
The Renin-Angiotensin System: A Key Role in SARS-CoV-2-Induced COVID-19


 ,
,  , and
, and 
Abstract
1. Introduction
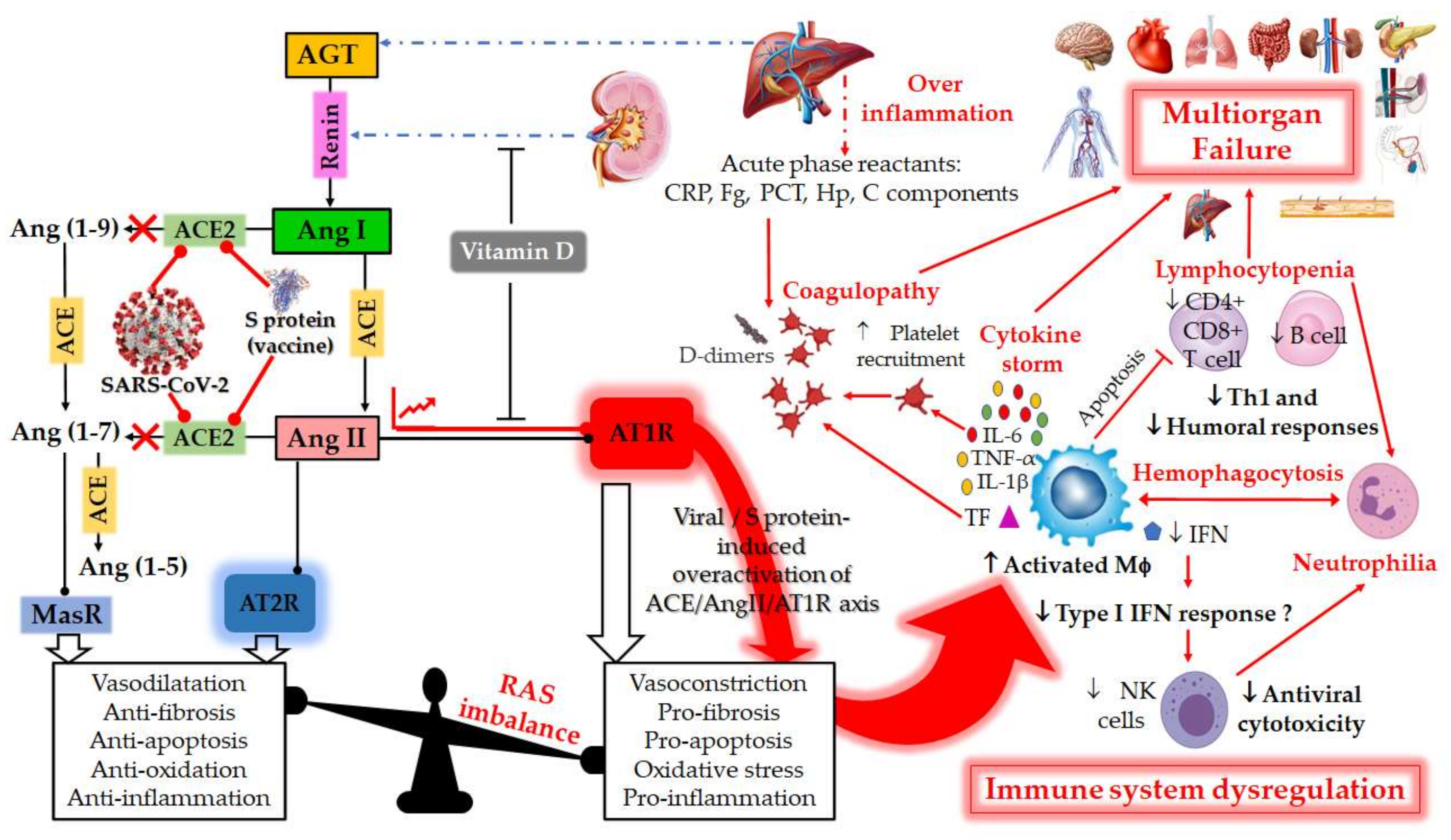
2. RAS and COVID-19
2.1. RAS: A Portal Entry for SARS-CoV-2
2.2. RAS Imbalance and Underlying Pathologies
2.3. RAS Component Polymorphism
2.3.1. ACE1 Polymorphism
2.3.2. ACE2 Polymorphism
2.3.3. AGT Polymorphism
2.3.4. AT1R and AT2R Polymorphisms
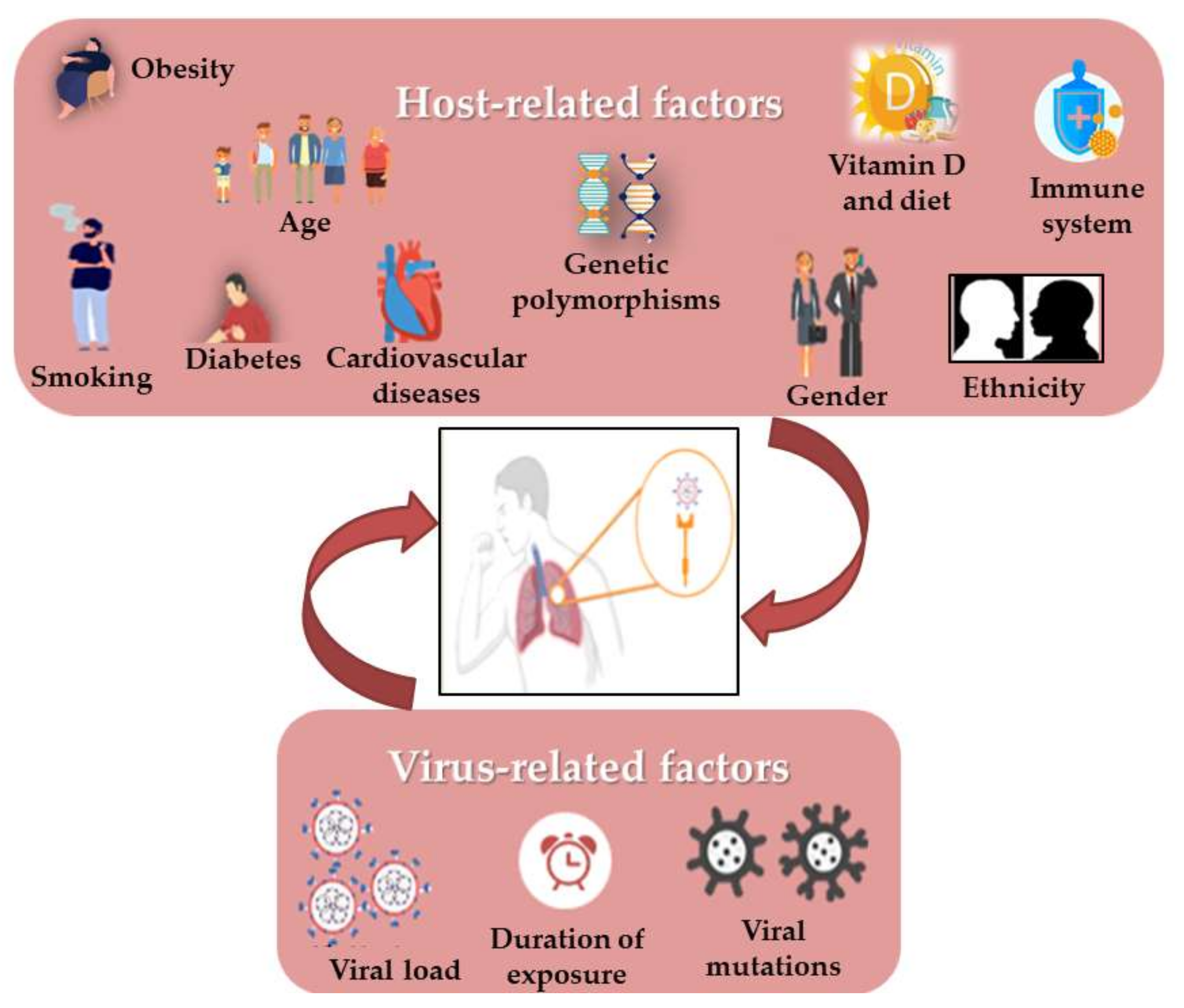
3. Correlation of Habits, Gender and COVID-19 with RAS Polymorphism
3.1. COVID-19 and Age
3.2. COVID-19 and Gender
3.3. COVID-19 and Smoking
3.4. COVID-19 and the Cardiovascular System
3.5. COVID-19 and Diabetes
3.6. COVID-19 and Obesity
3.7. COVID-19 and Vitamin D Deficiency
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Available online: https://www.who.int/westernpacific/health-topics/detail/coronavirus (accessed on 13 September 2021).
