
The Structural Determinants for α1-Adrenergic/Serotonin Receptors Activity among Phenylpiperazine-Hydantoin Derivatives

 ,
,  , and
, and
Abstract
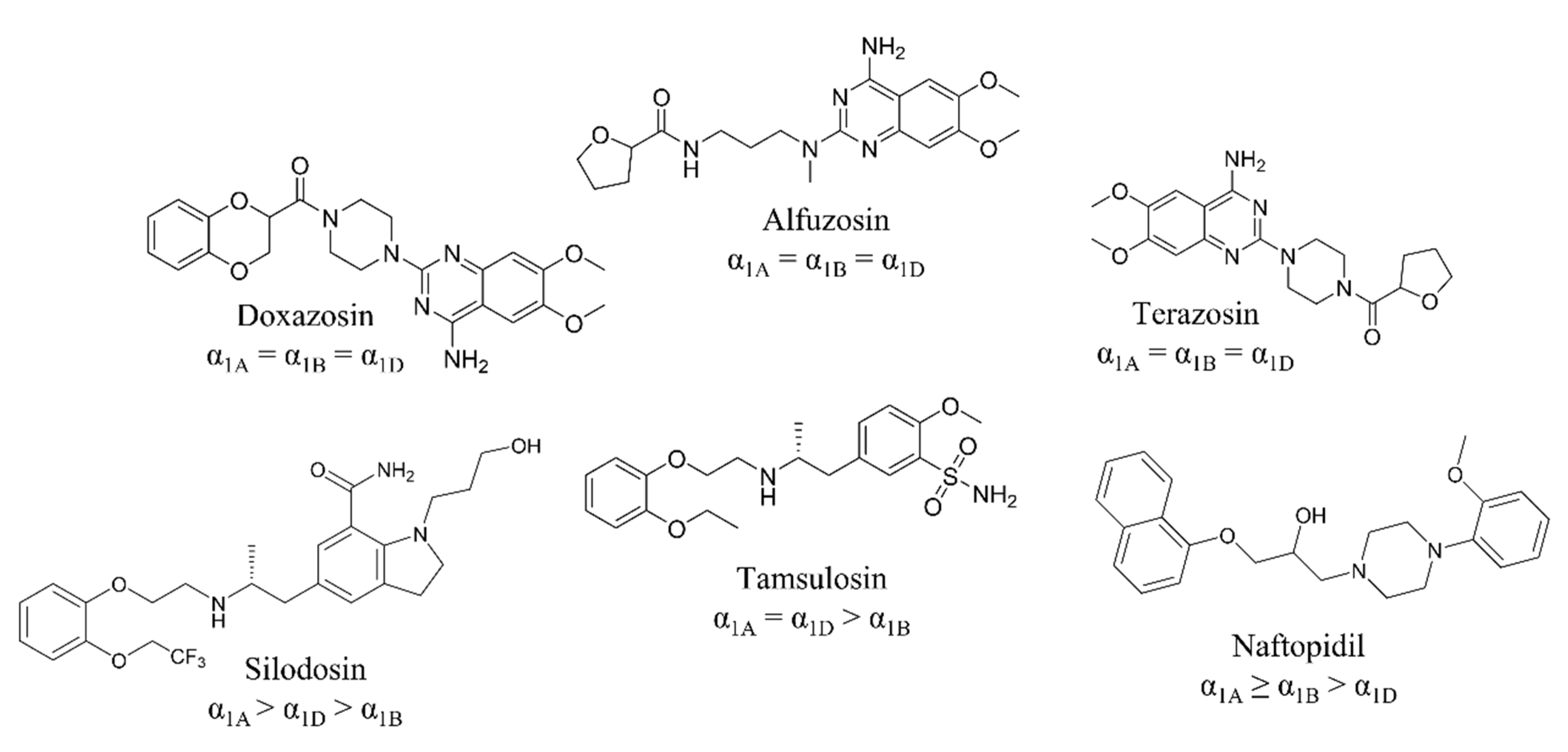
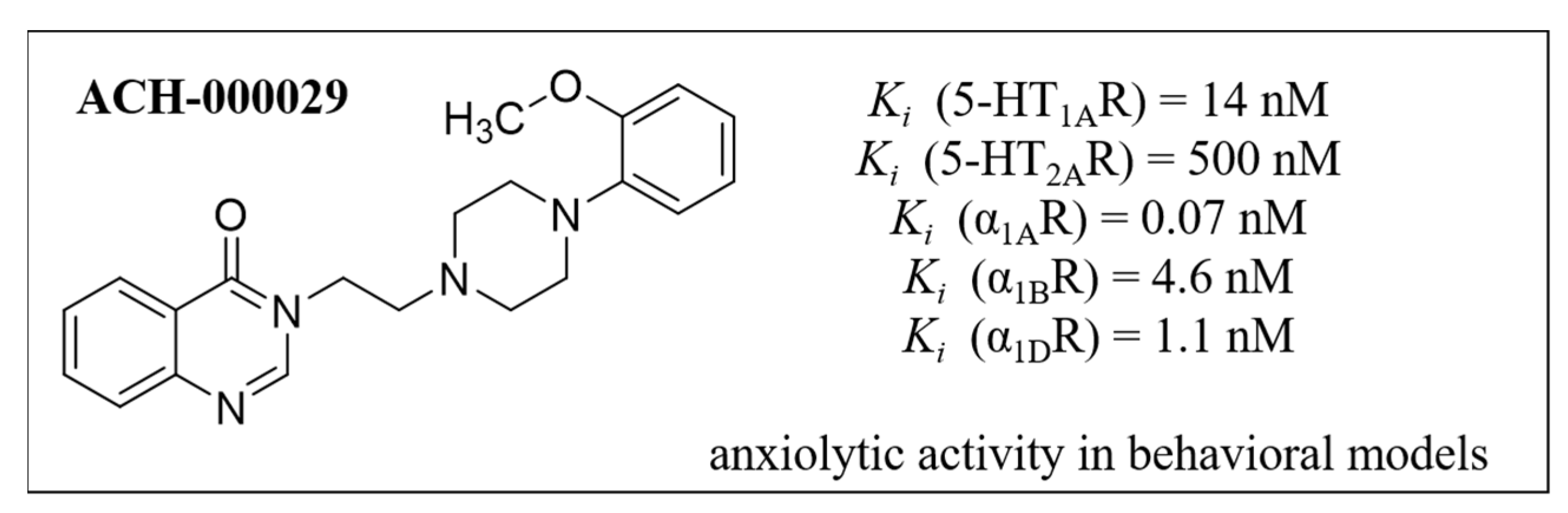
:1. Introduction
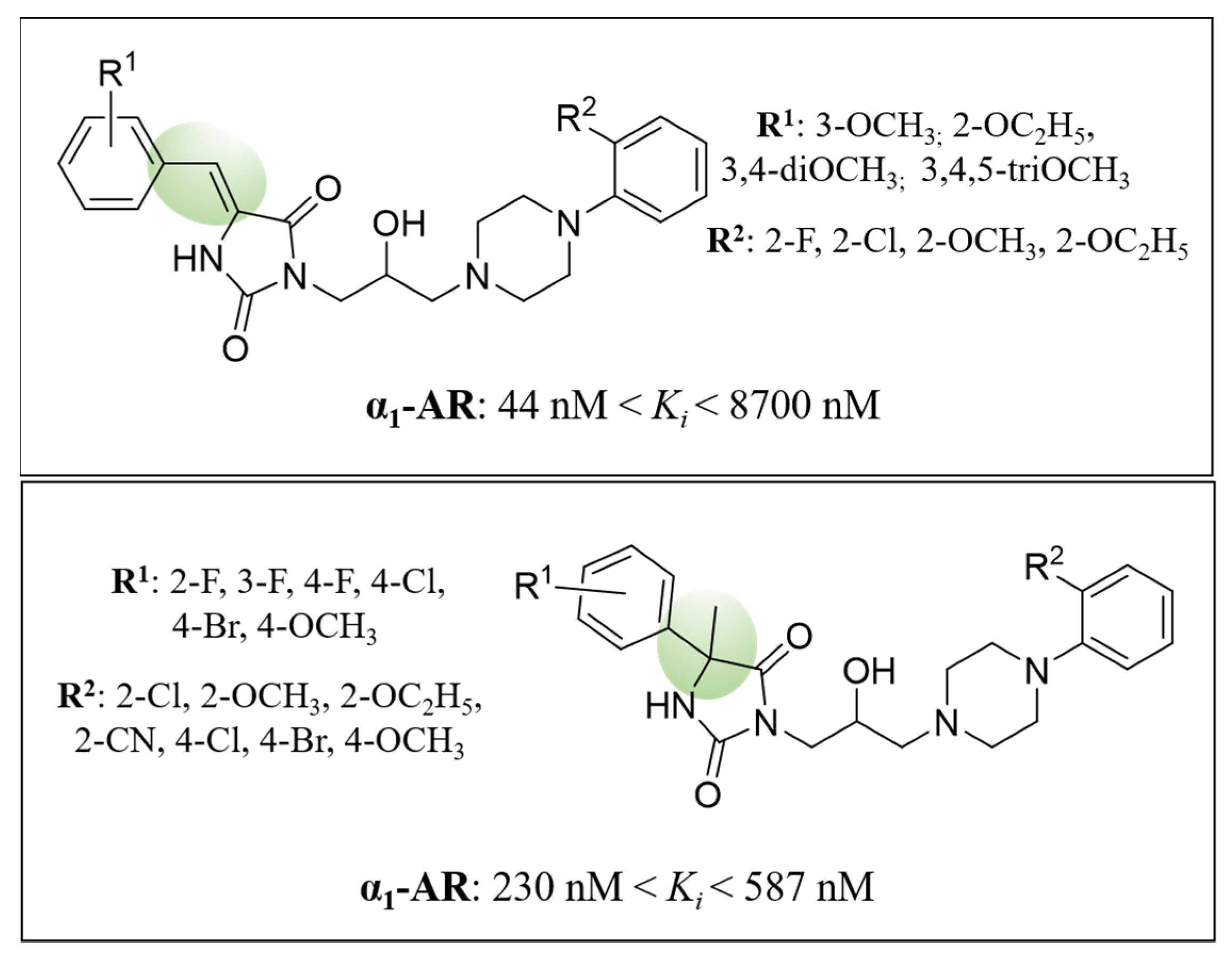
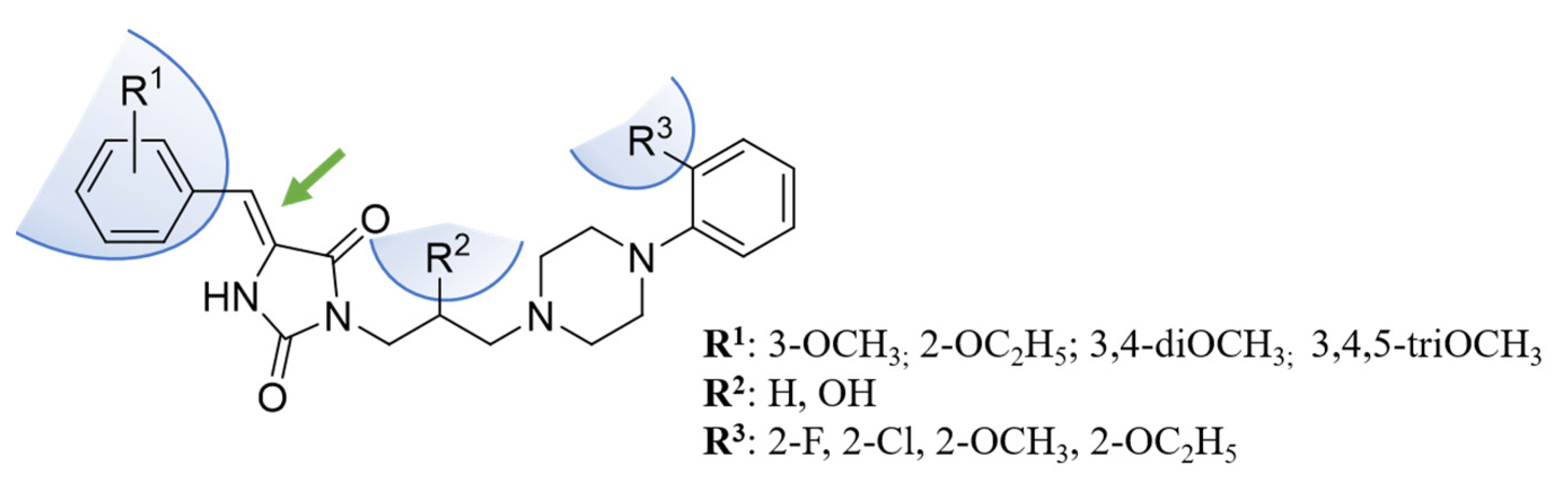
2. Results and Discussion
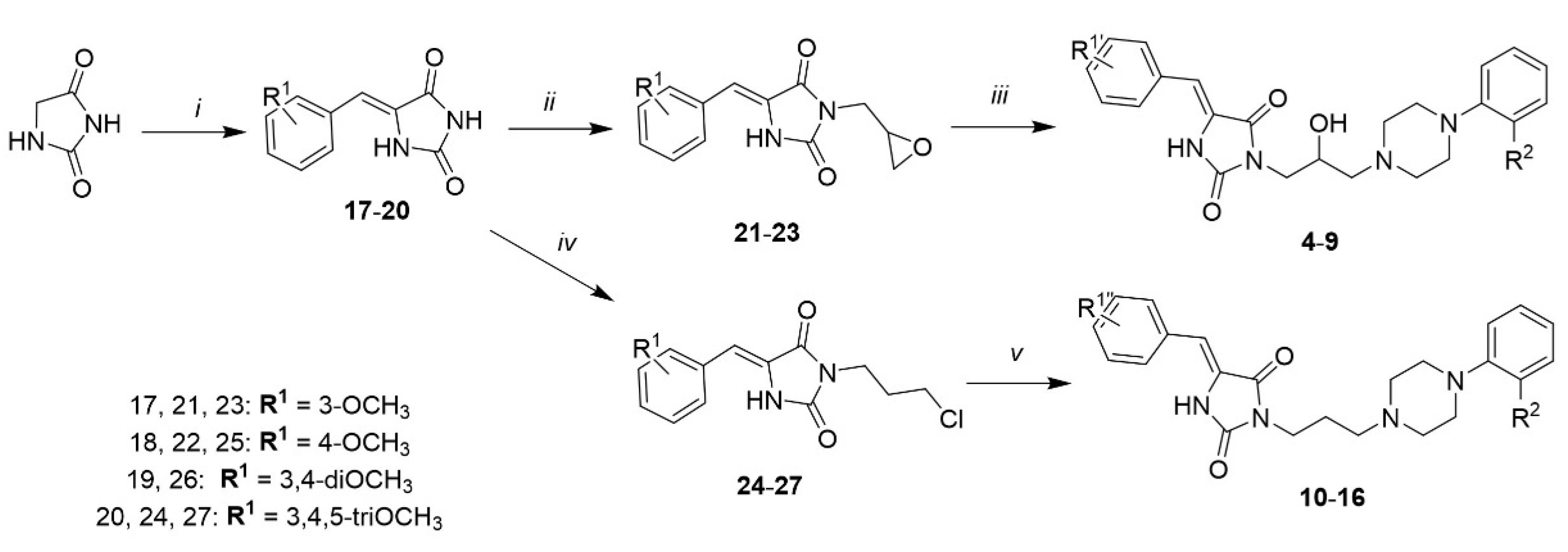
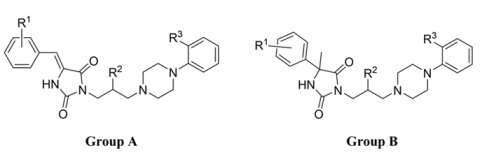
2.1. Chemical Synthesis
2.2. Pharmacology
2.2.1. Radioligand Binding Assays
2.2.2. Evaluation of Intrinsic Activity towards α1-AR Subtypes
2.3. Cheminformatic Analysis and Molecular Modeling
2.3.1. ChEMBL-Database-Oriented Searches for Structurally Similar Compounds
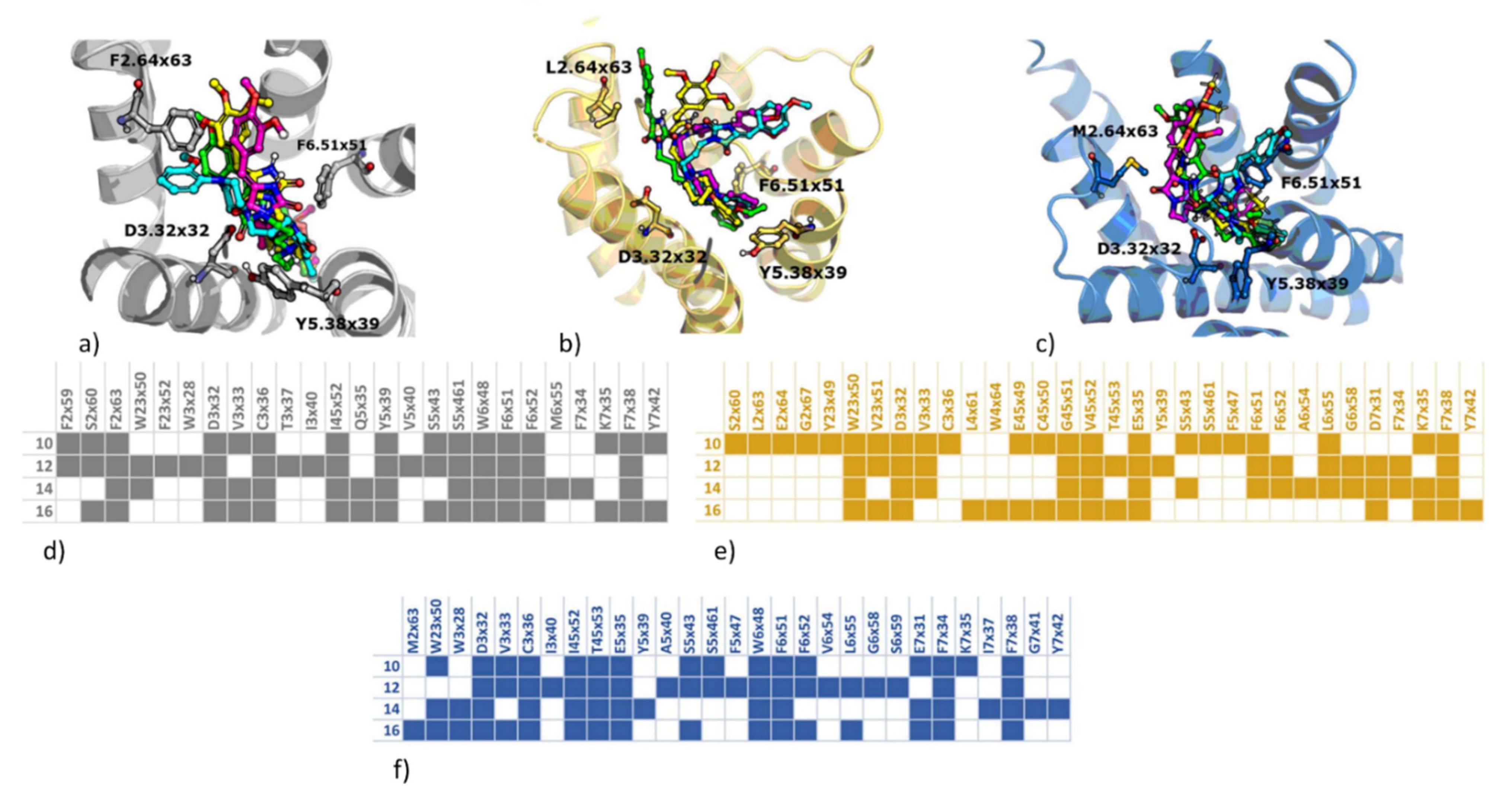
2.3.2. Computer-Aided Structure-Activity Relationship towards Adrenergic Receptors
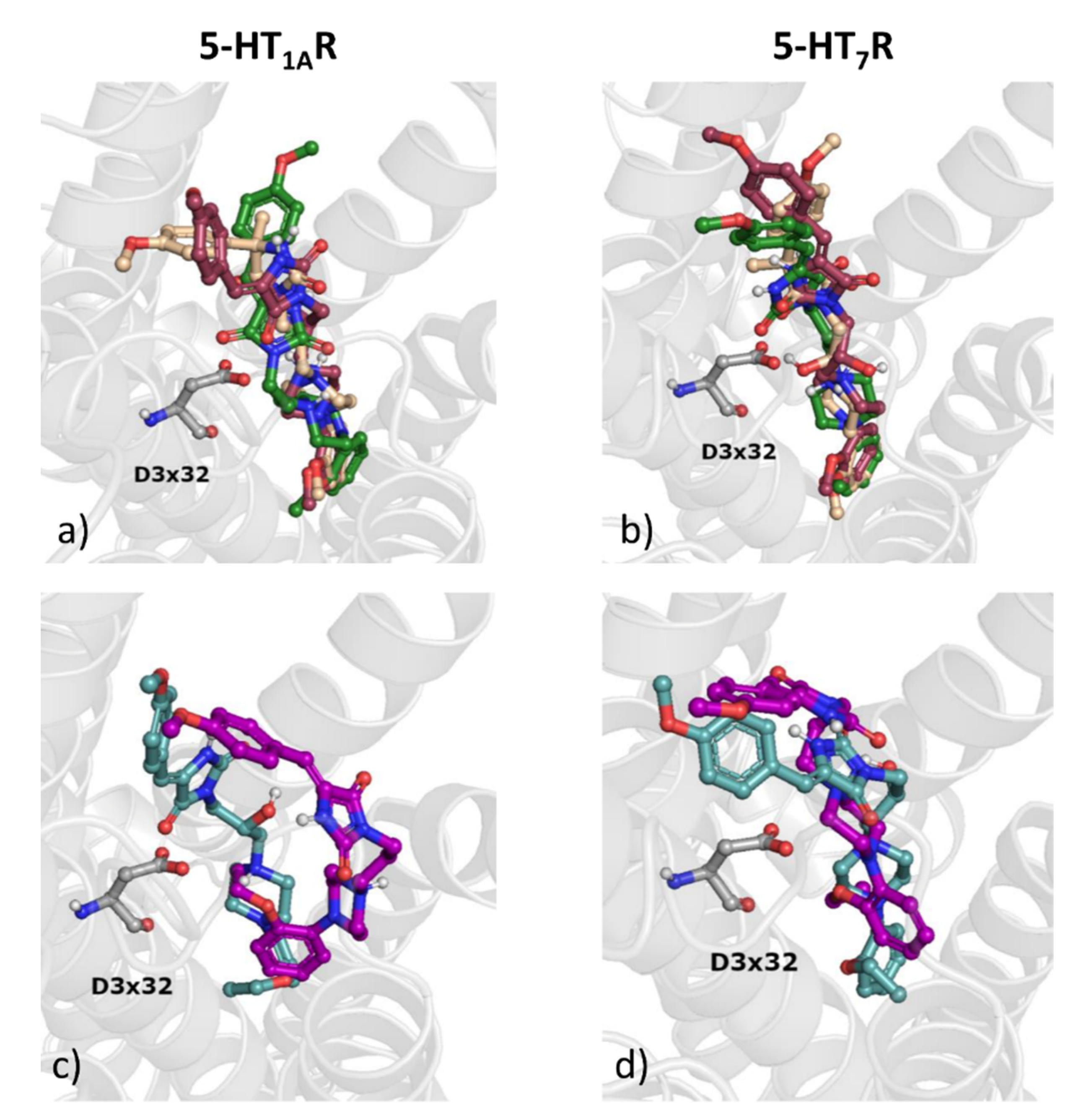
2.3.3. Examination of Activity Profiles towards Serotonin Receptor Subtypes
3. Materials and Methods
3.1. Chemical Synthesis
3.1.1. (Z)-3-(2-hydroxy-3-(4-(2-methoxyphenyl)piperazin-1-yl)propyl)-5-(3-methoxybenzylidene)imidazolidine-2,4-dione (4)
3.1.2. (Z)-3-(3-(4-(2-ethoxyphenyl)piperazin-1-yl)-2-hydroxypropyl)-5-(3-methoxybenzylidene)imidazolidine-2,4-dione (5)
3.1.3. (Z)-3-(2-hydroxy-3-(4-(2-methoxyphenyl)piperazin-1-yl)propyl)-5-(4-methoxybenzylidene)imidazolidine-2,4-dione (6)
3.1.4. (Z)-3-(3-(4-(2-ethoxyphenyl)piperazin-1-yl)-2-hydroxypropyl)-5-(4-methoxybenzylidene)imidazolidine-2,4-dione (7)
3.1.5. (Z)-3-(3-(4-(2-fluorophenyl)piperazin-1-yl)-2-hydroxypropyl)-5-(4-methoxybenzylidene)imidazolidine-2,4-dione (8)
3.1.6. (Z)-3-(3-(4-(2-ethoxyphenyl)piperazin-1-yl)-2-hydroxypropyl)-5-(3,4,5-trimethoxybenzylidene)imidazolidine-2,4-dione (9)
3.1.7. (Z)-3-(3-(4-(2-ethoxyphenyl)piperazin-1-yl)propyl)-5-(3-methoxybenzylidene)imidazolidine-2,4-dione (10)
