
Determination of the Protein-Protein Interactions within Acyl Carrier Protein (MmcB)-Dependent Modifications in the Biosynthesis of Mitomycin

 ,
, 
 and
and
Abstract
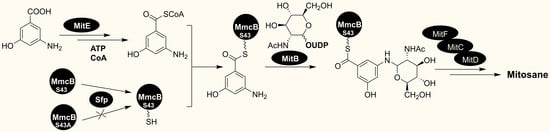
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. MitE Catalyzed the CoA Ligation of AHBA by the Genetic and Biochemcal Analysis
2.2. MmcB-Tethering AHBA Triggered the Biosynthesis of Mitomycin C
2.3. The Completed MitB Catalyzed the Glycosylation of ACP-Bound AHBA
2.4. The Protein–Protein Interactions Occurred between MmcB and MitB
2.5. MmcB Interacted with MitF during Mitomycin C Assembly
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains, Plasmids, and Culture Conditions
4.2. Gene Deletion and Complementation
4.2.1. Gene MitE In-Frame Deletion and Complementation
4.2.2. Gene MmcB In-Frame Deletion and Complementation
4.2.3. Gene MitB In-Frame Deletion and Complementation
4.2.4. Gene MitF In-Frame Deletion and Complementation
4.3. Production and Detection of Mitomycin C in Strain NRRL2564
4.4. Protein Expression and Purification
4.5. Enzyme Activity Analysis
4.6. Determination of the Protein Molecular Weight by Q-TOF Mass Spectrometry
4.7. Protein–Protein Interaction Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Remers, W.A. The mitomycins. Anticancer Agents Nat. Prod. 2005, 23, 475–493. [Google Scholar]
- Bass, P.D.; Gubler, D.A.; Judd, T.C.; Williams, R.M. Mitomycinoid alkaloids: Mechanism of action, biosynthesis, total syntheses, and synthetic approaches. Chem. Rev. 2013, 113, 6816–6863. [Google Scholar] [CrossRef]
- Kang, Q.; Shen, Y.; Bai, L. Biosynthesis of 3,5-AHBA-derived natural products. Nat. Prod. Rep. 2012, 29, 243–263. [Google Scholar] [CrossRef] [PubMed]
- Verweij, J.; Sparreboom, A.; Nooter, K. Mitomycins. Cancer. Chemother. Biol. Response. Modif. 1999, 18, 46–58. [Google Scholar] [PubMed]
- Abraham, L.M.; Selva, D.; Casson, R.; Leibovitch, I. Mitomycin: Clinical applications in ophthalmic practice. Drugs 2006, 66, 321–340. [Google Scholar] [CrossRef]
- Chen, R.; Zhang, D.; Mao, Y.; Zhu, J.; Ming, H.; Wen, J.; Ma, J.; Cao, Q.; Lin, H.; Tang, Q.; et al. A human Fab-based immunoconjugate specific for the LMP1 extracellular domain inhibits nasopharyngeal carcinoma growth in vitro and in vivo. Mol. Cancer. Ther. 2012, 11, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Frøysnes, I.S.; Andersson, Y.; Larsen, S.G.; Davidson, B.; Øien, J.T.; Julsrud, L.; Fodstad, Ø.; Dueland, S.; Flatmark, K. ImmunoPeCa trial: Long-term outcome following intraperitoneal MOC31PE immunotoxin treatment in colorectal peritoneal metastasis. Eur. J. Surg. Oncol. 2021, 47, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Wang, S.; Wang, Y.; Zhang, F.; Shao, T. Targeted delivery of mitomycin C-loaded and LDL-conjugated mesoporous silica nanoparticles for inhibiting the proliferation of pterygium subconjunctival fibroblasts. Exp. Eye. Res. 2020, 197, 108124. [Google Scholar] [CrossRef]
- Anderson, M.G.; Kibby, J.J.; Rickards, R.W.; Rothschild, J.M. Biosynthesis of the mitomycin antibiotics from 3-amino-5-hydroxybenzoic acid. J. Chem. Soc. Chem. Commun. 1980, 24, 1277–1278. [Google Scholar] [CrossRef]
- Floss, H.G.; Yu, T.W.; Arakawa, K. The biosynthesis of 3-amino-5-hydroxybenzoic acid (AHBA), the precursor of mC(7)N units in ansamycin and mitomycin antibiotics: A review. J. Antibiot. (Tokyo) 2011, 64, 35–44. [Google Scholar] [CrossRef]
