; Toth, G. Cleavage of the acetal rings in bis (methyl 4,6-O-benzylidene-α-d-glucopyranosido)-18-crown-6. Carbohydr. Res. 1986, 147, 31–37. [Google Scholar] [CrossRef]
- Tőke, L.; Bakó, P.; Keserű, G.M.; Albert, M.; Fenichel, L. Asymmetric Michael Addition and Deracemization of Enolate by Chiral Crown Ether. Tetrahedron 1998, 54, 213–222. [Google Scholar] [CrossRef]
- Tőke, L.; Fenichel, L.; Albert, M. Asymmetric Michael Addition. Deracemization of Enolate by Chiral Crown Ether. Tetrahedron Lett. 1995, 33, 5951–5954. [Google Scholar] [CrossRef]
- Alonso-Lopez, M.; Bernabe, M.; Fernandez-Mayoralas, A.; Jimenez-Barbero, J.; Martin-Lomas, M.; Penadés, S. Chiral macrocyclic compounds from lactose derivatives. Carbohydr. Res. 1986, 150, 103–109. [Google Scholar] [CrossRef]
- Fernandez-Mayoralas, A.; Martin-Lomas, M. 4-O-β-d-Galactopyranosyl-3-O-methyl-d-glucose: A new synthesis and application to the evaluation of intestinal lactase. Carbohydr. Res. 1985, 140, 81–91. [Google Scholar] [CrossRef]
- Fernandez-Mayoralas, A.; Martin-Lomas, M. Synthesis of 3- and 2′-fucosyl-lactose and 3,2′-difucosyl-lactose from partially benzylated lactose derivatives. Carbohydr. Res. 1986, 154, 95–101. [Google Scholar] [CrossRef]
- Alonso-Lopez, M.; Barbat, J.; Fanton, E.; Fernandez-Mayoralas, A.; Gelas, J.; Horton, D.; Martin-Lomas, M.; Penadés, S. The acetonation of lactose and benzyl β-lactoside with 2-methoxypropene. Tetrahedron 1987, 43, 1169–1176. [Google Scholar] [CrossRef]
- Alonso-Lopez, M.; Jimenez-Barbero, J.; Martin-Lomas, M.; Penadés, S. Synthesis, complexing properties and applications in asymmetric synthesis of bis-lacto-18-crown-6 compounds. Tetrahedron 1988, 44, 1535–1543. [Google Scholar] [CrossRef]
- Alonso-Lopez, M.; Martin-Lomas, M.; Penadés, S. Asymmetric Michael reaction using macrocyclic lactose derivatives as chiral catalysts. Tetrahedron Lett. 1986, 27, 3551–3554. [Google Scholar] [CrossRef]
- Dondoni, A.; Marra, A. Transformation of linear oligoketosides into macrocyclic neoglycoconjugates. Tetrahedron Lett. 2009, 50, 3593–3596. [Google Scholar] [CrossRef]
- Dondoni, A.; Marra, A.; Scherrmann, M.-C.; Bertolasi, V. Chemical Synthesis of Linear and Cyclic Unnatural Oligosaccharides by Iterative Glycosidation of Ketoses. Chem. Eur. J. 2001, 7, 1371–1382. [Google Scholar] [CrossRef]
- Dondoni, A.; Scherrmann, M.-C. Thiazole-Based Synthesis of Formyl C-Glycosides. J. Org. Chem. 1994, 59, 6404–6412. [Google Scholar] [CrossRef]
- For a review on thiazolylketoses, see: Dondoni, A.; Marra, A. Thiazolylketoses: A new class of versatile intermediates for glycoside synthesis. Chem. Commun. 1999, 1999, 2133–2145. [Google Scholar] [CrossRef]
