
Photoreactivity of an Exemplary Anthracene Mixture Revealed by NMR Studies, including a Kinetic Approach


Abstract
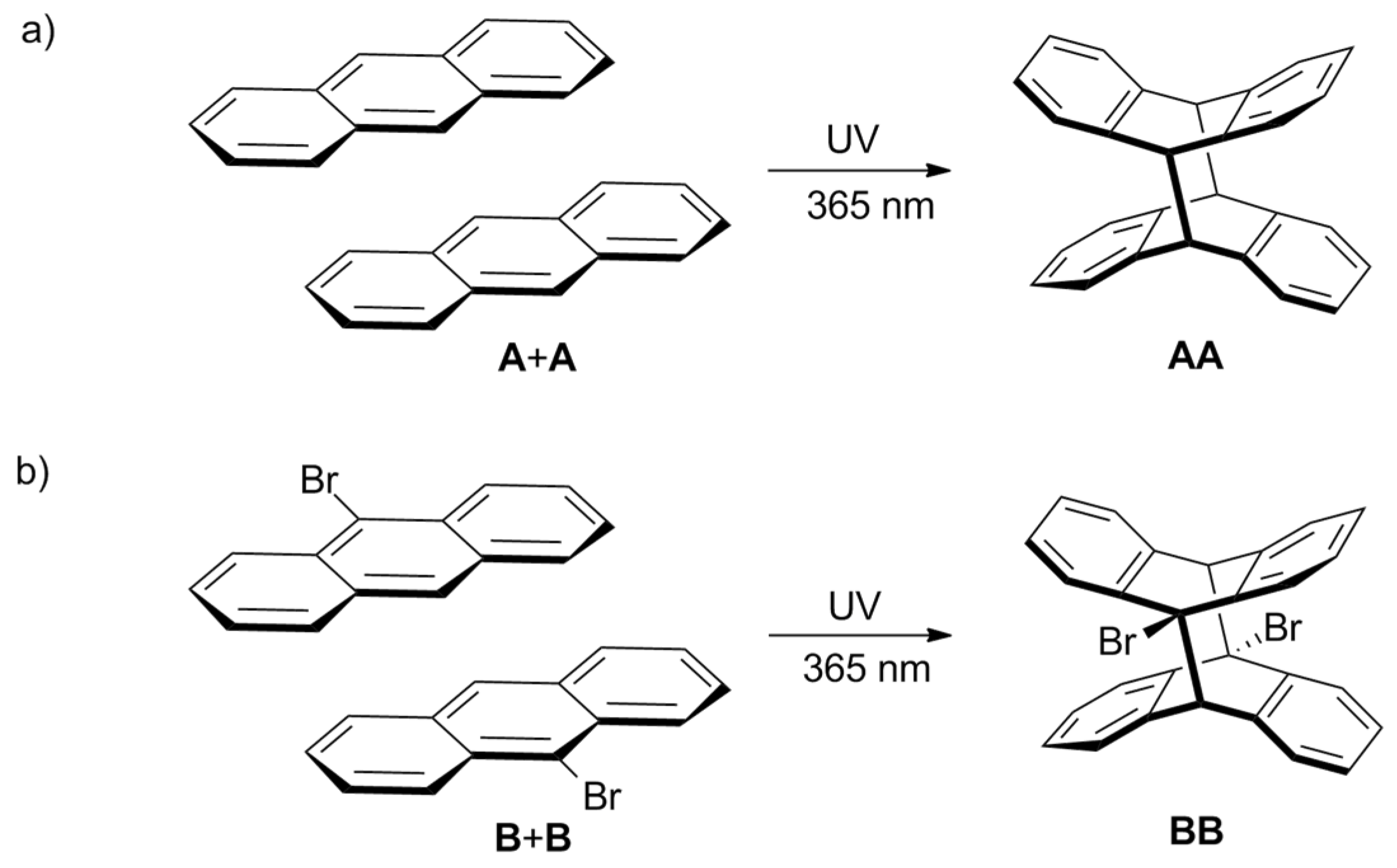
:1. Introduction
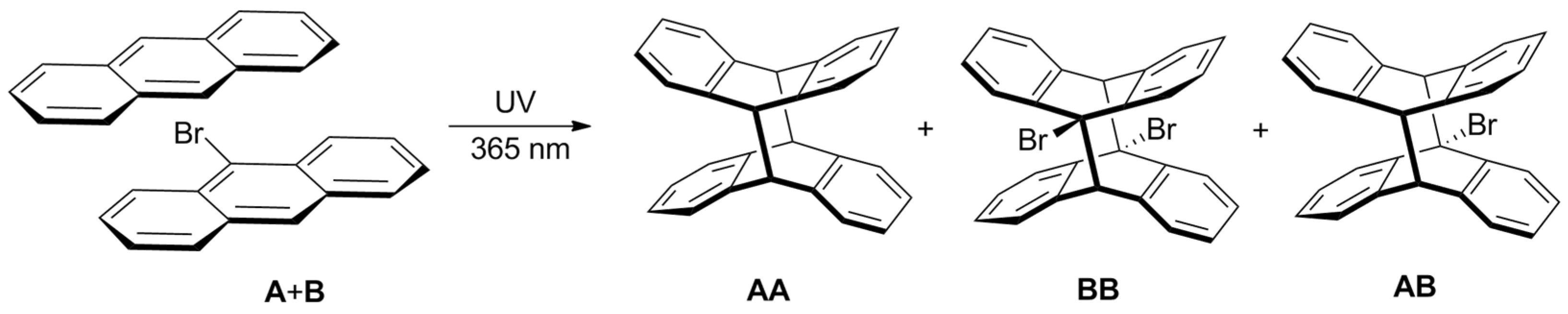
2. Results and Discussion
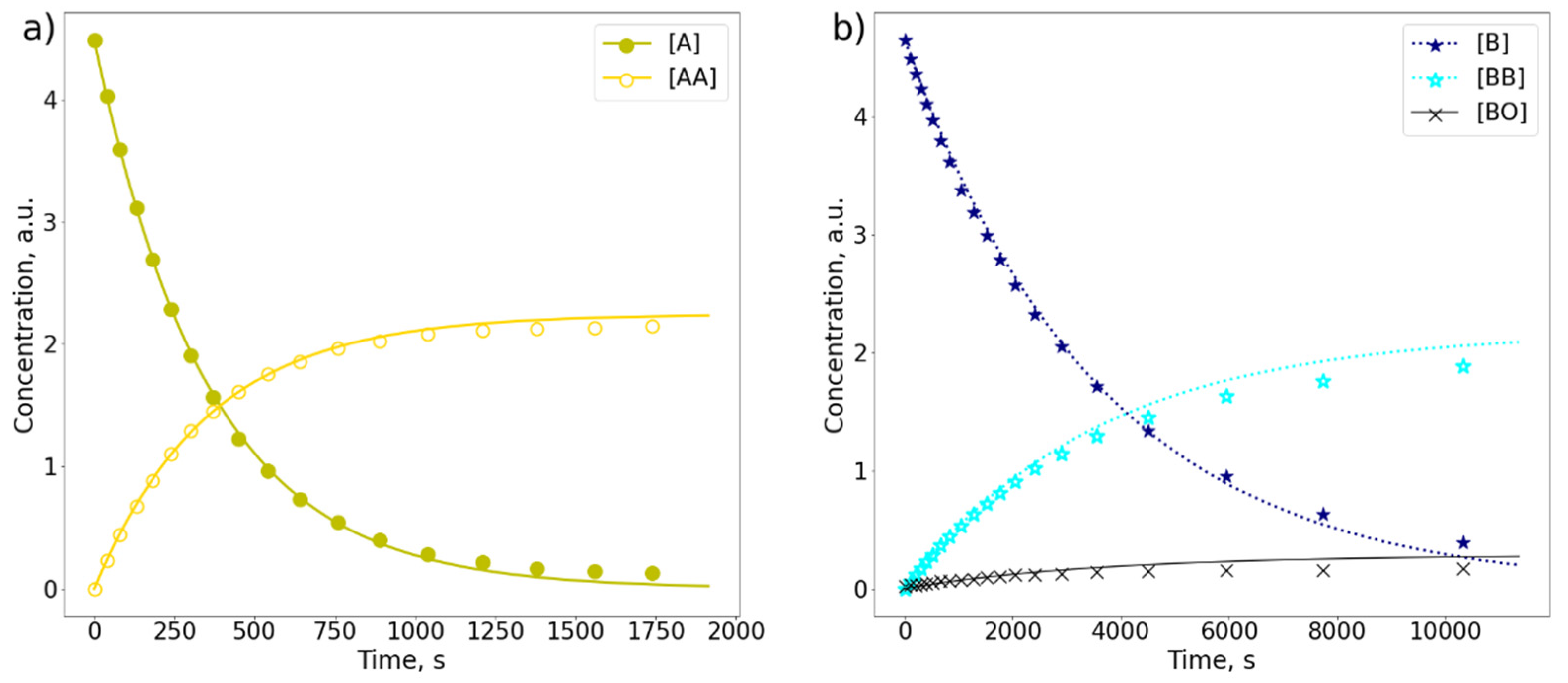
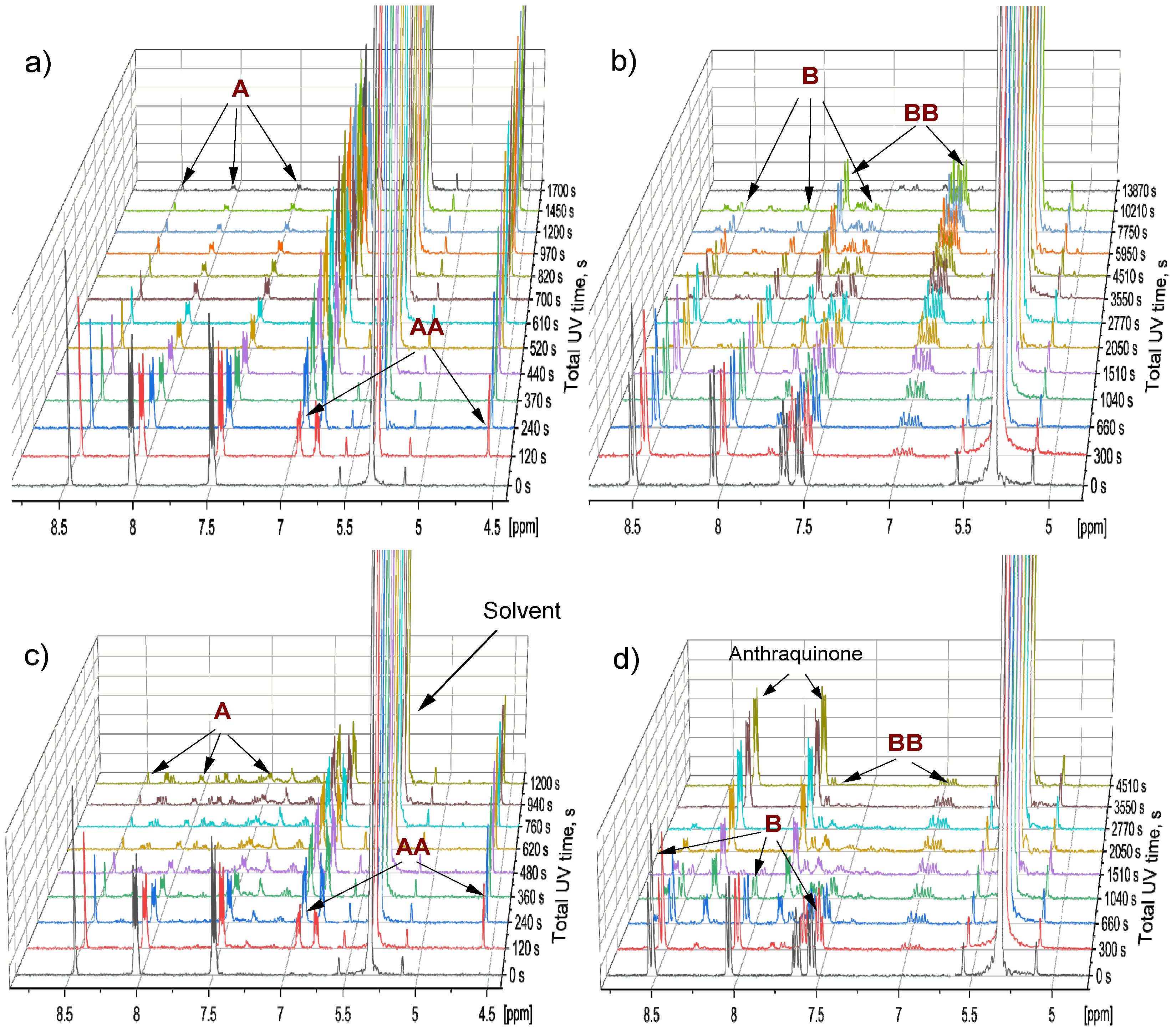
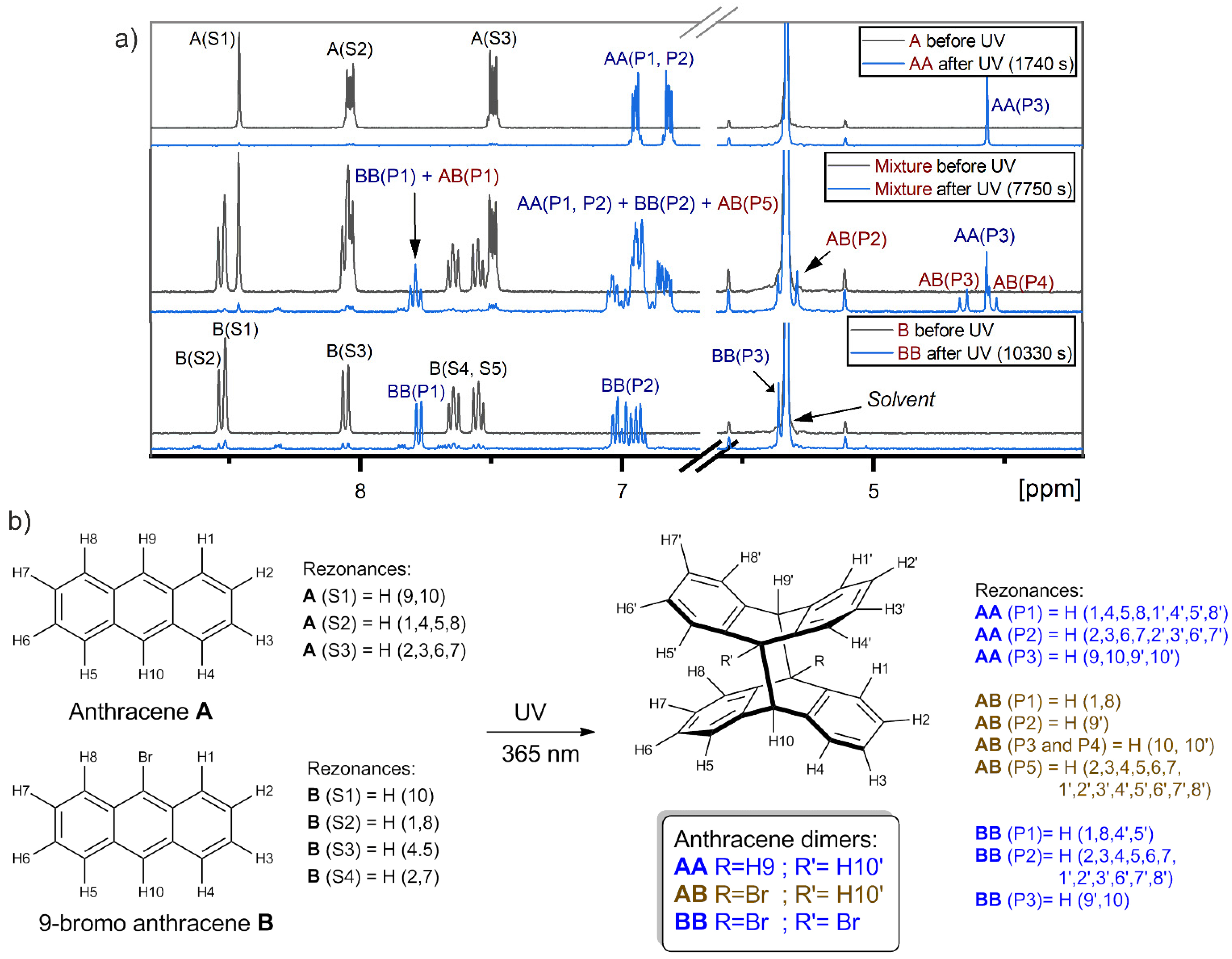
2.1. Comparison of A and B in Separate Samples
2.2. The Effect of Experimental Conditions
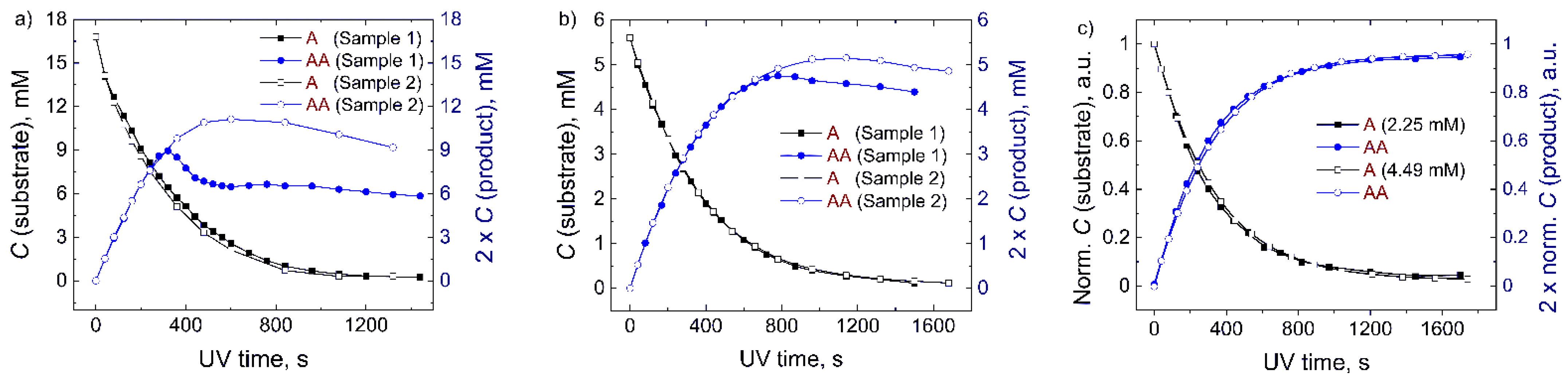
2.2.1. The Effect of the Concentration of Anthracenes
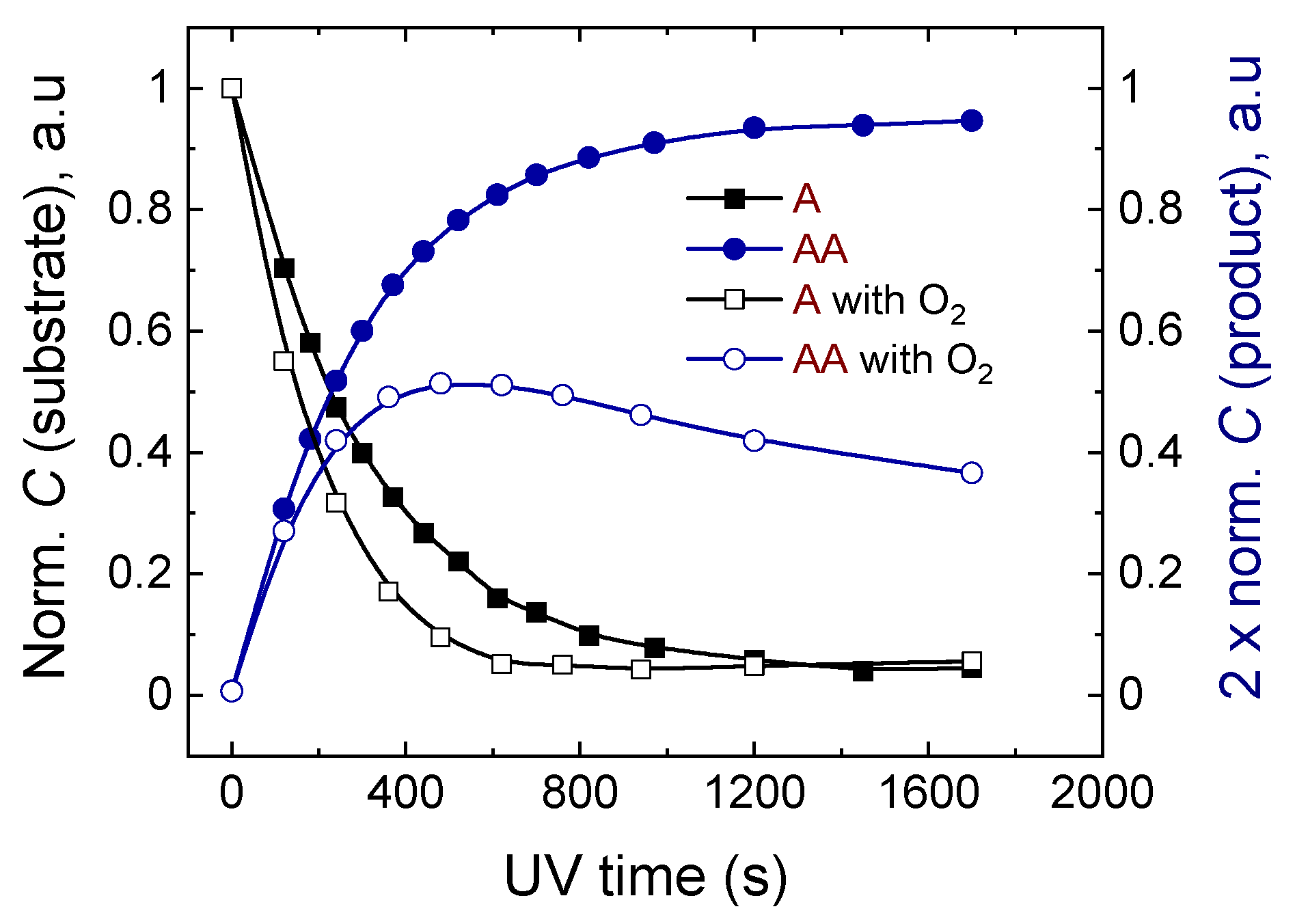
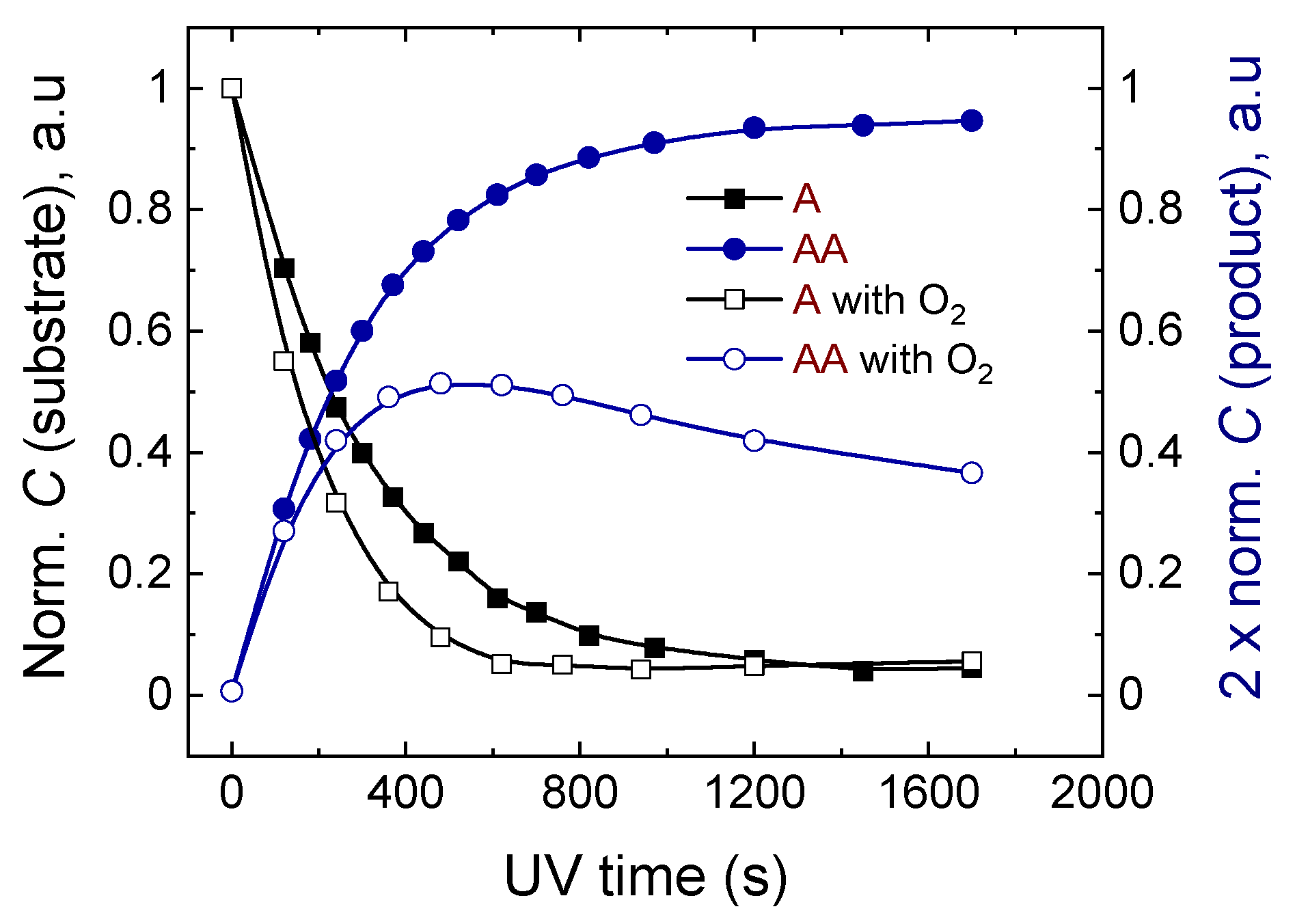
2.2.2. The Effect of Oxygenation of the Sample
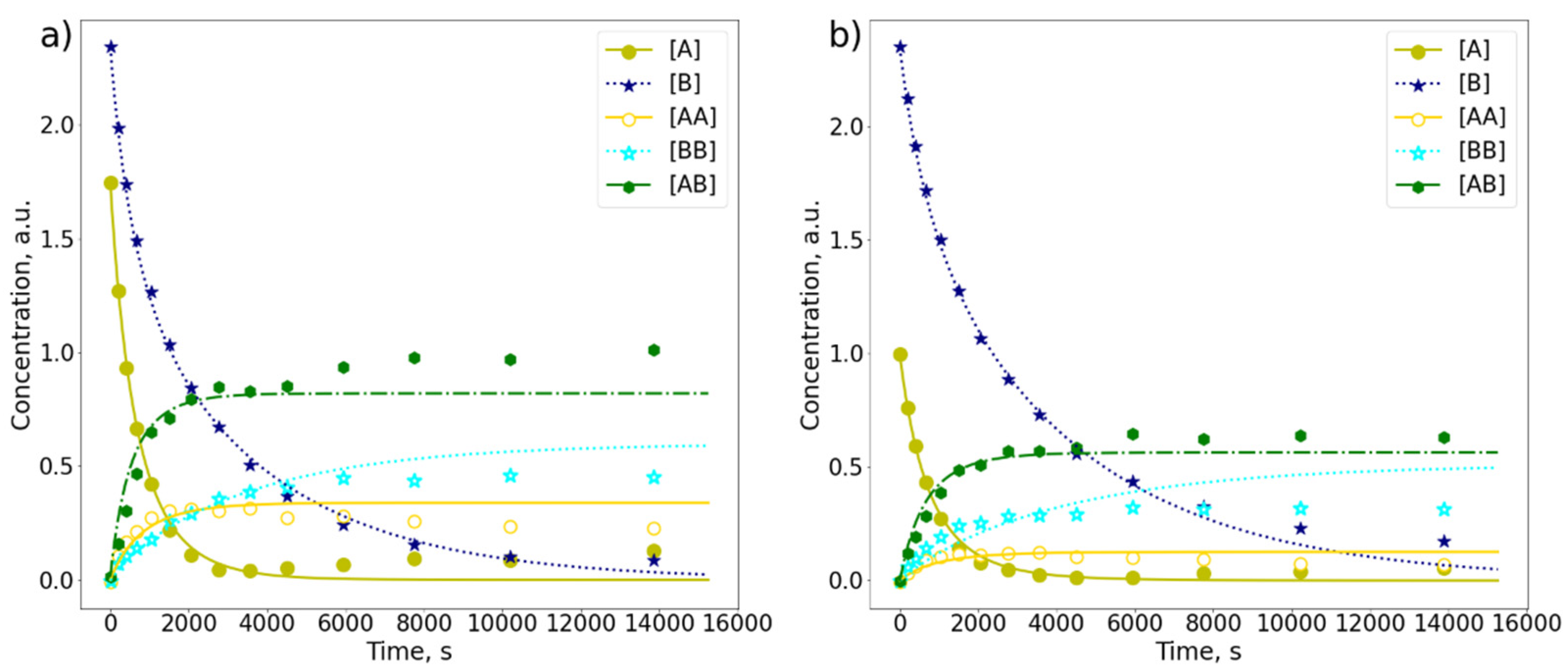
2.3. Comparison of A and B in the Mixture
3. Materials and Methods
3.1. Samples
3.2. UV Illumination
3.3. NMR Experiment
3.4. Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Clar, E. Vorschläge Zur Nomenklatur Kondensierter Ringsysteme (Aromatische Kohlenwasserstoffe, XXVI. Mitteil.). Ber. Dtsch. Chem. Ges. 1939, 72, 2137–2139. [Google Scholar] [CrossRef]
- Tönshoff, C.; Bettinger, H.F. Pushing the Limits of Acene Chemistry: The Recent Surge of Large Acenes. Chem. Eur. J. 2021, 27, 3193–3212. [Google Scholar] [CrossRef]
- Brancart, J.; Van Damme, J.; Du Prez, F.; Van Assche, G. Substituent Effect on the Thermophysical Properties and Thermal Dissociation Behaviour of 9-Substituted Anthracene Derivatives. Phys. Chem. Chem. Phys. 2021, 23, 2252–2263. [Google Scholar] [CrossRef]
- Müller, M.; Ahrens, L.; Brosius, V.; Freudenberg, J.; Bunz, U.H.F. Unusual Stabilization of Larger Acenes and Heteroacenes. J. Mater. Chem. C 2019, 7, 14011–14034. [Google Scholar] [CrossRef]
- Ye, Q.; Chi, C. Recent Highlights and Perspectives on Acene Based Molecules and Materials. Chem. Mater. 2014, 26, 4046–4056. [Google Scholar] [CrossRef]
- Dong, S.; Ong, A.; Chi, C. Photochemistry of Various Acene Based Molecules. J. Photochem. Photobiol. C Photochem. Rev. 2019, 38, 27–46. [Google Scholar] [CrossRef]