- Bezanson, G.S.; Vining, L.C. Studies on the biosynthesis of mitomycin C by Streptomyces verticillatus. Can. J. Biochem. 1971, 49, 911–918. [Google Scholar] [CrossRef] [PubMed]
- Hornemann, U.; Cloyd, J.C. Biosynthesis of the mitomycin antibiotics by Streptomyces verticillatus. J. Chem. Soc. Chem. Commun. 1971, 7, 301–302. [Google Scholar] [CrossRef]
- Hornemann, U.; Eggert, J.H. Utilization of the intact carbamoyl group of L-(NH2CO-13C,15N) citrulline in mitomycin biosynthesis by Streptomyces verticillatus. J. Antibiot. (Tokyo) 1975, 28, 841–843. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hornemann, Y.; Kehrer, J.P.; Nunez, C.S.; Ranieri, R.L. D-glucosamine and L-citrulline, precursors in mitomycin biosynthesis by Streptomyces verticillatus. J. Am. Chem. Soc. 1974, 96, 320–322. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Varoglu, M.; Sherman, D.H. Molecular characterization and analysis of the biosynthetic gene cluster for the antitumor antibiotic mitomycin C from Streptomyces lavendulae NRRL 2564. Chem. Biol. 1999, 6, 251–263. [Google Scholar] [CrossRef]
- Varoglu, M.; Mao, Y.; Sherman, D.H. Mapping the mitomycin biosynthetic pathway by functional analysis of the MitM aziridine N-methyltransferase. J. Am. Chem. Soc. 2001, 123, 6712–6713. [Google Scholar] [CrossRef] [PubMed]
- Gruschow, S.; Chang, L.C.; Mao, Y.; Sherman, D.H. Hydroxyquinone O-methylation in mitomycin biosynthesis. J. Am. Chem. Soc. 2007, 129, 6470–6476. [Google Scholar] [CrossRef]
- Nguyen, H.P.; Yokoyama, K. Characterization of acyl carrier protein-dependent glycosyltransferase in mitomycin C biosynthesis. Biochemistry. 2019, 58, 2804–2808. [Google Scholar] [CrossRef] [PubMed]
- Ogasawara, Y.; Nakagawa, Y.; Maruyama, C.; Hamano, Y.; Dairi, T. In vitro characterization of MitE and MitB: Formation of N-acetylglucosaminyl-3-amino-5-hydroxybenzoyl-MmcB as a key intermediate in the biosynthesis of antitumor antibiotic mitomycins. Bioorg. Med. Chem. Lett. 2019, 29, 2076–2078. [Google Scholar] [CrossRef]
- Hu, X.; Li, X.; Sheng, Y.; Wang, H.; Li, X.; Ou, Y.; Deng, Z.; Bai, L.; Kang, Q. p-aminophenylalanine involved in the biosynthesis of antitumor dnacin b1 for quinone moiety formation. Molecules. 2020, 25, 4186. [Google Scholar] [CrossRef]
- Sheng, Y.; Ou, Y.; Hu, X.; Deng, Z.; Bai, L.; Kang, Q. Generation of tetramycin B derivative with improved pharmacological property based on pathway engineering. Appl. Microbiol. Biotechnol. 2020, 104, 2561–2573. [Google Scholar] [CrossRef]
- Milligan, J.C.; Lee, D.J.; Jackson, D.R.; Schaub, A.J.; Beld, J.; Barajas, J.F.; Hale, J.J.; Luo, R.; Burkart, M.D.; Tsai, S.C. Molecular basis for interactions between an acyl carrier protein and a ketosynthase. Nat. Chem. Biol. 2019, 15, 669–671. [Google Scholar] [CrossRef] [PubMed]
- Barajas, J.F.; Finzel, K.; Valentic, T.R.; Shakya, G.; Gamarra, N.; Martinez, D.; Meier, J.L.; Vagstad, A.L.; Newman, A.G.; Townsend, C.A.; et al. Structural and biochemical analysis of protein-protein interactions between the acyl-carrier protein and product template domain. Angew. Chem. Int. Ed. Engl. 2016, 55, 13005–13009. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic. Acids. Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.M.; Wu, B.; Zheng, J.; Rock, C.O. Key residues responsible for acyl carrier protein and beta-ketoacyl-acyl carrier protein reductase (FabG) interaction. J. Biol. Chem. 2003, 278, 52935–52943. [Google Scholar] [CrossRef]
