
Lack of Neuroprotective Effects of High-Density Lipoprotein Therapy in Stroke under Acute Hyperglycemic Conditions

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
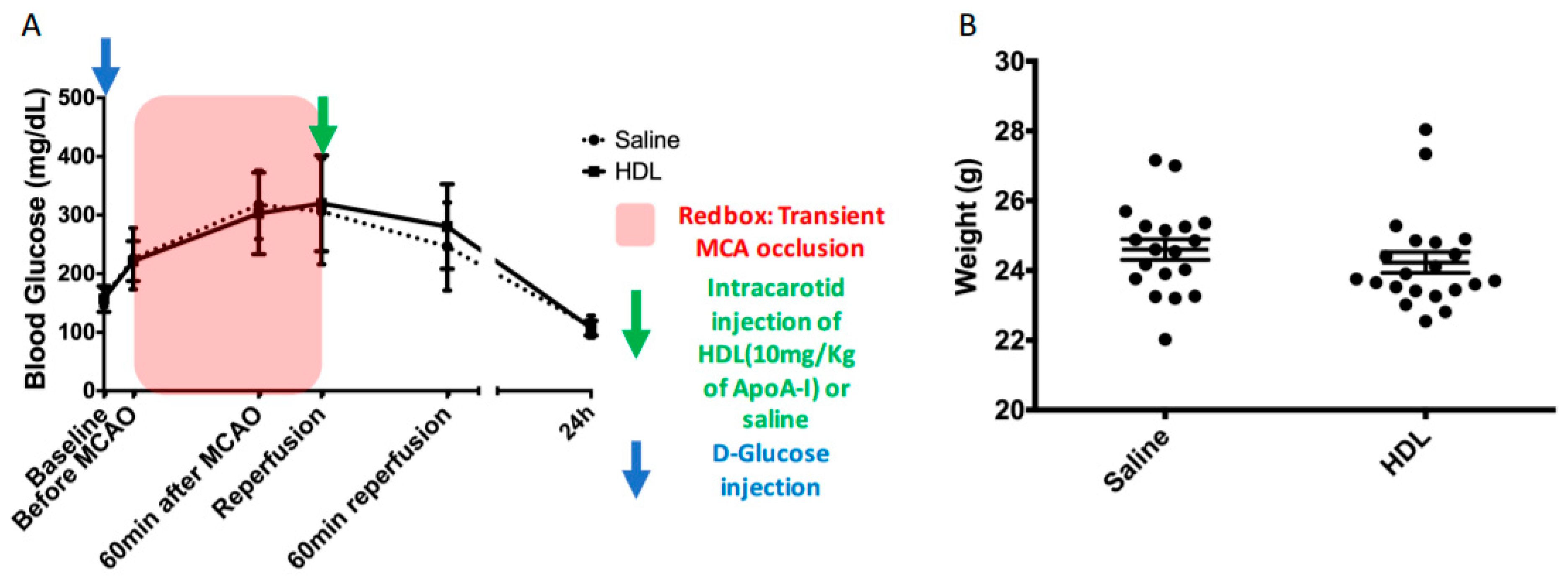
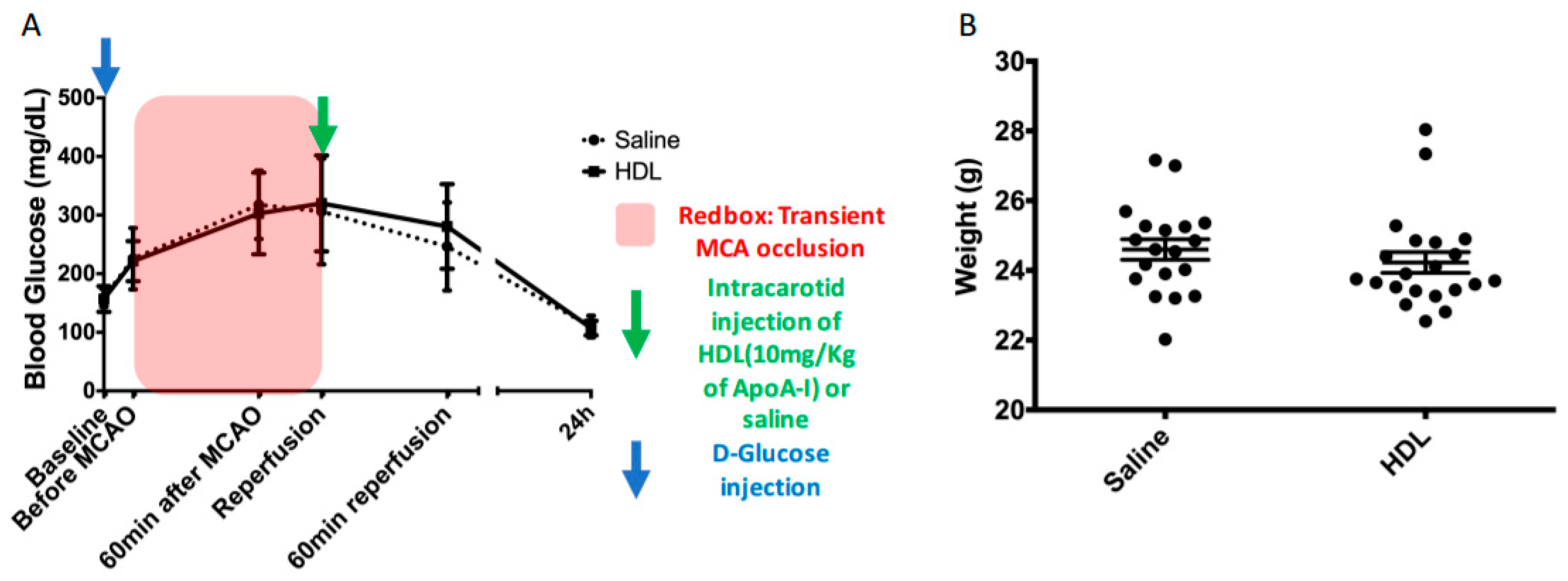
2.1. Acute Hyperglycemia
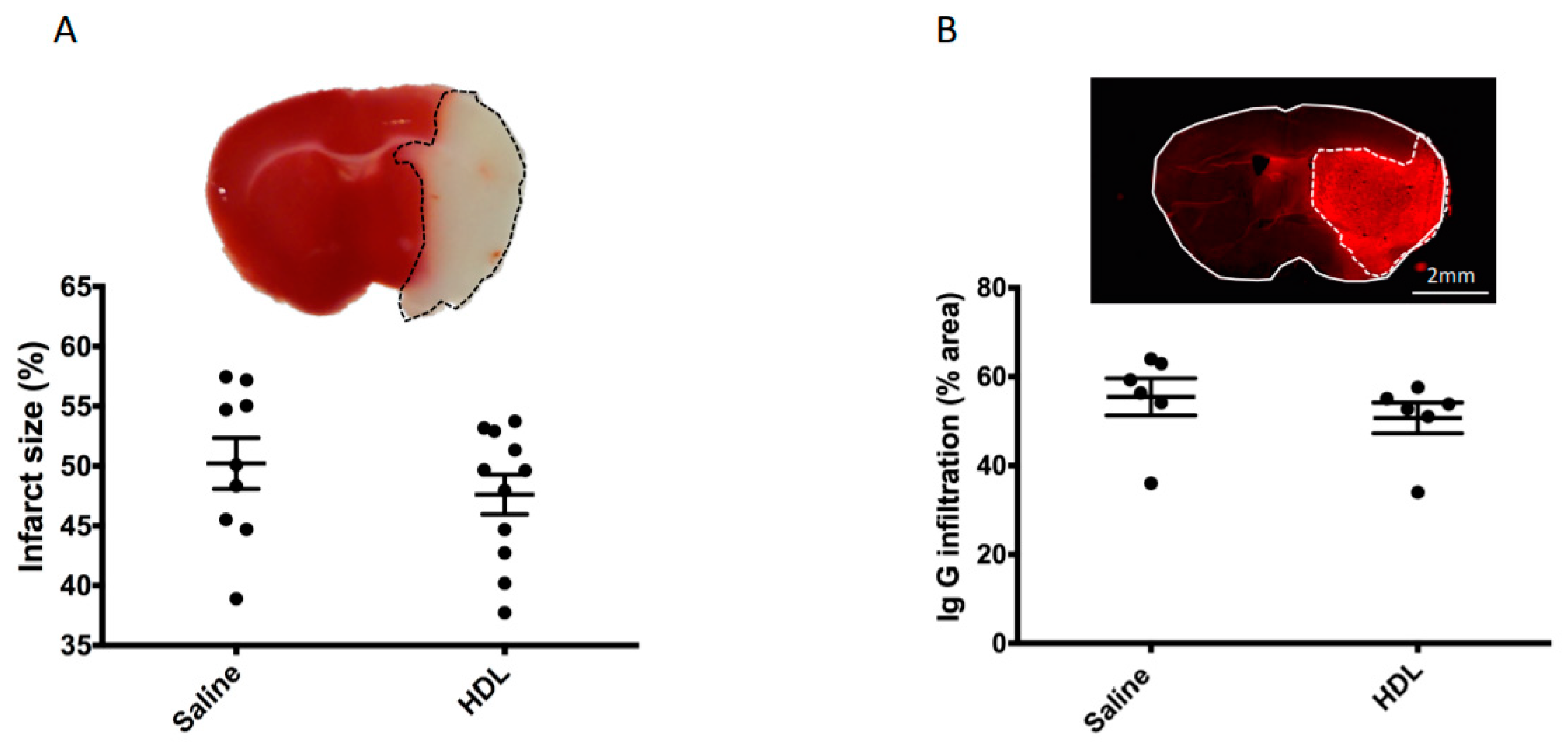
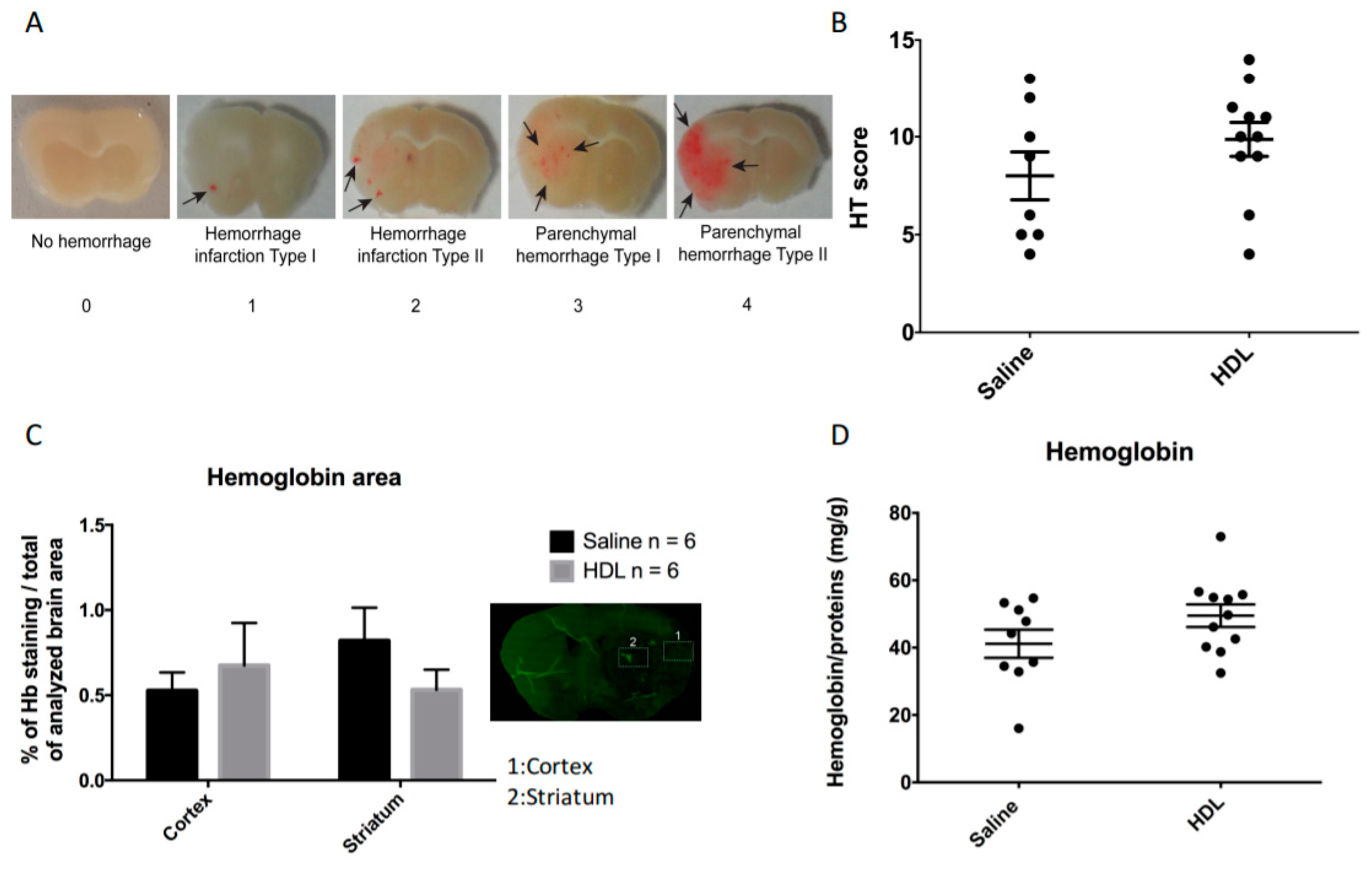
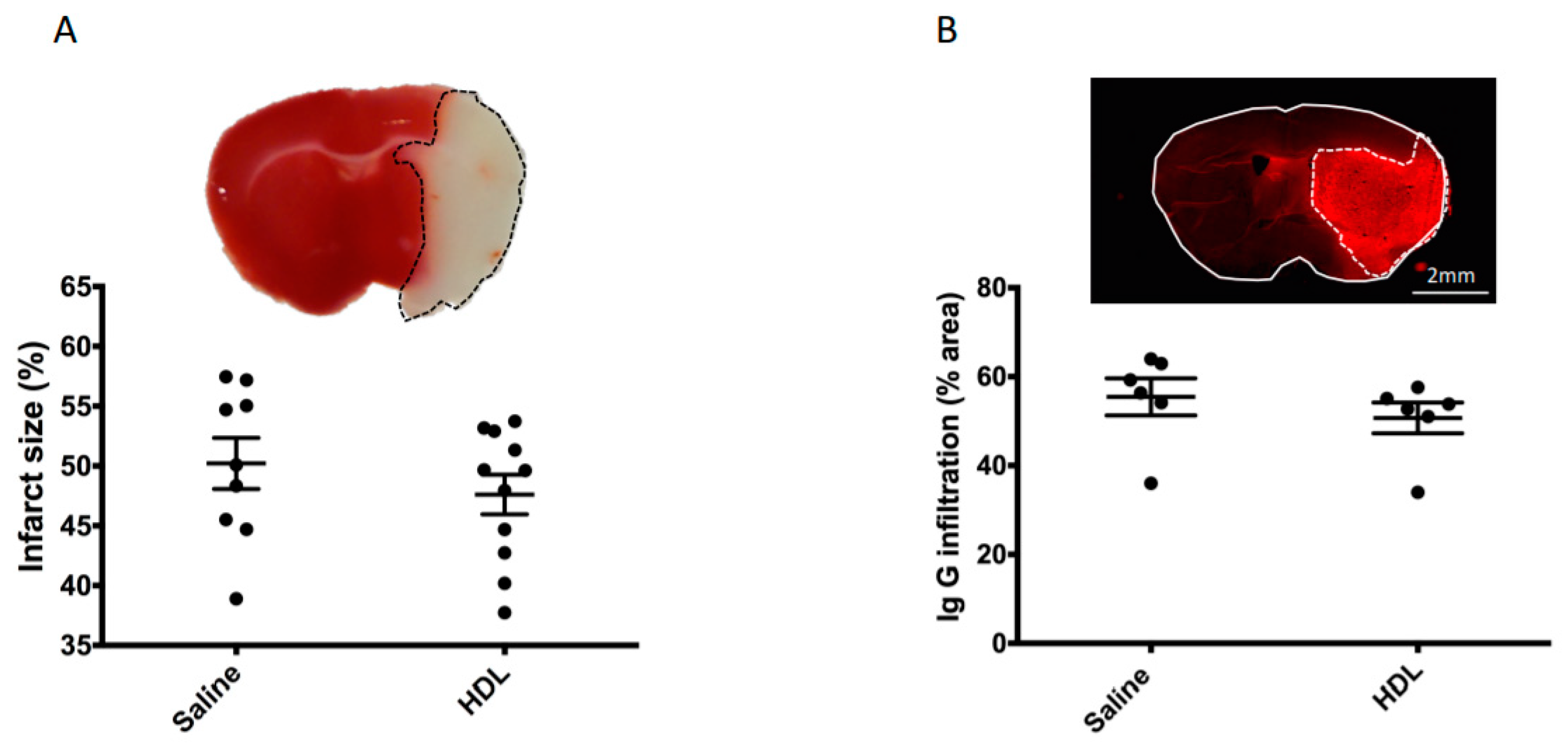
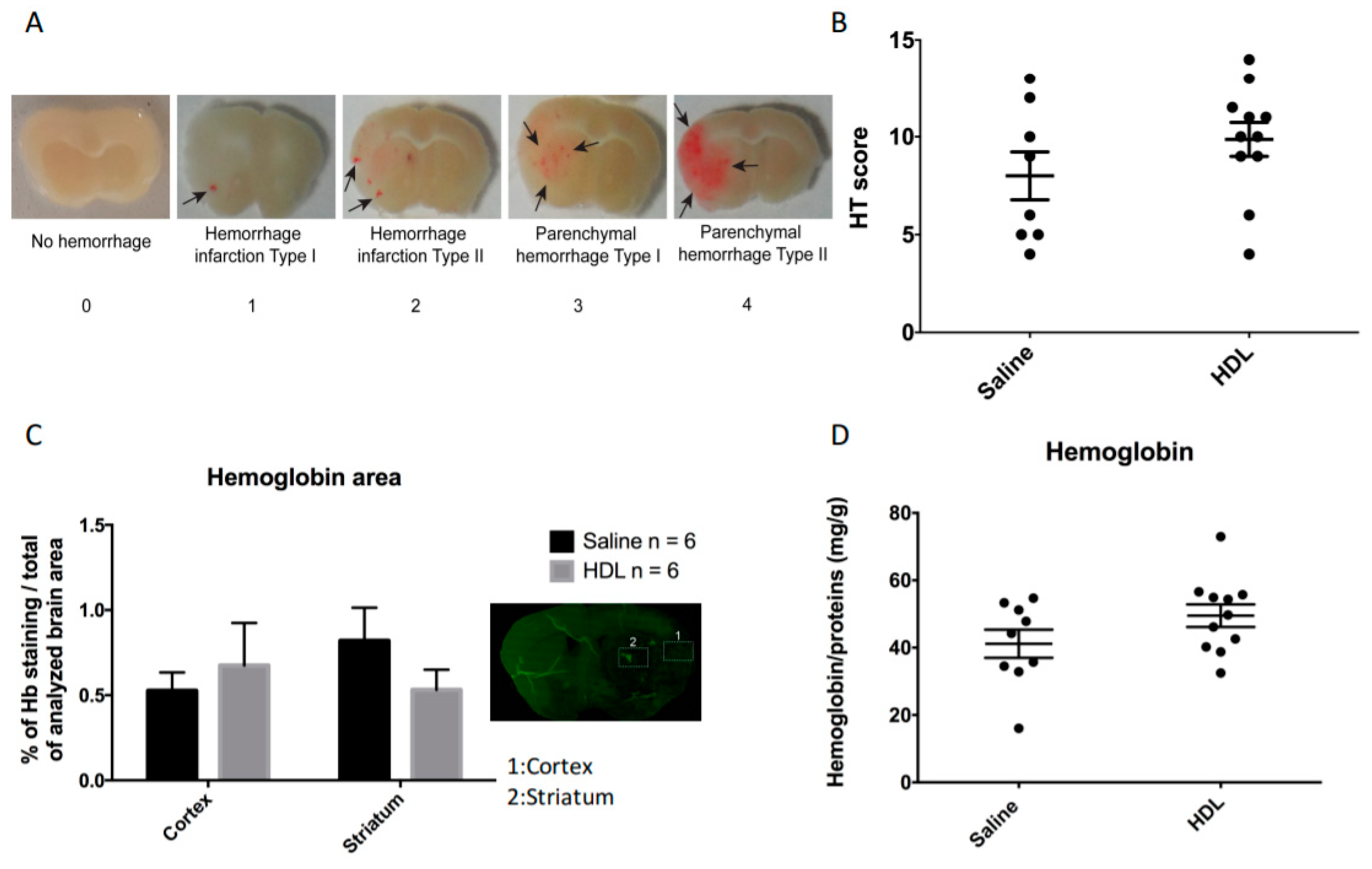
2.2. Mortality, Infarct Size, Hemorrhagic Transformation and BBB Leakage
2.3. Neurological Evolution
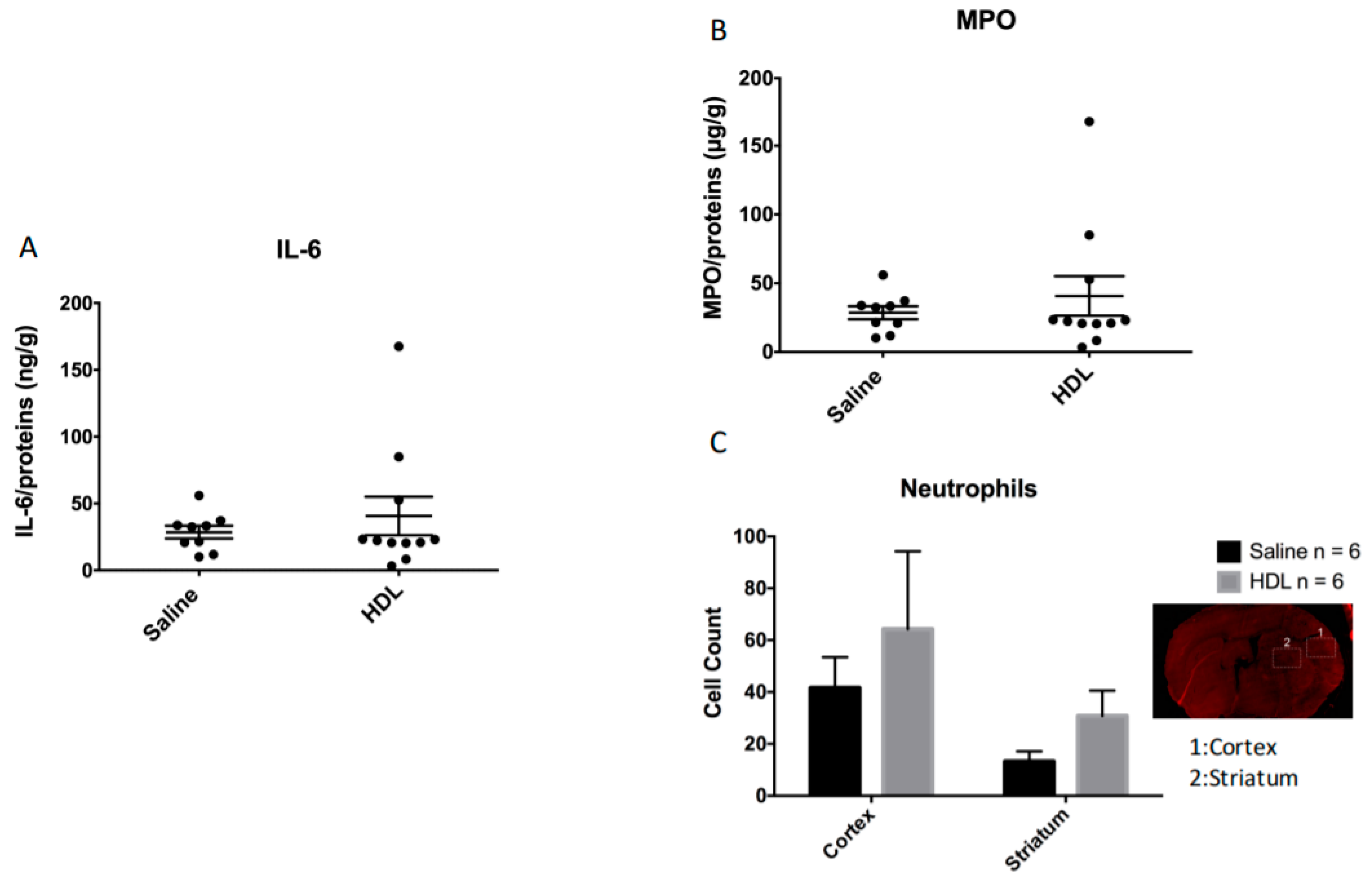
2.4. Polymorphonulcear Cell Infiltration, MPO and IL-6 Quantification
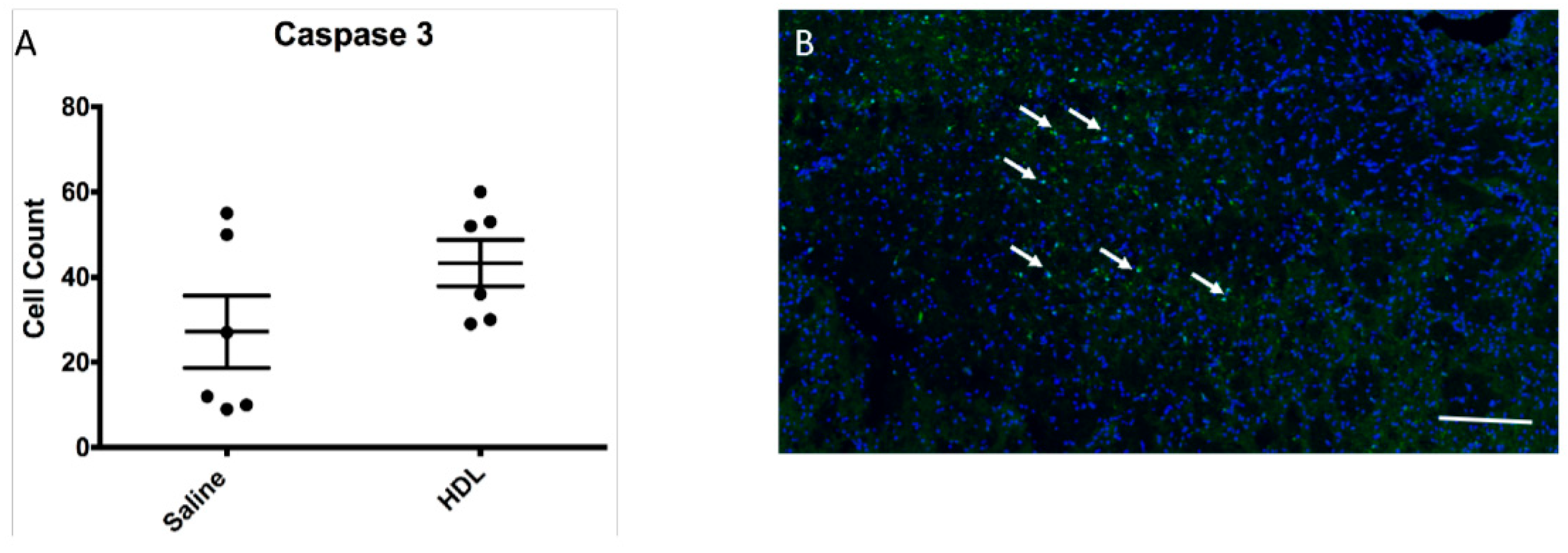
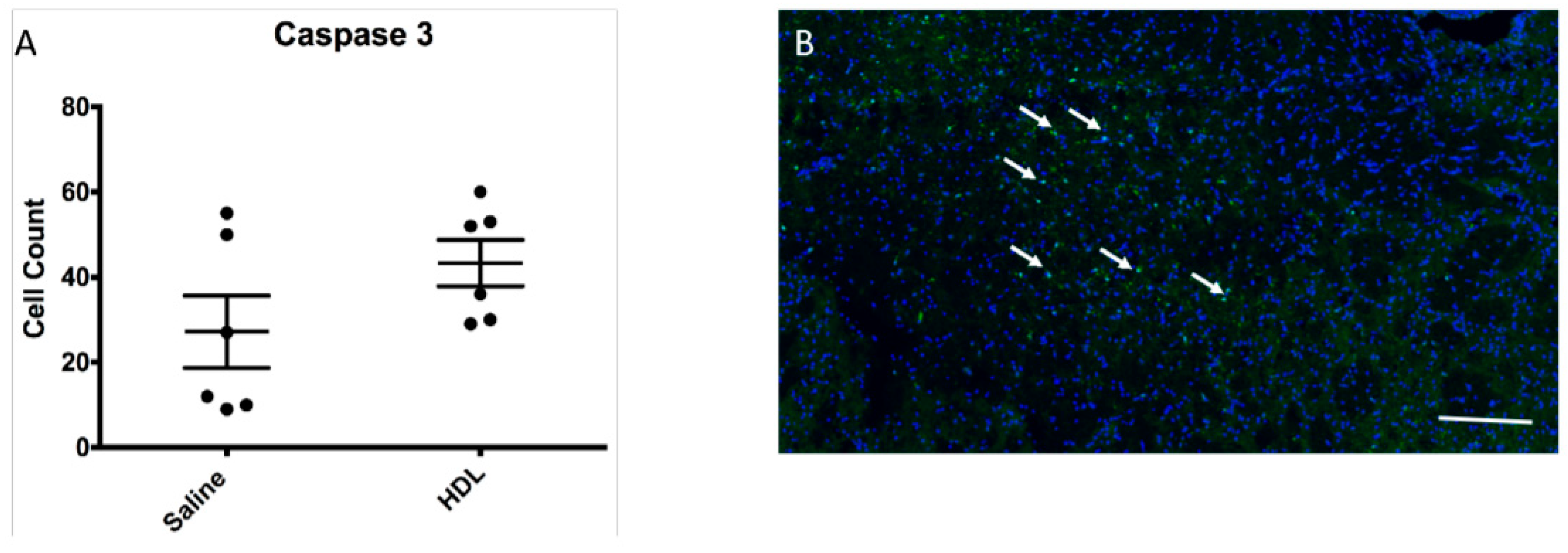
2.5. Apoptosis Activity
3. Discussion
4. Materials and Methods
4.1. Acute Hyperglycemia
4.2. Isolation of High-Density Lipoproteins
4.3. Mouse Model of Focal Cerebral Ischemia and Experimental Protocol
4.4. Infarct Size and Hemorrhagic Transformation Score Evaluation
4.4.1. Infarct Volume Assessment
4.4.2. Hemorrhagic Transformation Score Assessment
4.5. Neurological Evaluation and Mortality
4.6. Immunofluorescence Assay
4.6.1. Histology
4.6.2. Hemoglobin, Neutrophil Detection, IgG and Caspase-3 Detection
4.7. Enzyme-Linked Immunosorbent Assay for Hemoglobin in the Brain
4.8. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
Abbreviations
| AIS | acute ischemic stroke |
| BBB | Blood–brain barrier |
| HDL | High Density Lipoprotein |
| HG | Hyperglycemia |
| HT | Hemorrhagic Transformation |
| IL-6 | Interleukin 6 |
| MCAO | Middle Cerebral Artery Occlusion |
| MPO | Myeloperoxidase |
| PMN | Polymorphonuclear |
| TTC | Triphenyltetrazolium chloride. |
References
- Moller, D.E.; Kaufman, K.D. Metabolic syndrome: A clinical and molecular perspective. Annu. Rev. Med. 2005, 56, 45–62. [Google Scholar] [CrossRef]
- Hossain, P.; Kawar, B.; El Nahas, M. Obesity and diabetes in the developing world--a growing challenge. N. Engl. J. Med. 2007, 356, 213–215. [Google Scholar] [CrossRef] [Green Version]