- Baviera, G.S.; Donate, P.M. Recent Advances in the Syntheses of Anthracene Derivatives. Beilstein J. Org. Chem. 2021, 17, 2028–2050. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa, M.; Klosterman, J.K. Molecular Architectures of Multi-Anthracene Assemblies. Chem. Soc. Rev. 2014, 43, 1885–1898. [Google Scholar] [CrossRef]
- Bouas-Laurent, H.; Castellan, A.; Desvergne, J.-P.; Lapouyade, R. Photodimerization of Anthracenes in Fluid Solutions: (Part 2) Mechanistic Aspects of the Photocycloaddition and of the Photochemical and Thermal Cleavage. Chem. Soc. Rev. 2001, 30, 248–263. [Google Scholar] [CrossRef]
- Breton, G.W.; Vang, X. Photodimerization of Anthracene. J. Chem. Educ. 1998, 75, 81. [Google Scholar] [CrossRef]
- Fidder, H.; Lauer, A.; Freyer, W.; Koeppe, B.; Heyne, K. Photochemistry of Anthracene-9,10-Endoperoxide. J. Phys. Chem. A 2009, 113, 6289–6296. [Google Scholar] [CrossRef]
- Zhu, J.; Zou, J.; Zhang, J.; Sun, Y.; Dong, X.; Zhang, Q. An Anthracene Functionalized BODIPY Derivative with Singlet Oxygen Storage Ability for Photothermal and Continuous Photodynamic Synergistic Therapy. J. Mater. Chem. B 2019, 7, 3303–3309. [Google Scholar] [CrossRef]
- Fudickar, W.; Linker, T. Reversible Photooxygenation of Alkynylanthracenes: Chemical Generation of Singlet Oxygen under Very Mild Conditions. Chem. Eur. J. 2011, 17, 13661–13664. [Google Scholar] [CrossRef]
- Geroldinger, G.; Tonner, M.; Fudickar, W.; De Sarkar, S.; Dighal, A.; Monzote, L.; Staniek, K.; Linker, T.; Chatterjee, M.; Gille, L. Activation of Anthracene Endoperoxides in Leishmania and Impairment of Mitochondrial Functions. Molecules 2018, 23, 1680. [Google Scholar] [CrossRef] [Green Version]
- Klaper, M.; Wessig, P.; Linker, T. Base Catalysed Decomposition of Anthracene Endoperoxide. Chem. Commun. 2016, 52, 1210–1213. [Google Scholar] [CrossRef] [Green Version]
