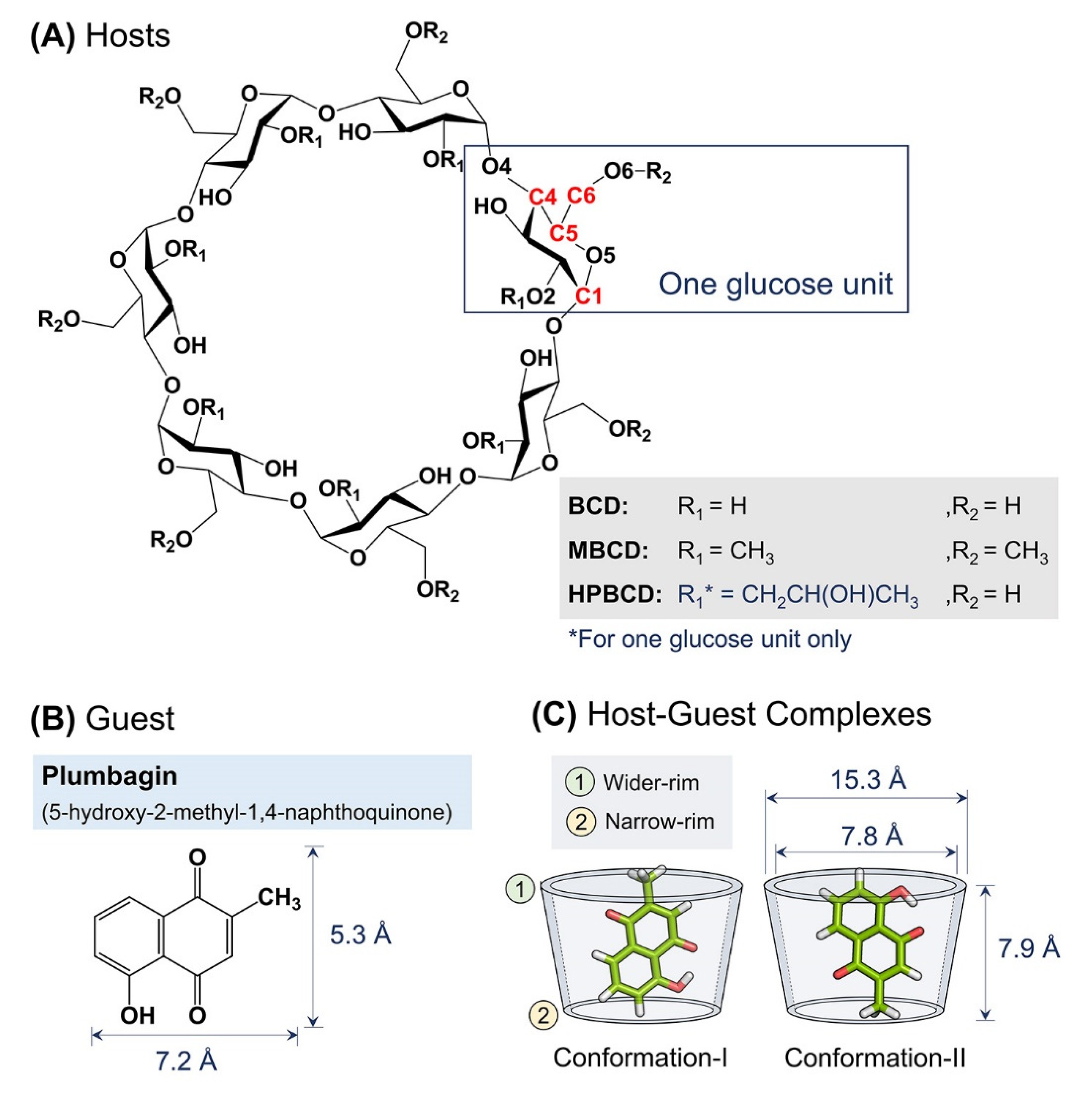
2.1. Solvated Inclusion Complexes Equilibrium and Stability
The total system energy of all equilibrated inclusion complexes in an aqueous solution under a storage temperature of 4 °C was observed throughout 200 ns MD simulations. For both conformations (I and II) of plumbagin–BCD and plumbagin–HPBCD inclusion complexes (BCD-I, BCD-II, HPBCD-I, and HPBCD-II), the energies were steady at −7000 kcal/mol. For plumbagin–MBCD inclusion complexes, in both conformations (MBCD-I and MBCD-II), the energies were steady at −9000 kcal/mol. Thus, all solvated inclusion complexes fully reached equilibrium and were considered to be stable isothermal systems.
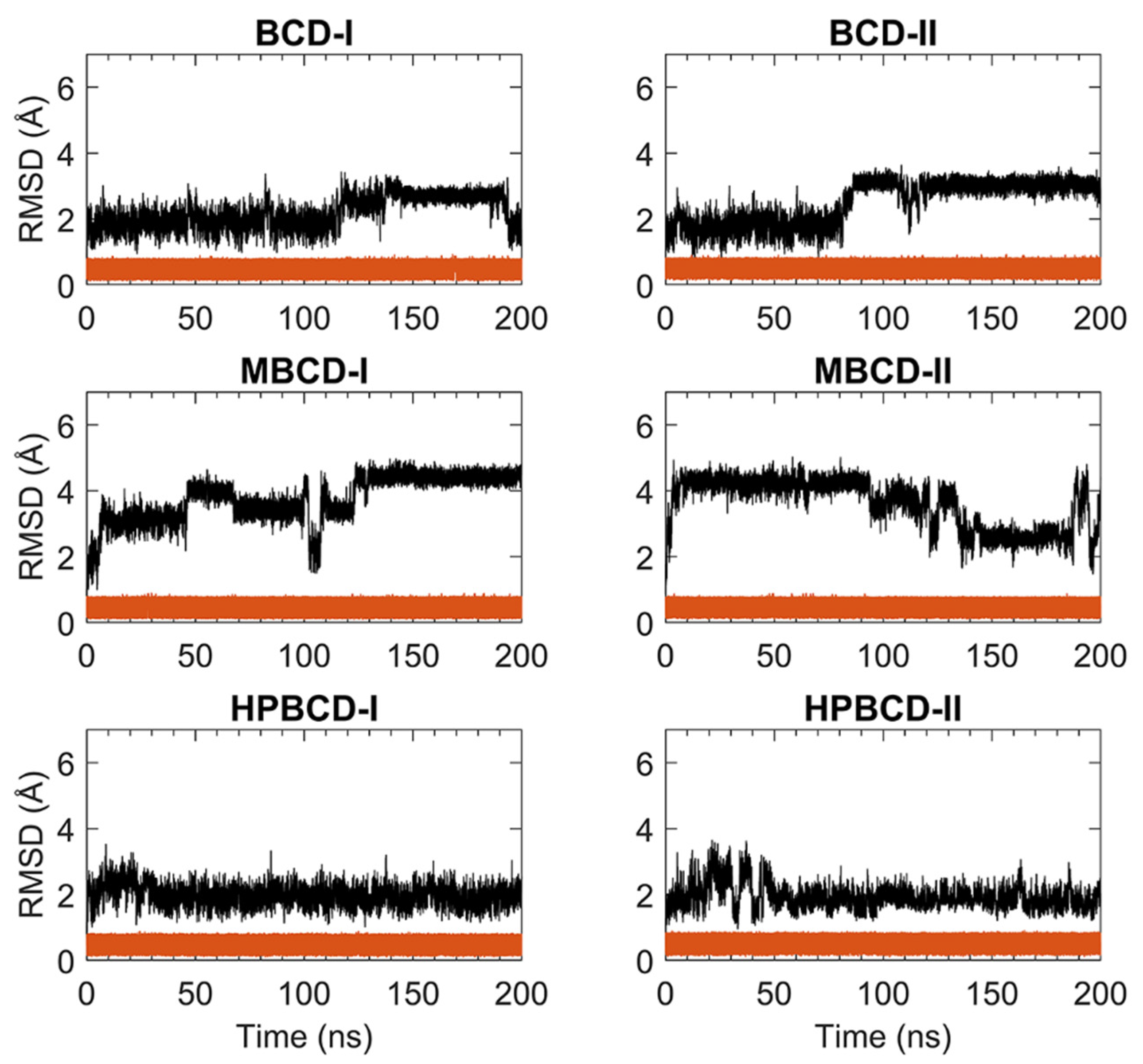
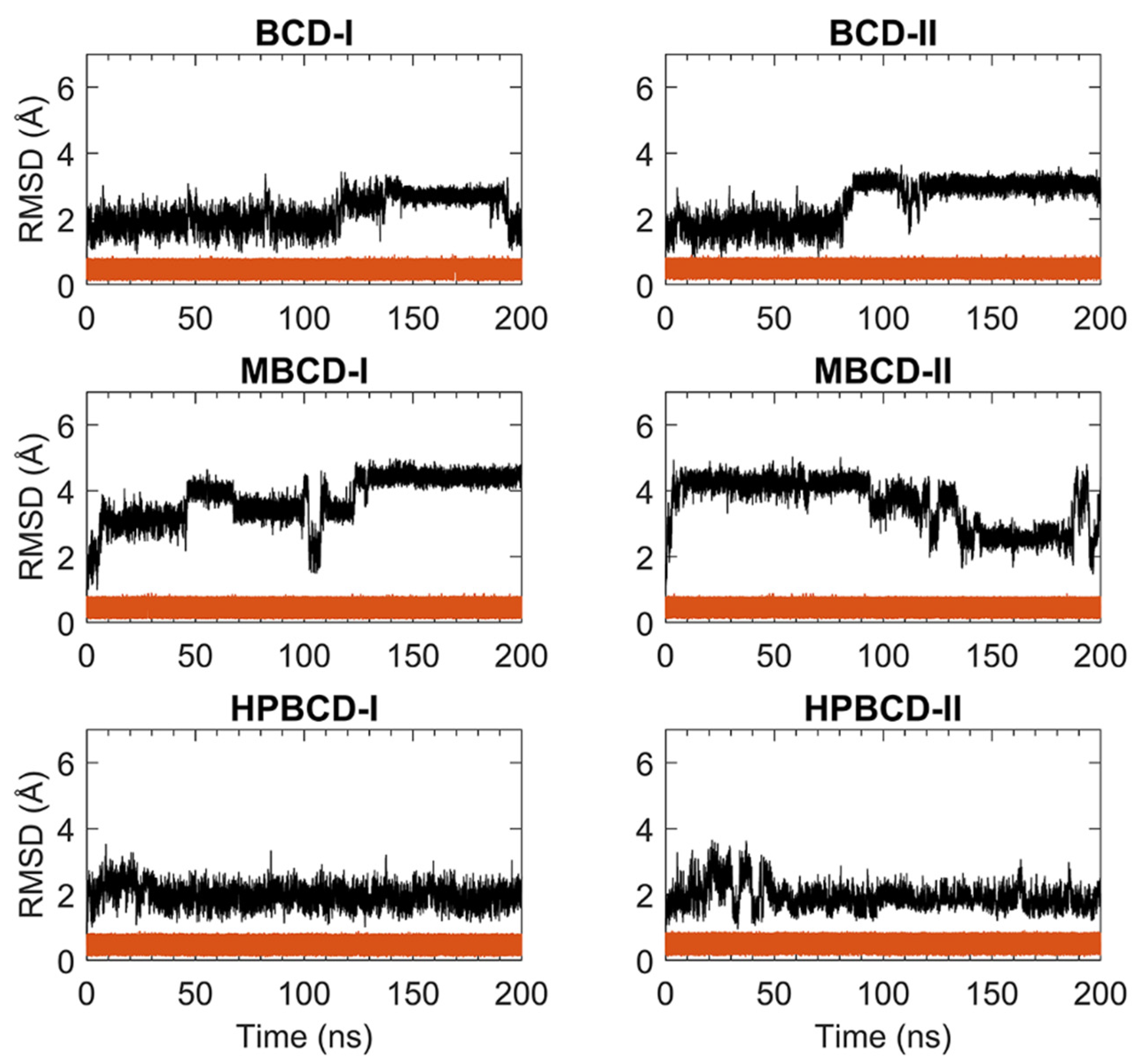
For stability analysis of plumbagin and hosts molecules, the all-atom root-mean-square deviation (RMSD) of plumbagin and BCDs were plotted using red and black lines, respectively, as shown in
Figure 1. The all-atom RMSD of the host’s molecules presented higher deviation than plumbagin molecule, as the consequence of the difference in molecular size and chemical structure. Plumbagin is a naphthoquinone that has limited structural flexibility; thus, its structural deviation throughout the whole MD simulation was small and steady (RMSD of 0.1 to 0.9 Å); meanwhile, hosts molecules—which are cyclic oligosaccharides with seven glucose units—lead to a higher degree of structural motions, especially when there are substituted functional groups on the rims of a truncated cone (e.g., MBCD and HPBCD).
The BCD structure of BCD-I was considerably similar to the reference’s orientation with low RMSD values (1.2 to 2.5 Å) during the first 115 ns. Afterward, the structural deviation of BCD started to increase and then re-stabilized within the next 30 ns. This lead to another stable interval with steady RMSD fluctuation between 2.8 and 3.1 Å until 190 ns. Then, they were probably reformed back to the reference’s orientation again, which is indicated by low RMSD. While the BCD structure in BCD-II stayed in the reference’s structure until 82 ns, its RMSD value then increased to be around 3 Å and steady until the end of simulations.