- Boehme, A.K.; Esenwa, C.; Elkind, M.S. Stroke Risk Factors, Genetics, and Prevention. Circ. Res. 2017, 120, 472–495. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; de Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation 2017, 135, e146–e603. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, L.B.; Adams, R.; Alberts, M.J.; Appel, L.J.; Brass, L.M.; Bushnell, C.D.; Culebras, A.; Degraba, T.J.; Gorelick, P.B.; Guyton, J.R.; et al. Primary prevention of ischemic stroke: A guideline from the American Heart Association/American Stroke Association Stroke Council: Cosponsored by the Atherosclerotic Peripheral Vascular Disease Interdisciplinary Working Group; Cardiovascular Nursing Council; Clinical Cardiology Council; Nutrition, Physical Activity, and Metabolism Council; and the Quality of Care and Outcomes Research Interdisciplinary Working Group: The American Academy of Neurology affirms the value of this guideline. Stroke 2006, 37, 1583–1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hacke, W.; Kaste, M.; Bluhmki, E.; Brozman, M.; Davalos, A.; Guidetti, D.; Larrue, V.; Lees, K.R.; Medeghri, Z.; Machnig, T.; et al. Thrombolysis with alteplase 3 to 4.5 h after acute ischemic stroke. N. Engl. J. Med. 2008, 359, 1317–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jauch, E.C.; Saver, J.L.; Adams, H.P., Jr.; Bruno, A.; Connors, J.J.; Demaerschalk, B.M.; Khatri, P.; McMullan, P.W., Jr.; Qureshi, A.I.; Rosenfield, K.; et al. Guidelines for the early management of patients with acute ischemic stroke: A guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2013, 44, 870–947. [Google Scholar] [CrossRef] [PubMed]
- Jovin, T.G.; Chamorro, A.; Cobo, E.; de Miquel, M.A.; Molina, C.A.; Rovira, A.; San Roman, L.; Serena, J.; Abilleira, S.; Ribo, M.; et al. Thrombectomy within 8 h after symptom onset in ischemic stroke. N. Engl. J. Med. 2015, 372, 2296–2306. [Google Scholar] [CrossRef] [Green Version]