- Khurana, B.; Gierlich, P.; Meindl, A.; Gomes-da-Silva, L.C.; Senge, M.O. Hydrogels: Soft Matters in Photomedicine. Photochem. Photobiol. Sci. 2019, 18, 2613–2656. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.S.; Chorociejus, G.; Angeli, J.P.F.; Vila Verde, G.; Aquino, G.L.B.; Ronsein, G.E.; De Oliveira, M.C.B.; Barbosa, L.F.; Medeiros, M.H.G.; Greer, A.; et al. Heck Reaction Synthesis of Anthracene and Naphthalene Derivatives as Traps and Clean Chemical Sources of Singlet Molecular Oxygen in Biological Systems. Photochem. Photobiol. Sci. 2020, 19, 1590–1602. [Google Scholar] [CrossRef]
- Van Damme, J.; Du Prez, F. Anthracene-Containing Polymers toward High-End Applications. Prog. Polym. Sci. 2018, 82, 92–119. [Google Scholar] [CrossRef]
- Kislyak, A.; Frisch, H.; Gernhardt, M.; Van Steenberge, P.H.M.; D’Hooge, D.R.; Barner-Kowollik, C. Time-Dependent Differential and Integral Quantum Yields for Wavelength-Dependent [4+4] Photocycloadditions. Chem. Eur. J. 2020, 26, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Slifkin, M.A. Charge Transfer and Excimer Formation. Nature 1963, 200, 766–767. [Google Scholar] [CrossRef]
- Das, A.; Danao, A.; Banerjee, S.; Raj, A.M.; Sharma, G.; Prabhakar, R.; Srinivasan, V.; Ramamurthy, V.; Sen, P. Dynamics of Anthracene Excimer Formation within a Water-Soluble Nanocavity at Room Temperature. J. Am. Chem. Soc. 2021, 143, 2025–2036. [Google Scholar] [CrossRef] [PubMed]
- Applequist, D.E.; Litle, R.L.; Friedrich, E.; Wall, R.E. Anthracene Photodimers. I. Elimination and Substitution Reactions of the Photodimer of 9-Bromoanthracene1. J. Am. Chem. Soc. 1959, 81, 452–456. [Google Scholar] [CrossRef]
- Arumugam, S.; Vutukuri, D.R.; Thayumanavan, S.; Ramamurthy, V. A Styrene Based Water Soluble Polymer as a Reaction Medium for Photodimerization of Aromatic Hydrocarbons in Water. J. Photochem. Photobiol. A Chem. 2007, 185, 168–171. [Google Scholar] [CrossRef]