For plumbagin–MBCD inclusion complexes (MBCD-I and MBCD-II), the structural deviation of MBCD molecules was the highest among other inclusion complexes (RMSD ranging from 1.8 to 4.8 Å). The high fluctuation could be the result of additional dynamic motions of substituted methyl groups on both rims of the truncated cone. Nevertheless, several steady intervals were found during 200 ns MD simulations. In MBCD-I, the MBCD structure was gradually deviated until reaching a maximum distance of 3.95 Å within the first 10 ns. Then, there were three short stable intervals, 20 to 40 ns each, until 100 ns, with 2.85 to 4.35 Å fluctuation. Interestingly, the RMSD profile was highly fluctuated, ranging from 1.8 to 4.8 Å during 100 to 130 ns, which could refer to large structural motions within this specific period. However, after 130 ns, the RMSD fluctuation was stabilized within the 4.0 to 4.8 Å range until the end of simulation, which indicated the restoration of MBCD’s structural stability. Inversely, the RMSD of the MBCD molecule in MBCD-II revealed a large structural deviation during the first 90 ns with steady fluctuation (3.9 to 4.6 Å). Then, the deviation of MBCD structure gradually declined during 90 to 130 ns to the minimum distance of 1.9 Å. Afterward, the structural fluctuation was kept steady between 2.5 and 3.1 Å until 185 ns. However, the high degree of structural deviation arose again toward the end of simulation, with RMSD fluctuating between 1.8 to 4.8 Å. This was an interesting structural motion of MBCD from two inclusion complex conformations because the RMSD profiles appeared to be in reversed trend.
