
Application of Skyline for Analysis of Protein–Protein Interactions In Vivo


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
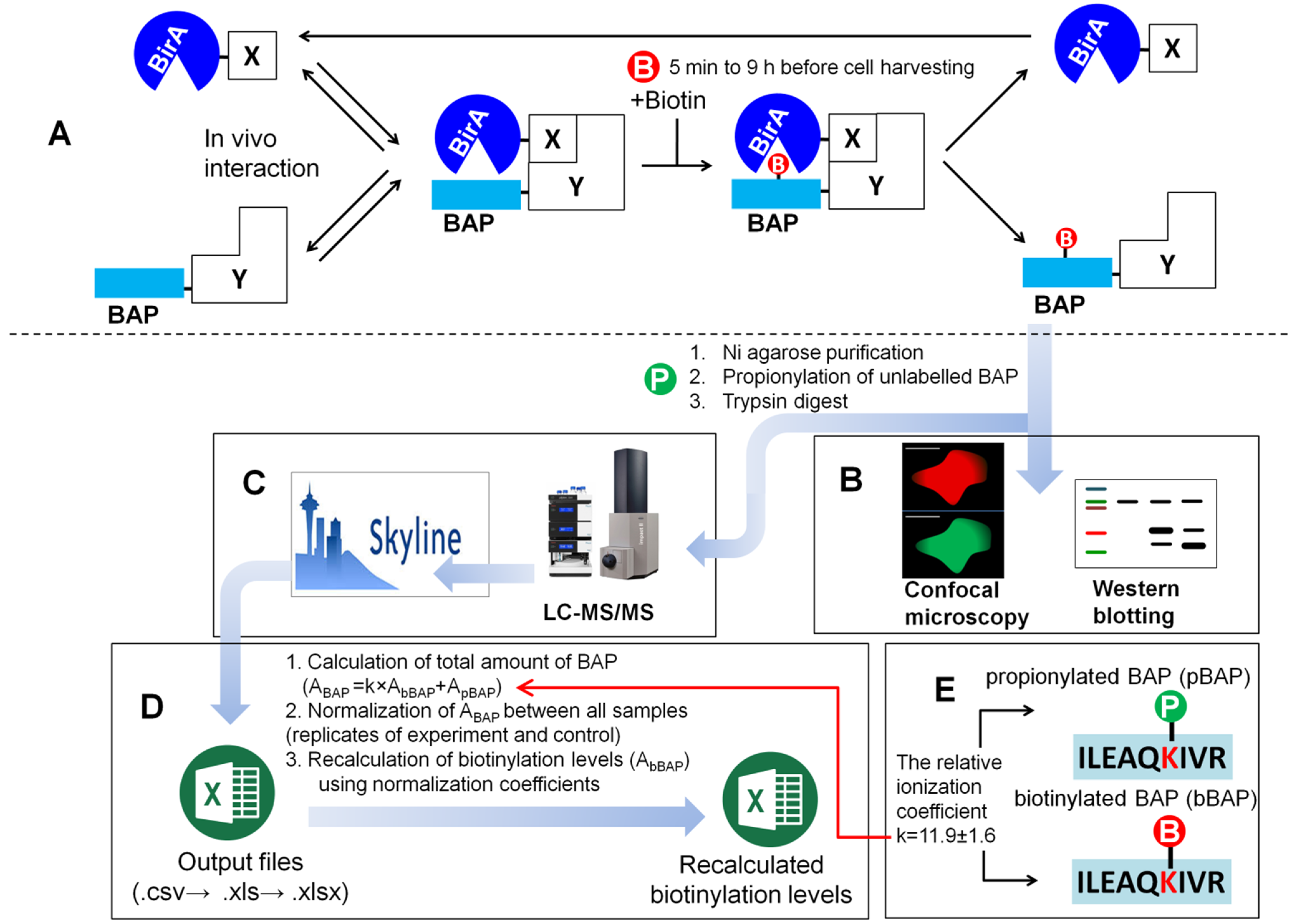
2.1. Overview of the Method and Experimental Workflow of the PUB Protocol
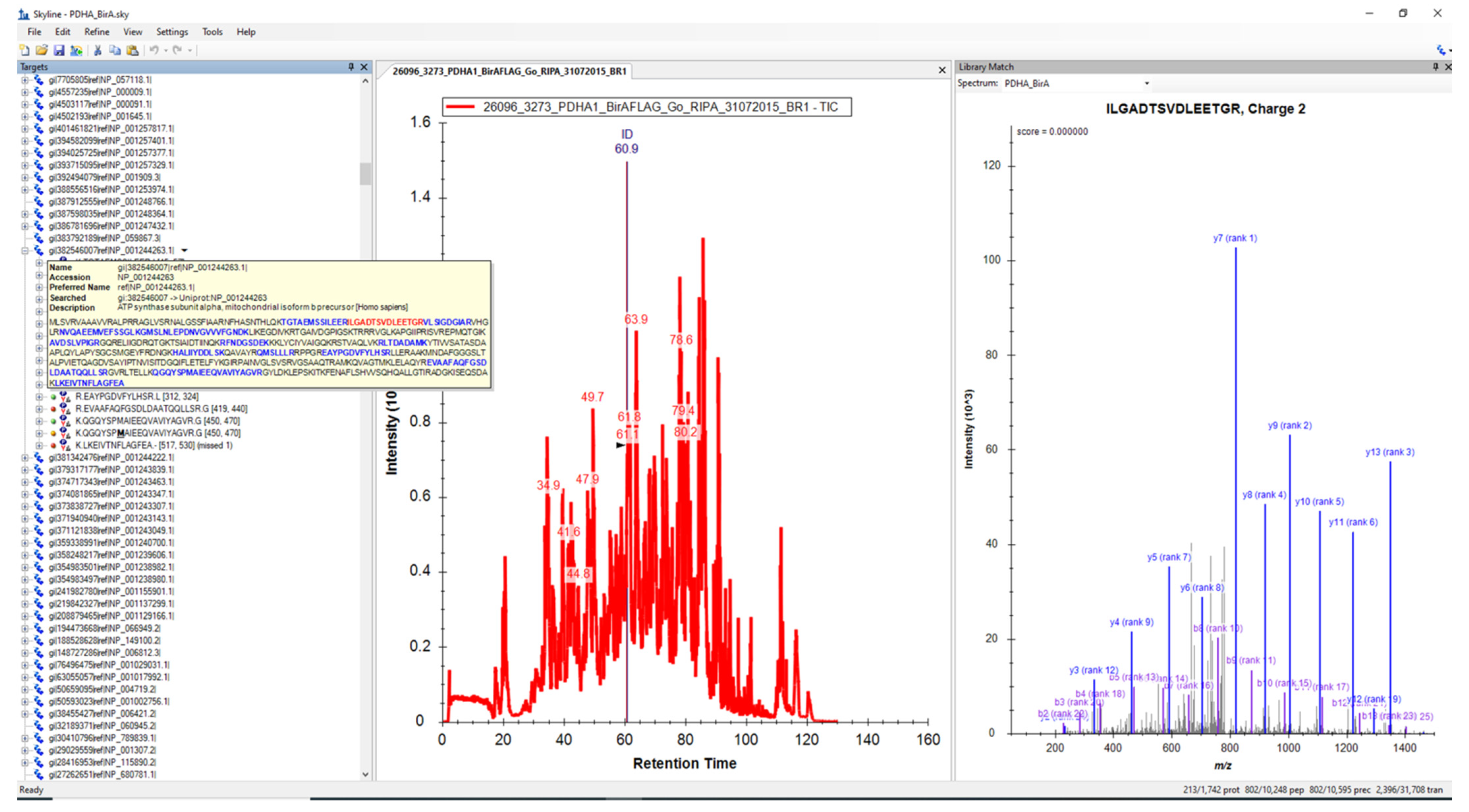
2.1.1. Creation of MRM Method and LC-MS/MS Analysis of the Samples
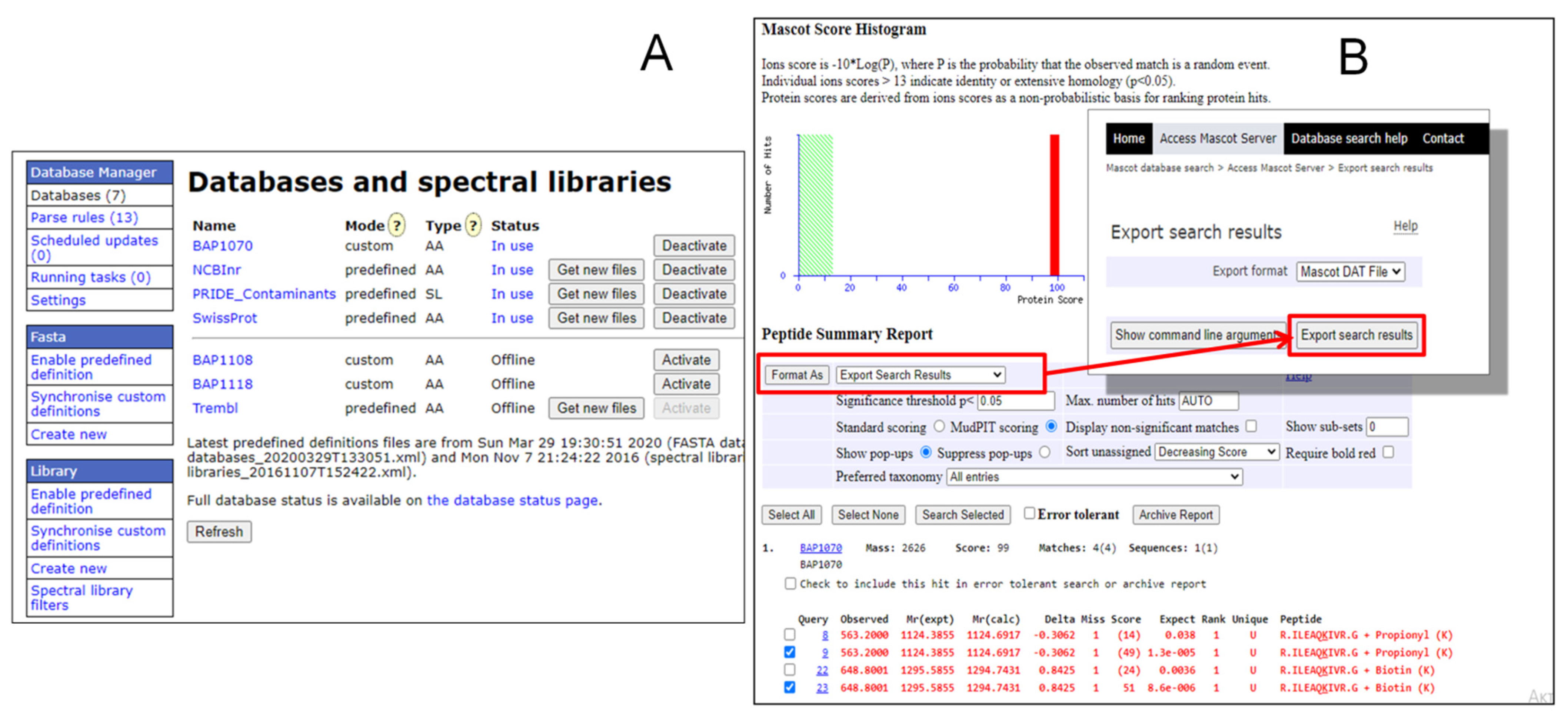
2.1.2. Creation of BAP1070 Database on Mascot Search Server Using Database Manager
2.1.3. MRM Analysis and Post-Processing of Data
2.2. DNA Dependent Interaction of Sox2 and Oct4
