
Assessing HDL Metabolism in Subjects with Elevated Levels of HDL Cholesterol and Coronary Artery Disease


Abstract
1. Introduction
2. Results
2.1. Demographic Data, Lipids, and Apolipoproteins
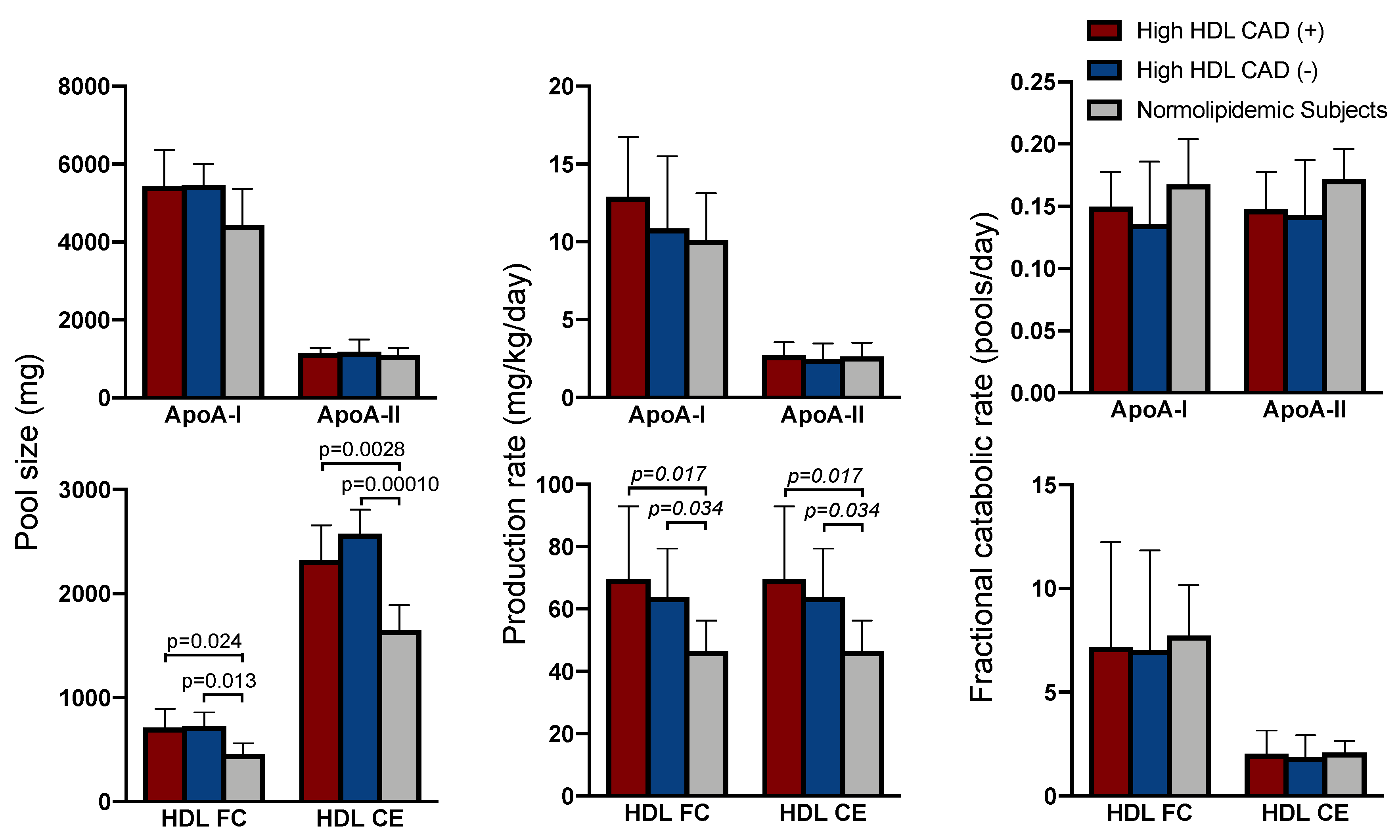
2.2. Apolipoprotein Kinetics
2.3. HDL Cholesterol Kinetics
2.4. Cholesterol Efflux
2.5. Correlations between HDL Apolipoprotein and Cholesterol Kinetics and Efflux
3. Discussion
4. Materials and Methods
4.1. Subjects
4.2. Lipid and Apolipoprotein Measurements
4.3. Kinetic Study
4.4. Cholesterol Efflux
4.5. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Gordon, D.J.; Rifkind, B.M. High-density lipoprotein--the clinical implications of recent studies. N. Engl. J. Med. 1989, 321, 1311–1316. [Google Scholar] [CrossRef]
- Goldbourt, U.; Yaari, S.; Medalie, J.H. Isolated low HDL cholesterol as a risk factor for coronary heart disease mortality. A 21-year follow-up of 8000 men. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 107–113. [Google Scholar] [CrossRef]
- Emerging Risk Factors, C.; Di Angelantonio, E.; Sarwar, N.; Perry, P.; Kaptoge, S.; Ray, K.K.; Thompson, A.; Wood, A.M.; Lewington, S.; Sattar, N.; et al. Major lipids, apolipoproteins, and risk of vascular disease. JAMA 2009, 302, 1993–2000. [Google Scholar] [CrossRef] [PubMed]
- Genest, J., Jr.; Bard, J.M.; Fruchart, J.C.; Ordovas, J.M.; Schaefer, E.J. Familial hypoalphalipoproteinemia in premature coronary artery disease. Arterioscler. Thromb. 1993, 13, 1728–1737. [Google Scholar] [CrossRef]
- Casula, M.; Colpani, O.; Xie, S.; Catapano, A.L.; Baragetti, A. HDL in Atherosclerotic Cardiovascular Disease: In Search of a Role. Cells 2021, 10, 1869. [Google Scholar] [CrossRef] [PubMed]
- Qasim, A.; Rader, D.J. Human genetics of variation in high-density lipoprotein cholesterol. Curr. Atheroscler. Rep. 2006, 8, 198–205. [Google Scholar] [CrossRef]
- Frikke-Schmidt, R.; Nordestgaard, B.G.; Stene, M.C.; Sethi, A.A.; Remaley, A.T.; Schnohr, P.; Grande, P.; Tybjaerg-Hansen, A. Association of loss-of-function mutations in the ABCA1 gene with high-density lipoprotein cholesterol levels and risk of ischemic heart disease. JAMA 2008, 299, 2524–2532. [Google Scholar] [CrossRef] [PubMed]
- Voight, B.F.; Peloso, G.M.; Orho-Melander, M.; Frikke-Schmidt, R.; Barbalic, M.; Jensen, M.K.; Hindy, G.; Holm, H.; Ding, E.L.; Johnson, T.; et al. Plasma HDL cholesterol and risk of myocardial infarction: A mendelian randomisation study. Lancet 2012, 380, 572–580. [Google Scholar] [CrossRef]
- Haase, C.L.; Tybjaerg-Hansen, A.; Qayyum, A.A.; Schou, J.; Nordestgaard, B.G.; Frikke-Schmidt, R. LCAT, HDL cholesterol and ischemic cardiovascular disease: A Mendelian randomization study of HDL cholesterol in 54,500 individuals. J. Clin. Endocrinol. Metab. 2012, 97, E248–E256. [Google Scholar] [CrossRef] [PubMed]