The finest dynamics system occurred in both conformations of plumbagin–HPBCD inclusion complexes (HPBCD-I and HPBCD-II). However, the slight RMSD fluctuation (1.0 to 3 Å) can still be found during the first 30 and 50 ns for HPBCD-I and HPBCD-II, respectively. These structural deviations at the beginning of simulations might have come from motions of a hydroxypropyl group attached to the wider rim of HPBCD. The RMSD profiles of HPBCD were noticeably steady (RMSD of 1.0 to 2.2 Å) during the last 170 and 150 ns of the simulation time for HPBCD-I and HPBCD-II, respectively.
2.2. Dynamics Behavior of Inclusion Complexes
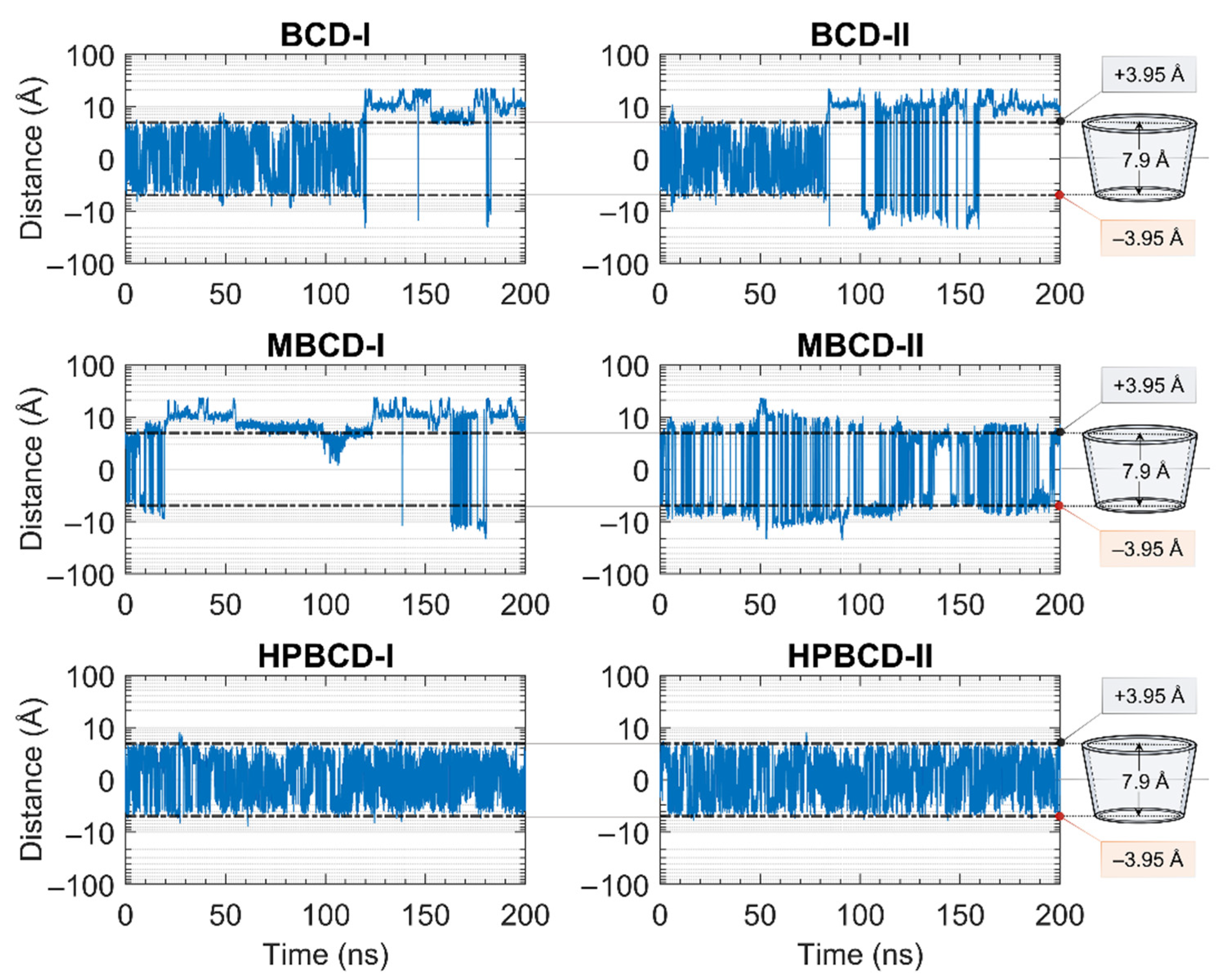
The stability of inclusion complexes was extensively analyzed based on the structural deviation of the BCDs molecule the using all-atoms RMSD profiles in the previous section. However, the RMSD value is not a sufficient parameter to fully represent plumbagin movement inside the simulated space. Therefore, the distance between the centers of mass of plumbagin and BCDs were measured to investigate the position of plumbagin during MD simulations, as shown in
Figure 2. However, to avoid defects in calculation due to asymmetrical structure, the substituted functional groups of BCD derivatives were not taken into consideration. The center of mass of BCD’s core structure was considered to be the origin; thus, positive and negative distances indicated whether the plumbagin molecule was located above or below that origin. The black horizontal dashed lines on each subplot represent half of the vertical distance of BCD core structure in the positive and negative directions. Therefore, the positive and negative distances above or below these horizontal lines indicate the release of the plumbagin molecule from BCDs inner cavity.
The plumbagin molecule bounced upward and downward within the BCD cavity for the first 115 and 82 ns in BCD-I and BCD-II conformations, respectively. Then, the plumbagin molecule started to migrate out from the binding cavity with several attempts to migrate back inside the cavity space. However, the plumbagin could not be stabilized inside the inner cavity of BCD anymore, which could be a consequence of structural deviation of BCD, as mentioned in the stability analysis.
Similarly, the relationship between the structural deviation of MBCD and dynamics of plumbagin could be identified as follows. For MBCD-I conformation, the MBCD structure deviation could be roughly divided into three stable intervals during the first 100 ns, which also affects the movement of plumbagin in the same way. In other words, plumbagin bounced up and down inside the cavity during the first 20 ns and then migrated out and kept a constant distance with MBCD until 50 ns. Afterward, it shifted back and stayed closer to the MBCD again until 100 ns. During 100 and 130 ns, in which RMSD indicated the high structural fluctuation of MBCD, plumbagin was attracted back into MBCD cavity. However, the highly fluctuated movement was found after 130 ns and the plumbagin mostly stayed away from MBCD inner cavity, which corresponds to the final stable interval of MBCD structural deviation. For MBCD-II conformation, the relationship between MBCD structural deviation and plumbagin movement interpretation is difficult due to the high fluctuation of plumbagin movement: it intensively bounced upward and downward both inside and outside the MBCD cavity. Thus, the trend of plumbagin dynamics could not be clearly seen. However, the distance profile could be divided into three main intervals with different plumbagin movements, and they are roughly comparable to the RMSD profile of MBCD. In the first interval, the highest fluctuation of plumbagin movement could be observed during the first 130 ns, which represented the interchange between its bound and released states. The second interval was defined between 130 and 185 ns and the fluctuation of plumbagin movement was reduced, which resulted in a bouncing motion within MBCD cavity only. In the third interval, the fluctuation of plumbagin movement increased until the end of the simulation, which represented the revival of interchange between bound and released states.
