
A Study on Synthesis and Upscaling of 2′-O-AECM-5-methyl Pyrimidine Phosphoramidites for Oligonucleotide Synthesis

 , and
, and
Abstract
:
1. Introduction
2. Results and Discussion
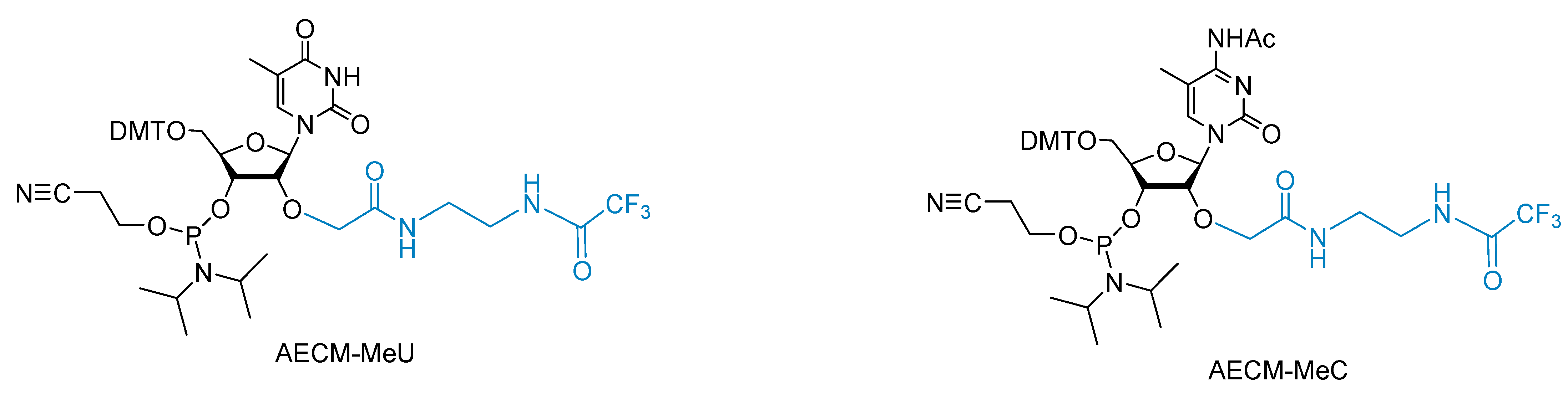
2.1. Synthesis of AECM-MeU Phosphoramidite
2.2. Synthesis of AECM-MeC Phosphoramidite
3. Materials and Methods
3.1. General Information
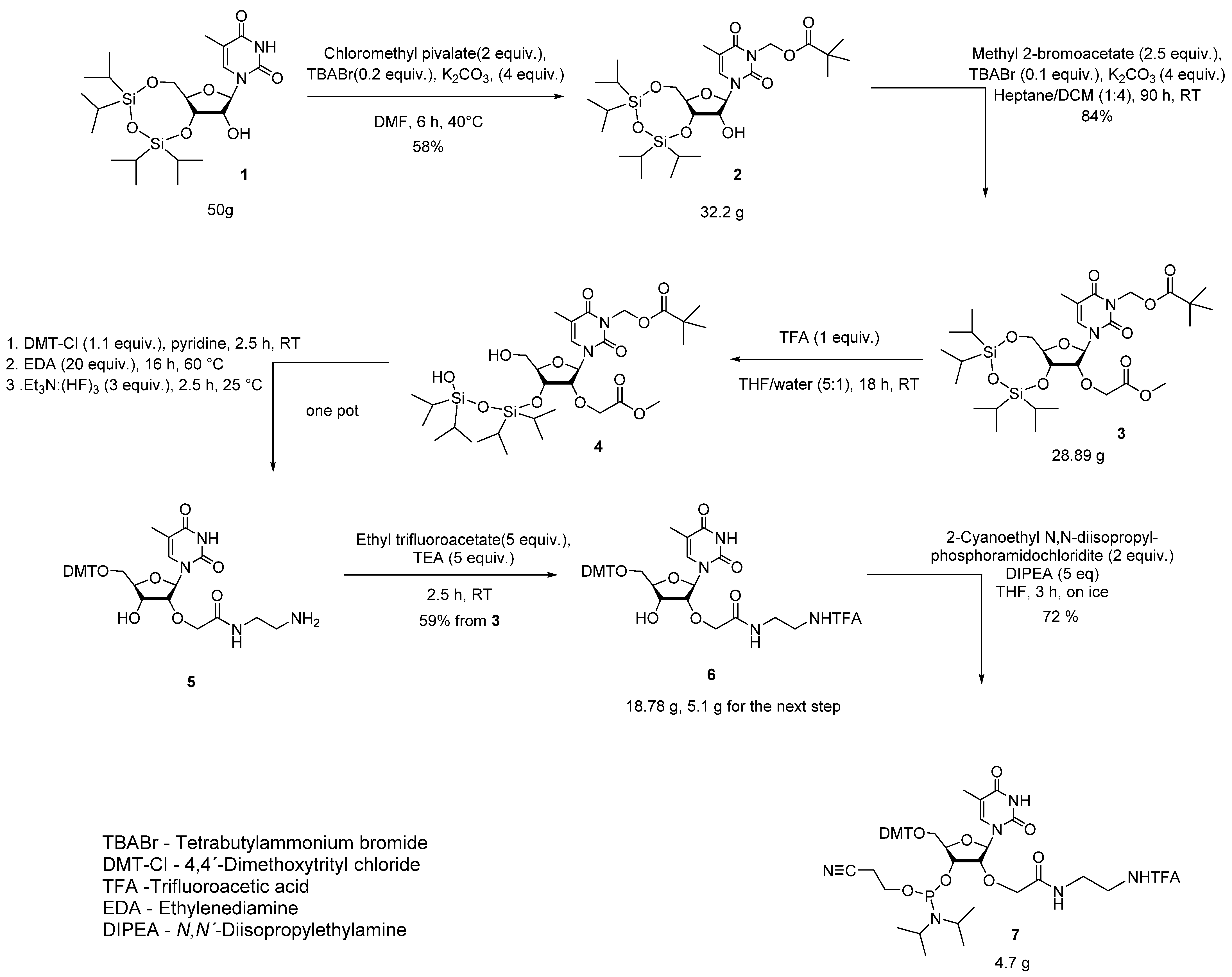
3.2. Synthesis of Compounds 2–7
- N3-Pivaloyloxymethyl-3′,5′-O-[(tetraisopropyldisiloxan-1,3-diyl)]-5-methyl-uridine (2).
- 2′-O-(O-Methylcarboxymethyl)-N3-pivaloyloxymethyl-3′,5′-O-[(1,1,3,3-tetraisopropyl-1,3-disiloxanediyl)methyl]-5-methyl-uridine (3).
- 5′-O-(4,4′-Dimethoxytrityl)-2′-O-[(N-(trifluoroacetamidoethyl)carbamoyl)methyl]-5-methyl-uridine (6).