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus–Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef]
- Suryamohan, K.; Diwanji, D.; Stawiski, E.W.; Gupta, R.; Miersch, S.; Liu, J.; Chen, C.; Jiang, Y.P.; Fellouse, F.A.; Sathirapongsasuti, J.F.; et al. Human ACE2 receptor polymorphisms and altered susceptibility to SARS-CoV-2. Commun. Biol. 2021, 4, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Yuen, K.S.; Ye, Z.W.; Fung, S.Y.; Chan, C.P.; Jin, D.Y. SARS-CoV-2 and COVID-19: The most important research questions. Cell Biosci. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Bialek, S.; Boundy, E.; Bowen, V.; Chow, N.; Cohn, A.; Dowling, N.; Ellington, S.; Gierke, R.; Hall, A.; MacNeil, J. Severe Outcomes Among Patients with Coronavirus Disease 2019 (COVID-19)—United States. CDC 2021, 69, 343–346. [Google Scholar] [CrossRef]
- Jirjees, F.; Saad, A.K.; Al Hano, Z.; Hatahet, T.; Al Obaidi, H.; Dallal Bashi, Y.H. COVID-19 Treatment Guidelines: Do They Really Reflect Best Medical Practices to Manage the Pandemic? Infect. Dis. Rep. 2021, 13, 29. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Chu, Y.; Wang, Y. HIV protease imhibitors: A review of molecular selectivity and toxicity. HIV/AIDS-Res. Palliat. Care 2014, 7, 95–104. [Google Scholar] [CrossRef]
- Yu, B.; Li, C.; Chen, P.; Zhou, N.; Wang, L.; Li, J.; Jiang, H.; Wang, D.W. Low dose of hydroxychloroquine reduces fatality of critically ill patients with COVID-19. Sci. China Life Sci. 2020, 63, 1515–1521. [Google Scholar] [CrossRef]
- Hadjadj, J.; Yatim, N.; Barnabel, L.; Corneau, A. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef]
- Miao, G.; Zhao, H.; Li, Y.; Bi, Y.; Wang, P.; Zhang, H. ORF3a of the COVID-19 virus SARS-CoV-2 blocks HOPS complex-mediated assembly of the SNARE complex required for autolysosome formation. Dev. Cell 2020, 56, 427–442. [Google Scholar] [CrossRef]
- Liu, P.P.; Blet, A.; Smyth, D. The science underlying COVID-19, Implications for the cardiovascular system. Circulation 2020, 142, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Barré, J.; Sabatier, J.M.; Annweiler, C. Montelukast Drug May Improve COVID-19 Prognosis: A Review of Evidence. Front Pharmacol. 2020, 11, 1344–1349. [Google Scholar] [CrossRef] [PubMed]
- Annweiler, C.; Papon, N.; Sabatier, J.M.; Barré, J. DAMPening Severe COVID-19 with Dexamethasone. Infect. Disord. Drug Targets 2021, 21. [Google Scholar] [CrossRef] [PubMed]
- Vann, K.R.; Tencer, A.H.; Kutateladze, T.G. Inhibition of translation and immune responses by the virulence factor Nsp1 of SARS-CoV-2. Signal Transduct. Target. Ther. 2020, 5, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020, 18, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Chen, Z.; Xia, Y.; Lin, W.; Li, H. Investigation of the genetic variation in ACE2 on the structural recognition by the novel coronavirus (SARS-CoV-2). J. Transl. Med. 2020, 18, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Rysz, S.; Al-Saadi, J.; Sjöström, A.; Farm, M.; Jalde, F.C.; Plattén, M.; Eriksson, H.; Klein, M.; Vargas-Paris, R.; Nyrén, S. COVID-19 pathophysiology may be driven by an imbalance in the renin-angiotensin-aldosterone system. Nat. Commun. 2021, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Perlot, T.; Penninger, J.M. ACE2–From the renin–angiotensin system to gut microbiota and malnutrition. Microbes Infect. 2013, 15, 866–873. [Google Scholar] [CrossRef] [PubMed]
- Annweiler, C.; Cao, Z.; Wu, Y.; Faucon, F.; Mouhat, S.; Kovacic, H.; Sabatier, J.M. Counter-regulatory ‘Renin-Angiotensin’ System-based Candidate Drugs to Treat COVID-19 Diseases in SARS-CoV-2-infected Patients. Infect. Disord. Drug Targets 2020, 20, 407–408. [Google Scholar] [CrossRef]
- Coto, E.; Avanzas, P.; Gómez, J. The Renin–Angiotensin–Aldosterone System and Coronavirus Disease 2019. Eur. Cardiol. Rev. 2021, 16, e07. [Google Scholar] [CrossRef]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, e00127-20. [Google Scholar] [CrossRef]
- Cao, Z.; Wu, Y.; Faucon, E.; Sabatier, J.M. SARS-CoV-2 & Covid-19: Key-Roles of the ‘Renin-Angiotensin’ System/Vitamin D Impacting Drug and Vaccine Developments. Infect. Disord. Drug Targets 2020, 20, 348–349. [Google Scholar] [CrossRef]
- Lei, Y.; Zhang, J.; Schiavon, C.R.; He, M.; Chen, L.; Shen, H.; Zhang, Y.; Yin, Q.; Cho, Y.; Andrade, L. SARS-CoV-2 Spike Protein Impairs Endothelial Function via Downregulation of ACE2. Circ. Res. 2021, 128, 1323–1326. [Google Scholar] [CrossRef] [PubMed]
- Bezabih, Y.M.; Bezabih, A.; Alamneh, E.; Peterson, G.M.; Bezabhe, W. Comparison of renin–angiotensin–aldosterone system inhibitors with other antihypertensives in association with coronavirus disease-19 clinical outcomes. BMC Infect. Dis. 2021, 21, 527. [Google Scholar] [CrossRef] [PubMed]
- Vaduganathan, M.; Vardeny, O.; Michel, T.; McMurray, J.J.V.; Pfeffer, M.A.; Solomon, S.D. Renin–Angiotensin–Aldosterone System Inhibitors in Patients with Covid-19. N. Engl. J. Med. 2020, 382, 1653–1659. [Google Scholar] [CrossRef]
- Rafiullah, M. Can a Combination of AT1R Antagonist and Vitamin D Treat the Lung Complication of COVID-19? Am. J. Med Sci. 2020, 360, 338–341. [Google Scholar] [CrossRef]
- Feng, Y.; Hans, C.; Mcllwain, E.; Varner, K.J.; Lazartigues, E. Angiotensin-Converting Enzyme 2 Over-Expression in the Central Nervous System Reduces Angiotensin-II- Mediated Cardiac Hypertrophy. PLoS ONE 2012, 7, e48910. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Ghafil, F.; Hadi, N.R.; Mohammad, B.I.; Al-Aubaidy, H.A. Genetic Polymorphism of Angiotensin II Type 1 Receptors and Their Effect on the Clinical Outcome of Captopril Treatment in Arab Iraqi Patients with Acute Coronary Syndrome (Mid Euphrates). Ind. J. Clin. Biochem. 2019, 36, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.H.; Zhang, Y.H.; Dong, X.F.; Hao, Q.Q.; Zhou, X.M.; Yu, Q.Y.; Li, S.Y.; Chen, X.; Tengbeh, A.F.; Dong, B.; et al. ACE2 and Ang-(1–7) protect endothelial cell function and prevent early atherosclerosis by inhibiting inflammatory response. Inflamm. Res. 2015, 64, 253–260. [Google Scholar] [CrossRef]