The movement of plumbagin in HPBCD-I and HPBCD-II conformations was stable, as expected, because the high stability of HPBCD structural deviation was previously observed. Moreover, the plumbagin molecule bounced up and down inside the binding cavity with no evidence of migration to the solvated space.
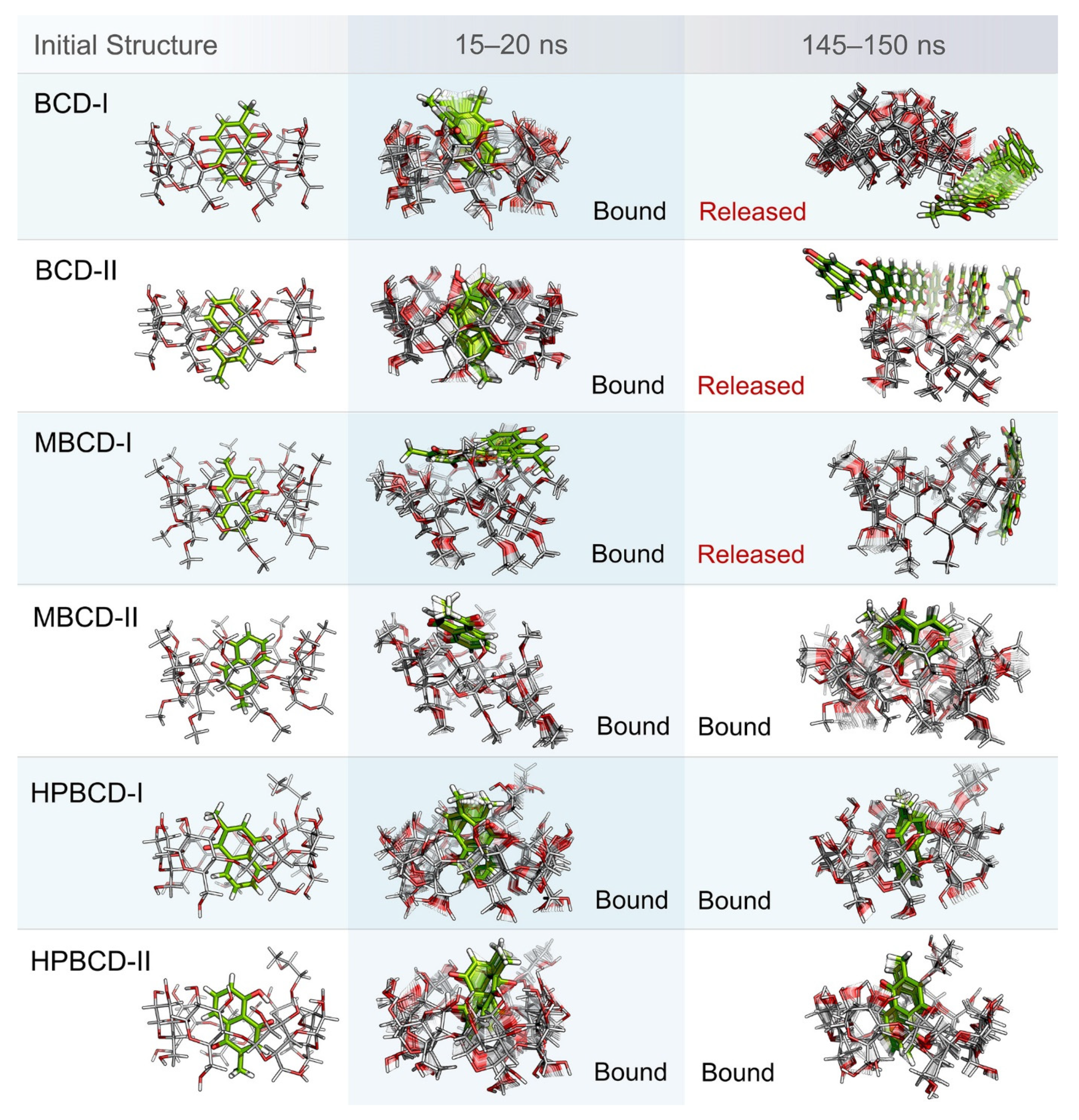
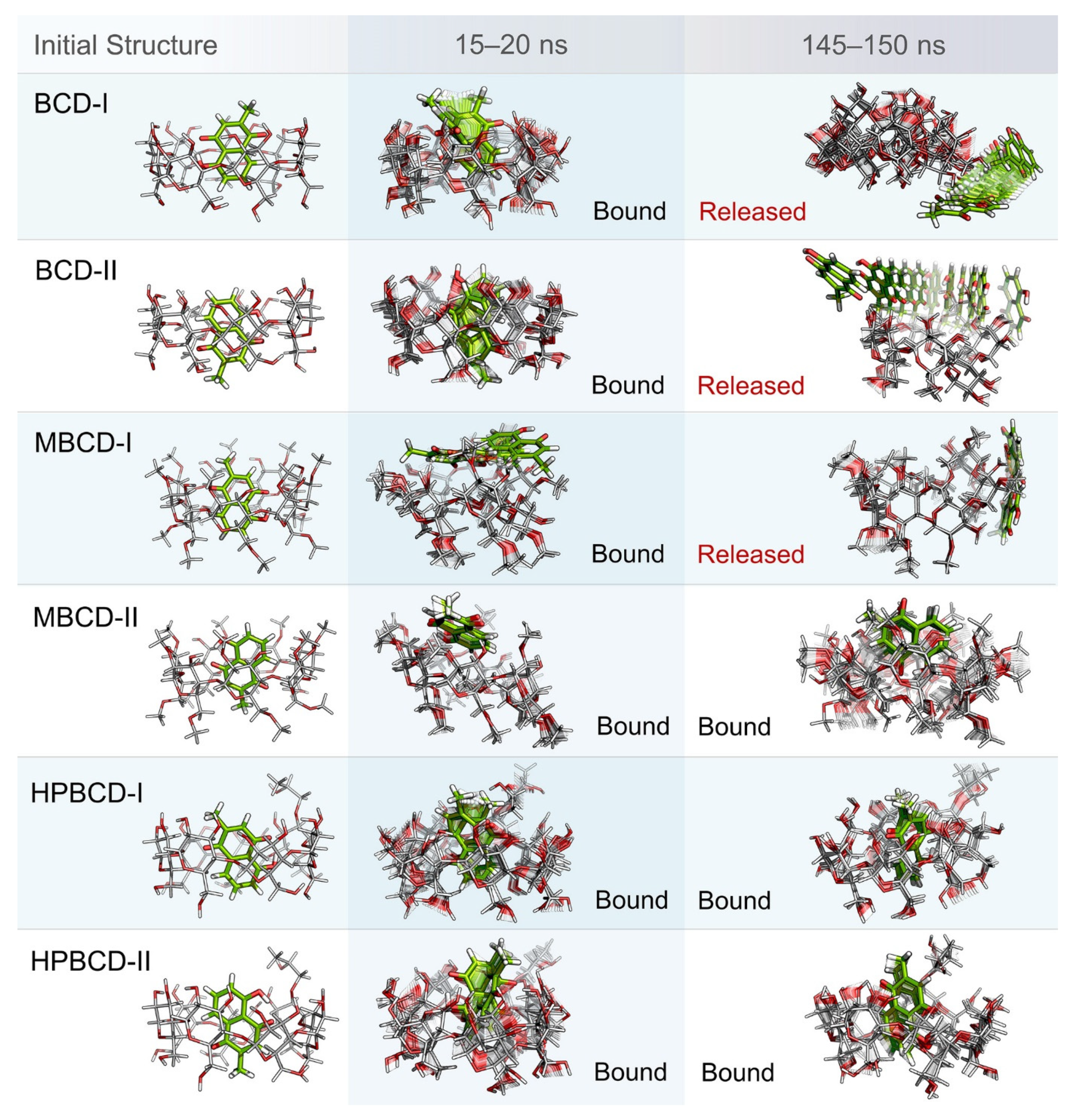
Even though the structural movement of BCDs and dynamics of plumbagin can be predicted using all-atom RMSD and distance profile, the visualization of inclusion complexes is also important for behavior analysis. According to previously analyzed results, there are multiple stable intervals during MD simulations that could be selected for further analysis. After careful consideration based on RMSD profiles and distance profiles, two stable intervals were selected. The first stable interval (15 to 20 ns) was chosen to represent the initial bound state during MD simulations. The second stable interval (145 to 150 ns) was used to represent the dynamics behavior of inclusion complexes during the latter part of simulations.