- Koto, S.; Morishima, N.; Miyata, Y.; Zen, S. Preparation of 2,3,4,6-Tetra-O-benzyl-d-mannose. Bull. Chem. Soc. Jpn. 1976, 49, 2639–2640. [Google Scholar] [CrossRef] [Green Version]
- For a review on the use of thiazole in carbohydrate chemistry, see: Dondoni, A.; Marra, A. Thiazole-mediated synthetic methodology. Chem. Rev. 2004, 104, 2557–2599. [Google Scholar] [CrossRef]
- Gokel, G.W. Lariat ethers: From Simple Side-arms to Supramolecular System. Chem. Soc. Rev. 1992, 21, 39–47. [Google Scholar] [CrossRef]
- Bakó, P.; Bakó, T.; Bisztray, K.; Szöllősy, A.; Nagy, K.; Tőke, L. Synthesis, Extraction Ability and Application in an Asymmetric Synthesis of Azacrown Ethers Derived from d-Mannitol. J. Incl. Phenom. Macrocycl. Chem. 2001, 39, 247–251. [Google Scholar] [CrossRef]
- Bakó, T.; Bakó, P.; Keglevich, G.; Báthori, N.; Czugler, M.; Tatai, J.; Novák, T.; Parlagh, G.; Tőke, L. Enantioselective Michael addition of 2-nitropropane to chalcone analogues catalyzed by chiral azacrown ethers based on d-glucose and d-mannitol. Tetrahedron Asymmetry 2003, 14, 1917–1923. [Google Scholar] [CrossRef]
- Bakó, T.; Bakó, P.; Bombicz, P.; Kubinyi, M.; Pál, K.; Bodor, S.; Makó, A.; Tőke, L. Phase-transfer catalyzed asymmetric epoxidation of chalcones using chiral crown ethers derived from d-glucose, d-galactose, and d-mannitol. Tetrahedron Asymmetry 2004, 15, 1589–1595. [Google Scholar] [CrossRef]
- Nemcsok, T.; Rapi, Z.; Bagi, P.; Keglevich, G.; Bakó, P. Synthesis of xylal- and arabinal-based crown ethers and their application as asymmetric phase transfer catalysts. Chirality 2020, 32, 107–119. [Google Scholar] [CrossRef] [PubMed]
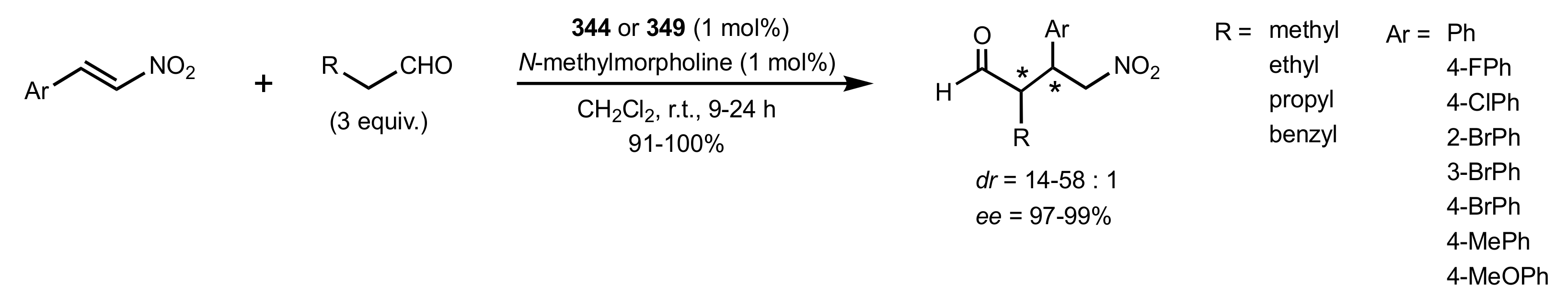
- Bakó, P.; Kiss, T.; Tőke, L. Chiral Azacrown Ethers Derived from d-glucose as Catalysts for Enantioselective Michael Addition. Tetrahedron Lett. 1997, 38, 7259–7262. [Google Scholar] [CrossRef]
- Bakó, P.; Novák, T.; Ludányi, K.; Pete, B.; Tőke, L.; Keglevich, G. d-Glucose-based azacrown ethers with a phosphonoalkyl side chain: Application as enantioselective phase transfer catalysts. Tetrahedron Asymmetry 1999, 10, 2373–2380. [Google Scholar] [CrossRef]
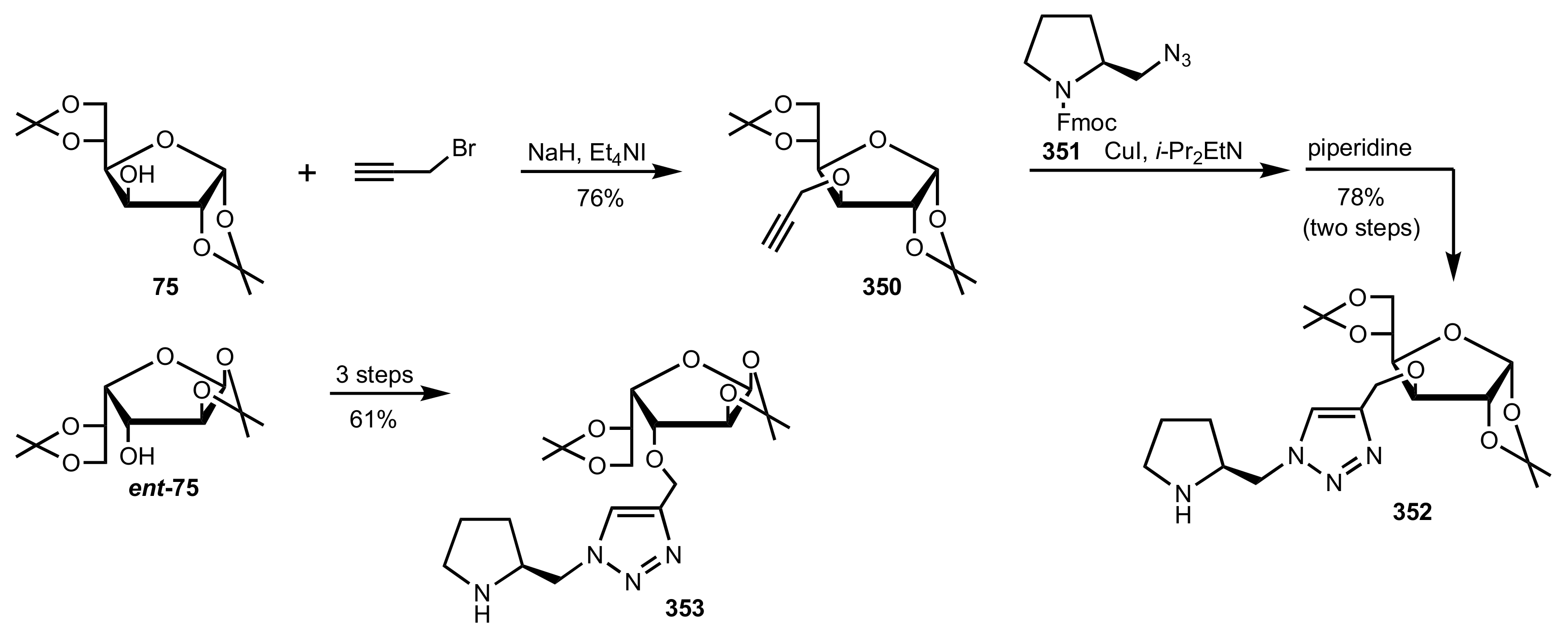
- Bakó, P.; Czinege, E.; Bakó, T.; Czugler, M.; Tőke, L. Asymmetric C–C bond forming reactions with chiral crown catalysts derived from d-glucose and d-galactose. Tetrahedron Asymmetry 1999, 10, 4539–4551. [Google Scholar] [CrossRef]
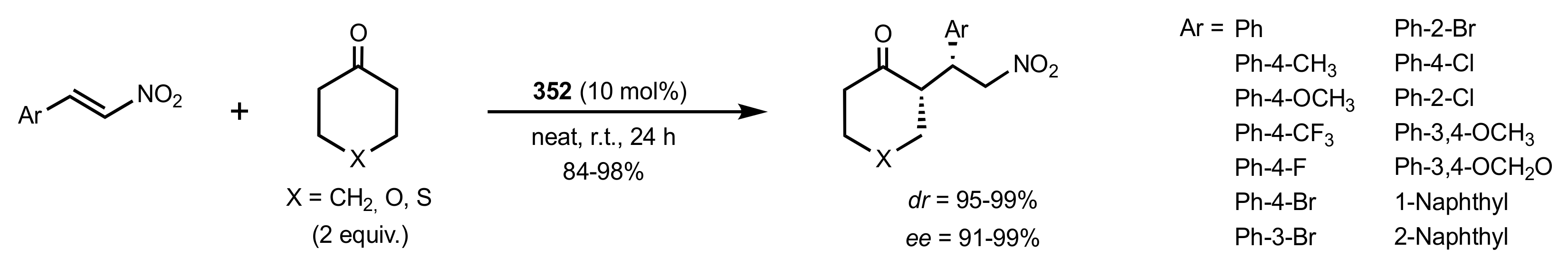
- Bakó, P.; Tőke, L. Novel Monoaza- and Diazacrown Ethers Incorporating Sugar Units and Their Extraction Ability towards Picrate Salts. J. Incl. Phenom. 1995, 23, 195–201. [Google Scholar] [CrossRef]