- Rao, M.; Wu, W.; Yang, C. Effects of Temperature and Host Concentration on the Supramolecular Enantiodifferentiating [4 + 4] Photodimerization of 2-Anthracenecarboxylate through Triplet-Triplet Annihilation Catalyzed by Pt-Modified Cyclodextrins. Molecules 2019, 24, 1502. [Google Scholar] [CrossRef] [Green Version]
- Rao, M.; Wu, W.; Yang, C. Recent Progress on the Enantioselective Excited-State Photoreactions by Pre-Arrangement of Photosubstrate(s). Green Synth. Catal. 2021, 2, 131–144. [Google Scholar] [CrossRef]
- Geiger, T.; Haupt, A.; Maichle-Mössmer, C.; Schrenk, C.; Schnepf, A.; Bettinger, H.F. Synthesis and Photodimerization of 2- and 2,3-Disubstituted Anthracenes: Influence of Steric Interactions and London Dispersion on Diastereoselectivity. J. Org. Chem. 2019, 84, 10120–10135. [Google Scholar] [CrossRef]
- Takaguchi, Y.; Tajima, T.; Yanagimoto, Y.; Tsuboi, S.; Ohta, K.; Motoyoshiya, J.; Aoyama, H. Self-Assembly and Regioselective Photodimerization of Anthracene Having a Dendritic Substituent. Org. Lett. 2003, 5, 1677–1679. [Google Scholar] [CrossRef]
- Maturi, M.M.; Fukuhara, G.; Tanaka, K.; Kawanami, Y.; Mori, T.; Inoue, Y.; Bach, T. Enantioselective [4+4] Photodimerization of Anthracene-2,6-Dicarboxylic Acid Mediated by a C2-Symmetric Chiral Template. Chem. Commun. 2016, 52, 1032–1035. [Google Scholar] [CrossRef] [Green Version]
- Lamm, J.-H.; Glatthor, J.; Weddeling, J.-H.; Mix, A.; Chmiel, J.; Neumann, B.; Stammler, H.-G.; Mitzel, N.W. Polyalkynylanthracenes–Syntheses, Structures and Their Behaviour towards UV Irradiation. Org. Biomol. Chem. 2014, 12, 7355–7365. [Google Scholar] [CrossRef]
- Kan, L.; Cheng, H.; Li, B.; Zhang, X.; Wang, Q.; Wei, H.; Ma, N. Anthracene Dimer Crosslinked Polyurethanes as Mechanoluminescent Polymeric Materials. New J. Chem. 2019, 43, 2658–2664. [Google Scholar] [CrossRef]