Ten snapshots during the selected stable intervals were illustrated in comparison with the initial structure from structural minimization as shown in
Figure 3. During 15 to 20 ns, plumbagin molecule in all inclusion complexes was located inside or near the rim of BCDs cavity, which is consistent with the distance profiles. However, the motion of the plumbagin molecule was elevated during the latter stable interval (145 to 150 ns) in three inclusion complexes, which are the BCD-I, BCD-II, and MBCD-I conformations. Thus, this resulted in the release of a plumbagin molecule from the inner cavity of BCD and MBCD into the solvated space. According to the all-atom RMSD profiles of these three inclusion complexes, the structural deviation of BCD and MBCD was also increased during the latter part of the simulations, which can be used to support the observation from molecular visualization. On the other hand, the plumbagin molecule was not released from the inner cavities of the MBCD-II, HPBCD-I, and HPBCD-II conformations during the latter stable interval. Therefore, these three inclusion complexes should provide a stronger binding between the hosts (BCDs) and the guest (plumbagin) molecule than the previous three systems.
In addition, the visualization of MD snapshots can also reveal the interesting behavior of the plumbagin molecule in each inclusion complex. For the BCD-I conformation, the plumbagin molecule was released from the narrow rim, which was enlarged due to the structural deformation of BCD, and its methyl group was rotated down approximately 90 degrees. For the BCD-II conformation, the alignment of plumbagin was kept at the original orientation, even during the migration from BCD’s inner cavity. For MBCD-I and MBCD-II conformations, the plumbagin molecule floated up to the wider rim of MBCD with changing in orientation by pointing its methyl group toward the side of MBCD cavity. However, after plumbagin was released in MBCD-I conformation, its alignment seemed to be random and clung on the outer surface of MBCD with some weak interaction. For the HPBCD-I conformation, the orientation of plumbagin inside the HPBCD cavity did not change at all. Inversely, the plumbagin molecule flipped its methyl group toward the wider rim of HPBCD in the HPBCD-II conformation, and this meant that plumbagin preferred to orient as conformation-I inside HPBCD’s cavity.
2.3. Binding Energies and Intermolecular Interactions
To gain more understanding about the binding interactions between BCDs and plumbagin in each inclusion complex, molecular mechanics–generalized born surface area continuum solvation (MM/GBSA) approach [
19] was used to estimate the binding energy. The calculations were based on 5,000 frames from the two selected stable intervals (15 to 20 ns and 145 to 150 ns). The binding energy and all energetic contributions were plotted and listed in
Figure 4 and
Table 1, respectively.
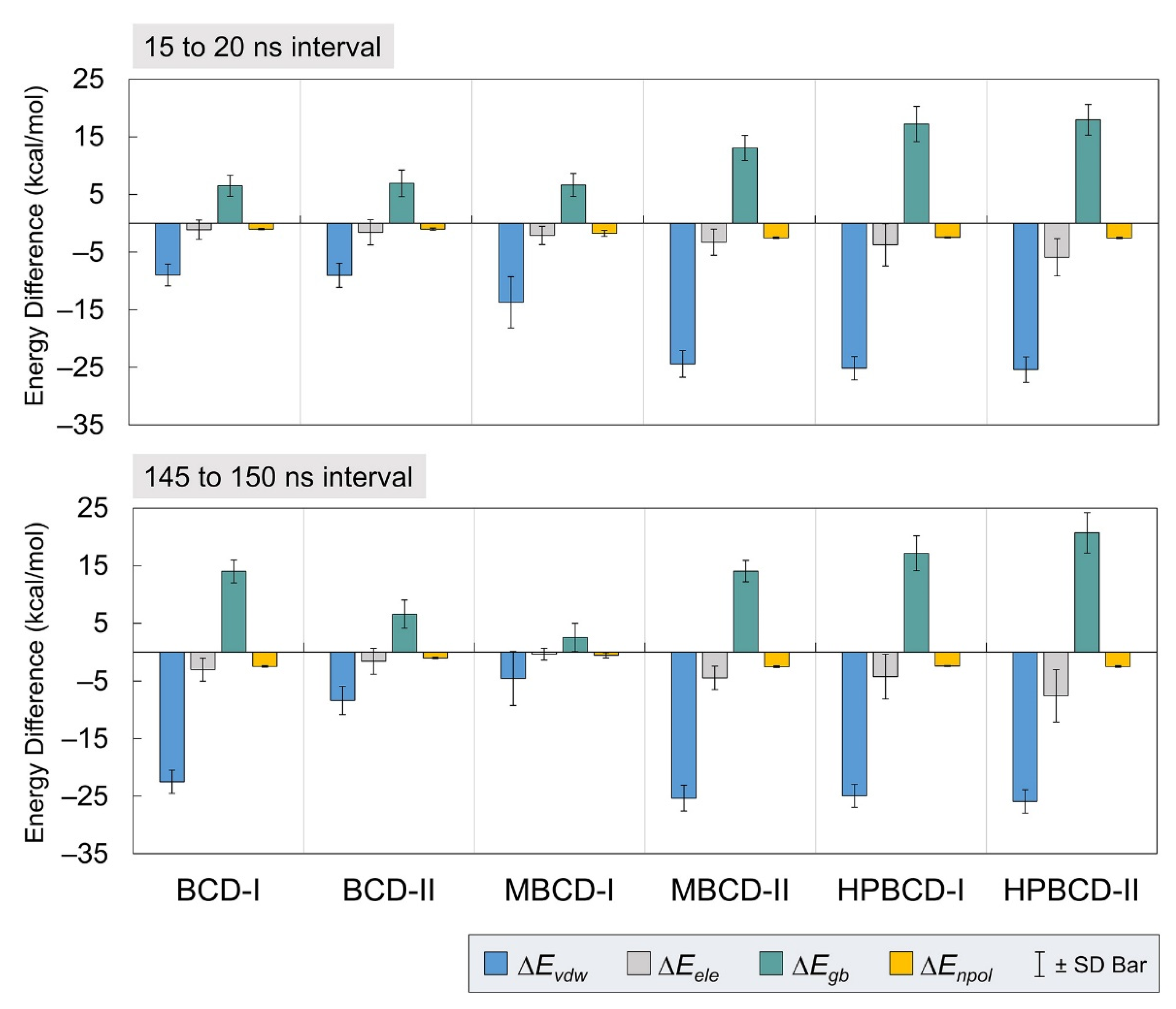
The average interaction energy in the gas phase (ΔGgas) was the summation of van der Waals (ΔEvdw) and electrostatic (ΔEele) energies. The solvation-free energy in the implicit aqueous phase (ΔGsol) was the summation of electrostatic (ΔEgb) and nonpolar (ΔEnpol) energies. Therefore, the total energy difference (ΔGTotal) from the binding was the summation of ΔGgas and ΔGsol. Then, the entropy change (TΔS) of plumbagin from the host–guest complexation at a storage temperature was subtracted from ΔGTotal to obtain the binding energy (ΔGbind(MM/GBSA)).