- Novák, T.; Bakó, P.; Keglevich, G.; Dobó, A.; Vékey, K.; Tőke, L. Synthesis of d-Glucose-based Azacrown Ethers with Phosphinoxidoalkyl Side Chains and Their Application to an Enantioselective Reaction. J. Incl. Phenom. Macrocycl. Chem. 2001, 40, 207–212. [Google Scholar] [CrossRef]
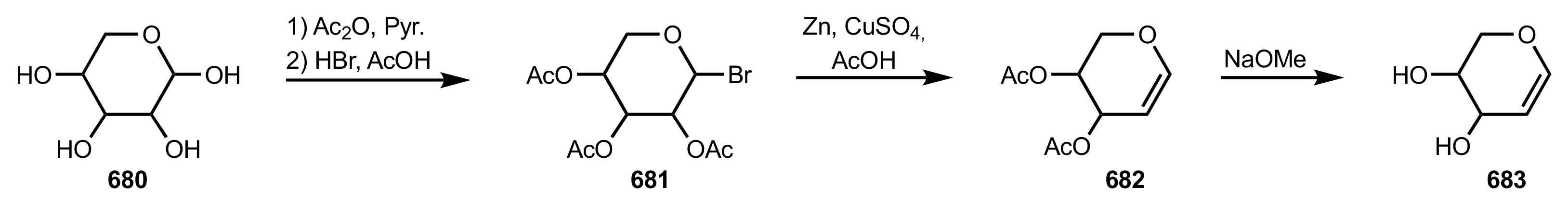
- Rapi, Z.; Bakó, P.; Drahos, L.; Keglevich, G. Side-Arm Effect of a Methyl α-d-Glucopyranoside Based Lariat Ether Catalysts in Asymmetric Syntheses. Heteroat. Chem. 2015, 26, 63–71. [Google Scholar] [CrossRef]
- Itoh, T.; Shirakami, S. Synthesis of chiral azacrown ethers derived from α-d-glucose and their catalytic properties on the asymmetric Michael addition. Heterocycles 2001, 55, 37–43. [Google Scholar] [CrossRef]
- Bakó, P.; Makó, A.; Keglevich, G.; Menyhárt, D.K.; Sefcsik, T.; Fekete, J. Alkali Metal- and Ammonium Picrate Extraction and Complex Forming Capabilities of d-Glucose and d-Mannose-based Lariat Ethers. J. Incl. Phenom. Macrocycl. Chem. 2006, 55, 295–302. [Google Scholar] [CrossRef]
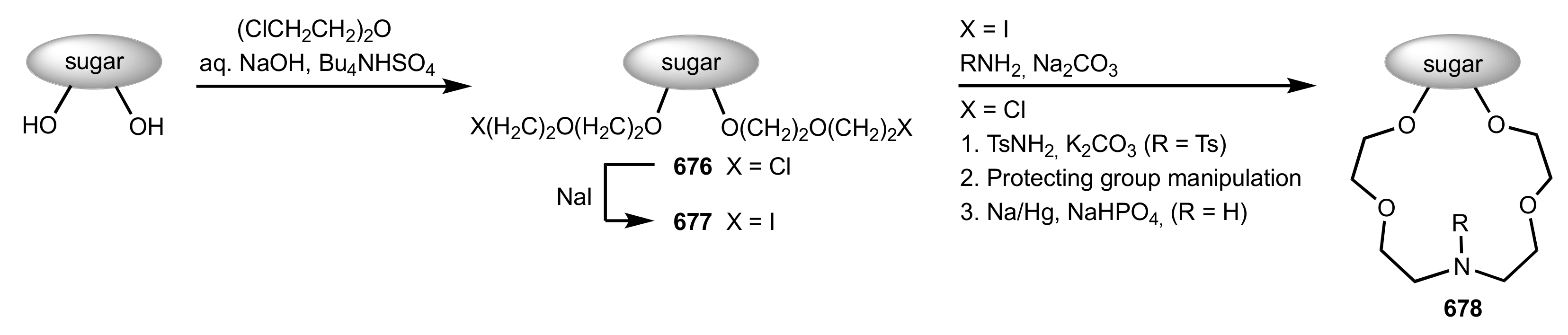
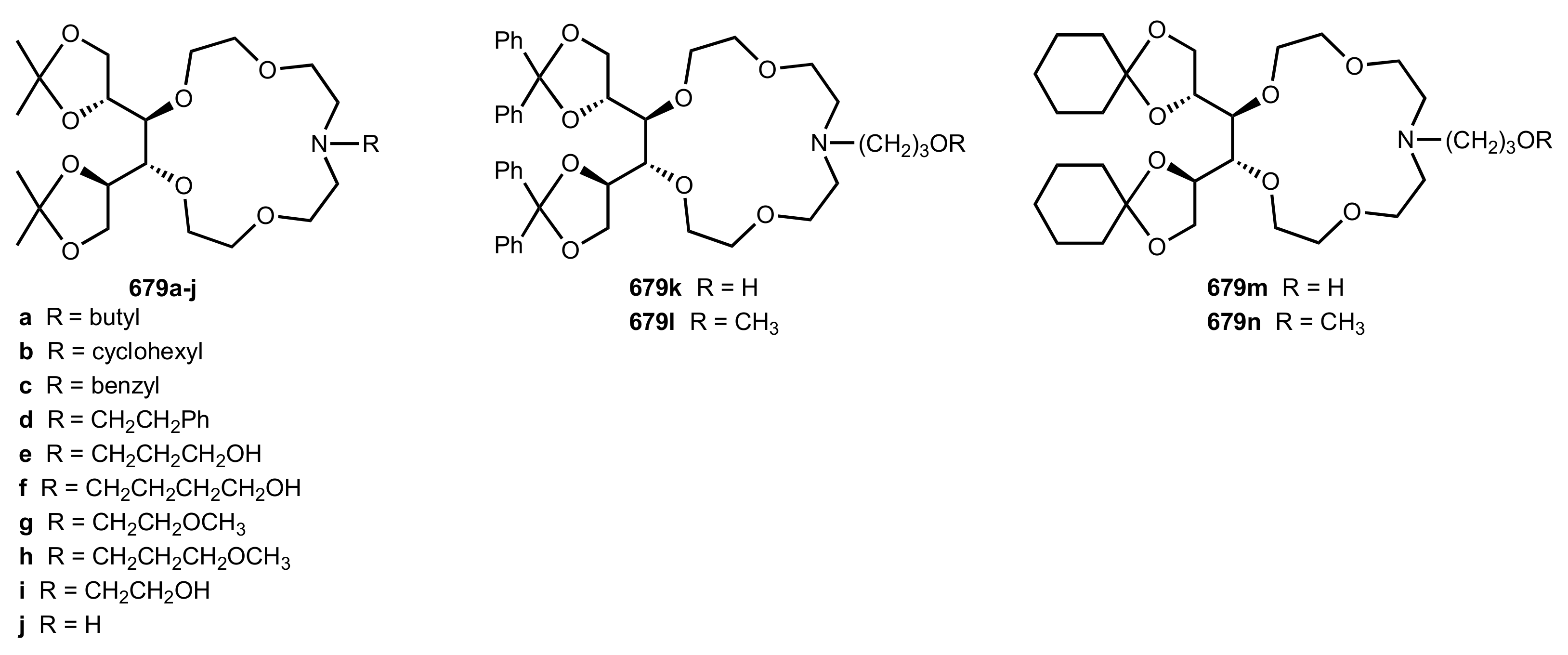
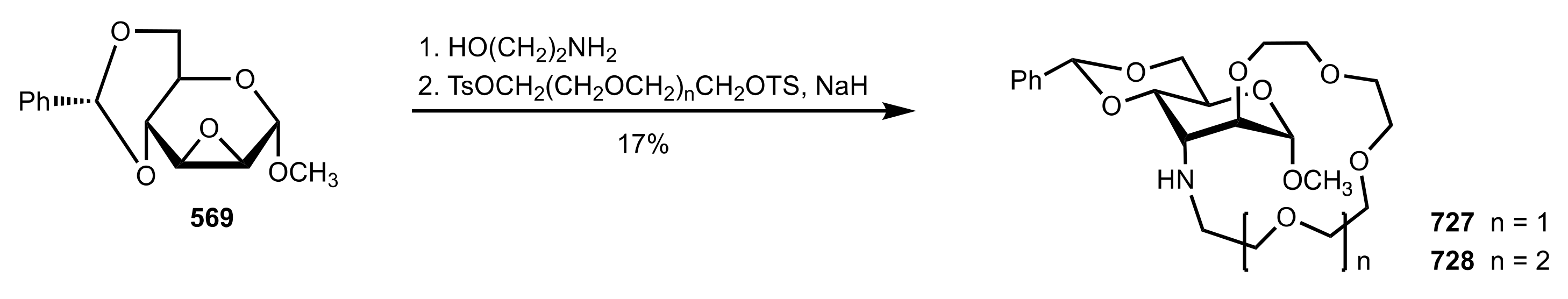
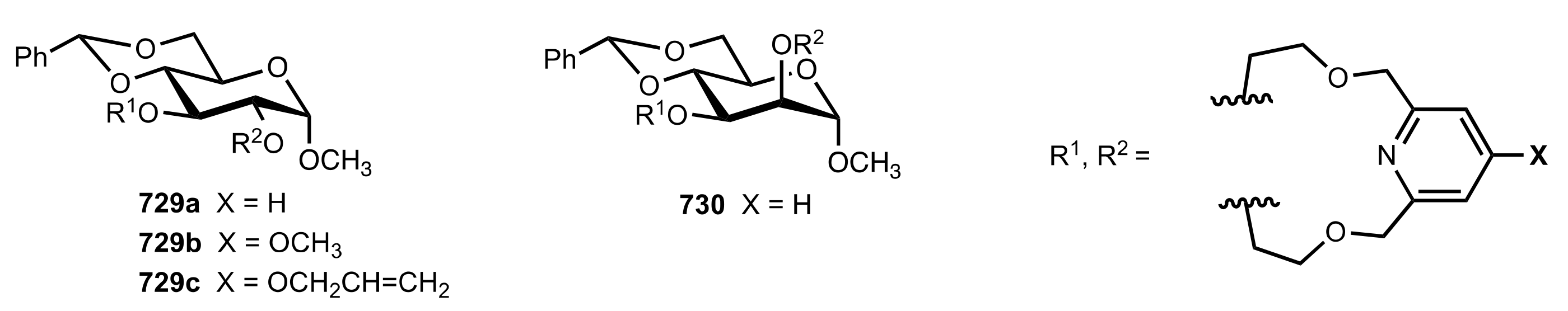
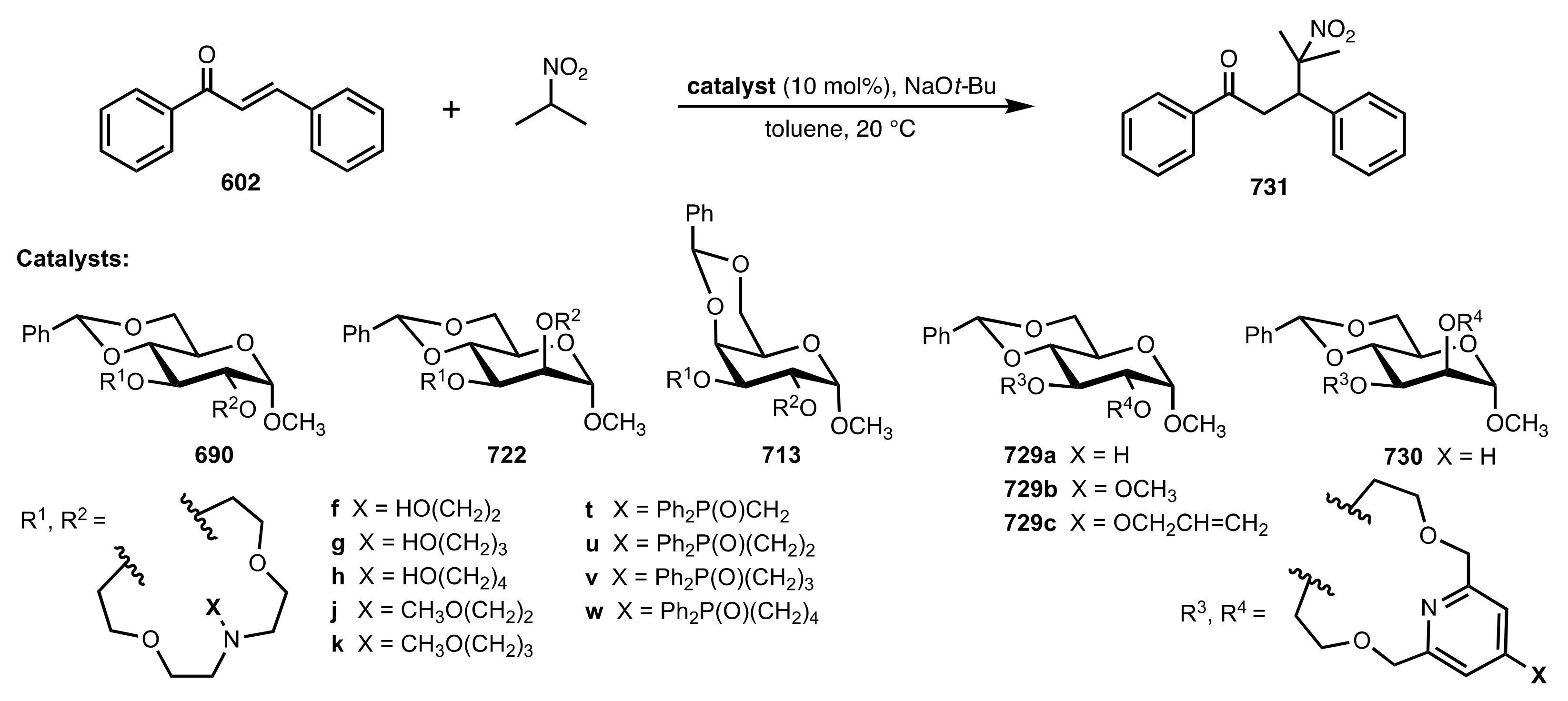
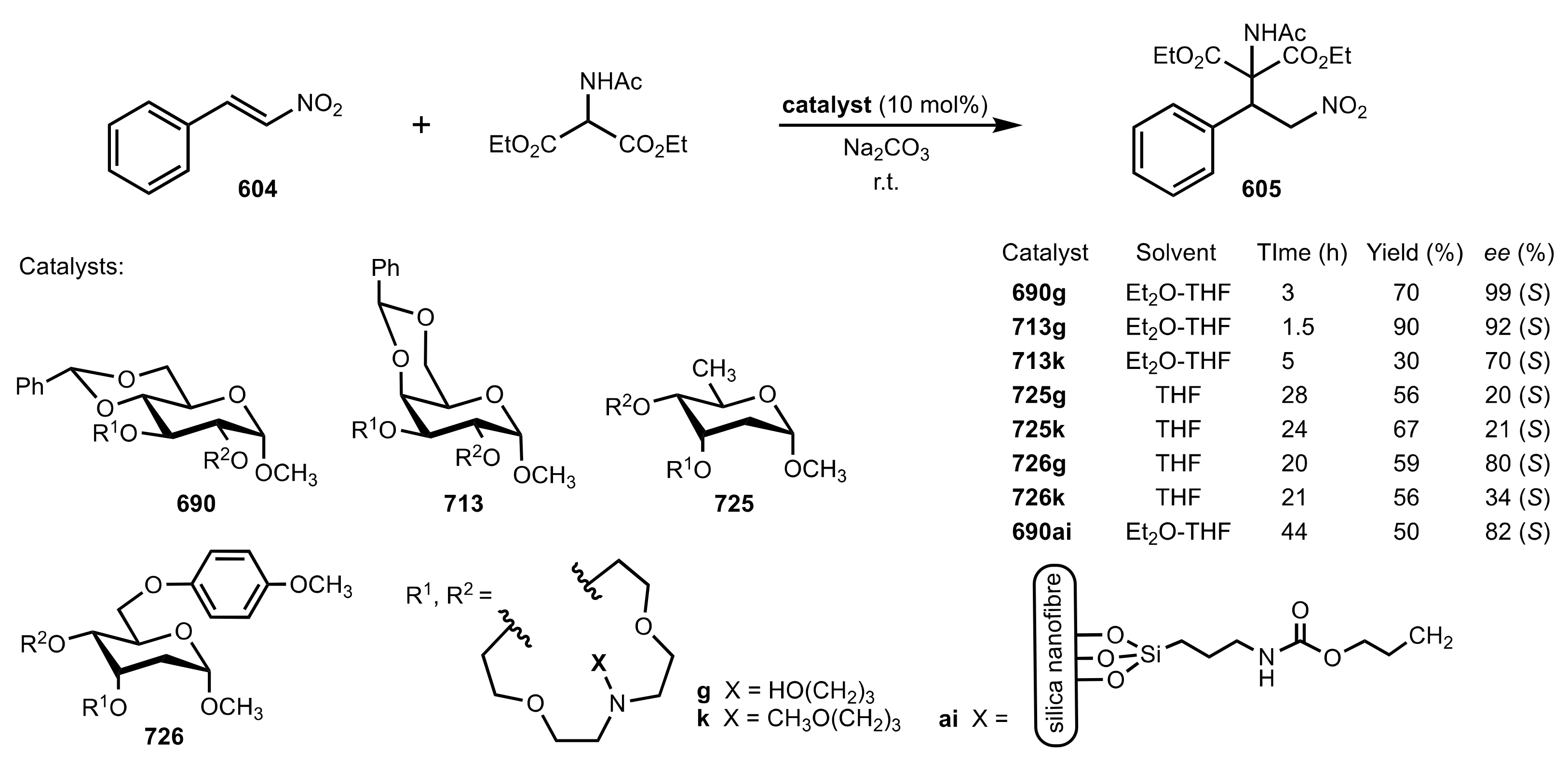
- Sóti, P.L.; Telkes, L.; Rapi, Z.; Tóth, A.; Vigh, T.; Nagy, Z.K.; Bakó, P.; Marosi, G. Synthesis of an Aza Chiral Crown Ether Grafted to Nanofibrous Silica Support and Application in Asymmetric Michael Addition. J. Inorg. Organomet. Polym. 2014, 2, 713–721. [Google Scholar] [CrossRef]
- Makó, A.; Szöllősy, Á.; Keglevich, G.; Menyhárd, D.K.; Bakó, P.; Tőke, L. Synthesis of methyl α-d-glucopyranoside-based azacrown ethers and their application in enantioselective reactions. Monatsh Chem. 2008, 139, 525–535. [Google Scholar] [CrossRef]
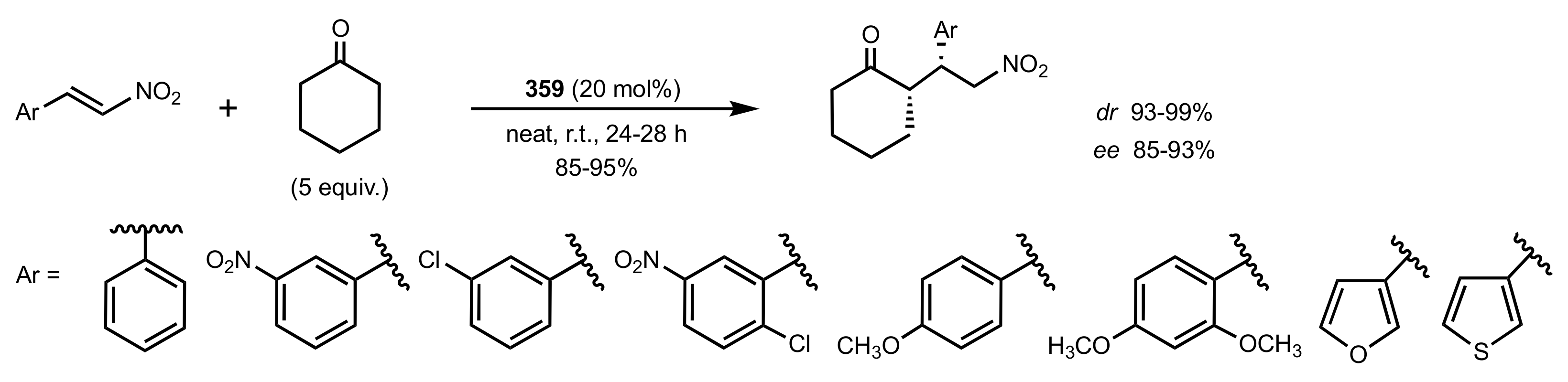
- Bakó, P.; Bajor, Z.; Tőke, L. Synthesis of novel chiral crown ethers derived from d-glucose and their application to an enantioselective Michael reaction. J. Chem. Soc. Perkin Trans. 1 1999, 1999, 3651–3655. [Google Scholar] [CrossRef]
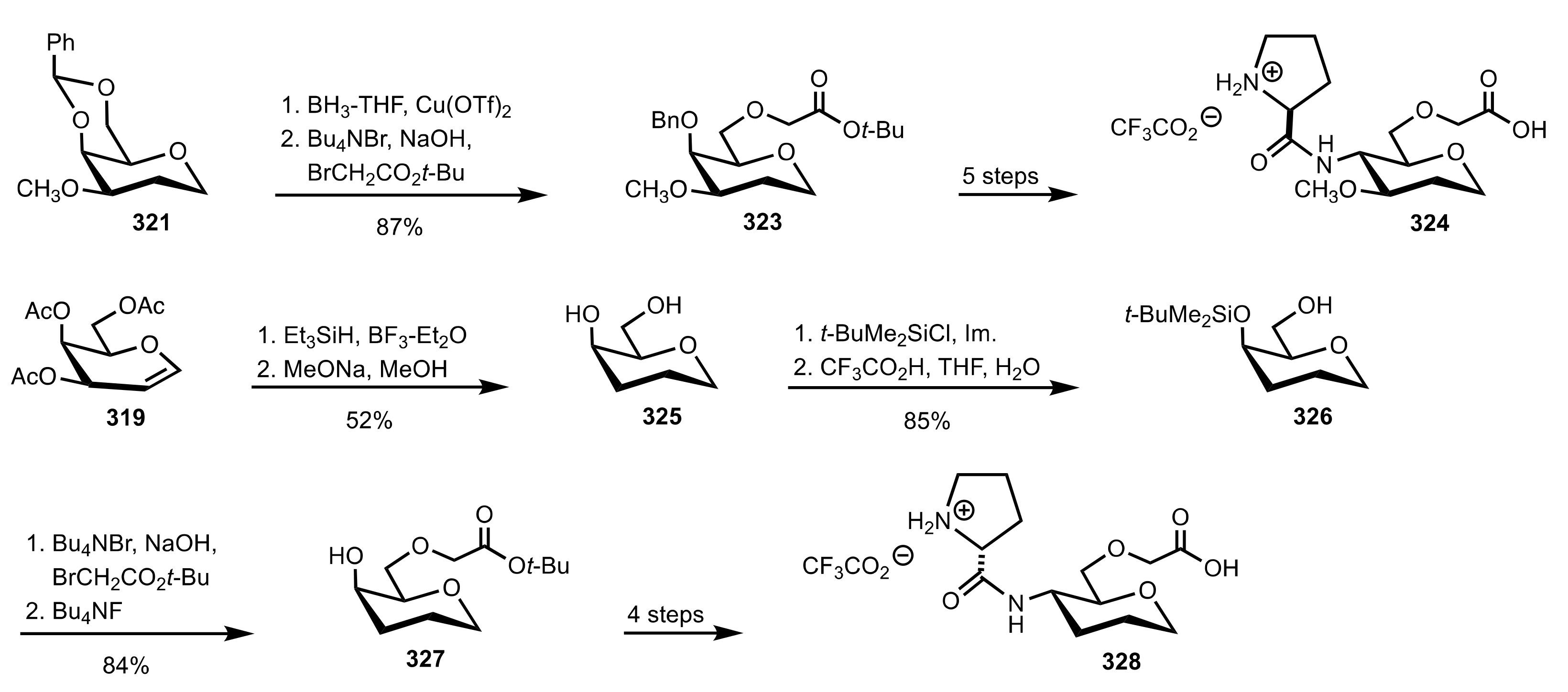
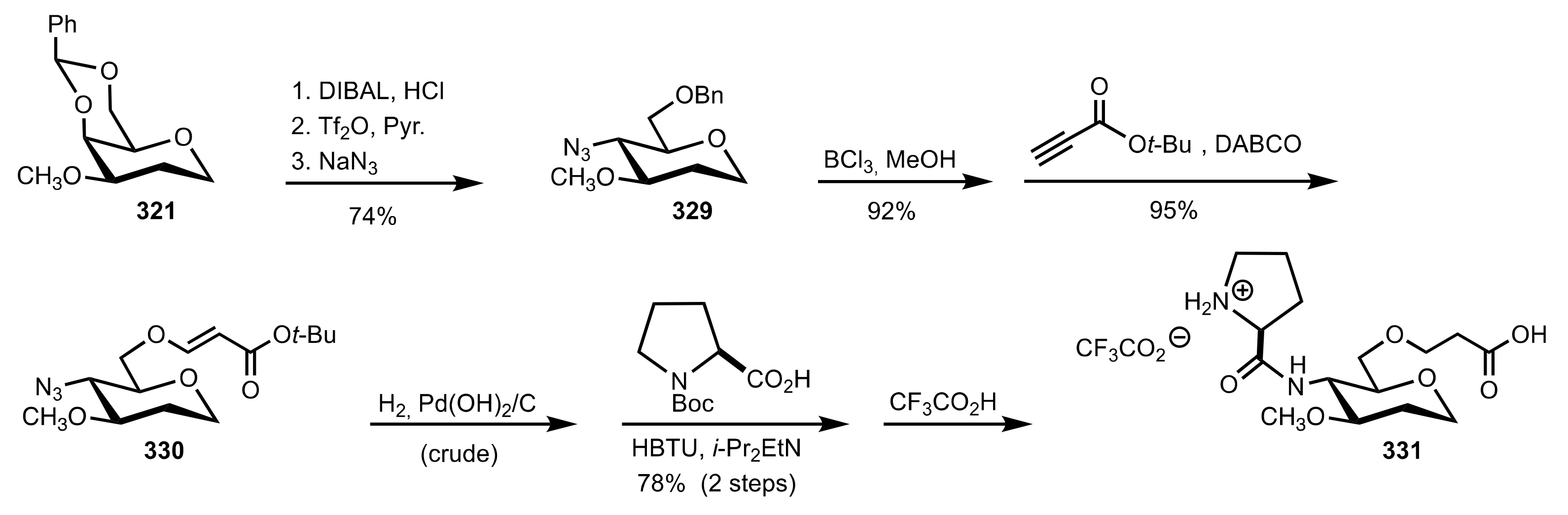
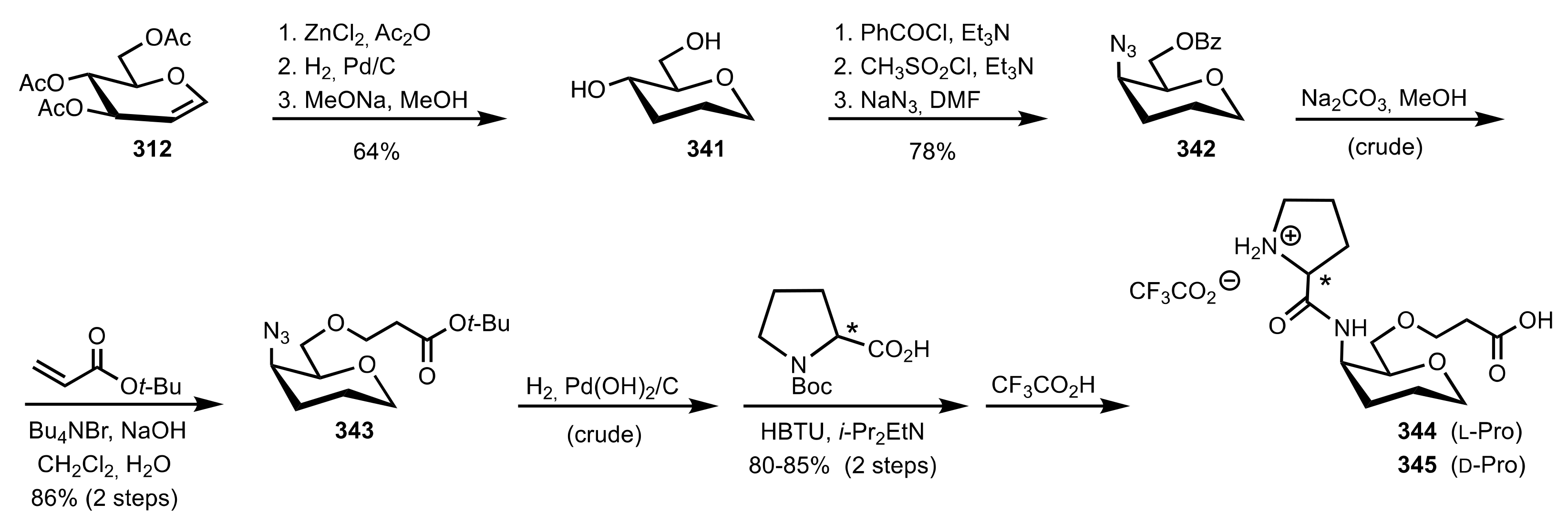
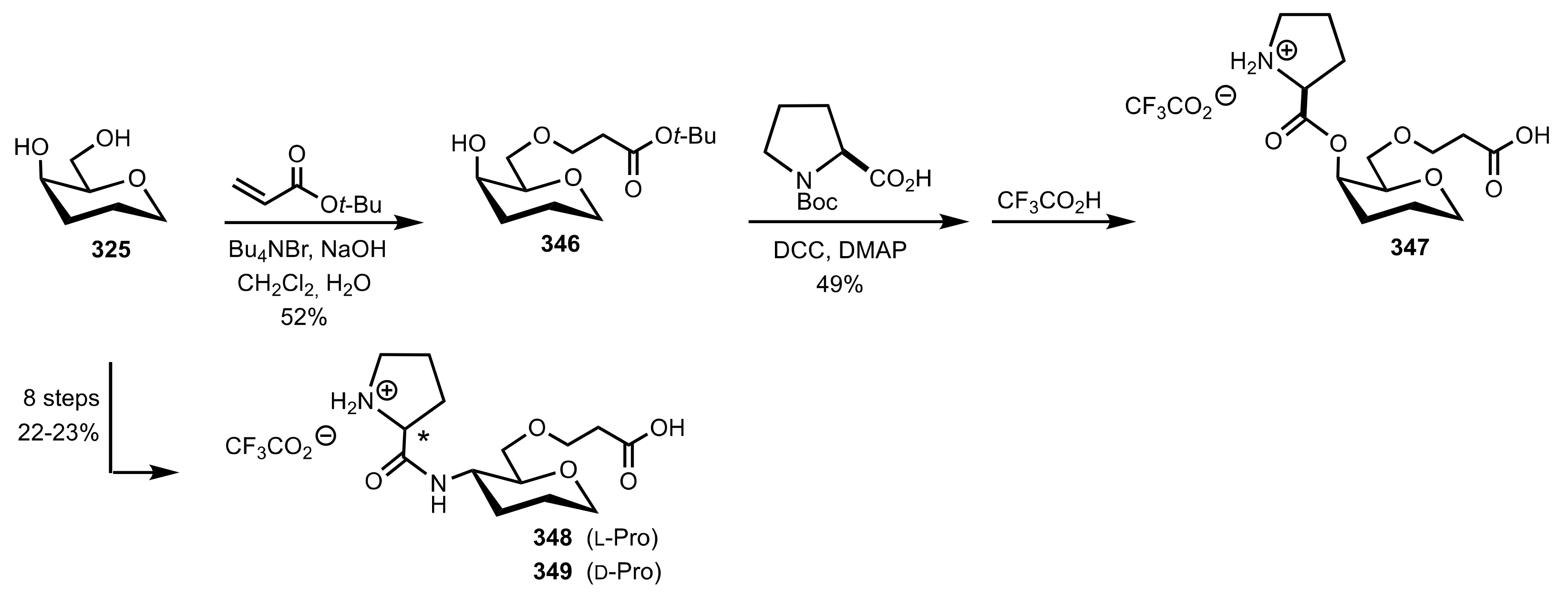
- Murphy, P.V.; O’Brien, J.L.; Gorey-Feret, L.J.; Smith, A.B., III. Synthesis of novel HIV-1 protease inhibitors based on carbohydrate scaffolds. Tetrahedron 2003, 59, 2259–2271. [Google Scholar] [CrossRef]
- Coppola, C.; Saggiomo, V.; Di Fabio, G.; De Napoli, L.; Montesarchio, D. Novel Amphiphilic Cyclic Oligosaccharides: Synthesis and Self-Aggregation Properties. J. Org. Chem. 2007, 72, 9679–9689. [Google Scholar] [CrossRef]
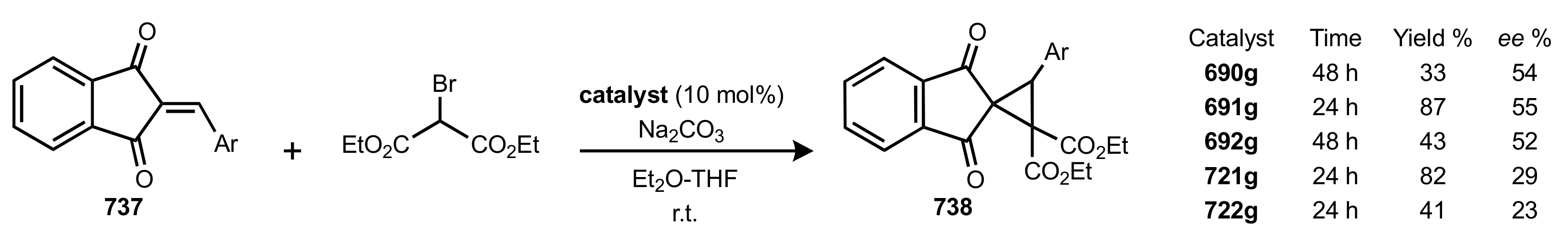
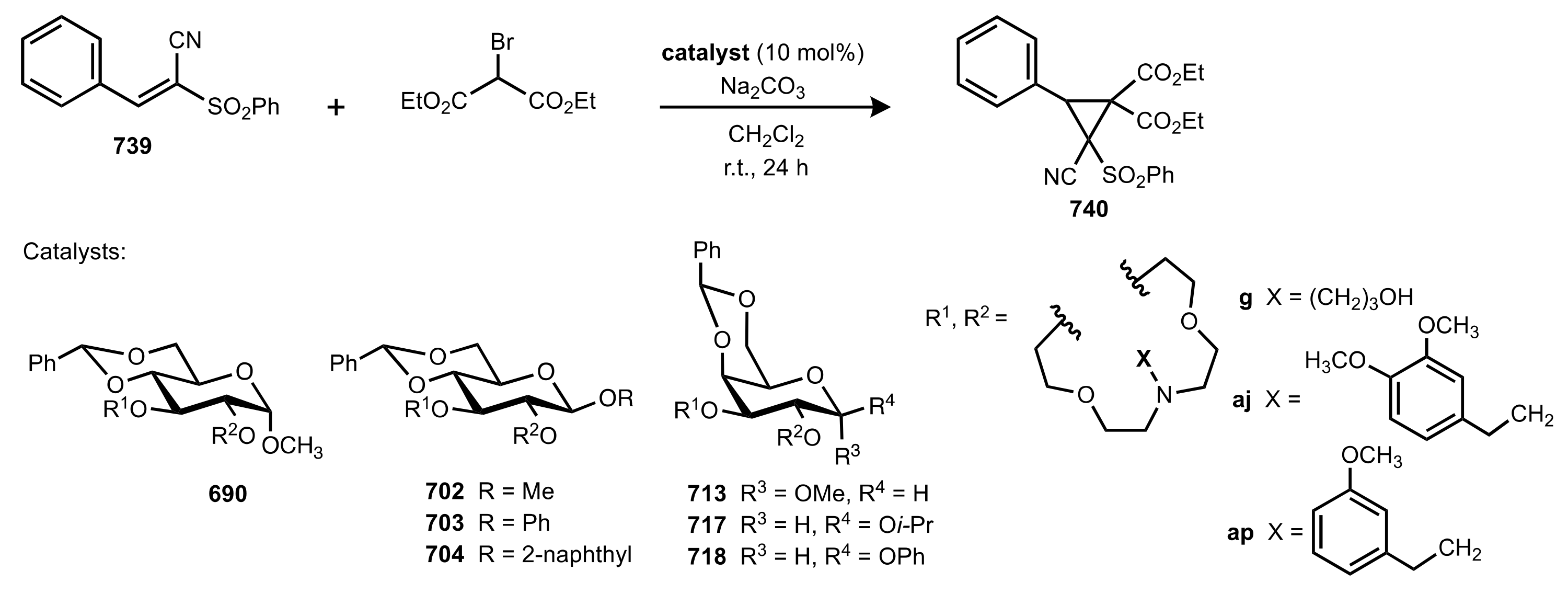