3.1.8. (Z)-3-(3-(4-(2-chlorophenyl)piperazin-1-yl)propyl)-5-(3-methoxybenzylidene)imidazolidine-2,4-dione (11)
3.1.9. (Z)-5-(4-methoxybenzylidene)-3-(3-(4-(2-methoxyphenyl)piperazin-1-yl)propyl)imidazolidine-2,4-dione (12)
3.1.10. (Z)-3-(3-(4-(2-ethoxyphenyl)piperazin-1-yl)propyl)-5-(4-methoxybenzylidene)imidazolidine-2,4-dione (13)
3.1.11. (Z)-3-(3-(4-(2-methoxyphenyl)piperazin-1-yl)propyl)-5-(3,4,5-trimethoxybenzylidene)imidazolidine-2,4-dione (15)
3.1.12. (Z)-3-(3-(4-(2-ethoxyphenyl)piperazin-1-yl)propyl)-5-(3,4,5-trimethoxybenzylidene)imidazolidine-2,4-dione (16)
3.2. Pharmacology In Vitro
3.2.1. Radioligand Binding Assays: Affinity for α1-Receptor
3.2.2. Radioligand Binding Assays: Binding Affinities for 5-HT1A, 5-HT6 and 5-HT7 Receptors
3.3. Functional Tests
3.3.1. Determination of the Intrinsic Activity for the α1A-Ars
3.3.2. Determination of the Intrinsic Activity for the α1B-ARs and α1D-Ars
3.4. Molecular Modeling and Cheminformatic Analysis
3.4.1. Searches for Structurally Similar Compounds in the ChEMBL Database
3.4.2. Docking
3.4.3. Molecular Dynamics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- O’Connell, T.D.; Jensen, B.C.; Baker, A.J.; Simpson, P.C. Cardiac alpha1-adrenergic receptors: Novel aspects of expression, signaling mechanisms, physiologic function, and clinical importance. Pharmacol. Rev. 2014, 66, 308–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrow, A.L.; Creese, I. Characterization of alpha 1-adrenergic receptor subtypes in rat brain: A reevaluation of [3H]WB4104 and [3H]prazosin binding. Mol. Pharmacol. 1986, 29, 321–330. [Google Scholar] [PubMed]
- Schwinn, D.A.; Roehrborn, C.G. α1-Adrenoceptor subtypes and lower urinary tract symptoms. Int. J. Urol. 2008, 15, 193–199. [Google Scholar] [CrossRef] [Green Version]
