
Brownmillerites CaFeO2.5 and SrFeO2.5 as Catalyst Support for CO Oxidation

 ,
,
Abstract
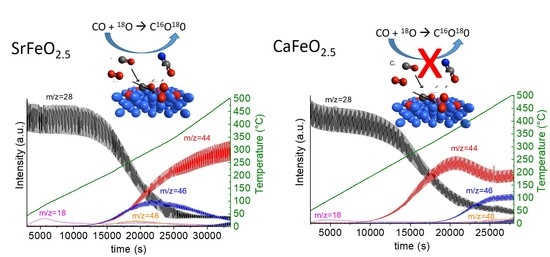
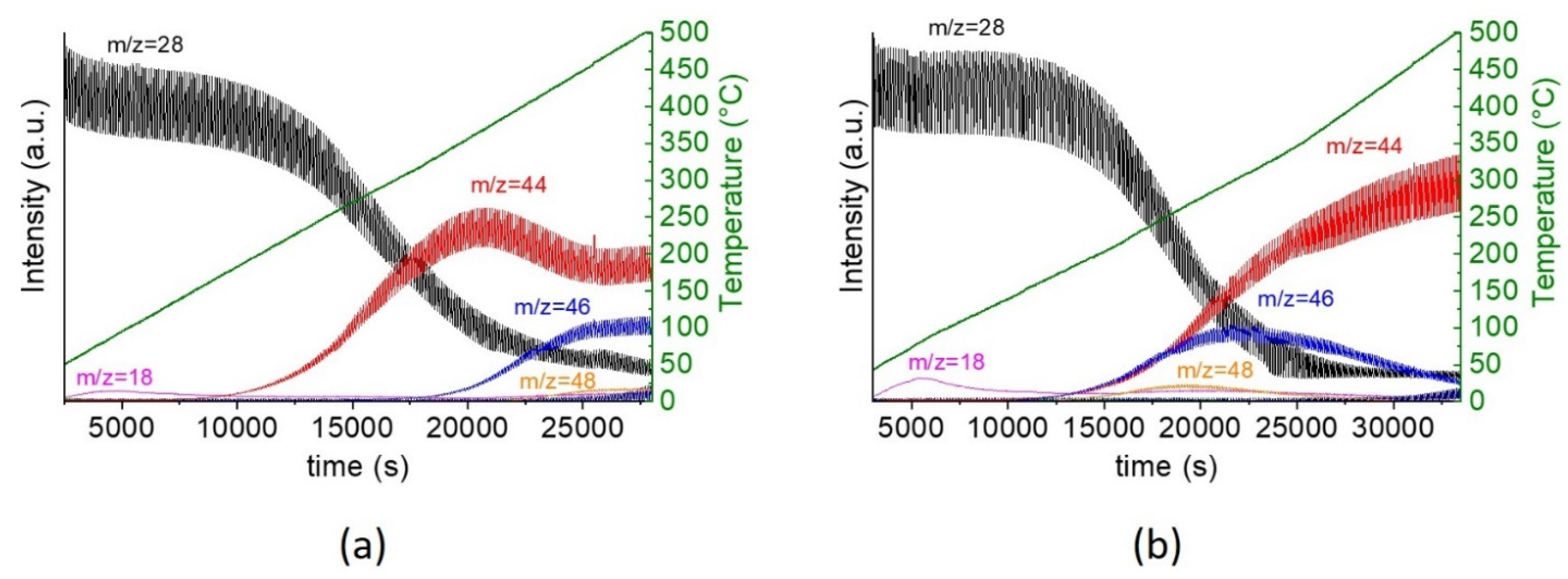
:
1. Introduction
2. Results
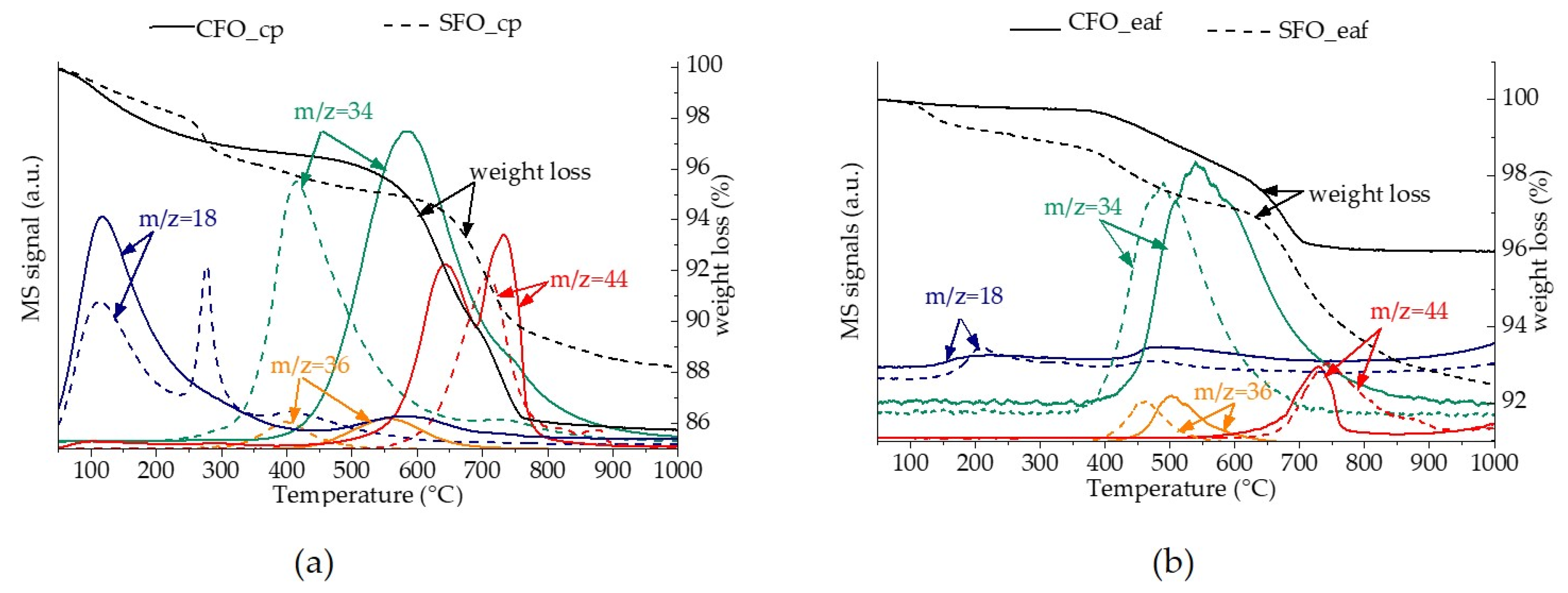
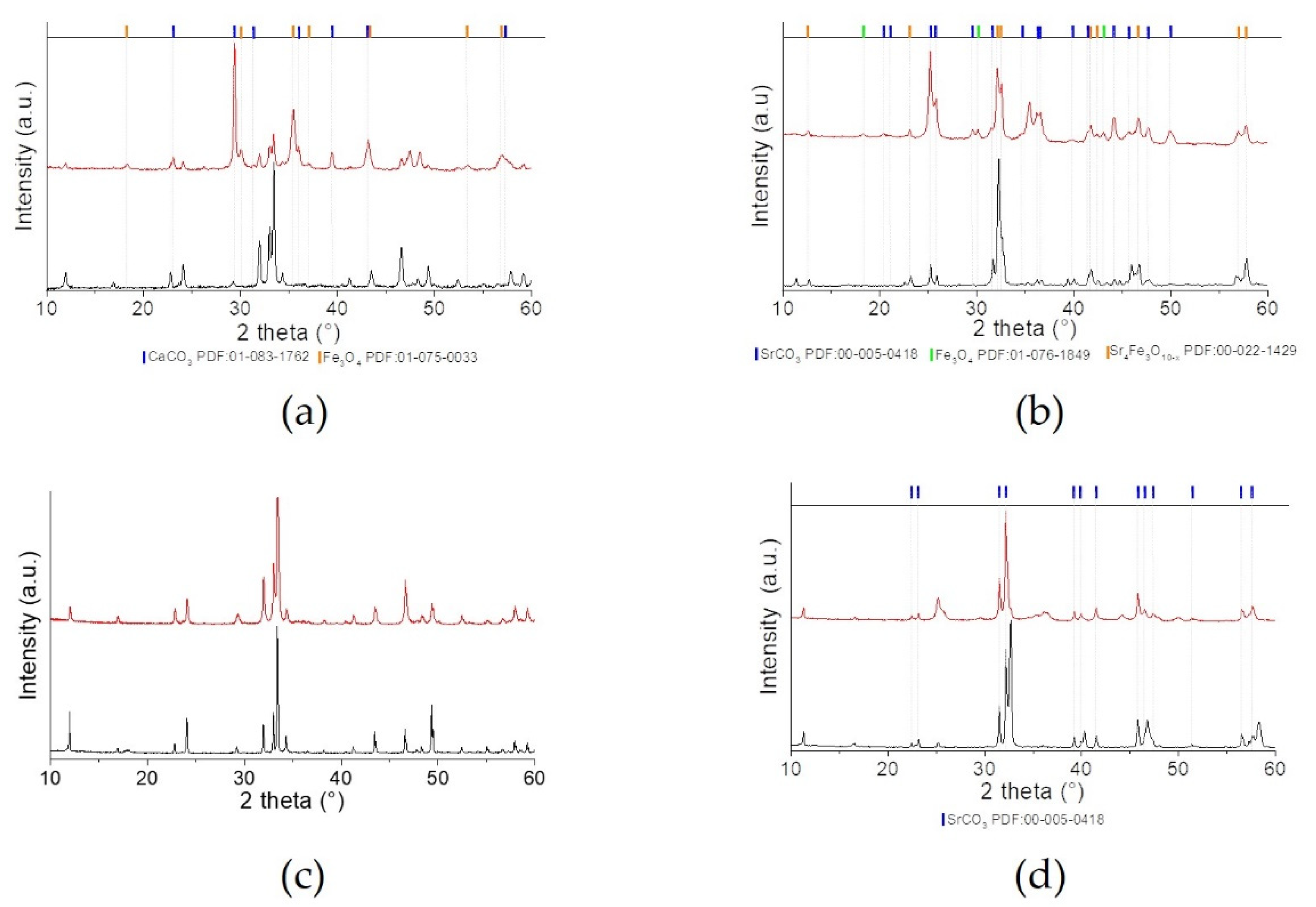
2.1. Physico-Chemical and Structural Properties
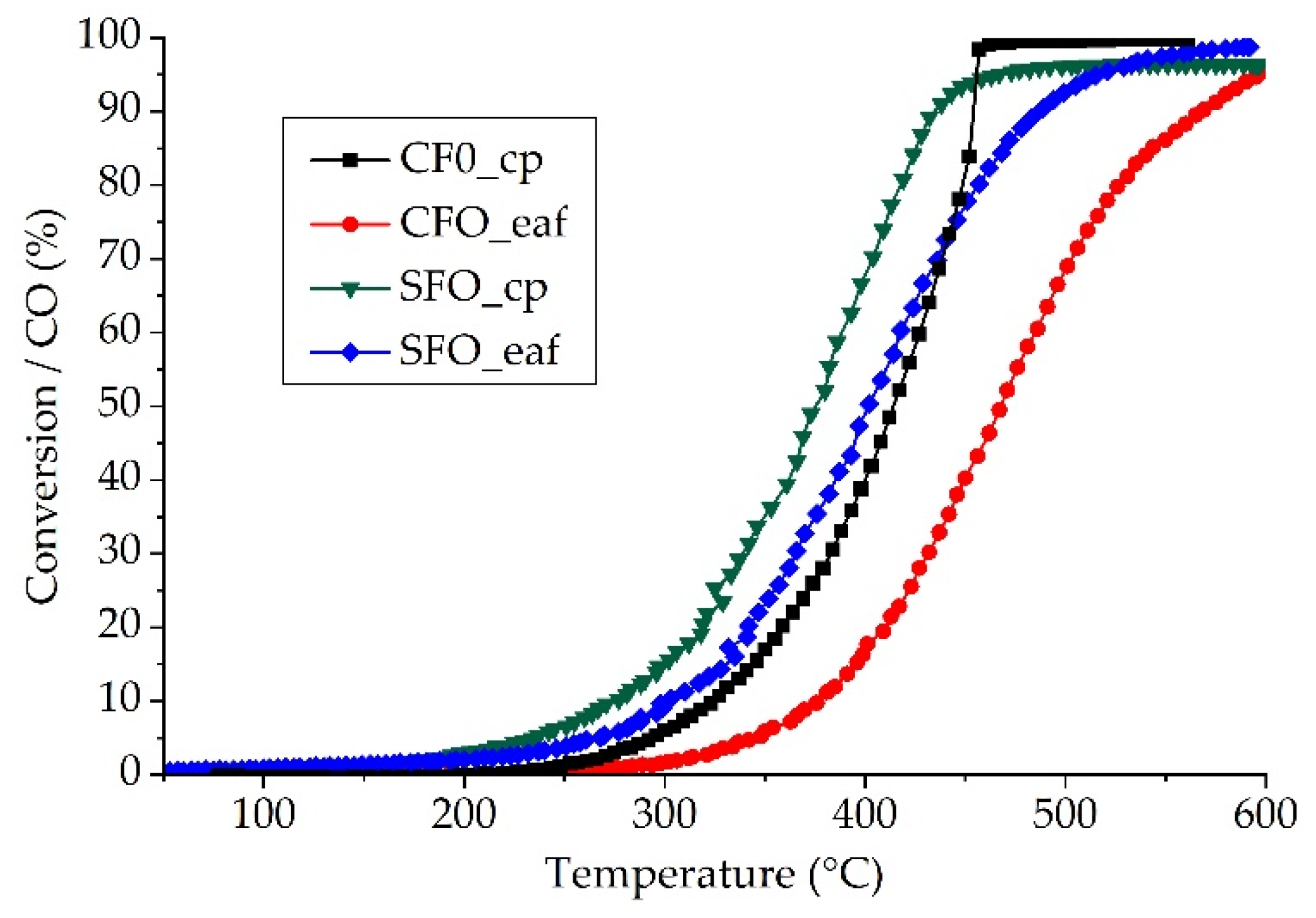
2.2. Catalytic Performance for CO Oxidation
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Royer, S.; Duprez, D.; Can, F.; Courtois, X.; Batiot-Dupeyrat, C.; Laassiri, S.; Alamdari, H. Perovskites as Substitutes of Noble Metals for Heterogeneous Catalysis: Dream or Reality. Chem. Rev. 2014, 114, 10292–10368. [Google Scholar] [CrossRef] [PubMed]
- Royer, S.; Duprez, D.; Kaliaguine, S. Role of bulk and grain boundary oxygen mobility in the catalytic oxidation activity of LaCo1−xFexO3. J. Catal. 2005, 234, 364–375. [Google Scholar] [CrossRef]
- Jeen, H.; Choi, W.S.; Biegalski, M.D.; Folkman, C.M.; Tung, I.-C.; Fong, D.D.; Freeland, J.W.; Shin, D.; Ohta, H.; Chisholm, M.F.; et al. Reversible redox reactions in an epitaxially stabilized SrCoO(x) oxygen sponge. Nat. Mater. 2013, 12, 1057–1063. [Google Scholar] [CrossRef] [PubMed]
