
Adaptive Modelling of Mutated FMO3 Enzyme Could Unveil Unexplored Scenarios Linking Variant Haplotypes to TMAU Phenotypes


 ,
,  ,
,  ,
,  and
and 
Abstract
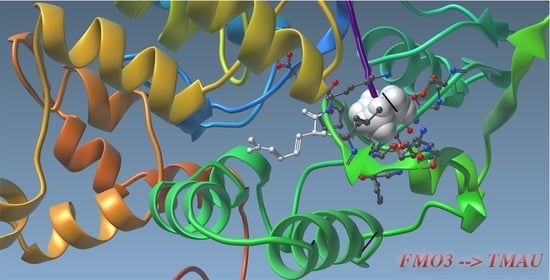
1. Introduction
2. Results
2.1. Genotyping of TMAU Patients Revealed Different Haplotypes Made of Causative or Associated Variants Carried by FMO3
2.2. Primary Structural and Biochemical Analyses Highlighted Possible Altered Chemical-Physical Features in Mutated FMO3
2.3. Tertiary Structure Prediction of Mutated FMO3 Showed That Mutated Amino Acids Changed Their Distance from Surrounding Amino Acids
2.4. Docking Analysis of FMO3 Forms Unveiled the Most Probable Binding Sites of TMA and How Considered Variants Could Impair This Binding
2.5. The Unbinding Pathway Analyses of Mutated FMO3 Revealed an Impaired Interaction between TMA and Enzyme Active Site
2.6. NMR Spectra Functionally Confirmed an Altered Catalytic Activity of Mutated FMO3
3. Discussion
4. Methods
4.1. Clinical Data
4.2. Mutational Analysis of FMO3 Gene
4.3. In Silico Structural Analyses of Wild-Type and Muted Forms of FMO3
4.4. Molecular Dynamics Analyses of FMO3/TMA Complex
4.5. Urine Sample Preparation and 1H NMR Spectroscopy
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Doyle, S.; O’Byrne, J.J.; Nesbitt, M.; Murphy, D.N.; Abidin, Z.; Byrne, N.; Pastores, G.; Kirk, R.; Treacy, E.P. The genetic and biochemical basis of trimethylaminuria in an Irish cohort. JIMD Rep. 2019, 47, 35–40. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Miller, N.B.; Beigelman, A.; Utterson, E.; Shinawi, M. Transient massive trimethylaminuria associated with food protein-induced enterocolitis syndrome. JIMD Rep. 2014, 12, 11–15. [Google Scholar] [CrossRef]
- Chhibber-Goel, J.; Gaur, A.; Singhal, V.; Parakh, N.; Bhargava, B.; Sharma, A. The complex metabolism of trimethylamine in humans: Endogenous and exogenous sources. Expert Rev. Mol. Med. 2016, 18, e8. [Google Scholar] [CrossRef] [PubMed]
- Phillips, I.R.; Shephard, E.A. Primary Trimethylaminuria. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Donato, L.; Alibrandi, S.; Scimone, C.; Castagnetti, A.; Rao, G.; Sidoti, A.; D’Angelo, R. Gut-Brain Axis Cross-Talk and Limbic Disorders as Biological Basis of Secondary TMAU. J. Pers. Med. 2021, 11, 87. [Google Scholar] [CrossRef] [PubMed]
- Roddy, D.; McCarthy, P.; Nerney, D.; Mulligan-Rabbitt, J.; Smith, E.; Treacy, E.P. Impact of trimethylaminuria on daily psychosocial functioning. JIMD Rep. 2021, 57, 67–75. [Google Scholar] [CrossRef]
- Ramos, N.; Wystrach, C.; Bolton, M.; Shaywitz, J.; IsHak, W.W. Delusional disorder, somatic type: Olfactory reference syndrome in a patient with delusional trimethylaminuria. J. Nerv. Ment. Dis. 2013, 201, 537–538. [Google Scholar] [CrossRef]
- Schmidt, A.C.; Leroux, J.C. Treatments of trimethylaminuria: Where we are and where we might be heading. Drug Discov. Today 2020, 25, 1710–1717. [Google Scholar] [CrossRef] [PubMed]