- Capes, S.E.; Hunt, D.; Malmberg, K.; Pathak, P.; Gerstein, H.C. Stress hyperglycemia and prognosis of stroke in nondiabetic and diabetic patients: A systematic overview. Stroke J. Cereb. Circ. 2001, 32, 2426–2432. [Google Scholar] [CrossRef] [Green Version]
- Baird, T.A.; Parsons, M.W.; Phan, T.; Butcher, K.S.; Desmond, P.M.; Tress, B.M.; Colman, P.G.; Chambers, B.R.; Davis, S.M. Persistent poststroke hyperglycemia is independently associated with infarct expansion and worse clinical outcome. Stroke 2003, 34, 2208–2214. [Google Scholar] [CrossRef] [Green Version]
- Shimoyama, T.; Kimura, K.; Uemura, J.; Saji, N.; Shibazaki, K. Elevated glucose level adversely affects infarct volume growth and neurological deterioration in non-diabetic stroke patients, but not diabetic stroke patients. Eur. J. Neurol 2014, 21, 402–410. [Google Scholar] [CrossRef]
- Masrur, S.; Cox, M.; Bhatt, D.L.; Smith, E.E.; Ellrodt, G.; Fonarow, G.C.; Schwamm, L. Association of Acute and Chronic Hyperglycemia With Acute Ischemic Stroke Outcomes Post-Thrombolysis: Findings From Get With The Guidelines-Stroke. J. Am. Heart Assoc. 2015, 4, e002193. [Google Scholar] [CrossRef] [Green Version]
- Williams, L.S.; Rotich, J.; Qi, R.; Fineberg, N.; Espay, A.; Bruno, A.; Fineberg, S.E.; Tierney, W.R. Effects of admission hyperglycemia on mortality and costs in acute ischemic stroke. Neurology 2002, 59, 67–71. [Google Scholar] [CrossRef]
- MacDougall, N.J.; Muir, K.W. Hyperglycaemia and infarct size in animal models of middle cerebral artery occlusion: Systematic review and meta-analysis. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2011, 31, 807–818. [Google Scholar] [CrossRef] [Green Version]
- Powers, W.J.; Rabinstein, A.A.; Ackerson, T.; Adeoye, O.M.; Bambakidis, N.C.; Becker, K.; Biller, J.; Brown, M.; Demaerschalk, B.M.; Hoh, B.; et al. 2018 Guidelines for the Early Management of Patients With Acute Ischemic Stroke: A Guideline for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke 2018, 49, e46–e110. [Google Scholar] [CrossRef]