2.3. Protein Oligomerization HP1γ-HP1γ
2.4. Analysis of Shotgun Proteomics Data (Mutant Biotin Ligase BioID Application)
2.5. Perspectives of Enzymatically Catalyzed Labeling for Biological Research
3. Materials and Methods
- Nanoflow HPLC system (Thermo Dionex Ultimate 3000, ThermoScientific) with Acclaim PepMap100 C18 pre-column, 5 mm × 300 μm; 5 μm particles (Thermo Scientific, #160454) and Acclaim Pep-Map RSLC column 15 cm × 75μm, 2 μm particles (Thermo Scientific, #164534) coupled via CaptiveSpray to the QTOF Impact II mass spectrometer (Bruker). Raw LC-MS/MS data were interpreted with the Bruker Compass DataAnalysis (version 4.3) software. The separation gradient was 48 min from 2% to 50% acetonitrile. Flow rates—300 nL/min.
- Nano-HPLC (Agilent Technologies 1200) was coupled to an ion-trap mass spectrometer (Bruker 6300 series) equipped with a nanoelectrospray source via protein HPLC Chip (Agilent Technologies, G4240-62001) with 40 nL trap 75 um × 43 mm 5 um 300SB-C18-ZX and analytical column packed with ZORBAX 300SB-C18, 5 µm particle size. The separation gradient was 7 min from 5% to 90% acetonitrile. Flow rates—300 nL/min.