- Manning, N.J.; Allen, E.K.; Kirk, R.J.; Sharrard, M.J.; Smith, E.J. Riboflavin-responsive trimethylaminuria in a patient with homocystinuria on betaine therapy. JIMD Rep. 2012, 5, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Fujieda, M.; Togashi, M.; Saito, T.; Preti, G.; Cashman, J.R.; Kamataki, T. Effects of the dietary supplements, activated charcoal and copper chlorophyllin, on urinary excretion of trimethylamine in Japanese trimethylaminuria patients. Life Sci. 2004, 74, 2739–2747. [Google Scholar] [CrossRef]
- Wang, Z.; Bergeron, N.; Levison, B.S.; Li, X.S.; Chiu, S.; Jia, X.; Koeth, R.A.; Li, L.; Wu, Y.; Tang, W.H.W.; et al. Impact of chronic dietary red meat, white meat, or non-meat protein on trimethylamine N-oxide metabolism and renal excretion in healthy men and women. Eur. Heart J. 2019, 40, 583–594. [Google Scholar] [CrossRef]
- Fennema, D.; Phillips, I.R.; Shephard, E.A. Trimethylamine and Trimethylamine N-Oxide, a Flavin-Containing Monooxygenase 3 (FMO3)-Mediated Host-Microbiome Metabolic Axis Implicated in Health and Disease. Drug Metab. Dispos. 2016, 44, 1839–1850. [Google Scholar] [CrossRef] [PubMed]
- Scimone, C.; Alibrandi, S.; Donato, L.; Giofre, S.V.; Rao, G.; Sidoti, A.; D’Angelo, R. Antiretroviral treatment leading to secondary trimethylaminuria: Genetic associations and successful management with riboflavin. J. Clin. Pharm. Ther. 2021, 46, 304–309. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, R.; Scimone, C.; Esposito, T.; Bruschetta, D.; Rinaldi, C.; Ruggeri, A.; Sidoti, A. Fish odor syndrome (trimethylaminuria) supporting the possible FMO3 down expression in childhood: A case report. J. Med. Case Rep. 2014, 8, 328. [Google Scholar] [CrossRef]
- Esposito, T.; Varriale, B.; D’Angelo, R.; Amato, A.; Sidoti, A. Regulation of flavin-containing mono-oxygenase (Fmo3) gene expression by steroids in mice and humans. Horm. Mol. Biol. Clin. Investig. 2014, 20, 99–109. [Google Scholar] [CrossRef]
- Scimone, C.; Donato, L.; Rinaldi, C.; Sidoti, A.; D’Angelo, R. First case of Currarino syndrome and trimethylaminuria: Two rare diseases for a complex clinical presentation. J. Dig. Dis. 2016, 17, 628–632. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, R.; Esposito, T.; Calabro, M.; Rinaldi, C.; Robledo, R.; Varriale, B.; Sidoti, A. FMO3 allelic variants in Sicilian and Sardinian populations: Trimethylaminuria and absence of fish-like body odor. Gene 2013, 515, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Shephard, E.A. Mutation, polymorphism and perspectives for the future of human flavin-containing monooxygenase 3. Mutat. Res. 2006, 612, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Krueger, S.K.; Williams, D.E.; Yueh, M.F.; Martin, S.R.; Hines, R.N.; Raucy, J.L.; Dolphin, C.T.; Shephard, E.A.; Phillips, I.R. Genetic polymorphisms of flavin-containing monooxygenase (FMO). Drug Metab. Rev. 2002, 34, 523–532. [Google Scholar] [CrossRef]