- Johnston, K.C.; Bruno, A.; Pauls, Q.; Hall, C.E.; Barrett, K.M.; Barsan, W.; Fansler, A.; Van de Bruinhorst, K.; Janis, S.; Durkalski-Mauldin, V.L.; et al. Intensive vs. Standard Treatment of Hyperglycemia and Functional Outcome in Patients With Acute Ischemic Stroke: The SHINE Randomized Clinical Trial. JAMA 2019, 322, 326–335. [Google Scholar] [CrossRef]
- Besler, C.; Luscher, T.F.; Landmesser, U. Molecular mechanisms of vascular effects of High-density lipoprotein: Alterations in cardiovascular disease. EMBO Mol. Med. 2012, 4, 251–268. [Google Scholar] [CrossRef] [Green Version]
- Navab, M.; Hama, S.Y.; Hough, G.P.; Subbanagounder, G.; Reddy, S.T.; Fogelman, A.M. A cell-free assay for detecting HDL that is dysfunctional in preventing the formation of or inactivating oxidized phospholipids. J. Lipid Res. 2001, 42, 1308–1317. [Google Scholar] [CrossRef]
- Ortiz-Munoz, G.; Couret, D.; Lapergue, B.; Bruckert, E.; Meseguer, E.; Amarenco, P.; Meilhac, O. Dysfunctional HDL in acute stroke. Atherosclerosis 2016, 253, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Tran-Dinh, A.; Levoye, A.; Couret, D.; Galle-Treger, L.; Moreau, M.; Delbosc, S.; Hoteit, C.; Montravers, P.; Amarenco, P.; Huby, T.; et al. High-Density Lipoprotein Therapy in Stroke: Evaluation of Endothelial SR-BI-Dependent Neuroprotective Effects. Int. J. Mol. Sci. 2020, 22, 106. [Google Scholar] [CrossRef] [PubMed]
- Menzies, S.A.; Hoff, J.T.; Betz, A.L. Middle cerebral artery occlusion in rats: A neurological and pathological evaluation of a reproducible model. Neurosurgery 1992, 31, 100–106, discussion 106–107. [Google Scholar] [CrossRef]
- Lapergue, B.; Dang, B.Q.; Desilles, J.P.; Ortiz-Munoz, G.; Delbosc, S.; Loyau, S.; Louedec, L.; Couraud, P.O.; Mazighi, M.; Michel, J.B.; et al. High-density lipoprotein-based therapy reduces the hemorrhagic complications associated with tissue plasminogen activator treatment in experimental stroke. Stroke J. Cereb. Circ. 2013, 44, 699–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapergue, B.; Moreno, J.A.; Dang, B.Q.; Coutard, M.; Delbosc, S.; Raphaeli, G.; Auge, N.; Klein, I.; Mazighi, M.; Michel, J.B.; et