- Boden, W.E.; Probstfield, J.L.; Anderson, T.; Chaitman, B.R.; Desvignes-Nickens, P.; Koprowicz, K.; McBride, R.; Teo, K.; Weintraub, W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N. Engl. J. Med. 2011, 365, 2255–2267. [Google Scholar] [CrossRef] [PubMed]
- Lincoff, A.M.; Nicholls, S.J.; Riesmeyer, J.S.; Barter, P.J.; Brewer, H.B.; Fox, K.A.A.; Gibson, C.M.; Granger, C.; Menon, V.; Montalescot, G.; et al. Evacetrapib and Cardiovascular Outcomes in High-Risk Vascular Disease. N. Engl. J. Med. 2017, 376, 1933–1942. [Google Scholar] [CrossRef]
- Rosenson, R.S. Clinical trials of HDL cholesterol-raising therapy: What have we learned about the HDL hypothesis from AIM-HIGH? Curr. Atheroscler. Rep. 2012, 14, 190–192. [Google Scholar] [CrossRef] [PubMed]
- Movva, R.; Rader, D.J. Laboratory assessment of HDL heterogeneity and function. Clin. Chem. 2008, 54, 788–800. [Google Scholar] [CrossRef] [PubMed]
- Rothblat, G.H.; Phillips, M.C. High-density lipoprotein heterogeneity and function in reverse cholesterol transport. Curr. Opin. Lipidol. 2010, 21, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Rached, F.H.; Chapman, M.J.; Kontush, A. HDL particle subpopulations: Focus on biological function. Biofactors 2015, 41, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Davidson, W.S.; Cooke, A.L.; Swertfeger, D.K.; Shah, A.S. The Difference Between High Density Lipoprotein Subfractions and Subspecies: An Evolving Model in Cardiovascular Disease and Diabetes. Curr. Atheroscler. Rep. 2021, 23, 23. [Google Scholar] [CrossRef] [PubMed]
- Rader, D.J.; Hovingh, G.K. HDL and cardiovascular disease. Lancet 2014, 384, 618–625. [Google Scholar] [CrossRef]
- Rohatgi, A.; Khera, A.; Berry, J.D.; Givens, E.G.; Ayers, C.R.; Wedin, K.E.; Neeland, I.J.; Yuhanna, I.S.; Rader, D.R.; de Lemos, J.A.; et al. HDL cholesterol efflux capacity and incident cardiovascular events. N. Engl. J. Med. 2014, 371, 2383–2393. [Google Scholar] [CrossRef]
- Tangirala, R.K.; Tsukamoto, K.; Chun, S.H.; Usher, D.; Pure, E.; Rader, D.J. Regression of atherosclerosis induced by liver-directed gene transfer of apolipoprotein A-I in mice. Circulation 1999, 100, 1816–1822. [Google Scholar] [CrossRef]
- Rubin, E.M.; Krauss, R.M.; Spangler, E.A.; Verstuyft, J.G.; Clift, S.M. Inhibition of early atherogenesis in transgenic mice by human apolipoprotein AI. Nature 1991, 353, 265–267. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Tsunoda, T.; Tuzcu, E.M.; Schoenhagen, P.; Cooper, C.J.; Yasin, M.; Eaton, G.M.; Lauer, M.A.; Sheldon, W.S.; Grines, C.L.; et al. Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: A randomized controlled trial. JAMA 2003, 290, 2292–2300. [Google Scholar] [CrossRef] [PubMed]