- 3′-O-(N,N-Diisopropylamino-(2-cyanoethoxy)phosphinyl)-5′-O-(4-methoxytrityl)-2′-O-[(N-(trifluoroacetamidoethyl)carbamoyl)methyl]-5-methyl-uridine (7).
3.3. Synthesis of Compounds 9–16
- N4-Dimethylformamidono-3′,5′-O-[(1,1,3,3-tetraisopropyl-1,3-disiloxanediyl)]-5-methyl-cytidine (9).
- N4-Dimethylformamidino-2′-O-(O-methylcarboxymethyl)-3′,5′-O-[(1,1,3,3-tetraisopropyl-1,3-disiloxanediyl)]-5-methyl-cytidine (10).
- 2′-O-(N-(Ethyl)carbamoyl)methyl-3′,5′-O-[(1,1,3,3-tetraisopropyl-1,3-disiloxanediyl])-5-methyl-cytidine (11).
- 2′-O-(N-(Trifluoroacetamidoethyl)carbamoyl)methyl-3′,5′-O-[(1,1,3,3-tetraisopropyl-1,3-disiloxanediyl])-5-methyl-cytidine (12).
- N4-Acetyl-2′-O-[(N-(trifluoroacetamidoethyl)carbamoyl)methyl-3′,5′-O-[(1,1,3,3-tetraisopropyl-1,3-disiloxanediyl])-5-methyl-cytidine (13).
- N4-Acetyl-2′-O-[(N-(trifluoroacetamidoethyl)carbamoyl)methyl]-5-methyl-cytidine (14).