al. Protective effect of high-density lipoprotein-based therapy in a model of embolic stroke. Stroke J. Cereb. Circ. 2010, 41, 1536–1542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couret, D.; Bourane, S.; Catan, A.; Nativel, B.; Planesse, C.; Dorsemans, A.C.; Ait-Arsa, I.; Cournot, M.; Rondeau, P.; Patche, J.; et al. A hemorrhagic transformation model of mechanical stroke therapy with acute hyperglycemia in mice. J. Comp. Neurol 2018, 526, 1006–1016. [Google Scholar] [CrossRef] [PubMed]
- Ribo, M.; Molina, C.; Montaner, J.; Rubiera, M.; Delgado-Mederos, R.; Arenillas, J.F.; Quintana, M.; Alvarez-Sabin, J. Acute hyperglycemia state is associated with lower tPA-induced recanalization rates in stroke patients. Stroke 2005, 36, 1705–1709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.T.; Jahan, R.; Saver, J.L.; Investigators, S. Impact of Glucose on Outcomes in Patients Treated With Mechanical Thrombectomy: A Post Hoc Analysis of the Solitaire Flow Restoration With the Intention for Thrombectomy Study. Stroke 2016, 47, 120–127. [Google Scholar] [CrossRef] [Green Version]
- Elgebaly, M.M.; Ogbi, S.; Li, W.; Mezzetti, E.M.; Prakash, R.; Johnson, M.H.; Bruno, A.; Fagan, S.C.; Ergul, A. Neurovascular injury in acute hyperglycemia and diabetes: A comparative analysis in experimental stroke. Transl. Stroke Res. 2011, 2, 391–398. [Google Scholar] [CrossRef] [Green Version]
- Hafez, S.; Coucha, M.; Bruno, A.; Fagan, S.C.; Ergul, A. Hyperglycemia, acute ischemic stroke, and thrombolytic therapy. Transl. Stroke Res. 2014, 5, 442–453. [Google Scholar] [CrossRef] [Green Version]
- Jickling, G.C.; Liu, D.; Stamova, B.; Ander, B.P.; Zhan, X.; Lu, A.; Sharp, F.R. Hemorrhagic transformation after ischemic stroke in animals and humans. J. Cereb Blood Flow Metab. 2014, 34, 185–199. [Google Scholar] [CrossRef] [Green Version]