- Razmazma, H.; Ebrahimi, A. The effects of cation-pi and anion-pi interactions on halogen bonds in the [Ncdots, three dots, centeredXcdots, three dots, centeredN](+) complexes: A comprehensive theoretical study. J. Mol. Graph. Model. 2018, 84, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.Y.; Yang, J.R. Analysis and prediction of highly effective antiviral peptides based on random forests. PLoS ONE 2013, 8, e70166. [Google Scholar] [CrossRef]
- Ikai, A. Thermostability and aliphatic index of globular proteins. J. Biochem. 1980, 88, 1895–1898. [Google Scholar]
- Zhang, J.; Tran, Q.; Lattard, V.; Cashman, J.R. Deleterious mutations in the flavin-containing monooxygenase 3 (FMO3) gene causing trimethylaminuria. Pharmacogenetics 2003, 13, 495–500. [Google Scholar] [CrossRef]
- Lambert, D.M.; Mamer, O.A.; Akerman, B.R.; Choiniere, L.; Gaudet, D.; Hamet, P.; Treacy, E.P. In vivo variability of TMA oxidation is partially mediated by polymorphisms of the FMO3 gene. Mol. Genet. Metab. 2001, 73, 224–229. [Google Scholar] [CrossRef]
- Donato, L.; Scimone, C.; Nicocia, G.; Denaro, L.; Robledo, R.; Sidoti, A.; D’Angelo, R. GLO1 gene polymorphisms and their association with retinitis pigmentosa: A case-control study in a Sicilian population. Mol. Biol. Rep. 2018, 45, 1349–1355. [Google Scholar] [CrossRef]
- Chen, Y.; Patel, N.A.; Crombie, A.; Scrivens, J.H.; Murrell, J.C. Bacterial flavin-containing monooxygenase is trimethylamine monooxygenase. Proc. Natl. Acad. Sci. USA 2011, 108, 17791–17796. [Google Scholar] [CrossRef] [PubMed]
- Bortolussi, S.; Catucci, G.; Gilardi, G.; Sadeghi, S.J. N- and S-oxygenation activity of truncated human flavin-containing monooxygenase 3 and its common polymorphic variants. Arch. Biochem. Biophys. 2021, 697, 108663. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, M.R.; Gasteiger, E.; Bairoch, A.; Sanchez, J.C.; Williams, K.L.; Appel, R.D.; Hochstrasser, D.F. Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 1999, 112, 531–552. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Bitzek, E.; Koskinen, P.; Gähler, F.; Moseler, M.; Gumbsch, P. Structural Relaxation Made Simple. Phys. Rev. Lett. 2006, 97, 170201. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Nguyen, M.K.; Jaillet, L.; Redon, S. ART-RRT: As-Rigid-As-Possible search for protein conformational transition paths. J. Comput. Aided Mol. Des. 2019, 33, 705–727. [Google Scholar] [CrossRef] [PubMed]
- Zuppi, C.; Messana, I.; Forni, F.; Rossi, C.; Pennacchietti, L.; Ferrari, F.; Giardina, B. 1H NMR spectra of normal urines: Reference ranges of the major metabolites. Clin. Chim. Acta 1997, 265, 85–97. [Google Scholar] [CrossRef]