- N4-Acetyl-5′-O-(4,4′-dimethoxytrityl)-2′-O-[(N-(trifluoroacetamidoethyl)carbamoyl)methyl]-5-methyl-cytidine (15).
- N4-Acetyl-3′-O-(N,N-diisopropylamino-(2-cyanoethoxy)phosphinyl)-5′-O-(4-methoxytrityl)-2′-O-[(N-(trifluoroacetamidoethyl)carbamoyl)methyl]methyl-cytidine (16).
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smith, C.E.; Zain, R. Therapeutic Oligonucleotides: State of the Art. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 605–630. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.F.; Baker, B.F.; Pham, N.; Swayze, E.; Geary, R.S. Pharmacology of Antisense Drugs. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 81–105. [Google Scholar] [CrossRef] [PubMed]
- Walder, J.A.; Walder, R.Y. Nucleic Acid Hybridization and Amplification Method for Detection of Specific Sequences in Which a Complementary Labeled Nucleic Acid Probe Is Cleaved. U.S. Patent 5,403,711, 4 April 1995. [Google Scholar]
- Carthew, R.W.; Sontheimer, E.J. Origins and Mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schirle, N.T.; Kinberger, G.A.; Murray, H.F.; Lima, W.F.; Prakash, T.P.; MacRae, I.J. Structural Analysis of Human Argonaute-2 Bound to a Modified siRNA Guide. J. Am. Chem. Soc. 2016, 138, 8694–8697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Fougerolles, A.; Manoharan, M.; Meyers, R.; Vornlocher, H.-P. RNA Interference In Vivo: Toward Synthetic Small Inhibitory RNA-Based Therapeutics. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2005; Volume 392, pp. 278–296. [Google Scholar]
- Eichhorn, S.W.; Guo, H.; McGeary, S.E.; Rodriguez-Mias, R.A.; Shin, C.; Baek, D.; Hsu, S.-H.; Ghoshal, K.; Villen, J.; Bartel, D.P. mRNA Destabilization Is the Dominant Effect of Mammalian MicroRNAs by the Time Substantial Repression Ensues. Mol. Cell 2014, 56, 104–115. [Google Scholar] [CrossRef] [Green Version]
- Anderson, B.R.; Muramatsu, H.; Nallagatla, S.R.; Bevilacqua, P.C.; Sansing, L.H.; Weissman, D.; Karikó, K. Incorporation of pseudouridine into mRNA enhances translation by diminishing PKR activation. Nucleic Acids Res. 2010, 38, 5884–5892. [Google Scholar] [CrossRef] [Green Version]
- Andries, O.; Mc Cafferty, S.; De Smedt, S.C.; Weiss, R.; Sanders, N.N.; Kitada, T. N1-methylpseudouridine-incorporated mRNA outperforms pseudouridine-incorporated mRNA by providing enhanced protein expression and reduced immunogenicity in mammalian cell lines and mice. J. Control. Release 2015, 217, 337–344. [Google Scholar] [CrossRef]
- Svitkin, Y.V.; Cheng, Y.M.; Chakraborty, T.; Presnyak, V.; John, M.; Sonenberg, N. N1-methyl-pseudouridine in mRNA enhances translation through eIF2α-dependent and independent mechanisms by increasing ribosome density. Nucleic Acids Res. 2017, 45, 6023–6036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karikó, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA Recognition by Toll-like Receptors: The Impact of Nucleoside Modification and the Evolutionary Origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef] [Green Version]
- Prakash, T.P. An Overview of Sugar-Modified Oligonucleotides for Antisense Therapeutics. Chem. Biodivers. 2011, 8, 1616–1641. [Google Scholar] [CrossRef]
