
Photoinduced Atom Transfer Radical Addition/Cyclization Reaction between Alkynes or Alkenes with Unsaturated α-Halogenated Carbonyls


Abstract
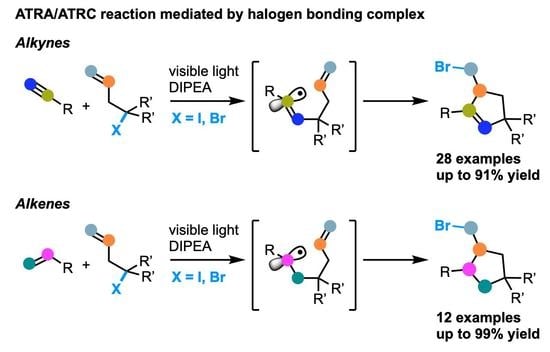
:
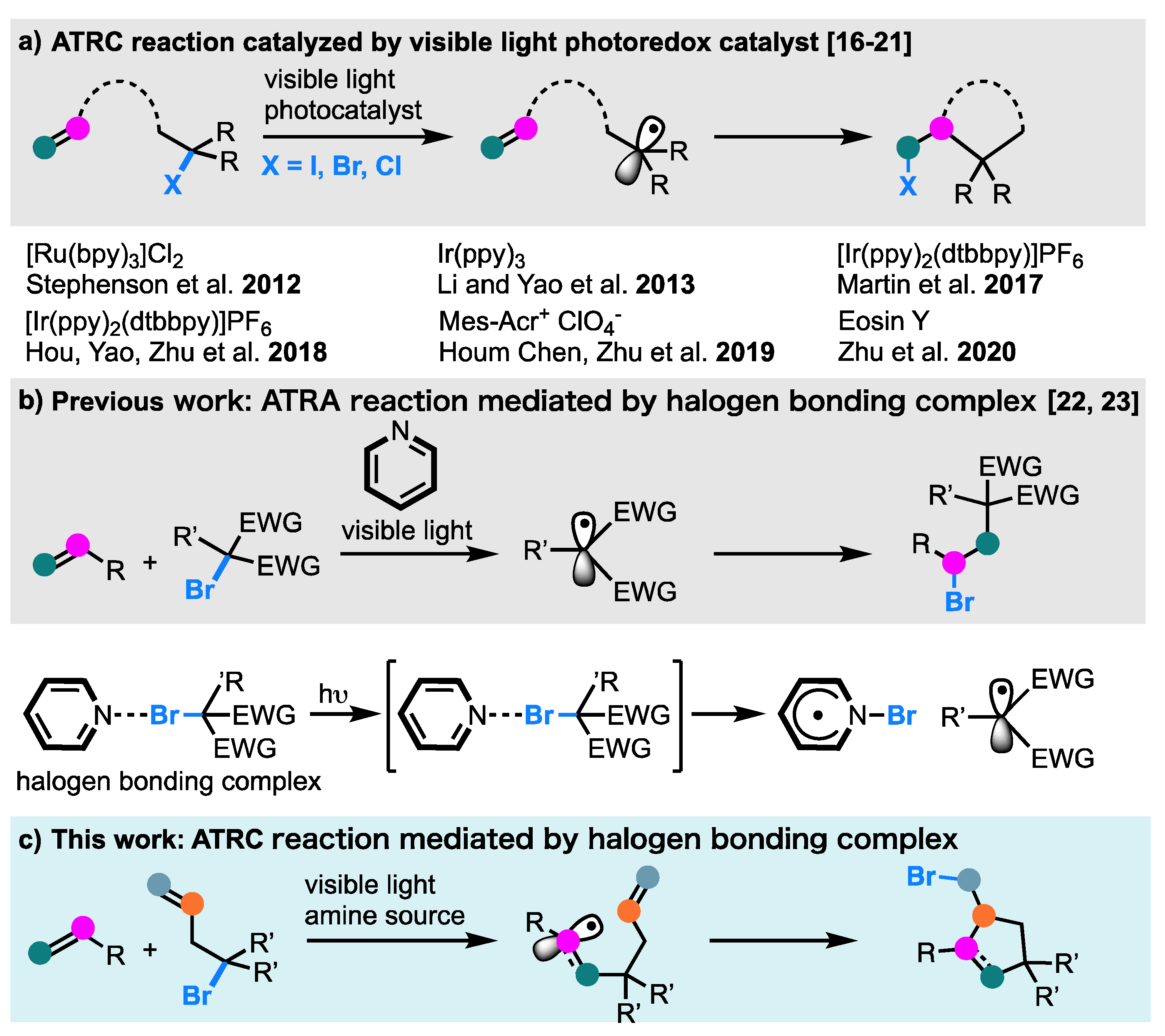
1. Introduction
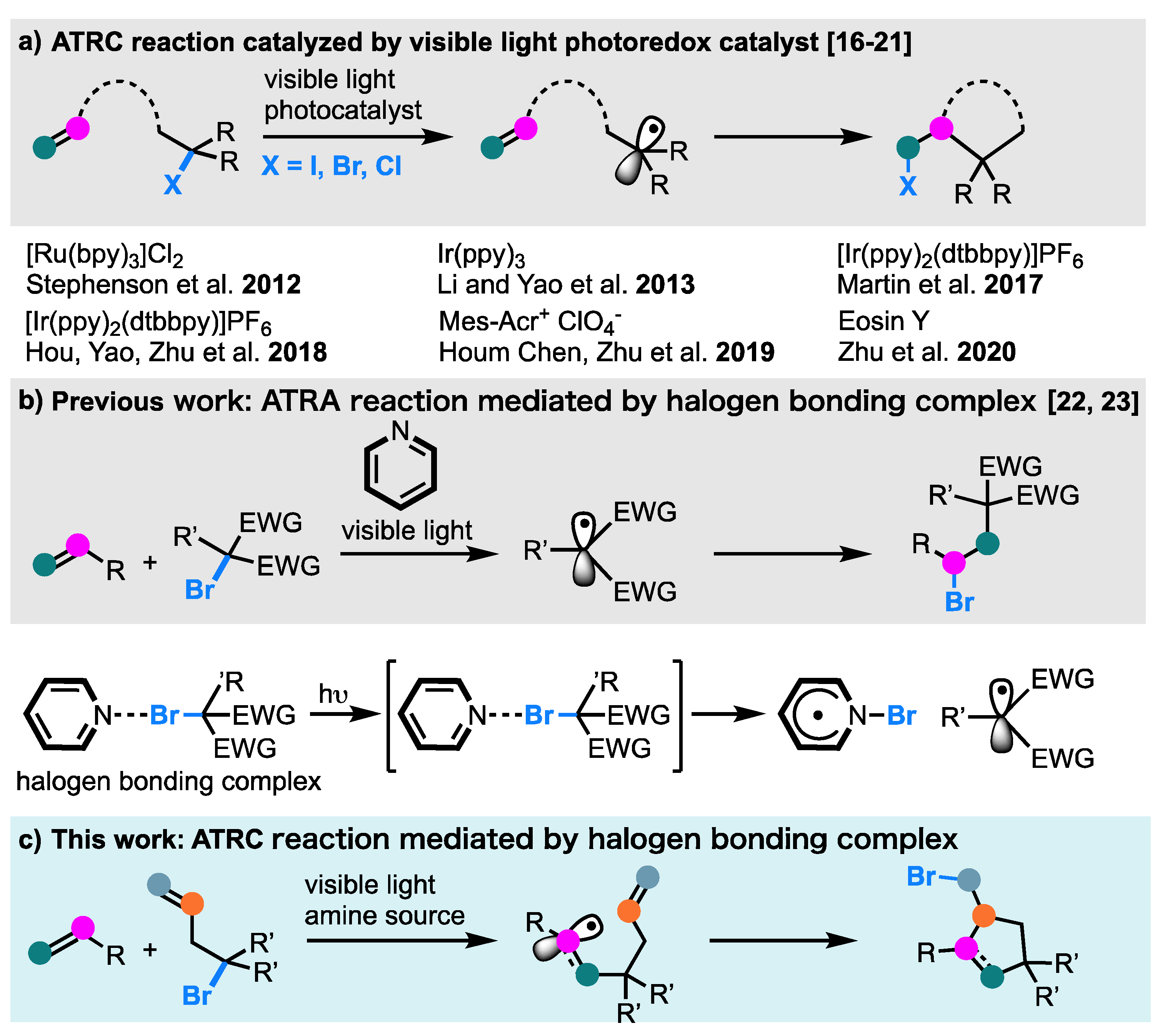
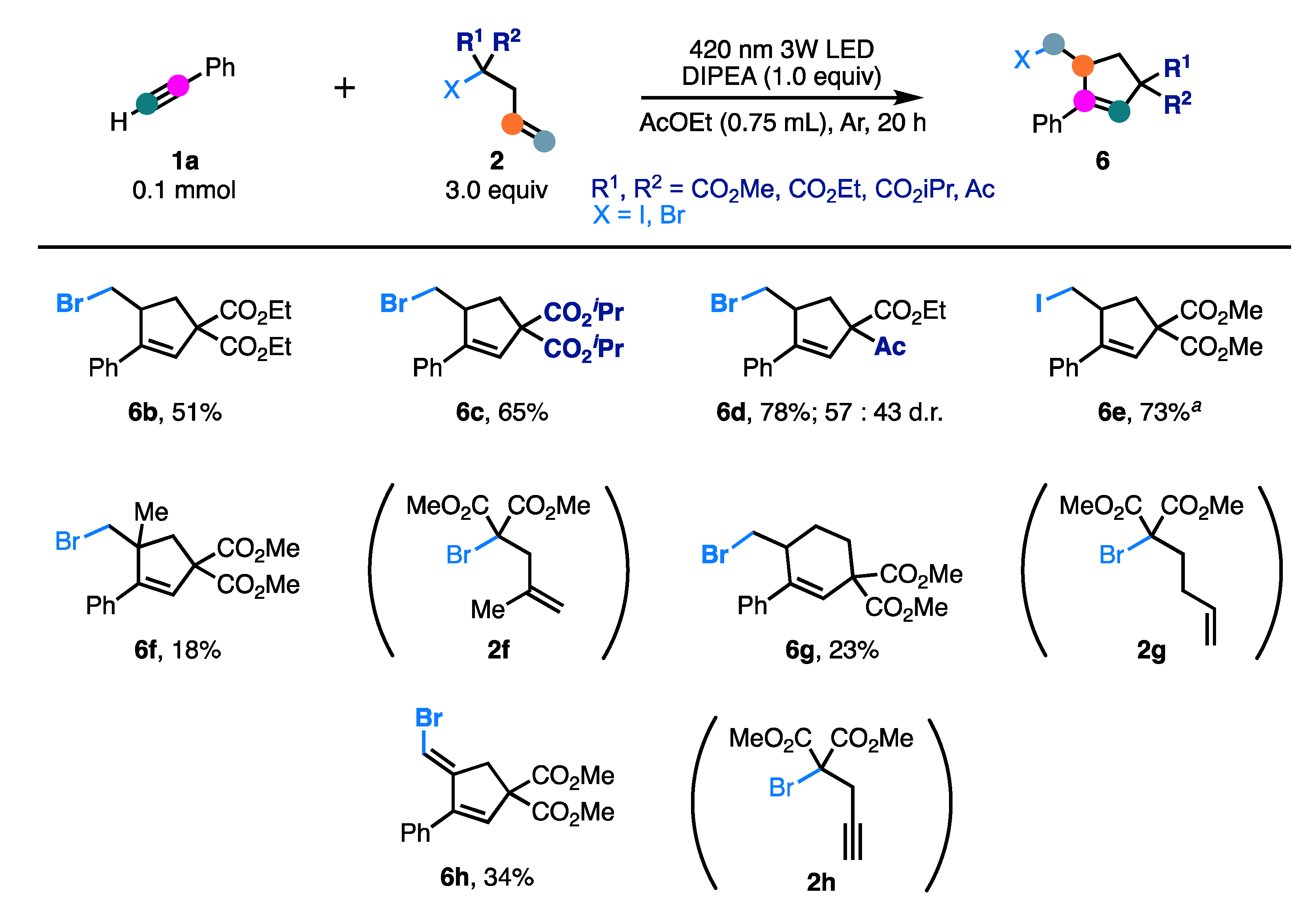
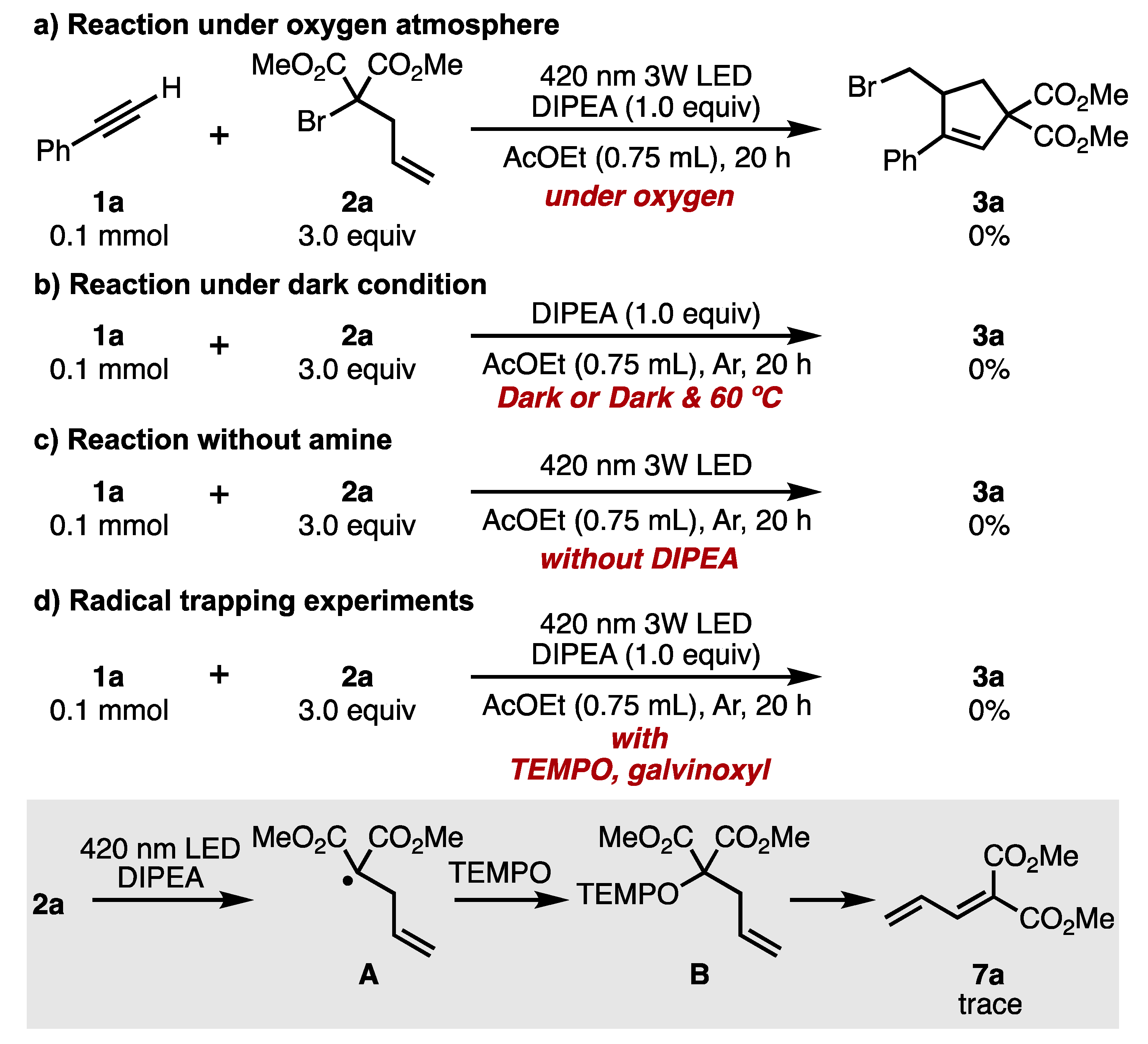
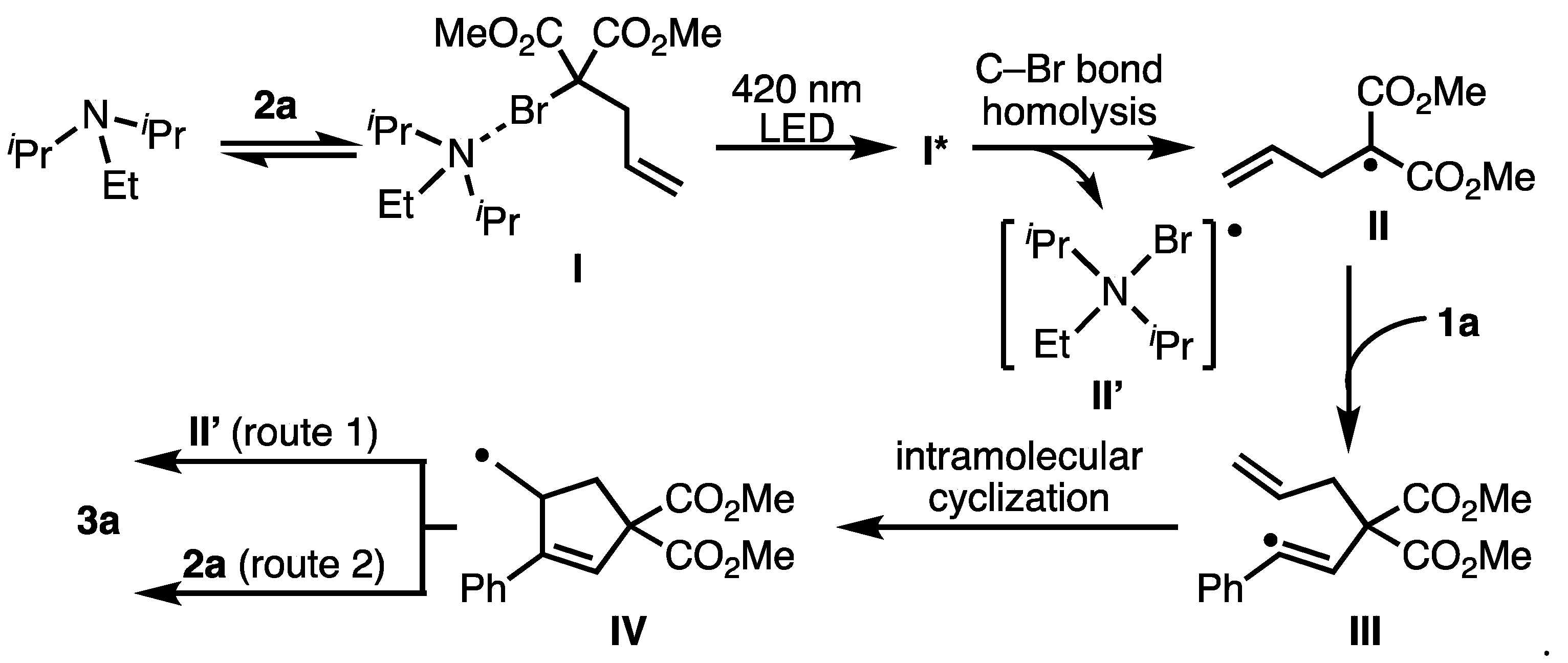
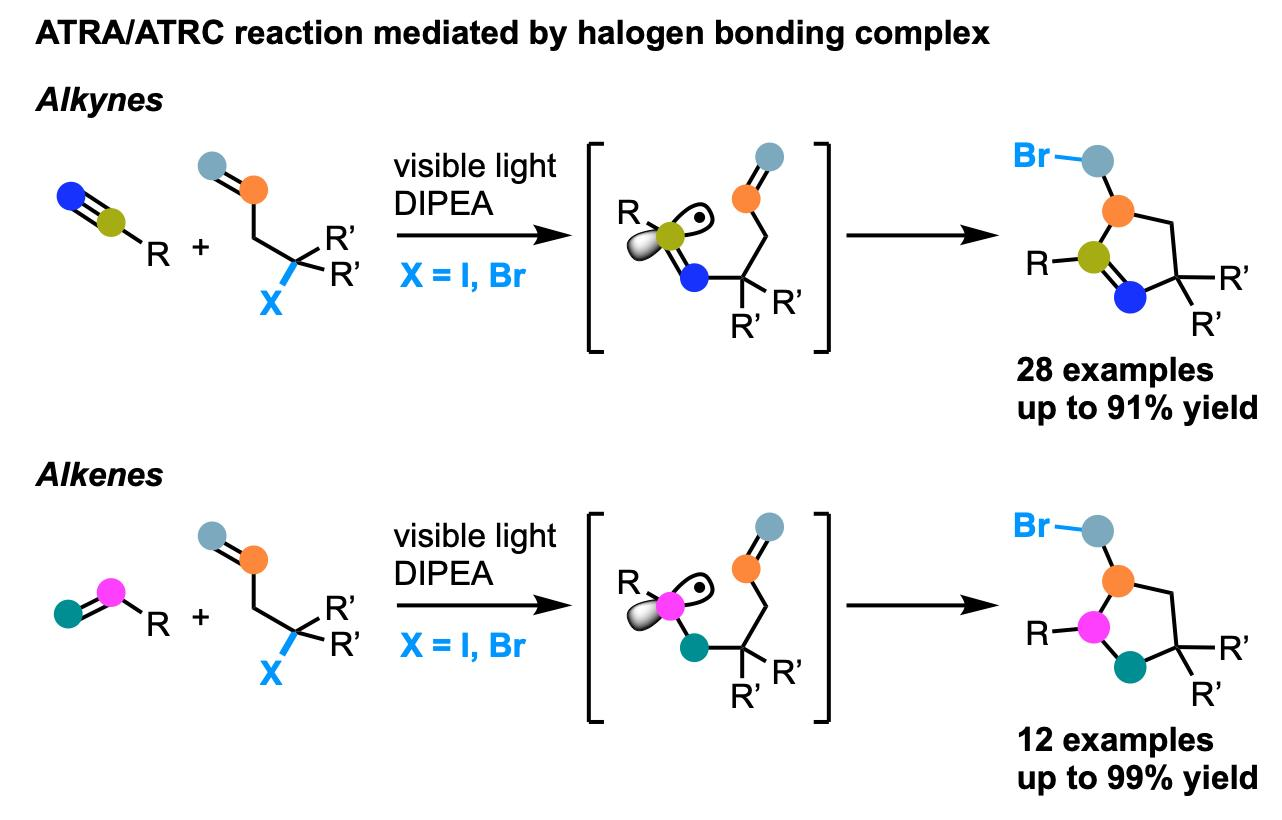
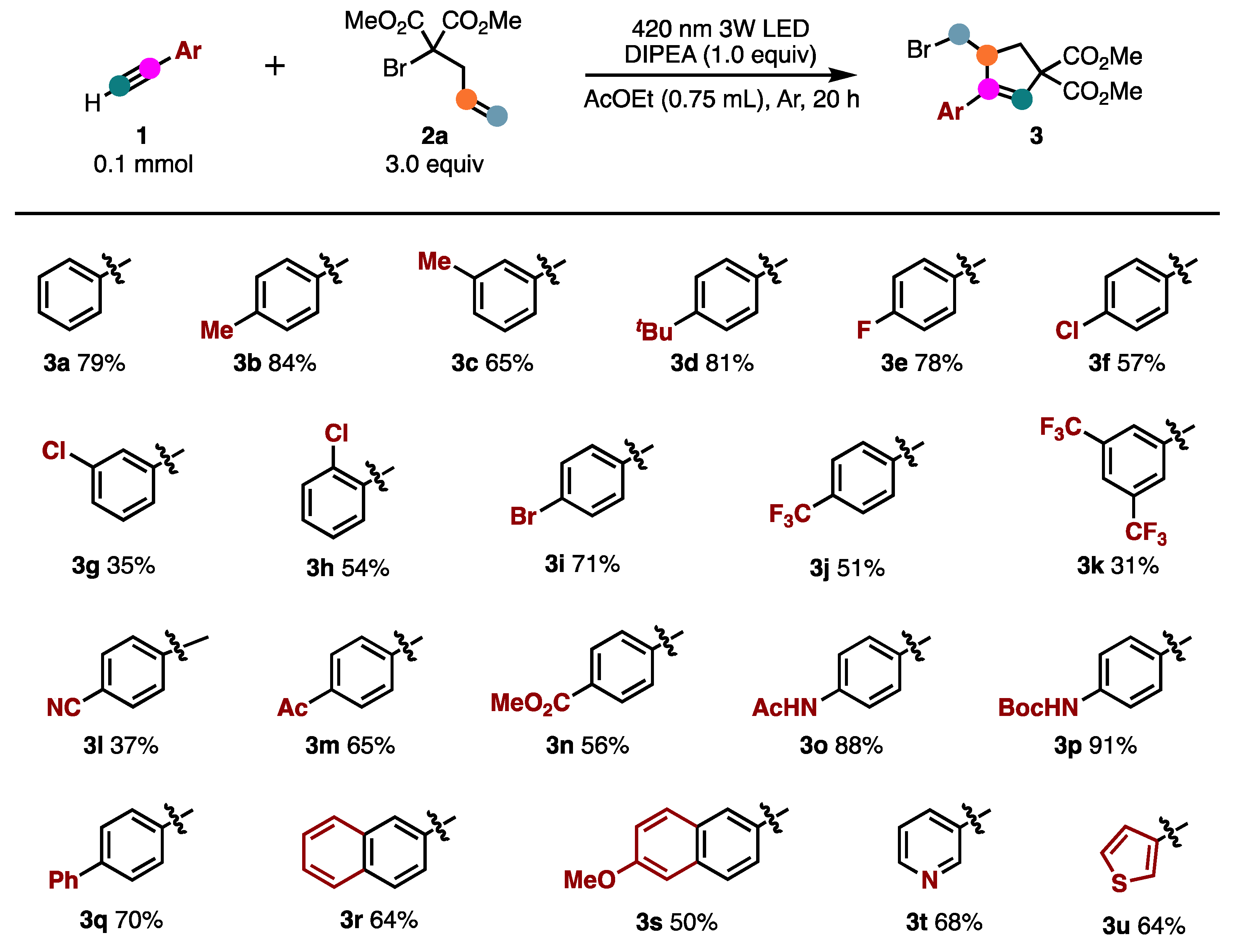
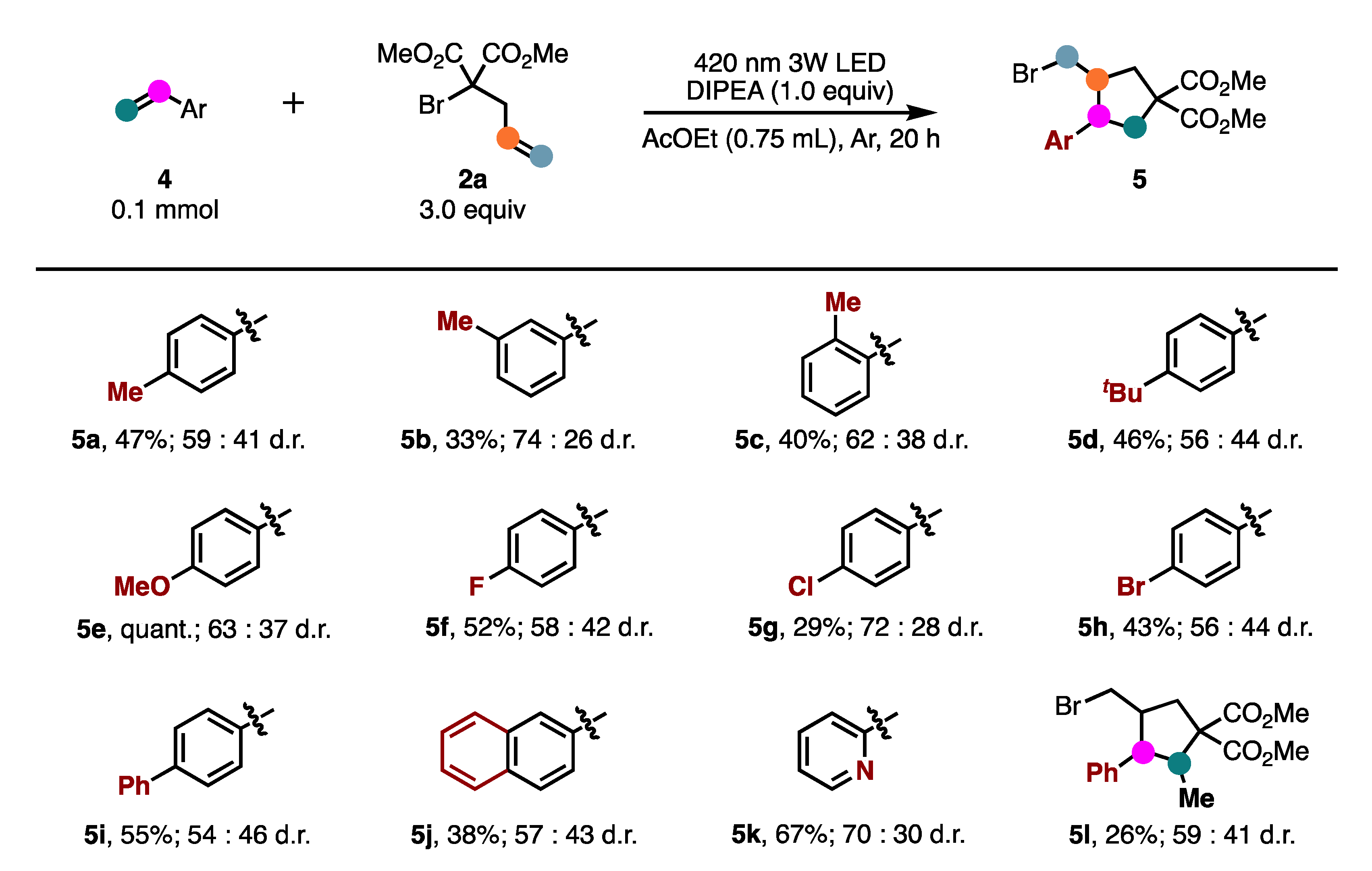
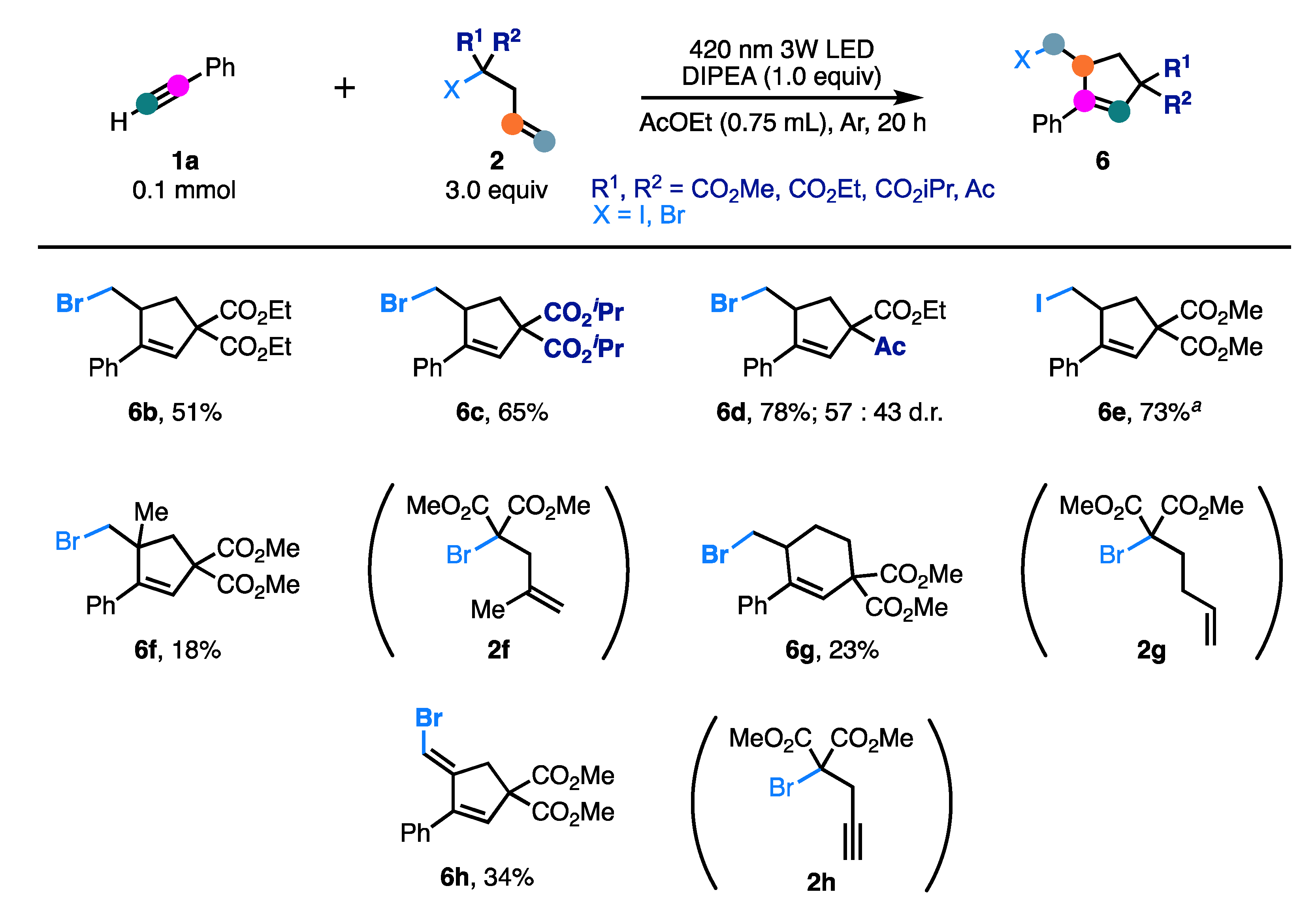
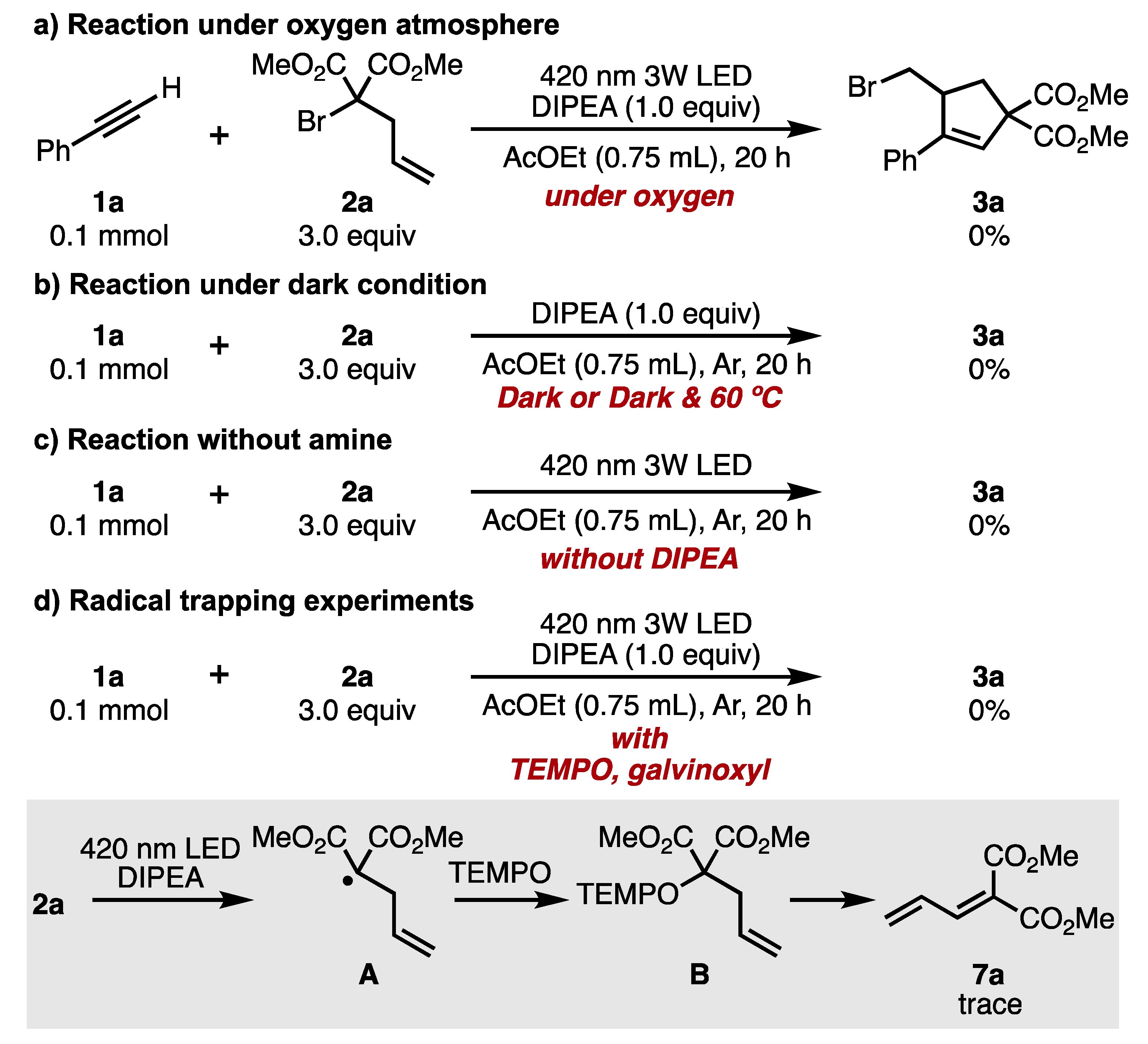
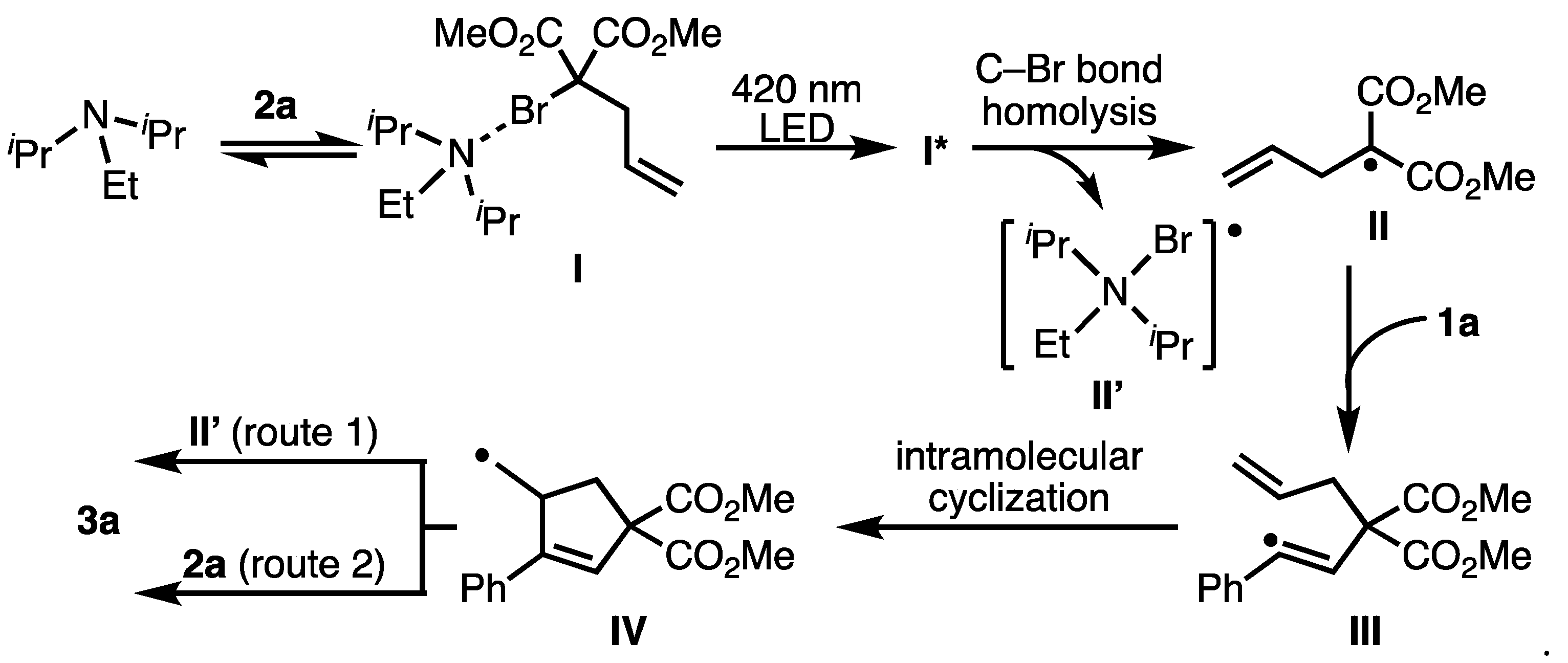
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. General Procedure for ATRA/ATRC Reaction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References and Notes
- Vince, R.; Hua, M. Synthesis and anti-HIV Activity of Carbocyclic 2,3-didehydro-2,3-dideoxy 2,6-disubstituted Purine Nucleosides. J. Med. Chem. 1990, 33, 17–21. [Google Scholar] [CrossRef]