- Nemcsok, T.; Rapi, Z.; Bagi, P.; Guan, Y.H.; Orbán, I.; Keglevich, G.; Bakó, P. Enantioselective cyclopropanation of conjugated cyanosulfones using carbohydrate-based crown ether catalysts. Tetrahedron 2020, 76, 130965. [Google Scholar] [CrossRef]
- Bakó, P.; Vizvárdi, K.; Bajor, Z.; Tőke, L. Synthesis and application in asymmetric synthesis of azacrown ethers derived from d-glucose. Chem. Commun. 1998, 1998, 1193–1194. [Google Scholar] [CrossRef]
- Bakó, P.; Vízvárdi, K.; Toppet, S.; Van der Eycken, E.; Hoornaert, G.J.; Tőke, L. Synthesis, extraction ability and application in asymmetric synthesis of azacrown ethers derived from d-glucose. Tetrahedron 1998, 54, 14975–14988. [Google Scholar] [CrossRef]
- Jászay, Z.; Pham, T.S.; Németh, G.; Bakó, P.; Petneházy, I.; Tőke, L. Asymmetric Synthesis of Substituted α-Amino Phosphonates with Chiral Crown Ethers as Catalysts. Synlett 2009, 2009, 1429–1432. [Google Scholar] [CrossRef]
- Pham, T.S.; Rapi, Z.; Bakó, Z.; Petneházy, I.; Stirling, A.; Jászay, Z. Enantioselective synthesis of substituted α-aminophosphonates catalysed by d-glucose-based crown ethers: Pursuit of the origin of stereoselectivity. New J. Chem. 2017, 41, 14945–14953. [Google Scholar] [CrossRef] [Green Version]
- Rapi, Z.; Nemcsok, T.; Bagi, P.; Keglevich, G.; Bakó, P. Synthesis of chiral crown ethers derived from d-galactose and their application in enantioselective reactions. Tetrahedron 2019, 75, 3993–4004. [Google Scholar] [CrossRef]
- Rapi, Z.; Grün, A.; Keglevich, G.; Stirling, A.; Bakó, P. Synthesis of α-d-galactose-based azacrown ethers and their application as enantioselective catalysts in Michael reactions. New J. Chem. 2016, 40, 7856–7865. [Google Scholar] [CrossRef]
- Makó, A.; Menyhárd, D.K.; Bakó, P.; Keglevich, G.; Tőke, L. Theoretical study of the asymmetric phase-transfer mediated epoxidation of chalcone catalyzed by chiral crown ethers derived from monosaccharides. J. Mol. Struct. 2008, 892, 336–342. [Google Scholar] [CrossRef]
- Bakó, P.; Makó, A.; Keglevich, G.; Kubinyi, M.; Pál, K. Synthesis of d-mannose-based azacrown ethers and their application in enantioselective reactions. Tetrahedron Asymmetry 2005, 16, 1861–1871. [Google Scholar] [CrossRef]
- Szabó, T.; Rapi, Z.; Keglevich, G.; Szöllősy, Á.; Drahos, L.; Bakó, P. Synthesis of l-arabinose-based crown ethers and their application as enantioselective phase transfer catalysts. Arkivoc 2012, 8, 36–48. [Google Scholar] [CrossRef]
- Rapi, Z.; Bakó, P.; Keglevich, G.; Szöllősy, Á.; Drahos, L.; Hegedűs, L. Synthesis of ribose and altrose based azacrown ethers and their application in an asymmetric Michael addition. Carbohydr. Res. 2013, 365, 61–68. [Google Scholar] [CrossRef]