- Hou, L.; Quan, X.; Li, X.; Su, X. Correlation between gene polymorphism in angiotensin II type 1 receptor and type 2 diabetes mellitus complicated by hypertension in a population of Inner Mongolia. BMC Med Genet. 2020, 21, 83. [Google Scholar] [CrossRef] [PubMed]
- Lovren, F.; Pan, Y.; Quan, A.; Teoh, H.; Wang, G.; Shukla, P.C.; Levitt, K.S.; Oudit, G.; Al-Omran, M.; Stewart, D.J.; et al. Angiotensin converting enzyme-2 confers endothelial protection and attenuates atherosclerosis. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Li, L.; Xu, M.; Wu, J.; Luo, D.; Zhu, Y.; Li, B.; Song, X.Y.; Zhou, X. Prognostic value of interleukin-6, C-reactive protein, and procalcitonin in patients with COVID-19. J. Clin. Virol. 2020, 127, 104370. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Bandopadhyay, A.; Das, D.; Pandey, R.; Singh, V.; Khanam, N.; Srivastava, N.; Singh, P.P.; Dubey, P.K.; Pathak, A.; et al. Genetic Association of ACE2 rs2285666 Polymorphism With COVID-19 Spatial Distribution in India. Front. Genet. 2020, 11, 1163. [Google Scholar] [CrossRef]
- Cafiero, C.; Rosapepe, F.; Palmirotta, R.; Re, A.; Ottaiano, M.P.; Benincasa, G.; Perone, R.; Varriale, E.; D’Amato, G.; Cacciamani, A.; et al. Angiotensin System Polymorphisms’ in SARS-CoV-2 Positive Patients: Assessment Between Symptomatic and Asymptomatic Patients: A Pilot Study. Pharm. Pers. Med. 2021, 14, 621–629. [Google Scholar] [CrossRef]
- I’ñiguez, M.; Pe´rez-Matute, P.; Villoslada-Blanco, P.; Recio-Fernandez, E.; Ezquerro-Pe´rez, D.; Alba, J.; Ferreira-Laso, M.L.; Oteo, J.A. ACE Gene Variants Rise the Risk of Severe COVID-19 in Patients with Hypertension, Dyslipidemia or Diabetes: A Spanish Pilot Study. Front. Endocrinol. 2021, 12. [Google Scholar] [CrossRef]
- Abdollahi, M.R.; Gaunt, T.R.; Syddall, H.E.; Cooper, C.; Phillips, D.I.; Ye, S.; Day, I.N.M. Angiotensin II type I receptor gene polymorphism: Anthropometric and metabolic syndrome traits. J. Med. Genet. 2005, 42, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Narang, R.; Sreenivas, V.; Bhatia, J.; Saluja, D.; Srivastava, K. Association of Angiotensin II Type 1 Receptor (A1166C) Gene Polymorphism and Its Increased Expression in Essential Hypertension: A Case-Control Study. PLoS ONE 2014, 9, e101502. [Google Scholar] [CrossRef]
- Li, W.; Moore, M.J.; Vasilieva, V.N.; Sui, S.J.; Wong, S.K.; Berne, M.A.; Somasundaran, S.M.; Sullivan, J.L.; Luzuriaga, L.K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L. Pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–286. [Google Scholar] [CrossRef] [PubMed]
- Mousavizadeh, L.; Ghasemi, S. Genotype and phenotype of COVID-19: Their roles in pathogenesis. J. Microbiol. Immunol. Infect. 2021, 54, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Parton, R.G.; Joggerst, B.; Simons, K. Regulated Internalization of Caveolae. J. Ceil Biol. 1994, 127, 1199–1215. [Google Scholar] [CrossRef]
- Inoue, Y.; Tanaka, N.; Tanaka, Y.; Inoue, S.; Morita, K.; Zhuang, M.; Hattori, T.; Sugamura, K. Clathrin-Dependent Entry of Severe Acute Respiratory Syndrome Coronavirus into Target Cells Expressing ACE2 with the Cytoplasmic Tail Deleted. J. Virol. 2007, 81, 8722–8729. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Zhao, J.; Martin, W.; Kallianpur, A.; Chung, M.K.; Jehi, L.; Sharifi, N.; Erzurum, S.; Eng, C.; Cheng, F. New insights into genetic susceptibility of COVID-19: An ACE2 and TMPRSS2 polymorphism analysis. BMC Med. 2020, 18, 216. [Google Scholar] [CrossRef] [PubMed]
- Asselta, R.; Paraboschi, E.V.; Mantovani, A.; Stefano Duga, S. ACE2 and TMPRSS2 variants and expression as candidates to sex and country differences in COVID-19 severity in Italy. Aging 2020, 12, 10087–10098. [Google Scholar] [CrossRef]
- Devaux, C.A.; Pinault, L.; Osman, I.O.; Raoult, D. Can ACE2 Receptor Polymorphism Predict Species Susceptibility to SARS-CoV-2? Front. Public Health 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Ariumi, Y.; Nishida, N.; Yamamoto, R.; Bauer, G.; Gojobori, T.; Shimotohno, K.; Mizokami, M. SARS-CoV-2 infections and COVID-19 mortalities strongly correlate with ACE1 I/D genotype. Gene 2020, 758, 144944. [Google Scholar] [CrossRef] [PubMed]
- Adamzik, M.; Frey, U.; Sixt, S.; Knemeyer, L.; Beiderlinden, M.; Peters, J.; Siffert, W. ACE I/D but not AGT (-6)A/G polymorphismis a risk factor for mortality in ARDS. Eur. Respir. J. 2007, 29, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Richardson, S.; Hirsch, J.S.; Narasimhan, M.; Crawford, J.M.; McGinn, T.; Davidson, K.W. Presenting Characteristics, Comorbidities, and OutcomesAmong 5700 Patients Hospitalized WithCOVID-19 in the New York City Area. JAMA 2020, 323, 2052–2059. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus–induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Ruan, Q.; Yang, K.; Wang, W.; Jiang, L.; Song, J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 2020, 46, 846–848. [Google Scholar] [CrossRef]
- Li, M.Y.; Li, L.; Zhang, Y.; Wang, X.S. Expression of the SARS-CoV-2 cell receptor gene ACE2 in a wide variety of human tissues. Infect. Dis. Poverty 2020, 9, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Xia, J.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet Respir. Med. 2020, 8, 475–481. [Google Scholar] [CrossRef]
- Sandoval, J.; Valle-Mondragón, L.D.; Masso, F.; Zayas, N.; Pulido, T.; Teijeiro, R.; Gonzalez-Pacheco, H.; Olmedo-Ocampo, R.; Sisniega, C.; Paez-Arenas, A.; et al. Angiotensin Converting Enzyme 2 and Angiotensin (1–7) axis in Pulmonary Arterial Hypertension. Eur. Respir. J. 2020, 56, 1902416. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, C.; Annunziata, A.; Coppola, A.; Pafundi, P.C.; Guarino, S.; Di Spirito, V.; Maddaloni, V.; Pepe, N.; Fiorentino, G. ACE Gene I/D Polymorphism and Acute Pulmonary Embolism in COVID19 Pneumonia: A Potential Predisposing Role. Front. Med. 2021, 7, 1136. [Google Scholar] [CrossRef]
- Wang, H.X.; Teng, Y.; Wang, K.; Xia, Z.W.; Tian, Y.Y.; Li, C.J. The M235T polymorphism in the angiotensinogen gene and atrial fibrillation: A meta-analysis. J. Renin-Angiotensin-Aldosterone Syst. 2015, 16, 647–652. [Google Scholar] [CrossRef]