- De Mey, C.; Michel, M.C.; McEwen, J.; Moreland, T. A double-blind comparison of terazosin and tamsulosin on their differential effects on ambulatory blood pressure and nocturnal orthostatic stress testing. Eur. Urol. 1998, 33, 481–488. [Google Scholar] [CrossRef]
- Newton, T.F.; De La Garza, R.; Brown, G.; Kosten, T.R.; Mahoney, J.J.; Haile, C.N. Noradrenergic α1 Receptor Antagonist Treatment Attenuates Positive Subjective Effects of Cocaine in Humans: A Randomized Trial. PLoS ONE. 2012, 7, e30854. [Google Scholar] [CrossRef]
- Shorter, D.I.; Zhang, X.; Domingo, C.B.; Nielsen, E.M.; Kosten, T.R.; Nielsen, D.A. Doxazosin treatment in cocaine use disorder: Pharmacogenetic response based on an alpha-1 adrenoreceptor subtype D genetic variant. Am. J. Drug Alcohol Abuse 2020, 46, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Qian, Y.; Jia, H.; Yu, Y.; Zhang, H.; Zhao, S.; Shen, J. Synthesis and pharmacological evaluation of naftopidil-based arylpiperazine derivatives containing the bromophenol moiety. Pharmacol. Rep. 2020, 72, 1058–1068. [Google Scholar] [CrossRef]
- Perez, D.M.; Doze, V.A. Cardiac and neuroprotection regulated by α1-adrenergic receptor subtypes. J. Recept. Signal Transduct. 2011, 31, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breen, A.; Blankley, K.; Fine, J. The efficacy of prazosin for the treatment of posttraumatic stress disorder nightmares in U.S. military veterans. J. Am. Assoc. Nurse Pract. 2017, 29, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Norton, J.; Carrière, I.; Ritchie, K.; Chaudieu, I.; Ryan, J.; Ancelin, M.L. Preliminary evidence for a role of the adrenergic nervous system in generalized anxiety disorder. Sci. Rep. 2017, 7, 42676. [Google Scholar] [CrossRef] [Green Version]
