
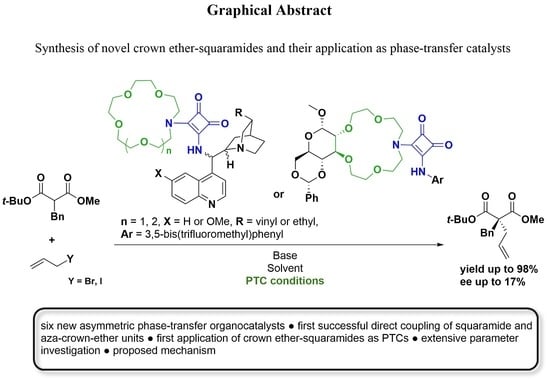
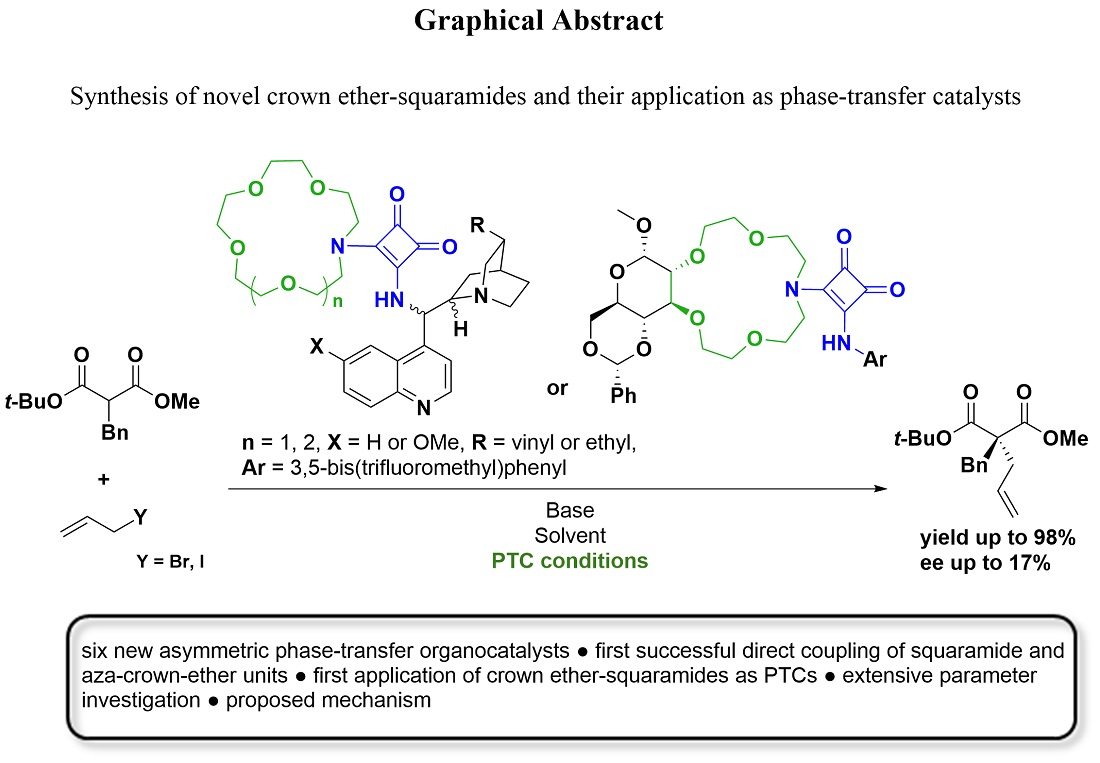
Synthesis of Novel Crown Ether-Squaramides and Their Application as Phase-Transfer Catalysts

 , ,
, ,  , ,
, ,  ,
,  ,
, 
Abstract
:
1. Introduction
2. Results and Discussion
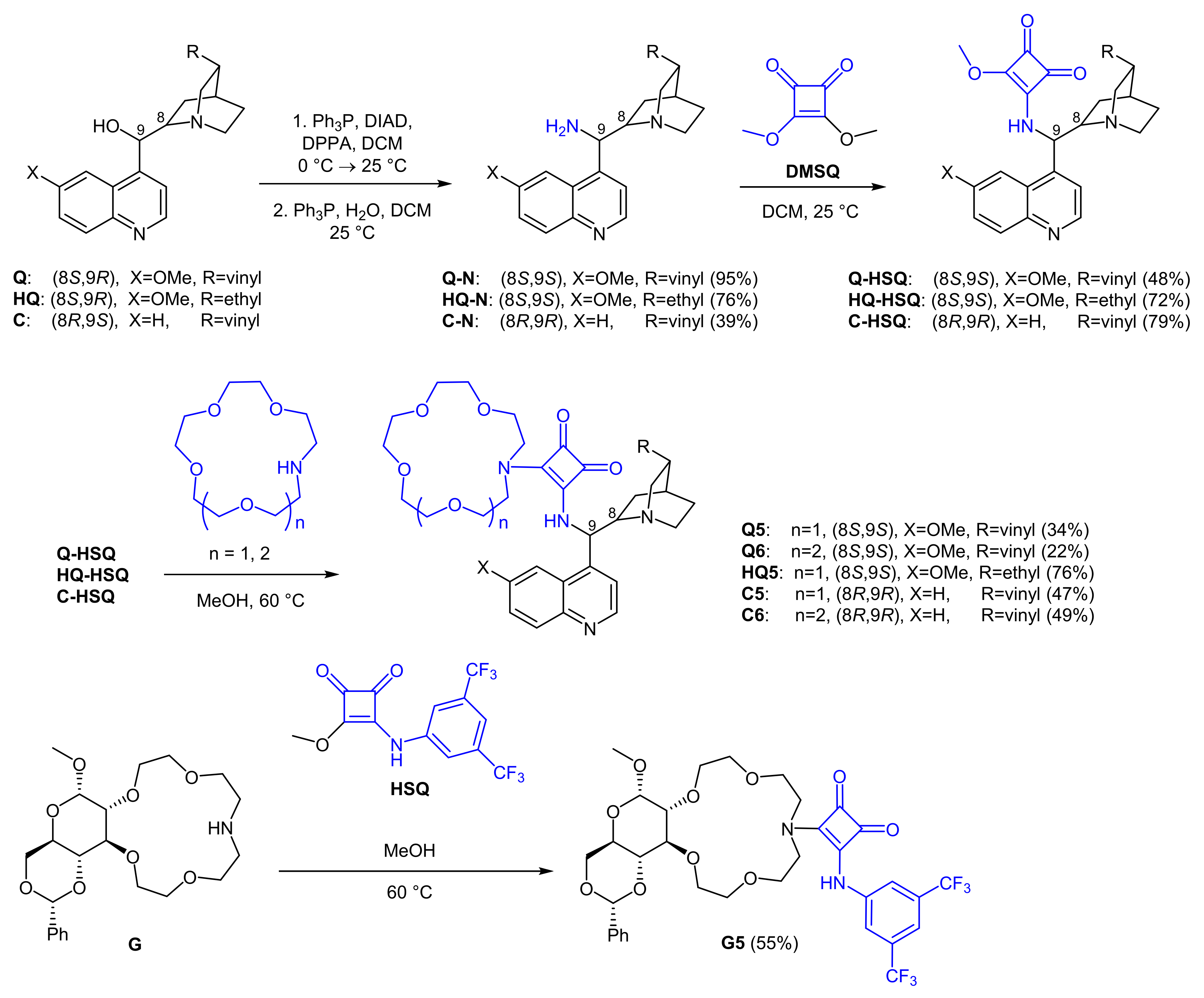
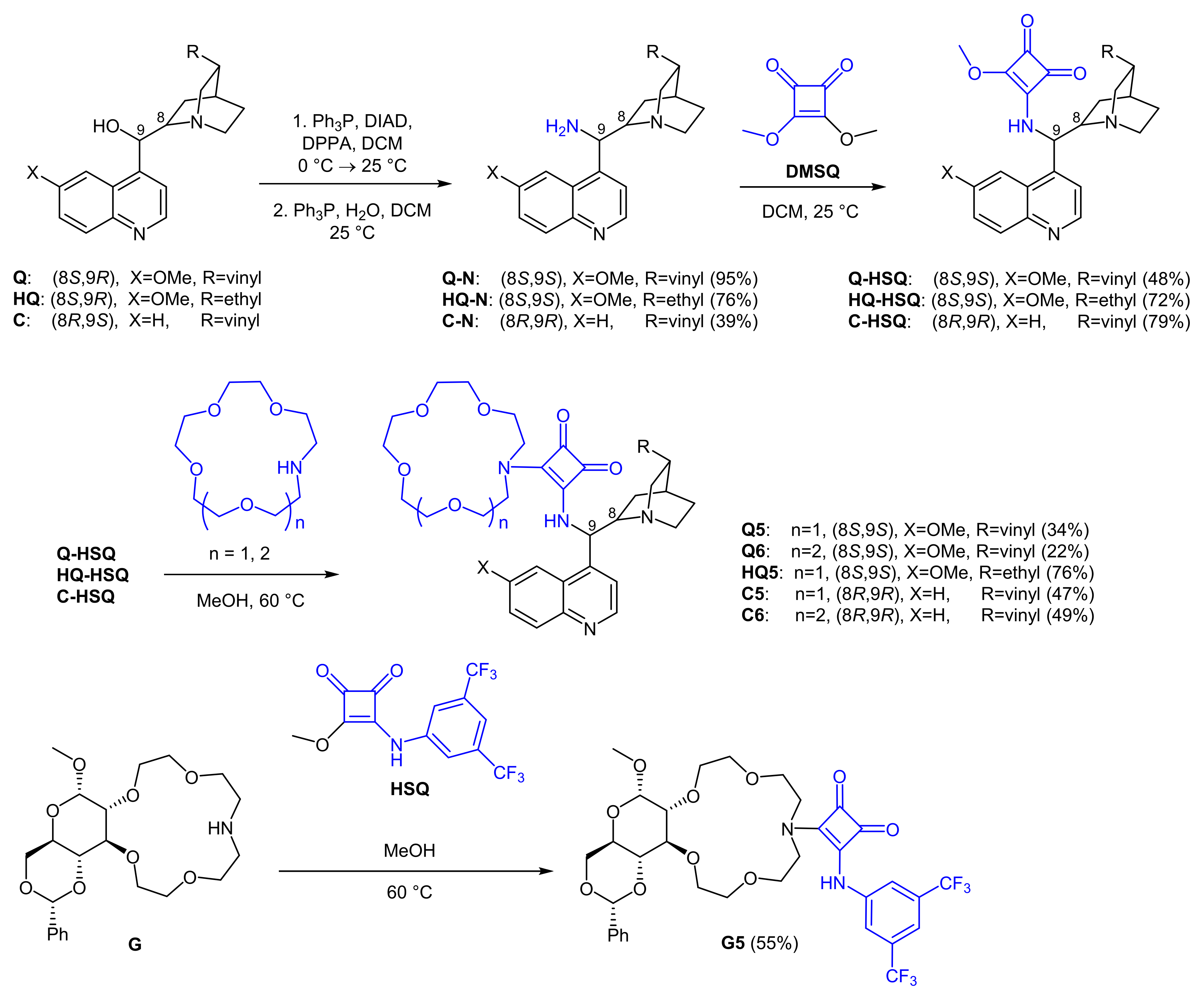
2.1. Synthesis of Crown Ether-Squaramide Phase-Transfer Catalysts
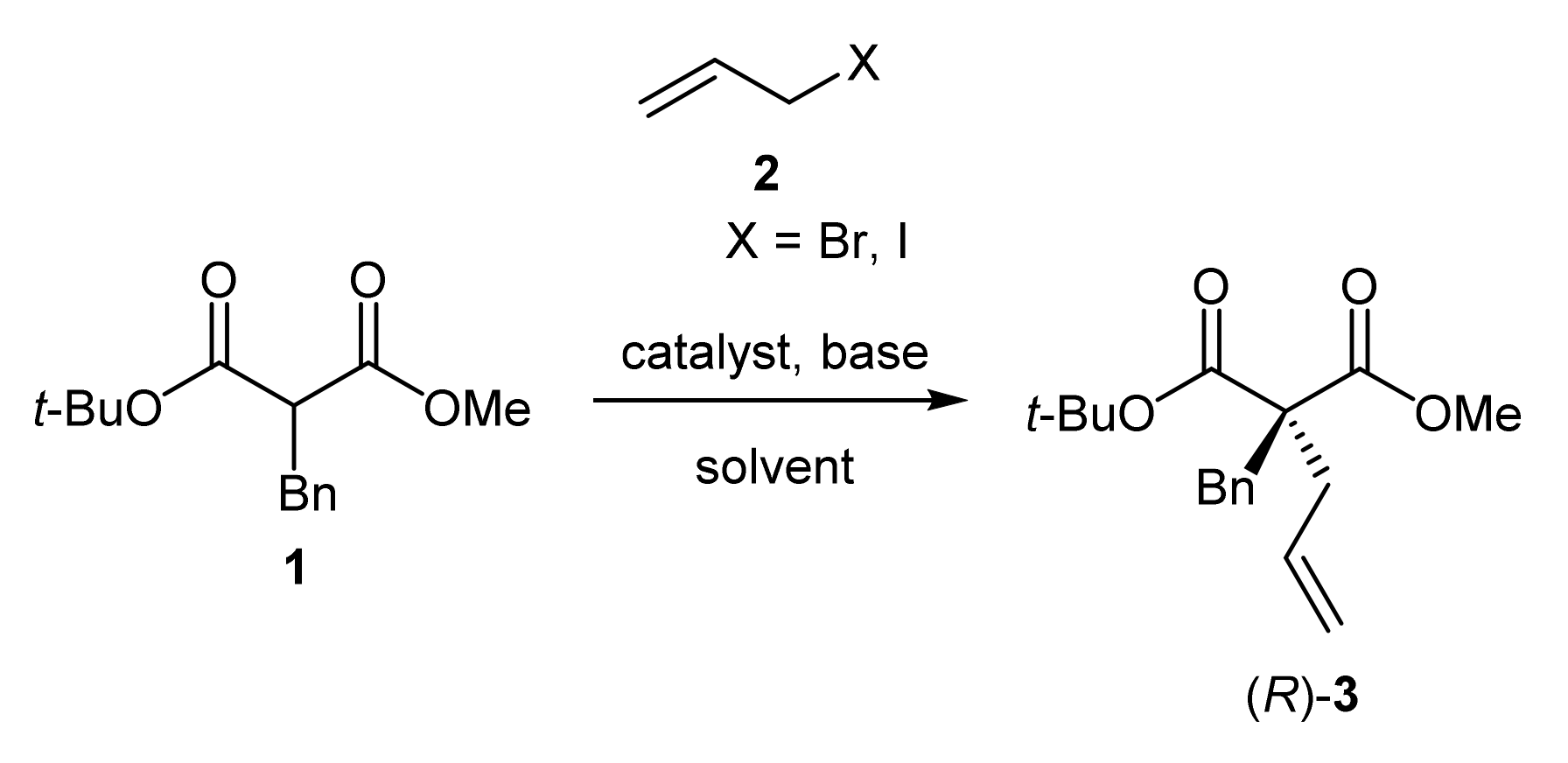
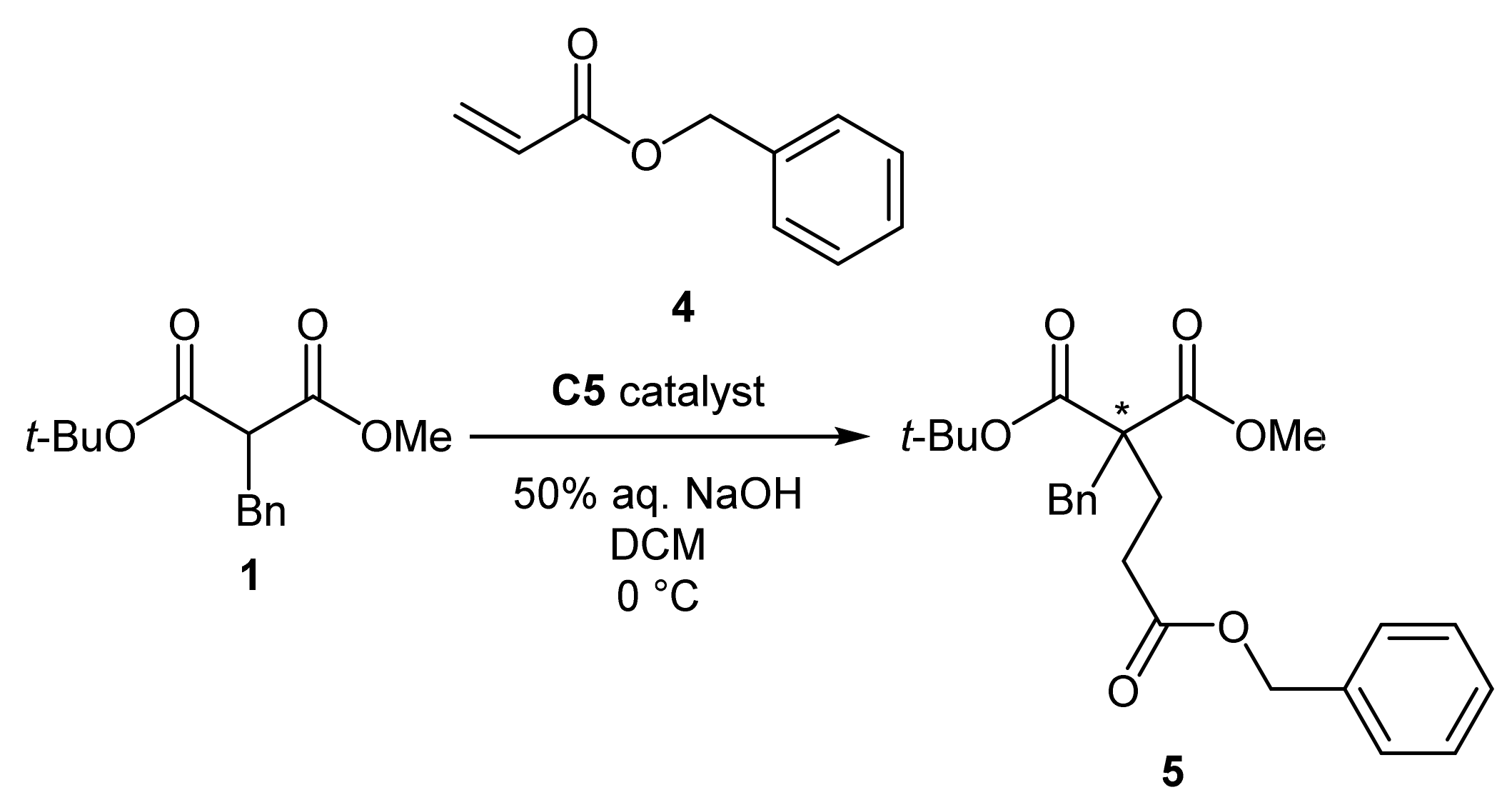
2.2. Application of the Catalysts
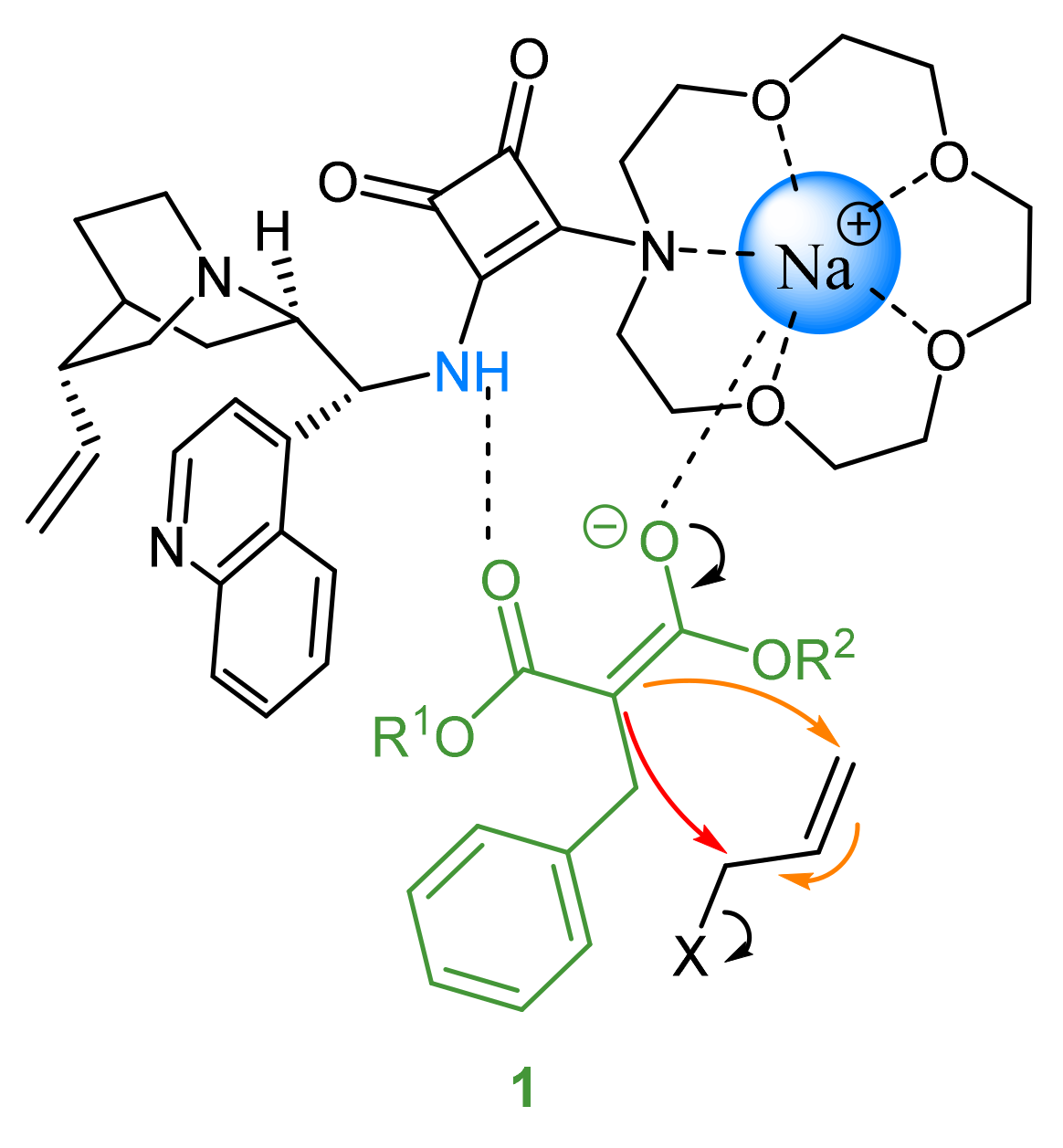
2.3. Proposed Mechanism
3. Materials and Methods
3.1. General
3.2. Procedures
3.2.1. 3-(+)-Methoxy-4-(((R)-quinolin-4-yl((1S,2R,4S,5R)-5-vinylquinuclidin-2-yl)methyl)amino)cyclobut-3-ene-1,2-dione (C-HSQ)
3.2.2. General Procedure for the Preparation of Cinchona Alkaloid-Based Catalysts
3.2.3. 3-(+)-(1,4,7,10-Tetraoxa-13-azacyclopentadecan-13-yl)-4-(((S)-(6-methoxyquinolin-4-yl)((1S,2S,4S,5R)-5-vinylquinuclidin-2-yl)methyl)amino)cyclobut-3-ene-1,2-dione (Q5)
3.2.4. 3-(+)-(1,4,7,10,13-Pentaoxa-16-azacyclooctadecan-16-yl)-4-(((S)-(6-methoxyquinolin-4-yl)((1S,2S,4S,5R)-5-vinylquinuclidin-2-yl)methyl)amino)cyclobut-3-ene-1,2-dione (Q6)
3.2.5. 3-(+)-(1,4,7,10-Tetraoxa-13-azacyclopentadecan-13-yl)-4-(((S)-((1S,2S,4S,5R)-5-ethylquinuclidin-2-yl)(6-methoxyquinolin-4-yl)methyl)amino)cyclobut-3-ene-1,2-dione (HQ5)
3.2.6. 3-(+)-(1,4,7,10-Tetraoxa-13-azacyclopentadecan-13-yl)-4-(((R)-quinolin-4-yl((1S,2R,4S,5R)-5-vinylquinuclidin-2-yl)methyl)amino)cyclobut-3-ene-1,2-dione (C5)
3.2.7. 3-(+)-(1,4,7,10,13-Pentaoxa-16-azacyclooctadecan-16-yl)-4-(((R)-quinolin-4-yl((1S,2R,4S,5R)-5-vinylquinuclidin-2-yl)methyl)amino)cyclobut-3-ene-1,2-dione (C6)