- Bisacchi, G.S.; Chao, S.T.; Bachard, C.; Daris, J.P.; Innaimo, S.; Jacobs, G.A.; Kocy, O.; Lapointe, P.; Martel, A.; Merchant, Z.; et al. BMS-200475, A Novel Carbocyclic 2-deoxyguanosine analog with Potent and Selective Anti-hepatitis B Virus Qctivity in vitro. Biol. Med. Chem. Lett. 1997, 7, 127–132. [Google Scholar] [CrossRef]
- Meijere, A.; Kozhushkov, S.I.; Schill, H. Three-Membered-Ring-Based Molecular Architectures. Chem. Rev. 2006, 106, 4926–4996. [Google Scholar] [CrossRef] [PubMed]
- Bowman, W.R.; Bridge, C.F.; Brookes, P. Synthesis of Heterocycles by Radical Cyclisation. J. Chem. Soc. Perkin Trans. 2000, 1, 1–14. [Google Scholar] [CrossRef]
- Kamigaito, M.; Ando, T.; Sawamoto, M. Metal-Catalyzed Living Radical Polymerization. Chem. Rev. 2001, 101, 3689–3745. [Google Scholar] [CrossRef]
- Kharasch, M.S.; Jensen, E.V.; Urry, W.H. Addition of Carbon Tetrachloride and Chloroform to Olefines. Science 1945, 102, 128. [Google Scholar] [CrossRef]
- Kharasch, M.S.; Skell, P.S.; Fisher, P. Reactions of Atoms and Free Radicals in Solution. XII. The Addition of Bromo Esters to Olefins. J. Am. Chem. Soc. 1948, 70, 1055–1059. [Google Scholar] [CrossRef]