- Penkala, B.; Gatla, S.; Aubert, D.; Ceretti, M.; Tardivat, C.; Paulus, W.; Kaper, H. In situ generated catalyst: Copper(II) oxide and copper (I) supported on Ca2Fe2O5 for CO oxidation. Catal. Sci. Technol. 2018, 8, 5236–5243. [Google Scholar] [CrossRef]
- Penkala, B.; Aubert, D.; Kaper, H.; Tardivat, C.; Conder, K.; Paulus, W. The role of lattice oxygen in CO oxidation over Ce18O2-based catalysts revealed under operando conditions. Catal. Sci. Technol. 2015, 5, 4839–4848. [Google Scholar] [CrossRef]
- Shin, S.; Hatakeyama, Y.; Ogawa, K.; Shimomura, K. Catalytic decomposition of NO over Brownmillerite-like compounds, Ca2Fe2O5 and Sr2Fe2O5. Mater. Res. Bull. 1979, 14, 133–136. [Google Scholar] [CrossRef]
- Hirabayashi, D.; Yoshikawa, T.; Mochizuki, K.; Suzuki, K.; Sakai, Y. Formation of Brownmillerite type calcium ferrite (Ca2Fe2O5) and catalytic properties in propylene combustion. Catal. Lett. 2006, 110, 269–274. [Google Scholar] [CrossRef]