- Al-Samkari, H.; Leaf, R.S.K.; Dzik, W.D.; Carlson, J.C.T.; Fogerty, A.E.; Waheed, W.A.; Goodarzi, K.; Bendapudi, P.K.; Bornikova, L.; Gupta, S.; et al. COVID-19 and coagulation: Bleeding and thrombotic manifestations of SARS-CoV-2 infection. Blood 2020, 136, 489–500. [Google Scholar] [CrossRef]
- Tang, N.; Li, D.; Wang, X.; Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 844–847. [Google Scholar] [CrossRef]
- Annweiler, C.; Bourgeais, A.; Faucon, E.; Cao, Z.; Wu, Y.; Sabatier, J.M. Neurological, Cognitive, and Behavioral Disorders during COVID-19: The Nitric Oxide Track. J. Am. Geriatr. Soc. 2020, 68, 1922–1923. [Google Scholar] [CrossRef]
- Zucker, I.H.; Xiao, L.; Haack, K.K.V. The central renin-angiotensin system and sympathetic nerve activity in chronic heart failure. Clin. Sci. 2014, 126, 695–706. [Google Scholar] [CrossRef]
- Writing Group Members; Roger, V.L.; Go, A.S.; Lloyd-Jones, D.; Benjamin, E.; Berry, J.D.; Borden, W.B.; Bravata, D.M.; Dai, S.; Ford, E.S.; et al. Heart disease and stroke statistics—2012 update: A report from the American Heart Association. Circulation 2012, 125, e2–e220. [Google Scholar] [CrossRef]
- Perticone, M.; Zito, R.; Miceli, S.; Pinto, A.; Suraci, E.; Greco, M.; Guigliotti, S.; Hribal, M.L.; Corrao, S.; Sesti, G.; et al. Immunity, Inflammtion and Heart Failure: Their role on Cardiac Function and Iron Status. Front Immunol. 2019, 10, 2315. [Google Scholar] [CrossRef] [PubMed]
- Hartupee, J.; Mann, D.L. Neurohormonal activation in heart failure with reduced ejection fraction. Nat. Rev. Cardiol. 2017, 14, 30–38. [Google Scholar] [CrossRef]
- Piazza, G.; Seddighzadeh, A.; Goldhaber, S.Z. Heart failure in patients with deep vein thrombosis. Am. J. Cardiol. 2008, 101, 1056–1059. [Google Scholar] [CrossRef] [PubMed]
- Perticonne, F.; Ceravolo, R.; Pujia, A.; Ventura, G.; Lacopino, S.; Scozzafava, A.; Ferraro, A.; Chello, M.; Mastroroberto, P.; Verdecchia, P.; et al. Prognostic significance of endothelial dysfunction in hypertensive patients. Circulation 2001, 104, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Quyyumj, A.A. Endothelial function in health and disease: New insights into the genesis of cardiovascular disease. Am. J. Med. 1998, 105, 32–39. [Google Scholar] [CrossRef]
- Hatami, N.; Ahi, S.; Fereydoni, M.; Sadeghinikoo, A.; Keshavarz, P.; Foroughian, M.; Osseini, A. Worldwide ACE (I/D) polymorphism may affect COVID-19 recovery rate: An ecological meta-regression. Endocrine 2020, 68, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Abbas, M.; Verma, S.; Khand, F.H.; Raza, S.T.; Siddiqi, Z.; Ahmada, I.; Mahdi, F. Impact of I/D polymorphism of angiotensin-converting enzyme 1 (ACE1) gene on the severity of COVID-19 patients. Infect. Genet. Evol. 2021, 91. [Google Scholar] [CrossRef] [PubMed]
- Patia, A.; Mahtob, H.; Padhia, S.; Panda, A.K. ACE deletion allele is associated with susceptibility to SARS-CoV-2 infection and mortality rate: An epidemiological study in the Asian population. Clin. Chim. Acta 2020, 15, 455–458. [Google Scholar] [CrossRef]
- Nie, W.; Zang, Y.; Chen, J.; Liu, T.; Xiao, L.; Xiu, Q. Angiotensin-converting enzyme I/D polymorphism is associated with pneumonia risk: A meta-analysis. J. Renin-Angiotensin-Aldosterone Syst. 2014, 15, 585–592. [Google Scholar] [CrossRef]
- Sarangarajan, R.; Winn, R.; Kiebish, M.A.; Bountra, C.; Granger, E.; Narain, N.R. Ethnic Prevalence of Angiotensin-Converting Enzyme Deletion (D) Polymorphism and COVID-19 Risk: Rationale for Use of Angiotensin-Converting Enzyme Inhibitors/Angiotensin Receptor Blockers. J. Racial Ethn. Health Disparities 2020, 8, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Marshall, R.P.; Webb, S.; Bellingan, G.J.; Montgomery, H.E.; Chaudhari, B.; McAnulty, R.J.; Humphries, S.E.; Hill, M.R.; Laurent, G.J. Angiotensin Converting Enzyme Insertion/Deletion Polymorphism Is Associated with Susceptibility and Outcome in Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2002, 166, 646–650. [Google Scholar] [CrossRef]
- Szolnoki, Z.; Havasi, V.; Talián, G.; Bene, J.; Komlósi, K.; Somogyvári, F.; Kondacs, A.; Szabó, M.; Fodor, L.; Bodor, A.; et al. Angiotensin II Type-1 Receptor A1166C Polymorphism is Associated with Increased Risk of Ischemic Stroke in Hypertensive Smokers. J. Mol. Neurosci. 2006, 28, 285–290. [Google Scholar] [CrossRef]
- Jerng, J.S.; Yu, C.J.; Wang, H.C.; Chen, K.Y.; Cheng, S.L.; Yang, P.C. Polymorphism of the angiotensin-converting enzyme gene affects the outcome of acute respiratory distress syndrome. Crit. Care Med. 2006, 34, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Mathew, J.; Basheeruddin, K.; Prabhaka, S. Differences in Frequency of the Deletion Polymorphism of the Angiotensin-Converting Enzyme Gene in Different Ethnic Groups. Angiology 2001, 52, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Saab, Y.B.; Gard, P.R.; Overall, A.D.J. The geographic distribution of the ACE II genotype: A novel finding. Genet. Res. 2007, 89, 259–267. [Google Scholar] [CrossRef]
- Rani, B.; Kumar, A.; Bahl, A.; Sharma, R.; Prasad, R.; Khullar, M. Renin–angiotensin system gene polymorphisms as potential modifiers of hypertrophic and dilated cardiomyopathy phenotypes. Mol. Cell Biochem. 2017, 427, 1–11. [Google Scholar] [CrossRef]
- Pouladi, N.; Abdolahi, S. Investigating the ACE2 polymorphisms in COVID- 19 susceptibility: An in silico analysis. Mol. Genet. Genomic. Med. 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Imen, T.; Grissa, M.H.; Boubaker, H.; Beltaief, K.; Messous, S.; Tounsi, N.; Slimani, A.; Khouloud, C.; Bouida, W.; Boukef, R.; et al. AGT M235t polymorphism and heart failure in a cohort of Tunisian population: Diagnostic and prognostic value. Int. J. Clin. Exp. Med. 2015, 8, 16346–16351. [Google Scholar] [PubMed]
- Tran, T.T.; Mai, T.P.; Tran, H.C.B.; Le, L.H.G.; Vu, H.A.; Tran, T.K.; Hoang, S.V.; Chau, H.N.; Do, M.D. Association Between AGT M235T and Left Ventricular Mass in Vietnamese Patients Diagnosed with Essential Hypertension. Front. Cardiovasc. Med. 2021, 8, 16. [Google Scholar] [CrossRef]