- Clark, A.J. Atom Transfer Radical Cyclisation Reactions Mediated by Copper Complexes. Chem. Soc. Rev. 2002, 31, 1–11. [Google Scholar] [CrossRef]
- Pintauer, T.; Matyjaszewski, K. Atom Transfer Radical Addition and Polymerization Reactions Catalyzed by ppm Amounts of Copper Complexes. Chem. Soc. Rev. 2008, 37, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, H.; Wakamatsu, H.; Itoh, K.; Tomo, Y.; Tsuji, J. New Regio and Stereoselective Preparation of Trichlorinated γ-Butyrolactones by Copper Catalyzed Cyclization of Allyl Trichloroacetates. Tetrahedron Lett. 1983, 24, 2395–2398. [Google Scholar] [CrossRef]
- Lee, G.M.; Parvez, M.; Weinreb, S.M. Intramolecular Metal Catalyzed Kharasch Cyclizations of Olefinic α-Halo Esters and Acids. Tetrahedron 1988, 44, 4671–4678. [Google Scholar] [CrossRef]
- Quayle, P.; Fengas, D.; Richards, S. Atom Transfer Radical Cyclisations Mediated by the Grubbs Ruthenium Metathesis Catalyst. Synlett 2003, 12, 1797–1800. [Google Scholar] [CrossRef]