- Chalmers, R.A.; Bain, M.D.; Michelakakis, H.; Zschocke, J.; Iles, R.A. Diagnosis and management of trimethylaminuria (FMO3 deficiency) in children. J. Inherit. Metab. Dis. 2006, 29, 162–172. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | FMO3 Variants | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| c. G472A (p.E158K) (DFP) | c.627+10 C>G (DM?) | c.485-22G>A | c.1184-32_1184-31insT | c.923A>G (p.E308G) (DFP) | c.769G>A (p.V257M) (FP) | c.458C>T (p.P153L) (DM) | c.441C>T (p.S147=) (DP) | c.993_994delTA (p.Tyr331Stop) | c.1474C>Tp.R492W (DM) | c.1139_1140del (p.Pro380fs) | c.713G>A (p.R238Q) (DM) | c.1424G>A (G475D) (DM) | c.713G>C (p.R238P) (DM) | c.855C>T (Asn285=) | c.422A>T (Asp141Val) | c.539G>T (Gly180Val) | |
| 1 | HET | ||||||||||||||||
| 2 | HET | HET | HET | ||||||||||||||
| 3 | HET | ||||||||||||||||
| 4 | HET | HET | |||||||||||||||
| 5 | HET | HET | HET | HET | HET | ||||||||||||
| 6 | HET | HET | |||||||||||||||
| 7 | HET | HET | HET | ||||||||||||||
| 8 | HET | HET | HET | HET | HET | ||||||||||||
| 9 | HET | HET | HET | ||||||||||||||
| 10 | HET | HET | HET | ||||||||||||||
| 11 | HET | HET | |||||||||||||||
| 12 | HOM | HOM | HOM | HOM | |||||||||||||
| 13 | HET | HET | |||||||||||||||
| 14 | HET | HET | HET | ||||||||||||||
| 15 | HET | HET | HET | ||||||||||||||
| 16 | HET | HET | |||||||||||||||
| 17 | HOM | HOM | HOM | ||||||||||||||
| 18 | HET | HET | HET | ||||||||||||||
| 19 | HET | HET | |||||||||||||||
| 20 | HOM | HOM | |||||||||||||||
| 21 | HOM | HET | HET | ||||||||||||||
| 22 | HET | HET | HET | ||||||||||||||
| 23 | HET | ||||||||||||||||
| 24 | HET | ||||||||||||||||
| 25 | HET | HET | |||||||||||||||
| 26 | HET | HET | HET | HET | |||||||||||||
| WILD | P153L_E158K | V267M | E158K | E158K_E308G | E158K_R492W | E158K_R238Q | E158K_G475D | D141V_G180V | Y331Stop | R238P_G475D | P380fs_P153L_E158K | P380fs | P380fs_E308G | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Residues | 532 | 532 | 532 | 532 | 532 | 532 | 532 | 532 | 532 | 330 | 532 | 381 | 381 | 381 |
| MW (g/mol) | 60,033 | 60,048 | 60,065 | 60,032 | 59,960 | 60,062 | 60,004 | 60,090 | 60,059 | 36,917 | 60,032 | 42,556 | 42,541 | 42,469 |
| 1 μg to pmol | 16.6 | 16.65 | 16.65 | 16.66 | 16.68 | 16.65 | 16.67 | 16.64 | 16.65 | 27.09 | 16.66 | 23.5 | 23.51 | 23.55 |
| Net charge (pH = 7) | 2.17 | 4.17 | 2.17 | 4.17 | 5.17 | 3.17 | 3.17 | 3.17 | 3.17 | −2.74 | 0.17 | −0.65 | −2.65 | −1.65 |
| pI | 7.9 | 8.33 | 7.9 | 8.33 | 8.47 | 8.15 | 8.15 | 8.15 | 8.16 | 6.26 | 7.09 | 6.76 | 6.33 | 6.52 |
| Avg. hydropathy (GRAVY) | −0.08 | −0.07 | −0.08 | −0.08 | −0.07 | −0.07 | −0.08 | −0.09 | −0.06 | −0.21 | −0.08 | −0.10 | −0.11 | −0.10 |
| Aliphatic index | 82.07 | 82.8 | 81.52 | 82.07 | 82.07 | 82.07 | 82.07 | 82.07 | 83.16 | 73.21 | 82.07 | 80.05 | 79.03 | 79.03 |
| Instability index | 34.24 | 33.64 | 35.12 | 33.87 | 33.19 | 33.19 | 33.59 | 33.75 | 34.37 | 33.02 | 34.48 | 33.48 | 34.31 | 33.36 |