- Kirchgessner, T.G.; Martin, R.; Sleph, P.; Grimm, D.; Liu, X.; Lupisella, J.; Smalley, J.; Narayanan, R.; Xie, Y.; Ostrowski, J.; et al. Pharmacological characterization of a novel liver X receptor agonist with partial LXRalpha activity and a favorable window in nonhuman primates. J. Pharmacol. Exp. Ther. 2015, 352, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Rader, D.J.; Alexander, E.T.; Weibel, G.L.; Billheimer, J.; Rothblat, G.H. The role of reverse cholesterol transport in animals and humans and relationship to atherosclerosis. J. Lipid Res. 2009, 50, S189–S194. [Google Scholar] [CrossRef]
- Braun, A.; Trigatti, B.L.; Post, M.J.; Sato, K.; Simons, M.; Edelberg, J.M.; Rosenberg, R.D.; Schrenzel, M.; Krieger, M. Loss of SR-BI expression leads to the early onset of occlusive atherosclerotic coronary artery disease, spontaneous myocardial infarctions, severe cardiac dysfunction, and premature death in apolipoprotein E-deficient mice. Circ. Res. 2002, 90, 270–276. [Google Scholar] [CrossRef]
- Liao, J.; Guo, X.; Wang, M.; Dong, C.; Gao, M.; Wang, H.; Kayoumu, A.; Shen, Q.; Wang, Y.; Wang, F.; et al. Scavenger Receptor Class B Type 1 Deletion Led to Coronary Atherosclerosis and Ischemic Heart Disease in Low-density Lipoprotein Receptor Knockout Mice on Modified Western-type Diet. J. Atheroscler. Thromb. 2017, 24, 133–146. [Google Scholar] [CrossRef]
- Zhang, Y.; Da Silva, J.R.; Reilly, M.; Billheimer, J.T.; Rothblat, G.H.; Rader, D.J. Hepatic expression of scavenger receptor class B type I (SR-BI) is a positive regulator of macrophage reverse cholesterol transport in vivo. J. Clin. Investig. 2005, 115, 2870–2874. [Google Scholar] [CrossRef] [PubMed]
- Zanoni, P.; Khetarpal, S.A.; Larach, D.B.; Hancock-Cerutti, W.F.; Millar, J.S.; Cuchel, M.; DerOhannessian, S.; Kontush, A.; Surendran, P.; Saleheen, D.; et al. Rare variant in scavenger receptor BI raises HDL cholesterol and increases risk of coronary heart disease. Science 2016, 351, 1166–1171. [Google Scholar] [CrossRef]
- Khera, A.V.; Cuchel, M.; de la Llera-Moya, M.; Rodrigues, A.; Burke, M.F.; Jafri, K.; French, B.C.; Phillips, J.A.; Mucksavage, M.L.; Wilensky, R.L.; et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N. Engl. J. Med. 2011, 364, 127–135. [Google Scholar] [CrossRef]
- Brownell, N.; Rohatgi, A. Modulating cholesterol efflux capacity to improve cardiovascular disease. Curr. Opin. Lipidol. 2016, 27, 398–407. [Google Scholar] [CrossRef]
- Schwartz, C.C.; VandenBroek, J.M.; Cooper, P.S. Lipoprotein cholesteryl ester production, transfer, and output in vivo in humans. J. Lipid Res. 2004, 45, 1594–1607. [Google Scholar] [CrossRef]