- Takamura, N.; Masuda, T.; Inoue, T.; Nakagawa, S.; Koyama, T. Progress in Neuro-Psychopharmacology & Biological Psychiatry The effects of the co-administration of the α 1-adrenoreceptor antagonist prazosin on the anxiolytic effect of citalopram in conditioned fear stress in the rat. Prog. Neuropsychopharmacol. Biol. Psychiatry 2012, 39, 107–111. [Google Scholar] [PubMed]
- Azevedo, H.; Ferreira, M.; Costa, R.W.; Russo, V.; Russo, E.; Mascarello, A.; Guimarães, C.R.W. Preclinical characterization of ACH-000029, a novel anxiolytic compound acting on serotonergic and alpha-adrenergic receptors. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 95, 109707. [Google Scholar] [CrossRef] [PubMed]
- Celada, P.; Bortolozzi, A.; Artigas, F. Serotonin 5-HT1A receptors as targets for agents to treat psychiatric disorders: Rationale and current status of research. CNS Drugs 2013, 27, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Overstreet, D.H.; Commissaris, R.C.; De la Garza, R.; File, S.E.; Knapp, D.J.; Seiden, L.S. Involvement of 5-HT1A receptors in animal tests of anxiety and depression: Evidence from genetic models. Stress 2003, 6, 101–110. [Google Scholar] [CrossRef]
- Weisstaub, N.V.; Zhou, M.; Lira, A.; Lambe, E.; González-Maeso, J.; Hornung, J.P.; Sibille, E.; Underwood, M.; Itohara, S.; Dauer, W.T.; et al. Cortical 5-HT2A receptor signaling modulates anxiety-like behaviors in mice. Science 2006, 313, 536–540. [Google Scholar] [CrossRef] [Green Version]
- Wesołowska, A.; Nikiforuk, A.; Stachowicz, K.; Tatarczyńska, E. Effect of the selective 5-HT7 receptor antagonist SB 269970 in animal models of anxiety and depression. Neuropharmacology 2006, 51, 578–586. [Google Scholar] [CrossRef]
- Nikiforuk, A. Targeting the Serotonin 5-HT 7 Receptor in the Search for Treatments for CNS Disorders: Rationale and Progress to Date. CNS Drugs 2015, 29, 265–275. [Google Scholar] [CrossRef] [Green Version]