| 1 mg/mL to A₂₈₀ | 1.46 | 1.46 | 1.46 | 1.46 | 1.46 | 1.55 | 1.46 | 1.46 | 1.46 | 1.23 | 1.46 | 1.14 | 1.14 | 1.14 |
| ε₂₈₀ (M⁻¹ cm⁻¹) | 87,485 | 87,485 | 87,485 | 87,485 | 87,485 | 92,85 | 87,485 | 87,485 | 87,485 | 45,420 | 87,485 | 48,400 | 48,400 | 48,400 |
| 1 A₂₈₀ to mg/mL | 0.69 | 0.69 | 0.69 | 0.69 | 0.69 | 0.65 | 0.69 | 0.69 | 0.69 | 0.81 | 0.69 | 0.88 | 0.88 | 0.88 |
| 1 mg/mL to A₂₈₀ (red.) | 1.45 | 1.45 | 1.45 | 1.45 | 1.45 | 1.54 | 1.45 | 1.45 | 1.45 | 1.22 | 1.45 | 1.13 | 1.13 | 1.13 |
| ε₂₈₀ (M⁻¹ cm⁻¹) (red.) | 86,860 | 86,860 | 86,860 | 86,860 | 86,860 | 92,360 | 86,860 | 86,860 | 86,860 | 44,920 | 86,860 | 47,900 | 47,900 | 47,900 |
| 1 A₂₈₀ to mg/mL (red.) | 0.69 | 0.69 | 0.69 | 0.69 | 0.69 | 0.65 | 0.69 | 0.69 | 0.69 | 0.82 | 0.69 | 0.89 | 0.89 | 0.89 |
| Features | NAP, FAD, WP5, OXY, ADP, IND, NA, 23 helices, 22 strands | NAP, FAD, WP5, OXY, ADP, IND, NA, 46 helices, 44 strands | NAP, FAD, WP5, OXY, ADP, IND, NA, 46 helices, 44 strands | NAP, FAD, WP5, OXY, ADP, IND, 46 helices, 44 strands | NAP, FAD, WP5, OXY, ADP, IND, NA, 46 helices, 44 strands | NAP, FAD, WP5, OXY, ADP, IND, NA, 46 helices, 44 strands | NAP, FAD, WP5, OXY, ADP, IND, NA, 46 helices, 44 strands | NAP, FAD, WP5, OXY, ADP, IND, NA, 46 helices, 44 strands | NAP, FAD, WP5, OXY, ADP, IND, NA, 46 helices, 44 strands | NAP, FAD, OXY, MG, IND, MMZ, CYH, 11 helices, 20 strands | NAP, FAD, WP5, OXY, ADP, IND, NA, 46 helices, 44 strands | NAP, FAD, WP5, OXY, ADP, IND, NA, 28 helices, 52 strands | NAP, FAD, OXY, IND, MMZ, CYH, 14 helices, 26 strands | NAP, FAD, WP5, OXY, ADP, IND, NA, 28 helices, 52 strands |
| ID | Variant | Wt | Nearest a.a. | a.a. Distance (Å) | Mut | Nearest a.a. | a.a. Distance (Å) |
|---|---|---|---|---|---|---|---|
| 2 | P153L | Pro153 (CG) | Arg174 (NE) | 4.33 | Leu153 (CD1) | Arg174 (NE) | 5.20 |
| Pro153 © | Leu155 (N) | 3.20 | Leu153 (CD2) | Leu155 (CD2) | 1.71 | ||
| Pro153 (CB) | His172 (CE1) | 3.32 | Leu153 (CB) | His172 (CG) | 4.00 | ||
| E158K | Glu158 (N) | Asn164 (OD1) | 5.94 | Lys158 (CA) | Asn164 (ND2) | 5.71 | |
| 4 | E308G | Glu308 | / | / | Gly308 | / | / |
| 5 | P153L | Pro153 (CG) | Arg174 (NE) | 4.33 | Leu153 (CD1) | Arg174 (NE) | 5.20 |
| Pro153 (C) | Leu155 (N) | 3.20 | Leu153 (CD2) | Leu155 (CD2) | 1.71 | ||
| Pro153 (CB) | His172 (CE1) | 3.32 | Leu153 (CB) | His172 (CG) | 4.00 | ||
| E158K | Glu158 (N) | Asn164 (OD1) | 5.94 | Lys158 (CA) | Asn164 (ND2) | 5.71 | |
| 6 | E158K | Glu158 (N) | Asn164 (OD1) | 5.94 | Lys158 (CA) | Asn164 (ND2) | 5.71 |
| 7 | V257M | Val257 (N) | Tyr256 (C) | 1.35 | Met257 (N) | Tyr256 (C) | 1.2 |
| Asp253 (O) | 2.97 | Asp253 (O) | 3.13 | ||||
| Val257 (CG2) | Val277 (N) | 4.03 | Met257 (CE) | Val277 (N) | 4.09 | ||
| 8 | E158K | Glu158 (N) | Asn164 (OD1) | 5.94 | Lys158 (CA) | Asn164 (ND2) | 5.71 |
| E308G | Glu308 | / | / | Gly308 | / | / | |
| 9 | E158K | Glu158 (N) | Asn164 (OD1) | 5.94 | Lys158 (CA) | Asn164 (ND2) | 5.71 |