- Rader, D.J.; Schaefer, J.R.; Lohse, P.; Ikewaki, K.; Thomas, F.; Harris, W.A.; Zech, L.A.; Dujovne, C.A.; Brewer, H.B., Jr. Increased production of apolipoprotein A-I associated with elevated plasma levels of high-density lipoproteins, apolipoprotein A-I, and lipoprotein A-I in a patient with familial hyperalphalipoproteinemia. Metabolism 1993, 42, 1429–1434. [Google Scholar] [CrossRef]
- Ikewaki, K.; Rader, D.J.; Sakamoto, T.; Nishiwaki, M.; Wakimoto, N.; Schaefer, J.R.; Ishikawa, T.; Fairwell, T.; Zech, L.A.; Nakamura, H.; et al. Delayed catabolism of high density lipoprotein apolipoproteins A-I and A-II in human cholesteryl ester transfer protein deficiency. J. Clin. Investig. 1993, 92, 1650–1658. [Google Scholar] [CrossRef] [PubMed]
- Morton, A.M.; Koch, M.; Mendivil, C.O.; Furtado, J.D.; Tjonneland, A.; Overvad, K.; Wang, L.; Jensen, M.K.; Sacks, F.M. Apolipoproteins E and CIII interact to regulate HDL metabolism and coronary heart disease risk. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Sacks, F.M.; Jensen, M.K. From High-Density Lipoprotein Cholesterol to Measurements of Function: Prospects for the Development of Tests for High-Density Lipoprotein Functionality in Cardiovascular Disease. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.J.; Probstfield, J.L.; Garrison, R.J.; Neaton, J.D.; Castelli, W.P.; Knoke, J.D.; Jacobs, D.R., Jr.; Bangdiwala, S.; Tyroler, H.A. High-density lipoprotein cholesterol and cardiovascular disease. Four prospective American studies. Circulation 1989, 79, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Rosenson, R.S.; Brewer, H.B., Jr.; Ansell, B.J.; Barter, P.; Chapman, M.J.; Heinecke, J.W.; Kontush, A.; Tall, A.R.; Webb, N.R. Dysfunctional HDL and atherosclerotic cardiovascular disease. Nat. Rev. Cardiol. 2016, 13, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; DiDonato, J.A.; Levison, B.S.; Schmitt, D.; Li, L.; Wu, Y.; Buffa, J.; Kim, T.; Gerstenecker, G.S.; Gu, X.; et al. An abundant dysfunctional apolipoprotein A1 in human atheroma. Nat. Med. 2014, 20, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Agarwala, A.P.; Rodrigues, A.; Risman, M.; McCoy, M.; Trindade, K.; Qu, L.; Cuchel, M.; Billheimer, J.; Rader, D.J. High-Density Lipoprotein (HDL) Phospholipid Content and Cholesterol Efflux Capacity Are Reduced in Patients With Very High HDL Cholesterol and Coronary Disease. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1515–1519. [Google Scholar] [CrossRef]
- Hancock-Cerutti, W.; Lhomme, M.; Dauteuille, C.; Lecocq, S.; Chapman, M.J.; Rader, D.J.; Kontush, A.; Cuchel, M. Paradoxical coronary artery disease in humans with hyperalphalipoproteinemia is associated with distinct differences in the high-density lipoprotein phosphosphingolipidome. J. Clin. Lipidol. 2017, 11, 1192–1200. [Google Scholar] [CrossRef] [PubMed]