- Monks, B.M.; Cook, S.P. Palladium-catalyzed Intramolecular Iodine-transfer Reactions in the Presence of β-Hydrogen Atoms. Angew. Chem. Int. Ed. 2013, 52, 14214–14218. [Google Scholar] [CrossRef] [PubMed]
- Boivin, J.; Yousfi, M.; Zard, S.Z. A Versatile Radical Based Synthesis of γ–Lactams using Nickel Powder/Acetic acid. Tetrahedron Lett. 1994, 35, 5629–5632. [Google Scholar] [CrossRef]
- Bag, D.; Kour, H.; Sawant, S.D. Photo-induced 1,2-carbohalofunctionalization of C-C Multiple Bonds via ATRA Pathway. Org. Biomol. Chem. 2021, 18, 8278–8293. [Google Scholar] [CrossRef]
- Wallentin, C.J.; Nguyen, J.D.; Finkbeiner, P.; Stephenson, C.R.J. Visible Light-Mediated Atom Transfer Radical Addition via Oxidative and Reductive Quenching of Photocatalysts. J. Am. Chem. Soc. 2012, 134, 8875–8884. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Cornella, J.; Julia-Hernandez, F.; Martin, R. Visible-Light-Promoted Atom Transfer Radical Cyclization of Unactivated Alkyl Iodides. ACS Catal. 2017, 7, 409–412. [Google Scholar] [CrossRef]