3.2.8. Procedure for the Preparation of the Glucose-Based Catalyst: 3-(+)-((3,5-bis(trifluoromethyl)phenyl)amino)-4-((2R,4aR,6S,6aR,19aS,19bR)-6-methoxy-2-phenyltetradecahydro-13H-[1,3]dioxino[4′,5′:5,6]pyrano[3,4-e][1,4,7,10]tetraoxa[13]azacyclopentadecin-13-yl)cyclobut-3-ene-1,2-dione (G5)
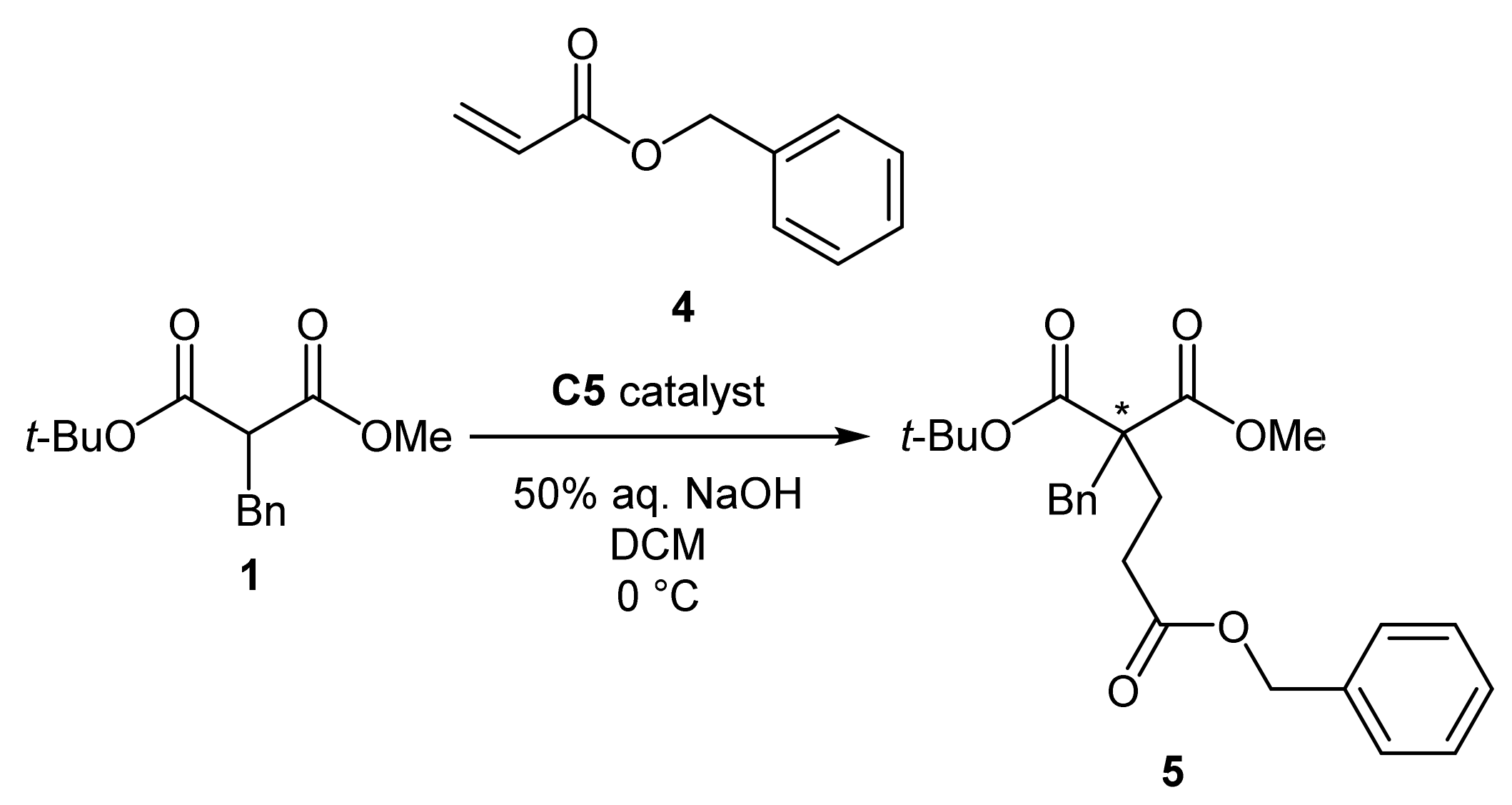
3.2.9. General Procedure for the α-alkylation of tert-butyl methyl α-benzylmalonate with allyl halides (3)
3.2.10. General Procedure for the Michael Addition of tert-butyl methyl α-benzylmalonate to benzyl acrylate (5)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Hong, S.; Lee, J.; Kim, M.; Park, Y.; Park, C.; Kim, M.; Jew, S.; Park, H. Highly Enantioselective Synthesis of α,α-Dialkylmalonates by Phase-Transfer Catalytic Desymmetrization. J. Am. Chem. Soc. 2011, 133, 4924–4929. [Google Scholar] [CrossRef]
- Odanaka, Y.; Kanemitsu, T.; Iwasaki, K.; Mochizuki, Y.; Miyazaki, M.; Nagata, K.; Kato, M.; Itoh, T. Asymmetric Michael addition of malonic diesters to acrylates by phase-transfer catalysis toward the construction of quaternary stereogenic α-carbons. Tetrahedron 2019, 75, 209–219. [Google Scholar] [CrossRef]
- Kanemitsu, T.; Koga, S.; Nagano, D.; Miyazaki, M.; Nagata, K.; Itoh, T. Asymmetric Alkylation of Malonic Diester Under Phase-Transfer Conditions. ACS Catal. 2011, 1, 1331–1335. [Google Scholar] [CrossRef]
- Ha, M.W.; Lee, H.; Yi, H.Y.; Park, Y.; Kim, S.; Hong, S.; Lee, M.; Kim, M.; Kim, T.; Park, H. Enantioselective Phase-Transfer Catalytic α-Benzylation and α-Allylation of α-tert-Butoxycarbonyllactones. Adv. Synth. Catal. 2013, 355, 637–642. [Google Scholar] [CrossRef]