- Wróbel, M.Z.; Chodkowski, A.; Marciniak, M.; Dawidowski, M.; Maksymiuk, A.; Siwek, A.; Nowak, G.; Turło, J. Synthesis of new 4-butyl-arylpiperazine-3-(1H-indol-3-yl)pyrrolidine-2,5-dione derivatives and evaluation for their 5-HT1A and D2 receptor affinity and serotonin transporter inhibition. Bioorg. Chem. 2020, 97, 103662. [Google Scholar] [CrossRef]
- Shimoda, Y.; Yui, J.; Xie, L.; Fujinaga, M.; Yamasaki, T.; Ogawa, M.; Nengaki, N.; Kumata, K.; Hatori, A.; Kawamura, K.; et al. Synthesis and evaluation of 1-[2-(4-[11C]methoxyphenyl)phenyl] piperazine for imaging of the serotonin 5-HT7 receptor in the rat brain. Bioorganic Med. Chem. 2013, 21, 5316–5322. [Google Scholar] [CrossRef]
- Szczepańska, K.; Karcz, T.; Siwek, A.; Kuder, K.J.; Latacz, G.; Bednarski, M.; Szafarz, M.; Hagenow, S.; Lubelska, A.; Olejarz-Maciej, A.; et al. Structural modifications and in vitro pharmacological evaluation of 4-pyridyl-piperazine derivatives as an active and selective histamine H3 receptor ligands. Bioorg. Chem. 2019, 91, 103071. [Google Scholar] [CrossRef]
- Handzlik, J.; Bojarski, A.J.; Satała, G.; Kubacka, M.; Sadek, B.; Ashoor, A.; Siwek, A.; Więcek, M.; Kucwaj, K.; Filipek, B.; et al. SAR-studies on the importance of aromatic ring topologies in search for selective 5-HT 7 receptor ligands among phenylpiperazine hydantoin derivatives. Eur. J. Med. Chem. 2014, 78, 324–339. [Google Scholar] [CrossRef] [PubMed]
- Löber, S.; Aboul-Fadl, T.; Hübner, H.; Gmeiner, P. Di- and trisubstituted pyrazolo[1,5-a]pyridine derivatives: Synthesis, dopamine receptor binding and ligand efficacy. Bioorganic Med. Chem. Lett. 2012, 12, 633–636. [Google Scholar] [CrossRef]