3.1. Data Preparation and Creation of MGF File Using DataAnalysis
3.2. Creation of BAP1070 Fasta Database on Mascot Search Server
4. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Aebersold, R.; Mann, M. Mass-spectrometric exploration of proteome structure and function. Nature 2016, 537, 347–355. [Google Scholar] [CrossRef]
- Sobsey, C.A.; Ibrahim, S.; Richard, V.R.; Gaspar, V.; Mitsa, G.; Lacasse, V.; Zahedi, R.P.; Batist, G.; Borchers, C.H. Targeted and Untargeted Proteomics Approaches in Biomarker Development. Proteomics 2020, 20, e1900029. [Google Scholar] [CrossRef]
- Domenick, T.M.; Gill, E.L.; Vedam-Mai, V.; Yost, R.A. Mass Spectrometry-Based Cellular Metabolomics: Current Approaches, Applications, and Future Directions. Anal. Chem. 2021, 93, 546–566. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Riano, C.; Dudzik, D.; Garcia, A.; Gil-de-la-Fuente, A.; Gradillas, A.; Godzien, J.; Lopez-Gonzalvez, A.; Rey-Stolle, F.; Rojo, D.; Ruperez, F.J.; et al. Recent Developments along the Analytical Process for Metabolomics Workflows. Anal. Chem. 2020, 92, 203–226. [Google Scholar] [CrossRef]
- Ezan, E.; Dubois, M.; Becher, F. Bioanalysis of recombinant proteins and antibodies by mass spectrometry. Analyst 2009, 134, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Hickey, J.M.; Sahni, N.; Toth, R.T., IV; Kumru, O.S.; Joshi, S.B.; Middaugh, C.R.; Volkin, D.B. Challenges and opportunities of using liquid chromatography and mass spectrometry methods to develop complex vaccine antigens as pharmaceutical dosage forms. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2016, 1032, 23–38. [Google Scholar] [CrossRef]
- Ebhardt, H.A.; Root, A.; Sander, C.; Aebersold, R. Applications of targeted proteomics in systems biology and translational medicine. Proteomics 2015, 15, 3193–3208. [Google Scholar] [CrossRef] [PubMed]
- Schubert, O.T.; Rost, H.L.; Collins, B.C.; Rosenberger, G.; Aebersold, R. Quantitative proteomics: Challenges and opportunities in basic and applied research. Nat. Protoc. 2017, 12, 1289–1294. [Google Scholar] [CrossRef]
- Carr, S.A.; Abbatiello, S.E.; Ackermann, B.L.; Borchers, C.; Domon, B.; Deutsch, E.W.; Grant, R.P.; Hoofnagle, A.N.; Huttenhain, R.; Koomen, J.M.; et al. Targeted peptide measurements in biology and medicine: Best practices for mass spectrometry-based assay development using a fit-for-purpose approach. Mol. Cell Proteom. 2014, 13, 907–917. [Google Scholar] [CrossRef] [Green Version]
- Kulyyassov, A.; Fresnais, M.; Longuespee, R. Targeted liquid chromatography-tandem mass spectrometry analysis of proteins: Basic principles, applications, and perspectives. Proteomics 2021, e2100153. [Google Scholar] [CrossRef]
- Berggard, T.; Linse, S.; James, P. Methods for the detection and analysis of protein-protein interactions. Proteomics 2007, 7, 2833–2842. [Google Scholar] [CrossRef]
- Braun, P.; Gingras, A.C. History of protein-protein interactions: From egg-white to complex networks. Proteomics 2012, 12, 1478–1498. [Google Scholar] [CrossRef]
- Fang, X.; Yoon, J.G.; Li, L.; Tsai, Y.S.; Zheng, S.; Hood, L.; Goodlett, D.R.; Foltz, G.; Lin, B. Landscape of the SOX2 protein-protein interactome. Proteomics 2011, 11, 921–934. [Google Scholar] [CrossRef]
- Huang, X.; Wang, J. The extended pluripotency protein interactome and its links to reprogramming. Curr. Opin. Genet. Dev. 2014, 28, 16–24. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.; Cox, J.L.; Gilmore, J.M.; Ormsbee, B.D.; Mallanna, S.K.; Washburn, M.P.; Rizzino, A. Determination of Protein Interactome of Transcription Factor Sox2 in Embryonic Stem Cells Engineered for Inducible Expression of Four Reprogramming Factors. J. Biol. Chem. 2012, 287, 11384–11397. [Google Scholar] [CrossRef] [Green Version]