| R492W | Arg492 (CD) | Asp76 (OD2) | 3.07 | Tyr492 (NE1) | ASp76 (OD2) | 2.53 | |
| Arg492 (NH2) | Thr488 (OG1) | 2.6 | Trp492 (CH2) | Thr488 (O) | 3.11 | ||
| Arg492 (NH1) | Ala485 (O) | 2.72 | Trp492 (N) | Ala485 (CB) | 4.3 | ||
| Arg492 (NE) | Glu65 (OE2) | 2.68 | Trp492 (CE3) | Glu65 (OE2) | 2.92 | ||
| 10 | E158K | Glu158 (N) | Asn164 (OD1) | 5.94 | Lys158 (CA) | Asn164 (ND2) | 5.71 |
| R238Q | Arg238 (NH2) | Pro445 (O) | 4.95 | Gln238 (OE1) | Pro445 (O) | 7.59 | |
| Asn446 (N) | 7.15 | Asn446 (N) | 9.81 | ||||
| Arg238 (CG) | Val261 (O) | 3.12 | Gln238 (CB) | Val461 (O) | 3.59 | ||
| Arg238 (CB) | Tyr462 (O) | 3.06 | Tyr462 (O) | 2.59 | |||
| 11 | E158K | Glu158(N) | Asn164 (OD1) | 5.94 | Lys158 (CA) | Asn164 (ND2) | 5.7 |
| G475D | Gly475 (N) | Pro445 (N) | 7.07 | Asp475 (OD2) | Pro445 (N) | 5.90 | |
| Gly475 (CA) | Lys444 (N) | 6.18 | Lys444 (N) | 3.83 | |||
| Ala443 (CA) | 4.30 | Ala443 (CA) | 3.02 | ||||
| Gly442 (O) | 4.52 | Gly442 (O) | 1.41 | ||||
| 12 | D141V | D141 (N) | Lys4 (O) | 2.87 | Val141 (N) | Lys4 (O) | 3.02 |
| Val139 (C) | 4.52 | Val139 (C) | 4.50 | ||||
| Phe140 (C) | 1.34 | Phe140 (C) | 1.25 | ||||
| D141 (OD1) | Lys3 (CA) | 3.52 | Val141 (CG1) | Lys3 (CG) | 3.88 | ||
| D141 (C) | Trp125 (CD1) | 4.59 | Val141 (C) | Trp125 (CD1) | 4.44 | ||
| G180V | Gly180 (N) | Leu203 (CD2) | 6.28 | Val180 (N) | Leu203 (CD2) | 6.22 | |
| Ala207 (N) | 8.42 | Ala207 (N) | 8.28 | ||||
| Thr206 (OG1) | 5.63 | Thr206 (OG1) | 5.49 | ||||
| 13 | R238P | Arg238 (NH1) | Ile447 (CD) | 3.62 | Pro238 (CB) | Ile447 (CD) | 6.78 |
| Arg238 (NH2) | Pro445 (O) | 4.95 | Pro238 (CG) | Pro445 (O) | 9.01 | ||
| Arg238 (N) | Gly464 (O) | 3.44 | Pro238 (CD) | Gly464 (O) | 2.74 | ||
| Arg238 (N) | Cys466 (N) | 4.56 | Pro238 (CD) | Cys466(N) | 3.65 | ||
| G475D | Gly475 (N) | Pro445 (N) | 7.07 | Asp475 (OD2) | Pro445 (N) | 5.90 | |
| Gly475 (CA) | Lys444 (N) | 6.18 | Lys444 (N) | 3.83 | |||
| Ala443 (CA) | 4.30 | Ala443 (CA) | 3.02 | ||||
| Gly442 (O) | 4.52 | Gly442 (O) | 1.41 |
| Protein | Affinity (kcal/mol) | Ki (µmol) | Lower Bound of the RMSD from This Ligand’s Best Mode (A) | Upper Bound of the RMSD from This Ligand’s Best Mode (A) |
|---|---|---|---|---|
| WILD-TYPE | −2.103 | 28,756.2 | 0.000 | 0.000 |
| D141V_G180V | −2.160 | 26,096.6 | 0.000 | 0.000 |
| E158K_E308G | −2.242 | 22,732.4 | 0.000 | 0.000 |
| E158K_G475D | −2.239 | 22,844.0 | 0.000 | 0.000 |
| E158K_R238Q | −2.240 | 22,789.5 | 0.000 | 0.000 |
| E158K_R492W | −2.242 | 22,739.5 | 0.000 | 0.000 |
| E158K | −2.240 | 22,791.2 | 0.000 | 0.000 |
| P153L_E158K | −2.240 | 22,807.7 | 0.000 | 0.000 |
| P380fs | −2.079 | 29,951.3 | 0.000 | 0.000 |
| P380fs_P153L_E158K | −2.082 | 29,760.4 | 0.000 | 0.000 |
| P380fs_E308G | −2.082 | 29,789.6 | 0.000 | 0.000 |
| R238P_G475D | −2.242 | 22,736.2 | 0.000 | 0.000 |
| V257M | −2.242 | 22,749.4 | 0.000 | 0.000 |