- Mutharasan, R.K.; Thaxton, C.S.; Berry, J.; Daviglus, M.L.; Yuan, C.; Sun, J.; Ayers, C.; Lloyd-Jones, D.M.; Wilkins, J.T. HDL efflux capacity, HDL particle size, and high-risk carotid atherosclerosis in a cohort of asymptomatic older adults: The Chicago Healthy Aging Study. J. Lipid Res. 2017, 58, 600–606. [Google Scholar] [CrossRef]
- Seckler, H.D.S.; Fornelli, L.; Mutharasan, R.K.; Thaxton, C.S.; Fellers, R.; Daviglus, M.; Sniderman, A.; Rader, D.; Kelleher, N.L.; Lloyd-Jones, D.M.; et al. A Targeted, Differential Top-Down Proteomic Methodology for Comparison of ApoA-I Proteoforms in Individuals with High and Low HDL Efflux Capacity. J. Proteome Res. 2018, 17, 2156–2164. [Google Scholar] [CrossRef]
- Chan, D.C.; Watts, G.F. Pharmacological regulation of dyslipoproteinaemia in insulin resistant states. Curr. Vasc. Pharm. 2008, 6, 67–77. [Google Scholar] [CrossRef]
- Millar, J.S.; Reyes-Soffer, G.; Jumes, P.; Dunbar, R.L.; deGoma, E.M.; Baer, A.L.; Karmally, W.; Donovan, D.S.; Rafeek, H.; Pollan, L.; et al. Anacetrapib lowers LDL by increasing ApoB clearance in mildly hypercholesterolemic subjects. J. Clin. Investig. 2016, 126, 1603–1604. [Google Scholar] [CrossRef] [PubMed]
- National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 2002, 106, 3143–3421. [Google Scholar] [CrossRef]
- Millar, J.S.; Duffy, D.; Gadi, R.; Bloedon, L.T.; Dunbar, R.L.; Wolfe, M.L.; Movva, R.; Shah, A.; Fuki, I.V.; McCoy, M.; et al. Potent and selective PPAR-alpha agonist LY518674 upregulates both ApoA-I production and catabolism in human subjects with the metabolic syndrome. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 140–146. [Google Scholar] [CrossRef]
- Ouguerram, K.; Krempf, M.; Maugeais, C.; Maugere, P.; Darmaun, D.; Magot, T. A new labeling approach using stable isotopes to study in vivo plasma cholesterol metabolism in humans. Metabolism 2002, 51, 5–11. [Google Scholar] [CrossRef]
- Cohn, J.S.; Wagner, D.A.; Cohn, S.D.; Millar, J.S.; Schaefer, E.J. Measurement of very low density and low density lipoprotein apolipoprotein (Apo) B-100 and high density lipoprotein Apo A-I production in human subjects using deuterated leucine. Effect of fasting and feeding. J. Clin. Investig. 1990, 85, 804–811. [Google Scholar] [CrossRef] [PubMed]
- Kharroubi, A.T.; Masterson, T.M.; Aldaghlas, T.A.; Kennedy, K.A.; Kelleher, J.K. Isotopomer spectral analysis of triglyceride fatty acid synthesis in 3T3-L1 cells. Am. J. Physiol. 1992, 263, E667–E675. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| High HDL CAD (+) | High HDL CAD (−) | Normolipidemic Reference | |
|---|---|---|---|
| Age (years) | 58 ± 8 ** | 65 ± 5 ** | 38 ± 14 |