- Hata, T.; Hoshi, T.; Kanamori, K.; Matsumae, A.; Sano, Y.; Shima, T.; Sugawara, R. Mitomycin, a new antibiotic from Streptomyces. I. J. Antibiot. (Tokyo) 1956, 9, 141–146. [Google Scholar]
- Wakaki, S.; Marumo, H.; Tomioka, K.; Shimizu, G.; Kato, E.; Kamada, H.; Kudo, S.; Fujimoto, Y. Isolation of new fractions of antitumor mitomycins. Antibiot. Chemother. 1958, 8, 228–240. [Google Scholar]
- Li, Y.; Zhao, P.; Kang, Q.; Ma, J.; Bai, L.; Deng, Z. Dual carbamoylations on the polyketide and glycosyl moiety by asm21 result in extended ansamitocin biosynthesis. Chem. Biol. 2011, 18, 1571–1580. [Google Scholar] [CrossRef]
- Izore, T.; Cryle, M.J. The many faces and important roles of protein-protein interactions during non-ribosomal peptide synthesis. Nat. Prod. Rep. 2018, 35, 1120–1139. [Google Scholar] [CrossRef]
- Bartholow, T.G.; Sztain, T.; Patel, A.; Lee, D.J.; Young, M.A.; Abagyan, R.; Burkart, M.D. Elucidation of transient protein-protein interactions within carrier protein-dependent biosynthesis. Commun. Biol. 2021, 4, 340. [Google Scholar] [CrossRef]
- Roujeinikova, A.; Simon, W.J.; Gilroy, J.; Rice, D.W.; Rafferty, J.B.; Slabas, A.R. Structural studies of fatty acyl-(acyl carrier protein) thioesters reveal a hydrophobic binding cavity that can expand to fit longer substrates. J. Mol. Biol. 2007, 365, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Mindrebo, J.T.; Patel, A.; Kim, W.E.; Davis, T.D.; Chen, A.; Bartholow, T.G.; La Clair, J.J.; McCammon, J.A.; Noel, J.P.; Burkart, M.D. Gating mechanism of elongating β-ketoacyl-ACP synthases. Nat. Commun. 2020, 11, 1727. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhang, F.; Liu, W. A KAS-III heterodimer in lipstatin biosynthesis nondecarboxylatively condenses C(8) and C(14) fatty acyl-coa substrates by a variable mechanism during the establishment of a C(22) aliphatic skeleton. J. Am. Chem. Soc. 2019, 141, 3993–4001. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leng, D.; Sheng, Y.; Wang, H.; Wei, J.; Ou, Y.; Deng, Z.; Bai, L.; Kang, Q. Determination of the Protein-Protein Interactions within Acyl Carrier Protein (MmcB)-Dependent Modifications in the Biosynthesis of Mitomycin. Molecules 2021, 26, 6791. https://doi.org/10.3390/molecules26226791
Leng D, Sheng Y, Wang H, Wei J, Ou Y, Deng Z, Bai L, Kang Q. Determination of the Protein-Protein Interactions within Acyl Carrier Protein (MmcB)-Dependent Modifications in the Biosynthesis of Mitomycin. Molecules. 2021; 26(22):6791. https://doi.org/10.3390/molecules26226791
Chicago/Turabian StyleLeng, Dongjin, Yong Sheng, Hengyu Wang, Jianhua Wei, Yixin Ou, Zixin Deng, Linquan Bai, and Qianjin Kang. 2021. "Determination of the Protein-Protein Interactions within Acyl Carrier Protein (MmcB)-Dependent Modifications in the Biosynthesis of Mitomycin" Molecules 26, no. 22: 6791. https://doi.org/10.3390/molecules26226791
APA StyleLeng, D., Sheng, Y., Wang, H., Wei, J., Ou, Y., Deng, Z., Bai, L., & Kang, Q. (2021). Determination of the Protein-Protein Interactions within Acyl Carrier Protein (MmcB)-Dependent Modifications in the Biosynthesis of Mitomycin. Molecules, 26(22), 6791. https://doi.org/10.3390/molecules26226791