- Grossmann, L.; King, B.T.; Reichlmaier, S.; Hartmann, N.; Rosen, J.; Heckl, W.M.; Björk, J.; Lackinger, M. On-Surface Photopolymerization of Two-Dimensional Polymers Ordered on the Mesoscale. Nat. Chem. 2021, 13, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Goswami, M.; Zhang, Y.; Liu, T.; Zhang, J.; Kessler, M.R.; Wang, L.; Rios, O. Combined Light- and Heat-Induced Shape Memory Behavior of Anthracene-Based Epoxy Elastomers. Sci. Rep. 2020, 10, 20214. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fang, D.; Fu, T.; Ali, M.U.; Shi, Y.; He, Y.; Hu, Z.; Yan, C.; Mei, Z.; Meng, H. Anthracene Derivative Based Multifunctional Liquid Crystal Materials for Optoelectronic Devices. Mater. Chem. Front. 2020, 4, 3546–3555. [Google Scholar] [CrossRef]
- Chen, M.; Yan, L.; Zhao, Y.; Murtaza, I.; Meng, H.; Huang, W. Anthracene-Based Semiconductors for Organic Field-Effect Transistors. J. Mater. Chem. C 2018, 6, 7416–7444. [Google Scholar] [CrossRef]
- Tümay, S.O.; Irani-Nezhad, M.H.; Khataee, A. Multi-Anthracene Containing Fluorescent Probe for Spectrofluorimetric Iron Determination in Environmental Water Samples. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2021, 248, 119250. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Moscoso, A.; Ballester, P. Light-Responsive Molecular Containers. Chem. Commun. 2017, 53, 4635–4652. [Google Scholar] [CrossRef]
- Saura, A.V.; Marín, M.J.; Burguete, M.I.; Russell, D.A.; Galindo, F.; Luis, S.V. The Synthesis of New Fluorescent Bichromophoric Compounds as Ratiometric PH Probes for Intracellular Measurements. Org. Biomol. Chem. 2015, 13, 7736–7749. [Google Scholar] [CrossRef] [PubMed]
- Duprey, J.-L.H.A.; Bassani, D.M.; Hyde, E.I.; Jonusauskas, G.; Ludwig, C.; Rodger, A.; Spencer, N.; Vyle, J.S.; Wilkie, J.; Zhao, Z.-Y.; et al. Rationalisation of a Mechanism for Sensing Single Point Variants in Target DNA Using Anthracene-Tagged Base Discriminating Probes. Org. Biomol. Chem. 2018, 16, 6576–6585. [Google Scholar] [CrossRef] [Green Version]
- Soylemez, S.; Goker, S.; Toppare, L. A Newly Designed Anthracene and Isoindigo Based Polymer: Synthesis{,} Electrochemical Characterization and Biosensor Applications. New J. Chem. 2019, 43, 13979–13984. [Google Scholar] [CrossRef]