- Lesnik, E.A.; Guinosso, C.J.; Kawasaki, A.M.; Sasmor, H.; Zounes, M.; Cummins, L.L.; Ecker, D.J.; Cook, P.D.; Freier, S.M. Oligodeoxynucleotides containing 2’-O-modified adenosine: Synthesis and effects on stability of DNA:RNA duplexes. Biochemistry 1993, 32, 7832–7838. [Google Scholar] [CrossRef]
- Cuenoud, B.; Casset, F.; Hüsken, D.; Natt, F.; Wolf, R.M.; Altmann, K.-H.; Martin, P.; Moser, H.E. Dual Recognition of Double-Stranded DNA by 2′-Aminoethoxy-Modified Oligonucleotides. Angew. Chem. Int. Ed. 1998, 37, 1288–1291. [Google Scholar] [CrossRef]
- Buchini, S.; Leumann, C.J. 2′-O-Aminoethyl Oligoribonucleotides Containing Novel Base Analogues: Synthesis and Triple-Helix Formation At Pyrimidine/Purine Inversion Sites. Eur. J. Org. Chem. 2006, 2006, 3152–3168. [Google Scholar] [CrossRef]
- Grøtli, M.; Beijer, B.; Sproat, B. 2′-O-(carbamoylmethyl)oligoribonucleotides. Tetrahedron 1999, 55, 4299–4314. [Google Scholar] [CrossRef]
- Milton, S.; Ander, C.; Yeheskiely, E.; Strömberg, R. Stability of a 2′-O-(Carbamoylmethyl)adenosine-Containing Dinucleotide. Eur. J. Org. Chem. 2012, 2012, 539–543. [Google Scholar] [CrossRef]
- Prakash, T.P.; Kawasaki, A.M.; Wancewicz, E.V.; Shen, L.; Monia, B.P.; Ross, B.S.; Bhat, B.; Manoharan, M. Comparing In Vitro and In Vivo Activity of 2′-O-[2-(Methylamino)-2-oxoethyl]- and 2′-O-Methoxyethyl-Modified Antisense Oligonucleotides. J. Med. Chem. 2008, 51, 2766–2776. [Google Scholar] [CrossRef]
- Keller, T.H.; Häner, R. A General Method for the Synthesis of 2′-O-Modified Ribonucleosides. Helv. Chim. Acta 1993, 76, 884–892. [Google Scholar] [CrossRef]
- Pattanayek, R.; Sethaphong, L.; Pan, C.; Prhavc, M.; Prakash, T.P.; Manoharan, A.M.; Egli, M. Structural Rationalization of a Large Difference in RNA Affinity Despite a Small Difference in Chemistry between Two 2‘-O-Modified Nucleic Acid Analogues. J. Am. Chem. Soc. 2004, 126, 15006–15007. [Google Scholar] [CrossRef]
- Ozaki, H.; Nomura, S.; Momiyama, S.; Yokotsuka, K.; Kuwahara, M.; Sawai, H. Synthesis and Properties of Oligodeoxyribonucleotides Bearing a Polyamino Group at the 2′ Position via 2′-O-Carbamoylmethyl and 2′-S-Carbamoylmethyl groups. Nucleosides Nucleotides Nucleic Acids 2009, 28, 943–952. [Google Scholar] [CrossRef]
- Teplova, M.; Wallace, S.T.; Tereshko, V.; Minasov, G.; Symons, A.M.; Cook, P.D.; Manoharan, M.; Egli, M. Structural origins of the exonuclease resistance of a zwitterionic RNA. Proc. Natl. Acad. Sci. USA 1999, 96, 14240–14245. [Google Scholar] [CrossRef] [Green Version]
- Prakash, T.P.; Kawasaki, A.M.; Lesnik, E.A.; Owens, A.S.R.; Manoharan, M. 2‘-O-[2-(Amino)-2-oxoethyl] Oligonucleotides. Org. Lett. 2003, 5, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Noe, C.R.; Winkler, J.; Urban, E.; Gilbert, M.; Haberhauer, G.; Brunar, H. Zwitterionic Oligonucleotides: A Study on Binding Properties of 2′-O-Aminohexyl Modifications. Nucleosides Nucleotides Nucleic Acids 2005, 24, 1167–1185. [Google Scholar] [CrossRef]
- Milton, S.; Honcharenko, D.; Rocha, C.S.J.; Moreno, P.M.D.; Smith, C.I.E.; Strömberg, R. Nuclease resistant oligonucleotides with cell penetrating properties. Chem. Commun. 2015, 51, 4044–4047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honcharenko, D.; et al. 2′-O-Aminoethylcarbamoylmethyl (AECM) modification allows for lower phosphorothioate content in splice-switching oligonucleotides with retained activity. Nucl. Acid. Ther. submitted.