- Demchuk, A.M.; Morgenstern, L.B.; Krieger, D.W.; Linda Chi, T.; Hu, W.; Wein, T.H.; Hardy, R.J.; Grotta, J.C.; Buchan, A.M. Serum glucose level and diabetes predict tissue plasminogen activator-related intracerebral hemorrhage in acute ischemic stroke. Stroke J. Cereb. Circ. 1999, 30, 34–39. [Google Scholar] [CrossRef] [Green Version]
- Tanne, D.; Kasner, S.E.; Demchuk, A.M.; Koren-Morag, N.; Hanson, S.; Grond, M.; Levine, S.R. Markers of increased risk of intracerebral hemorrhage after intravenous recombinant tissue plasminogen activator therapy for acute ischemic stroke in clinical practice: The Multicenter rt-PA Stroke Survey. Circulation 2002, 105, 1679–1685. [Google Scholar] [CrossRef] [PubMed]
- Grisotto, C.; Taile, J.; Planesse, C.; Diotel, N.; Gonthier, M.P.; Meilhac, O.; Couret, D. High-Fat Diet Aggravates Cerebral Infarct, Hemorrhagic Transformation and Neuroinflammation in a Mouse Stroke Model. Int. J. Mol. Sci 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Azcutia, V.; Abu-Taha, M.; Romacho, T.; Vazquez-Bella, M.; Matesanz, N.; Luscinskas, F.W.; Rodriguez-Manas, L.; Sanz, M.J.; Sanchez-Ferrer, C.F.; Peiro, C. Inflammation determines the pro-adhesive properties of high extracellular d-glucose in human endothelial cells in vitro and rat microvessels in vivo. PLoS ONE 2010, 5, e10091. [Google Scholar] [CrossRef] [PubMed]
- Tran-Dinh, A.; Diallo, D.; Delbosc, S.; Varela-Perez, L.M.; Dang, Q.B.; Lapergue, B.; Burillo, E.; Michel, J.B.; Levoye, A.; Martin-Ventura, J.L.; et al. HDL and endothelial protection. Br. J. Pharmacol. 2013, 169, 493–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, R.A.K. Dysfunctional HDL in diabetes mellitus and its role in the pathogenesis of cardiovascular disease. Mol. Cell Biochem. 2018, 440, 167–187. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, E.; Barbosa, D.S.; Mazzuco, T.L.; Nunes, V.S.; Passarelli, M.; Nakandakare, E.R.; Carrilho, A.J. Impaired antioxidant action of high density lipoprotein in patients with type 1 diabetes with normoalbuminuria and microalbuminuria. Diabetes Res. Clin. Pract. 