- Zhou, W.; Fang, Y.; Wu, X.; Han, Y.; Yang, J.; Shen, L.; Song, Y. Anthracene Derivatives as Broadband Nonlinear Optical Materials: Nonlinear Absorption and Excited-State Dynamics Analysis. RSC Adv. 2020, 10, 19974–19981. [Google Scholar] [CrossRef]
- Zhou, W.; Chen, Y.; Yu, Q.; Li, P.; Chen, X.; Liu, Y. Photo-Responsive Cyclodextrin/Anthracene/Eu3+ Supramolecular Assembly for a Tunable Photochromic Multicolor Cell Label and Fluorescent Ink. Chem. Sci. 2019, 10, 3346–3352. [Google Scholar] [CrossRef] [Green Version]
- Pitzer, L.; Schäfers, F.; Glorius, F. Rapid Assessment of the Reaction-Condition-Based Sensitivity of Chemical Transformations. Angew. Chem. Int. Ed. 2019, 58, 8572–8576. [Google Scholar] [CrossRef] [PubMed]
- Paululat, T.; Rabe, M.; Berdnikova, D.V. Modification of an NMR Probe for Monitoring of Photoreactions. J. Magn. Reson. 2021, 327, 106990. [Google Scholar] [CrossRef]
- Dobkowski, J.; Gorski, A.; Kijak, M.; Pietrzak, M.; Redeckas, K.; Vengris, M. Combined Picosecond Time-Resolved UV–Vis and NMR Techniques Used for Investigation of the Excited State Intramolecular Triplet–Triplet Energy Transfer. J. Phys. Chem. A 2019, 123, 6978–6985. [Google Scholar] [CrossRef] [PubMed]
- Gołowicz, D.; Kazimierczuk, K.; Urbańczyk, M.; Ratajczyk, T. Monitoring Hydrogenation Reactions Using Benchtop 2D NMR with Extraordinary Sensitivity and Spectral Resolution. ChemistryOpen 2019, 8, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Gomes, B.F.; Lobo, C.M.S.; Colnago, L.A. Monitoring Electrochemical Reactions in Situ with Low Field NMR: A Mini-Review. Appl. Sci. 2019, 9, 498. [Google Scholar] [CrossRef] [Green Version]
- Skubi, K.L.; Swords, W.B.; Hofstetter, H.; Yoon, T.P. LED-NMR Monitoring of an Enantioselective Catalytic [2 + 2] Photocycloaddition. ChemPhotoChem 2020, 4, 685–690. [Google Scholar] [CrossRef]