- Makó, A.; Bakó, P.; Szöllősy, Á.; Bakó, T.; Peltz, C.; Keglevich, P. Synthesis of chiral pyridino-15-crown-5 type ligands containing α-d-hexapyranoside unit and their application in asymmetric synthesis. Arkivoc 2009, 7, 165–179. [Google Scholar] [CrossRef] [Green Version]
- Makó, A.; Rapi, Z.; Drahos, L.; Szöllősy, Á.; Keglevich, G.; Bakó, P. Enantioselective Michael Addition of 2-Nitropropane to Substituted Chalcones and Chalcone Analogues Catalyzed by Chiral Crown Ethers Incorporating an α-d-Glucose or an α-d-Mannose Unit. Lett. Org. Chem. 2010, 7, 424–431. [Google Scholar] [CrossRef]
- Bakó, T.; Bakó, P.; Szöllősy, Á.; Czugler, M.; Keglevich, G.; Tőke, L. Enantioselective Michael reaction of 2-nitropropane with substituted chalcones catalysed by chiral azacrown ethers derived from α-d-glucose. Tetrahedron Asymmetry 2002, 13, 203–209. [Google Scholar] [CrossRef]
- Novák, T.; Tatai, J.; Bakó, P.; Czugler, M.; Keglevich, G.; Tőke, L. Asymmetric Michael Addition Catalyzed by d-Glucose-based Azacrown Ethers. Synlett 2001, 3, 424–426. [Google Scholar] [CrossRef]
- Bakó, P.; Tőke, L.; Szöllősy, A.; Bombicz, P. Asymmetric Michael Addition of 2-Nitropropane to a Chalcone Catalyzed by Chiral Crown Ethers Incorporating a d-glucose Unit. Heteroat. Chem. 1997, 8, 333–337. [Google Scholar] [CrossRef]
- Bakó, P.; Szöllősy, A.; Bombicz, P.; Tőke, L. Asymmetric C-C Bond Forming Reactions by Chiral Crown Catalysts; Darzens Condensation and Nitroalkane Addition to the Double Bond. Synlett 1997, 1997, 291–292. [Google Scholar] [CrossRef]
- Rapi, Z.; Démuth, B.; Keglevich, G.; Grűn, G.; Drahos, L.; Sóti, P.L.; Bakó, P. Enantioselective Michael addition of malonates to aromatic nitroalkenes catalyzed by monosaccharide-based chiral crown ethers. Tetrahedron Asymmetry 2014, 25, 141–147. [Google Scholar] [CrossRef]
- Bakó, P.; Zsolt, R.; Grün, A.; Nemcsok, T.; Hegedűs, L.; Keglevich, G. Asymmetric Michael Addition of Malonates to Enones Catalyzed by an α-d-Glucopyranoside-Based Crown Ether. Synlett 2015, 26, 1847–1851. [Google Scholar] [CrossRef] [Green Version]
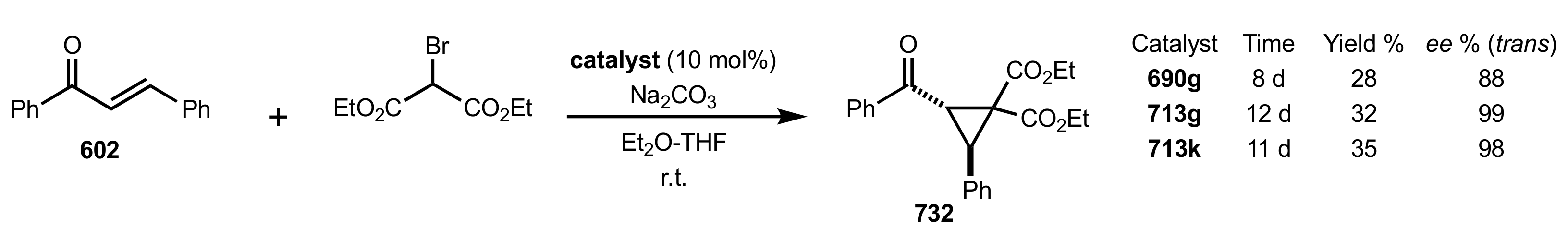
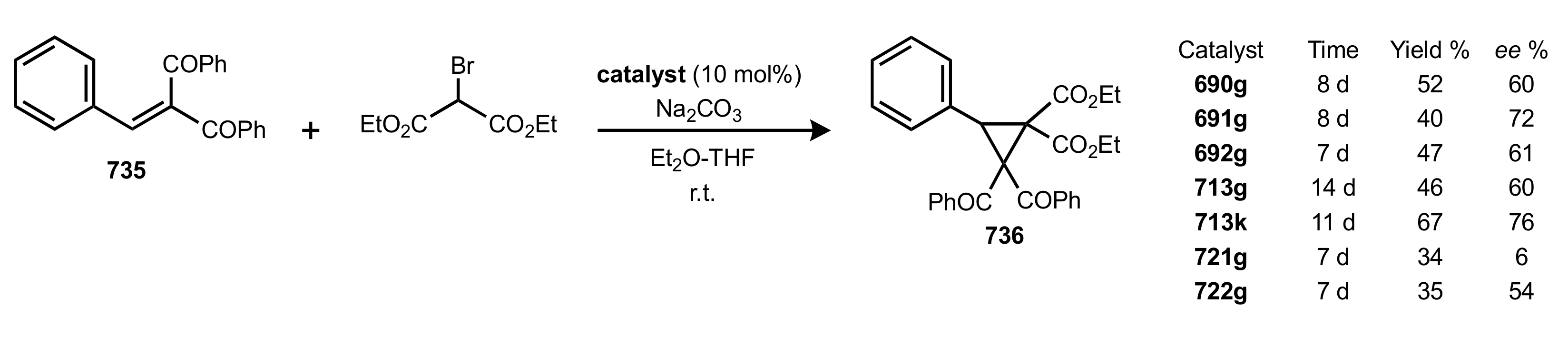
- Rapi, Z.; Nemcsok, T.; Grün, A.; Pálvölgyi, Á.; Samu, G.; Hessz, D.; Kubinyi, M.; Kállay, M.; Keglevich, G.; Bakó, P. Asymmetric cyclopropanation reactions catalyzed by carbohydrate-based crown ethers. Tetrahedron 2018, 74, 3512–3526. [Google Scholar] [CrossRef]
- Bakó, P.; Bakó, T.; Mészáros, A.; Keglevich, G.; Szöllősy, Á.; Bodor, S.; Makó, A.; Tőke, L. Phase Transfer Catalysed Asymmetric Epoxidation of Chalcones Using Chiral Crown Ethers Derived from d-Glucose and d-Mannose. Synlett 2004, 4, 643–646. [Google Scholar] [CrossRef]