2013, 99, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Kotani, K.; Sakane, N.; Ueda, M.; Mashiba, S.; Hayase, Y.; Tsuzaki, K.; Yamada, T.; Remaley, A.T. Oxidized high-density lipoprotein is associated with increased plasma glucose in non-diabetic dyslipidemic subjects. Clin. Chim. Acta 2012, 414, 125–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verges, B. Lipid modification in type 2 diabetes: The role of LDL and HDL. Fundam Clin. Pharmacol. 2009, 23, 681–685. [Google Scholar] [CrossRef] [PubMed]
- Calvo, C.; Ponsin, G.; Berthezene, F. Characterization of the non enzymatic glycation of high density lipoprotein in diabetic patients. Diabete Metab. 1988, 14, 264–269. [Google Scholar]
- Yano, M.; Hasegawa, G.; Ishii, M.; Yamasaki, M.; Fukui, M.; Nakamura, N.; Yoshikawa, T. Short-term exposure of high glucose concentration induces generation of reactive oxygen species in endothelial cells: Implication for the oxidative stress associated with postprandial hyperglycemia. Redox Rep. 2004, 9, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Mussa, S.; Gastaldi, D.; Sadowski, J.; Ratnatunga, C.; Pillai, R.; Channon, K.M. Mechanisms of increased vascular superoxide production in human diabetes mellitus: Role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation 2002, 105, 1656–1662. [Google Scholar] [CrossRef] [Green Version]
- Arcambal, A.; Taile, J.; Rondeau, P.; Viranaicken, W.; Meilhac, O.; Gonthier, M.P. Hyperglycemia modulates redox, inflammatory and vasoactive markers through specific signaling pathways in cerebral endothelial cells: Insights on insulin protective action. Free Radic Biol. Med. 2019, 130, 59–70. [Google Scholar] [CrossRef]
- Shao, B.; Bayraktutan, U. Hyperglycaemia promotes human brain microvascular endothelial cell apoptosis via induction of protein kinase C-ssI and prooxidant enzyme NADPH oxidase. Redox Biol. 2014, 2, 694–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arcambal, A.; Taile, J.; Couret, D.; Planesse, C.; Veeren, B.; Diotel, N.; Gauvin-Bialecki, A.; Meilhac, O.; Gonthier, M.P. Protective Effects of Antioxidant Polyphenols against Hyperglycemia-Mediated Alterations in Cerebral Endothelial Cells and a Mouse Stroke Model. Mol. Nutr. Food Res. 2020, 64, e1900779. [Google Scholar] [CrossRef] [PubMed]