In
Figure 4, all inclusion complexes during both stable intervals showed negative van der Waals and electrostatic contributions in gas phase energy; however, they showed small negative electrostatic and highly positive nonpolar contributions in solvation-free energy. Therefore, all inclusion complexes complied with the typical phenomena, which are the favorable interaction energy in the gas phase and unfavorable solvation-free energy. The reason that most systems would have unfavorable solvation-free energy is that this energetic contribution refers to the de-solvation process of bound molecules from solvated space and restoring the binding interface. It was also very clear that the leading contribution for host–guest binding in all plumbagin–BCDs inclusion complexes was van der Waals interactions.
From
Table 1, the negative total energy difference and binding energy of all inclusion complexes, during both stable intervals, indicated the favorable host–guest complexations. However, the binding affinity among all inclusion complexes is different, with several interesting perspectives. For the 15 to 20 ns interval, the total energy difference suggests the binding affinity rank without the effect of entropy change, as follows: MBCD-II > HPBCD-II > HPBCD-I > MBCD-I > BCD-II > BCD-I. For the 145 to 150 ns interval, the ranking is MBCD-II > HPBCD-II > HPBCD-I > BCD-I > BCD-II > MBCD-I. Comparison between the results from two stable intervals showed the switching of binding affinity ranking of BCD-I and MBCD-I, which are the inclusion complexes with evidence of plumbagin release. Thus, this leads to one important question, which is that of why the binding affinity between plumbagin and BCD in BCD-I conformation was stronger, with evidence of plumbagin leaving the binding cavity. Then, a hypothesis was proposed as follows: the stronger binding between plumbagin and BCD may occur at the rim, which consists of hydroxyl groups. If this hypothesis is true, this means that the release of plumbagin in BCD-I conformation is not the complete release.
However, the binding affinity analysis, based solely on the total energy difference, was not comprehensive because the entropy effect was excluded. Therefore, the binding affinity based on MM/GBSA binding energy was investigated as well. For 15 to 20 ns, the binding affinity ranking is MBCD-II > MBCD-I > BCD-I > BCD-II > HPBCD-II > HPBCD-I. For 145 to 150 ns, the binding affinity ranking is MBCD-I > BCD-II > BCD-I > MBCD-II > HPBCD-I > HPBCD-II. By considering the entropy change listed in
Table 1, there were both positive and negative entropy changes. As generally known, a negative entropy change implies that the system is in high order and positive entropy implies the elevation is in disorder. Therefore, BCD-II, BCD-II, and MBCD-I conformations tend to progress toward disorder, with respect to time, because the entropy changes are positive and elevated, especially in BCD-II and MBCD-I conformations. For the MBCD-II conformation, the entropy change increased from a small negative value to a small positive value, which can suggest that this system is not stable because the motion of molecules in this system tends to generate disorder as well. On the other hand, HPBCD-I and HPBCD-II conformations produced the negative entropy change, which supports the immobility of plumbagin inside HPBCD cavity. Thus, plumbagin–HPBCD inclusion complexes tend to produce the highest stability of host–guest binding, even though the binding energy (Δ
Gbind(MM/GBSA)) is not strong compared with others.
To validate the calculated binding energies, the data from other literature have been listed for comparison in
Table 2. The results show that the calculated energy difference of inclusion complexes, without entropic contribution, is comparable with published data from molecular docking and semi-empirical PM6 method. However, the reason that the result from HPBCD-II shows a high deviation from the published value was a consequence of the changing of plumbagin alignment to conformation I. Thus, our calculated energy difference of the HPBCD-II conformation during both stable intervals was represented in the inclusion complex conformation I. From this comparison, the calculated results were considered to be reliable.
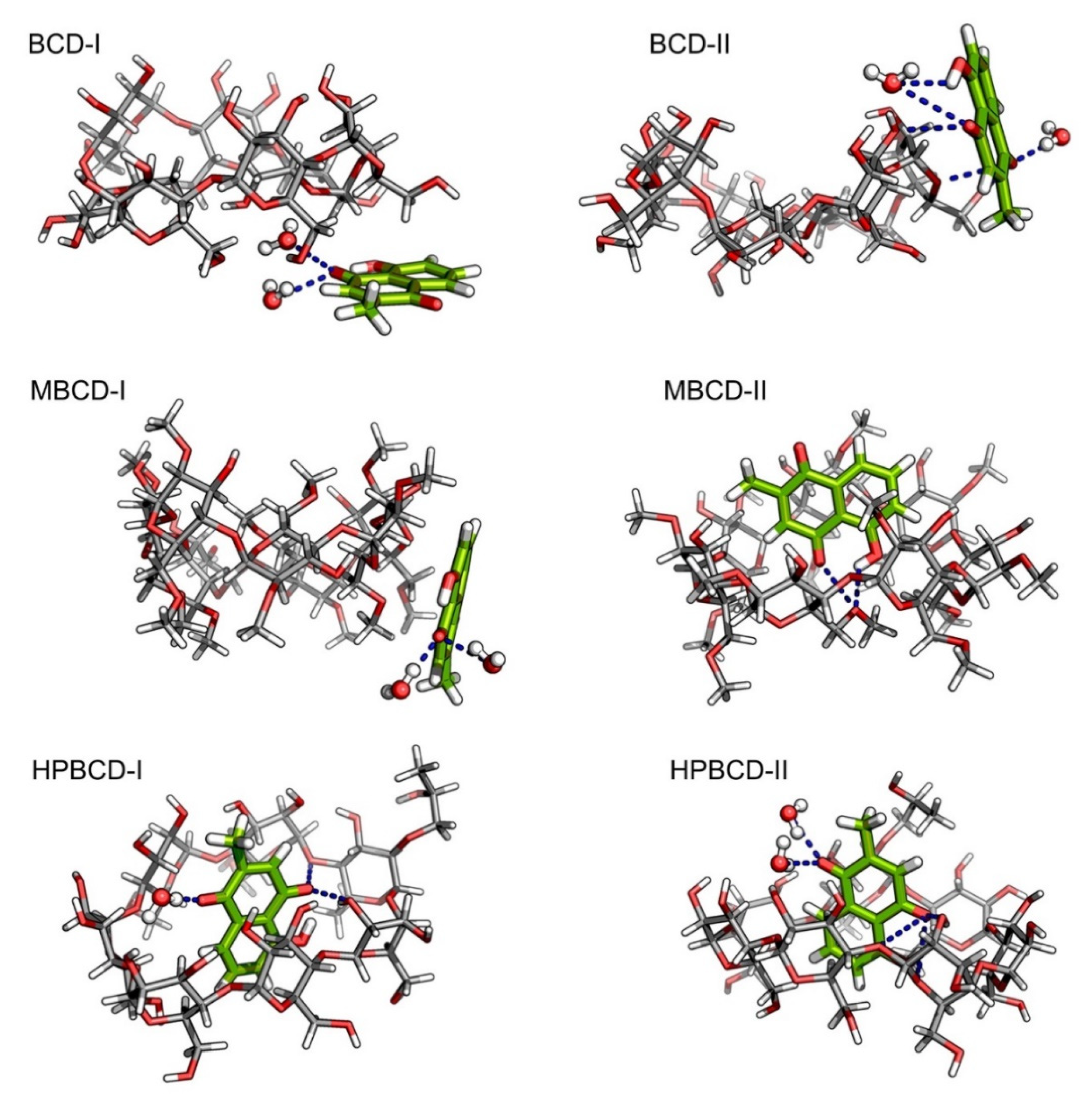
Despite the fact that the plumbagin binding behavior can be obtained based on interpretation from binding energy and entropy change, the intermolecular interaction between plumbagin and BCDs is not fully understood. Consequently, the intermolecular bonding of the inclusion complexes was investigated from snapshots during the latter stable intervals (145 to 150 ns) in order to find the key factor that facilitates the binding or release mechanisms. The interaction that could be clearly visualized was hydrogen bonding, which was related to electrostatic contribution, as shown in
Figure 4. However, it is not necessary that the hydrogen bond will always be found within the inclusion complexes because the small ligand might be attracted to the BCDs solely on hydrophobic interaction. The representative snapshots (145 ns) from each conformation were illustrated in
Figure 5 with hydrogen bonding shown as the blue dash line.