| Sex (M/F) | 2/4 | 2/4 | 2/4 |
| BMI | 23.7 ± 2.0 | 25.7 ± 3.4 | 25.2 ± 2.7 |
| Total Cholesterol | 230 ± 46 | 236 ± 41 | 198 ± 47 |
| Triglyceride | 75 ± 18 | 70 ± 16 | 103 ± 51 |
| HDL-C | 108 ± 27 * | 110 ± 29 * | 64 ± 11 |
| LDL-C | 107 ± 39 | 107 ± 18 | 105 ± 31 |
| apoA-I | 230 ± 49 | 245 ± 46 * | 171 ± 35 |
| apoA-II | 46 ± 8 | 43 ± 7 | 40 ± 11 |
| apoB | 82 ± 25 | 81 ± 13 | 76 ± 25 |
| High HDLCAD (+) | High HDL CAD (−) | Normolipidemic Reference | |
|---|---|---|---|
| VLDL ApoB100 PS (mg) | 194 (117–283) | 401 (135–562) | 288 (255–396) |
| VLDL ApoB100 PR (mg/kg per day) | 17.3 (11.5–18.2) ** | 13.1 (7.85–24.2) * | 28.3 (23.4–35.0) |
| VLDL ApoB100 FCR (pools/day) | (5.44 ± 2.24) | 4.03 ± 2.20 | 6.32 ± 2.93 |
| High HDL CAD (+) | High HDL CAD (−) | Normolipidemic Reference | |
|---|---|---|---|
| % Total Efflux | 6.35 ± 2.19 | 6.39 ± 0.78 | 5.47 ± 0.96 |
| % Non-ABCA1-Mediated Efflux | 4.85 ± 1.93 | 5.29 ± 1.03 | 3.88 ± 0.87 |
| % ABCA1-mediated Efflux | 1.50 ± 0.43 | 1.10 ± 0.32 | 1.59 ± 0.82 |
| Total Efflux | Non-ABCA1 Mediated Efflux | ABCA1 Mediated Efflux | |
|---|---|---|---|
| apoA-I PS | 0.393 | 0.503 * | −0.052 |
| apoA-I PR | 0.348 | 0.552 * | −0.113 |
| apoA-I FCR | −0.197 | −0.057 | 0.037 |
| HDL FC PS | 0.836 *** | 0.939 *** | −0.244 |
| HDL FC PR | −0.056 | 0.157 | −0.238 |
| HDL FC FCR | −0.839 *** | −0.759 *** | 0.146 |
| HDL CE PS | 0.726 ** | 0.796 *** | −0.358 |
| HDL CE PR | −0.056 | 0.157 | −0.238 |
| HDL CE FCR | −0.779 *** | −0.652 ** | 0.112 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hancock-Cerutti, W.; Millar, J.S.; Valentini, S.; Liu, J.; Billheimer, J.T.; Rader, D.J.; Cuchel, M. Assessing HDL Metabolism in Subjects with Elevated Levels of HDL Cholesterol and Coronary Artery Disease. Molecules 2021, 26, 6862. https://doi.org/10.3390/molecules26226862
Hancock-Cerutti W, Millar JS, Valentini S, Liu J, Billheimer JT, Rader DJ, Cuchel M. Assessing HDL Metabolism in Subjects with Elevated Levels of HDL Cholesterol and Coronary Artery Disease. Molecules. 2021; 26(22):6862. https://doi.org/10.3390/molecules26226862
Chicago/Turabian StyleHancock-Cerutti, William, John S. Millar, Silvia Valentini, Jason Liu, Jeffrey T. Billheimer, Daniel J. Rader, and Marina Cuchel. 2021. "Assessing HDL Metabolism in Subjects with Elevated Levels of HDL Cholesterol and Coronary Artery Disease" Molecules 26, no. 22: 6862. https://doi.org/10.3390/molecules26226862
APA StyleHancock-Cerutti, W., Millar, J. S., Valentini, S., Liu, J., Billheimer, J. T., Rader, D. J., & Cuchel, M. (2021). Assessing HDL Metabolism in Subjects with Elevated Levels of HDL Cholesterol and Coronary Artery Disease. Molecules, 26(22), 6862. https://doi.org/10.3390/molecules26226862