- Cheng, J.; Cheng, Y.; Xie, J.; Zhu, C. Photoredox Divergent 1,2-Difunctionalization of Alkenes with gem-Dibromides. Org. Lett. 2017, 19, 6452–6455. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Xu, G.; Xu, J.; Wang, Z.; Xu, P. A lutidine-promoted Photoredox Catalytic Atom-transfer Radical Cyclization Reaction for the Synthesis of 4-Bromo-3,3-dialkyl-octahydro-indol-2-ones. Chem. Commun. 2019, 56, 2206–2209. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.; Tang, D.; Li, H.; Xu, Y.; Yan, C.; Shi, Y.; Chen, X.; Zhu, S. Visible-Light-Driven Chlorotrifluoromethylative and Chlorotrichloromethylative Cyclizations of Enynes. J. Org. Chem. 2019, 84, 7509–7517. [Google Scholar] [CrossRef]
- Hou, H.; Xu, Y.; Yang, H.; Chen, X.; Yan, C.; Shi, Y.; Zhu, S. Visible-Light Mediated Hydrosilylative and Hydrophosphorylative Cyclizations of Enynes and Dienes. Org. Lett. 2020, 22, 1748–1753. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Yamaguchi, E.; Itoh, A. In Situ-Generated Halogen-Bonding Complex Enables Atom Transfer Radical Addition (ATRA) Reactions of Olefins. J. Org. Chem. 2020, 85, 10574–10583. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Kondo, T.; Yamaguchi, E.; Itoh, A. Photoinduced Atom Transfer Radical Addition Reaction of Olefins with α-Bromo Carbonyls. Chem. Pharm. Bull 2021, 69, 796–801. [Google Scholar] [CrossRef] [PubMed]
- The reaction temperature rose to about 30 °C after 20 h of LED light irradiation.
- At low concentrations, hydrogen radicals were abstracted from the amine or solvent before the intermolecular addition (ATRA) reaction proceeded, and the hydrogenated form of the malonate ester was preferentially formed. (Table 1, entry 5. 88% hydrogenated malonate was recovered.)
- Wilkinson, F.; Abdel-Shafi, A.A. Mechanism of Quenching of Triplet States by Oxygen: Biphenyl Derivatives in Acetonitrile. J. Phys. Chem. A 1997, 101, 5509–5516. [Google Scholar] [CrossRef]
- Sylla, M.; Joseph, D.; Chevallier, E.; Camara, C.; Dumas, F. A Simple and Direct Access to Ethylidene Malonates. Synthesis 2006, 6, 1045–1049. [Google Scholar]