According to our hypothesis about the interaction between plumbagin and BCD in BCD-I conformation, after the detailed visualization, there was no hydrogen bonding between plumbagin and the hydroxyl group of BCDs, as was expected. However, the plumbagin molecule formed two hydrogen bonds between its oxygen atom and two water molecules instead, which could facilitate the formation of an interaction network near BCD cavity. In addition, the enlarged narrow rim of BCD that has been observed earlier occurred because some of the primary hydroxyl groups are bent toward the inner cavity of BCD. Interestingly, even though the plumbagin molecule already migrated out from BCD’s cavity in the BCD-II conformation, it formed several hydrogen bonds with secondary hydroxyl groups and O4 atom of BCD, along with another two water molecules. This interaction should be the consequence of high distortion of BCD structure which is similar to the dynamic motion of BCD in BCD-I conformation. Therefore, this should be the reason that the binding energy of these two inclusion complexes turned out to be stronger than expected.
For MBCD-I conformation, the plumbagin molecule formed two hydrogen bonds between its oxygen atom and two water molecule, which is exactly the same as the interaction found in BCD-I conformations. On the other hand, the plumbagin molecule formed two hydrogen bonds between its oxygen atom and hydroxyl group with O4 atom of MBCD in MBCD-II conformation, which facilitates the binding of plumbagin inside MBCD inner cavity. The visualization also revealed another interesting motion of MBCD which turn out to be similar to BCD. Several methyl groups on narrow rim of MBCD migrated inside the hydrophobic cavity and made the inner cavity of MBCD shallow. Therefore, this could be the reason behind the floating of the plumbagin molecule to the wider rim of MBCD.
As expected, the plumbagin molecule formed several hydrogen bonds between its oxygen atoms with secondary hydroxyl groups and O4 atom of HPBCD, along with water molecules in both conformations. Therefore, this can confirm that the plumbagin molecule tends to stay inside the encapsulated cavity of HPBCD.
2.4. Hydrogen Bonding Lifetime of Inclusion Complexes and the Dynamic of Water Molecules
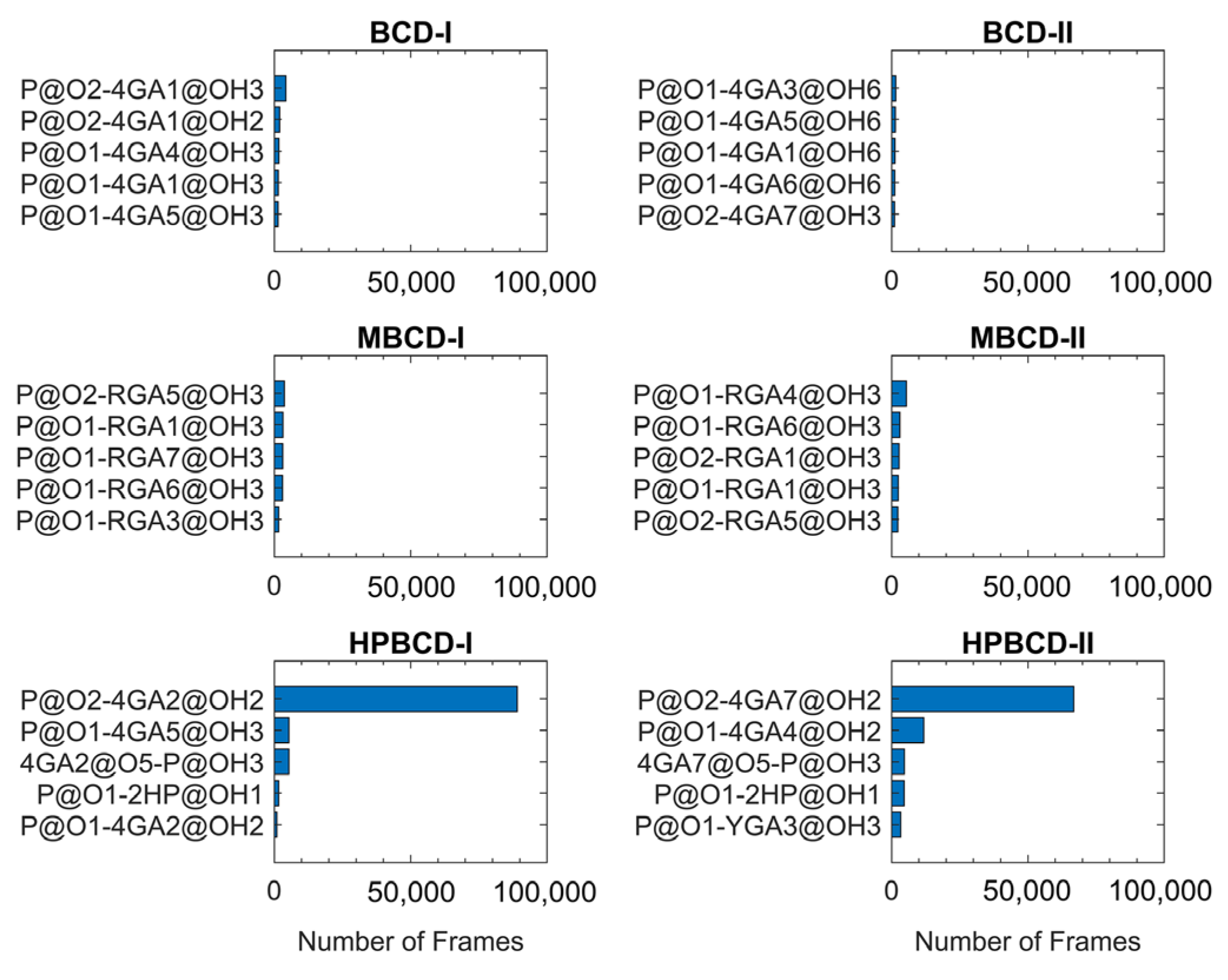
The lifetime analysis of hydrogen bonding between BCDs and plumbagin throughout 200 ns MD simulations (200,000 frames) can provide more detailed information about the time-dependent behavior of the hydrogen bonds. The distance and angle cutoff for the hydrogen bond used in the calculation were 3.0 Å and 135°, respectively. The highest five ranks of hydrogen bonding frequency from each inclusion complex are illustrated in
Figure 6.