- Raygan, F.; Karimian, M.; Rezaeian, A.; Bahmani, B.; Behjati, M. Angiotensinogen-M235T as a risk factor for myocardial infarction in Asian populations: A genetic association study and a bioinformatics approach. Croat. Med. J. 2016, 57, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Pilbrow, A.P.; Palmer, B.R.; Frampton, C.M.; Yandle, T.G.; Troughton, R.W.; Campbell, E.; Skelton, L.; Lainchbury, J.G.; Richards, A.M.; Cameron, V.A. Angiotensinogen M235T and T174M Gene Polymorphisms in Combination Doubles the Risk of Mortality in Heart Failure. Hypertension 2007, 49, 322–327. [Google Scholar] [CrossRef]
- Abd El-Aziz, T.A.; Mohamed, R.H.; Rezk, N.A. Association of angiotensin II type I and type II receptor genes polymorphisms with the presence of premature coronary disease and metabolic syndrome. Mol. Biol. Rep. 2014, 41, 1027–1033. [Google Scholar] [CrossRef] [PubMed]
- Shahin, D.S.; Irshaid, Y.M.; Abu Saleh, A.S. The A1166C polymorphism of the AT1R gene is associated with an early onset of hypertension and high waist circumference in Jordanian males attending the Jordan University Hospital. Clin. Exp. Hypertens. 2014, 36, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Alavi-Shahri, J.; Behravan, J.; Hassany, M.; Tatari, F.; Kasaian, J.; Ganjali, R.; Tavallaie, S.; Sabouri, S.; Sahebkar, A.; Oladi, M.; et al. Association Between Angiotensin II Type 1 Receptor Gene Polymorphism and Metabolic Syndrome in a Young Female Iranian Population. Arch. Med. Res. 2010, 41, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Ali Hashemi, S.N.; Thijssen, M.; Hosseini, S.Y.; Tabarraei, A.; Pourkarim, M.R.; Sarvari, J. Human gene polymorphisms and their possible impact on the clinical outcome of SARS-CoV-2 infection. Arch. Virol. 2021, 166, 2089–2108. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, Y.; Guan, T.; Lai, Y.; Shen, Y.; Zeyaweiding, A.; Zhao, H.; Li, F.; Maimaiti, T. ACE2 polymorphisms associated with cardiovascular risk in Uygurs with type 2 diabetes mellitus. Cardiovasc. Diabetol. 2018, 17, 127. [Google Scholar] [CrossRef]
- Fan, Z.; Wu, G.; Yue, M.; Ye, J.; Chen, Y.; Xu, B.; Shu, Z.; Zhu, J.; Lu, N.; Tan, X. Hypertension and hypertensive left ventricular hypertrophy are associated with ACE2 genetic polymorphism. Life Sci. 2019, 225, 39–45. [Google Scholar] [CrossRef]
- Hamet, P.; Pausova, Z.; Attaoua, R.; Hishmih, C.; Haloui, M.; Jean Shin, J.; Paus, T.; Abrahamowicz, M.; Gaudet, D.; Santucci, L.; et al. SARS-COV-2 receptor ACE2 gene is associated with hypertension and severity of COVID 19: Interaction with sex, obesity and smoking. Am. J. Hypertens. 2021, 34. [Google Scholar] [CrossRef]
- Yao, R.; Du, Y.Y.; Zhang, Y.Z.; Chen, Q.H.; Zhao, L.S.; Li, L. Association between G-217A polymorphism in the AGT gene and essential hypertension: A meta-analysis. Genet. Mol. Res. 2015, 14, 5527–5534. [Google Scholar] [CrossRef]
- Zeng, R.; Wang, Q.P.; Fang, M.X.; Zhuang, J.; Fan, R.X. Association of A-20C polymorphism in the angiotensinogen gene with essential hypertension: A meta-analysis. Genet. Mol. Res. 2015, 14, 12984–12992. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.Y.; Wang, Y.W.; Pang, X.H.; Wang, H.W. T174M polymorphism in the angiotensinogen gene and risk of myocardial infarction: A meta-analysis. Genet. Mol. Res. 2015, 14, 3767–3774. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.Z. Association between T174M polymorphism in the angiotensinogen gene and risk of coronary artery disease: A meta-analysis. J. Geriatr. Cardiol. 2013, 10, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Szolnoki, Z.; Maasz, A.; Magyari, L.; Horvatovich, K.; Farago, B.; Somogyvari, F.; Kondacs, A.; Szabo, M.; Fodor, L.; Bodor, A.; et al. Coexistence of Angiotensin II Type-1 Receptor A1166C and Angiotensin-Converting Enzyme D/D Polymorphism Suggests Susceptibility for Small-Vessel-Associated Ischemic Stroke. NeuroMolecular Med. 2006, 8, 353–360. [Google Scholar] [CrossRef]
- Agachan, B.; Isbir, T.; Yilmaz, H.; Akoglu, E. Angiotensin converting enzyme I/D, angiotensinogen T174M-M235T and angiotensin II type 1 receptor A1166C gene polymorphisms in Turkish hypertensive patients. Exp. Mol. Med. 2003, 35, 545–549. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dzida, G.; Biłan, A.; Golon-Siekierska, P.; Strzemecka, J.; Sobstyl, J.; Puźniak, A.; Mosiewicz, J.; Hanzlik, J. Methylenetetrahydrofolate reductase gene polymorphism in patients with type 2 diabetes. Pol. Arch. Med. Wewn. 2001, 106, 543–549. [Google Scholar]
- Kainulainen, K.; Perola, M.; Terwilliger, J.; Kaprio, J.; Koskenvuo, M.; Syvänen, A.C.; Vartiainen, E.; Peltonen, L.; Kontula, K. Evidence for Involvement of the Type 1 Angiotensin II Receptor Locus in Essential Hypertension. Hypertension 1999, 33, 844–849. [Google Scholar] [CrossRef] [PubMed]
- Bayramoglu, A.; Kurt, H.; Gunes, H.V.; Ata, N.A.; Birdane, A.; Dikmen, M.; Ustuner, M.C.; Colak, E.; Degirmenci, I. Angiotensin II Type 1 Receptor (AT1) Gene A1166C Is Associated with the Risk of Hypertension. Genet. Test. Mol. Biomarkers 2015, 19, 14–17. [Google Scholar] [CrossRef]
- Miller, D.S.; Kok, T.; Li, P. The virus inoculum volume influences outcome of influenza A infection in mice. Lab. Anim. 2013, 47, 74–77. [Google Scholar] [CrossRef]
- Li, X.; Xu, S.; Yu, M.; Wang, K.; Tao, Y.; Zhou, Y.; Shi, Z.M.; Zhou, M.; Wu, B.; Yang, Z.; et al. Risk factors for severity and mortality in adult COVID-19 in patients in Wuhan. J. Allergy Clin. Immunol. 2020, 146, 110–118. [Google Scholar] [CrossRef]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef]
- Crafaa, A.; Cannarellaa, R.; Condorellia, R.A.; Mongioìa, L.M.; Barbagalloa, F.; Aversab, A.; La Vigneraa, S.; Calogero, A.E. Influence of 25-hydroxy-cholecalciferol levels on SARS-CoV-2 infection and COVID-19 severity: A systematic review and meta-analysis. EClinicalMedicine 2021, 37, 100967. [Google Scholar] [CrossRef] [PubMed]