- Bakó, P.; Rapi, Z.; Keglevich, G.; Szabó, T.; Sóti, P.L.; Vígh, T.; Grün, A.; Holczbauer, T. Asymmetric C–C bond formation via Darzens condensation and Michael addition using monosaccharide-based chiral crown ethers. Tetrahedron Lett. 2011, 52, 1473–1476. [Google Scholar] [CrossRef]
- Rapi, Z.; Bakó, P.; Keglevich, G.; Szöllősy, A.; Drahos, L.; Botyánszki, L.; Holczbauer, T. Asymmetric phase transfer Darzens reactions catalyzed by d-glucose- and d-mannose-based chiral crown ethers. Tetrahedron Asymmetry 2012, 23, 489–496. [Google Scholar] [CrossRef]
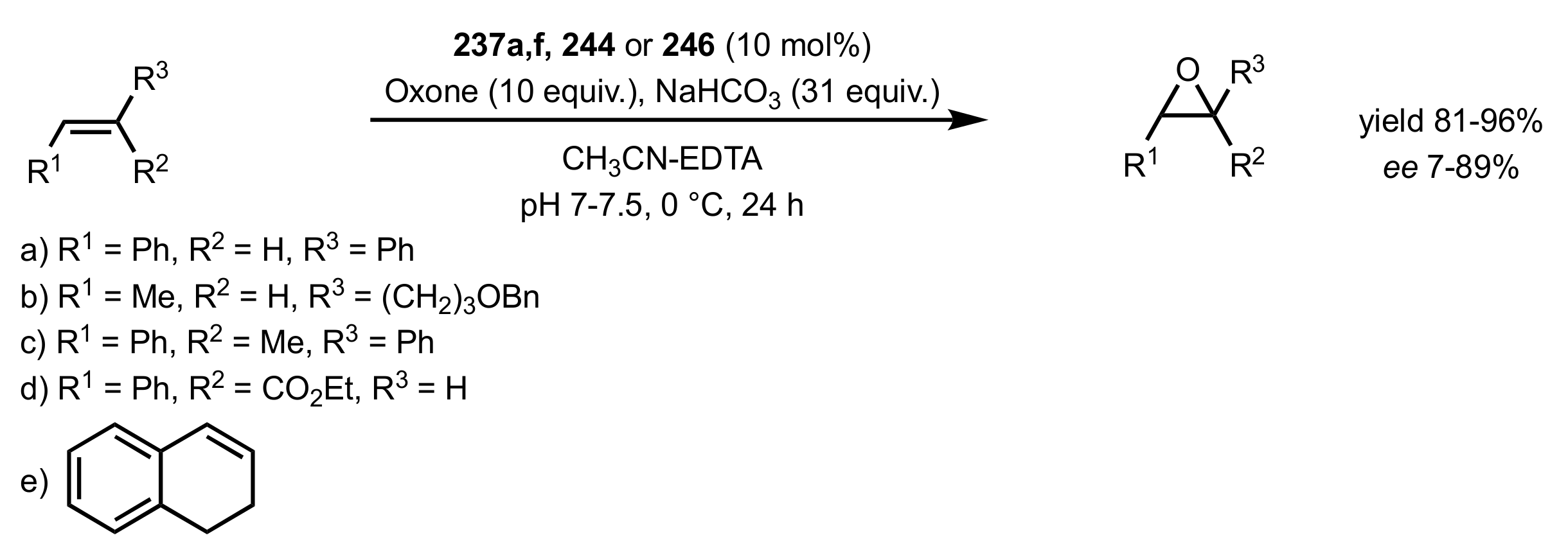
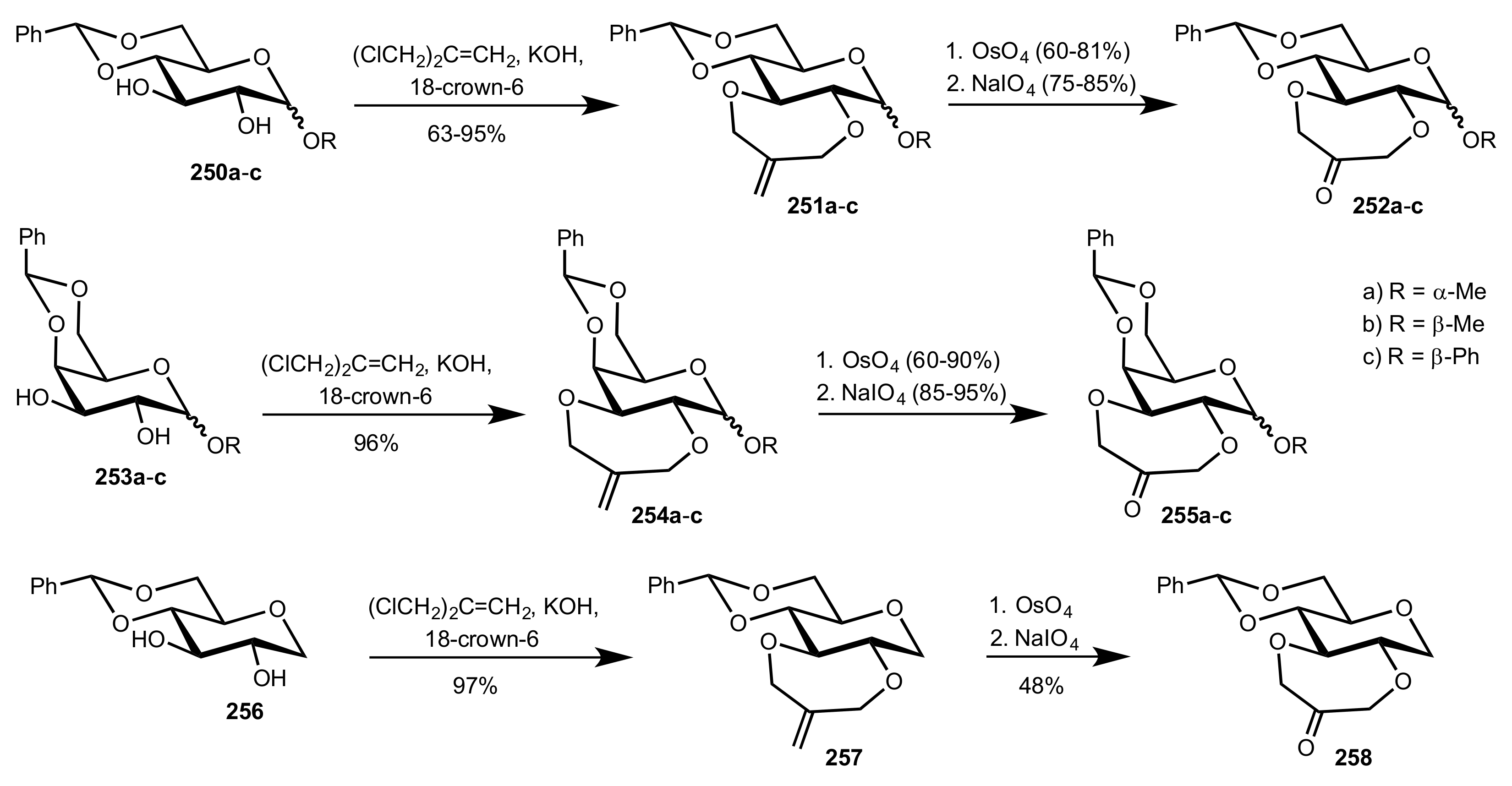
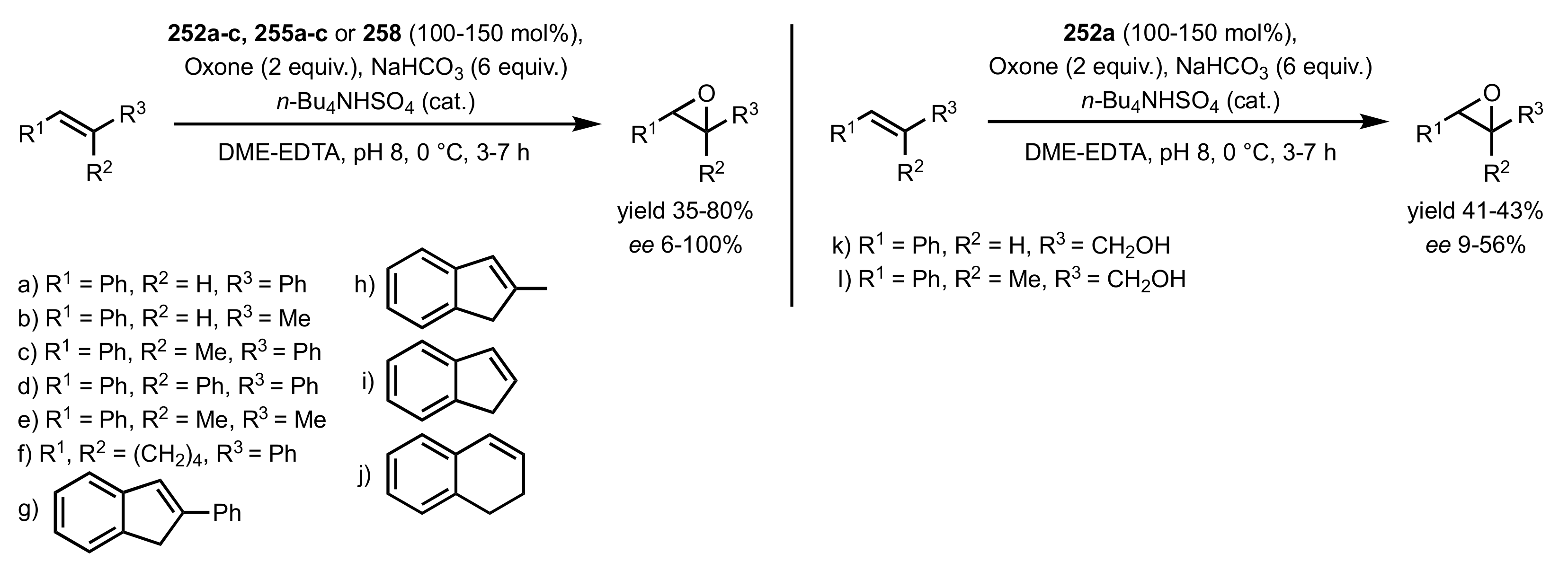
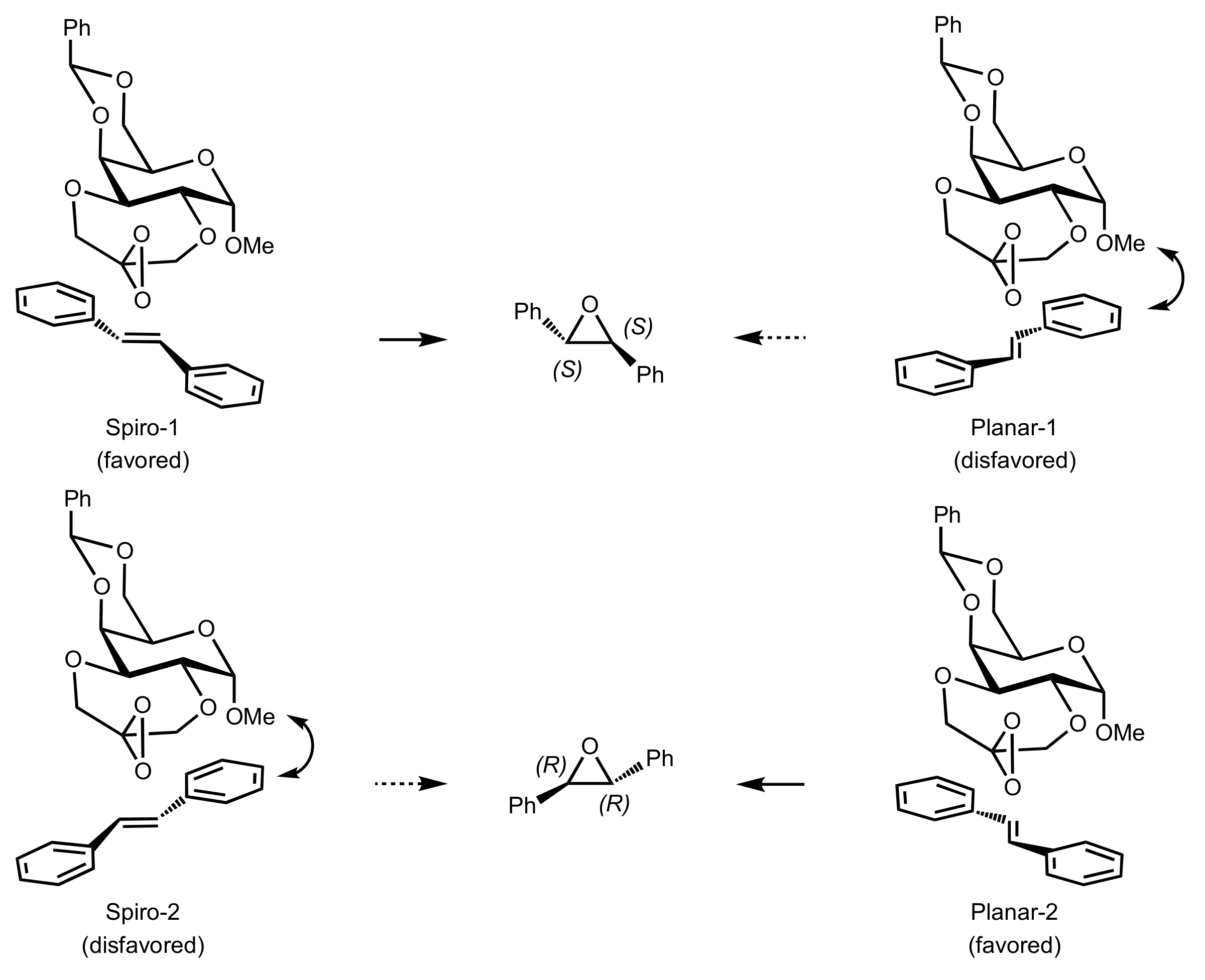
- Rapi, Z.; Szabó, T.; Keglevich, G.; Szöllősy, A.; Drahos, L.; Bakó, P. Enantioselective synthesis of heteroaromatic epoxyketones under phase-transfer catalysis using d-glucose- and d-mannose-based crown ethers. Tetrahedron Asymmetry 2011, 22, 1189–1196. [Google Scholar] [CrossRef]









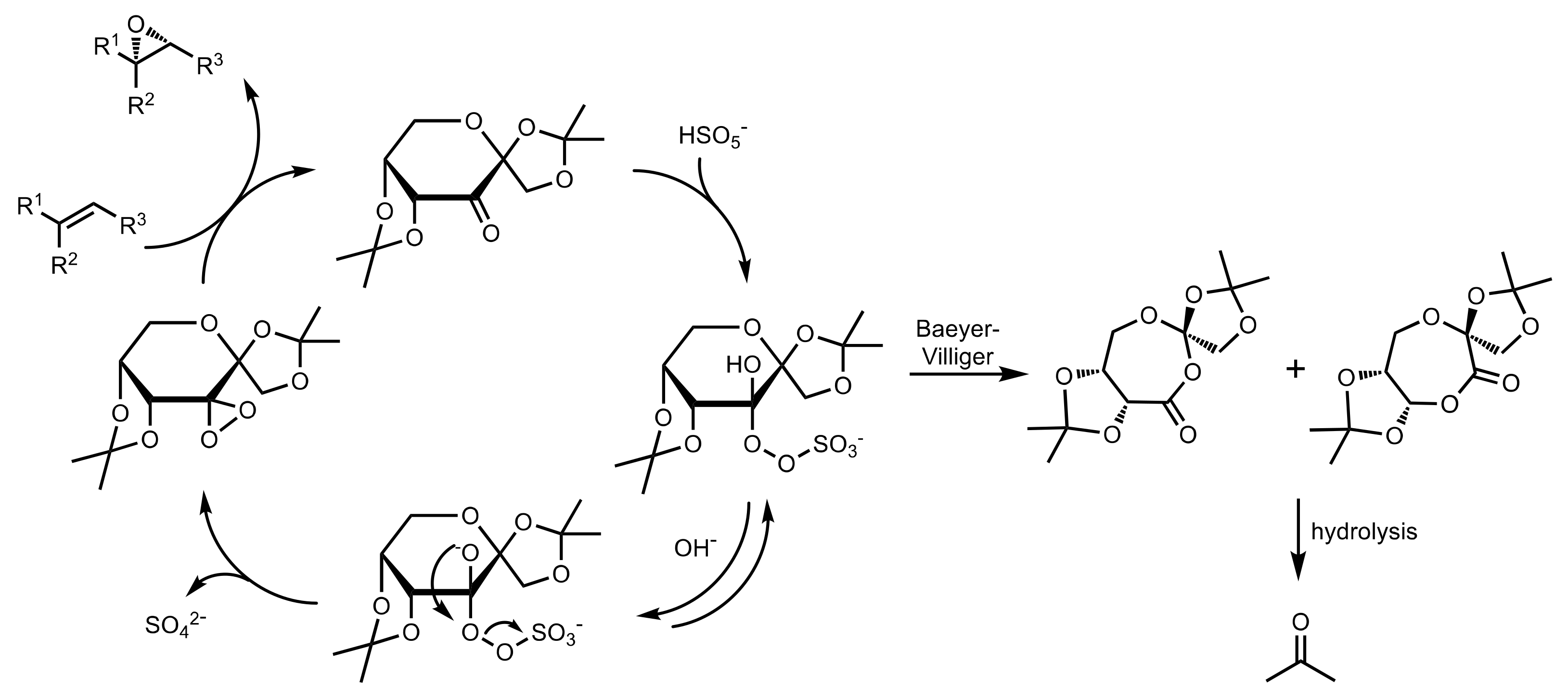
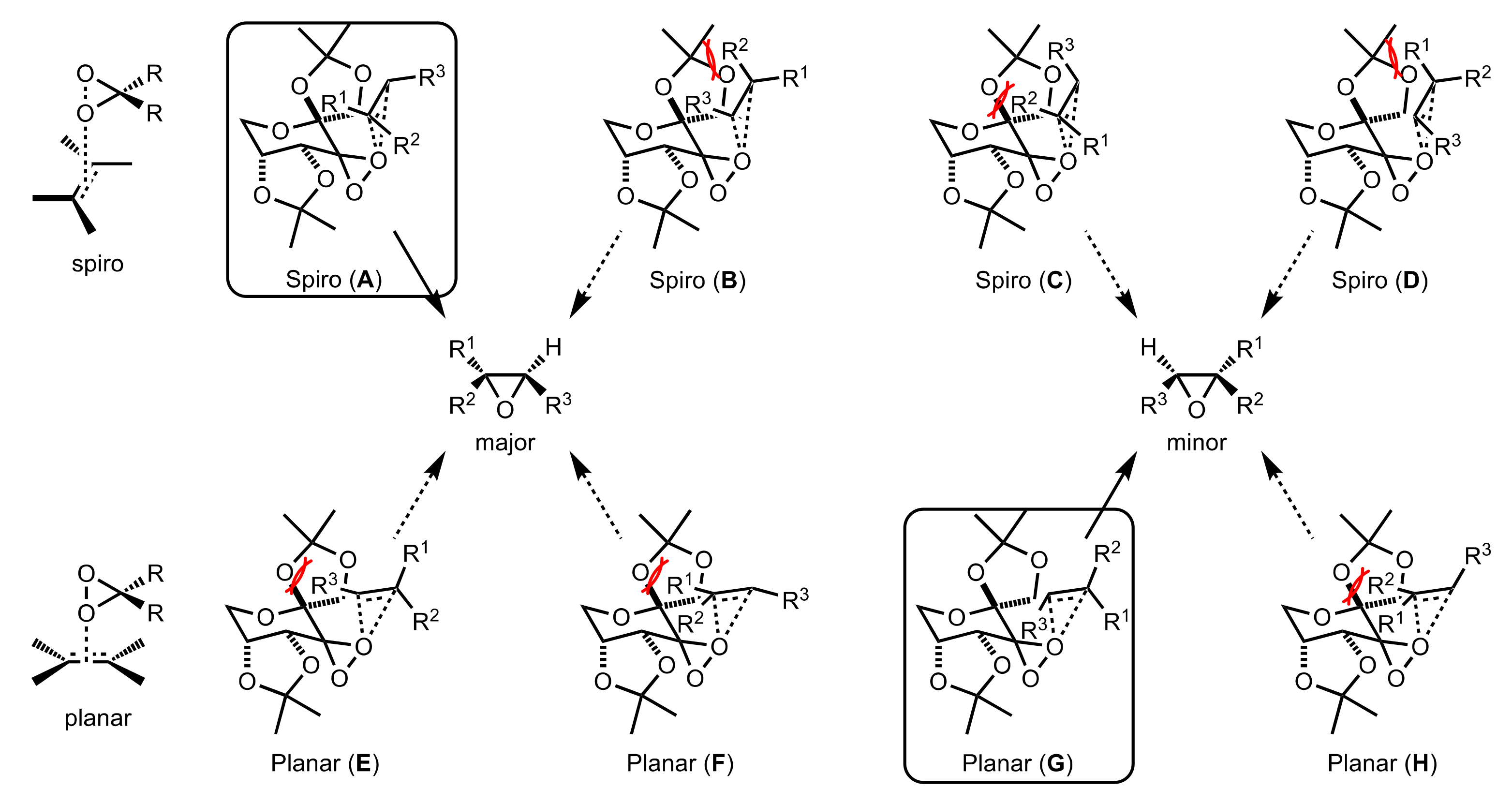
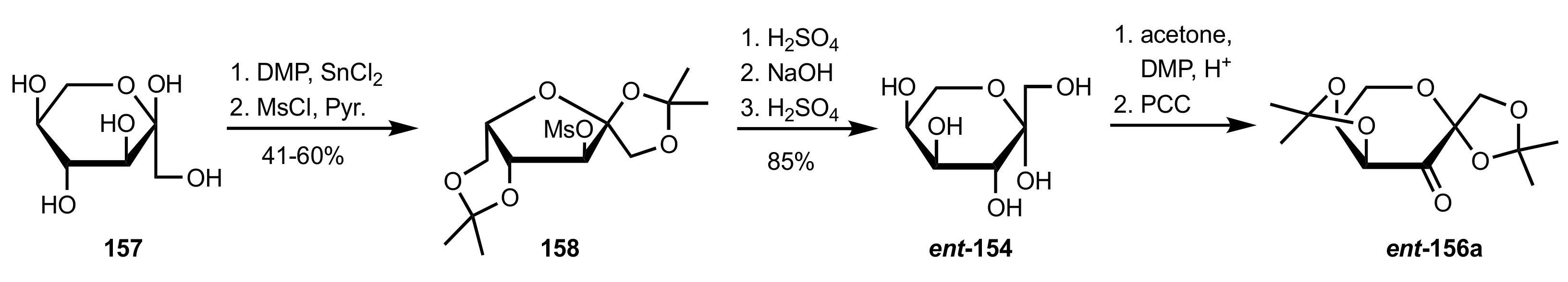
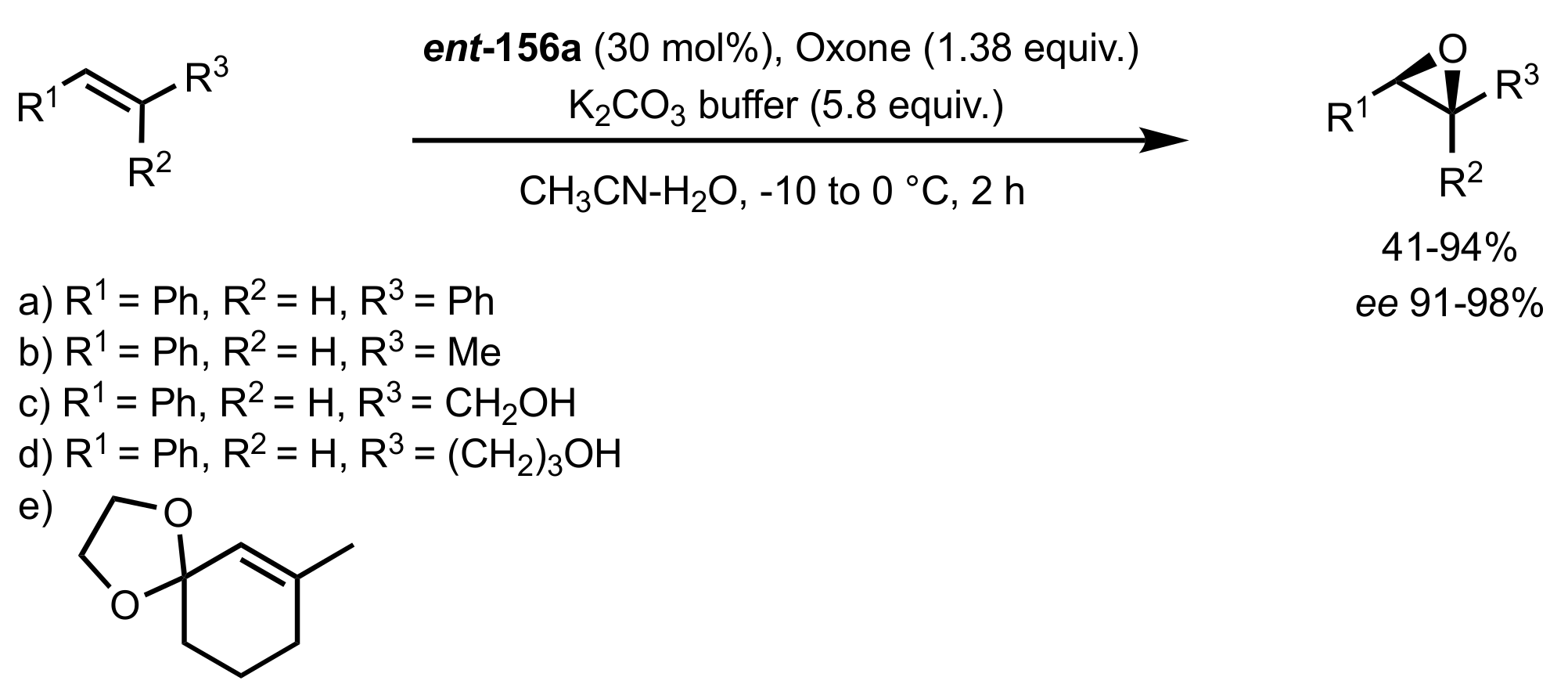
















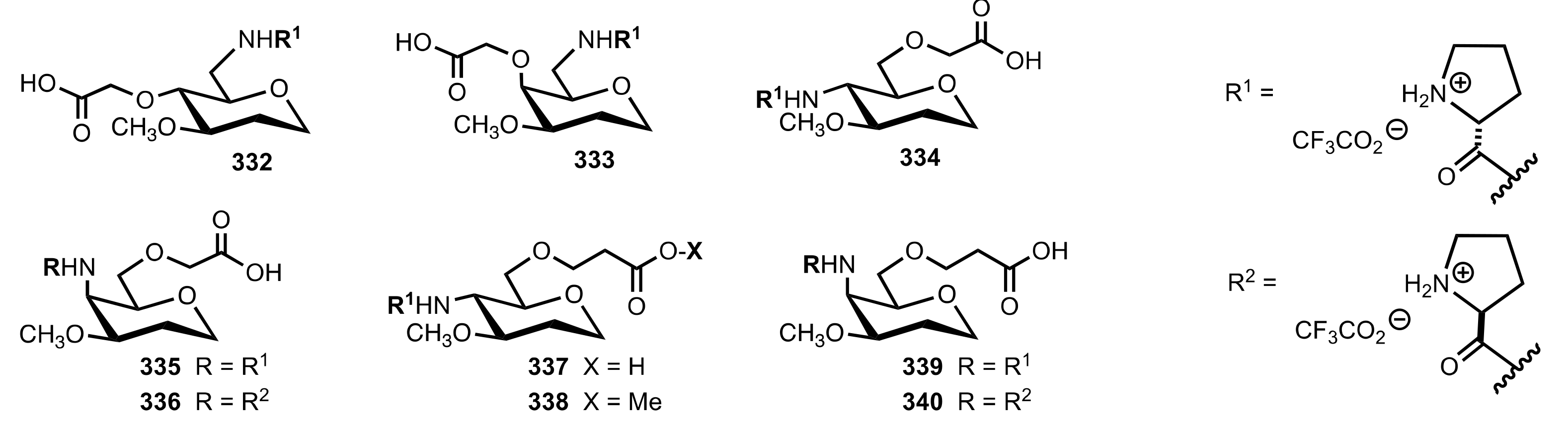
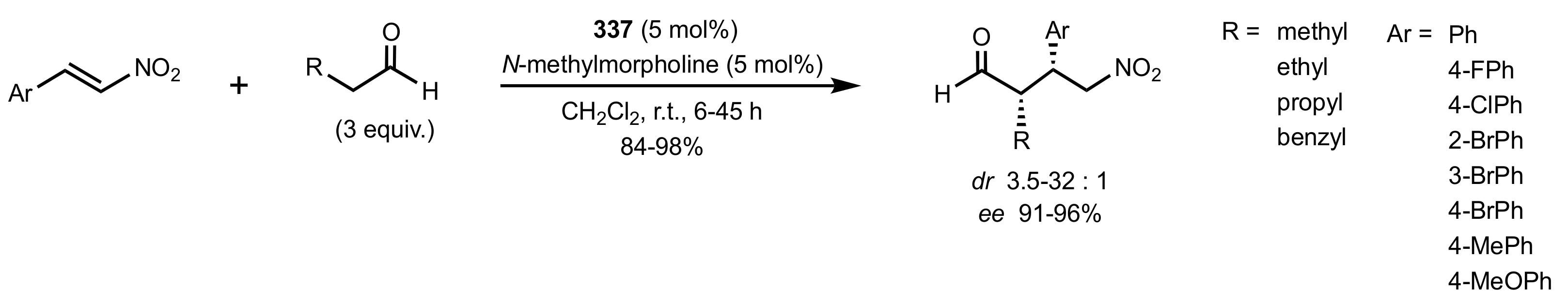


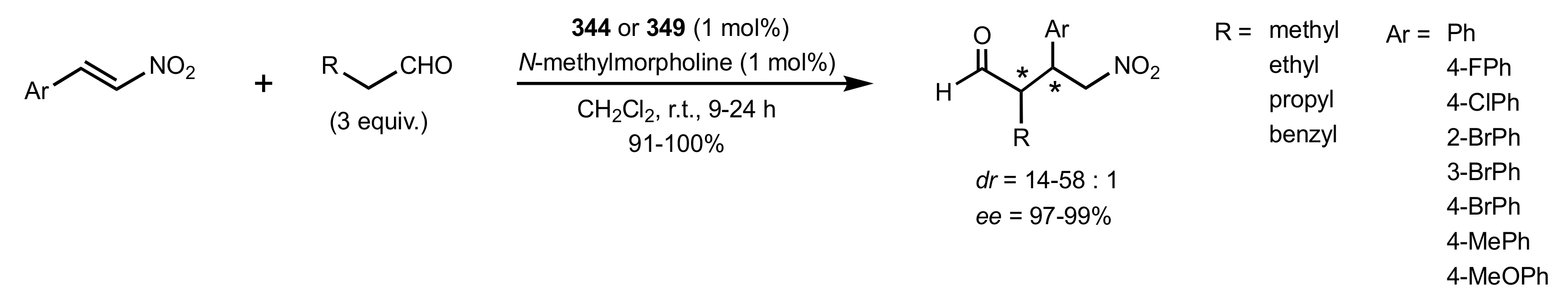
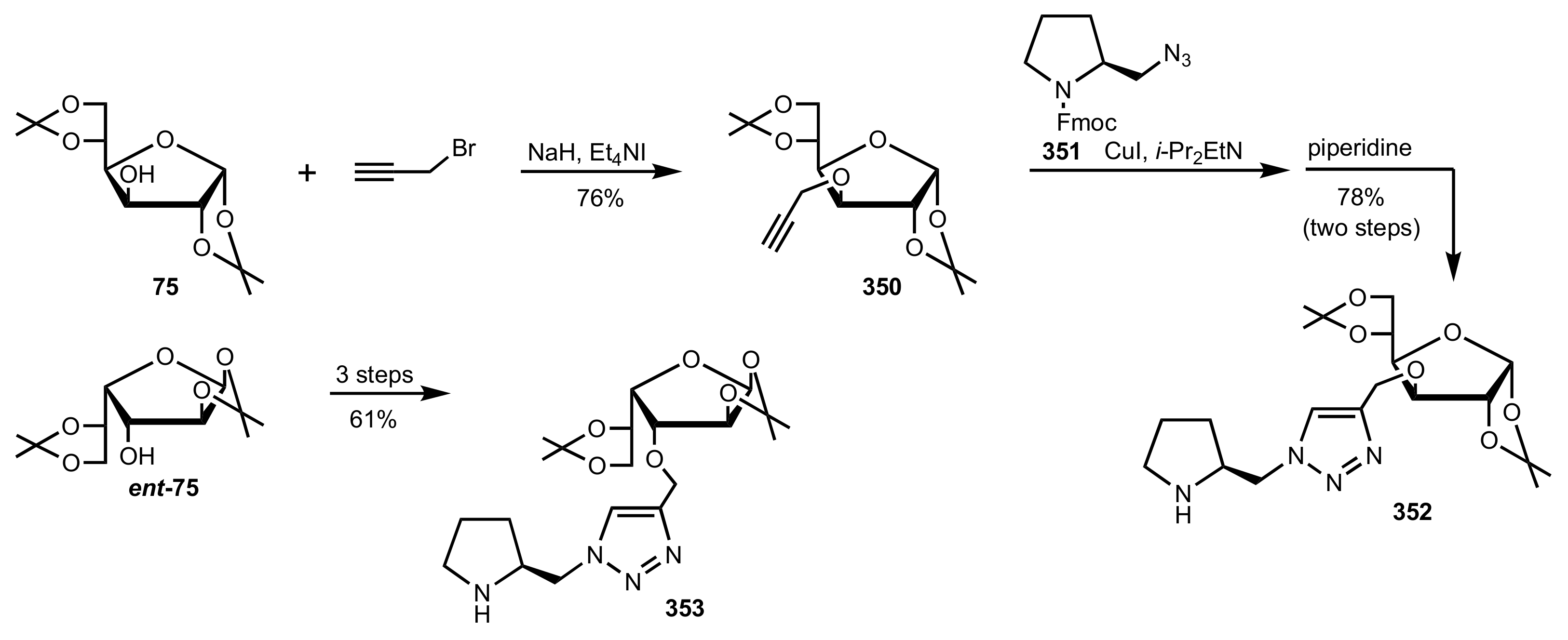











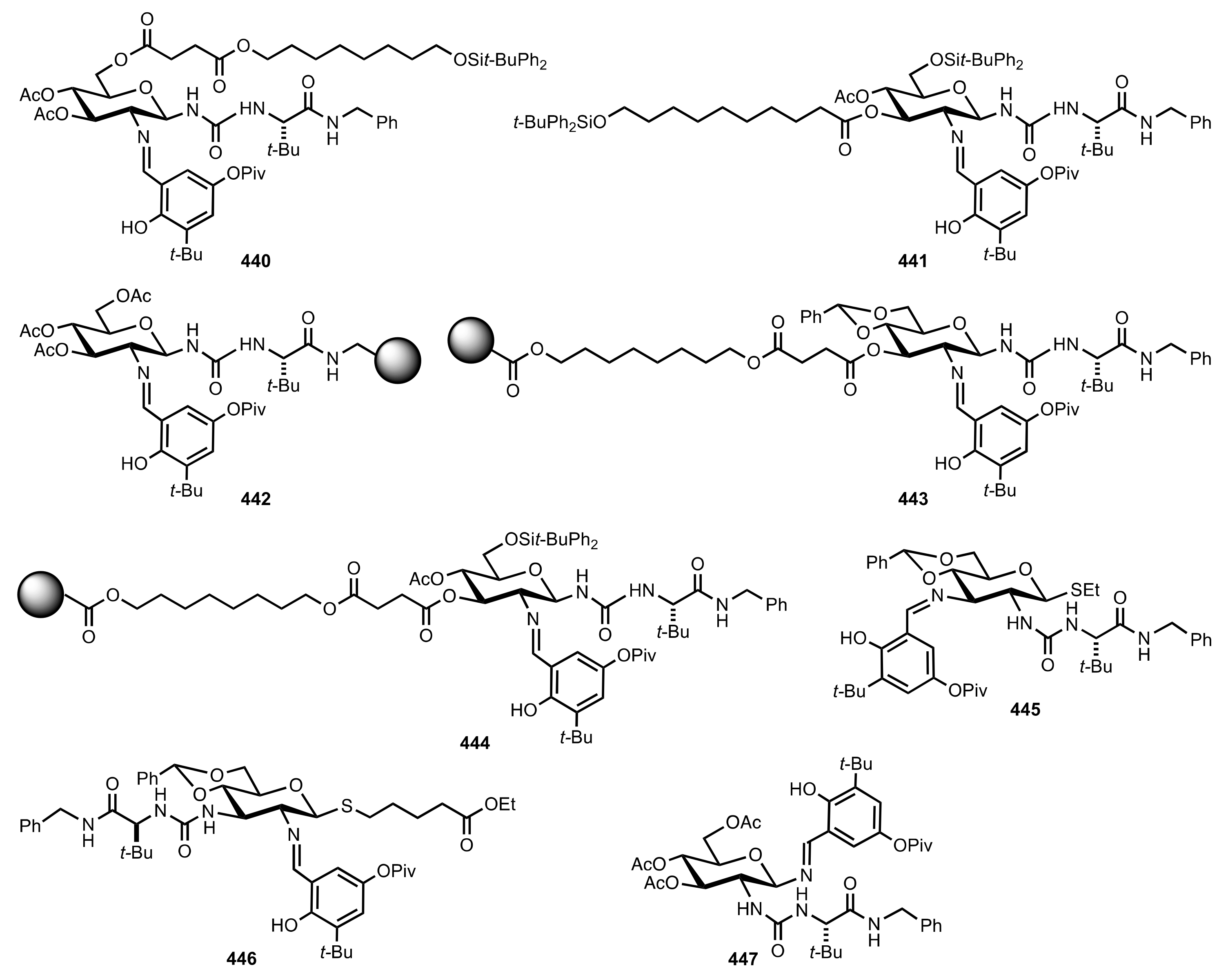

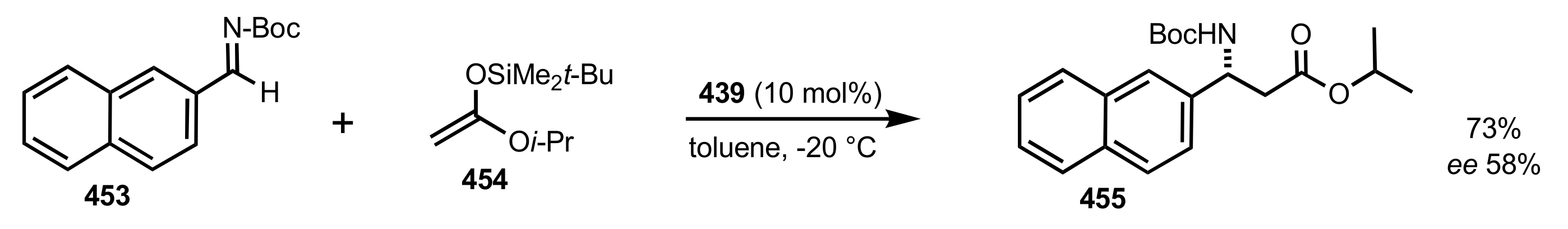
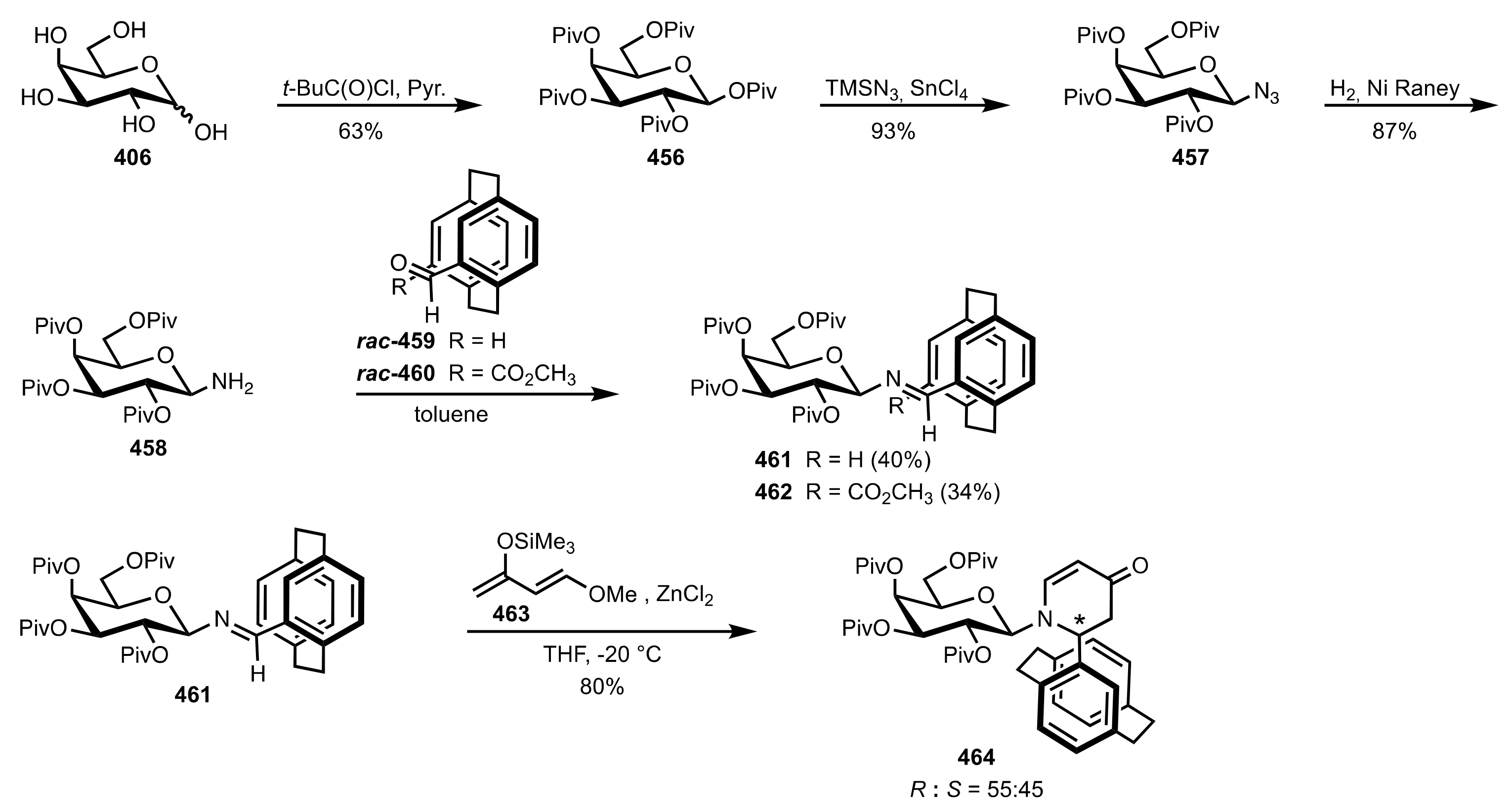
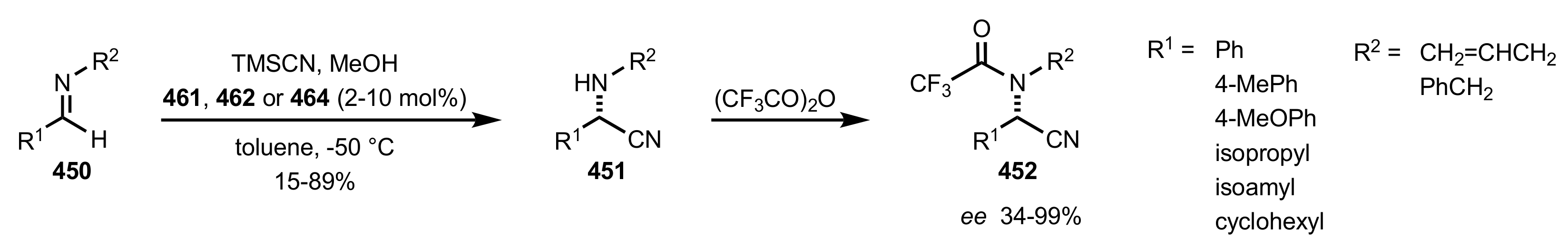
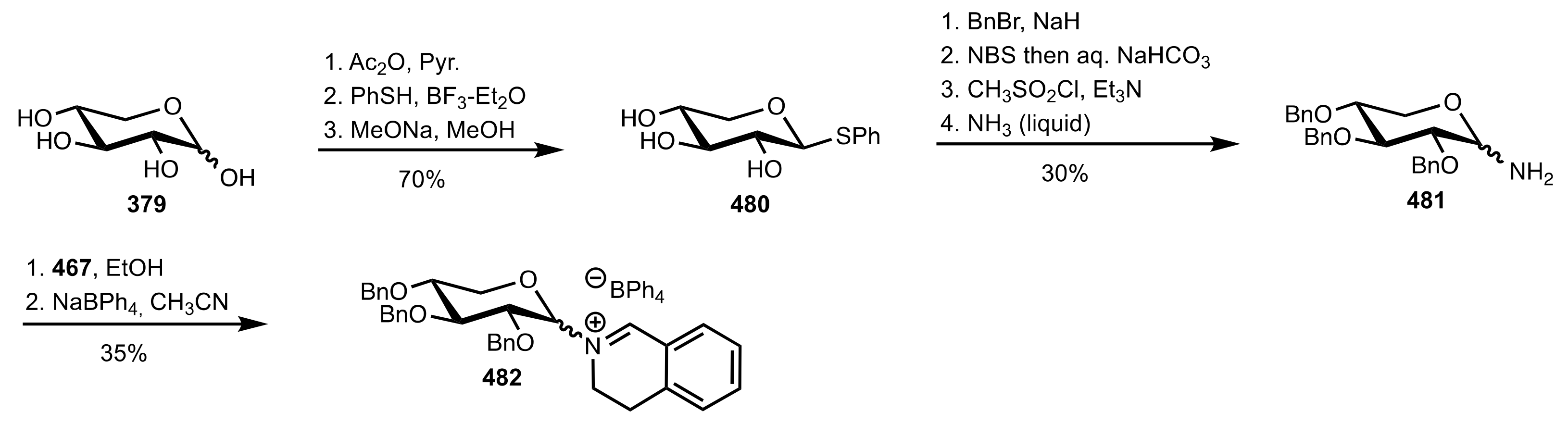

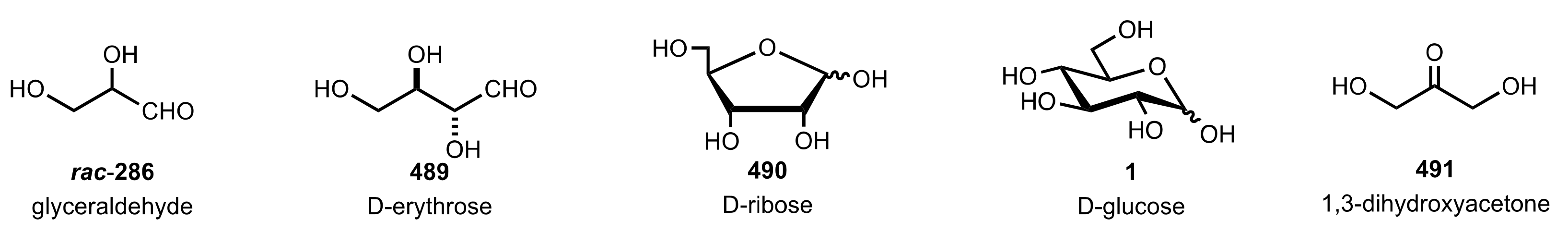
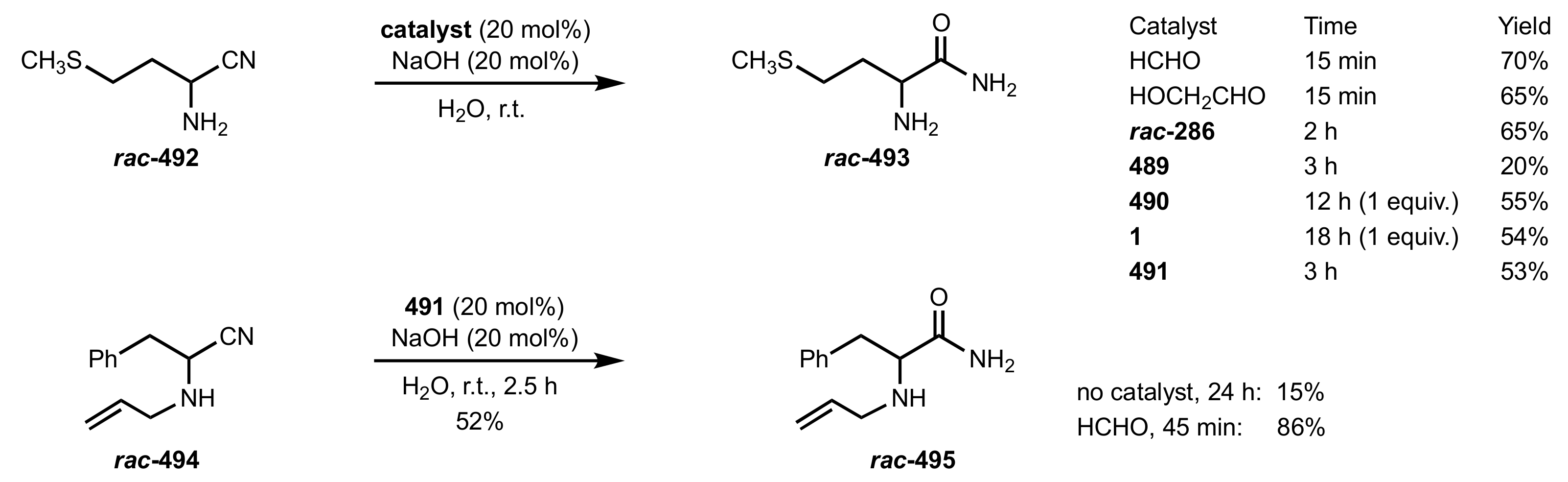


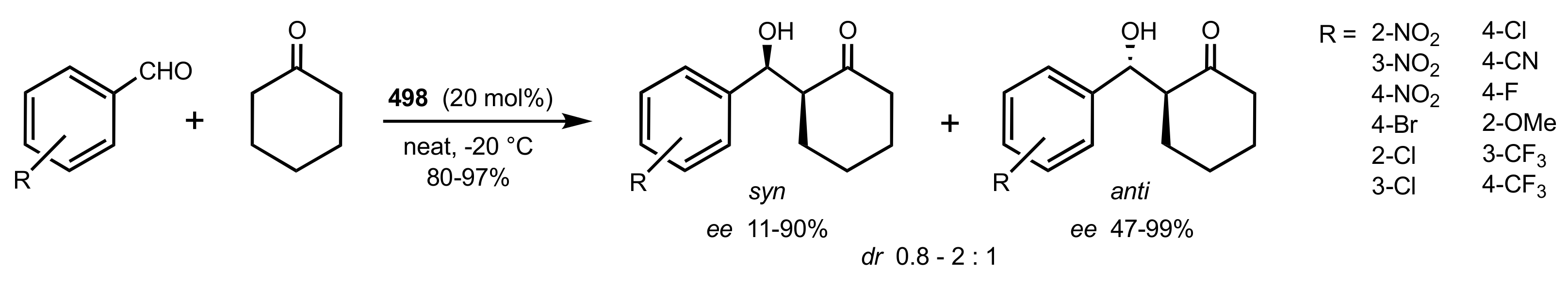










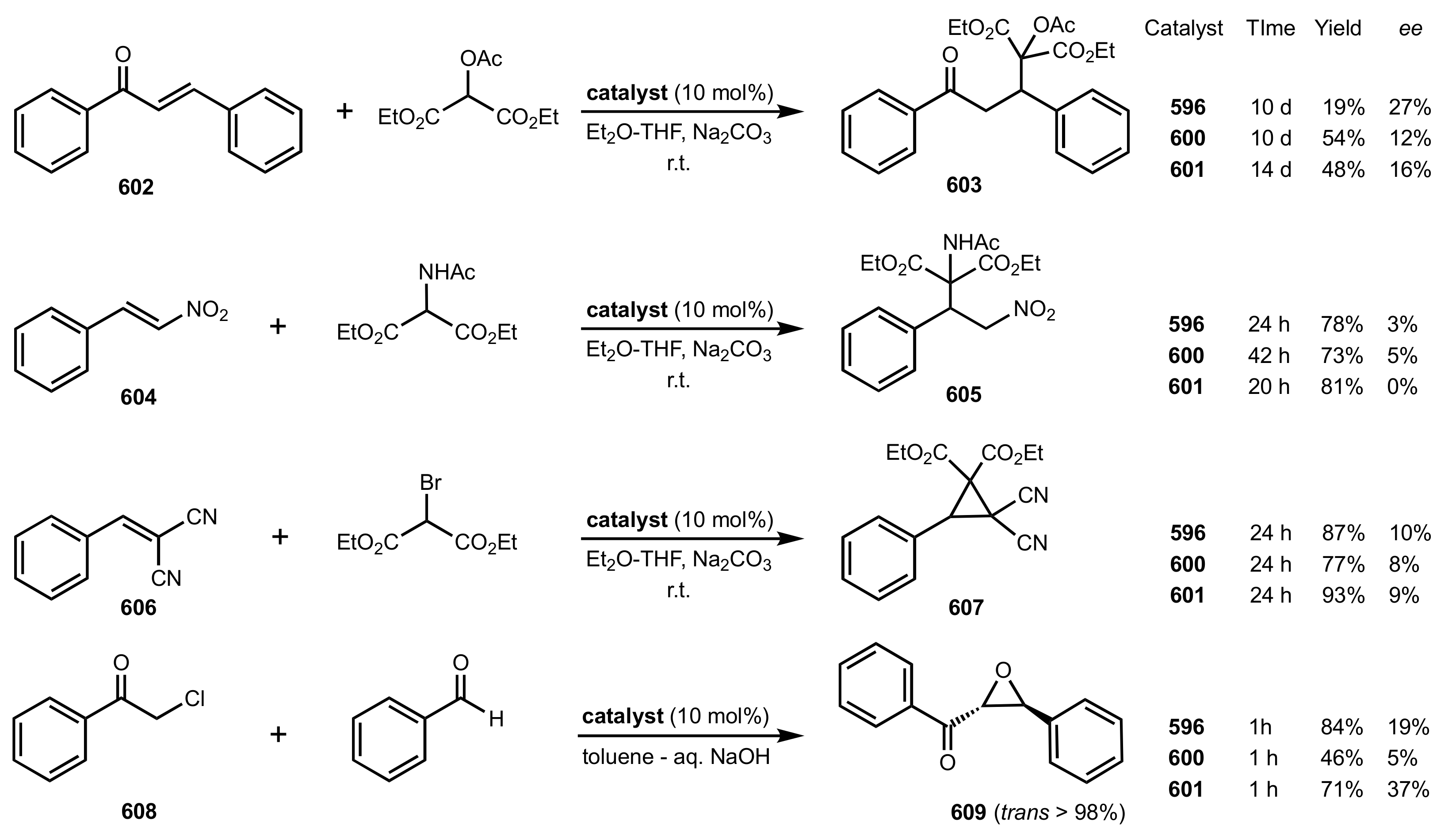


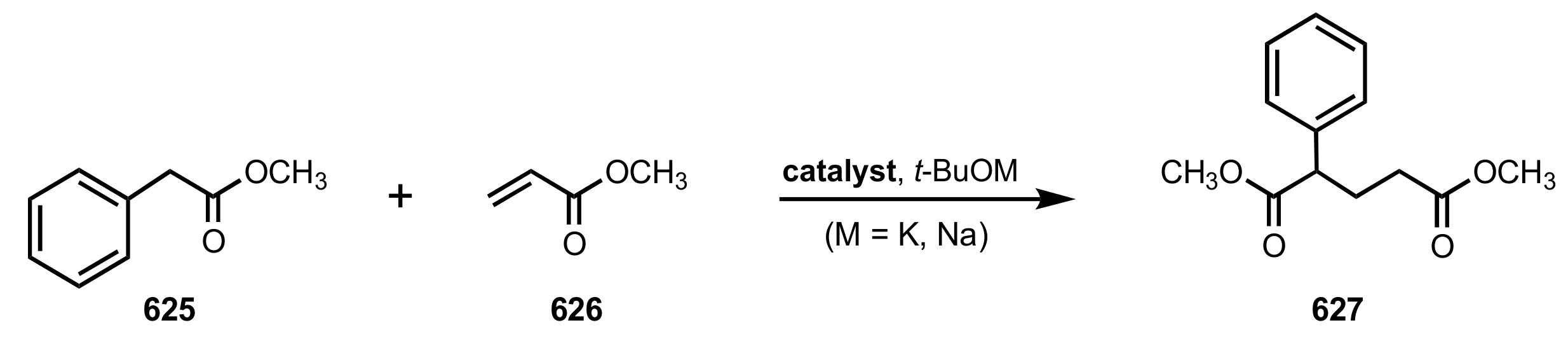
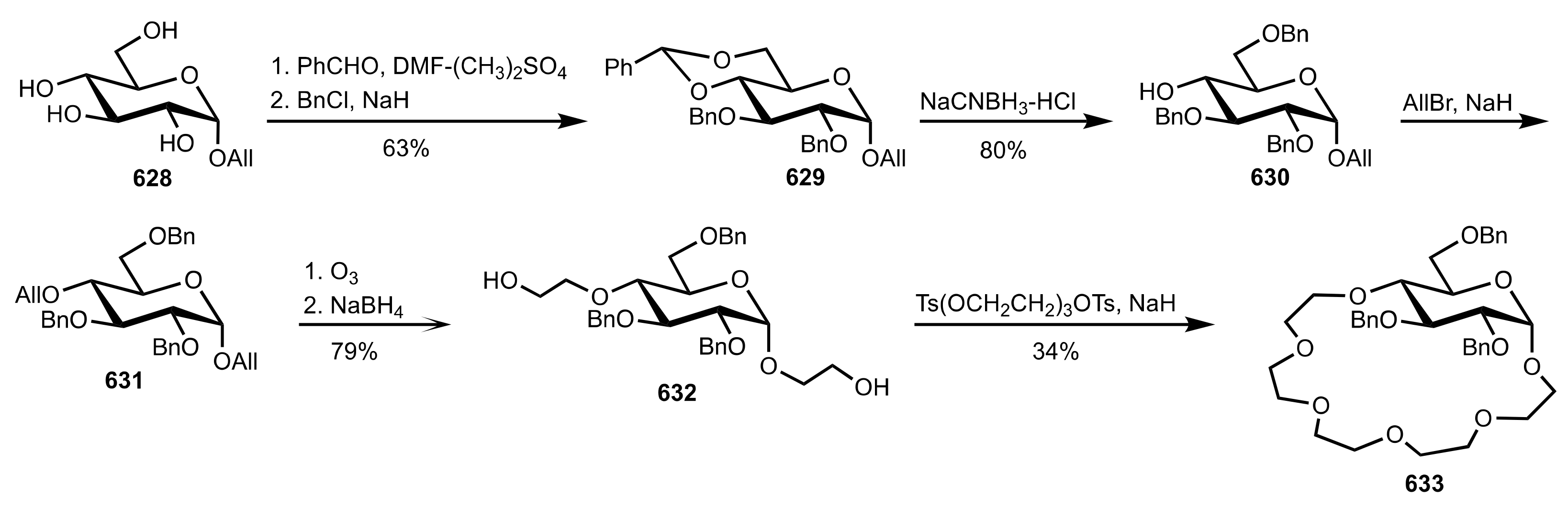
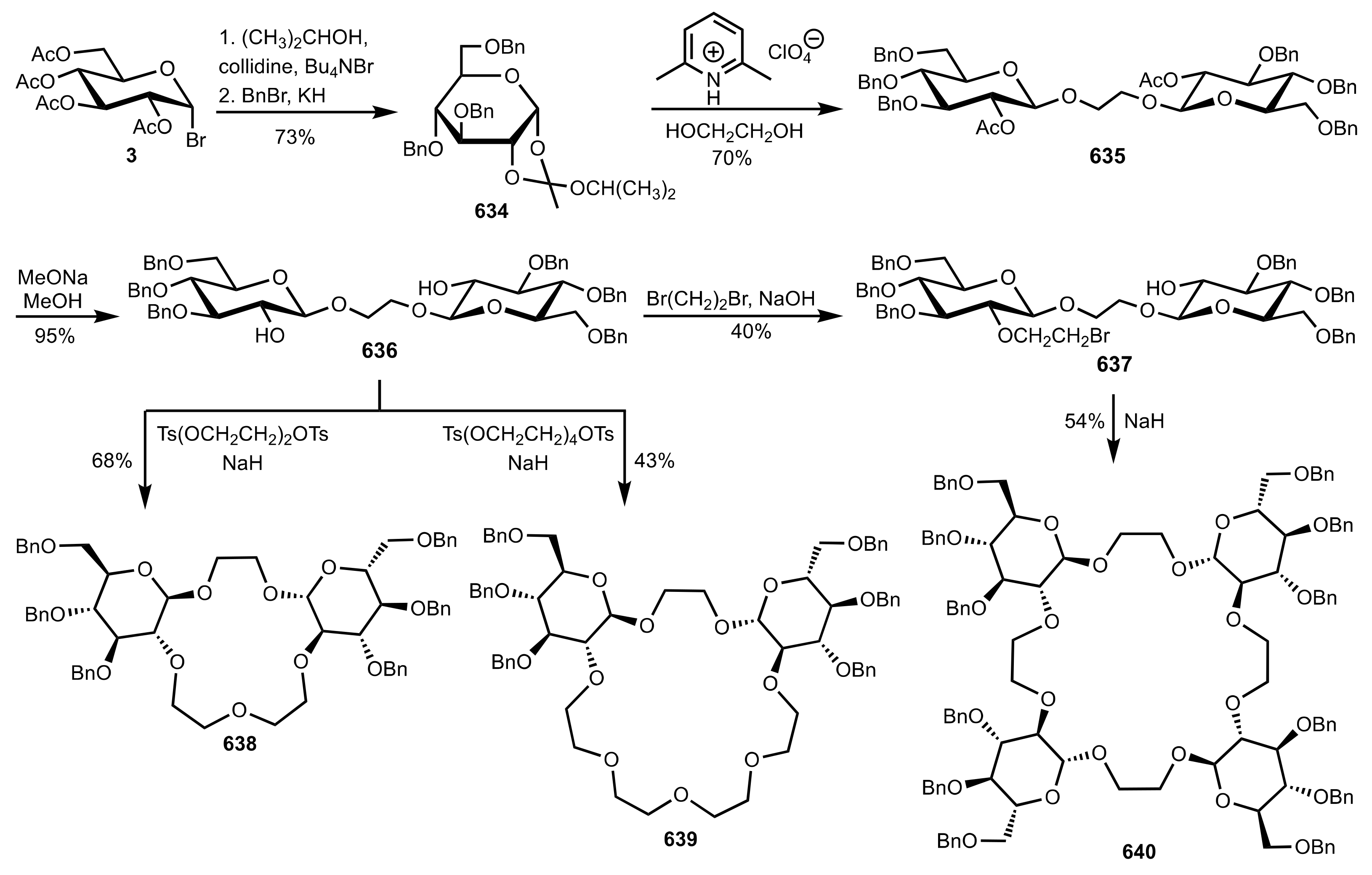




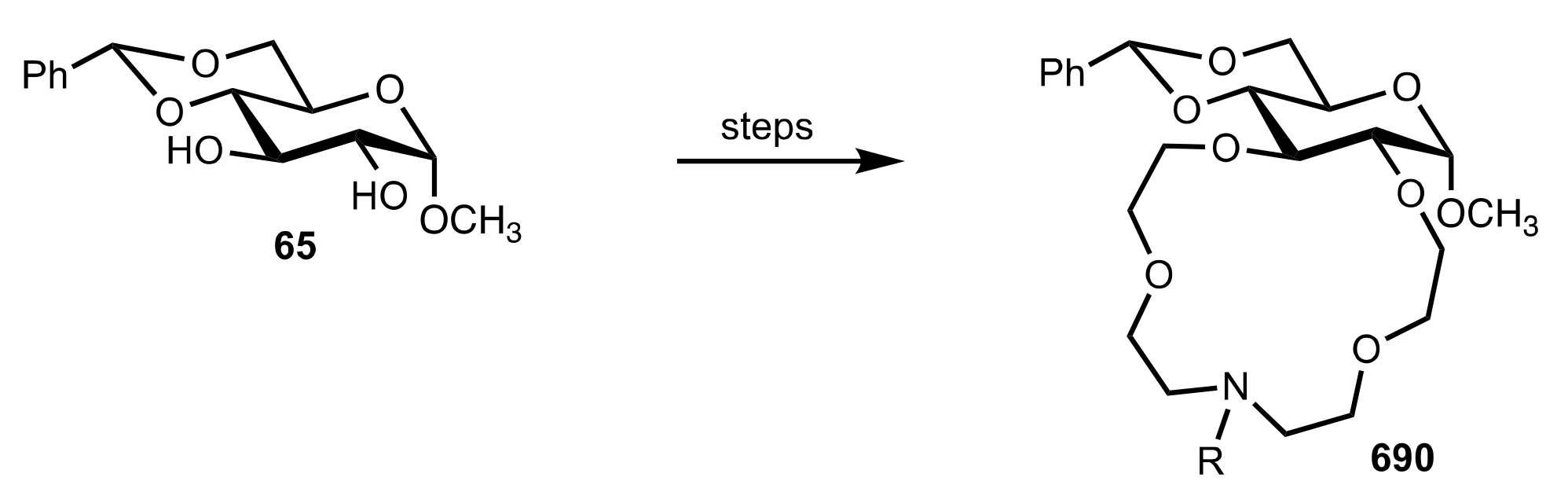

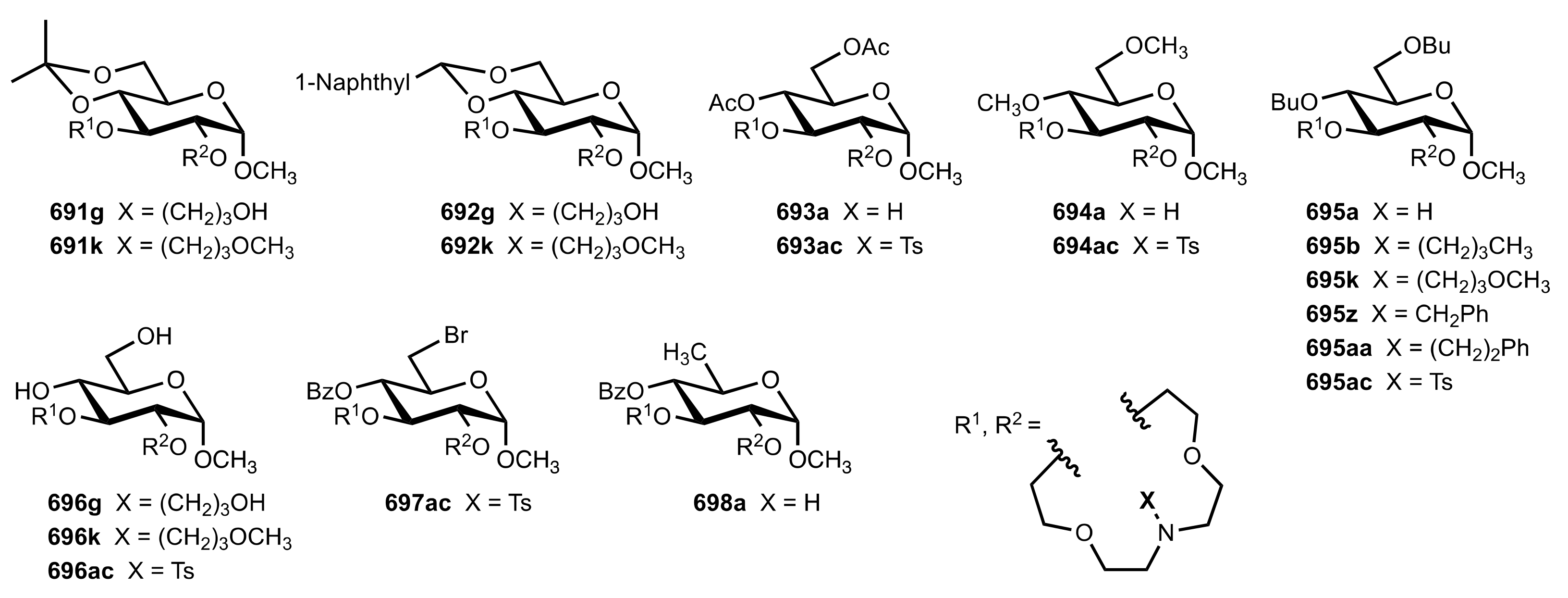
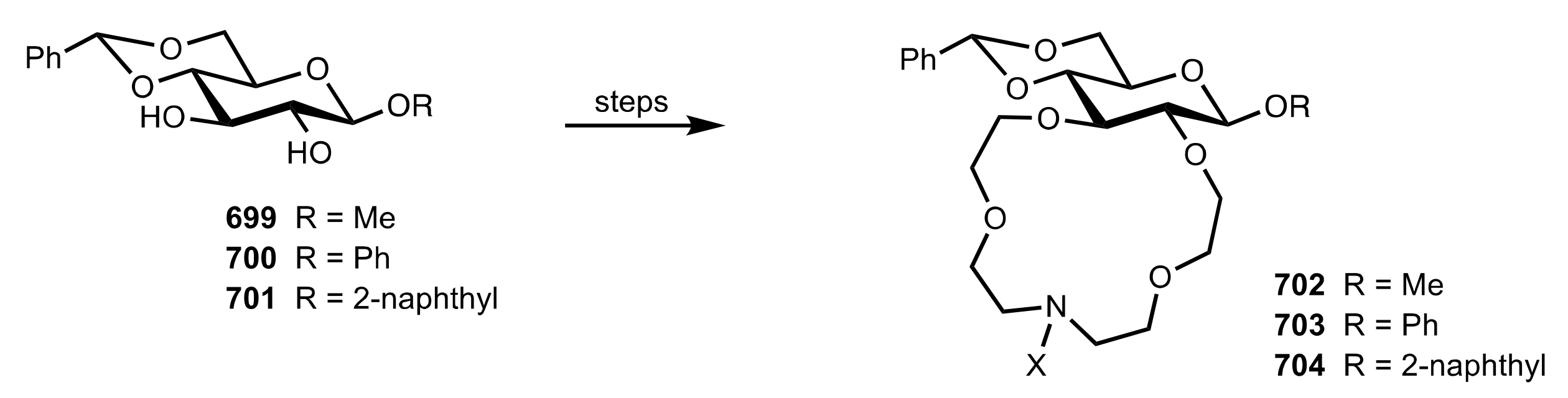

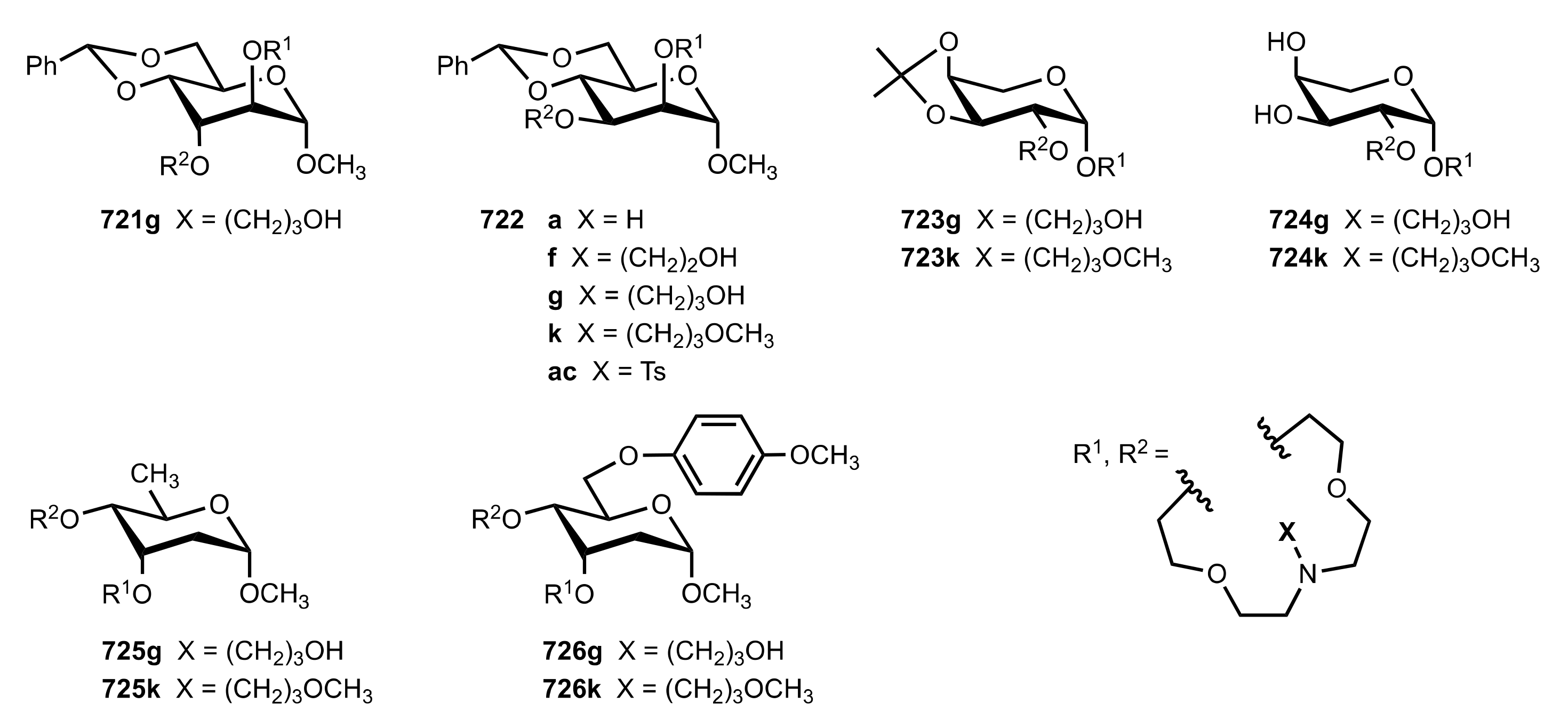


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
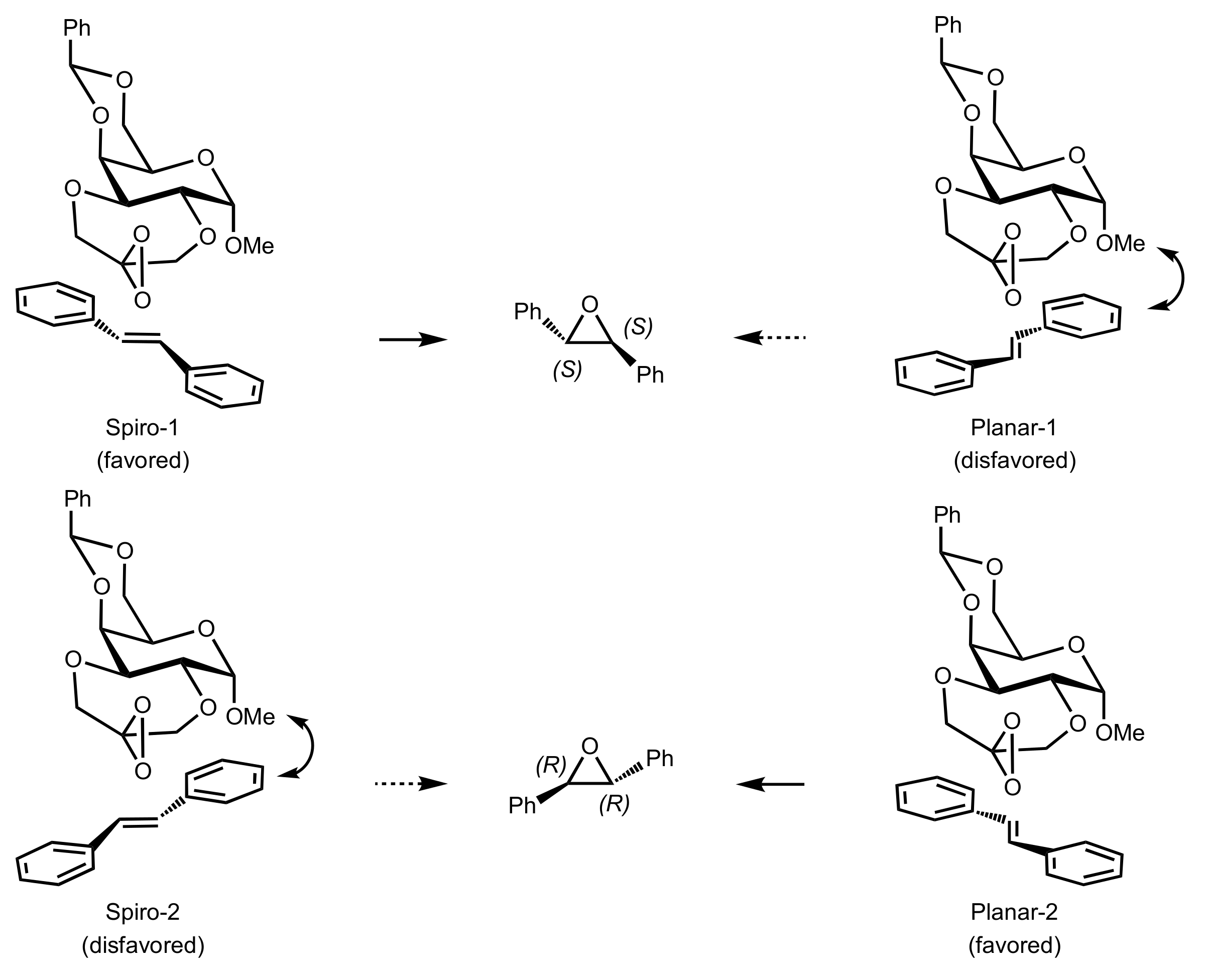{kind=link}
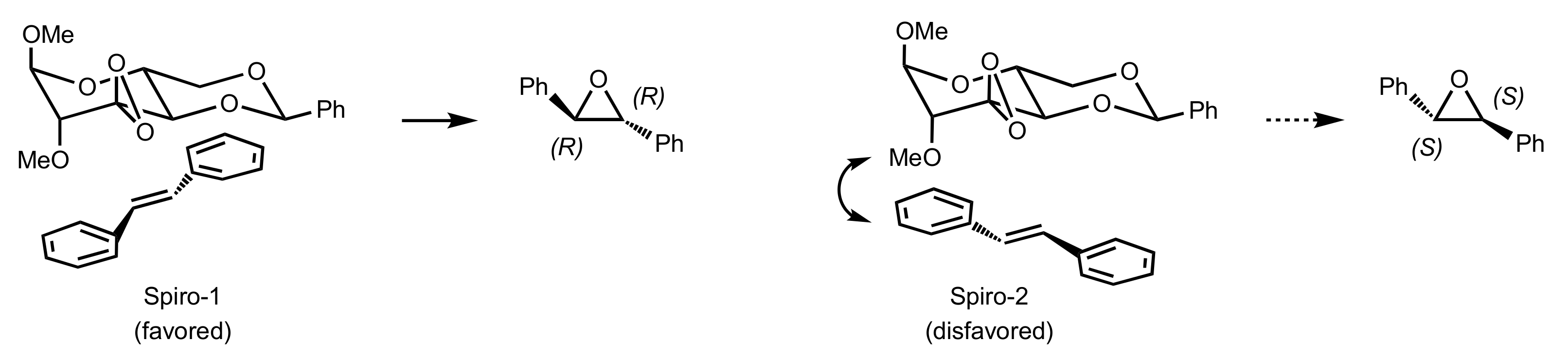{kind=link}
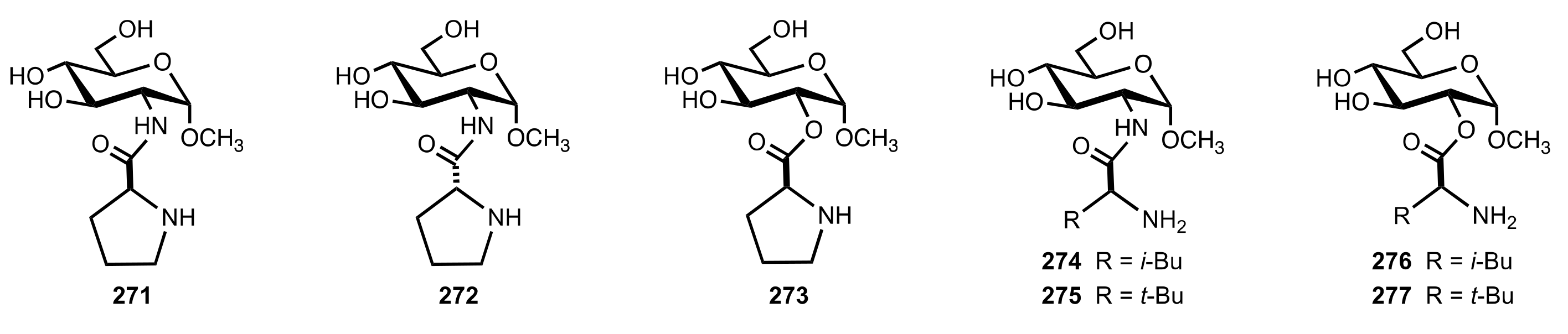{kind=link}
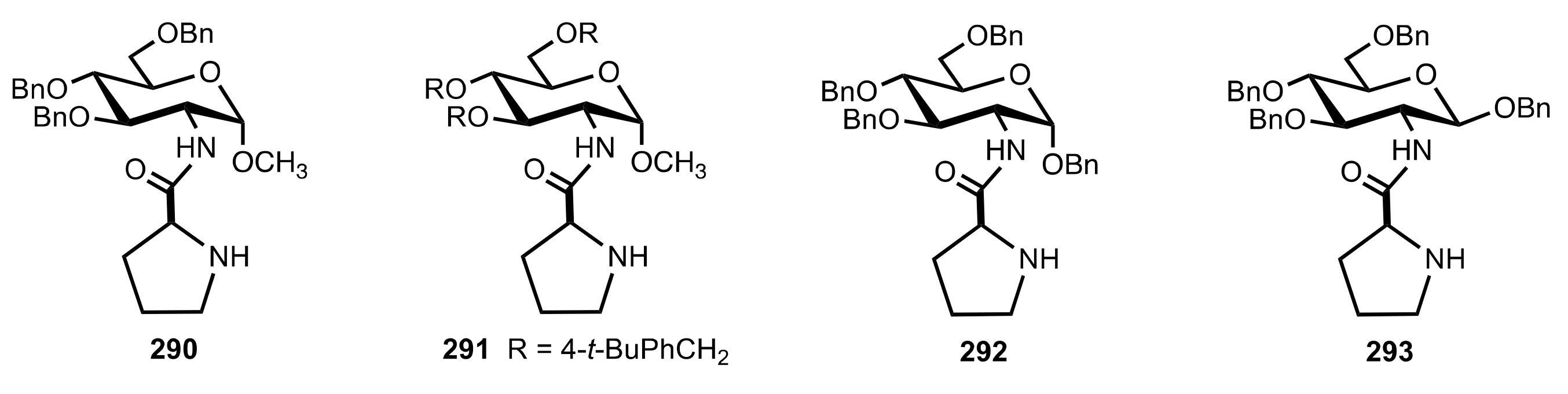{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
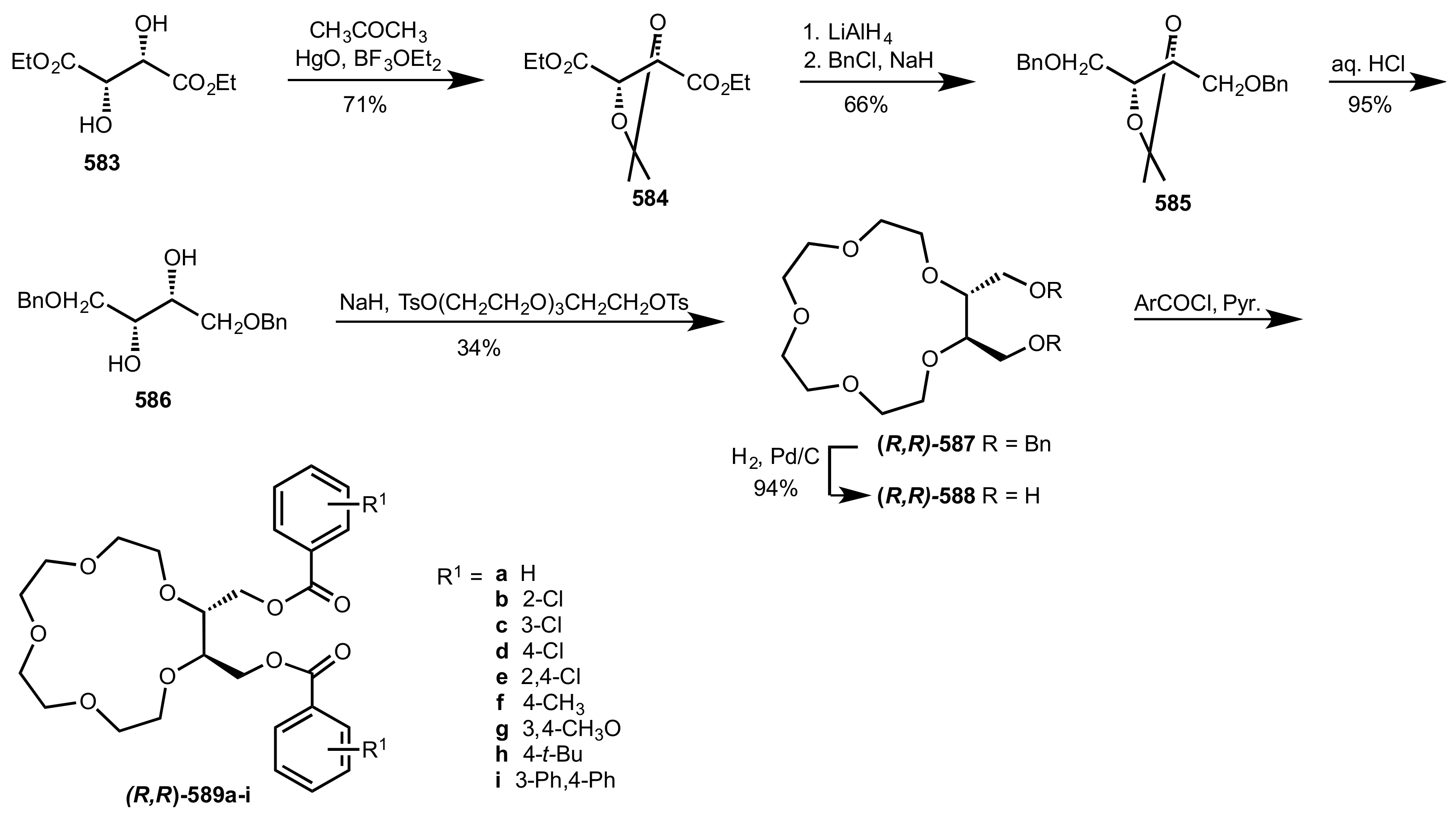{kind=link}
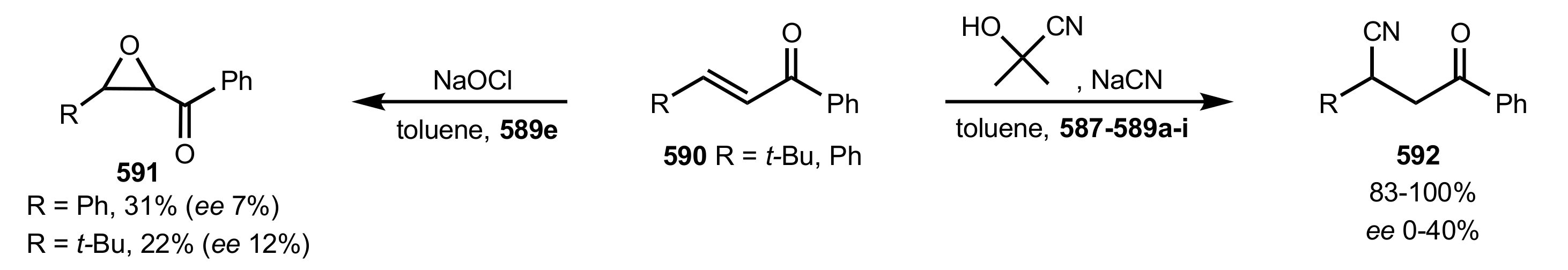{kind=link}
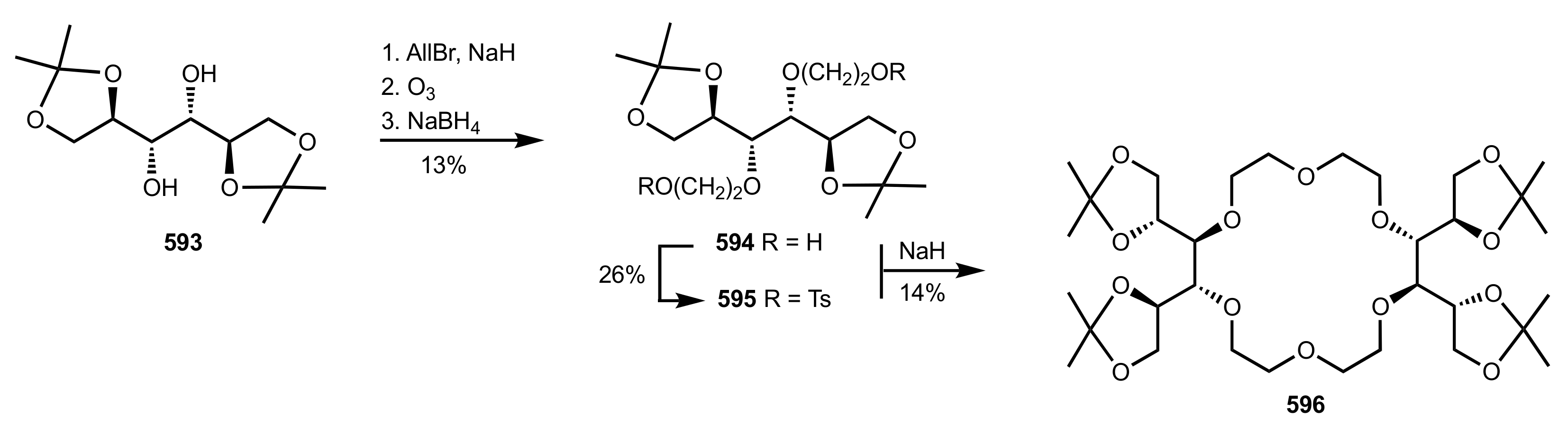{kind=link}
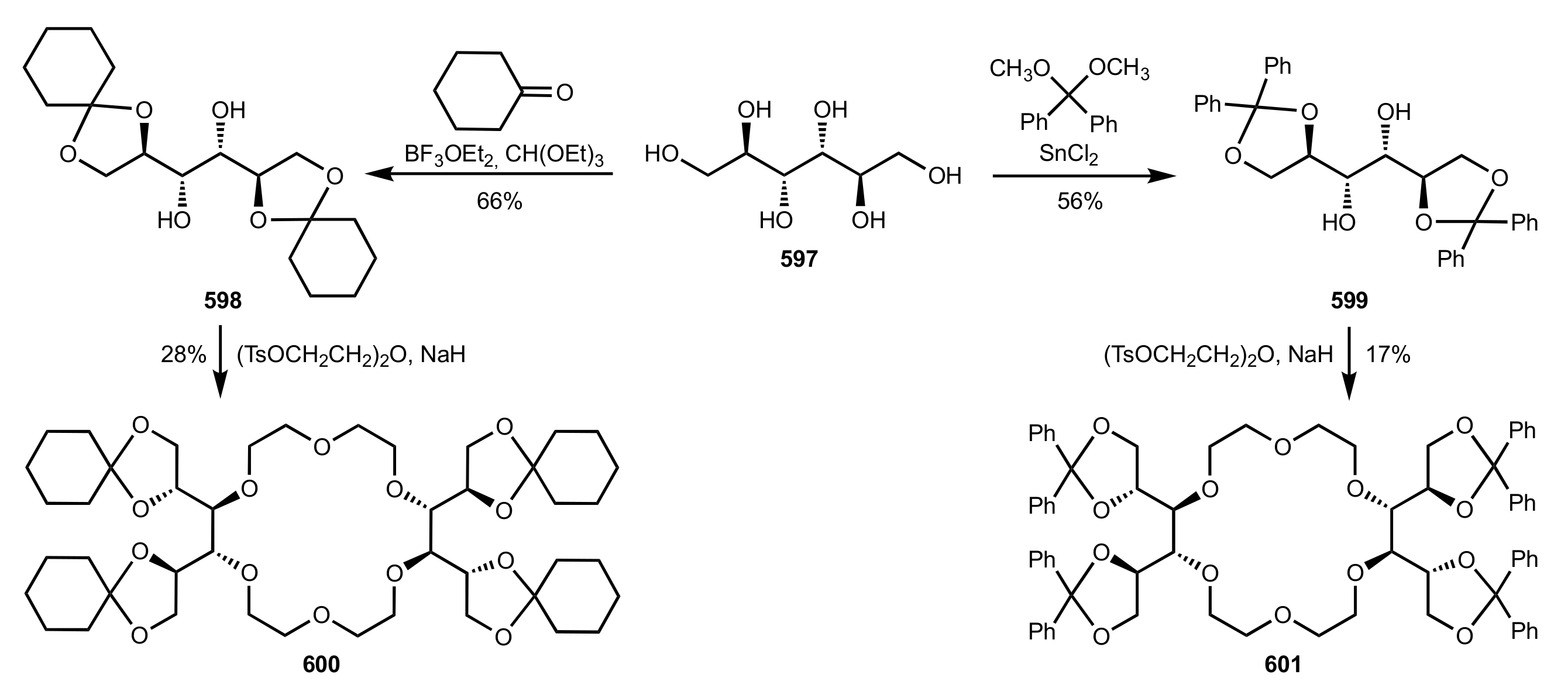{kind=link}
{kind=link}
{kind=link}
{kind=link}
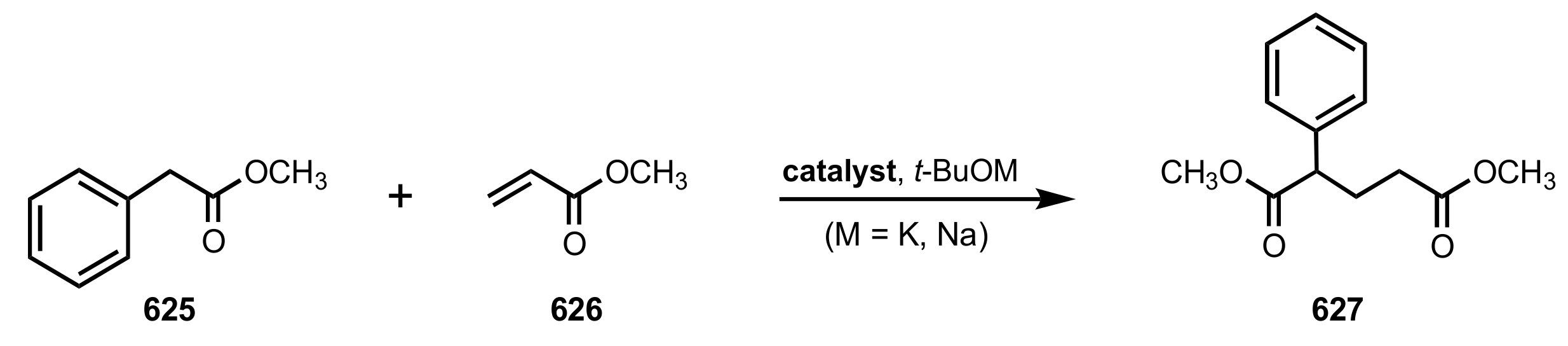{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