- Ha, M.W.; Hong, S.; Park, C.; Park, Y.; Lee, J.; Kim, M.; Lee, J.; Park, H. Enantioselective phase-transfer catalytic α-alkylation of 2-methylbenzyl tert-butyl malonates. Org. Biomol. Chem. 2013, 11, 4030–4039. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Ha, M.W.; Kim, B.; Hong, S.; Kim, D.; Park, Y.; Kim, M.; Lee, J.K.; Lee, J.; Park, H. Enantioselective α-Alkylation of Benzylideneamino tert-Butyl Malonates by Phase-Transfer Catalysis. Adv. Synth. Catal. 2015, 357, 2841–2848. [Google Scholar] [CrossRef]
- Kanemitsu, T.; Sato, M.; Yoshida, M.; Ozasa, E.; Miyazaki, M.; Odanaka, Y.; Nagata, K.; Itoh, T. Enantioselective α-Benzoyloxylation of Malonic Diesters by Phase-Transfer Catalysis. Org. Lett. 2016, 18, 5484–5487. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Yasuda, N. Contemporary Asymmetric Phase Transfer Catalysis: Large-Scale Industrial Applications. Org. Process Res. Dev. 2015, 19, 1731–1746. [Google Scholar] [CrossRef]
- Hashimoto, T.; Maruoka, K. Recent Development and Application of Chiral Phase-Transfer Catalysts. Chem. Rev. 2007, 107, 5656–5682. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, S.; Maruoka, K. Recent Developments in Asymmetric Phase-Transfer Reactions. Angew. Chem. Int. Ed. 2013, 52, 4312–4348. [Google Scholar] [CrossRef]
- Majdecki, M.; Niedbała, P.; Tyszka-Gumkowska, A.; Jurczak, J. Assisted by Hydrogen-Bond Donors: Cinchona Quaternary Salts as Privileged Chiral Catalysts for Phase-Transfer Reactions. Synthesis 2021, 53, 2777–2786. [Google Scholar] [CrossRef]
- Brak, K.; Jacobsen, E.N. Asymmetric Ion-Pairing Catalysis. Angew. Chem. Int. Ed. 2013, 52, 534–561. [Google Scholar] [CrossRef] [Green Version]