- Sharif-Askari, N.A.; Sharif-Askari, F.S.; Alabed, M.; Temsah, M.H.; Al Heialy, S.; Hamid, Q.; Halwani, R. Airways Expressionof SARS-CoV-2Receptor, ACE2, and TMPRSS2 Is Lower in Children Than Adults and Increases with Smoking and COPD. Mol. Ther. Methods Clin. Dev. 2020, 18, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Olson, D.R.; Simonsen, L.; Edelson, P.J.; Morse, S.S. Epidemiological evidence of an early wave of the 1918 influenza pandemic in New York City. PNAS 2005, 102, 11059–11063. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.H.; Su, M.C.; Tien, N.; Huang, C.J.; Lan, Y.C.; Lin, C.S.; Chen, C.H.; Lin, C.H. Epidemiology of human coronavirus NL63 infection among hospitalized patients with pneumonia in Taiwan. J. Microbiol. Immunol. Infect. 2017, 50, 763–770. [Google Scholar] [CrossRef]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk Factors Associated WithAcute Respiratory Distress Syndrome and Death in Patients with Coronavirus Disease 2019 Pneumoniain Wuhan, China. JAMA Intern. Med. 2020, 180, 934–943. [Google Scholar] [CrossRef]
- Xudong, X.; Junzhu, C.; Xingxiang, W.; Furong, Z.; Yanrong, L. Age- and gender-related difference of ACE2 expression in rat lung. Life Sci. 2006, 78, 2166–2171. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Ling, Y.; Bai, T.; Huang, J.; Li, J.; Xiong, W.; Yang, D.; Chen, R.; Lu, F.; Lu, Y.; et al. COVID-19 with Different Severities: A Multicenter Study of Clinical Features. Am. J. Respir. Crit. Care Med. 2020, 201, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Pearman, A.; Hughes, M.L.; Smith, E.L.; Neupert, S.D. Age Differences in Risk and Resilience Factors in COVID-19-Related Stress. J. Gerontol. B Psychol. Sci. Soc. Sci. 2021, 76, 38–44. [Google Scholar] [CrossRef]
- Greenglass, E.; Schwarzer, R.; Jakubiec, D.; Fiksenbaum, L.; Taubert, S. The Proactive Coping Inventory (PCI): A Multidimensional Research Instrument. In Proceedings of the 20th International Conference of the Stress and Anxiety Research Society (STAR), Cracow, Poland, 12–14 July 1999. [Google Scholar]
- Gemmati, D.; Bramanti, B.; Serino, M.L.; Secchiero, P.; Zauli, G.; Tisato, V. COVID-19 and Individual Genetic Susceptibility/Receptivity: Role of ACE1/ACE2 Genes, Immunity, Inflammation and Coagulation. Might the Double X-chromosome in Females Be Protective against SARS-CoV-2 Compared to the Single X-Chromosome in Males? Int. J. Mol. Sci. 2020, 21, 3474. [Google Scholar] [CrossRef]
- Smith, J.S.; Sausville, E.L.; Girish, V.; Yuan, M.L.; John, K.M.; Sheltzer, J.M. Cigarette smoke exposure and inflammatory signaling increase the expression of the SARS-CoV-2 receptor ACE2 in the respiratory tract. Dev. Cell 2020, 53, 514–529. [Google Scholar] [CrossRef] [PubMed]
- Yilin, Z.; Yandong, N.; Faguang, J. Role of angiotensin-converting enzyme (ACE) and ACE2 in a rat model of smoke inhalation induced acute respiratory distress syndrome. Burns 2015, 41, 1468–1477. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; He, X.; Zhang, L.; Ran, Q.; Wang, J.; Xiong, A.; Wu, D.; Chen, F.; Sun, J.; Chang, C. Assessing ACE2 expression patterns in lung tissues in the pathogenesis of COVID-19. J. Autoimmun. 2020, 112, 102463. [Google Scholar] [CrossRef]
- Cai, G.; Bossé, Y.; Xiao, F.; Kheradmand, F.; Amos, C.I. Tobacco Smoking Increases the Lung Gene Expression of ACE2, the Receptor of SARS-CoV-2. AJRCCM 2020, 201, 1557–1559. [Google Scholar] [CrossRef]
- Arias-Vásquez, A.; Sayed-Tabatabaei, F.A.; Schut, A.F.C.; Hofman, A.; Bertolli-Avella, A.M.; Vergeer, J.M.; Aulchenko, Y.S.; Witteman, J.C.M.; Van Duijn, C.M. Angiotensin converting enzyme gene, smoking and mortality in a population-based study Blackwell Publishing, Ltd. Eur. J. Clin. Investig. 2005, 35, 444–449. [Google Scholar] [CrossRef]
- Xie, Y.; You, Q.; Wu, C.; Cao, S.; Qu, G.; Yan, X.; Han, X.; Wang, C.; Zhang, H. Impact of Cardiovascular Disease on Clinical Characteristics and Outcomes of Coronavirus Disease 2019 (COVID-19). Circ. J. 2020, 84, 1277–1283. [Google Scholar] [CrossRef]
- Chen, L.; Li, X.; Chen, M.; Feng, Y.; Xiong, C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc. Res. 2020, 116, 1097–1100. [Google Scholar] [CrossRef] [PubMed]
- Sekuri, C.; Cam, F.S.; Eser, E.; Berdeli, A.; Ercan, E.; Akin, M.; Tengiz, I.; Sagcan, A. Renin-angiotensin system gene polymorphisms and premature coronary heart disease. J. Renin-Angiotensin-Aldosterone Syst. 2005, 6. [Google Scholar] [CrossRef]
- Oudit, G.Y.; Kassiri, Z.; Jiang, C.; Liu, P.P.; Poutanen§, S.M.; Penninger, J.M.; Butany, J. SARS-coronavirus modulation of myocardial ACE2 expression and inflammation in patients with SARS. Eur. J. Clin. Invest. 2009, 39, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Amara, A.; Mrad, M.; Sayeh, A.; Lahideb, D.; Layouni, S.; Haggui, A.; Fekih-Mrissa, N.; Haouala, H.; Nsiri, B. The Effect of ACE I/D Polymorphisms Alone and With Concomitant Risk Factors on Coronary Artery Disease. Clin. Appl. Thromb. Hemost. 2018, 24, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Freitas, A.I.; Mendonça, I.; Brión, M.; MSequeira, M.; Reis, R.P.; Carracedo, A.; Brehm, A. RAS gene polymorphisms, classical risk factors and the advent of coronary artery disease in the Portuguese population. BMC Cardiovasc. Disord. 2008, 8, 15. [Google Scholar] [CrossRef] [PubMed]
- Vladeanu, M.; Bojan, I.B.; Bojan, A.; Iliescu, D.; Badescu, M.C.; Badulescu, O.V.; Badescu, M.; Georgescu, C.A.; Ciocoiu, M. Angiotensin-converting enzyme gene D-allele and the severity of coronary artery disease. Exp. Ther. Med. 2020, 20, 3407–3411. [Google Scholar] [CrossRef]
- Niemiec, P.; Zak, I.; Wita, K. The Risk of Coronary Artery Disease Associated with Cigarette Smoking and Hypercholesterolemia Is Additionally Increased by the Presence of the AT1R Gene 1166C Allele. Biochem. Genet. 2008, 46, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Wallentin, L.; Lindback, J.; Eriksson, N.; Hijazi, Z.; Eikelboom, J.W.; Ezekowitz, M.D.; Granger, C.B.; Lopes, R.D.; Yusuf, S.; Oldgren, J.; et al. Prevention and epidemiology Angiotensin-converting enzyme 2 (ACE2) levels in relation to risk factors forCOVID-19 in two large cohorts of patientswith atrial fibrillation. Eur. Heart J. 2020, 41, 4037–4046. [Google Scholar] [CrossRef] [PubMed]