- Andrade, M.A.; Martins, L.M.D.R.S. New Trends in C–C Cross-Coupling Reactions: The Use of Unconventional Conditions. Molecules 2020, 25, 5506. [Google Scholar] [CrossRef] [PubMed]
- Chanmungkalakul, S.; Ervithayasuporn, V.; Boonkitti, P.; Phuekphong, A.; Prigyai, N.; Kladsomboon, S.; Kiatkamjornwong, S. Anion Identification Using Silsesquioxane Cages. Chem. Sci. 2018, 9, 7753–7765. [Google Scholar] [CrossRef] [Green Version]
- Kislyak, A.; Kodura, D.; Frisch, H.; Feist, F.; Van Steenberge, P.H.M.; Barner-Kowollik, C.; D’Hooge, D.R. A Holistic Approach for Anthracene Photochemistry Kinetics. Chem. Eng. J. 2020, 402, 126259. [Google Scholar] [CrossRef]
- Rutnakornpituk, M.; Ngamdee, P. Surface and Mechanical Properties of Microporous Membranes of Poly(Ethylene Glycol)–Polydimethylsiloxane Copolymer/Chitosan. Polymer 2006, 47, 7909–7917. [Google Scholar] [CrossRef]
- Han, D.; Lu, H.; Li, W.; Li, Y.; Feng, S. Light- and Heat-Triggered Reversible Luminescent Materials Based on Polysiloxanes with Anthracene Groups. RSC Adv. 2017, 7, 56489–56495. [Google Scholar] [CrossRef] [Green Version]
- Brancart, J.; Van Damme, J.; Du Prez, F.; Van Assche, G. Thermal Dissociation of Anthracene Photodimers in the Condensed State: Kinetic Evaluation and Complex Phase Behaviour. Phys. Chem. Chem. Phys. 2020, 22, 17306–17313. [Google Scholar] [CrossRef]
- Huynh, V.N.; Leitner, M.; Bhattacharyya, A.; Uhlstein, L.; Kreitmeier, P.; Sakrausky, P.; Rehbein, J.; Reiser, O. Diels–Alder Reactions and Electrophilic Substitutions with Atypical Regioselectivity Enable Functionalization of Terminal Rings of Anthracene. Commun. Chem. 2020, 3, 158. [Google Scholar] [CrossRef]
- Nitschke, P.; Lokesh, N.; Gschwind, R.M. Combination of Illumination and High Resolution NMR Spectroscopy: Key Features and Practical Aspects, Photochemical Applications, and New Concepts. Prog. Nucl. Magn. Reson. Spectrosc. 2019, 114–115, 86–134. [Google Scholar] [CrossRef] [PubMed]