- Schettini, R.; Sicignano, M.; De Riccardis, F.; Izzo, I.; Della Sala, G. Macrocyclic Hosts in Asymmetric Phase-Transfer Catalyzed Reactions. Synthesis 2018, 50, 4777–4795. [Google Scholar] [CrossRef]
- Wang, H. Chiral Phase-Transfer Catalysts with Hydrogen Bond: A Powerful Tool in the Asymmetric Synthesis. Catalysts 2019, 9, 244. [Google Scholar] [CrossRef] [Green Version]
- Majdecki, M.; Niedbala, P.; Jurczak, J. Amide-Based Cinchona Alkaloids as Phase-Transfer Catalysts: Synthesis and Potential Application. Org. Lett. 2019, 21, 8085–8090. [Google Scholar] [CrossRef] [PubMed]
- Majdecki, M.; Grodek, P.; Jurczak, J. Stereoselective α-Chlorination of β-Keto Esters in the Presence of Hybrid Amide-Based Cinchona Alkaloids as Catalysts. J. Org. Chem. 2021, 86, 995–1001. [Google Scholar] [CrossRef]
- Wang, B.; Liu, Y.; Sun, C.; Wei, Z.; Cao, J.; Liang, D.; Lin, Y.; Duan, H. Asymmetric Phase-Transfer Catalysts Bearing Multiple Hydrogen-Bonding Donors: Highly Efficient Catalysts for Enantio-and Diastereoselective Nitro-Mannich Reaction of Amidosulfones. Org. Lett. 2014, 16, 6432–6435. [Google Scholar] [CrossRef] [PubMed]
- Craig, R.; Litvajova, M.; Cronin, S.A.; Connon, S.J. Enantioselective acyl-transfer catalysis by fluoride ions. Chem. Commun. 2018, 54, 10108–10111. [Google Scholar] [CrossRef]
- Lu, N.; Fang, Y.; Gao, Y.; Wei, Z.; Cao, J.; Liang, D.; Lin, Y.; Duan, H. Bifunctional Thiourea–Ammonium Salt Catalysts Derived from Cinchona Alkaloids: Cooperative Phase-Transfer Catalysts in the Enantioselective Aza-Henry Reaction of Ketimines. J. Org. Chem. 2018, 83, 1486–1492. [Google Scholar] [CrossRef]
- Cui, D.; Li, Y.; Huang, P.; Tian, Z.; Jia, Y.; Wang, P. Bifunctional phase-transfer catalysts for synthesis of 2-oxazolidinones from isocyanates and epoxides. RSC Adv. 2020, 10, 12360–12364. [Google Scholar] [CrossRef]