- Milton, S.; Ander, C.; Honcharenko, D.; Honcharenko, M.; Yeheskiely, E.; Strömberg, R. Synthesis and Stability of a 2′-O-[N-(Aminoethyl)carbamoyl]methyladenosine-Containing Dinucleotide. Eur. J. Org. Chem. 2013, 2013, 7184–7192. [Google Scholar] [CrossRef]
- Strömberg, R.; Honcharenko, D.; Milton, S. Cell-Penetrating Oligonucleotides. U.S. Patent 9,463,200 B2, 11 October 2016. [Google Scholar]
- Lemaire, S.; Houpis, I.; Wechselberger, R.; Langens, J.; Vermeulen, W.A.A.; Smets, N.; Nettekoven, U.; Wang, Y.; Xiao, T.; Qu, H.; et al. Practical Synthesis of (2′R)-2′-Deoxy-2′-C-methyluridine by Highly Diastereoselective Homogeneous Hydrogenation. J. Org. Chem. 2011, 76, 297–300. [Google Scholar] [CrossRef]
- Kachalova, A.; Zubin, E.; Stetsenko, D.; Gait, M.; Oretskaya, T. Oligonucleotides with 2′-O-carboxymethyl group: Synthesis and 2′-conjugation via amide bond formation on solid phase. Org. Biomol. Chem. 2004, 2, 2793–2797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appell, R.B.; Duguid, R.J. New Synthesis of a Protected Ketonucleoside by a Non-Cryogenic Oxidation with TFAA/DMSOOrg. Process. Res. Dev. 2000, 4, 172–174. [Google Scholar] [CrossRef]
- McBride, L.J.; Kierzek, R.; Beaucage, S.L.; Caruthers, M.H. Nucleotide chemistry. Amidine protecting groups for oligonucleotide synthesis. J. Am. Chem. Soc. 1986, 108, 2040–2048. [Google Scholar] [CrossRef]
- Bhat, V.; Ugarkar, B.G.; Sayeed, V.A.; Grimm, K.; Kosora, N.; Domenico, P.A.; Stocker, E. A Simple and Convenient Method for the Selective N-Acylations of Cytosine Nucleosides. Nucleosides Nucleotides 1989, 8, 179–183. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Pom-Cl, Equiv. | Solvent, 5 Vol | Base, 3 Equiv. | (PTC) Catalyst, 0.2 Equiv. | HPLC Area%, Conver., 50 °C, 1 h | HPLC Area%, Conver., 50 °C, Overnight |
|---|---|---|---|---|---|---|
| 1. | 1.5 | Heptane | KHCO3 | TBABr | Product trace | 66% |
| 2. | 1.5 | Heptane | NaHCO3 | TBABr | Product trace | Product trace |
| 3. | 1.5 | DCM | NaHCO3 | TBABr | Product trace | 6% |
| 4. | 1.5 | Toluene | KHCO3 | TBABr | Product trace | 33% |
| 5. | 1.5 | Toluene | NaHCO3 | TBABr | Product trace | 6% |
| 6. | 1.5 | DMF | KHCO3 | TBABr | 23% | 50% |
| 7. | 1.5 | DMF | NaHCO3 | TBABr | 4% | 23% |
| 8. | 1.5 | DMF | K2CO3 | TBABr | 53% | 14% * |
| 9. | 1.5 | DMF | Na2CO3 | TBABr | 6% | 54% |
| No. | Methyl 2-Bromoacetate | Solvent, 5 Vol | Base | (PTC) Catalyst | HPLC Area%, Conver., RT, 1 h | HPLC Area%, Conver., RT, Overnight |
|---|---|---|---|---|---|---|
| 1. | 1.2 equiv. | MeCN | K2CO3, 2 equiv. | MeNOct3Cl, 0.2 equiv. | 7% | 50% |
| 2. | 1.2 equiv. | MeCN | K2CO3, 2 equiv. | - | Product trace | 16% |
| 3. | 1.2 equiv. | MeCN | K2CO3, 2 equiv. | Oct4NBr, 0.2 equiv. | 8% | 54% |
| 4. | 2 equiv. | MeCN | K2CO3, 2 equiv. | Oct4NBr, 0.2 equiv. | 15% | 86% |
| 5. | 2 equiv. | Toluene | K2CO3, 2 equiv. | Oct4NBr, 0.2 equiv. | Product trace | 11% |