- Hayatshahi, H.S.; Xu, K.; Griffin, S.A.; Taylor, M.; Mach, R.H.; Liu, J.; Luedtke, R.R. Analogues of Arylamide Phenylpiperazine Ligands to Investigate the Factors Influencing D3 Dopamine Receptor Bitropic Binding and Receptor Subtype Selectivity. ACS Chem. Neurosci. 2018, 9, 2972–2983. [Google Scholar] [CrossRef]
- Handzlik, J.; Szymańska, E.; Wójcik, R.; Dela, A.; Jastrzebska-Wiesek, M.; Karolak-Wojciechowska, J.; Fruziński, A.; Siwek, A.; Filipek, B.; Kieć-Kononowicz, K. Synthesis and SAR-study for novel arylpiperazine derivatives of 5-arylidenehydantoin with α 1-adrenoceptor antagonistic properties. Bioorganic Med. Chem. 2012, 20, 4245–4257. [Google Scholar] [CrossRef] [PubMed]
- Czopek, A.; Bucki, A.; Kołaczkowski, M.; Zagórska, A.; Drop, M.; Pawłowski, M.; Siwek, A.; Głuch-Lutwin, M.; Pękala, E.; Chrzanowska, A.; et al. Novel multitarget 5-arylidenehydantoins with arylpiperazinealkyl fragment: Pharmacological evaluation and investigation of cytotoxicity and metabolic stability. Bioorganic Med. Chem. 2019, 27, 4163–4173. [Google Scholar] [CrossRef] [PubMed]
- Kucwaj-Brysz, K.; Kurczab, R.; Jastrzębska-Więsek, M.; Żesławska, E.; Satała, G.; Nitek, W.; Partyka, A.; Siwek, A.; Jankowska, A.; Wesołowska, A.; et al. Computer-aided insights into receptor-ligand interaction for novel 5-arylhydantoin derivatives as serotonin 5-HT 7 receptor agents with antidepressant activity. Eur. J. Med. Chem. 2018, 147, 102–114. [Google Scholar] [CrossRef]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, 1100–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajusz, D.; Rácz, A.; Héberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminform. 2015, 7, 20. [Google Scholar] [CrossRef] [Green Version]
- Pándy-Szekeres, G.; Munk, C.; Tsonkov, T.M.; Mordalski, S.; Harpsøe, K.; Hauser, A.S.; Bojarski, A.J.; Gloriam, D.E. GPCRdb in 2018: Adding GPCR structure models and ligands. Nucleic Acids Res. 2018, 46, D440–D446. [Google Scholar] [CrossRef] [Green Version]