- Alemán, J.; Parra, A.; Jiang, H.; Jørgensen, K.A. Squaramides: Bridging from Molecular Recognition to Bifunctional Organocatalysis. Chem. Eur. J. 2011, 17, 6890–6899. [Google Scholar] [CrossRef]
- Frontera, A.; Orell, M.; Garau, C.; Quiñonero, D.; Molins, E.; Mata, I.; Morey, J. Preparation, Solid-State Characterization, and Computational Study of a Crown Ether Attached to a Squaramide. Org. Lett. 2005, 7, 1437–1440. [Google Scholar] [CrossRef]
- Zdanowski, S.; Piątek, P.; Romański, J. An ion pair receptor facilitating the extraction of chloride salt from the aqueous to the organic phase. New J. Chem. 2016, 40, 7190–7196. [Google Scholar] [CrossRef]
- Jagleniec, D.; Siennicka, S.; Dobrzycki, Ł.; Karbarz, M.; Romański, J. Recognition and Extraction of Sodium Chloride by a Squaramide-Based Ion Pair Receptor. Inorg. Chem. 2018, 57, 12941–12952. [Google Scholar] [CrossRef] [PubMed]
- Jagleniec, D.; Dobrzycki, Ł.; Karbarz, M.; Romański, J. Ion-pair induced supramolecular assembly formation for selective extraction and sensing of potassium sulfate. Chem. Sci. 2019, 10, 9542–9547. [Google Scholar] [CrossRef]
- Yu, X.-H.; Cai, X.-J.; Hong, X.-Q.; Tam, K.Y.; Zhang, K.; Chen, W.-H. Synthesis and biological evaluation of aza-crown ether-squaramide conjugates as anion/cation symporters. Future Med. Chem. 2019, 11, 1091–1106. [Google Scholar] [CrossRef]
- Zaleskaya, M.; Jagleniec, D.; Karbarz, M.; Dobrzycki, Ł.; Romański, J. Squaramide based ion pair receptors possessing ferrocene as a signaling unit. Inorg. Chem. Front. 2020, 7, 972–983. [Google Scholar] [CrossRef]