- Ng, P.M.; Lufkin, T. Embryonic stem cells: Protein interaction networks. Biomol. Concepts 2011, 2, 13–25. [Google Scholar] [PubMed] [Green Version]
- Titeca, K.; Lemmens, I.; Tavernier, J.; Eyckerman, S. Discovering cellular protein-protein interactions: Technological strategies and opportunities. Mass Spectrom. Rev. 2019, 38, 79–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Rao, S.; Chu, J.; Shen, X.; Levasseur, D.N.; Theunissen, T.W.; Orkin, S.H. A protein interaction network for pluripotency of embryonic stem cells. Nature 2006, 444, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Fan, X.; Hu, Y. In vivo interactome profiling by enzyme-catalyzed proximity labeling. Cell Biosci. 2021, 11, 27. [Google Scholar] [CrossRef]
- Roux, K.J.; Kim, D.I.; Raida, M.; Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 2012, 196, 801–810. [Google Scholar] [CrossRef] [Green Version]
- Branon, T.C.; Bosch, J.A.; Sanchez, A.D.; Udeshi, N.D.; Svinkina, T.; Carr, S.A.; Feldman, J.L.; Perrimon, N.; Ting, A.Y. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 2018, 36, 880–887. [Google Scholar] [CrossRef]
- Kwon, K.; Streaker, E.D.; Beckett, D. Binding specificity and the ligand dissociation process in the E. coli biotin holoenzyme synthetase. Protein Sci. 2002, 11, 558–570. [Google Scholar] [CrossRef]
- Kwon, K.; Beckett, D. Function of a conserved sequence motif in biotin holoenzyme synthetases. Protein Sci. 2000, 9, 1530–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckett, D.; Kovaleva, E.; Schatz, P.J. A minimal peptide substrate in biotin holoenzyme synthetase-catalyzed biotinylation. Protein Sci. 1999, 8, 921–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, I.; Howarth, M.; Lin, W.; Ting, A.Y. Site-specific labeling of cell surface proteins with biophysical probes using biotin ligase. Nat. Methods 2005, 2, 99–104. [Google Scholar] [CrossRef]
- Fernandez-Suarez, M.; Chen, T.S.; Ting, A.Y. Protein-protein interaction detection in vitro and in cells by proximity biotinylation. J. Am. Chem. Soc. 2008, 130, 9251–9253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slavoff, S.A.; Liu, D.S.; Cohen, J.D.; Ting, A.Y. Imaging protein-protein interactions inside living cells via interaction-dependent fluorophore ligation. J. Am. Chem. Soc. 2011, 133, 19769–19776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulyyassov, A.; Shoaib, M.; Pichugin, A.; Kannouche, P.; Ramanculov, E.; Lipinski, M.; Ogryzko, V. PUB-MS: A Mass Spectrometry-based Method to Monitor Protein-Protein Proximity in vivo. J. Proteome Res. 2011, 10, 4416–4427. [Google Scholar] [CrossRef]
- Chapman-Smith, A.; Cronan, J.E. The enzymatic biotinylation of proteins: A post-translational modification of exceptional specificity. Trends Biochem. Sci. 1999, 24, 359–363. [Google Scholar] [CrossRef]
- Tenzer, S.; Moro, A.; Kuharev, J.; Francis, A.C.; Vidalino, L.; Provenzani, A.; Macchi, P. Proteome-wide characterization of the RNA-binding protein RALY-interactome using the in vivo-biotinylation-pulldown-quant (iBioPQ) approach. J. Proteome Res. 2013, 12, 2869–2884. [Google Scholar] [CrossRef]
- Lectez, B.; Migotti, R.; Lee, S.Y.; Ramirez, J.; Beraza, N.; Mansfield, B.; Sutherland, J.D.; Martinez-Chantar, M.L.; Dittmar, G.; Mayor, U. Ubiquitin profiling in liver using a transgenic mouse with biotinylated ubiquitin. J. Proteome Res. 2014, 13, 3016–3026. [Google Scholar] [CrossRef]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef] [Green Version]