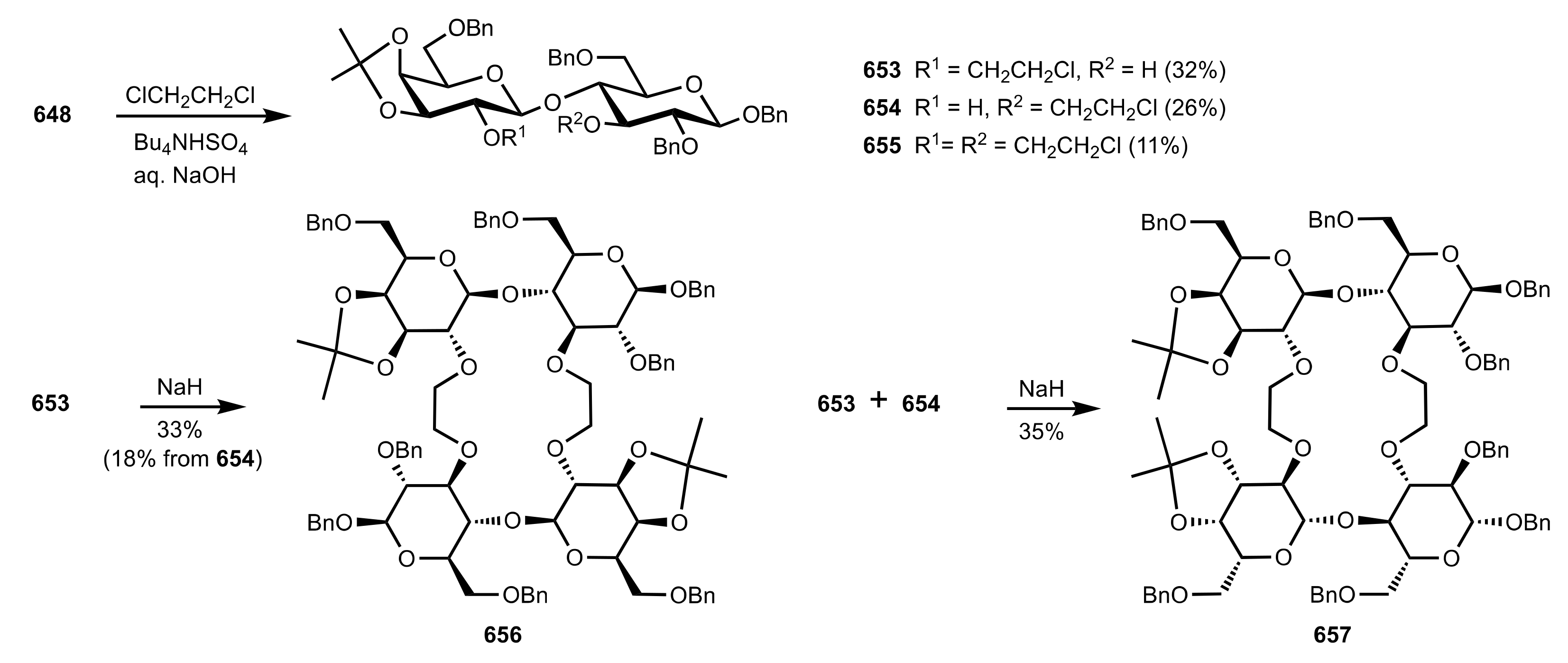{kind=link}
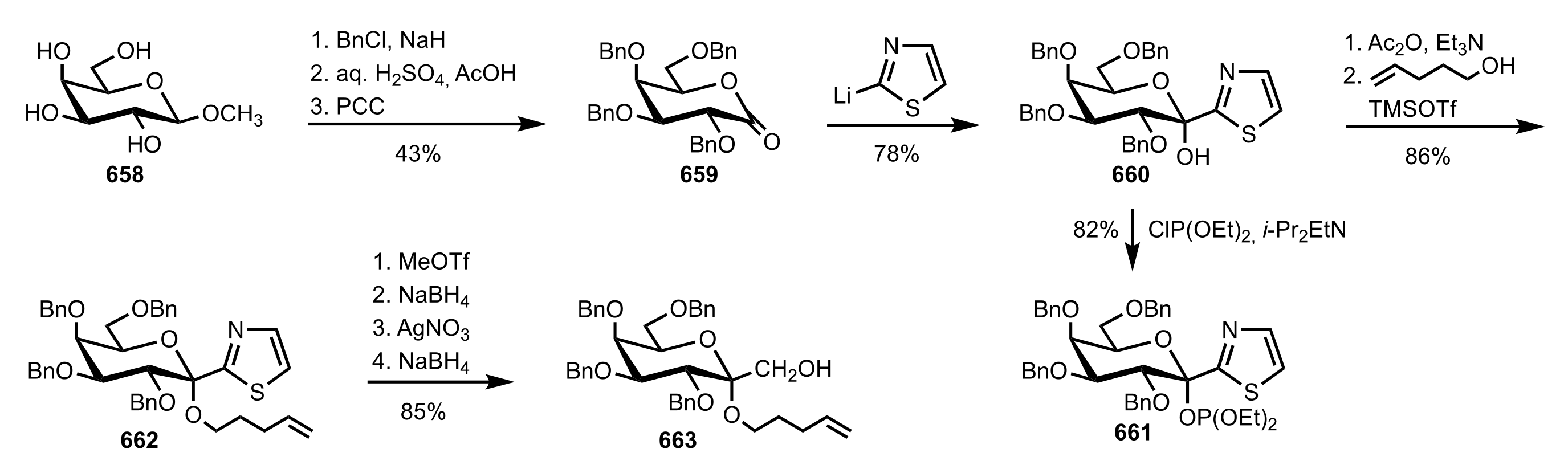{kind=link}
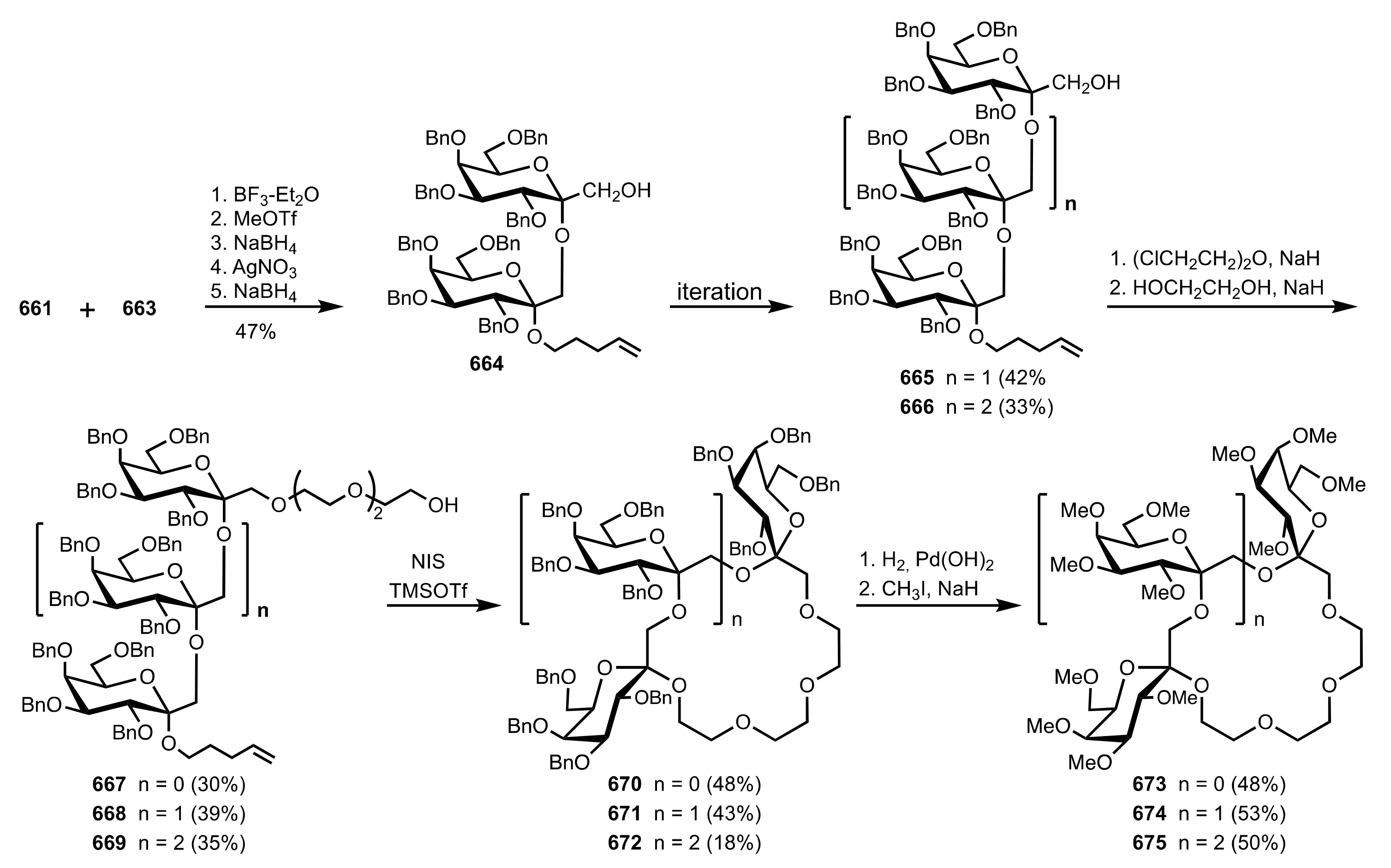{kind=link}
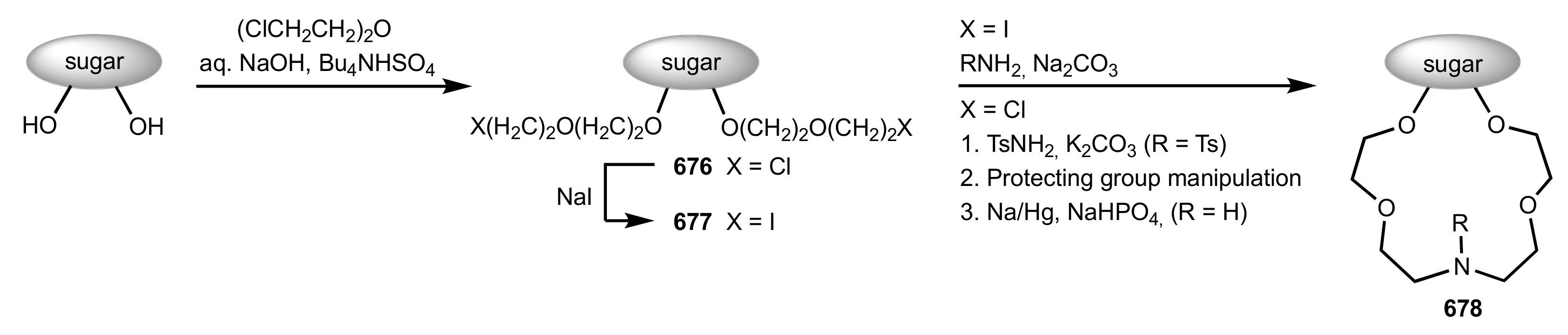{kind=link}
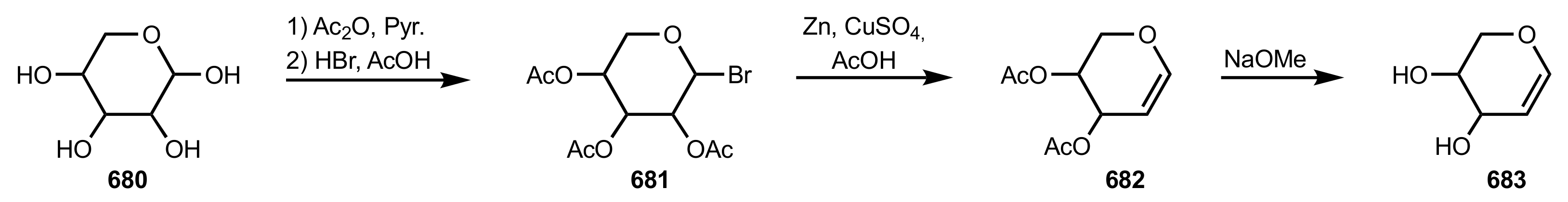{kind=link}
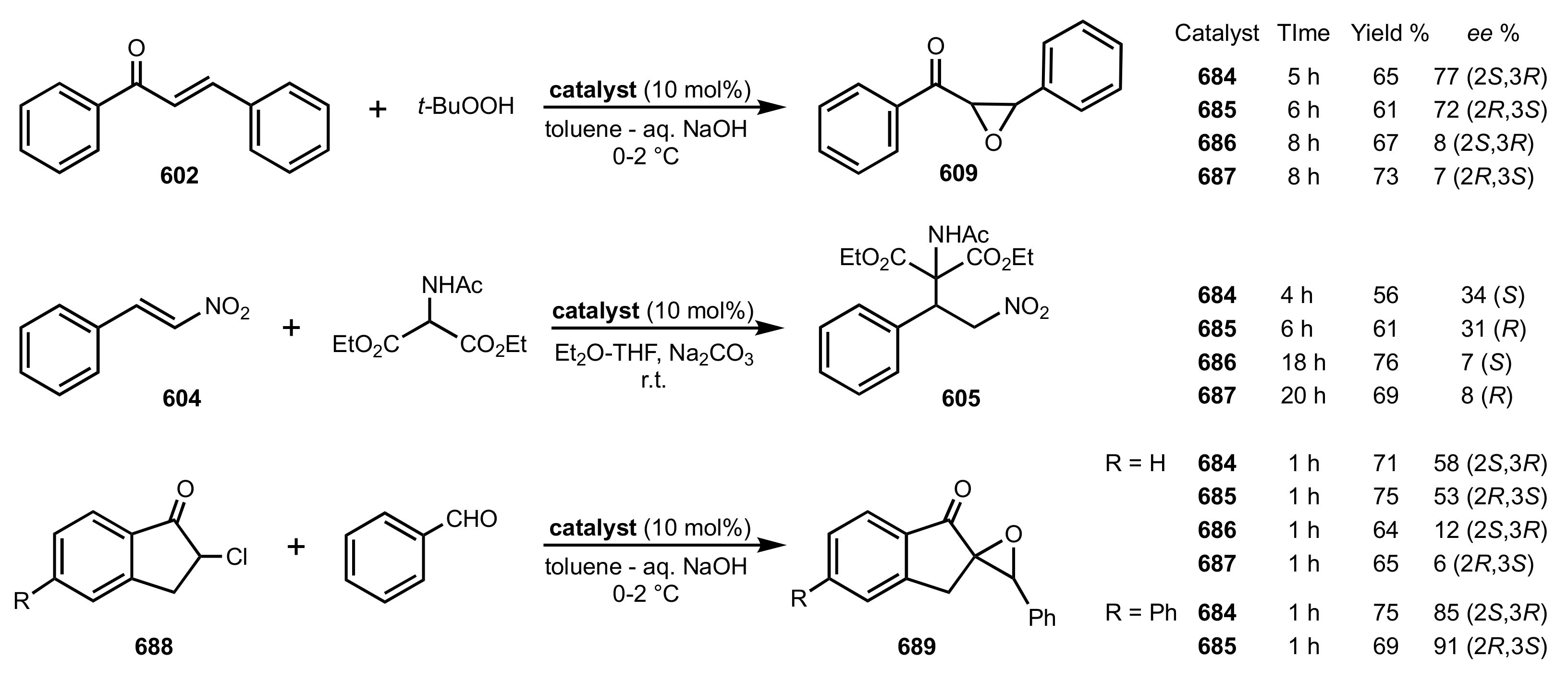{kind=link}
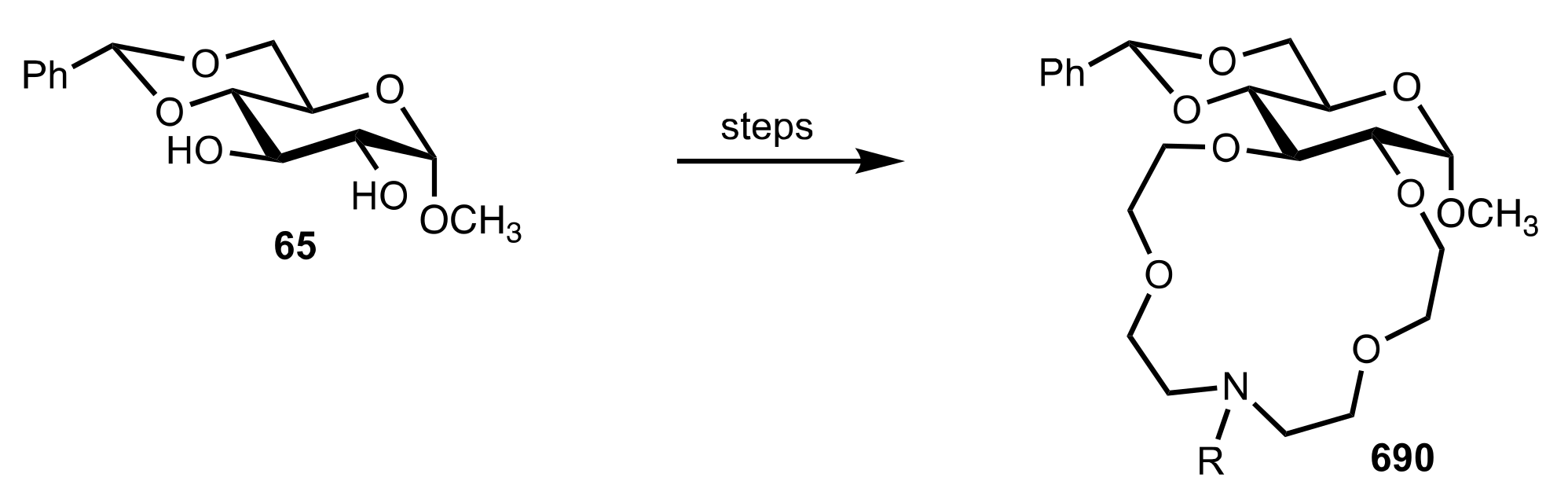{kind=link}
{kind=link}
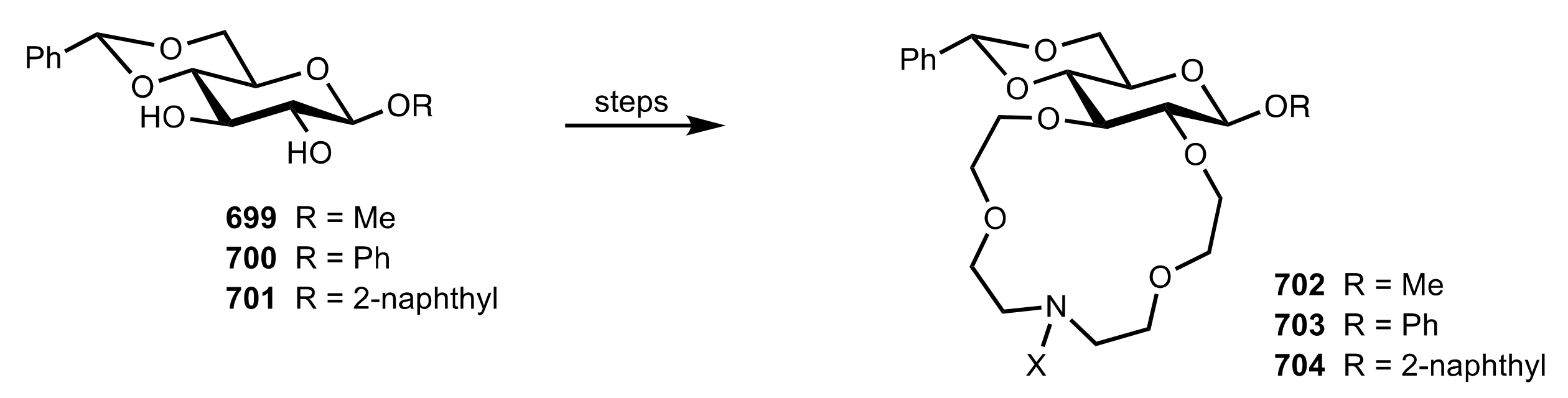{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
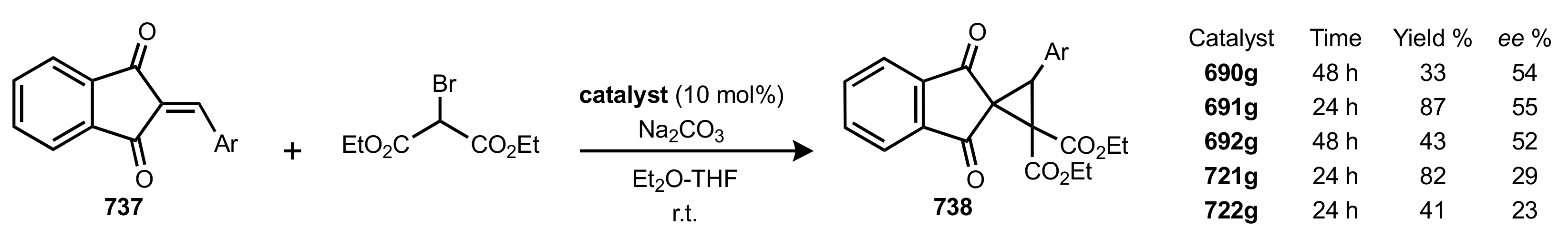{kind=link}
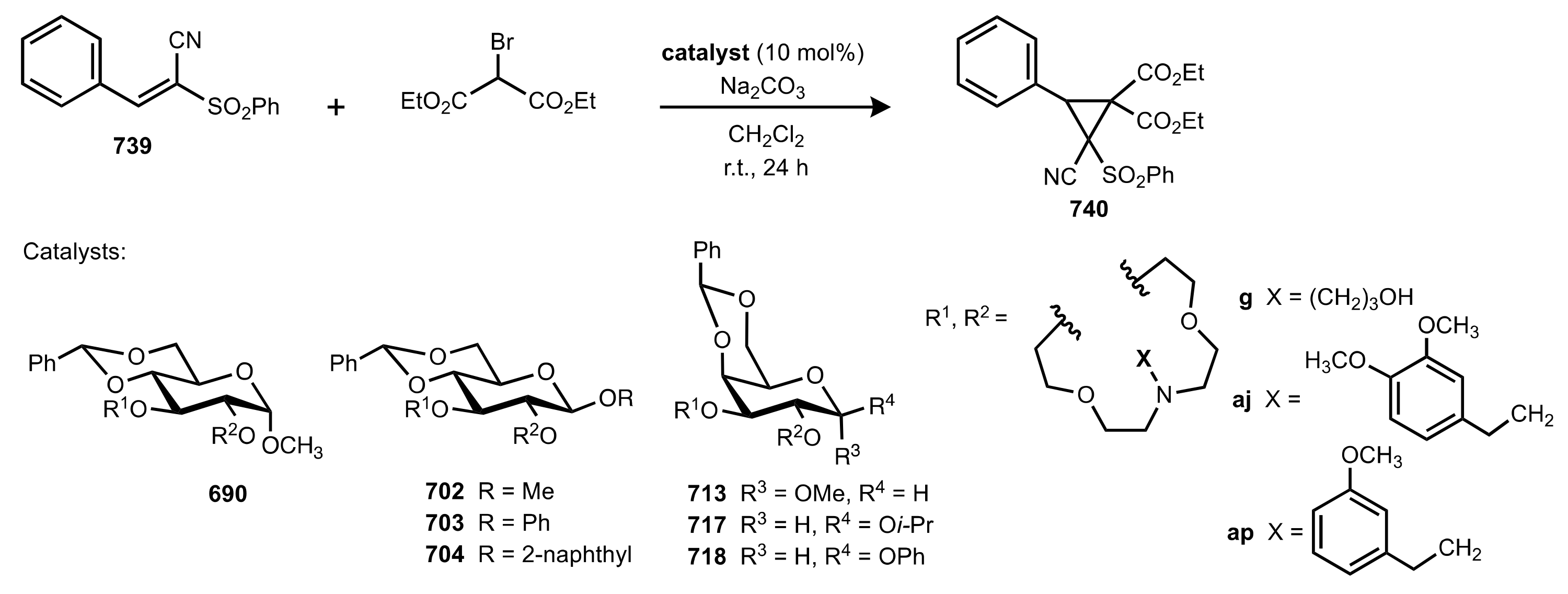{kind=link}
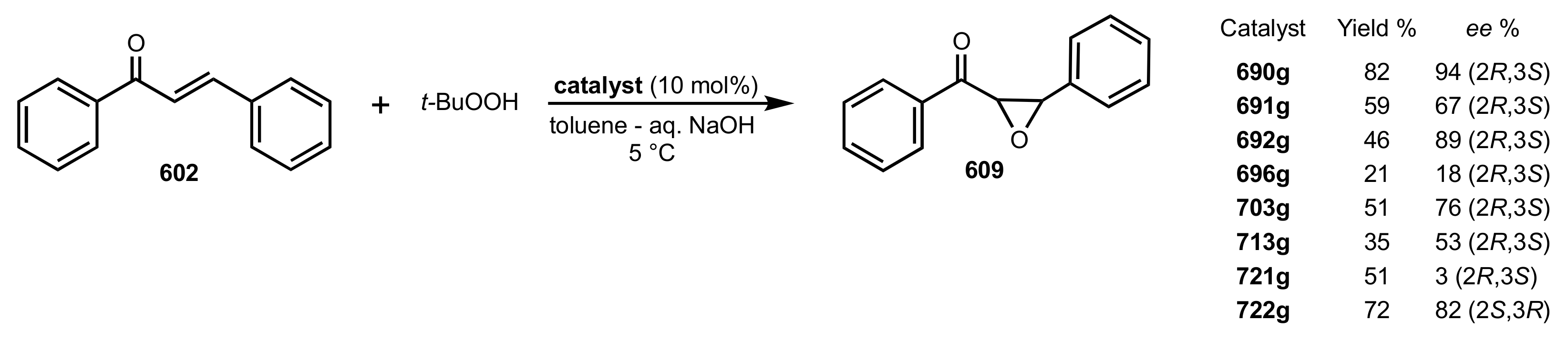{kind=link}
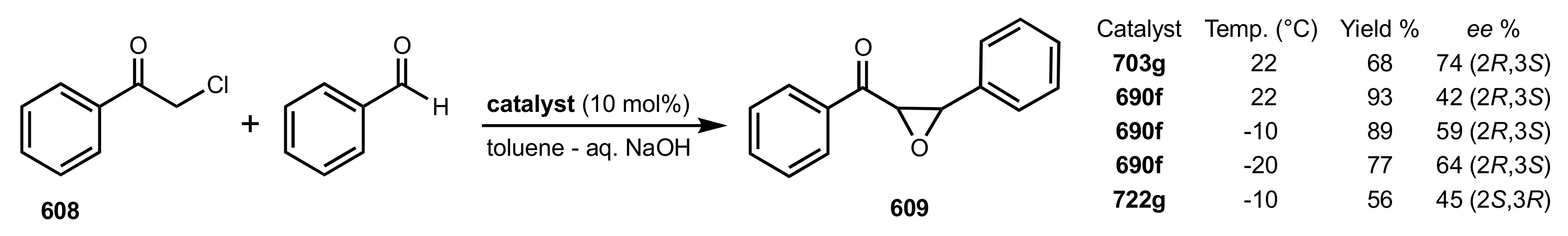{kind=link}
| Entry | Catalyst | Yield (%) | ee % (S) |
|---|---|---|---|
| 1 | 642 | 59 | 17 |
| 2 | 643a | 44 | 7 |
| 3 | 643ba | 82 | 84 |
| 4 | 643b | 100 | 80 |
| 5 | 643bb | 100 | 76 |
| 6 | 643c | 69 | 46 |
| 7 | 643d | 69 | 29 |
| 8 | 643e | 100 | 76 |
| 9 | 643f | 75 | 17 |
| 10 | 643g | 67 | 0.4 |
| 11 | 643h | 31 | 1.7 |
| 12 | 643i | 66 | 0.2 |
| Compound | R | Ref. | Compound | R | Ref. |
|---|---|---|---|---|---|
| 690a | H | [284] | 690r | (CH2)4P(O)(OEt)2 | [285] |
| 690b | butyl | [286] | 690s | (CH2)5P(O)(OEt)2 | [285] |
| 690c | decyl | [287] | 690t | CH2P(O)(Ph)2 | [288] |
| 690d | cyclohexyl | [287] | 690u | (CH2)2P(O)(Ph)2 | [288] |
| 690e | allyl | [289] | 690v | (CH2)3P(O)(Ph)2 | [288] |
| 690f | (CH2)2OH | [287] | 690w | (CH2)4P(O)(Ph)2 | [288] |
| 690g | (CH2)3OH | [286] | 690x | (CH2)5P(O)(Ph)2 | [288] |
| 690h | (CH2)4OH | [286] | 690y | C6H5 | [287] |
| 690i | CH2CH(OH)CH2OH | [289] | 690z | CH2C6H5 | [287] |
| 690j | (CH2)2OCH3 | [286] | 690aa | (CH2)2C6H5 | [286] |
| 690k | (CH2)3OCH3 | [289] | 690ab | (CH2)3C6H5 | [289] |
| 690l | (CH2)3O(CH2)2OCH3 | [289] | 690ac | Ts | [284] |
| 690m | (CH2)3N(CH3)2 | [289] | 690ad | (CH2)3NHC(S)NHPh | [289] |
| 690n | CH2CO2CH3 | [287] | 690ae a |  | [290] |
| 690o | CH2P(O)(OEt)2 | [285] | 690af |  | [291] |
| 690p | (CH2)2P(O)(OEt)2 | [285] | 690ag |  | [289] |
| 690q | (CH2)3P(O)(OEt)2 | [285] | 690ah a |  | [290] |
| Compound | X | Ref. | Compound | X | Ref. |
|---|---|---|---|---|---|
| 702aj |  | [297] | 703ak | CHi-Pr2 | [298,299] |
| 703a | H | [300] a | 703al | CH2-cyclohexyl | [298,299] |
| 703b | butyl | [298,299] | 703am | hexyl | [299] |
| 703d | cyclohexyl | [298] a | 703an | (CH2)2CH3 | [301] a |
| 703f | (CH2)2OH | [298,299] | 703ao | (CH2)4OCH3 | [301] a |
| 703g | (CH2)3OH | [298,299] | 703ap |  | [301] |
| 703j | (CH2)2OCH3 | [298,299] | 703aq |  | [299] |
| 703k | (CH2)3OCH3 | [299] | 703ar |  | [299] |
| 703z | CH2C6H5 | [298,299] | 703as | 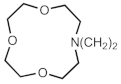 | [299] |