| Y331Stop | −2.033 | 32,327.0 | 0.000 | 0.000 |
| FMO3 Haplotypes | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Active site a.a. Residues | Wild-Type | Y331Stop | P153L + E158K | P380Fs | P380Fs + E308G | P153L + E158K + P380Fs | E158K | V257M | E308G + E158K | R492W + E158K | R238Q + E158K | G475D + E158K | D141V + G180V | R238P + G475D |
| Gly 9 | X | X | X | |||||||||||
| Ala 10 | X | X | X | |||||||||||
| Gly 11 | X | X | X | |||||||||||
| Val 12 | X | X | X | |||||||||||
| Ser 13 | X | X | X | |||||||||||
| Gly 14 | X | X | X | |||||||||||
| Glu 32 | X | X | X | |||||||||||
| Gly 38 | X | X | ||||||||||||
| Gly 39 | X | X | X | |||||||||||
| Leu 40 | X | X | X | |||||||||||
| Ala 52 | X | X | X | |||||||||||
| Cys 146 | X | X | X | |||||||||||
| Ser 147 | X | X | X | |||||||||||
| Gly 148 | X | X | X | |||||||||||
| Hys 149 | X | X | X | |||||||||||
| Val 151 | X | X | ||||||||||||
| Ser 216 | X | X | ||||||||||||
| Gly 217 | X | X | ||||||||||||
| Ser 218 | X | X | ||||||||||||
| Trp 219 | X | X | ||||||||||||
| Gly 240 | X | |||||||||||||
| Leu 243 | X | |||||||||||||
| Lys 244 | X | |||||||||||||
| Leu 247 | X | |||||||||||||
| Ile 251 | X | |||||||||||||
| Ser 252 | X | |||||||||||||
| Asp 253 | X | |||||||||||||
| Leu 255 | X | |||||||||||||
| Tyr 256 | X | |||||||||||||
| Gln 259 | X | |||||||||||||
| Pro 273 | X | X | ||||||||||||
| Asn 275 | X | X | ||||||||||||
| Gly 276 | X | X | ||||||||||||
| Leu 278 | X | X | ||||||||||||
| Arg 279 | X | X | X | X | X | X | X | X | X | X | ||||
| Lys 280 | X | X | ||||||||||||
| Glu 281 | X | X | ||||||||||||
| Pro 282 | X | X | ||||||||||||
| Leu 352 | X | |||||||||||||
| Phe 353 | X | |||||||||||||
| Lys 354 | X | |||||||||||||
| Gly 355 | X | |||||||||||||
| Phe 371 | X | |||||||||||||
| Val 372 | X | |||||||||||||
| Ser 381 | X | X | X | |||||||||||
| Lys 412 | X | X | X | X | X | X | X | X | ||||||
| Met 413 | X | |||||||||||||
| Lys 415 | X | X | X | X | X | X | X | |||||||
| Lys 416 | X | X | X | X | X | X | X | X | ||||||
| Arg 417 | X | X | X | X | X | X | ||||||||
| Trp 419 | X | X | X | X | X | X | X | |||||||
| Phe 420 | X | X | X | X | X | X | X | |||||||
| Lys 422 | X | X | X | X | X | |||||||||
| Thr 425 | X | X | X | X | X | X | X | |||||||
| Ile 426 | X | |||||||||||||
| Gln 427 | X | X | X | X | X | X | X | |||||||
| Thr 428 | X | X | X | X | X | X | X | X | ||||||
| Asp 429 | X | X | X | X | X | X | X | |||||||
| Tyr 433 | X | |||||||||||||
| ID | Age | Smell Description | Intestinal Disorders | Liver Diseases | Age of Onset | Drugs | Supplements/Probiotics |
|---|---|---|---|---|---|---|---|
| 1 | 9 | Rotten fish | / | / | 6 months | / | / |
| 2 | 42 | Rotten fish | Colitis | no | 7/8 years | no | no |
| 3 | 48 | Genital fishy odour, body garbage odour, and scalp acid/sulphur odour | Irritable colon | no | 10 years | Eutirox, Prisma 50 | no |
| 4 | Rotten fish | ||||||