- McCammon, C.A.; Becerro, A.I.; Langenhorst, F.; Angel, R.J.; Marion, S.; Seifert, F. Short-range ordering of oxygen vacancies in CaFexTi1−xO3−x/2 perovskites (0 <x <0.4). J. Phys. Condens. Matter 2000, 12, 2969–2984. [Google Scholar]
- Paulus, W.; Schober, H.; Eibl, S.; Johnson, M.; Berthier, T.; Hernandez, O.; Ceretti, M.; Plazanet, M. Lattice Dynamics To Trigger Low Temperature Oxygen Mobility in Solid Oxide Ion Conductors Werner. J. Am. Chem. Soc. 2008, 130, 16080–16085. [Google Scholar] [CrossRef] [PubMed]
- Ceretti, M.; Piovano, A.; Cousson, A.; Berthier, T.; Meven, M.; Agostini, G.; Schefer, J.; Hernandez, O.; Lamberti, C.; Paulus, W. Growth and characterization of large high quality Brownmillerite CaFeO2.5 single crystals. CrystEngComm 2012, 14, 5771. [Google Scholar] [CrossRef]
- Falcón, H.; Barbero, J.A.; Alonso, J.A.; Martínez-Lope, M.J.; Fierro, J.L.G. SrFeO3-δ perovskite oxides: Chemical features and performance for methane combustion. Chem. Mater. 2002, 14, 2325–2333. [Google Scholar] [CrossRef]