The plots in
Figure 6 clearly show that hydrogen bonding between the oxygen atom of the plumbagin molecule and the hydroxyl group from the glucose unit in both HPBCD-I and II conformations had a high frequency. These hydrogen bonds sustained 44.51% and 33.41% of total simulation time in HPBCD-I and HPBCD-II conformation, respectively. Therefore, the frequency of hydrogen bonding between plumbagin molecule and HPBCD was significantly higher than other systems. Moreover, the binding in HPBCD-I conformation tended to be stronger than HPBCD-II conformation, due to its higher degree of sustained hydrogen bonding. Interestingly, the oxygen atom of the plumbagin molecule formed the hydrogen bond with the hydroxyl group of the attached hydroxypropyl chain of HPBCD as well. Even though these bonds were not frequently presented during the simulations, their frequency was considerably higher than some interacting pairs in other systems. Thus, this single hydroxypropyl chain in HPBCD could still be considered as one of the factors that help improve the efficiency of plumbagin binding.
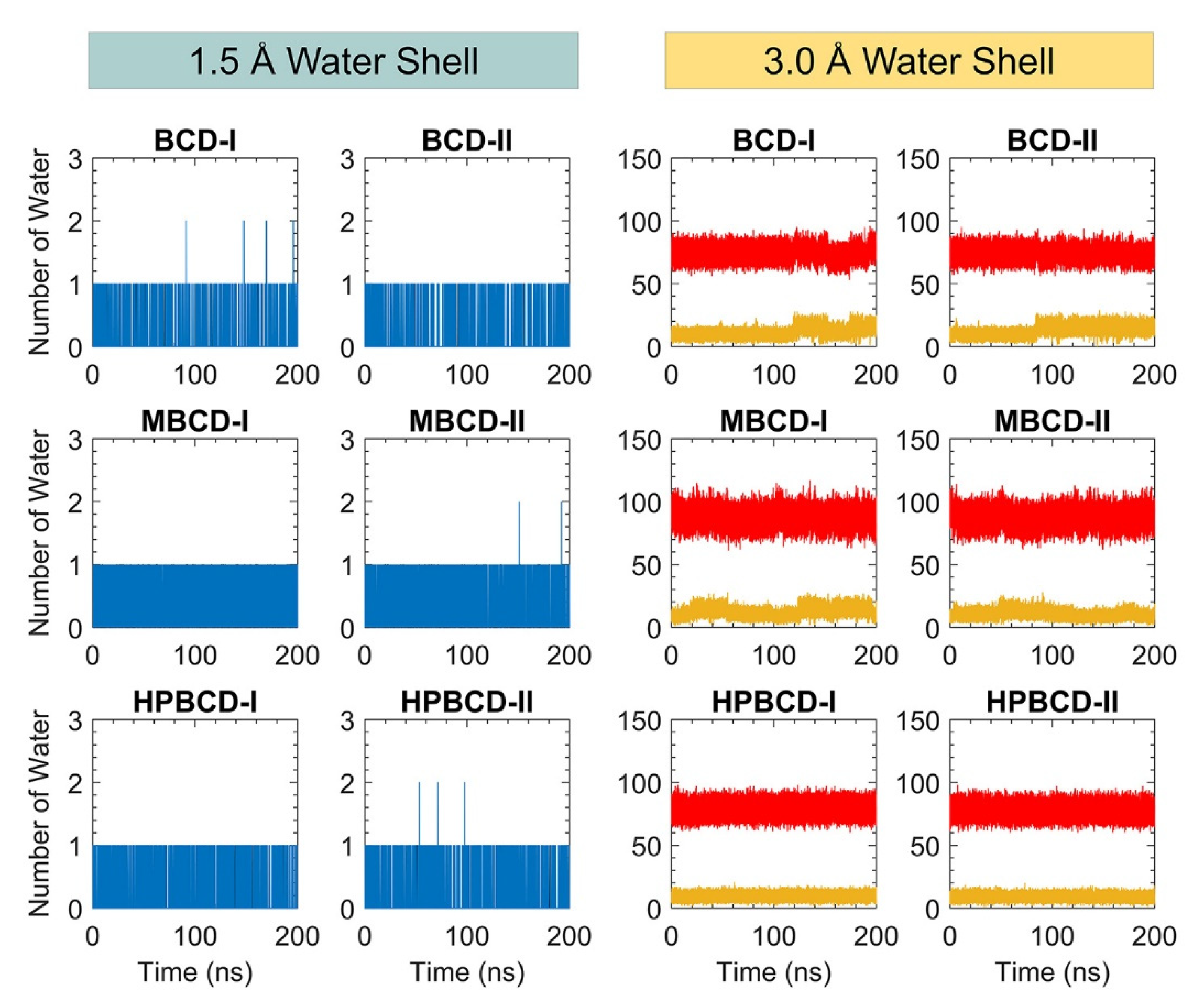
In addition, the dynamic of water molecules around inclusion complexes are investigated to gain more understanding about the role of the water molecule in binding and release states of plumbagin. The water molecule counting around inclusion complexes could represent the dynamic of water molecules and indicate the difference in the water network between binding and release states. However, the total number of water molecules inside the periodic box is large, and distant water molecules should not have significant interaction with the inclusion complexes. Therefore, two layers of spherical water shell were separately defined around BCDs and plumbagin molecules, with a radius of 1.5 and 3.0 Å. The number of water molecules inside both water shells was collected throughout 200 ns simulations, as shown in
Figure 7.
There are only one or two water molecules that stayed around BCDs and plumbagin structures within the 1.5 Å water shell, which are shown as blue and black lines in
Figure 7. These water counting profiles were consistent with the MD snapshot illustrations in
Figure 5, which indicates that the plumbagin molecule interacted with one or two water molecules for all inclusion complexes.
More water molecules were found inside the second water shell with a 3.0 Å radius. The red lines in
Figure 7 refer to the number of water molecules around BCDs structure and they are higher than the yellow lines that represent the number of water molecules around the plumbagin. The water molecules counting profiles around BCDs were quite stable, ranging from 60 to 90, 70 to 100, and 65 to 90 molecules for BCD-I/II, MBCD-I/II, and HPBCD-I/II conformations, respectively. The reason that number of water molecules were all stable around BCDs, even though plumbagin molecules migrated out for some systems, was that the hydrophobicity of BCDs inner cavities should not attract more water molecules to fulfill them.
On the other hand, the water molecules counting profiles around plumbagin are different among inclusion complexes. For BCD-I and BCD-II conformations, the number of water molecules noticeably increased at 120 ns and 90 ns, respectively, which were close to the time that plumbagin leaves the encapsulated cavity. Therefore, the water molecules were attracted by the plumbagin molecule after it migrated from BCD inner cavity. For MBCD-I and MBCD-II conformations, the water molecules counting profiles were the most fluctuated due to the abrupt motion of plumbagin molecule throughout the simulations, as discussed earlier. For HPBCD-I and HPBCD-II conformations, the water molecules counting profiles around plumbagin were very stable, which indicates that plumbagin never left the inner cavity of HPBCD and these were consistent with the results from previous sections.
Consequently, all this information can be used to support the superior stability of plumbagin encapsulation with HPBCD over other BCD derivatives.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}