- Desaphy, J.; Raimbaud, E.; Ducrot, P.; Rognan, D. Encoding protein-ligand interaction patterns in fingerprints and graphs. J. Chem. Inf. Model. 2013, 53, 623–637. [Google Scholar] [CrossRef]
- Fisher, R.A. Frequency Distribution of the Values of the Correlation Coefficient in Samples from an Indefinitely Large Population. Biometrika 1915, 10, 507–521. [Google Scholar] [CrossRef]
- Handzlik, J.; Szymańska, E.; Alibert, S.; Chevalier, J.; Otrȩbska, E.; Pȩkala, E.; Pagès, J.M.; Kieć-Kononowicz, K. Search for new tools to combat Gram-negative resistant bacteria among amine derivatives of 5-arylidenehydantoin. Bioorganic Med. Chem. 2013, 21, 135–145. [Google Scholar] [CrossRef] [Green Version]
- Marton, J.; Enisz, J.; Hosztafi, S.; Tímár, T. Preparation and Fungicidal Activity of 5-Substituted Hydantoins and Their 2-Thio Analogs. J. Agric. Food Chem. 1993, 41, 148–152. [Google Scholar] [CrossRef]
- Handzlik, J.; Kieć-Kononowicz, K.; Dela, A.; Otrębska, E.; Kaleta, M. Novel N3-aminoalkyl Derivatives of 5-arylidenehydantoin; Pharmaceutical Composition Containing the Above and Application thereof. WO/2015/065212, 27 May 2015. [Google Scholar]
- Cheng, Y.; Prusoff, W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [PubMed]
- Rogers, D.; Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef] [PubMed]
- JChem 18.3.0, 2018, ChemAxon. Available online: http://www.chemaxon.com (accessed on 30 September 2021).
- LigPrep, Schrödinger Release 2020-1, LLC, New York, NY, USA. 2020. Available online: https://www.schrodinger.com/products/ligprep (accessed on 30 September 2021).
- Glide, Schrödinger Release 2020-1, LLC, New York, NY, USA. 2020. Available online: https://www.schrodinger.com/products/glide (accessed on 30 September 2021).