- Borzyszkowska, J.; Stanislawska-Sachadyn, A.; Wirtwein, M.; Sobiczewski, W.; Ciecwierz, D.; Targonski, R.; Gruchala, M.; Rynkiewicz, A.; Limon, J. Angiotensin converting enzyme gene polymorphism is associated with severity of coronary artery disease in men with high total cholesterol levels. J. Appl. Genet. 2012, 53, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Akbariqomi, M.; Hosseini, M.S.; Rashidiani, J.; Sedighian, H.; Biganeh, H.; Heidari, R.; Moghaddam, M.M.; Farnoosh, G.; Kooshki, H. Clinical characteristics and outcome of hospitalized COVID-19 patients with diabetes: A single-center, retrospective study in Iran. Diabetes Res. Clin. Pract. 2020, 169, 108467. [Google Scholar] [CrossRef]
- McGurnaghan, S.J.; Weir, A.; Bishop, J.; Kennedy, S.; Blackbourn, L.A.K.; McAllister, D.A.; Hutchinson, S.; Caparrotta, T.M.; Mellor, J.; Jeyam, A. Risks of and risk factors for COVID-19 disease in people with diabetes: A cohort study of the total population of Scotland. Lancet Diabetes Endocrinol. 2021, 9, 82–93. [Google Scholar] [CrossRef]
- Clément, K.; Coupaye, M.; Laville, M.; Oppert, J.M.; Ziegler, O. COVID-19: A Lever for the Recognition of Obesity as a Disease? The French Experience. Obesity 2020, 28, 1584–1585. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Zhao, H.; Lu, X.; Wen, Y.; Zhou, Y.; Deng, B.; Wang, Y.; Wang, W.; Kang, J.; Liu, P. Factors associated with death in hospitalized pneumonia patients with 2009 H1N1 influenza in Shenyang, China. BMC Infect. Dis. 2010, 10, 145. [Google Scholar] [CrossRef] [PubMed]
- Moser, J.S.; Galindo-Fraga, A.; Ortiz-Hernández, A.A.; Gu, W.; Hunsberger, S.; Galán-Herrera, J.F.; Guerrero, M.L.; Ruiz-Palacios, G.M.; Beigel, J.H. Underweight, overweight, and obesity as independent risk factors for hospitalization in adults and children from influenza and other respiratory viruses. Influenza Other Repiratory Viruses 2018, 13, 3–9. [Google Scholar] [CrossRef]
- Theurich, S.; Tsaousidou, E.; Hanssen, R.; Lempradl, A.M.; Mauer, J.; Timper, K.; Schilbach, K.; Folz-Donahue, K.; Heilinger, C.; Sexl, V.; et al. IL-6/Stat3-Dependent Induction of a Distinct, Obesity-Associated NK Cell Subpopulation Deteriorates Energy and Glucose Homeostasis. Cell Metab. 2017, 26, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, T.A.; Barcala-Jorge, A.S.; Andrade, J.M.O.; Pinheiro, T.A.; Ferreira, E.C.N.; Crespo, T.S.; Batista-Jorge, G.C.; Vieira, C.A.; Lelis, D.d.F.; Paraíso, A.F.; et al. Obesity and malnutrition similarly alter the renin-angiotensin system and inflammation in mice and human adipose. J. Nutr. Biochem. 2017, 48, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Simonnet, A.; Chetboun, M.; Poissy, J.; Raverdy, V.; Noulette, J.; Duhamel, A.; Labreuche, J.; Mathieu, D.; Pattou, F.; Jourdain, M. High Prevalence of Obesity in Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2) Requiring Invasive Mechanical Ventilation. Obesity 2020, 28, 1195–1199. [Google Scholar] [CrossRef]
- Del Valle, D.M.; Kim-Schulze, S.; Huang, H.H.; Beckmann, N.D.; Nirenberg, S.; Wang, B.; Lavin, Y.; Swartz, T.H.; Madduri, D.; Stock, A. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 2020, 26, 1636–1643. [Google Scholar] [CrossRef]
- Cummings, M.J.; Baldwin, M.R.; Abrams, D.; Jacobson, S.D.; Meyer, B.J.; Balough, E.M.; Aaron, J.G.; Claassen, J.; Rabbani, L.E.; Hastie, J.; et al. Epidemiology, clinical course, and outcomes of critically ill adults with COVID-19 in New York City: A prospective cohort study. Lancet 2020, 395, 1763–1770. [Google Scholar] [CrossRef]
- Albini, A.; Guardo, G.D.; Noonan, D.M.; Lombardo, M. The SARS-CoV-2 receptor, ACE-2, is expressed on many different cell types: Implications for ACE-inhibitor- and angiotensin II receptor blocker-based cardiovascular therapies. Intern. Emerg. Med. 2020, 15, 759–766. [Google Scholar] [CrossRef]
- Lu, P.R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, P.W.; Song, H.; Huang, B.; Zhu, N. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef]
- Milner, J.M.; Rebeles, J.; Dhungana, S.; Stewart, D.A.; Sumner, S.C.J.; Meyers, M.H.; Mancuso, P.; Beck, M.A. Obesity Increases Mortality and Modulates the Lung Metabolome during Pandemic H1N1 Influenza Virus Infection in Mice. J. Immunol. 2015, 194, 4846–4859. [Google Scholar] [CrossRef]
- Moriconi, D.; Masi, S.; Rebelosa, E.; Virdis, A.; Manca, M.L.; De Marco, S.; Taddei, S.; Nannipieri, M. Obesity prolongs the hospital stay in patients affected by COVID-19, and may impact on SARS-CoV-2 shedding. Obes. Res. Clin. Pract. 2020, 14, 205–209. [Google Scholar] [CrossRef]
- Lighter, J.; Phillips, M.; Hochman, S.; Sterling, S.; Johnson, D.; Francois, F.; Stache, A. Obesity in Patients Younger Than 60 Years Is a Risk Factor for COVID-19 Hospital Admission. Clin. Infect. Dis. 2020, 71, 896–897. [Google Scholar] [CrossRef]
- Zhang, F.; Xiong, Y.; Wei, Y.; Hu, Y.; Wang, F.; Li, G.; Liu, K.; Du, R.; Wang, C.Y.; Zhu, W. Obesity predisposes to the risk of higher mortality in young COVID-19 patients. J. Med. Virol. 2020, 92, 2536–2542. [Google Scholar] [CrossRef]
- Dreher, M.; Kersten, A.; Bickenbach, J.; Balfanz, P.; Hartmann, B.; Cornelissen, C.; Daher, A.; Stöhr, R.; Kleines, M.; Lemmen, S.W. The Characteristics of 50 Hospitalized COVID-19 Patients with and Without ARDS. Dtsch. Arztebl. Int. 2020, 117, 271–278. [Google Scholar] [CrossRef]
- Nimavat, N.; Singh, S.; Singh, P.; Singh, S.K.; Sinha, N. Vitamin D deficiency and COVID-19: A case-control study at a tertiary care hospital in India. Ann. Med. Surg. 2021, 68, 102661. [Google Scholar] [CrossRef] [PubMed]
- Grant, W.B.; Baggerly, C.A.; Lahore, H. Reply: “Vitamin D Supplementation in Influenza and COVID-19 Infections. Comment on: Evidence That Vitamin D Supplementation Could Reduce Risk of Influenza and COVID-19 Infections and Deaths Nutrients 2020, 12(4), 988”. Nutrients 2020, 12, 1620. [Google Scholar] [CrossRef]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Tramontana, F.; Napoli, N.; Fuleihan, G.E.-H.; Strollo, R. The D-side of COVID-19: Musculoskeletal benefits of vitamin D and beyond. Endocrine 2020, 69, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Demir, M.; Demir, F.; Aygun, H. Vitamin D deficiency is associated with COVID-19 positivity and severity of the disease. J. Med. Virol. 2021, 93, 2992–2999. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Available online: https://www.cdc.gov/coronavirus/2019-nCoV/index.html (accessed on 15 September 2021).