- Richmond, J. Refinement, reduction, and replacement of animal use for regulatory testing: Future improvements and implementation within the regulatory framework. ILAR J. 2002, 43, S63–S68. [Google Scholar] [CrossRef] [Green Version]
- Fiorelli, M.; Bastianello, S.; von Kummer, R.; del Zoppo, G.J.; Larrue, V.; Lesaffre, E.; Ringleb, A.P.; Lorenzano, S.; Manelfe, C.; Bozzao, L. Hemorrhagic transformation within 36 h of a cerebral infarct: Relationships with early clinical deterioration and 3-month outcome in the European Cooperative Acute Stroke Study I (ECASS I) cohort. Stroke 1999, 30, 2280–2284. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Couret, D.; Planesse, C.; Patche, J.; Diotel, N.; Nativel, B.; Bourane, S.; Meilhac, O. Lack of Neuroprotective Effects of High-Density Lipoprotein Therapy in Stroke under Acute Hyperglycemic Conditions. Molecules 2021, 26, 6365. https://doi.org/10.3390/molecules26216365
Couret D, Planesse C, Patche J, Diotel N, Nativel B, Bourane S, Meilhac O. Lack of Neuroprotective Effects of High-Density Lipoprotein Therapy in Stroke under Acute Hyperglycemic Conditions. Molecules. 2021; 26(21):6365. https://doi.org/10.3390/molecules26216365
Chicago/Turabian StyleCouret, David, Cynthia Planesse, Jessica Patche, Nicolas Diotel, Brice Nativel, Steeve Bourane, and Olivier Meilhac. 2021. "Lack of Neuroprotective Effects of High-Density Lipoprotein Therapy in Stroke under Acute Hyperglycemic Conditions" Molecules 26, no. 21: 6365. https://doi.org/10.3390/molecules26216365
APA StyleCouret, D., Planesse, C., Patche, J., Diotel, N., Nativel, B., Bourane, S., & Meilhac, O. (2021). Lack of Neuroprotective Effects of High-Density Lipoprotein Therapy in Stroke under Acute Hyperglycemic Conditions. Molecules, 26(21), 6365. https://doi.org/10.3390/molecules26216365