- Gui, Q.; Hu, L.; Chen, X.; Liu, J.; Tan, Z. Stereoselective Synthesis of Vinylphosphonates and Phosphine Oxides via Silver-catalyzed Phosphorylation of Styrenes. Chem. Commun. 2015, 51, 13922–13924. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
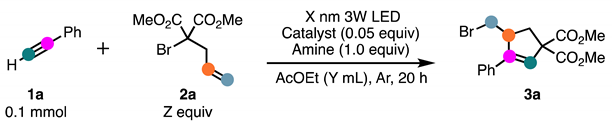
| Entry | Catalyst | Amine | X (nm) | Y (mL) | Z (equiv) | Yield (%) a |
|---|---|---|---|---|---|---|
| 1 | 4-Ph-pyridine | 380 | 1.0 | 2.5 | 53 | |
| 2 | 4-Ph-pyridine | 400 | 1.0 | 2.5 | 32 | |
| 3 | 4-Ph-pyridine | 420 | 1.0 | 2.5 | 57 (50) | |
| 4 | 4-Ph-pyridine | 450 | 1.0 | 2.5 | 0 | |
| 5 | 4-Ph-pyridine | 420 | 2.0 | 2.5 | 0 | |
| 6 | 4-Ph-pyridine | 420 | 0.75 | 2.5 | 60 (62) | |
| 7 | 4-Ph-pyridine | 420 | 0.25 | 2.5 | 47 | |
| 8 | 4-Ph-pyridine | DIPEA | 420 | 0.75 | 2.5 | 79 (71) |
| 9 | DIPEA | 420 | 0.75 | 2.5 | 89 (75) | |
| 10 | DIPEA | 420 | 0.75 | 2.0 | 69 | |