- Dancer, R.C.A.; Parekh, D.; Lax, S.; D’Souza, V.; Zheng, S.; Bassford, C.R.; Park, D.; Bartis, G.; Mahida, R.; Turner, A.M. Vitamin D deficiency contributes directly to the acute respiratory distress syndrome (ARDS). Thorax 2015, 70, 617–624. [Google Scholar] [CrossRef]
- Annweiler, C.; Hanotte, B.; Grandin de l’Eprevier, C.; Sabatier, J.M.; Lafaie, L.; Célarie, T. Vitamin D and survival in COVID-19 patients: A quasi-experimental study. J. Steroid Biochem. Mol. Biol. 2020, 204, 105771. [Google Scholar] [CrossRef] [PubMed]
- Ginde, A.A.; Mansbach, J.M.; Camargo, C.A., Jr. Association Between Serum 25-Hydroxyvitamin D Level and Upper Respiratory Tract Infection in the Third National Health and Nutrition Examination Survey. Arch. Intern. Med. 2009, 169, 384–390. [Google Scholar] [CrossRef]
- Martineau, A.R.; Jolliffe, D.A.; Hooper, R.L.; Greenberg, L.; Aloia, J.F.; Bergman, P.; Dubnov-Raz, G.; Esposito, S.; Ganmaa, D.; Ginde, A.A.; et al. Vitamin D supplementation to prevent acute respiratory tract infections: Systematic review and meta-analysis of individual participant data. BMJ 2017, 356. [Google Scholar] [CrossRef] [PubMed]
- Tsujino, I.; Ushikoshi-Nakayama, R.; Yamazaki, T.; Matsumoto, N.; Saito, I. Pulmonary activation of vitamin D3 and preventive effect against interstitial pneumonia. J. Clin. Biochem. 2019, 65, 245–251. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| RAS Component Gene | Chromosomal Location | Associated Disease/Phenotype | Mutations, Polymorphisms and rs Number | Allele/Genotype Frequencies in Populations and Ethnicities | References |
|---|---|---|---|---|---|
| ACE1 | 17q23.3 | CVD Kidney disease Autoimmune diseases Hypertension Hypercoagulability ARDS Type 2 diabetes Risk of obesity | Insertion/Deletion (I/D) of a 287-bp Alu repeat in intron 16 (rs1799752) | Lebanese I: 0.27, D: 0.73 | [77] |
| Indians I: 0.55, D: 0.45 Whites I: 0.5, D: 0.5 African Americans I: 0.41, D: 0.59 | [76] | ||||
| British I: 0.31, D: 0.69 (ARDS) I: 0.49, D: 0.51 (healthy population) | [73] | ||||
| Chinese I: 0.705, D: 0.295 | [75] | ||||
| Italians I: 0.342, D: 0.658 | [56] | ||||
| Italians I: 0.27, D: 0.73 | [35] | ||||
| Germans I/I: 0.27, I/D: 0.43, D/D: 30 | [48] | ||||
| Indians I: 0.575, D: 0.425 | [78] | ||||
| ACE2 | Xp22.2 | Cardiovascular risk, Retinopathy in type-2 Diabetes Mellitus, Hypertension and Hypertensive left ventricular hypertrophy | c.*1860-449C > T SNP (rs2074192) | Italians C: 0.56, T: 0.44 | [35] |
| c.*264+788T > C (rs2106809) | Italians A: 0.77, G: 0.33 | ||||
| c.2115-268A > T SNP (rs233574) | Africans C: 0.92, T: 0.08 Europeans C: 0.67, T: 0.33 East Asians C: 0.996, T: 0.004 South Asians C: 0.814, T: 0.814 Americans C: 0.767, T: 0.233 | [79] | |||
| c.1402A > G p.Ile468Val SNP (rs191860450) | East Asians with an allele frequency (AF) = 0.011 | [16] | |||
| c.1022A > G p.Lys341Arg SNP (rs138390800) | Africans AF = 4 × 10−3 | ||||
| c.2191C > T p.Leu731Phe SNP (rs147311723) | Africans AF = 0.014 | ||||
| c.631G > A p.Gly211Arg SNP (rs148771870) | Europeans AF = 2 × 10−3 South Asians AF = 1.9 × 10−3 | ||||
| c.2089A > G p.Arg697Gly SNP (rs751603885) | South Asians AF = 2.4 × 10−3 | ||||
| c.2074T > C p.Ser692Pro SNP (rs14903946) | Africans AF = 6 × 10−3 | ||||
| c.55T > C p.Ser19Pro SNP (rs73635825) | Africans AF = 3 × 10−3 | ||||
| AGT | 1q42.2 1q42–43 | Hypertension Heart failure Myocardial infraction | c.704T > C p.Met235Thr (aka Met268Thr) SNP (rs699) | Tunisians M/M: 0.291, M/T: 0.291 T/T:0.419 | [80] |
| Vietnamese T: 0.92, M: 0.08 | [81] | ||||
| Iranians T: 0.39, M: 0.61 | [82] | ||||
| Indians M: 0.52, D: 0.48 | [78] | ||||
| New Zealanders T/T: 0.19, T/M: 0.47, M/M: 0.34 | [83] | ||||
| c.521C > T p.Thr174Met SNP (rs4762) | New Zealanders T/T: 0.7, T/M: 0.2, M/M: 0.1 | [83] | |||
| AT1R | 3q21–q25 | Systolic blood pressure Left ventricular hypertrophy Hypertension Aortic stiffness Myocardial infarction Carotid intimal-medial thickening, CAD and stroke, Overweight, Diabetes | c.1166A > C in the 3′ UTR SNP (rs5186) | Egyptians C:0.24, A:0.76 (control group) C:0.34, G:0.66 (premature CAD patients) | [84] |
| Jordanians Higher frequency of A allele | [85] | ||||
| Iranians Higher frequency of A allele | [86] | ||||
| AT2R | Xq23–26 | Metabolic syndrome | −1332A > G SNP (rs1403543) | Egyptians A:0.55, G:0.45 (control group) A:0.41, G:0.50 (premature CAD patients) | [84] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Arif, G.; Farhat, A.; Khazaal, S.; Annweiler, C.; Kovacic, H.; Wu, Y.; Cao, Z.; Fajloun, Z.; Khattar, Z.A.; Sabatier, J.M. The Renin-Angiotensin System: A Key Role in SARS-CoV-2-Induced COVID-19. Molecules 2021, 26, 6945. https://doi.org/10.3390/molecules26226945
El-Arif G, Farhat A, Khazaal S, Annweiler C, Kovacic H, Wu Y, Cao Z, Fajloun Z, Khattar ZA, Sabatier JM. The Renin-Angiotensin System: A Key Role in SARS-CoV-2-Induced COVID-19. Molecules. 2021; 26(22):6945. https://doi.org/10.3390/molecules26226945
Chicago/Turabian StyleEl-Arif, George, Antonella Farhat, Shaymaa Khazaal, Cédric Annweiler, Hervé Kovacic, Yingliang Wu, Zhijian Cao, Ziad Fajloun, Ziad Abi Khattar, and Jean Marc Sabatier. 2021. "The Renin-Angiotensin System: A Key Role in SARS-CoV-2-Induced COVID-19" Molecules 26, no. 22: 6945. https://doi.org/10.3390/molecules26226945
APA StyleEl-Arif, G., Farhat, A., Khazaal, S., Annweiler, C., Kovacic, H., Wu, Y., Cao, Z., Fajloun, Z., Khattar, Z. A., & Sabatier, J. M. (2021). The Renin-Angiotensin System: A Key Role in SARS-CoV-2-Induced COVID-19. Molecules, 26(22), 6945. https://doi.org/10.3390/molecules26226945