| 703aa | (CH2)2C6H5 | [298,299] | 704g | (CH2)3OH | [297] |
| 703af | 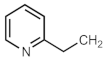 | [301] a | 704aj |  | [297] |
| 703aj | | [301] |
| Compound | X | Ref. | Compound | X | Ref. |
|---|---|---|---|---|---|
| 713b | butyl | [286] | 714g | (CH2)3OH | [302] |
| 713c | decyl | [287] | 715g | (CH2)3OH | [302] |
| 713g | (CH2)3OH | [303] | 716g | (CH2)3OH | [302] |
| 713j | (CH2)2OCH3 | [286] | 717g | (CH2)3OH | [302] |
| 713k | (CH2)3OCH3 | [303] | 718g | (CH2)3OH | [302] |
| 713z | CH2C6H5 | [287] | 718aj | | [297] |
| 713aa | (CH2)2C6H5 | [286] | 719g | (CH2)3OH | [302] |
| 713aj | | [297] | 720g | (CH2)3OH | [302] |
| 713as |  | [297] |
| Entry | Catalyst | Time (h) | Yield (%) | ee (%) | Ref. |
|---|---|---|---|---|---|
| 1 | 690f | 20 | 51 | 62 (R) a | [286,313] |
| 2 | 690g | 28 | 53 | 85 (R) a | [286,293,309] |
| 3 | 690j | 40 | 45 | 87 (R) | [286,310] |
| 4 | 690t | 48 | 39 | 60 (R) | [288] |
| 5 | 690u | 48 | 41 | 74 (R) | [288] |
| 6 | 690v | 48 | 32 | 77 (R) | [288] |
| 7 | 690w | 32 | 43 | 94 (R) | [285,288,310] |
| 8 | 722f | 44 | 32 | 70 (S) | [305] |
| 9 | 722g | 52 | 37 | 92 (S) | [305,309] |
| 10 | 722h | 58 | 40 | 63(S) | [305] |
| 11 | 722k | 50 | 37 | 77 (S) | [305] |
| 12 | 713j | 38 | 34 | 52 (R) | [286] |
| 13 | 729a | 24 | 48 | 72 (S) | [308] |
| 14 | 729b | 30 | 47 | 76 (S) | [308] |
| 15 | 729c | 30 | 51 | 67 (S) | [308] |
| 16 | 730 | 25 | 50 | 80 (R) | [308] |
| Entry | Catalyst | Yield (%) | ee trans (%) |
|---|---|---|---|
| 1 | 690g | 89 | 50 |
| 2 | 690aj | 94 | 73 |
| 3 | 702aj | 91 | 58 |
| 4 | 704g | 88 | 18 |
| 5 | 704aj | 90 | 35 |
| 6 | 713g | 85 | 62 |
| 7 | 713aj | 93 | 80 |
| 8 | 713ap | 95 | 76 |
| 9 | 717g | 93 | 61 |
| 10 | 718g | 91 | 43 |
| 11 | 718aj | 90 | 72 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wojaczyńska, E.; Steppeler, F.; Iwan, D.; Scherrmann, M.-C.; Marra, A. Synthesis and Applications of Carbohydrate-Based Organocatalysts. Molecules 2021, 26, 7291. https://doi.org/10.3390/molecules26237291
Wojaczyńska E, Steppeler F, Iwan D, Scherrmann M-C, Marra A. Synthesis and Applications of Carbohydrate-Based Organocatalysts. Molecules. 2021; 26(23):7291. https://doi.org/10.3390/molecules26237291
Chicago/Turabian StyleWojaczyńska, Elżbieta, Franz Steppeler, Dominika Iwan, Marie-Christine Scherrmann, and Alberto Marra. 2021. "Synthesis and Applications of Carbohydrate-Based Organocatalysts" Molecules 26, no. 23: 7291. https://doi.org/10.3390/molecules26237291
APA StyleWojaczyńska, E., Steppeler, F., Iwan, D., Scherrmann, M.-C., & Marra, A. (2021). Synthesis and Applications of Carbohydrate-Based Organocatalysts. Molecules, 26(23), 7291. https://doi.org/10.3390/molecules26237291