| 11 | DIPEA | 420 | 0.75 | 3.0 | 88 (79) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsuo, K.; Yoshitake, T.; Yamaguchi, E.; Itoh, A. Photoinduced Atom Transfer Radical Addition/Cyclization Reaction between Alkynes or Alkenes with Unsaturated α-Halogenated Carbonyls. Molecules 2021, 26, 6781. https://doi.org/10.3390/molecules26226781
Matsuo K, Yoshitake T, Yamaguchi E, Itoh A. Photoinduced Atom Transfer Radical Addition/Cyclization Reaction between Alkynes or Alkenes with Unsaturated α-Halogenated Carbonyls. Molecules. 2021; 26(22):6781. https://doi.org/10.3390/molecules26226781
Chicago/Turabian StyleMatsuo, Kazuki, Tadashi Yoshitake, Eiji Yamaguchi, and Akichika Itoh. 2021. "Photoinduced Atom Transfer Radical Addition/Cyclization Reaction between Alkynes or Alkenes with Unsaturated α-Halogenated Carbonyls" Molecules 26, no. 22: 6781. https://doi.org/10.3390/molecules26226781
APA StyleMatsuo, K., Yoshitake, T., Yamaguchi, E., & Itoh, A. (2021). Photoinduced Atom Transfer Radical Addition/Cyclization Reaction between Alkynes or Alkenes with Unsaturated α-Halogenated Carbonyls. Molecules, 26(22), 6781. https://doi.org/10.3390/molecules26226781