- Kuprov, I.; Hore, P.J. Uniform Illumination of Optically Dense NMR Samples. J. Magn. Reson. 2004, 171, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Pietrzak, M.; Dobkowski, J.; Gorski, A.; Gawinkowski, S.; Kijak, M.; Luboradzki, R.; Hansen, P.E.; Waluk, J. Arresting Consecutive Steps of a Photochromic Reaction: Studies of β-Thioxoketones Combining Laser Photolysis with NMR Detection. Phys. Chem. Chem. Phys. 2014, 16, 9128–9137. [Google Scholar] [CrossRef] [Green Version]
- Roelfs, M.; Kroon, P.C. TBuLi/Symfit: Symfit 0.5.3; Zenodo: Genève, Switzerland, 2020. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | kA, s−1 | kB, s−1 | kAB, s−1 |
|---|---|---|---|
| A (4.5 mM) | (2.8 ± 0.03) × 10−3 | - | - |
| B (4.5 mM) | - | (2.60 ± 0.06) × 10−4 | - |
| Mixture of A:B = 1:1.3 | (5.01 ± 0.83) × 10−4 | (2.15 ± 0.26) × 10−4 | (3.88 ± 0.41) × 10−4 |
| Mixture of A:B = 1:2.3 | (2.81 ± 1.04) × 10−4 | (1.32 ± 0.19) × 10−4 | (3.89 ± 0.59) × 10−4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kristinaityte, K.; Urbańczyk, M.; Mames, A.; Pietrzak, M.; Ratajczyk, T. Photoreactivity of an Exemplary Anthracene Mixture Revealed by NMR Studies, including a Kinetic Approach. Molecules 2021, 26, 6695. https://doi.org/10.3390/molecules26216695
Kristinaityte K, Urbańczyk M, Mames A, Pietrzak M, Ratajczyk T. Photoreactivity of an Exemplary Anthracene Mixture Revealed by NMR Studies, including a Kinetic Approach. Molecules. 2021; 26(21):6695. https://doi.org/10.3390/molecules26216695
Chicago/Turabian StyleKristinaityte, Kristina, Mateusz Urbańczyk, Adam Mames, Mariusz Pietrzak, and Tomasz Ratajczyk. 2021. "Photoreactivity of an Exemplary Anthracene Mixture Revealed by NMR Studies, including a Kinetic Approach" Molecules 26, no. 21: 6695. https://doi.org/10.3390/molecules26216695
APA StyleKristinaityte, K., Urbańczyk, M., Mames, A., Pietrzak, M., & Ratajczyk, T. (2021). Photoreactivity of an Exemplary Anthracene Mixture Revealed by NMR Studies, including a Kinetic Approach. Molecules, 26(21), 6695. https://doi.org/10.3390/molecules26216695