- Yan, L.; Huang, G.; Wang, H.; Xiong, F.; Peng, H.; Chen, F. Squaramide-Linked Chloramphenicol Base Hybrid Catalysts for the Asymmetric Michael Addition of 2,3-Dihydrobenzofuran-2-carboxylates to Nitroolefins. Eur. J. Org. Chem. 2018, 1, 99–103. [Google Scholar] [CrossRef]
- Kisszekelyi, P.; Alammar, A.; Kupai, J.; Huszthy, P.; Barabas, J.; Holtzl, T.; Szente, L.; Bawn, C.; Adams, R.; Szekely, G. Asymmetric synthesis with cinchona-decorated cyclodextrin in a continuous-flow membrane reactor. J. Catal. 2019, 371, 255–261. [Google Scholar] [CrossRef]
- Rapi, Z.; Bakó, P.; Drahos, L.; Keglevich, G. Side-Arm Effect of a Methyl α-D-Glucopyranoside Based Lariat Ether Catalysts in Asymmetric Syntheses. Heteroatom Chem. 2015, 26, 63–71. [Google Scholar] [CrossRef]
- McCooey, S.H.; Connon, S.J. Readily Accessible 9-epi-amino Cinchona Alkaloid Derivatives Promote Efficient, Highly Enantioselective Additions of Aldehydes and Ketones to Nitroolefins. Org. Lett. 2007, 9, 599–602. [Google Scholar] [CrossRef]
- Nagy, S.; Dargó, G.; Kisszékelyi, P.; Fehér, Z.; Simon, A.; Barabás, J.; Höltzl, T.; Mátravölgyi, B.; Kárpáti, L.; Drahos, L.; et al. New enantiopure binaphthyl-cinchona thiosquaramides: Synthesis and application for enantioselective organocatalysis. New J. Chem. 2019, 43, 5948–5959. [Google Scholar] [CrossRef] [Green Version]
- Nagy, S.; Fehér, Z.; Dargó, G.; Barabás, J.; Garádi, Z.; Mátravölgyi, B.; Kisszékelyi, P.; Dargó, G.; Huszthy, P.; Höltzl, T.; et al. Comparison of Cinchona Catalysts Containing Ethyl or Vinyl or Ethynyl Group at Their Quinuclidine Ring. Materials 2019, 12, 3034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakó, P.; Tőke, L.J. Novel monoaza-and diazacrown ethers incorporating sugar units and their extraction ability towards picrate salts. Inclusion Phenom. Mol. Recognit. Chem. 1995, 23, 195–201. [Google Scholar] [CrossRef]