- Schrödinger Release 2020-1: Desmond Molecular Dynamics System, D.E. Shaw Research, New York, NY, 2020. Maestro-Desmond Interoperability Tools; Schrödinger: New York, NY, USA, 2020. Available online: https://www.schrodinger.com/products/desmond (accessed on 30 September 2021).
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cmpd | Group | R 1 | R 2 | R 3 | Ki [nM] | |||
|---|---|---|---|---|---|---|---|---|
| α1 | 5-HT1A | 5-HT6 | 5-HT7 | |||||
| 1 * | A | 3,4-diOCH3 | OH | 2-OCH3 | 44.5 | nt | nt | nt |
| 2 * | B | 4-F | OH | 2-OCH3 | 230 | 121 | 10,790 | 3 |
| 3 * | B | 4-OCH3 | OH | 2-OCH3 | 530 | 99 | nt | 8 |
| 4 | A | 3-OCH3 | OH | 2-OCH3 | 187.0 ± 18.6 | 530 | 17,610 | 283 |
| 5 | A | 3-OCH3 | OH | 2-OC2H5 | 75.9 ± 2.3 | 556.7 | 19,220 | 79 |
| 6 | A | 4-OCH3 | OH | 2-OCH3 | 144.6 ± 13.3 | 282 | 20,420 | 197 |
| 7 | A | 4-OCH3 | OH | 2-OC2H5 | 99.1 ± 11.3 | 313 | 10,250 | 120 |
| 8 | A | 4-OCH3 | OH | 2-F | 143.7 ± 7.9 | 3532 | 11,730 | 1036 |
| 9 | A | 3,4,5-triOCH3 | OH | 2-OC2H5 | 89.5 ± 9.0 | 1870 | 2835 | 1253 |
| 10 | A | 3-OCH3 | H | 2-OC2H5 | 20.2 ± 4.1 | 103.0 | nt | nt |
| 11 | A | 3-OCH3 | H | 2-Cl | 44.8 ± 3.3 | 319 | 1720 | 152 |
| 12 | A | 4-OCH3 | H | 2-OCH3 | 34.6 ± 4.6 | 20.6 | 21,990 | 101 |
| 13 | A | 4-OCH3 | H | 2-OC2H5 | 19.5 ± 2.6 | 45.8 | >50,000 | 114 |
| 14 | A | 3,4-diOCH3 | H | 2-OCH3 | 11.9 ± 2.2 | 128 | 15,780 | 189 |
| 15 | A | 3,4,5-triOCH3 | H | 2-OCH3 | 25.8 ± 2.9 | 172 | 12,320 | 668 |
| 16 | A | 3,4,5-triOCH3 | H | 2-OC2H5 | 14.7 ± 1.1 | 164.5 | 15,240 | 138 |
| Ref | - | - | - | - | 15.7 ± 1.5 1 | 32 2 | 7 3 | 62 4 |
| Cmpd | IC50 ± SEM [nM] * | ||
|---|---|---|---|
| α1A-AR | α1B-AR | α1D-AR | |
| Phenylephrine | 30.0 ± 3.4 | 27.0 ± 4.2 | 26.2 ± 4.1 |
| Prazosin | 4.1 ± 0.5 | 3.82 ± 0.5 | n.d |
| Terazosin | 51.9 ± 0.8 | 1.73 ± 0.3 | 0.1 ± 0.02 |
| Tamsulosin | 0.07 ± 0.01 | 0.13 ± 0.02 | n.d. |
| 10 | 7.2 ± 1.1 | 9.2 ± 1.3 | 1.2 ± 0.2 |
| 12 | 12.8 ± 2.5 | 8.5 ± 1.1 | 1.5 ± 0.3 |
| 14 | 57.6 ± 7.2 | 182 ± 21.4 | 3.0 ± 0.5 |
| 16 | 19.1 ± 3.4 | 0.82 ± 0.1 | 4.0 ± 0.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kucwaj-Brysz, K.; Dela, A.; Podlewska, S.; Bednarski, M.; Siwek, A.; Satała, G.; Czarnota, K.; Handzlik, J.; Kieć-Kononowicz, K. The Structural Determinants for α1-Adrenergic/Serotonin Receptors Activity among Phenylpiperazine-Hydantoin Derivatives. Molecules 2021, 26, 7025. https://doi.org/10.3390/molecules26227025
Kucwaj-Brysz K, Dela A, Podlewska S, Bednarski M, Siwek A, Satała G, Czarnota K, Handzlik J, Kieć-Kononowicz K. The Structural Determinants for α1-Adrenergic/Serotonin Receptors Activity among Phenylpiperazine-Hydantoin Derivatives. Molecules. 2021; 26(22):7025. https://doi.org/10.3390/molecules26227025
Chicago/Turabian StyleKucwaj-Brysz, Katarzyna, Anna Dela, Sabina Podlewska, Marek Bednarski, Agata Siwek, Grzegorz Satała, Kinga Czarnota, Jadwiga Handzlik, and Katarzyna Kieć-Kononowicz. 2021. "The Structural Determinants for α1-Adrenergic/Serotonin Receptors Activity among Phenylpiperazine-Hydantoin Derivatives" Molecules 26, no. 22: 7025. https://doi.org/10.3390/molecules26227025
APA StyleKucwaj-Brysz, K., Dela, A., Podlewska, S., Bednarski, M., Siwek, A., Satała, G., Czarnota, K., Handzlik, J., & Kieć-Kononowicz, K. (2021). The Structural Determinants for α1-Adrenergic/Serotonin Receptors Activity among Phenylpiperazine-Hydantoin Derivatives. Molecules, 26(22), 7025. https://doi.org/10.3390/molecules26227025