- Répécaud, P.-A.; Kaper, H.; Richard, M.; Can, F.; Bion, N. Enhancement of oxygen activation and mobility in CaTixFe1−xO3-δ oxides. ChemCatChem 2017, 9, 2095–2098. [Google Scholar] [CrossRef]
- Gamboa-Rosales, N.K.K.; Ayastuy, J.L.L.; Boukha, Z.; Bion, N.; Duprez, D.; Pérez-Omil, J.A.A.; del Río, E.; Gutiérrez-Ortiz, M.A.A. Ceria-supported Au–CuO and Au–Co3O4 catalysts for CO oxidation: An 18O/16O isotopic exchange study. Appl. Catal. B Environ. 2015, 168–169, 87–97. [Google Scholar] [CrossRef]
- Gatla, S.; Aubert, D.; Agostini, G.; Mathon, O.; Pascarelli, S.; Lunkenbein, T.; Willinger, M.G.; Kaper, H. Room-Temperature CO Oxidation Catalyst: Low-Temperature Metal–Support Interaction between Platinum Nanoparticles and Nanosized Ceria. ACS Catal. 2016, 6, 6151–6155. [Google Scholar] [CrossRef]
- Poffe, E.; Kaper, H.; Ehrhardt, B.; Gigli, L.; Aubert, D.; Nodari, L.; Gross, S.; Mascotto, S. Understanding Oxygen Release from Nanoporous Perovskite Oxides and Its Effect on the Catalytic Oxidation of CH4 and CO. ACS Appl. Mater. Interfaces 2021, 13, 25483–25492. [Google Scholar] [CrossRef] [PubMed]