- Pino, L.K.; Searle, B.C.; Bollinger, J.G.; Nunn, B.; MacLean, B.; MacCoss, M.J. The Skyline ecosystem: Informatics for quantitative mass spectrometry proteomics. Mass Spectrom. Rev. 2020, 39, 229–244. [Google Scholar] [CrossRef] [PubMed]
- Kulyyassov, A.; Ogryzko, V. In Vivo Quantitative Estimation of DNA-Dependent Interaction of Sox2 and Oct4 Using BirA-Catalyzed Site-Specific Biotinylation. Biomolecules 2020, 10, 142. [Google Scholar] [CrossRef] [Green Version]
- Sharma, V.; Eckels, J.; Schilling, B.; Ludwig, C.; Jaffe, J.D.; MacCoss, M.J.; MacLean, B. Panorama Public: A Public Repository for Quantitative Data Sets Processed in Skyline. Mol. Cell Proteom. 2018, 17, 1239–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulyyassov, A.; Kalendar, R. In Silico Estimation of the Abundance and Phylogenetic Significance of the Composite Oct4-Sox2 Binding Motifs within a Wide Range of Species. Data 2020, 5, 111. [Google Scholar] [CrossRef]
- Eissenberg, J.C.; Elgin, S.C. The HP1 protein family: Getting a grip on chromatin. Curr. Opin. Genet. Dev. 2000, 10, 204–210. [Google Scholar] [CrossRef]
- Lomberk, G.; Wallrath, L.; Urrutia, R. The Heterochromatin Protein 1 family. Genome Biol. 2006, 7, 228. [Google Scholar] [CrossRef] [Green Version]
- Sanulli, S.; Trnka, M.J.; Dharmarajan, V.; Tibble, R.W.; Pascal, B.D.; Burlingame, A.L.; Griffin, P.R.; Gross, J.D.; Narlikar, G.J. HP1 reshapes nucleosome core to promote phase separation of heterochromatin. Nature 2019, 575, 390–394. [Google Scholar] [CrossRef]
- Puri, T.; Wendler, P.; Sigala, B.; Saibil, H.; Tsaneva, I.R. Dodecameric structure and ATPase activity of the human TIP48/TIP49 complex. J. Mol. Biol. 2007, 366, 179–192. [Google Scholar] [CrossRef]
- Ikura, T.; Ogryzko, V.V.; Grigoriev, M.; Groisman, R.; Wang, J.; Horikoshi, M.; Scully, R.; Qin, J.; Nakatani, Y. Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell 2000, 102, 463–473. [Google Scholar] [CrossRef] [Green Version]
- Martens, L.; Chambers, M.; Sturm, M.; Kessner, D.; Levander, F.; Shofstahl, J.; Tang, W.H.; Rompp, A.; Neumann, S.; Pizarro, A.D.; et al. mzML—A community standard for mass spectrometry data. Mol. Cell Proteom. 2011, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kessner, D.; Chambers, M.; Burke, R.; Agus, D.; Mallick, P. ProteoWizard: Open source software for rapid proteomics tools development. Bioinformatics 2008, 24, 2534–2536. [Google Scholar] [CrossRef] [PubMed]
- Adusumilli, R.; Mallick, P. Data Conversion with ProteoWizard msConvert. Methods Mol. Biol. 2017, 1550, 339–368. [Google Scholar] [PubMed]
- Go, C.D.; Knight, J.D.R.; Rajasekharan, A.; Rathod, B.; Hesketh, G.G.; Abe, K.T.; Youn, J.Y.; Samavarchi-Tehrani, P.; Zhang, H.; Zhu, L.Y.; et al. A proximity-dependent biotinylation map of a human cell. Nature 2021, 595, 120–124. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulyyassov, A. Application of Skyline for Analysis of Protein–Protein Interactions In Vivo. Molecules 2021, 26, 7170. https://doi.org/10.3390/molecules26237170
Kulyyassov A. Application of Skyline for Analysis of Protein–Protein Interactions In Vivo. Molecules. 2021; 26(23):7170. https://doi.org/10.3390/molecules26237170
Chicago/Turabian StyleKulyyassov, Arman. 2021. "Application of Skyline for Analysis of Protein–Protein Interactions In Vivo" Molecules 26, no. 23: 7170. https://doi.org/10.3390/molecules26237170
APA StyleKulyyassov, A. (2021). Application of Skyline for Analysis of Protein–Protein Interactions In Vivo. Molecules, 26(23), 7170. https://doi.org/10.3390/molecules26237170