- Bakó, P.; Kiss, T.; Tőke, L. Chiral azacrown ethers derived from D-glucose as catalysts for enantioselective Michael addition. Tetrahedron Lett. 1997, 38, 7259–7262. [Google Scholar] [CrossRef]
- Yang, W.; Du, D.-M. Highly Enantioselective Michael Addition of Nitroalkanes to Chalcones Using Chiral Squaramides as Hydrogen Bonding Organocatalysts. Org. Lett. 2010, 12, 5450–5453. [Google Scholar] [CrossRef] [PubMed]
- Makó, A.; Menyhárd, D.K.; Bakó, P.; Keglevich, G.; Tőke, L. Theoretical study of the asymmetric phase-transfer mediated epoxidation of chalcone catalyzed by chiral crown ethers derived from monosaccharides. J. Mol. Struct. 2008, 892, 336–342. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
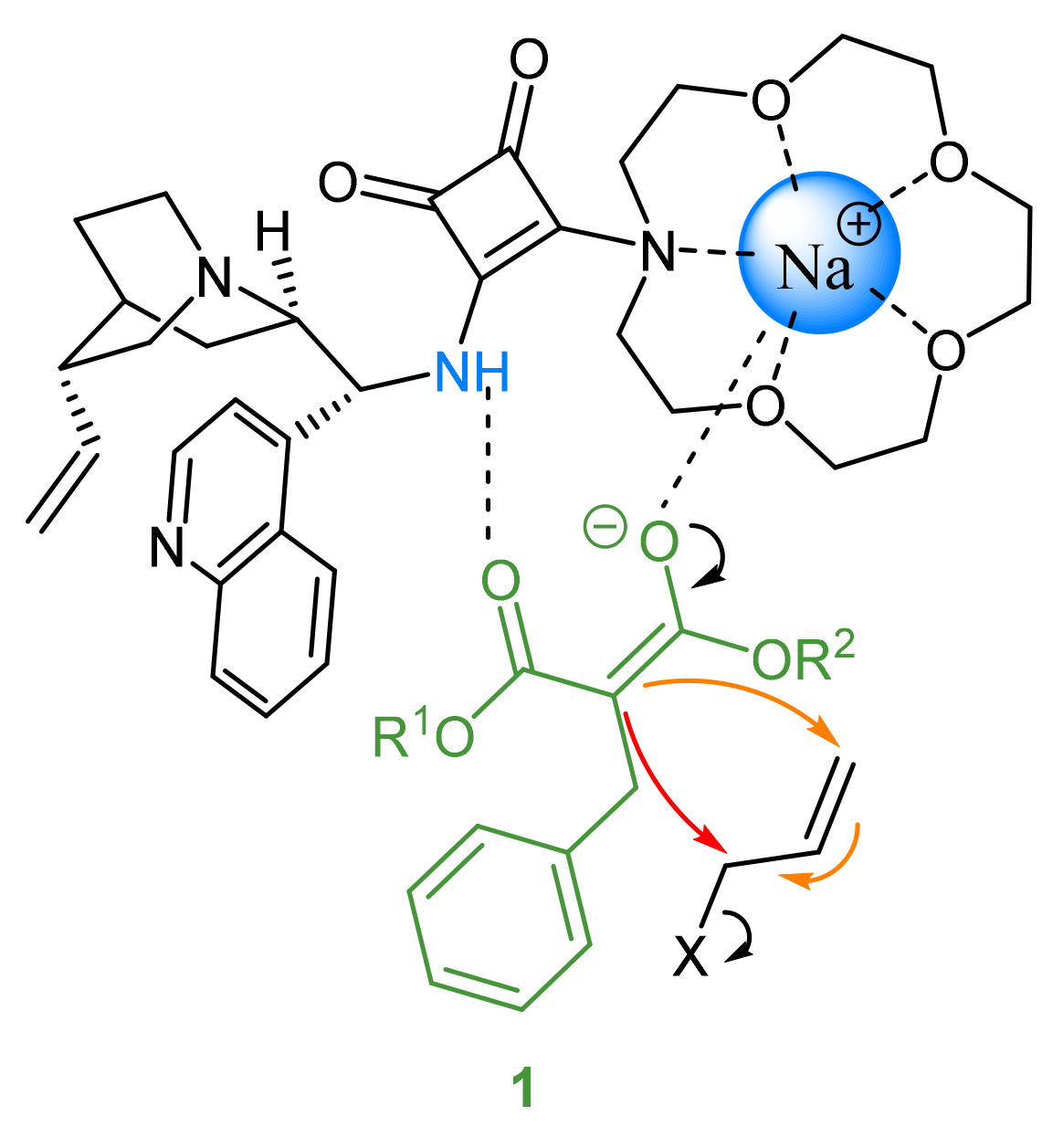{kind=link}
| Entry | Catalyst | Base b | Yield c (%) | ee d (%) |
|---|---|---|---|---|
| 1 | Q5 | 50% aq. NaOH | 98 | 5 |
| 2 | HQ5 | 50% aq. NaOH | 93 | <5 |
| 3 | C5 | 50% aq. NaOH | 73 | 9 |
| 4 | G5 | 50% aq. NaOH | 86 | 5 |
| 5 | Q6 | 50% aq. KOH | 59 | <5 |
| 6 | C6 | 50% aq. KOH | 37 | <5 |
| Entry | Solvent | Base | Base Quantity (eq) | Yield b (%) | ee c (%) |
|---|---|---|---|---|---|
| 1 | DCM | 50% aq. NaOH | 50 | 73 | 9 |
| 2 | toluene | 50% aq. NaOH | 50 | 60 | 7 |
| 3 | EtOAc | 50% aq. NaOH | 50 | 65 | 5 |
| 4 | MTBE | 50% aq. NaOH | 50 | 51 | <5 |
| 5 | CPME | 50% aq. NaOH | 50 | 49 | <5 |
| 6 | 2-Me-THF | 50% aq. NaOH | 50 | 51 | <5 |
| 7 | DCM | solid NaOH | 1 | 38 | 10 |
| 8 | MeCN | solid NaOH | 1 | 49 | <5 |
| 9 | THF | solid NaOH | 1 | 38 | <5 |