| 6. | 2 equiv. | Heptane | K2CO3, 2 equiv. | Oct4NBr, 0.2 equiv. | 5% | 51% |
| 7. | 2 equiv. | DCM | K2CO3, 2 equiv. | Oct4NBr, 0.2 equiv. | Product trace | 22% |
| 8. | 2 equiv. | DMF | K2CO3, 2 equiv. | Oct4NBr, 0.2 equiv. | 7% | 76% |
| 9. | 2 equiv. | MeCN | K3PO4, 2 equiv. | Oct4NBr, 0.2 equiv. | 51% | 83% |
| 10. | 2 equiv. | Toluene | K3PO4, 2 equiv. | Oct4NBr, 0.2 equiv. | 4% | 18% |
| 11. | 2 equiv. | Heptane | K3PO4, 2 equiv. | Oct4NBr, 0.2 equiv. | 9% | 50% |
| 12. | 2 equiv. | DCM | K3PO4, 2 equiv. | Oct4NBr, 0.2 equiv. | 5% | 37% |
| 13. | 2 equiv. | DMF | K3PO4, 2 equiv. | Oct4NBr, 0.2 equiv. | 9% | 32% |
| 14. | 2 equiv. | Heptane:DCM (4:1, v/v) | K2CO3, 4 equiv. | TBABr, 0.05 equiv. | Product trace | 76% (93% after 36 h) |
| No. | Methyl 2-Bromoacetate | Solvent, 5 Vol | Base | (PTC) Catalyst, 0.05 Equiv. | HPLC Area%, Conver., RT, 1 h | HPLC Area%, Conver., RT, Overnight | HPLC Area%, Conver., 50 °C, 3.5 h |
|---|---|---|---|---|---|---|---|
| 1. | 2 equiv. | Heptane | K2CO3, 4 equiv. | TBABr, 0.05 equiv. | 0% | - | 29% * |
| 2. | 2 equiv. | DCM | K2CO3, 4 equiv. | TBABr, 0.05 equiv. | 0% | 24% | 35% * |
| 3. | 2 equiv. | MeCN | K2CO3, 4 equiv. | TBABr, 0.05 equiv. | 8% | 63% | 62% * |
| 4. | 2 equiv. | Heptane | K3PO4, 2 equiv. | TBABr, 0.05 equiv. | 0% | - | 42% * |
| 5. | 2 equiv. | DCM | K3PO4, 2 equiv. | TBABr, 0.05 equiv. | 7% | 42% | 56% |
| 6. | 2 equiv. | MeCN | K3PO4, 2 equiv. | TBABr, 0.05 equiv. | 32% | 89% | 90% |
| 7. | 2 equiv. | DCM/heptane (5 vol), (1:4) (v/v) | K3PO4, 2 equiv. | TBABr, 0.02 equiv. | 0% | - | 89% |
| 8. | 2 equiv. | DCM/heptane (10 vol), (1:4) (v/v) | K3PO4, 2 equiv. | TBABr, 0.02 equiv. | 0% | - | 94% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karalė, K.; Bollmark, M.; Stulz, R.; Honcharenko, D.; Tedebark, U.; Strömberg, R. A Study on Synthesis and Upscaling of 2′-O-AECM-5-methyl Pyrimidine Phosphoramidites for Oligonucleotide Synthesis. Molecules 2021, 26, 6927. https://doi.org/10.3390/molecules26226927
Karalė K, Bollmark M, Stulz R, Honcharenko D, Tedebark U, Strömberg R. A Study on Synthesis and Upscaling of 2′-O-AECM-5-methyl Pyrimidine Phosphoramidites for Oligonucleotide Synthesis. Molecules. 2021; 26(22):6927. https://doi.org/10.3390/molecules26226927
Chicago/Turabian StyleKaralė, Kristina, Martin Bollmark, Rouven Stulz, Dmytro Honcharenko, Ulf Tedebark, and Roger Strömberg. 2021. "A Study on Synthesis and Upscaling of 2′-O-AECM-5-methyl Pyrimidine Phosphoramidites for Oligonucleotide Synthesis" Molecules 26, no. 22: 6927. https://doi.org/10.3390/molecules26226927
APA StyleKaralė, K., Bollmark, M., Stulz, R., Honcharenko, D., Tedebark, U., & Strömberg, R. (2021). A Study on Synthesis and Upscaling of 2′-O-AECM-5-methyl Pyrimidine Phosphoramidites for Oligonucleotide Synthesis. Molecules, 26(22), 6927. https://doi.org/10.3390/molecules26226927