- Bergeret, G. Characterization of Metallic Catalysts by X-Ray and Electron Microscopy Techniques. In Catalysis by Metals; Springer: Berlin/Heidelberg, Germany, 1997; pp. 167–180. [Google Scholar]
- Rodriguez-Carvajal, J. FULLPROF: A Program for Rietveld Refinement and Pattern Matching Analysis. In Abstracts of the Satellite Meeting on Powder Diffraction of the XV Congress of the IUCr; Toulouse, France, 1990; p. 127. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Phase | Wt% | Space Group | a | b | c |
|---|---|---|---|---|---|---|
| CFO_cp | CaFeO2.5 | 100 | Pnma | 5.433 (2) | 14.776 (7) | 5.604 (2) |
| CFO_eaf | CaFeO2.5 | 100 | Pnma | 5.430 (4) | 14.77 (1) | 5.597 (4) |
| SFO_cp | SrFeO2.5 | 47 | Icmm | 5.638 (6) | 15.55 (2) | 5.517 (5) |
| SrFeO2.75 | 41 | Cmmm | 11.10 (4) | 7.66 (1) | 5.52 (2) | |
| SrFeO2.875 | 12 | I4/mmm | 10.97 (2) | 10.97 (2) | 7.63 (2) | |
| SFO_eaf | SrFeO2.5 | 70 | Icmm | 5.638 (6) | 15.55 (2) | 5.517 (5) |
| SrFeO2.75 | 27 | Cmmm | 11.10 (4) | 7.66 (1) | 5.52 (2) | |