| Entry | Catalyst Quantity (mol%) | Allyl Bromide Quantity (eq) | Solvent Volume (mL) | Yield b (%) | ee c (%) |
|---|---|---|---|---|---|
| 1 | 10 | 1 | 2.4 | 73 | 9 |
| 2 | 20 | 1 | 2.4 | 79 | 8 |
| 3 | 10 | 1.2 | 2.4 | 83 | 11 |
| 4 | 10 | 1 | 1.2 | 92 | 6 |
| Entry | Temperature (°C) | Reagent Quantity (eq) | Base Quantity (eq) | Yield b (%) | ee c (%) |
|---|---|---|---|---|---|
| 1 | 25 | 1 | 50 | 98 | 12 |
| 2 | 0 | 1 | 50 | 94 | 15 |
| 3 | 0 | 1 | 25 | 79 | 15 |
| 4 | 0 | 1.2 | 50 | 97 | 15 |
| 5 | 0 | 1.2 | 25 | 74 | 15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fehér, Z.; Richter, D.; Nagy, S.; Bagi, P.; Rapi, Z.; Simon, A.; Drahos, L.; Huszthy, P.; Bakó, P.; Kupai, J. Synthesis of Novel Crown Ether-Squaramides and Their Application as Phase-Transfer Catalysts. Molecules 2021, 26, 6542. https://doi.org/10.3390/molecules26216542
Fehér Z, Richter D, Nagy S, Bagi P, Rapi Z, Simon A, Drahos L, Huszthy P, Bakó P, Kupai J. Synthesis of Novel Crown Ether-Squaramides and Their Application as Phase-Transfer Catalysts. Molecules. 2021; 26(21):6542. https://doi.org/10.3390/molecules26216542
Chicago/Turabian StyleFehér, Zsuzsanna, Dóra Richter, Sándor Nagy, Péter Bagi, Zsolt Rapi, András Simon, László Drahos, Péter Huszthy, Péter Bakó, and József Kupai. 2021. "Synthesis of Novel Crown Ether-Squaramides and Their Application as Phase-Transfer Catalysts" Molecules 26, no. 21: 6542. https://doi.org/10.3390/molecules26216542
APA StyleFehér, Z., Richter, D., Nagy, S., Bagi, P., Rapi, Z., Simon, A., Drahos, L., Huszthy, P., Bakó, P., & Kupai, J. (2021). Synthesis of Novel Crown Ether-Squaramides and Their Application as Phase-Transfer Catalysts. Molecules, 26(21), 6542. https://doi.org/10.3390/molecules26216542