| 5 | 36 | Rotten fish | Moderate degree of dysbiosis | no | 2/3 years | ||
| 6 | 71 | Rotten fish | Megacolon, anorectal stenosis, constipation, and congenital sacro-coccygeal malformation with fistulas | / | 2/3 years | / | / |
| 7 | 38 | Rotten fish | / | / | 37 years | Atazanavir, Abacavoir, and Lamivudin | Riboflavin |
| 8 | 45 | Rotten fish | / | / | 38 years | Oral contraceptives | / |
| 9 | 21 | Rotten fish | no | no | 6/7 years | no | no |
| 10 | 73 | Rotten fish | no | no | 63 years | / | no |
| 11 | 51 | Fish | no | no | 2/3 years | no | no |
| 12 | 20 | Rotten fish | no | no | 6 months | no | no |
| 13 | 28 | Fish | no | no | After weaning | no | no |
| 14 | 12 | Rotten fish | / | / | 5 years | / | / |
| 15 | 31 | Rotten fish, garbage, and garlic | Moderate dysbiosis | no | 17 years | / | / |
| 16 | 51 | Rotten fish | / | / | 44 years | Medicinal herbs | / |
| 17 | 26 | Rotten fish | no | no | 14 years | Psychotropic drugs and tranquilizers | |
| 18 | 48 | Rotten fish, faecal odour | Mild dysbiosis, slow oro-caecal transit | no | / | / | |
| 19 | 52 | Rotten fish | Mild dysbiosis | no | 6 years | / | Flebinic (carnitine-based supplement) |
| 20 | 24 | Rotten fish | no | no | After weaning | no | Activated carbon |
| 21 | 38 | Rotten fish | no | no | 34 years | no | no |
| 22 | 13 | Rotten fish | no | no | 10 years | no | no |
| 23 | 28 | Rotten fish | no | no | 28 years | no | no |
| 24 | 27 | Rotten fish, faecal odour, rot, and sulphur | Mild dysbiosis | no | 4 years | no | no |
| 25 | 47 | Rotten fish | / | / | 39 years | / | / |
| 26 | 71 | Rotten fish | / | / | 2/3 years | / | Assumption of a.a. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alibrandi, S.; Nicita, F.; Donato, L.; Scimone, C.; Rinaldi, C.; D’Angelo, R.; Sidoti, A. Adaptive Modelling of Mutated FMO3 Enzyme Could Unveil Unexplored Scenarios Linking Variant Haplotypes to TMAU Phenotypes. Molecules 2021, 26, 7045. https://doi.org/10.3390/molecules26227045
Alibrandi S, Nicita F, Donato L, Scimone C, Rinaldi C, D’Angelo R, Sidoti A. Adaptive Modelling of Mutated FMO3 Enzyme Could Unveil Unexplored Scenarios Linking Variant Haplotypes to TMAU Phenotypes. Molecules. 2021; 26(22):7045. https://doi.org/10.3390/molecules26227045
Chicago/Turabian StyleAlibrandi, Simona, Fabiana Nicita, Luigi Donato, Concetta Scimone, Carmela Rinaldi, Rosalia D’Angelo, and Antonina Sidoti. 2021. "Adaptive Modelling of Mutated FMO3 Enzyme Could Unveil Unexplored Scenarios Linking Variant Haplotypes to TMAU Phenotypes" Molecules 26, no. 22: 7045. https://doi.org/10.3390/molecules26227045
APA StyleAlibrandi, S., Nicita, F., Donato, L., Scimone, C., Rinaldi, C., D’Angelo, R., & Sidoti, A. (2021). Adaptive Modelling of Mutated FMO3 Enzyme Could Unveil Unexplored Scenarios Linking Variant Haplotypes to TMAU Phenotypes. Molecules, 26(22), 7045. https://doi.org/10.3390/molecules26227045