| SrFeO2.875 | 3 | I4/mmm | 10.97 (2) | 10.97 (2) | 7.63 (2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Répécaud, P.-A.; Ceretti, M.; Aouine, M.; Delwaulle, C.; Nonnet, E.; Paulus, W.; Kaper, H. Brownmillerites CaFeO2.5 and SrFeO2.5 as Catalyst Support for CO Oxidation. Molecules 2021, 26, 6413. https://doi.org/10.3390/molecules26216413
Répécaud P-A, Ceretti M, Aouine M, Delwaulle C, Nonnet E, Paulus W, Kaper H. Brownmillerites CaFeO2.5 and SrFeO2.5 as Catalyst Support for CO Oxidation. Molecules. 2021; 26(21):6413. https://doi.org/10.3390/molecules26216413
Chicago/Turabian StyleRépécaud, Pierre-Alexis, Monica Ceretti, Mimoun Aouine, Céline Delwaulle, Emmanuel Nonnet, Werner Paulus, and Helena Kaper. 2021. "Brownmillerites CaFeO2.5 and SrFeO2.5 as Catalyst Support for CO Oxidation" Molecules 26, no. 21: 6413. https://doi.org/10.3390/molecules26216413
APA StyleRépécaud, P.-A., Ceretti, M., Aouine, M., Delwaulle, C., Nonnet, E., Paulus, W., & Kaper, H. (2021). Brownmillerites CaFeO2.5 and SrFeO2.5 as Catalyst Support for CO Oxidation. Molecules, 26(21), 6413. https://doi.org/10.3390/molecules26216413